

Abstract
Mitotic division requires highly regulated morphological and biochemical changes to the cell. Upon commitment to exit mitosis, cells begin to remove mitotic regulators in a temporally and spatially controlled manner to bring about the changes that reestablish interphase. Ubiquitin-dependent pathways target these regulators to generate polyubiquitin-tagged substrates for degradation by the 26S proteasome. However, the lack of cell-based assays to investigate in vivo ubiquitination limits our knowledge of the identity of substrates of ubiquitin-mediated regulation in mitosis. Here we report an in vivo ubiquitin tagging system used in human cells that allows efficient purification of ubiquitin conjugates from synchronized cell populations. Coupling purification with mass spectrometry, we have identified a series of mitotic regulators targeted for polyubiquitination in mitotic exit. We show that some are new substrates of the anaphase-promoting complex/cyclosome and validate KIFC1 and RacGAP1/Cyk4 as two such targets involved respectively in timely mitotic spindle disassembly and cell spreading. We conclude that in vivo biotin tagging of ubiquitin can provide valuable information about the role of ubiquitin-mediated regulation in processes required for rebuilding interphase cells.
Ubiquitination has emerged as a major post-translational modification determining the fate of cellular proteins. One of these fates is proteolysis, whereby the assembly of polyubiquitin chains creates signatures on target proteins that specify delivery to the 26S proteasome for proteolytic destruction. Targeted proteolysis is critical to the control of cell division. For example, the universally conserved mechanism of mitotic exit depends upon rapid proteolysis of mitotic cyclins and securins to drive the transition from mitosis to interphase. This transition is under surveillance by the spindle assembly checkpoint (SAC),1 which controls the activity of a multi-subunit ubiquitin ligase, the anaphase-promoting complex/cyclosome (APC/C) (1, 2).
Much of the known specificity in the ubiquitin-proteasome system (UPS) is mediated at the level of substrate targeting by ubiquitin ligase (E3) enzymes, of which there are more than 600 in human cells. Given these facts, it is perhaps surprising that the APC/C is almost the only known engineer of the protein landscape after anaphase onset, targeting mitotic regulators for destruction with high temporal specificity (2–4). Some roles for nondegradative ubiquitination in regulating the localization of mitotic kinases Aurora B and Plk1 have been described (5–9), and a growing list of reported ubiquitin interactors can modulate ubiquitin-dependent events during mitosis (10). However, the majority of ubiquitination events that have so far been described as occurring at the transition from mitosis to interphase are APC/C-dependent.
Two co-activator subunits, Cdc20 and Cdh1, play vital roles in APC/C-dependent substrate recognition (11) by recognizing two widely characterized degrons, the D-box and the KEN motif (12, 13). Computational approaches that have been used to calculate the total number of APC/C substrates from the prevalence of degrons in the human proteome estimate that there are between 100 and 200 substrates (14), and experiments using in vitro ubiquitination of protein arrays have given rise to estimates in the same range (15). Most of the mitotic regulators targeted by the APC/C during mitotic exit in human cells have been identified via in vitro degradation assays or ubiquitination assays on in vitro–expressed pools of substrates (15–18). These approaches have identified several important substrates, but in the absence of in vivo parameters they may not identify substrates whose targeting depends on post-translational modifications or substrates that are only recognized in vivo as components of higher-order complexes. Not all substrates identified in this way have been validated as polyubiquitinated proteins in vivo. Multiple recent proteomic studies have identified large numbers of in vivo ubiquitin-modified sites from yeast (19–21) and human cells (22–29). None of these studies have used synchronized cell populations to provide information on the timing or regulation of substrate ubiquitination.
We reasoned that a better view of ubiquitin-mediated processes that regulate mitotic exit would come from identifying proteins that are ubiquitinated in vivo during mitotic exit. With this goal in mind we adopted a system for in vivo tagging of ubiquitin chains with biotin, previously used to identify ubiquitin-conjugated proteins from the Drosophila neural system (30), and applied it to a human cell line (U2OS) that can be tightly synchronized at mitosis. In contrast to several recent studies that employed antibodies specific to the diGly-Lys remnant that marks ubiquitination sites following trypsin digestion (19, 25), an in vivo ubiquitin tagging strategy allows direct validation of candidate ubiquitinated proteins (whether mono- or polyubiquitinated) through immunoblotting of samples. Moreover, in contrast to other methods for affinity tagging of ubiquitin, or affinity purification via ubiquitin-binding domains, the use of the biotin tag enables purification under highly denaturing conditions for stringent isolation of ubiquitin-conjugated material from higher eukaryotes. His6-tagged ubiquitin is also available for use under denaturing conditions, but it is not generally useful in higher eukaryotic cells, where a high frequency of proteins containing multiple histidine residues confounds the specificity of nickel-affinity pulldowns (as discussed in detail in Ref. 30). Therefore, in this paper we describe the reproducible identification and validation of mitoticphase-specific polyubiquitinated proteins via the in vivo biotinylation of ubiquitin. A large number of polyubiquitinated proteins that we identified are specific to mitotic exit, when the APC/C is active, and we expect that many of them are substrates for the APC/C. We formally identified KIFC1/HSET and Cyk4/RACGAP1 as targets of APC/C-dependent ubiquitin-mediated proteolysis after anaphase onset and investigated the role of their ubiquitination in the regulation of mitotic exit. Cell cycle phase-specific information on protein ubiquitination and the generation of ubiquitinated protein networks provides a framework for further investigation of ubiquitin-controlled processes occurring during the rebuilding of interphase cells.
EXPERIMENTAL PROCEDURES
Plasmids and siRNA
bioUb6-BirA and BirA sequences (30) were inserted in pTRE-tight vector (Invitrogen) to generate pTRE-bioUb6-BirA and pTRE-BirA plasmids. Venus was swapped for fluorescent proteins in pEYFP-N1-Aurora A (31) to make pVenus-N1-Aurora A. Human RacGAP1 cDNA (a kind gift from Masanori Mishima) was inserted into pVenus-C1. Human KIFC1 cDNA (a kind gift from Paola Coelho) was inserted into pVenus-N1. K292R, F289G, and ΔC versions of RacGAP1 and the R5A,L8A version of KIFC1 were generated using standard mutagenesis techniques. Full cloning details are available upon request. mCherry-MKLP1 was a gift from Masanori Mishima. siRNA sequences targeting APC3, Cdh1, and RacGAP1 3′UTR were as previously described (31, 32). Plasmids and siRNAs were electroporated into cells using the Neon□ transfection system (Invitrogen).
Cell Culture and Synchronization
U2OS tet-OFF cells were cultured in high-glucose DMEM (Invitrogen). Cell culture medium was supplemented with FBS (10%), penicillin-streptomycin, amphotericin B, 500 μg/ml geneticin (all from PAA Laboratories, Pasching, Austria), and 1 μg/ml tetracycline hydrochloride (Calbiochem). U2OS-bioUb cells and U2OS-BirA cells were obtained via clonal selection with 170 μg/ml Hygromycin B (Calbiochem) after transfection of U2OS tet-OFF cells with pTRE-bioUb6-BirA or pTRE-BirA in a ratio of 10:1 with pCMVhygro and were supplied with 5 μm biotin in culture. The hTERT-RPE1 (RPE) cell line stably expressing α-actinin-Venus was described previously (33). Cells were synchronized to prometaphase by sequential blocks with 25 mm thymidine and 10 μm S-trityl-l-cysteine (STLC, Tocris Bioscience, Bristol, UK) for 20 h and 16 h, respectively, and collected via mitotic shake-off. Mitotic exit cells were obtained through treatment with 10 μm Aurora B inhibitor ZM447439 (ZM) (Tocris Bioscience) or 300 nm CDK I/II inhibitor III (Calbiochem) for the indicated time.
Purification of in Vivo–ubiquitinated Material
For each sample, about 3 × 108 cells were lysed in 10 ml lysis buffer (8 M urea, 1% (v/v) SDS, 1× EDTA-free protease inhibitor mixture (Roche), 50 mm N-ethylmaleimide (Sigma) in 1× PBS). The lysate was then diluted with 17.5 ml dilution buffer (1.43 M NaCl, 1× EDTA-free protease inhibitor mixture, 50 mm N-ethylmaleimide in 1× PBS) and applied to 1 ml High Capacity NeutrAvidin Agarose Resin (Thermo Scientific) in a binding step incubated on rollers for 1 h at room temperature and 2 h at 4 °C. The beads were washed as described in Ref. 30. Material on beads was eluted in 4× Laemmli buffer + 100 mm DTT (95 °C, 10 min) for immunoblotting, or else processed directly for mass spectrometry.
