

Abstract
Chemical cross-linking combined with mass spectrometry (MS) is powerful to provide protein three-dimensional structure information but difficulties in identifying cross-linked peptides and elucidating their structures limit its usefulness. To tackle these challenges, this study presents a novel cross-linking MS in conjunction with electrochemistry using disulfide-bond-containing dithiobis[succinimidyl propionate] (DSP) as the cross-linker. In our approach, electrolysis of DSP-bridged protein/peptide products, as online monitored by desorption electrospray ionization mass spectrometry is highly informative. First, as disulfide bonds are electrochemically reducible, the cross-links are subject to pronounced intensity decrease upon electrolytic reduction, suggesting a new way to identify cross-links. Also, mass shift before and after electrolysis suggests the linkage pattern of cross-links. Electrochemical reduction removes disulfide bond constraints, possibly increasing sequence coverage for tandem MS analysis and yielding linear peptides whose structures are more easily determined than their cross-linked precursor peptides. Furthermore, this cross-linking electrochemical MS method is rapid, due to the fast nature of electrochemical conversion (much faster than traditional chemical reduction) and no need for chromatographic separation, which would be of high value for structural proteomics research.
Studies of protein three-dimensional (3D) structures and protein–protein interactions (PPIs) help understand protein biological functions. Traditional methods, such as X-ray crystallography1 and NMR,2 have been used for this purpose for decades due to their high resolution. However, a large amount of sample is required, which becomes a major limitation.3 In addition, X-ray crystallography requires crystals, and NMR is limited to small protein analysis. Protein structural analysis by mass spectrometry (MS) is highly sensitive. It can be coupled with different techniques, such as hydrogen–deuterium exchange (H/D exchange),4,5 hydroxyl radical footprinting,6−8 and chemical cross-linking,9−15 as a low-resolution method for probing protein 3D structures. The advantage of chemical cross-linking is that cross-linking reactions can be performed in a native buffering environment to obtain covalently modified proteins that can be analyzed by MS subsequently (typically in conjunction with chromatographic separation16). By utilizing a bottom-up approach, protein sizes are unlimited, since enzymatic digestion can be used to produce small peptides for detection.
However, several experimental obstacles limit the usefulness of chemical cross-linking. One major challenge is the identification of cross-linked peptides in the complex mixture produced by enzymatic digestion of the modified protein or protein complex. Another difficulty is the complexity of the fragmentation patterns of cross-linked peptides, which makes the tandem MS-based structural determination to be problematic. A cross-linked precursor may display a mixed fragmentation series owing to the presence of two peptide sequences, and further complexity is created by the presence of cross-linked fragments and amino acids with partial cross-linking reagent attached. This complexity can be greatly reduced by cleaving the cross-link prior to MS/MS identification, yielding easily identifiable linear peptides. Therefore, a cross-link reagent, which is cleavable and also allows ease identification of cross-links, would be ideal for the cross-linking MS. Great efforts were made to solve these problems. Successful approaches for rapid identification of cross-links from complex mixtures include using isotope,17,18 fluorescence,19 and affinity tag-labeled cross-linkers20 or using characteristic reporter ions that could be generated by MS/MS ion dissociation.21,22 For releasing cross-linked peptides, photocleavable groups were introduced to cross-linkers that could be cleaved upon light irradiation.11 Chemically cleavable cross-linkers were also prepared and regarded helpful to the identification of cross-linking products with their characteristic mass shifts after chemical reduction.23 Alternatively, collision-induced dissociation (CID) cleavable cross-linkers contributed to the identification process by producing specific fragmentation patterns in addition to backbone cleavage.21,24,25
Dithiobis[succinimidyl propionate] (DSP, structure shown in Scheme S-1a, Supporting Information) is a disulfide-bond-containing cross-linking reagent with the characteristics of having selective reactivity toward amine groups of proteins and being chemically reducible. In addition, it is compatible with the use of various buffers for reaction, and the produced amide products are soluble in buffer.26 Furthermore, its membrane penetrable property offers utility for in vivo cross-linking study.27 Typically, chemical reductants such as dithiothreitol are used for reducing DSP-cross-linked products, but the reduction requires an excess amount of reductant and takes over 30 min.23 The disulfide bond of DSP cross-linked products is also cleavable upon CID in the negative ion mode.25,28−30 Unfortunately, the disulfide bond cleavage in the positive ion mode is suppressed by backbone dissociation and hard to be observed.25 Other novel approaches for cleaving disulfide bonds include laser-based ionization,31 ultraviolet photodissociation,32 electron-capture dissociation,33 electron-transfer dissociation,34 plasma-induced oxidation,35,36 reactive electrospray-assisted laser desorption/ionization,37 or using new ion chemistry.38,39
Herein, we present a novel cross-linking approach of using DSP as the cross-linking reagent in conjunction with electrochemical mass spectrometry (EMS; refers to the online combined electrochemistry EC with MS) for probing protein 3D structures and PPIs. The EMS technique is powerful in the identification of the electrochemical reaction products or intermediates,40,41 leading to extensive applications in bioanalysis and mechanistic studies of redox reactions.42−44 The advantage of adopting MS as an EC detector stems from the fact that MS is sensitive and can provide molecular weight information and that tandem MS can be also used for structural analysis based on ion dissociation. The EMS technique used in this work is the combined EC/MS method coupled with desorption electrospray ionization (DESI45), which was recently developed in our laboratory for studying electrochemical reaction mechanisms and protein/peptide electrochemistry.41,44,46−51 In this cross-linking study, because of the fact that a disulfide bond is electrochemically reducible,41,44,46−51 electrolytic reduction provides a fast and “clean” method to cleave DSP cross-linked products without using chemical reductants and to provide linear reduced peptide products, thus facilitating the obtaining of sequence information and linkage information on the cross-linked products. In addition, upon electrolysis, the large relative intensity drop of the cross-link ions suggests a new approach for the identification of cross-links. The concurrent formation of the reduced peptides helps confirm the assignment of the precursor cross-links. Furthermore, different from the negative ion CID or laser-based cleavage of disulfide bond-containing ions, the side reactions such as C–S bond cleavage is avoided in the electrochemical reduction. As the electrochemical reduction occurs in solution, there is also a freedom to ionize the resulting reduced peptides/proteins into either positive or negative ions for both MS and MS/MS analysis. Also, mass shift before and after electrolytic reduction for cross-link peptide ions indicates the type of cross-links. Furthermore, this cross-linking EC/DESI-MS method is rapid, due to the fast nature of electrochemical conversion.