Mass Spectrometry
The proteins were converted to peptides using a two-step digestion protocol adapted from de Godoy et al. (34). The peptides were separated on a 15-cm reverse phase column (packed in-house with 3-μm Reprosil beads (Dr. Maisch GmbH, Ammerbuch, Germany)) using a 5% to 50% acetonitrile gradient (Proxeon nano LC, Thermo Scientific). The peptides were ionized on a Proxeon ion source and sprayed directly into the mass spectrometer (Q-Exactive or Velos-Orbitrap, Thermo Scientific). The recorded spectra were analyzed using the MaxQuant software package (version 1.3.0.5) with a 1% false discovery rate for both peptides and proteins (35). Searches were performed using the Andromeda search engine against an IPI human database (version 3.84, 90,166 entries). Cysteine carbamidomethylation was selected as fixed modification, and methionine oxidation, protein N-terminal acetylation, and glygly-lysine were allowed as variable modifications. Two missed trypsin (full specificity) cleavages were allowed. The mass tolerance of precursor ions was set at 6 ppm, and for fragment ions it was 20 ppm. The raw data associated with this study can be found at the Proteomics DB repository (https://www.proteomicsdb.org/) with project I.D. 4156. Annotated spectra from single peptide identifications can be viewed online as supplemental data.
Gene Ontology Enrichment Analysis
p values of Gene Ontology (GO) enrichments in each dataset of protein hits were determined using g:Profiler (36). The GO terms were filtered according to the following attributes: (a) the p value in at least one protein-hit dataset was less than 10−3, (b) the GO term belonged to “cellular processes,” and (c) the depth of the GO term was less than 10. The significance of enrichment was presented as a heat map using the R environment (37) and the gplots package (38).
Imaging
Cells were seeded after electroporation onto eight-well plastic-bottom slides (Ibidi GmbH, München, Germany) at a density of 2 × 104 cells/cm2. The imaging medium was l-15 supplemented with FBS and antibiotics as described above. All time-lapse imaging was carried out on an Olympus CellR imaging platform comprising an Olympus IX81 motorized inverted microscope, an Orca-CCD camera (Hamamatsu Photonics), a motorized stage (Prior Scientific, Cambridge, UK), and a 37 °C incubation chamber (Solent Scientific, Segensworth, UK). Cells were imaged via epifluorescence and differential interference contrast (DIC) microscopy using a ×40 1.3 numerical aperture oil objective. Movies were recorded using Olympus CellR software, acquiring data from up to 20 fields per experiment at 2-min intervals and with no binning. Image sequences were exported as 12-bit TIFF files for analysis in ImageJ. For quantification of Venus levels in single cell degradation assays, fluorescence was measured as pixel values within a region of interest (ROI) selected to include the entire cell when applied to all images in a series, and from which background pixel values were subtracted. DIC images were used to determine the onset of anaphase.
Antibodies
RESULTS
Purification of a Mitotic Exit Ubiquitome
Polyubiquitinated conjugates are typically difficult to isolate in significant quantities. Therefore, we adopted a strategy for proteomic profiling of ubiquitin conjugates that would allow their isolation from cell extracts prepared under fully denaturing conditions, using biotin-avidin affinity purification (the strongest non-covalent interaction known, Kd < 10−14 m). Fully denaturing conditions would preserve polyubiquitin chains and ensure isolation of ubiquitin-conjugated material only, excluding from our studies the ubiquitin-binding proteins that can represent a large proportion of hits in studies carried out under native conditions (39, 40).
We further reasoned that highly synchronized cell populations would be required to identify substrates of the UPS specific to mitotic exit, so we generated a highly synchronizable human cell line expressing in vivo–biotinylated ubiquitin. bioUb is generated via expression of a synthetic linear construct of six ubiquitin moieties bearing a short biotinylatable tag in tandem with the Escherichia coli biotin ligase BirA. Each ubiquitin has an N-terminal 16-amino-acid tag that can be recognized and biotinylated by the BirA enzyme (30, 41). We inserted the bioUb6–BirA construct into human U2OS cells on a plasmid allowing tight control of expression by tetracycline (42). After clonal selection we chose a cell line in which the level of biotinylated ubiquitin was low relative to levels of endogenous ubiquitin in the cell. Total ubiquitin levels in the cell were unchanged after induction (Fig. 1A), and measurement of the ratio of bioUb:Ub-modified histone H2A indicated that less than 10% of ubiquitin in the cell carried the biotinylatable tag (supplemental Fig. S1). We expected that a low bioUb:Ub ratio would minimize potential perturbation of polyubiquitin chain topology and favor the isolation of polyubiquitinated conjugates that incorporate multiple ubiquitins. Indeed, we found that ∼50% of high-MW ubiquitin conjugates, which contain polyubiquitinated proteins, were cleared from cell extracts by biotin–NeutrAvidin pulldown, whereas the level of free ubiquitin was hardly perturbed (Fig. 1B). We confirmed that the in vivo degradation kinetics of a known APC/C-dependent substrate of the UPS, Aurora A kinase (31), was unaffected by the induction of bioUb (Fig. 1C). We also constructed a cell line expressing BirA alone at levels similar to those of bioUb6-BirA expressed in our bioUb cells (supplemental Fig. S1) to allow us to control for the nonspecific biotinylation of cellular proteins.
Fig. 1.

Experimental set-up of mitotic ubiquitome analysis using in vivo biotinylated ubiquitin. A, induction of biotinylated ubiquitin in bioUb cells. Tetracycline was withdrawn for 48 h. Immunoblots of whole cell extracts prepared under fully denaturing conditions were probed with antibodies against biotin or ubiquitin conjugates (FK2). Quantification of the ubiquitin blot showed no increase (100 ± 1% (S.D.), n = 3) in total cellular ubiquitin after induction of bioUb6–BirA. Histone 2A (H2A) was a major ubiquitinated band in these cells. B, efficient pulldown of cellular ubiquitin conjugates using biotinylated ubiquitin. Immunoblots of cell extracts induced for bioUb expression showed levels of free and conjugated ubiquitin before and after incubation with NeutrAvidin beads. Even though the ratio of bioUb:ubiquitin in the cell was low, polyubiquitinated conjugates were efficiently purified from extracts. See also supplemental Note S1 and supplemental Fig. S1. C, induction of biotinylated ubiquitin did not affect the degradation kinetics of a substrate of mitotic proteolysis. Biotinylated ubiquitin was induced (-tet) or repressed (+tet) following electroporation of bioUb cells with Aurora A-Venus, with parental U2OS cells electroporated as controls. After 40 h mitotic cells were imaged via fluorescence time-lapse microscopy. Aurora A-Venus levels were quantified and plotted as a function of anaphase onset. Left, degradation curves for individual cells. Right, degradation averaged for cells under each condition; error bars show standard deviations. D, synchronization outline. BirA cells (negative control) and bioUb cells were synchronized in prometaphase by sequential thymidine/STLC block, collected by means of mitotic shake-off, and split into three aliquots. One aliquot was harvested under mitotic arrest (bioUBM sample, also called M phase). The other two aliquots were released from prometaphase block into mitotic exit for 30 min and 70 min before harvesting (bioUB30 and bioUB70 samples, also referred to as C1 and C2 phases). ZM, 10 μm ZM447439. E, immunoblotted extracts and NeutrAvidin pulldowns from cells synchronized as in D but released from prometaphase block for 15 min (metaphase, M*) or 30 min (C1 phase) were probed with anti-cyclin B1 antibody to reveal specific pulldown of ubiquitinated cyclin B1 from samples in which the SAC was inactivated. F, silver stain of 1/160 of the purified protein from large-scale preparations of bioUb cells and control BirA cells, showing clear enrichment of high-MW material in pulldowns from bioUb cells.
We prepared cells synchronized at mitosis after 40 h of expression of bioUb using sequential thymidine release/Eg5 inhibitor (STLC) treatment to arrest cells in prometaphase (M phase population) with monopolar spindles that trigger the SAC. We then released cells from SAC-mediated arrest through inhibition of Aurora B kinase activity (using specific inhibitor ZM) to generate mitotic exit fractions at 30 and 70 min after ZM treatment (Fig. 1D). The 30-min time point (mitotic exit I) was that at which we saw extensive ubiquitination of cyclin B1 (Fig. 1E), and 70 min (mitotic exit II) was the time point at which we saw extensive ubiquitination of Aurora kinase (see Fig. 3B). In terms of the morphology of cells undergoing mitotic exit, these time points correspond to early and late time points during C-phase, the window of time during which cells remain competent to complete cytokinesis (43), and we refer to them respectively as C1 and C2 in Figs. 1–3. We chose a ZM-release protocol over a protocol for the release of SAC-arrested cells using mitotic cyclin-dependent kinase (Cdk1) inhibitor treatment, as described elsewhere (44, 45), because the anaphase degradation rate of APC/C substrates that we have tested is reduced when Cdk1 is inhibited (but not when Aurora B is inhibited), suggesting that the timing of Cdk1 inactivation during anaphase is critical for optimal targeting of substrates at this time.2 Our synchronization protocol gave us highly enriched mitotic exit fractions as evidenced by the presence of cyclin B1 conjugates after single-step pulldown on NeutrAvidin beads. Critically, ubiquitinated cyclin B1 was recovered only from cells in which the SAC had been inactivated (Fig. 1E, lanes M* and C1), the condition for cyclin B1 degradation in vivo (46). Moreover, pulldowns were highly specific for ubiquitin-modified material, as unmodified cyclin B1 was undetectable in the pulldown fractions, even though these were loaded in 400-fold excess relative to the polyubiquitinated fraction (Fig. 1E). Having validated both the specificity of our pulldowns and the robustness of our synchronization protocol, we carried out large-scale preparations of cell extracts from bioUb cells and control BirA cells for NeutrAvidin pulldown. Silver staining of purified samples showed a specific enrichment of high-MW material in bioUb samples relative to BirA control samples, corresponding to ubiquitin-conjugated proteins (Fig. 1F).