In the study, ubiquitin, insulin, and the calmodulin–melittin complex were chosen as model proteins or protein complexes for the validation of our approach. Our results reveal that the DSP cross-linking reaction truly targets on solvent accessible residues of the proteins/protein complexes (either the N-terminal amine or lysine residue) and the identified intrapeptide and interpeptide cross-links are in good agreement with the previously reported literature results and 3D structures,14,52,53 showing the feasibility of probing protein 3D structures or PPIs using this cross-linking electrochemical MS.
Materials and Methods
Chemicals
Peptide HCKWFF was purchased from Bachem AG. Phosphate-buffered saline (PBS), dimethyl sulfoxide (DMSO), and other peptides/proteins used were all purchased from Sigma–Aldrich. DSP (MW 404.4 Da) was purchased from Thermo Scientific. Formic acid (FA) was purchased from Spectrum Chemical Mfg. Corp (Gardena, CA). HPLC-grade methanol was purchased from Fisher Scientific (Fair Lawn, NJ). The deionized water used for sample preparation was obtained using a Nanopure Diamond Barnstead purification system (Barnstead International, Dubuque, IA).
DSP Cross-Linking Experiments
Typically, an aqueous solution containing peptides, proteins, or protein complexes in PBS buffer was mixed with DSP in DMSO for effecting cross-linking reactions. Then, NH4HCO3 was used to quench the reaction, and either a C18 Ziptip or a cutoff filter was used for desalting before the EC/DESI-MS analysis. The experimental procedure is detailed in the Supporting Information.
Apparatus
The home-built apparatus for coupling a thin-layer electrochemical flow cell with a mass spectrometer by liquid sample DESI (Scheme S-2, Supporting Information) was used and described in detail previously.47 Except the insulin data that was collected using a Thermo Finnigan LCQ DECA ion trap (San Jose, CA), all other data was acquired using a Waters Xevo QTOF mass spectrometer (Milford, MA). The sample syringe and μ-PrepCell electrochemical thin-layer flow cell were connected using a piece of PEEK capillary tube, and the sample solution was injected for electrolysis at a flow rate of 5 μL/min. The thin-layer flow cell equipped with a magic diamond electrode (12 mm × 30 mm, Antec BV, Netherlands) as the working electrode was employed, and a Roxy potentiostat (Antec BV, Netherlands) was used to apply a reduction potential to the cell. The reduced species flowed out of the thin-layer cell via a piece of fused silica capillary and then underwent ionization by DESI. Unless otherwise specified, the spray solvent for DESI was methanol/water (1:1 by volume) containing 1% FA at the injection flow rate of 5 μL/min, and a high voltage of +5 kV was applied to the DESI spray probe. CID was carried out for ion structural analysis.
This cross-linking EC/DESI-MS technique we used turns out to be sensitive and comparable to traditional cross-linking MS methods. The cross-links could be detected using the protein digests with an injection concentration of 20 μM (see the detailed experimental procedure in the Supporting Information), which has no significant difference from the injection concentrations of 10–25 μM used in traditional cross-linking MS methods.54,55
Results and Discussion
In cross-linking experiments, analyzing how the protein is modified by cross-linker provides rich information about the protein 3D structure. The modified protein typically undergoes enzymatic digestion to produce peptides and subsequent liquid chromatography (LC)/MS and LC/MS/MS analysis. Dead-end peptide cross-links in the digested protein originate from the reaction of protein with only one reactive group of the cross-linker, providing information about the solvent accessibility of the reacted protein residue. If both ends of cross-linking reagents react with two residues of the same protein molecule, intramolecular inter- or intrapeptide cross-links are formed after the reaction and enzymatic digestion, providing proximity information for the two cross-linked residues. Intermolecular interpeptide cross-links, formed through bridging protein and its binding substrate with the cross-linking reagent, offer the most valuable information about the distance between the protein and substrate during their interaction. In the case of DSP, the cross-linking reaction may yield different products, dead-end cross-links, intrapeptide cross-links, and interpeptide cross-links (Scheme S-1, Supporting Information). The dead-end cross-links (Scheme S-1b, Supporting Information) carry a modification tag of −C(O)CH2CH2SSCH2CH2COOH that increases molecular weight by 192 Da in comparison to unmodified peptides. The unreacted end of the cross-linker hydrolyzes into a carboxylic acid group. Upon electrochemical reduction, the dead-end cross-links will have a 104 Da mass decrement by loss of HSCH2CH2COOH. The intrapeptide cross-links (Scheme S-1c, Supporting Information) carry a modification tag of −C(O)CH2CH2SSCH2CH2C(O)– (174 Da mass increment in comparison to unmodified peptides) and will have 2 Da increment upon electrolysis. Similarly, interpeptide cross-links (Scheme S-1d, Supporting Information) also carry a tag of −C(O)CH2CH2SSCH2CH2C(O)– but will be reduced into two smaller linear peptides. These features are useful in recognizing the linkage patterns of cross-links.
To examine the feasibility of the proposed method, a lysine-containing peptide HCK3FWW was first chosen as a test sample. Figure 1a shows the DESI-MS spectrum of the peptide after reaction with DSP. Besides the doubly charged HCK3FWW seen at m/z 453.7, two doubly charged ions of peptides P1 (m/z 540.7) and P2 (m/z 549.7) are observed. On the basis of the measured masses, P1 has a mass increment of 174.0 Da in comparison to the intact HCK3FWW, which indicates that P1 is an intrapeptide cross-link. P2 has a mass shift of 192.0 Da compared to the intact peptide, suggesting it as a dead-end product. It is expected that cross-links would have decreased intensity after electrolysis. Indeed, after electrolytic reduction (Figure 1b), the relative intensities of the doubly charged ion of P1 (m/z 540.7) and doubly charged ion of P2 (m/z 549.7) dropped by 62% and 37% (using the disulfide-bond-free peptide ion [HCK3FWW + 2H]2+ at m/z 453.7 as the reference; Table S-1, Supporting Information), respectively. The pronounced reduction of ion intensity would be useful for the fast identification of cross-linked products in a complicated digestion mixture. Furthermore, a new peptide [H▲CK3FWW + 2H]2+ (▲ denotes one reduced tag of −C(O)CH2CH2SH) at m/z 497.7 appeared as a result of P2 reduction with mass loss of 104.0 Da (a characteristics for dead-end cross-link reduction, Scheme S-1b, Supporting Information) confirming the assignment of P2 as a cross-linked product. Likewise, there is another +2 ion observed at m/z 541.7 (Figure 1b) as a result of reduction of P1 with mass increment of 2.0 Da (a characteristic for intrapeptide cross-link reduction, Scheme S-1c, Supporting Information), also confirming the assignment of P1 as an intrapeptide product.
Figure 1.
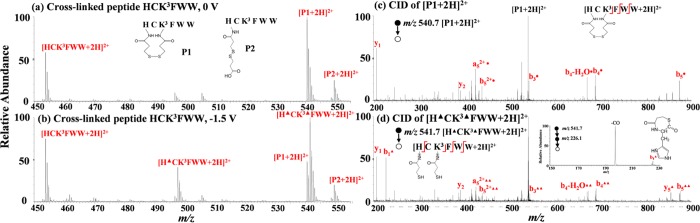
DESI-MS spectra of cross-linked peptide HCK3FWW (a) before and (b) after electrochemical reduction; CID MS/MS spectra of (c) [P1 + 2H]2+ (m/z 540.7) and (d) [H▲CK3▲FWW + 2H]2+ (m/z 541.7). Inset in (a) shows the structures of cross-linked P1 and P2, and inset in (d) shows the MS3 spectrum of the b1▲ ion with the proposed structure.
CID MS/MS analysis was applied to gain further structural information. Upon CID, the +2 ion of P1 (m/z 540.7) gave rise to unmodified y1 and y2 and modified fragment ions b3•, b4•, b4–H2O•, b5•, a52+•, and b52+• (• denotes the intrapeptide cross-linking modification tag of −C(O)CH2CH2SSCH2CH2C(O)–; Figure 1c). The presence of b3•–b5• indicates that the N-terminal and the Lys residue of P1 were cross-linked as they were the only possible cross-linking sites for this peptide. CID of the reduced P1 ion (m/z 541.7) yielded y1, y2, y5▲, b1▲, b3▲▲, b4▲▲, b4–H2O▲▲, b5▲▲, a52+▲▲, and b5–H2O 2+▲▲ (Figure 1d). The formation of fragment ions b1▲ and y5▲ results from the removal of the constraint of disulfide bond of P1 via electrolytic reduction, providing more backbone cleavage and facilitating sequencing the cross-link. Note that the b1 ion is typically not stable56−59 and the observation of the b1▲ ion in this case is probably due to the stabilization of the b1▲ ion with the reduced tag of −C(O)CH2CH2SH (see the proposed b1▲ structure in Figure 1d, inset). The MS3 of the b1▲ ion (Figure 1d, inset) shows the loss of carbon monoxide (CO), in agreement with the previously reported b1 ions.60 Furthermore, CID of the reduced P2 ion (m/z 497.7) gave rise to a complete set of fragment ions y1, y2, y3, y4, y5, b1▲, b2▲, b3▲, b4▲, and b5▲ (Figure S-1b, Supporting Information), clearly showing that dead-end modification site is on the peptide N terminus rather than the lysine residue.
The formation of intrapeptide product P1 (structure shown in Figure 1a, inset) suggests that both the N-terminal amine and the lysine residue of P1 are solvent accessible and close to each other, agreeing with the peptide structure. As a small peptide, the access to these two residues would have small steric hindrance. However, only one dead-end product P2 (structure shown in Figure 1a, inset) with the modified N terminus was produced, further suggesting that the N-terminal amino group is more reactive than the lysine residue. In this experiment, P1 and P2 are easily recognized by observing their intensity changes before and after electrolytic reduction. Furthermore, newly reduced products help obtain increased structural information by tandem MS analysis. To our knowledge, this result, for the first time, experimentally shows that the DSP disulfide bond can be electrochemically cleavable. With knowing the basic features of our EC/DESI-MS method, we further investigate its applications to the cross-linking study of ubiquitin, insulin proteins, and the calmodulin–melittin protein complex.