Fig. 3.

Immunoblot validation of specific ubiquitin conjugates identified via mass spectrometry. A–C, loading in pulldown lanes = 1000× loading in input lanes, except for LMNB2 (700×), UBE2R1 (700×), and UBE2C (550×). Ubiquitinated material in pulldowns appears either as up-shifted monoubiquitinated bands (*) or as polyubiquitinated proteins in high-MW smears (indicated by arrows). E2 enzymes purified as ubiquitin thiolester conjugates, which were reduced during elution because DTT was present, migrated at the same MW as the unmodified protein (indicated by arrowheads). A, Cdc20 and PSMC5 (Sug1) were strongly polyubiquitinated in all fractions. B, mitotic regulators KIF11, TPX2, AURKA, and KIFC1, as well as nuclear lamin LMNB2, were present as polyubiquitin conjugates specific to mitotic exit fractions. Where input and eluate are shown in separate panels, the eluate panel was taken from a longer exposure of the same blot. See supplemental Fig. S3 for full-size blots and additional validations. Mono-ubiquitination of Aurora A (*) has not previously been described. C, E2 enzymes UBE2R1 and UBE2C were strongly enriched in bioUb pulldowns, with UBE2C additionally polyubiquitinated late in mitotic exit (long exposure).
Fig. 2.

Identification of a mitotic ubiquitome. A, Venn diagram of hits identified from mitotic fractions. Hits were classified as follows, according to the peptide numbers present in each sample (classification using label-free quantification data gave very similar results; raw peptide numbers and intensities are in supplemental Table S1): 470 bioUb-specific hits were categorized as those either absent from the BirA sample or represented by at least 5-fold more peptides in the bioUb sample. 110 hits below the 5-fold threshold were classified as nonspecific. bioUb hits were further divided into mitotic-exit-specific (C-phase-specific), prometaphase-specific (M-phase-specific), and non-phase-specific groups. 170 mitotic-exit-specific hits were assigned as those either absent from the M phase sample or represented by at least 3-fold more peptides in either of the mitotic-exit-specific samples (C1, C2). 56 prometaphase-specific hits were those present only in the M phase sample or represented by at least 3-fold more peptides. 244 hits falling under these thresholds were categorized as non-phase-specific. Protein names in the corresponding categories are shown as word clouds with words sized according to the posterior error probability score of each hit across samples. A threshold of 1E-5 was applied to remove the low-scoring hits. See supplemental Fig. S2 for full word clouds and supplemental Table S2 for listed classified hits. B, Gene Ontology (GO) annotation enrichment analysis for each category of bioUb hits. The heat map shows the overrepresentation significance (−log10 of the p value) of GO biological process terms across different categories. The blue lines highlight GO terms specifically enriched in C-phase-specific hits, and the green line highlights those enriched in non-phase-specific hits.
We carried out LC-MS/MS analysis of purified ubiquitin-conjugated material on an LTQ-Orbitrap mass spectrometer. Output from M, C1, and C2 pulldowns was searched against the IPI database for peptides with up to two missed cleavages and modifications including ubiquitination. A total of 580 proteins were identified, with 470 of these specific to samples from bioUb cells (Fig. 2A, supplemental Fig. S2, supplemental Tables S1 and S2). Hits present in the BirA-expressing control samples were known endogenous biotinylated proteins (carboxylases) and potential contaminants also present in the bioUb samples, showing that pulldowns from the BirA cell line were valid technical controls. Among the bioUb-specific hits, 244 proteins were present in both M- and C-phase pulldowns (non-phase-specific), and 170 proteins were enriched or found exclusively in mitotic exit phases C1 and C2 (Fig. 2A, supplemental Table S2, Table I). Two repeats of the experiment gave overlapping datasets of a similar size (supplemental Tables S1 and S2, supplemental Fig. S2). Of the bioUb-specific hits, more than half of the hits from the first experiment were present in the second experiment, indicating the high reproducibility of our assay.
Table I. Mitotic exit-specific ubiquitome.
| Protein name | Posterior error probability (PEP) score | Peptide numbers C:M |
p value of C-phase enrichment | |
|---|---|---|---|---|
| Experiment 1 | Experiment 2 | |||
| Kinesin-like protein KIFC1 | 1.01E-78 | 7:2 | 13:0 | 0.000122 |
| Lamin-B2 | 3.09E-45 | 9:2 | 16:1 | 0.0001373 |
| Lamin-B1 | 5.65E-41 | 11:2 | 14:1 | 0.0004882 |
| Rac GTPase-activating protein 1 | 3.17E-147 | 10:0 | 10:0 | 0.0009765 |
| Cytoskeleton-associated protein 2 | 0.00066908 | 7:0 | 2:0 | 0.0078125 |
| Signal recognition particle 54 kDa protein | 4.01E-24 | 11:2 | 7:1 | 0.0112304 |
| Prelamin-A/C | 1.75E-26 | 6:1 | 10:2 | 0.0192871 |
| Cytoskeleton-associated protein 2-like | 0.012934 | 4:0 | 1:0 | 0.0625 |
| Importin subunit α-2 | 3.81E-20 | 4:0 | 2:0 | 0.0625 |
| d-3-phosphoglycerate dehydrogenase | 1.67E-05 | 3:0 | 3:1 | 0.125 |
| Kinesin-like protein KIF11 | 0.00021216 | 3:0 | 2:0 | 0.125 |
| Protein regulator of cytokinesis 1 | 6.59E-05 | 2:0 | 3:0 | 0.125 |
| 26S protease regulatory subunit 4 | 1.35E-12 | 1:0 | 4:1 | 0.1875 |
| G2/mitotic-specific cyclin-B1 | 5.66E-08 | 2:0 | 2:0 | 0.25 |
Protein hits from bioUb-specific pulldowns, ranked according to their enrichment in C phase samples. Only top-ranked proteins identified in two repeats of the experiment are included in the table. Peptide numbers from either C1 or C2 phase versus M phase samples are listed, and a binomial test was used to calculate the significance of the C phase enrichment in peptide numbers. The lesser of the two calculated p values is listed for each hit.
The non-phase-specific bioUb dataset showed strong enrichment for UPS components, evidenced by the overrepresentation of related GO annotations in this set (Fig. 2B), indicating that the mitotic arrest sample was a valid biological control. Indeed, many of the hits we expected to find in this dataset were present, in particular ubiquitin processing enzymes that carry ubiquitin as thioester conjugates—chiefly ubiquitin activating enzymes (UBA1, UBA6), multiple ubiquitin conjugating enzymes (UBEs), and HECT-family ubiquitin ligases (supplemental Table S3). Also present in this dataset were constitutively ubiquitinated substrates such as histones. The mitotic exit-specific (C-phase) dataset, in comparison, was enriched with mitosis-related GO terms (Fig. 2B), indicating that mitotic regulators targeted for ubiquitination were efficiently purified via this method. Indeed, this dataset contained 15 known substrates of the APC/C. We carried out an unbiased ranking of the C-phase dataset according to the degree of enrichment for ubiquitin-conjugated material (Table I). Of our 14 top-ranked hits, 2 (cyclin B1 and CKAP2) were previously described substrates of the APC/C, and others were putative substrates (PRC1, CKAP2L, RacGAP1, KIF11) or important mitotic regulators that are promising candidate substrates (KIFC1, Importin α2) (3, 47, 48).