Insulin
Insulin contains two chains A and B that are linked by two disulfide bonds, and there is an additional intrapeptide disulfide bond in the A chain (structure shown in Figure 2a, inset). For this protein, there are three possible reactive sites for DSP: N termini of two chains and the K29 residue of the B chain. Because of the small size of the protein, direct analysis of the cross-linked insulin by EC/DESI-MS was applied without using enzymatic digestion. Figure 2a shows the positive ion DESI-MS spectrum of native insulin cross-linked in neutral PBS buffer, in which +5 ions of unmodified and modified insulin molecules appear at m/z 1147.9 and m/z 1186.1, respectively. The 191.0 Da mass difference of the two protein ions is approximately equal to the mass of a dead-end modification tag (192 Da; the mass deviation might be due to the low mass accuracy for LCQ DECA ion trap instrument used for the +5 ion measurements) and indicates that the modified protein carries one dead-end cross-link tag (structure shown in Figure 2a, inset). The modified insulin ion is labeled as 5+′ (′ denotes a dead-end tag of −C(O)CH2CH2SSCH2CH2COOH). After electrolytic reduction (Figure 2b), in comparison to the +4 ion of the unmodified B chain at m/z 851.2, the +4 ion of the modified B chain at m/z 872.7 (labeled as B chain4+▲) has a mass increment of 86.0 Da, which is close to the mass of one reduced dead-end tag (88 Da). This modified B chain is the product of the modified insulin by reducing all three insulin inherent disulfide bonds and the disulfide bond of the dead-end tag. Interestingly, a +5 ion of the partial reduction product carrying one reduced dead-end tag from the modified insulin by only reducing the disulfide bond of the dead-end tag is seen at m/z 1165.3 in Figure 2b (labeled as 5+▲ ion), providing another example of disulfide bond partial electrochemical reduction.47 The preferred reduction of the dead-end cross-linking tag is probably due to its small steric hindrance (as shown below, the dead-end tag is on the N terminus of the B chain). However, in Figure 2b, the A chain was not detected in the positive ion mode, probably due to its low proton affinity, as electrolysis occurs in solution and ionization in the negative ion of the reduced protein would be favorable to detect A chain. In this regard, it is straightforward to use DESI-MS to generate negative ions from acidic protein solution (acid is in need for electrochemical reduction) as one can simply change the DESI spray solvent from methanol/water (containing 1% FA) to methanol/water (containing 1% NH4OH), which is a unique application of liquid sample DESI-MS44,61 for realizing “wrong-way-around” ionization.62 Indeed, with the negative ion DESI-MS detection, doubly charged anion of intact A chain is observed at m/z 1167.8 after electrolysis and modified A chain ion is not (Figure S-2b, Supporting Information), suggesting that the A chain was not modified. Again, doubly charged anion of the modified B chain carrying one reduced dead-end tag is observed m/z 1742.0 (Figure S-2b, Supporting Information), further confirming that the sole modified position of the native insulin was on its B chain.
Figure 2.

(+)-DESI-MS spectra of cross-linked insulin (a) before and (b) after electrochemical reduction (applied potential: −1.5 V); (−)-DESI-MS spectra of denatured and cross-linked insulin (c) before and (d) after electrochemical reduction (applied potential: −1.5 V); and (e) CID MS/MS spectra of B chain4+▲ (m/z 872.7). Inset in (a) shows the structures of intact insulin and insulin with one dead-end modification. Inset in (b) shows the structure of insulin with two dead-end modifications.
CID helped to further locate the modification site of the B chain, and the positive ion MS/MS mode was adopted as it provided more fragment ions than the negative ion mode. On the basis of the observation of unmodified fragment ions of y182+ and y192+ from CID MS/MS of the B chain4+▲ ion (Figure 2e), the lysine residue K29 is considered not to be modified. Furthermore, the observation of many modified fragment b ions such as b102+▲–b142+▲ (Figure 2e) points out that the modification position is at the N terminus of the B chain. This result agrees with the solvent accessibility (shown in Table S-2, Supporting Information) that the B chain N terminus is solvent accessible for native insulin while the lysine residue K29 is not. However, the failure of the reaction with the A chain N terminus might be due to the possibility that the N terminus of the A chain is involved in salt bridge formation63 so that it becomes less solvent accessible.
In addition to studying the cross-linking of native insulin, denatured insulin (by urea denaturation) was also interrogated. Figure 2c displays the negative ion DESI-MS spectrum of denatured and cross-linked insulin, in which −4 ions of intact insulin and modified insulin with one dead-end tag are observed at m/z 1432.6 and at m/z 1481.1, respectively. Besides, one additional −4 ion at m/z 1529.1 is seen, with mass increment of 386.0 Da from intact insulin that is roughly equivalent to two dead-end tags (the mass deviation might be due to the low mass accuracy for LCQ DECA ion trap instrument used in this experiment). After electrolytic reduction (Figure 2d), doubly charged anions of intact and modified B chains are detected at m/z 1698.9 and m/z 1742.4, respectively. In addition, doubly charged anions of the A chain and the modified A chain with one reduced dead-end tag are detected at m/z 1167.9 and m/z 1211.8, respectively. On the basis of the DSP reaction specificity, the cross-linking modified position of the A chain should be the N terminus, which is the only possible cross-linked site. Clearly, under the denaturing condition used in this experiment, the salt bridge of the A chain N terminus might be destroyed to release a free N terminus for cross-linking modification by DSP. However, the K29 of the B chain is still protected and not accessible for DSP modification.
Ubiquitin
Ubiquitin in neutral PBS buffer was reacted with DSP for cross-linking, followed with trypsin digestion and EC/DESI-MS analysis. Figure 3a shows the DESI-MS spectrum of the resulting digested and cross-linked protein, in which unmodified peptide ion [TITLEVEPSDTIENVK + 2H]2+ (m/z 894.4, from ubiquitin) and [LGEDNINVVEGNEQFISASK + 2H]2+ (m/z 1082.0, from autolytic digestion of trypsin) are observed. Upon electrolysis (Figure 3b), four peptide ions, [P3 + 2H]2+ (m/z 769.8), [P4 + 3H]3+ (m/z 775.0), [P5 + 2H]2+ (m/z 948.9), and [P6 + 3H]3+ (m/z 1217.2) experienced large relative intensity decrease (by 34–100% using disulfide bond-free peptide ion [LGEDNINVVEGNEQFISASK + 2H]2+ as the reference peak, Table S-3, Supporting Information), which suggests that P3–P6 are possible cross-links. The charge states of the four peptide ions can be determined based on their isotopic peak distributions as measured by the QTOF instrument used in this case. In contrast, the unmodified peptide ion [TITLEVEPSDTIENVK + 2H]2+ (m/z 894.4) underwent a much smaller intensity change (decreased by 7% probably due to MS signal fluctuation; Table S-3, Supporting Information), in line with the fact that it does not have electrochemically active disulfide bonds.
Figure 3.

DESI-MS spectra of tryptic digest of cross-linked ubiquitin (a) before and (b) after electrochemical reduction (applied potential: −1.5 V). Inset in (a) shows the structures of cross-links P3 to P6, and inset in (b) shows the X-ray 3D structure of ubiquitin (PDB: 1UBQ).
Furthermore, three new reduced peptide ions appear in Figure 3b and are assigned as [LIFAGK48▲QLEDGR + 2H]2+ (m/z 717.8), [TLSDYNIQK63▲ESTLHLVLR + 3H]3+ (m/z 740.0), and [AK29▲IQDK33▲EGIPPDQQR + 2H]2+ (m/z 949.9), based on their CID data and the known sequence of ubiquitin. CID of [LIFAGK48▲QLEDGR + 2H]2+ (m/z 717.8, Figure 4b) gave rise to y2, y3, y4, y5, y6, y82+▲, y92+▲, y7▲, y8▲, y9▲, y10▲, y11▲, b2, b3, b4, b7▲, b8▲, and b9▲, covering most of the backbone cleavage and pinpointing the modification position at the lysine residue K48. Likewise, the CID of reduced peptide [TLSDYNIQK63▲ESTLHLVLR + 3H]3+ (m/z 740.0, Figure 4c) generated y2, y3, y4, y5, y6, y7, y8, y9, y122+▲, y162+▲, y10▲, y11▲, y12▲, and b2, also covering most of the backbone cleavage and pinpointing the modification site at the lysine residue K63. Furthermore, as the sum of the masses of newly generated peptides LIFAGK48▲QLEDGR (measured mass: 1433.6 Da) and TLSDYNIQK63▲ESTLHLVLR (measured mass: 2217.0 Da) is larger than the mass of P6 (measured mass: 3648.6 Da) by 2.0 Da, P6 would be the interpeptide cross-linking product of two peptides LIFAGK48QLEDGR and TLSDYNIQK63ESTLHLVLR. This hypothesis is confirmed by CID data. CID of [P6 + 3H]3+ (m/z 1217.2, Figure 4a) yielded fragment ions A(y3), A(y4), A(y5), A(y6), B(b7), B(y3), B(y4), B(y5), B(y6), B(y7), B(y8), B(y9), B(y10)A2+, B(y11)A2+, B(y12)A2+, and B(y16)A2+. For the denotation, A and B represent peptide chains LIFAGK48QLEDGR and TLSDYNIQK63ESTLHLVLR, respectively. The B(y10)A2+ ion refers to the doubly charged y10 ion from the B chain carrying an intact A chain. From CID spectra shown in Figure 4, it can be seen that analysis of reduced linear peptide chains by MS/MS is easier than analysis of the cross-linked precursor peptide. The structure of the determined interpeptide cross-link P6 is shown in Figure 3a, inset.
Figure 4.

CID MS/MS spectra of (a) [P6 + 2H]2+ (m/z 1217.2); (b) [LIFAGK48▲QLEDGR + 2H]2+ (m/z 717.8); and (c) [TLSDYNIQK63▲ESTLHLVLR + 3H]3+ (m/z 740.0).
In addition, as the mass difference between P3 (measured mass: 1537.6 Da) and newly generated peptide LIFAGK48▲QLEDGR (measured mass: 1433.6 Da) is 104.0 Da; P3 might be the simple dead-end cross-link of LIFAGK48QLEDGR. This assumption is confirmed by CID of the +2 ion of P3 (m/z 769.8, Figure S-3a, Supporting Information) showing the formation of fragment ions y2, y3, y4, y5, y6, y7′, y8′, y82+′, y9′, y92+′, y10′, y102+′, y11′, b2, b3, b4, b5, b6′, b7′, b8′, b9′, and b11′. Likewise, P4 is shown to be the dead-end cross-link of TLSDYNIQK63ESTLHLVLR (Figure S-3c, Supporting Information). For product P5, the intensity of its +2 ion at m/z 948.9 dropped after electrolysis (Table S-3, Supporting Information) while an adjacent +2 ion [AK29▲IQDK33▲EGIPPDQQR + 2H]2+ at m/z 949.9 was generated. The mass difference of 2.0 Da suggests P5 as an intrapeptide cross-link of AK29IQDK33EGIPPDQQR and the only possible cross-linking is to bridge K29 and K33 by DSP. CID of [P5 + 2H]2+ (m/z 948.9) yielded y6, y8, b8•, and b9•, and CID of [AK29▲IQDK33▲EGIPPDQQR + 2H]2+ (m/z 949.9) produced y6, b8▲▲, and b9▲▲, (Figure S-3e and S-3f, Supporting Information), in line with their structure assignments.
In this case of ubiquitin cross-linking, it can be seen that four different cross-linking products are identified (structures shown in Figure 3a, inset), including two dead-end products (P3 and P4), one intrapeptide product (P5), and one interpeptide product (P6). The formation of P5 tells that K29 and K33 of ubiquitin are solvent accessible and the same intrapeptide cross-link was observed previously.25 In particular, identification of the interpeptide cross-link P6 further shows that K48 and K63 residues are within the distance that DSP can span (ca. 24 Å based on the structural similarity of DSP with DSS).64 Indeed, it was reported that the distance between the two residues is 17.9 Å52 (the proximity of two residues can also be seen in the X-ray 3D structure of ubiquitin, Figure 3b, inset).
Calmodulin–Melittin Complex
Besides individual proteins such as insulin and ubiquitin, we also testified our methodology for probing protein/substrate interaction, and the calmodulin–melittin complex was chosen as a test sample. In the DESI-MS spectrum of cross-linked and trypsin-digested calmodulin–melittin complex (Figure 5a), unmodified peptide ions from calmodulin and melittin are detected, including [VLTTGLPALISWIK + 2H]2+ (m/z 756.5), [HVMTNLGEKLTDEEVDEMIR + 3H]3+ (m/z 787.4), [EADIDGDGQVNYEEFVQMMTAK + 2H]2+ (m/z 1246.0) and [VLTTGLPALISW + H]+ (m/z 1270.7). Upon electrolytic reduction (Figure 5b), these unmodified peptide ions have a small intensity change of +3%∼−15% as summarized in Table S-4 (using disulfide-bond-free peptide ion [VLTTGLPALISWIK + 2H]2+ as the reference). In contrast, four other ions denoted as [P7 + 3H]3+ (m/z 744.7), [P8 + H]+ (m/z 849.4), [P9 + 2H]2+ (m/z 930.5), and [P10 + 2H]2+ (m/z 974.4) underwent a large intensity decrease by 38–60% (Table S-4, Supporting Information), indicating that they are possible cross-links. In addition, four newly reduced products were generated and assigned as peptide ions [K23▲R + H]+ (m/z 391.2), [G1▲IGAVLK + H]+ (m/z 745.4), [VLTTGLPALISWIK21▲R + 2H]2+ (m/z 878.5), and [VFDK94▲DGNGYISAAELR + 2H]2+ (m/z 922.4), based on CID data and the known sequences of calmodulin and melittin.