Validation of a Mitotic Exit Ubiquitome
We tested the reliability of our datasets by immunoblotting our samples with antibodies against a variety of hits chosen from both mitotic-exit-specific and non-phase-specific datasets to test for the presence of ubiquitin-conjugated material in our bioUb pulldowns. All of the 14 hits we tested were confirmed ubiquitin-conjugated proteins in our extracts, 12 candidate UPS substrates and 2 known ubiquitin conjugating enzymes (E2s). Of the 12 candidate substrates that we tested, 11 were polyubiquitinated, as evidenced by the presence of high-MW material in bioUb pulldowns, and just one, histone H3, was monoubiquitinated only, confirming the preferential purification of polyubiquitinated material from dividing bioUb cells (Figs. 3A and 3B, supplemental Fig. S3). All of the polyubiquitinated proteins chosen from the mitotic-exit-specific dataset (KIF11 (Eg5), TPX2, AURKA, KIFC1, RacGAP1 (Cyk4), and LMNB2 (Lamin B2)) showed mitotic-exit-specific or -increased ubiquitination (Fig. 3B, supplemental Fig. S3), whereas the proteins chosen from the non-phase-specific dataset, Cdc20, PSMC5 (ATPase of the proteasome 19S regulatory particle), spartin, and NPM1 (nucleophosmin), showed ubiquitination throughout mitosis (Fig. 3A, supplemental Fig. S3), in line with our classification of hits according to the peptide numbers observed via mass spectrometry (supplemental Table S2). Our classification was also consistent with the expected ubiquitination pattern of known substrates of mitotic proteolysis that appeared in our lists of hits (anillin, AURKA, AURKB, BUB1, cyclin B1, Cdc20, CKAP2, GTSE1/B99, KIF2C/MCAK, NUSAP1, PLK1, TPX2, TK1) or of homologues of known APC/C substrates in budding yeast (KIF11/Eg5, PRC1/Ase1) (3). We detected a small fraction of UBE2C as a polyubiquitinated protein in our C2 sample (Fig. 3C), consistent with its reported targeting for degradation in G1 phase (49, 50). However, most of the signal detected for the two E2s we validated by immunoblotting migrated in the gel at the same MW as the unmodified E2, consistent with purification via a thioester ubiquitin linkage disrupted under the reducing elution conditions used (Fig. 3C). Therefore the identified E2 enzymes (supplemental Table S3) were more likely purified through being charged with ubiquitin for conjugating to substrates, rather than as substrates themselves of the UPS.
In conclusion, our validated ubiquitome consists of a mixture of ubiquitinated proteins and ubiquitin-conjugated enzymes, and we predict that the majority of non-UPS hits in our mitotic-exit-specific dataset are novel polyubiquitinated substrates during mitotic exit.
KIFC1 and RacGAP1 Are Substrates of the APC/C at Anaphase Onset
We selected two of our top-ranked mitotic-exit-specific hits, KIFC1 and RacGAP1 (Table I), for further analysis. KIFC1/HSET (Kinesin-14 family) is a minus-end directed microtubule (MT) motor that contributes to mitotic spindle assembly, sliding parallel MTs to regulate spindle length (51) and acting to focus the minus ends of MTs at the spindle poles. Cancer cells that have multiple centrosomes depend on KIFC1 to cluster centrosomes in bipolar spindle formation (52, 53). During mitotic exit, KIFC1 has been shown to contribute to organization of the central spindle (54). It has not previously been described as a substrate of mitotic proteolysis. The GTPase activating protein RacGAP1, also known as MgcRACGAP or hsCYK4, is present in mitotic cells as a hetero-tetrameric complex known as centralspindlin, together with the kinesin-6 family member KIF23/MKLP1. Centralspindlin is recruited to and stabilizes the midzone of the anaphase spindle, where it serves as a critical regulator of cytokinesis and a major component of the compacted midbody after furrow ingression (55). RacGAP1 was previously reported as a putative APC/C substrate (48).
We tested for mitotic-exit-specific destruction of KIFC1 and RacGAP1 in vivo in single cell assays in which the quantification of Venus-tagged proteins could provide readout for substrate proteolysis. We found that both of these targets of mitotic-exit-specific ubiquitination underwent proteolysis during exit from unperturbed bipolar mitosis. KIFC1-Venus degradation started immediately and rapidly at anaphase onset, with a t1/2 of 35 ± 7 min (Fig. 4A). This degradation was entirely dependent on the presence of a D-box-like motif (R5SPL) in the unstructured N terminus of KIFC1, indicating that KIFC1 was probably a substrate of the APC/C. Venus-RacGAP1 disappeared from cells more slowly, with a t1/2 of 65 ± 5 min (Fig. 4B). We confirmed that the disappearance of Venus-RacGAP1 was proteolysis-dependent, as it did not occur in the presence of proteasome inhibitor MG132. However, although RacGAP1 contains five RxxL motifs, none are good candidates for a D-box (only one is present in an unstructured region, and mutation of this motif did not abrogate degradation (56)). Because Venus-RacGAP1 proteolysis was rather slow in this assay despite its high ranking in our screen for ubiquitinated proteins, we confirmed that proteolysis of Venus-RacGAP1 under the conditions used for our screen of endogenous substrates—that is, during monopolar cytokinesis or after treatment with ZM—was not significantly different from that seen in unperturbed bipolar mitosis (supplemental Fig. S4).
Fig. 4.

KIFC1 and RacGAP1 are mitotic exit-specific APC/C substrates. A–B, Venus-tagged KIFC1 or RacGAP1 were electroporated into U2OS cells, and after 12 to 24 h mitotic cells were imaged by means of fluorescence time-lapse microscopy. Venus levels in individual mitotic cells were quantified and plotted as a function of anaphase onset. In vivo degradation curves are shown as averaged results with error bars showing standard deviations. A, KIFC1-Venus is a substrate of mitotic-exit-specific proteolysis that depends on a putative D-box (R5xxL) in the unstructured N terminus. B, Venus-RacGAP1 is a substrate of mitotic-exit-specific proteolysis, as a decrease in Venus-RacGAP1 levels did not occur in cells treated at anaphase onset with 5 μm MG132. See also supplemental Fig. S4. C, depletion of APC3 stabilizes endogenous KIFC1 and RacGAP1 in mitotic exit. U2OS cells were transfected with siRNA against control (luciferase, GL2), APC3, or Cdh1 sequences, synchronized to prometaphase as described in Fig. 1D and released using CDKI/II inhibitor. Cells were harvested at the indicated time points, and whole cell lysate was examined via immunoblot with antibodies against known and putative APC/C substrates. APC3 immunoblot indicated progress of mitotic exit with reversal of marked mitotic phosphorylation shift. D, in vivo ubiquitination of KIFC1-Venus was sensitive to the depletion of APC3. KIFC1-Venus electroporated into bioUb cells synchronized in M and C phases as in C was subjected to GFP-Trap® pulldown as described in Ref. 57. Anti-GFP immunoblot revealed unmodified KIFC1-Venus (in green), whereas anti-biotin immunoblot revealed KIFC1-Venus-associated ubiquitin conjugates (in red).
Next, we tested whether proteolysis of these novel mitotic substrates depends on APC/C activity. We examined levels of endogenous KIFC1 and RacGAP1 in synchronized cell extracts after siRNA-mediated depletion of APC3 or Cdh1 (Fig. 4C). We found that the disappearance of both during mitotic exit was sensitive to APC3 siRNA but not to Cdh1 siRNA (like Cyclin B1 but unlike Cdh1-dependent substrate Aurora A (31)). We also examined the ubiquitination of these substrates in an in vivo ubiquitination assay to quantify the ubiquitin-modified fraction of Venus-tagged proteins purified using GFP-Trap® pulldown technology (57) from cells arrested in M phase or released into C phase by Cdk1 inhibition. We found that RacGAP1 ubiquitination could not be detected via this method, as most of this protein was not extracted from the C-phase nucleus under conditions optimized for GFP-Trap pulldown (data not shown). However, we could readily measure the ubiquitination of KIFC1 and found it to be highly C-phase specific and dependent on the APC/C (Fig. 4D). We concluded that KIFC1 and RacGAP1 are both likely to be direct substrates of APC/C-mediated proteolysis during mitotic exit and that, as has been found for several other characterized substrates, they can be targeted by either Cdc20- or Cdh1-activated APC/C.
New Functions for APC/C Targeting during Mitotic Exit
We set out to investigate how the identification of KIFC1 and RacGAP1 as substrates of the UPS at mitotic exit might add to our understanding of mitotic exit control. We compared wild-type and non-degradable Venus-tagged KIFC1 during mitotic exit in live cells (supplemental Fig. S5). From late anaphase onward, we found both wild-type and non-degradable KIFC1-Venus were strongly localized at the spindle poles and the central spindle. In cells expressing high levels of either version of KIFC1-Venus, but not in cells expressing low levels, the anaphase spindle was not correctly assembled, with reduced MT density in the central spindle relative to the spindle poles (supplemental Fig. S5). These observations suggest that control of KIFC1 levels is critical for correct regulation of MT dynamics. Indeed, it has previously been shown that ectopically expressed KIFC1 must be sequestered into the nucleus during interphase to prevent inappropriate bundling of cytoplasmic MTs during interphase (51). However, because the expression level of non-degradable KIFC1-Venus was always higher than that of wild-type KIFC1-Venus (supplemental Fig. S5), we were unable to draw conclusions about any specific role for mitotic degradation of KIFC1.
Therefore, we focused our subsequent studies on RacGAP1. As a key regulator of cytokinesis, RacGAP1 is required to recruit and activate Ect2, the guanine nucleotide exchange factor that activates RhoA to initiate contractile ring assembly and constriction (58). Depletion of RacGAP1 causes extensive cytokinesis failure (59, 60). RacGAP1 additionally plays a structural role in mediating interaction between the spindle midzone and the cell membrane through its C1 lipid-binding domain, an interaction that is required to maintain the connection of the plasma membrane to the midbody following furrow ingression, until abscission (61). The GAP activity of RacGAP1 is not thought to play a critical role in contractile ring assembly but is proposed to down-regulate Rac activity at the cell equator to reduce cell adhesion and allow furrow ingression (32). The corollary to this idea is that suppression of RacGAP1 function outside of the equatorial region would be required for the subsequent process of cell spreading that requires Rac activity (32, 62). Therefore, we tested whether ubiquitin-mediated proteolysis contributes to spatial regulation of RacGAP1 during mitotic exit.