Figure 5.

DESI-MS spectra of the tryptic digest of the cross-linked calmodulin–melittin complex (a) before and (b) after electrochemical reduction (applied potential: −1.7 V). Inset in (a) shows the structures of cross-links P7–P10, and inset in (b) illustrates the 3D structure of the calmodulin–melittin complex.
CID of [VFDK94▲DGNGYISAAELR + 2H]2+ (m/z 922.4, Figure S-4c, Supporting Information) produced y1, y2, y3, y4, y5, y6, y7, y9, y10, y11, y12, y13▲, y14▲, b2, b3, b4▲, b5▲▲, b6▲, b7▲, and b10▲, nearly covering all of backbone cleavages and pointing out the cross-linking site at K94 of calmodulin (i.e., the fourth residue of peptide VFDK94DGNGYISAAELR). CID of another newly generated peptide ion [K23▲R + H]+ (m/z 391.2) gave rise to y1 and y2▲ (Figure S-4b, Supporting Information), suggesting the sequence of the peptide as K23▲R (from melittin) and the modification position at K23 of melittin. Interestingly, among P7 to P10, the mass sum of peptides K23▲R (measured mass: 390.2 Da) and VFDK94▲DGNGYISAAELR (measured mass: 1842.8 Da) is higher than the mass of P7 (measured mass: 2231.1 Da) by 1.9 Da, which provides the clue that P7 is an interpeptide product consisting of the K23R chain from melittin and the VFDK94DGNGYISAAELR chain from calmodulin (see the structure of P7 in Figure 5a, inset). This hypothesis along with the ion structure assignment was confirmed by CID MS/MS analysis of [P7 + 3H]3+ (m/z 744.7) (Figure S-4a, Supporting Information) by observing A(y2), A(y5), A(y6), A(y7), A(y8), A(y9), A(y10), A(y11), A(y12), A(y13)B, and A(y14)B.
Upon CID, the newly resulting peptide ion [G1▲IGAVLK + H]+ (m/z 745.4) (Figure S-5b, Supporting Information) yielded y1, y2, y3, y4, b2▲, b3▲, b4▲, and b5▲, confirming its sequence and locating the modification site at the N terminus of G1IGAVLK (from melittin). According to the mass difference between P8 (measured mass: 848.4 Da) and the reduced product G1▲IGAVLK (measured mass: 744.4 Da), the mass decrease of 104.0 Da suggests that P8 is the dead-end cross-link precursor of GIGAVLK (see the structure of P8 in Figure 5a inset), which is further confirmed by CID of P8 (Figure S-5a, Supporting Information). Similarly, we assigned P9 and P10 as the dead-end cross-linking products of VLTTGLPALISWIK21R (modification site: K21) and VFDK94DGNGYISAAELR (modification site: K94), respectively (see Figure S-5 and discussion in Supporting Information).
From the cross-linking experiment of the calmodulin–melittin complex with subsequent EC/DESI-MS analysis, four cross-linking products P7–P10 are identified (structures shown in Figure 5a, inset). The formation of P8–P10 indicates that the N terminus of melittin (G1) and the K21 of melittin are also solvent accessible. The formation of P7 manifests that the K23 of melittin and the K94 of calmodulin are close to each other during the protein–substrate interaction process (Figure 5b illustrates the 3D structure of the complex). Therefore, they can be linked by DSP to form an interpeptide cross-linking product. This conclusion is in line with the literature reports14,53,65 and also with the previously reported MS-based chemical cross-linking results in which sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and LC separation were used to separate the protein complex digest before MS analysis of cross-links. In our experiment, no use of SDS-PAGE and LC separation and no use of chemical reductants contribute to the fast speed of our EC/DESI-MS method.
Conclusions
This study suggests a new, effective, and fast approach using electrochemical MS to analyze chemically cross-linked products resulting from the cross linking reactions of proteins/protein complexes with electrochemically reducible cross-linkers such as DSP. The online electrolytic reduction provides rich information for the structural analysis. First, cross-links present in the complicated digest mixtures could be quickly identified based on their large relative intensity decrease. The newly formed peptides from electrolysis could be related to their cross-linkers, based on their mass relationships. The electrochemical reduction gives rise to linear reduced peptides, facilitating the subsequent MS/MS analysis to provide sequence information and to pinpoint the modification sites of the cross-links. Furthermore, the electrolytic reduction takes place in seconds, and the EC/DESI-MS appears to be compatible with both top-down and bottom-up analyses, which would be of value for high-throughput proteomics applications. For the future work to probe 3D structures of protein samples with unknown sequences, the differentiation of native disulfide bonds with artificial ones introduced by cross-linking derivatization may be in need, as native disulfide-bond-containing peptides would also undergo electrolytic reduction. There are several possible approaches to address this issue. First, there are characteristic mass tags retained in the cross-links after reduction, which can be used for the differentiation purpose. In the case of insulin presented in this paper, there are two B chain peptides generated from electrolysis, one is intact B chain from reduction of two native disulfide bonds and the other one is the modified B chain with a reduced DSP tag. The presence of two B chains with the mass difference of one reduced dead-end tag identifies the cross-linked B chain, although the B chain carries two native disulfide bonds in the protein. Second, one could use a diselenide bond Se–Se to replace the disulfide bond S–S in the cross-linking reagent. Such a cross-linking reagent will have characteristic selenium isotopes, which help differentiate cross-link products from native disulfide-bond-containing peptides. Furthermore, the diselenide bond is easier to be electrochemically reduced than disulfide bonds.66 Thus, selective reduction of cross-links in the presence of native disulfide-bond-containing peptides is possible. Third, one could use isotope-labeled cross-linking reagents. If a mixture of DSP and deuterated DSP is used for performing cross-linking reactions,67 each cross-link will have dual peaks, which makes it distinct from native disulfide-bond-containing peptides. Such investigations are under the way.
Acknowledgments
This work is supported by NSF Career Award (CHE-1149367). H.Z. is from the laboratory of Prof. Michael L. Gross and thanks the support from the National Institute of General Medical Sciences (grant no. 8 P41 GM103422-35 to M.L.G.).
Supporting Information Available
Schemes showing the reduction of DSP cross-linked products and the experimental apparatus; detailed DSP cross-linking experimental procedures; and additional tables, DESI-MS data, and MS/MS data as noted in the text. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Kendrew J. C.; Bodo G.; Dintzis H. M.; Parrish R. G.; Wyckoff H.; Phillips D. C. Nature 1958, 181, 662–666. [DOI] [PubMed] [Google Scholar]
- Wuethrich K. J. Biol. Chem. 1990, 265, 22059–22062. [PubMed] [Google Scholar]
- Hyung S.-J.; Ruotolo B. T. Proteomics 2012, 12, 1547–1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Englander S. W. J. Am. Soc. Mass Spectrom. 2006, 17, 1481–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engen J. R. Anal. Chem. 2009, 81, 7870–7875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiselar J. G.; Chance M. R. J. Mass Spectrom. 2010, 45, 1373–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gau B. C.; Chen H.; Zhang Y.; Gross M. L. Anal. Chem. 2010, 82, 7821–7827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H.; Chen J.; Huang R. Y. C.; Weisz D.; Gross M. L.; Pakrasi H. B. J. Biol. Chem. 2013, 288, 14212–14220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramya T. N. C.; Weerapana E.; Liao L.; Zeng Y.; Tateno H.; Liao L.; Yates J. R. III; Cravatt B. F.; Paulson J. C. Mol. Cell. Proteomics 2010, 9, 1339–1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinz A. Mass Spectrom. Rev. 2006, 25, 663–682. [DOI] [PubMed] [Google Scholar]
- Sinz A. J. Mass Spectrom. 2003, 38, 1225–1237. [DOI] [PubMed] [Google Scholar]
- Sinz A. Anal. Bioanal. Chem. 2010, 397, 3433–3440. [DOI] [PubMed] [Google Scholar]
- Petrotchenko E. V.; Borchers C. H. Mass Spectrom. Rev. 2010, 29, 862–876. [DOI] [PubMed] [Google Scholar]
- Chavez J. D.; Liu N. L.; Bruce J. E. J. Proteome Res. 2011, 10, 1528–1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu H.; Wang Y. J. Proteome Res. 2009, 8, 1983–1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Löster K.; Josić D. J. Chromatogr. B: Biomed. Sci. Appl. 1997, 699, 439–461. [DOI] [PubMed] [Google Scholar]
- Müller D. R.; Schindler P.; Towbin H.; Wirth U.; Voshol H.; Hoving S.; Steinmetz M. O. Anal. Chem. 2001, 73, 1927–1934. [DOI] [PubMed] [Google Scholar]
- Fischer L.; Chen Z. A.; Rappsilber J. J. Proteomics 2013, 88, 120–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinz A.; Wang K. Anal. Biochem. 2004, 331, 27–32. [DOI] [PubMed] [Google Scholar]
- Alley S. C.; Ishmael F. T.; Jones A. D.; Benkovic S. J. J. Am. Chem. Soc. 2000, 122, 6126–6127. [Google Scholar]
- Sohn C. H.; Agnew H. D.; Lee J. E.; Sweredoski M. J.; Graham R. L. J.; Smith G. T.; Hess S.; Czerwieniec G.; Loo J. A.; Heath J. R.; Deshaies R. J.; Beauchamp J. L. Anal. Chem. 2012, 84, 2662–2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang X.; Munske G. R.; Siems W. F.; Bruce J. E. Anal. Chem. 2005, 77, 311–318. [DOI] [PubMed] [Google Scholar]
- Bennett K. L.; Kussmann M.; Bjork P.; Godzwon M.; Mikkelsen M.; Sorensen P.; Roepstorff P. Protein Sci. 2000, 9, 1503–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller M. Q.; Dreiocker F.; Ihling C. H.; Schaefer M.; Sinz A. Anal. Chem. 2010, 82, 6958–6968. [DOI] [PubMed] [Google Scholar]
- Calabrese A.; Good N.; Wang T.; He J.; Bowie J.; Pukala T. J. Am. Soc. Mass Spectrom. 2012, 23, 1364–1375. [DOI] [PubMed] [Google Scholar]
- Back J. W.; Sanz M. A.; de Jong L.; de Koning L. J.; Nijtmans L. G. J.; de Koster C. G.; Grivell L. A.; van der Spek H.; Muijsers A. O. Protein Sci. 2002, 11, 2471–2478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisbrod C. R.; Chavez J. D.; Eng J. K.; Yang L.; Zheng C.; Bruce J. E. J. Proteome Res. 2013, 12, 1569–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilusich D.; Bowie J. H. Mass Spectrom. Rev. 2009, 28, 20–34. [DOI] [PubMed] [Google Scholar]
- Andreazza H. J.; Bowie J. H. Phys. Chem. Chem. Phys. 2010, 12, 13400–13407. [DOI] [PubMed] [Google Scholar]