In support of the idea that RacGAP1 would be targeted for proteolysis outside of the equatorial region of the cell, we found that we could measure more rapid proteolysis of Venus-RacGAP1 if we excluded the midbody from our analysis of degradation in single cells (supplemental Fig. S6). However, our in vivo proteolysis assay relied on overexpressed RacGAP1 that may have been in excess relative to its centralspindlin partner MKLP1. Because the available antibody against RacGAP1 did not allow us to measure levels of the endogenous protein (because of nonspecific nuclear staining), we tested instead for spatial regulation of proteolysis of GFP-RacGAP1 in cell lines in which low-level expression of GFP-RacGAP1 complements siRNA-mediated knockdown of the endogenous protein (61) (Fig. 5A). In this experiment we measured RacGAP1 levels in late mitotic cells after fixation and processing for immunofluorescence analysis. We found that the ratio of RacGAP1 outside of the midbody was increased after treatment of cells with a proteasome inhibitor, suggesting that RacGAP1 is preferentially targeted for destruction outside of the midbody. We confirmed that Venus-RacGAP1Δ1–52, which is unable to bind MKLP1 and therefore defective in midbody localization (60, 61), was degraded more rapidly than Venus-RacGAP1-WT, and that overexpression of mCherry-MKLP1 conferred stabilization of Venus-RacGAP1, most dramatically at later time points during mitotic exit (supplemental Fig. S6). We concluded that incorporation of RacGAP1 into centralspindlin protects this pool from APC/C-mediated degradation.
Fig. 5.

APC/C suppresses cytoplasmic RacGAP1 to promote cell spreading after mitosis. A, RacGAP1-GFP is targeted for proteolysis outside of the midbody. Cells expressing low levels of RacGAP1-GFP and in which endogenous RacGAP1 was depleted by siRNA for 48 h were fixed after 15 min of treatment with 5 μm MG132 and stained with GFP antibody. Fluorescence intensities in cells undergoing mitotic exit were quantified and expressed as the ratio of total fluorescence in cytoplasm (non-midbody) versus midbody. Arrowheads indicate midbodies of cells where cytoplasmic GFP staining was not visible. Scale bar = 20 μm. B, fluorescence time-lapse images of cells expressing Venus-tagged RacGAP1 show that whereas in low-expressing cells all RacGAP1 was visible in the midbody (arrowheads), in high-expressing cells excess protein was clearly visible on the polar cell cortex (arrows) and in the nucleus. Scale bar = 10 μm. C, lysine residue K292 mediates mitotic-exit-specific polyubiquitination on RacGAP1. bioUb cells were electroporated with indicated Venus-RacGAP1 constructs and synchronized to M phase or C2 phase as described in Fig. 1D. Ubiquitinated proteins were purified via NeutrAvidin pulldown and probed with GFP antibody (that recognized the Venus tag). ΔC, Δ599–632. D, K292 mediates mitotic-exit-specific proteolysis of RacGAP1. Degradation of Venus-tagged RacGAP1 wild-type or K292R or K292R,K296R (2KR) mutant was measured as described in Fig. 4B. E, non-degradable RacGAP1 interfered with cell spreading after mitosis. mCherry-tagged RacGAP1 was electroporated into RPE α-actinin-Venus cells. After 8 to 20 h cells were imaged via fluorescence time-lapse microscopy. Cell area was visualized by α-actinin-Venus signal, measured at 10-min time intervals during mitotic exit and normalized to the cell area at metaphase for each cell. Spreading curves of individual cells (bottom panel) are averaged results with error bars to show standard deviations. Student's t test of cell spreading at the 70-min time point showed that the delay in the presence of K292R was significant (p = 0.0016), whereas in F289G it was not (p = 0.44). Scale bar in E = 20 μm.
RacGAP1 Ubiquitination Contributes to Cell Spreading after Mitosis
From these results we formed the idea that APC/C activity “titrates” RacGAP1 levels to ensure that all non-MKLP1-bound RacGAP1 is removed from the cytoplasm during mitotic exit. Our movies of Venus-RacGAP1 indicated that overexpressed protein was ectopically recruited to the cell cortex and associated with persistent contractility and blebbing (Fig. 5B, supplemental Movies S1 and S2) in a manner incompatible with post-mitotic cell spreading and in line with previous work showing that Drosophila RacGAP1 deliberately targeted to the cell cortex outside of the equatorial region promotes widespread ectopic furrowing (63). Therefore we reasoned that APC/C-mediated targeting of RacGAP1 would contribute to relaxation of the polar cortex for cell spreading after mitosis.
We wanted to test this hypothesis by generating a non-degradable version of RacGAP1. Inspection of proteomic datasets revealed that one ubiquitinated peptide (containing lysines K292 and K296) appears more frequently than any other (22–24, 26, 28), and moreover K292 is present in a context that fits our recently published description of APC/C ubiquitination site flanking region preferences with a serine at the -1 position (57). We mutated these lysines and confirmed that Venus-RacGAP1-K292R shows strongly reduced ubiquitination in mitotic exit (Fig. 5C) and that both lysines K292 and K296 contribute to in vivo destruction of RacGAP1 during mitotic exit (Fig. 5D). Because K292 has also been shown to be essential for the function of the RacGAP1 C1 domain in binding to the plasma membrane, we also tested a second C1 domain mutant, Venus-RacGAP1-F289G (61). This version was ubiquitinated as efficiently as wild-type Venus-RacGAP1, confirming that reduced ubiquitination of Venus-RacGAP1-K292R in vivo was not the result of altered RacGAP1 topology at the plasma membrane (Fig. 5C).
During the preparation of this manuscript, a newly published study described an APC/C-dependent degron for RacGAP1 in its C-terminal region (56). Our own study showed that a C-terminal truncation did reduce mitotic ubiquitination of RacGAP1 (Fig. 5C). However, we also found that when tagged with GFP, this version of RacGAP1 did not localize like the wild-type protein.3 Therefore, we confined our subsequent studies to the non-ubiquitinable version of RacGAP1 generated by mutating ubiquitin acceptor lysine K292.
We then compared cell spreading in cells expressing wild-type and non-ubiquitinable versions of mCherry-RacGAP1 using hTERT-RPE α-actinin-Venus cells in which cell spreading can readily be visualized and quantified (33). Cells electroporated with RacGAP1 constructs were recorded through mitosis by means of fluorescence time-lapse imaging, and the areas of daughter cells, normalized for the area at metaphase, were plotted against time. We found that the expression of wild-type mCherry-RacGAP1 had no effect on cell spreading relative to that in non-mCherry expressing control cells, whereas cell spreading was significantly defective in cells expressing mCherry-RacGAP1-K292R but not in cells expressing mCherry-RacGAP1-F289G (Fig. 5E; supplemental Movies S3 and S4). These results are consistent with our proposed model that delay in cell spreading is a consequence of reduced ubiquitin-mediated degradation of RacGAP1 (Fig. 6).
Fig. 6.
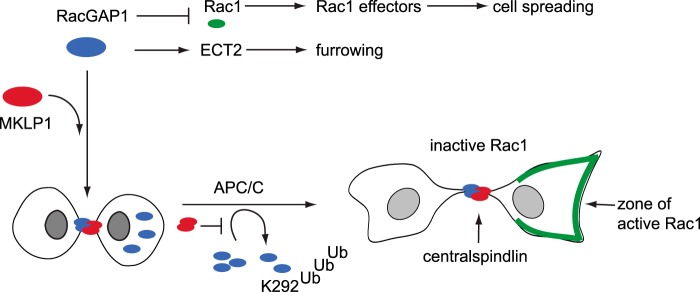
A model for spatial regulation of RacGAP1 by APC/C-mediated ubiquitination. Most RacGAP1 is recruited to the midbody as a heterotetrameric complex with MKLP1, where it recruits ECT and suppresses Rac1 activity to bring about cleavage furrow ingression. Any non-midbody-associated RacGAP1 can potentially interfere with cytokinesis through inappropriate recruitment of ECT2 or suppression of Rac1 (32, 63) and is targeted for ubiquitination on K292 by the APC/C.