- Zhang M.; Kaltashov I. A. Anal. Chem. 2006, 78, 4820–4829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson S. D.; Katta V. Anal. Chem. 1994, 66, 3727–3732. [DOI] [PubMed] [Google Scholar]
- Eva Fung Y. M.; Kjeldsen F.; Silivra O. A.; Dominic Chan T. W.; Zubarev R. A. Angew. Chem., Int. Ed. 2005, 44, 6399–6403. [DOI] [PubMed] [Google Scholar]
- Zubarev R. A.; Kruger N. A.; Fridriksson E. K.; Lewis M. A.; Horn D. M.; Carpenter B. K.; McLafferty F. W. J. Am. Chem. Soc. 1999, 121, 2857–2862. [Google Scholar]
- Gunawardena H. P.; Gorenstein L.; Erickson D. E.; Xia Y.; McLuckey S. A. Int. J. Mass Spectrom. 2007, 265, 130–138. [Google Scholar]
- Xia Y.; Cooks R. G. Anal. Chem. 2010, 82, 2856–2864. [DOI] [PubMed] [Google Scholar]
- Stinson C. A.; Xia Y. Analyst 2013, 138, 2840–2846. [DOI] [PubMed] [Google Scholar]
- Peng I. X.; Ogorzalek Loo R. R.; Shiea J.; Loo J. A. Anal. Chem. 2008, 80, 6995–7003. [DOI] [PubMed] [Google Scholar]
- Gunawardena H. P.; O’Hair R. A. J.; McLuckey S. A. J. Proteome Res. 2006, 5, 2087–2092. [DOI] [PubMed] [Google Scholar]
- Kim H. I.; Beauchamp J. L. J. Am. Soc. Mass Spectrom. 2009, 20, 157–166. [DOI] [PubMed] [Google Scholar]
- Zhou F.; Van Berkel G. J. Anal. Chem. 1995, 67, 3643–3649. [Google Scholar]
- Lu M.; Wolff C.; Cui W.; Chen H. Anal. Bioanal. Chem. 2012, 403, 355–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karst U. Angew. Chem., Int. Ed. 2004, 43, 2476–2478. [DOI] [PubMed] [Google Scholar]
- Diehl G.; Karst U. Anal. Bioanal. Chem. 2002, 373, 390–398. [DOI] [PubMed] [Google Scholar]
- Li J.; Dewald H. D.; Chen H. Anal. Chem. 2009, 81, 9716–9722. [DOI] [PubMed] [Google Scholar]
- Wiseman J. M.; Ifa D. R.; Song Q.; Cooks R. G. Angew. Chem., Int. Ed. 2006, 45, 7188–7192. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Dewald H. D.; Chen H. J. Proteome Res. 2011, 10, 1293–1304. [DOI] [PubMed] [Google Scholar]
- Zheng Q.; Zhang H.; Chen H. Int. J. Mass Spectrom. 2013, 353, 84–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P.; Lu M.; Zheng Q.; Zhang Y.; Dewald H. D.; Chen H. Analyst 2013, 138, 5519–5539. [DOI] [PubMed] [Google Scholar]
- Liu P.; Lanekoff I. T.; Laskin J.; Dewald H. D.; Chen H. Anal. Chem. 2012, 84, 5737–5743. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Cui W.; Zhang H.; Dewald H. D.; Chen H. Anal. Chem. 2012, 84, 3838–3842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.; Yuan Z.; Dewald H. D.; Chen H. Chem. Commun. 2011, 47, 4171–4173. [DOI] [PubMed] [Google Scholar]
- Kruppa G. H.; Schoeniger J.; Young M. M. Rapid Commun. Mass Spectrom. 2003, 17, 155–162. [DOI] [PubMed] [Google Scholar]
- Schulz D. M.; Ihling C.; Clore G. M.; Sinz A. Biochemistry 2004, 43, 4703–4715. [DOI] [PubMed] [Google Scholar]
- Olson A. L.; Liu F.; Tucker A. T.; Goshe M. B.; Cavanagh J. Biochem. Biophys. Res. Commun. 2013, 431, 253–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes A. F.; Gozzo F. C. J. Mass Spectrom. 2010, 45, 892–899. [DOI] [PubMed] [Google Scholar]
- O’Hair R. A. J.; Reid G. E. Rapid Commun. Mass Spectrom. 2000, 14, 1220–1225. [DOI] [PubMed] [Google Scholar]
- Paizs B.; Suhai S. Mass Spectrom. Rev. 2005, 24, 508–548. [DOI] [PubMed] [Google Scholar]
- Summerfield S. G.; Steen H.; O’Malley M.; Gaskell S. J. Int. J. Mass Spectrom. 1999, 188, 95–103. [Google Scholar]
- Yalcin T.; Csizmadia I. G.; Peterson M. R.; Harrison A. G. J. Am. Soc. Mass Spectrom. 1996, 7, 233–242. [DOI] [PubMed] [Google Scholar]
- Summerfield S. G.; Bolgar M. S.; Gaskell S. J. J. Mass Spectrom. 1997, 32, 225–231. [Google Scholar]
- Cai Y.; Liu Y.; Helmy R.; Chen H.. J. Am. Soc. Mass Spectrom. 2014, in press [DOI] [PubMed]
- Zhou S.; Cook K. D. J. Am. Soc. Mass Spectrom. 2000, 11, 961–966. [DOI] [PubMed] [Google Scholar]
- Millican R. L.; Brems D. N. Biochemistry 1994, 33, 1116–1124. [DOI] [PubMed] [Google Scholar]
- Kao A.; Chiu C.-l.; Vellucci D.; Yang Y.; Patel V. R.; Guan S.; Randall A.; Baldi P.; Rychnovsky S. D.; Huang L. Mol. Cell. Proteomics 2011, 10, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaloni A.; Miraglia N.; Orru S.; Amodeo P.; Motta A.; Marino G.; Pucci P. J. Mol. Biol. 1998, 277, 945–958. [DOI] [PubMed] [Google Scholar]
- Killa H. M. A.; Rabenstein D. L. Anal. Chem. 1988, 60, 2283–2287. [DOI] [PubMed] [Google Scholar]
- Singh P.; Panchaud A.; Goodlett D. R. Anal. Chem. 2010, 82, 2636–2642. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.