Novel Roles for the APC/C and Other Ubiquitin Ligases in Mitotic Exit Pathways
Having validated our mitotic-exit-specific hits KIFC1 and RacGAP1 as bona fide APC/C substrates contributing to mitotic exit, we then asked how many of our other 170 mitotic-exit-specific hits could be APC/C substrates. We compared the enrichment of APC/C degrons (D-box and KEN box) in our list of hits and in a list compiled from validated APC/C substrates (57). We found very strong enrichment for both types of motifs in the list of known APC/C substrates, as well as significant enrichment in our experimental dataset (Fig. 7A). From this analysis we estimated that one-quarter of our hits could be new APC/C substrates. However, given that non-canonical degrons have been identified in several APC/C substrates (3), it is possible that our dataset includes many more such APC/C substrates that lack D-boxes or KEN motifs. Alternatively, our result might indicate a role for other ubiquitin ligases in mitotic-exit-specific ubiquitination. Indeed, our purification of E2 enzymes and HECT-type E3 ligases specifically from the mitotic exit fractions suggests the activity of novel ubiquitination pathways at this point of the cell cycle (supplemental Table S3).
Fig. 7.

Ubiquitination of cellular components during mitotic exit. A, enrichment analysis of the D-box and the KEN box in mitotic-exit-specific hits compared with non-phase-specific hits and a dataset of known APC/C substrates curated from the literature (57). The sequence of each protein was scanned for the presence of potential D-box (RXXL) and KEN motifs. The significance of enrichments was plotted as −log10(p values) calculated using a binomial test (null hypothesis: the frequency of observing D-box and KEN degrons is not greater in our dataset than in the whole human proteome) and indicated the presence of new APC/C substrates in our dataset, as even after removing known APC/C substrates from our dataset of mitotic-exit-specific hits we observed enrichment of degrons relative to the nonspecific dataset. B, interactions among proteins specifically ubiquitinated during mitotic exit. Protein interaction data were obtained using PSICQUIC (64) and plotted using Cytoscape (71).
Finally, we searched our mitotic exit ubiquitome using PSICQUIC (64) for protein interactions that might indicate cellular processes regulated by ubiquitination during mitotic exit. As expected, one prominent cluster was the mitotic machinery, many components of which are known targets of APC/C-mediated proteolysis after mitosis. We also found other hits clustering to organelles known to undergo mitosis-specific regulatory processes, such as the nuclear envelope and the ribosome (65, 66) (Fig. 7B). We concluded that widespread ubiquitination of cellular components during mitotic exit contributes to the extensive reorganization required for the mitosis–interphase transition.
DISCUSSION
We have presented for the first time a ubiquitinated proteome from mammalian cells that contains cell cycle phase-specific information. Our identification of a large number of UPS substrates targeted specifically during mitotic exit, and even at specific times during mitotic exit (for example, Aurora A, Lamin B2), indicates the wealth of information that can be gathered from using synchronized cell populations. Indeed, 17 of our mitotic-exit-specific hits had never been identified as in vivo ubiquitinated proteins before, including APC/C substrates anillin, sororin, and TOME-1. This suggests that our highly synchronized samples enabled the identification of rare ubiquitinated substrates whose ubiquitination only happens at a specific moment in the cell cycle.
Our proteomic strategy based on the purification of intact ubiquitinated material allowed us to validate 100% of our candidate ubiquitinated proteins using immunoblotting with substrate-specific antibodies. This validation strategy provided significantly more information about identified ubiquitin conjugates than available from diGly-based proteomics, for example, by revealing the dramatic switch from mono- to (mono+poly)-ubiquitinated Aurora A kinase that occurs between C1 and C2 phases (Fig. 3B), a switch likely to be of biological significance. Unlike diGly-based proteomics, however, our strategy did not find many sites of ubiquitination on individual substrates (that is, only a small fraction of assigned peptides contained diGly- modified lysine residues). This limitation in our strategy could reflect a pattern of mitotic polyubiquitination on multiple adjacent lysine residues, a situation that precludes identification by mass spectrometry, or it could simply result from the complexity of the purified material presenting a challenge for analysis by mass spectrometry. Indeed, the peptide coverage of most substrates identified, including several of those that we subsequently validated, was very low. Future studies in which bioUb purification could be combined with a diGly purification step to (a) reduce the complexity of the samples and (b) identify ubiquitinated lysine residues would provide a valuable further resource for the investigation of substrate ubiquitination.
Our strategy also provided quantitative comparisons of the fraction of modified substrates and the extent of modification, enabling us to identify Cdc20, spartin, and PSMC5 as substrates with very high polyubiquitinated fractions. In the case of Cdc20, polyubiquitination plays a crucial function in SAC signaling (67), and we can speculate that spartin and PSMC5 might also require functionally significant constitutive polyubiquitination. Multiple monoubiquitination of spartin has previously been described (68). The same study identified potential K48-dependent polyubiquitination of spartin, but no functional significance has yet been attached to this modification.
We believe our two overlapping datasets identify a significant fraction of the mitotic polyubiquitinated proteome, although the absence of some critical APC/C substrates (such as securin) indicates that they are not complete. This could reflect the complexity of the purified material, as discussed above, or the fact that we did not treat cells with proteasome inhibitors, as is commonly done to enrich ubiquitinated material destined for degradation. We avoided conditions of proteasome inhibition because we wanted a global view of mitotic exit ubiquitination that would include non-degradative ubiquitination events. Proteasome inhibition can also give rise to stress-induced changes to UPS function (69).
A significant fraction of proteins specifically ubiquitinated in mitotic exit are likely to be substrates of the APC/C (and destined for the 26S proteasome). Indeed, the candidate hits that we selected for further study, kinesin 14 family member KIFC1 and centralspindlin component RacGAP1, were two such substrates. However, the enrichment of typical APC/C degron motifs in our mitotic exit ubiquitinated proteome was less than in the list of known APC/C substrates, indicating potential roles for other ubiquitin ligases in mitotic-exit-specific ubiquitination. Indeed, preliminary investigations of our novel targets of mitotic-exit-specific ubiquitination showed that some were not significantly degraded during mitotic exit,4 indicating roles for non-degradative ubiquitination. Our assay could be developed to test the roles of other ubiquitin ligases during mitotic exit.
We investigated further why RacGAP1 would be targeted to the 26S proteasome specifically during mitotic exit. Our findings indicated that any RacGAP1 not localized to the midbody (where it is required for different steps in cytokinesis) becomes a target for the APC/C after anaphase onset, and therefore APC/C-mediated targeting of RacGAP1 contributes to its exclusive localization at the equatorial region of cells exiting mitosis. Further studies with a stabilized version of RacGAP1 confirmed our hypothesis that ubiquitination of RacGAP1 in daughter cells is required for normal cell spreading as cells return to interphase. These results are consistent with the observation that Rac1 activity after telophase is increased in the polar regions of dividing cells (62), where it can be predicted to play critical roles in cell spreading via effectors that regulate actin dynamics (70). Cell spreading is just one of the major reorganizations of the cell that occurs at this time. The hits from our study generated functional clusters indicating some of the processes regulated by ubiquitination during mitotic exit—in particular via the proteasome lid, nuclear lamins, and mitotic spindle machinery—that remain to be explored in further detail.
In conclusion, we have described an important novel resource, human cells expressing in vivo–biotinylatable ubiquitin, than can be applied to the problem of identifying and quantifying endogenous polyubiquitinated targets of the UPS. In this study we used bioUb cells to elucidate the mitotic exit ubiquitome, thus furthering our understanding of UPS-mediated events regulating the assembly of daughter cells after mitosis.
Supplementary Material
Acknowledgments
We thank the labs of Paolo D'Avino, Anne Bertolotti, Evan Reid, Jon Pines, Steve Jackson, Fanni Gergely, and Viji Draviam for antibodies. Masanori Mishima kindly donated RacGAP1 and MKLP1 plasmids, and Mark Petronczki provided RacGAP1-GFP cell lines. We are grateful to Rhys Grant for help with mitotic shake-offs, Rebecca Migotti for assistance with LC-MS/MS, Masanori Mishima and Mark Petronczki for discussions about RacGAP1, and Paolo D'Avino for comments on the manuscript. We are grateful to David Glover and Marisa Segal for their support.
Footnotes
Author contributions: C.L. designed research; M.M. and C.L. performed research; U.M. and G.D. contributed new reagents or analytic tools; M.M., U.M., G.D., and C.L. analyzed data; M.M. and C.L. wrote the paper.
* This work was supported by Cancer Research UK (Grant No. C3/A10239 to C.L. and David M. Glover) and a Sun Hung Kai-Kwoks' Foundation Scholarship to M.M.
 This article contains supplemental material.
This article contains supplemental material.
2 C. Lindon, unpublished data.
3 M. Min and C. Lindon, unpublished data.
4 S. Wood, M. Min, and C. Lindon, unpublished data.
1 The abbreviations used are:
- SAC
- spindle assembly checkpoint
- APC/C
- anaphase-promoting complex/cyclosome
- UPS
- ubiquitin-proteasome system
- D-box
- destruction box degron
- STLC
- S-trityl-l-cysteine
- DIC
- differential interference contrast
- GO
- Gene Ontology
- MT
- microtubule
- MW
- molecular weight
- bioUb
- in vivo–biotinylated ubiquitin
- UBE
- ubiquitin conjugating enzyme
- ZM
- ZM447439.
REFERENCES
- 1. Barford D. (2011) Structure, function and mechanism of the anaphase promoting complex (APC/C). Q Rev. Biophys. 44, 153–190 [DOI] [PubMed] [Google Scholar]
- 2. Pines J. (2011) Cubism and the cell cycle: the many faces of the APC/C. Nat. Rev. Mol. Cell Biol. 12, 427–438 [DOI] [PubMed] [Google Scholar]
- 3. Meyer H. J., Rape M. (2011) Processive ubiquitin chain formation by the anaphase-promoting complex. Semin. Cell Dev. Biol. 22, 544–550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Min M., Lindon C. (2012) Substrate targeting by the ubiquitin-proteasome system in mitosis. Semin. Cell Dev. Biol. 23, 482–491 [DOI] [PubMed] [Google Scholar]
- 5. Vong Q. P., Cao K., Li H. Y., Iglesias P. A., Zheng Y. (2005) Chromosome alignment and segregation regulated by ubiquitination of survivin. Science 310, 1499–1504 [DOI] [PubMed] [Google Scholar]
- 6. Sumara I., Quadroni M., Frei C., Olma M. H., Sumara G., Ricci R., Peter M. (2007) A Cul3-based E3 ligase removes Aurora B from mitotic chromosomes, regulating mitotic progression and completion of cytokinesis in human cells. Dev. Cell 12, 887–900 [DOI] [PubMed] [Google Scholar]
- 7. Ramadan K., Bruderer R., Spiga F. M., Popp O., Baur T., Gotta M., Meyer H. H. (2007) Cdc48/p97 promotes reformation of the nucleus by extracting the kinase Aurora B from chromatin. Nature 450, 1258–1262 [DOI] [PubMed] [Google Scholar]
- 8. Maerki S., Olma M. H., Staubli T., Steigemann P., Gerlich D. W., Quadroni M., Sumara I., Peter M. (2009) The Cul3-KLHL21 E3 ubiquitin ligase targets aurora B to midzone microtubules in anaphase and is required for cytokinesis. J. Cell Biol. 187, 791–800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Beck J., Maerki S., Posch M., Metzger T., Persaud A., Scheel H., Hofmann K., Rotin D., Pedrioli P., Swedlow J. R., Peter M., Sumara I. (2013) Ubiquitylation-dependent localization of PLK1 in mitosis. Nat. Cell Biol. 15, 430–439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fournane S., Krupina K., Kleiss C., Sumara I. (2013) Decoding ubiquitin for mitosis. Genes Cancer 3, 697–711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Visintin R., Prinz S., Amon A. (1997) CDC20 and CDH1: a family of substrate-specific activators of APC-dependent proteolysis. Science 278, 460–463 [DOI] [PubMed] [Google Scholar]
- 12. Pfleger C. M., Kirschner M. W. (2000) The KEN box: an APC recognition signal distinct from the D box targeted by Cdh1. Genes Dev. 14, 655–665 [PMC free article] [PubMed] [Google Scholar]
- 13. Glotzer M., Murray A. W., Kirschner M. W. (1991) Cyclin is degraded by the ubiquitin pathway. Nature 349, 132–138 [DOI] [PubMed] [Google Scholar]
- 14. Michael S., Trave G., Ramu C., Chica C., Gibson T. J. (2008) Discovery of candidate KEN-box motifs using cell cycle keyword enrichment combined with native disorder prediction and motif conservation. Bioinformatics 24, 453–457 [DOI] [PubMed] [Google Scholar]
- 15. Merbl Y., Kirschner M. W. (2009) Large-scale detection of ubiquitination substrates using cell extracts and protein microarrays. Proc. Natl. Acad. Sci. U.S.A. 106, 2543–2548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ayad N. G., Rankin S., Ooi D., Rape M., Kirschner M. W. (2005) Identification of ubiquitin ligase substrates by in vitro expression cloning. Methods Enzymol. 399, 404–414 [DOI] [PubMed] [Google Scholar]
- 17. McGarry T. J., Kirschner M. W. (1998) Geminin, an inhibitor of DNA replication, is degraded during mitosis. Cell 93, 1043–1053 [DOI] [PubMed] [Google Scholar]
- 18. Song L., Rape M. (2010) Regulated degradation of spindle assembly factors by the anaphase-promoting complex. Mol. Cell 38, 369–382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Peng J., Schwartz D., Elias J. E., Thoreen C. C., Cheng D., Marsischky G., Roelofs J., Finley D., Gygi S. P. (2003) A proteomics approach to understanding protein ubiquitination. Nat. Biotechnol. 21, 921–926 [DOI] [PubMed] [Google Scholar]
- 20. Beltrao P., Albanese V., Kenner L. R., Swaney D. L., Burlingame A., Villen J., Lim W. A., Fraser J. S., Frydman J., Krogan N. J. (2012) Systematic functional prioritization of protein posttranslational modifications. Cell 150, 413–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Starita L. M., Lo R. S., Eng J. K., von Haller P. D., Fields S. (2012) Sites of ubiquitin attachment in Saccharomyces cerevisiae. Proteomics 12, 236–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Povlsen L. K., Beli P., Wagner S. A., Poulsen S. L., Sylvestersen K. B., Poulsen J. W., Nielsen M. L., Bekker-Jensen S., Mailand N., Choudhary C. (2012) Systems-wide analysis of ubiquitylation dynamics reveals a key role for PAF15 ubiquitylation in DNA-damage bypass. Nat. Cell Biol. 14, 1089–1098 [DOI] [PubMed] [Google Scholar]
- 23. Udeshi N. D., Mani D. R., Eisenhaure T., Mertins P., Jaffe J. D., Clauser K. R., Hacohen N., Carr S. A. (2012) Methods for quantification of in vivo changes in protein ubiquitination following proteasome and deubiquitinase inhibition. Mol. Cell. Proteomics 11, 148–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wagner S. A., Beli P., Weinert B. T., Nielsen M. L., Cox J., Mann M., Choudhary C. (2011) A proteome-wide, quantitative survey of in vivo ubiquitylation sites reveals widespread regulatory roles. Mol. Cell. Proteomics 10, M111.013284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Xu G., Paige J. S., Jaffrey S. R. (2010) Global analysis of lysine ubiquitination by ubiquitin remnant immunoaffinity profiling. Nat. Biotechnol. 28, 868–873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kim W., Bennett E. J., Huttlin E. L., Guo A., Li J., Possemato A., Sowa M. E., Rad R., Rush J., Comb M. J., Harper J. W., Gygi S. P. (2011) Systematic and quantitative assessment of the ubiquitin-modified proteome. Mol. Cell 44, 325–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Danielsen J. M., Sylvestersen K. B., Bekker-Jensen S., Szklarczyk D., Poulsen J. W., Horn H., Jensen L. J., Mailand N., Nielsen M. L. (2011) Mass spectrometric analysis of lysine ubiquitylation reveals promiscuity at site level. Mol. Cell. Proteomics 10, M110.003590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Emanuele M. J., Elia A. E., Xu Q., Thoma C. R., Izhar L., Leng Y., Guo A., Chen Y. N., Rush J., Hsu P. W., Yen H. C., Elledge S. J. (2011) Global identification of modular cullin-RING ligase substrates. Cell 147, 459–474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lee M. J., Lee B. H., Hanna J., King R. W., Finley D. (2011) Trimming of ubiquitin chains by proteasome-associated deubiquitinating enzymes. Mol. Cell. Proteomics 10, R110.003871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Franco M., Seyfried N. T., Brand A. H., Peng J., Mayor U. (2011) A novel strategy to isolate ubiquitin conjugates reveals wide role for ubiquitination during neural development. Mol. Cell. Proteomics 10, M110.002188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Floyd S., Pines J., Lindon C. (2008) APC/C Cdh1 targets aurora kinase to control reorganization of the mitotic spindle at anaphase. Curr. Biol. 18, 1649–1658 [DOI] [PubMed] [Google Scholar]
- 32. Bastos R. N., Penate X., Bates M., Hammond D., Barr F. A. (2012) CYK4 inhibits Rac1-dependent PAK1 and ARHGEF7 effector pathways during cytokinesis. J. Cell Biol. 198, 865–880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Floyd S., Whiffin N., Gavilan M. P., Kutscheidt S., De Luca M., Marcozzi C., Min M., Watkins J., Chung K., Fackler O. T., Lindon C. (2013) Spatiotemporal organization of Aurora-B by APC/CCdh1 after mitosis coordinates cell spreading through FHOD1. J. Cell Sci. 126, 2845–2856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. de Godoy L. M., Olsen J. V., Cox J., Nielsen M. L., Hubner N. C., Frohlich F., Walther T. C., Mann M. (2008) Comprehensive mass-spectrometry-based proteome quantification of haploid versus diploid yeast. Nature 455, 1251–1254 [DOI] [PubMed] [Google Scholar]
- 35. Cox J., Mann M. (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 [DOI] [PubMed] [Google Scholar]
- 36. Reimand J., Arak T., Vilo J. (2011) g:Profiler—a Web server for functional interpretation of gene lists (2011 update). Nucleic Acids Res. 39, W307–W315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. R Core Team (2012) R: A Language and Environment for Statistical Computing, R Foundation for Statistical Computing, Vienna, Austria [Google Scholar]
- 38. Warnes G. R. (2011) gplots: Various R Programming Tools for Plotting Data http://cran.r-project.org/web/packages/gplots/index.html [Google Scholar]
- 39. Matsumoto M., Hatakeyama S., Oyamada K., Oda Y., Nishimura T., Nakayama K. I. (2005) Large-scale analysis of the human ubiquitin-related proteome. Proteomics 5, 4145–4151 [DOI] [PubMed] [Google Scholar]
- 40. Lopitz-Otsoa F., Rodriguez-Suarez E., Aillet F., Casado-Vela J., Lang V., Matthiesen R., Elortza F., Rodriguez M. S. (2012) Integrative analysis of the ubiquitin proteome isolated using Tandem Ubiquitin Binding Entities (TUBEs). J. Proteomics 75, 2998–3014 [DOI] [PubMed] [Google Scholar]
- 41. Beckett D., Kovaleva E., Schatz P. J. (1999) A minimal peptide substrate in biotin holoenzyme synthetase-catalyzed biotinylation. Protein Sci. 8, 921–929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gossen M., Bujard H. (1992) Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc. Natl. Acad. Sci. U.S.A. 89, 5547–5551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Canman J. C., Hoffman D. B., Salmon E. D. (2000) The role of pre- and post-anaphase microtubules in the cytokinesis phase of the cell cycle. Curr. Biol. 10, 611–614 [DOI] [PubMed] [Google Scholar]
- 44. Hu C. K., Coughlin M., Field C. M., Mitchison T. J. (2008) Cell polarization during monopolar cytokinesis. J. Cell Biol. 181, 195–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ozlu N., Monigatti F., Renard B. Y., Field C. M., Steen H., Mitchison T. J., Steen J. J. (2010) Binding partner switching on microtubules and aurora-B in the mitosis to cytokinesis transition. Mol. Cell. Proteomics 9, 336–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Clute P., Pines J. (1999) Temporal and spatial control of cyclin B1 destruction in metaphase. Nat. Cell Biol. 1, 82–87 [DOI] [PubMed] [Google Scholar]
- 47. Jiang W., Jimenez G., Wells N. J., Hope T. J., Wahl G. M., Hunter T., Fukunaga R. (1998) PRC1: a human mitotic spindle-associated CDK substrate protein required for cytokinesis. Mol. Cell 2, 877–885 [DOI] [PubMed] [Google Scholar]
- 48. Zhao W. M., Fang G. (2005) MgcRacGAP controls the assembly of the contractile ring and the initiation of cytokinesis. Proc. Natl. Acad. Sci. U.S.A. 102, 13158–13163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yamanaka A., Hatakeyama S., Kominami K.-i., Kitagawa M., Matsumoto M., Nakayama K.-i. (2000) Cell cycle-dependent expression of mammalian E2-C regulated by the anaphase-promoting complex/cyclosome. Mol. Biol. Cell 11, 2821–2831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Walker A., Acquaviva C., Matsusaka T., Koop L., Pines J. (2008) UbcH10 has a rate-limiting role in G1 phase but might not act in the spindle checkpoint or as part of an autonomous oscillator. J. Cell Sci. 121, 2319–2326 [DOI] [PubMed] [Google Scholar]
- 51. Cai S., Weaver L. N., Ems-McClung S. C., Walczak C. E. (2009) Kinesin-14 family proteins HSET/XCTK2 control spindle length by cross-linking and sliding microtubules. Mol. Biol. Cell 20, 1348–1359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kleylein-Sohn J., Pöllinger B., Ohmer M., Hofmann F., Nigg E. A., Hemmings B. A., Wartmann M. (2012) Acentrosomal spindle organization renders cancer cells dependent on the kinesin HSET. J. Cell Sci. 125, 5391–5402 [DOI] [PubMed] [Google Scholar]
- 53. Kwon M., Godinho S. A., Chandhok N. S., Ganem N. J., Azioune A., Thery M., Pellman D. (2008) Mechanisms to suppress multipolar divisions in cancer cells with extra centrosomes. Genes Dev. 22, 2189–2203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Cai S., Weaver L. N., Ems-McClung S. C., Walczak C. E. (2010) Proper organization of microtubule minus ends is needed for midzone stability and cytokinesis. Curr. Biol. 20, 880–885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. White E. A., Glotzer M. (2012) Centralspindlin: at the heart of cytokinesis. Cytoskeleton 69, 882–892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Nishimura K., Oki T., Kitaura J., Kuninaka S., Saya H., Sakaue-Sawano A., Miyawaki A., Kitamura T. (2013) APC(CDH1) targets MgcRacGAP for destruction in the late M phase. PLoS One 8, e63001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Min M., Mayor U., Lindon C. (2013) Ubiquitination site preferences in anaphase promoting complex/cyclosome (APC/C) substrates. Open Biol. 3, 130097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Yuce O., Piekny A., Glotzer M. (2005) An ECT2-centralspindlin complex regulates the localization and function of RhoA. J. Cell Biol. 170, 571–582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Jantsch-Plunger V., Gonczy P., Romano A., Schnabel H., Hamill D., Schnabel R., Hyman A. A., Glotzer M. (2000) CYK-4: a Rho family gtpase activating protein (GAP) required for central spindle formation and cytokinesis. J. Cell Biol. 149, 1391–1404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Mishima M., Kaitna S., Glotzer M. (2002) Central spindle assembly and cytokinesis require a kinesin-like protein/RhoGAP complex with microtubule bundling activity. Dev. Cell 2, 41–54 [DOI] [PubMed] [Google Scholar]
- 61. Lekomtsev S., Su K. C., Pye V. E., Blight K., Sundaramoorthy S., Takaki T., Collinson L. M., Cherepanov P., Divecha N., Petronczki M. (2012) Centralspindlin links the mitotic spindle to the plasma membrane during cytokinesis. Nature 492, 276–279 [DOI] [PubMed] [Google Scholar]
- 62. Yoshizaki H., Ohba Y., Kurokawa K., Itoh R. E., Nakamura T., Mochizuki N., Nagashima K., Matsuda M. (2003) Activity of Rho-family GTPases during cell division as visualized with FRET-based probes. J. Cell Biol. 162, 223–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. D'Avino P. P., Savoian M. S., Capalbo L., Glover D. M. (2006) RacGAP50C is sufficient to signal cleavage furrow formation during cytokinesis. J. Cell Sci. 119, 4402–4408 [DOI] [PubMed] [Google Scholar]
- 64. Aranda B., Blankenburg H., Kerrien S., Brinkman F. S. L., Ceol A., Chautard E., Dana J. M., De Las Rivas J., Dumousseau M., Galeota E., Gaulton A., Goll J., Hancock R. E. W., Isserlin R., Jimenez R. C., Kerssemakers J., Khadake J., Lynn D. J., Michaut M., O'Kelly G., Ono K., Orchard S., Prieto C., Razick S., Rigina O., Salwinski L., Simonovic M., Velankar S., Winter A., Wu G., Bader G. D., Cesareni G., Donaldson I. M., Eisenberg D., Kleywegt G. J., Overington J., Ricard-Blum S., Tyers M., Albrecht M., Hermjakob H. (2011) PSICQUIC and PSISCORE: accessing and scoring molecular interactions. Nat. Methods 8, 528–529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Guttinger S., Laurell E., Kutay U. (2009) Orchestrating nuclear envelope disassembly and reassembly during mitosis. Nat. Rev. Mol. Cell Biol. 10, 178–191 [DOI] [PubMed] [Google Scholar]
- 66. Sivan G., Kedersha N., Elroy-Stein O. (2007) Ribosomal slowdown mediates translational arrest during cellular division. Mol. Cell Biol. 27, 6639–6646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Musacchio A., Ciliberto A. (2012) The spindle-assembly checkpoint and the beauty of self-destruction. Nat. Struct. Mol. Biol. 19, 1059–1061 [DOI] [PubMed] [Google Scholar]
- 68. Bakowska J. C., Jupille H., Fatheddin P., Puertollano R., Blackstone C. (2007) Troyer syndrome protein spartin is mono-ubiquitinated and functions in EGF receptor trafficking. Mol. Biol. Cell 18, 1683–1692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Leidecker O., Matic I., Mahata B., Pion E., Xirodimas D. P. (2012) The ubiquitin E1 enzyme Ube1 mediates NEDD8 activation under diverse stress conditions. Cell Cycle 11, 1142–1150 [DOI] [PubMed] [Google Scholar]
- 70. Ridley A. J., Paterson H. F., Johnston C. L., Diekmann D., Hall A. (1992) The small GTP-binding protein rac regulates growth factor-induced membrane ruffling. Cell 70, 401–410 [DOI] [PubMed] [Google Scholar]
- 71. Smoot M. E., Ono K., Ruscheinski J., Wang P.-L., Ideker T. (2011) Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics 27, 431–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.