

Abstract
Homeostasis, the dominant explanatory framework for physiological regulation, has undergone significant revision in recent years, with contemporary models differing significantly from the original formulation. Allostasis, an alternative view of physiological regulation, goes beyond its homeostatic roots, offering novel insights relevant to our understanding and treatment of several chronic health conditions. Despite growing enthusiasm for allostasis, the concept remains diffuse, due in part to ambiguity as to how the term is understood and used, impeding meaningful translational and clinical research on allostasis. Here we provide a more focused understanding of homeostasis and allostasis by explaining how both play a role in physiological regulation, and a critical analysis of regulation suggests how homeostasis and allostasis can be distinguished. Rather than focusing on changes in the value of a regulated variable (e.g., body temperature, body adiposity, or reward), research investigating the activity and relationship among the multiple regulatory loops that influence the value of these regulated variables may be the key to distinguishing homeostasis and allostasis. The mechanisms underlying physiological regulation and dysregulation are likely to have important implications for health and disease.
Keywords: Thermoregulation, Opioid-Induced Hyperalgesia, Sign-Reversal, Addiction, Obesity
Our goal in this article is to evaluate and compare the influential concepts of homeostasis and allostasis as they relate to physiological regulation. A particular focus is on whether either of these two models adequately explains the regulatory process. We begin by orienting the reader to the organization, definitions and positions taken in our analysis, and subsequent sections discuss the issues that we contend distinguish homeostasis and allostasis. We conclude that homeostasis and allostasis each has an important role and that research and theorizing on them has been hampered by lack of a clear operational definition of allostasis.
Introduction
Overview of Homeostasis
Homeostasis, the ongoing maintenance and defense of vital physiological variables such as blood pressure and blood sugar, was defined by Walter Cannon in 1929 as the major principle underlying physiological regulation. Homeostasis has a rich history, and although its understanding has evolved since its original formulation, the core concept of homeostasis as captured by Cannon’s phrase the ‘Wisdom of the Body’ (Cannon, 1932) remains its defining property. The wisdom of the body describes how an array of physiological and behavioral responses is elicited in a seemingly coordinated way to stabilize or defend critical physiological parameters.
Negative feedback was the first underlying process used to explain how homeostasis works. This was a reactive strategy whereby the perturbation of a regulated variable away from its optimal value was detected and consequently elicited corrective responses that served to return the variable back toward pre-perturbation levels. For example, a sudden drop in the oxygen content of the blood is detected by sensors that are synaptically linked to brain areas that control respiratory rate, and breathing and blood oxygen are consequently increased.
Cannon maintained that homeostasis is a reaction to an ongoing perturbation, never discussing the possibility that organisms might learn to anticipate regular or likely perturbations and make responses to mitigate or circumvent them altogether. As discussed later, many contemporary scholars consider learned anticipatory responses that minimize or preclude pending perturbations to be a general property of homeostasis. The realization that learning (e.g., classical and instrumental conditioning) plays a key role in homeostatic regulation placed the study of homeostasis firmly in the domain of psychology in addition to its traditional discipline of physiology. Nonetheless, others continue to consider ‘homeostasis’ per se to be restricted to reactive responding as Cannon proposed.
A fundamental tenet of homeostatic regulation is that responses work together in a coordinated fashion to wisely defend bodily parameters critical to an animal’s well being; i.e., that homeostatic regulation is purposeful. Cannon (1945, p. 108) stated this clearly, “My first article of belief is based on the observation, almost universally confirmed in present knowledge, that what happens in our bodies is directed toward a useful end. … The view that there are organic adjustments which promote bodily welfare, and consequently are useful, involves the conception that these activities are directed, …” As we discuss later, the coordination of responses was considered the result of a central controller or command center that directed the harmonious activation or deactivation of responses subserving the defense of one or another regulated parameter.
Over the last fifty or more years, a number of physiologically plausible models have been proposed that operationalize wise homeostatic control via a central command center, and in so doing, most of these models relied upon constructs taken from engineering control theory such as centrally regulated variables, set points, comparators and/or error signals or their equivalent. Cannon (1945) himself acknowledged that his advocation of purposeful coordination of responses to serve an animal’s best interest by defending critical regulated variables rendered homeostasis teleological, but the intuitive appeal of this tenet undoubtedly contributed to the enormous success of his model in generating hypotheses, stimulating research and interpreting experimental findings. However, limitations resulting from this perspective, how they have been considered by others, and how new data have resulted in novel insights about regulation, conspired to foster dissatisfaction with Cannon’s original view of homeostatic regulation.
The point is that in spite of the widespread influence of the homeostatic model, both theorizing as well as empirical research on physiological regulation have identified situations that appear inconsistent with its basic tenets. These seeming inconsistencies have spawned numerous alternative explanatory models (and a plethora of new terms) in an effort to address apparent gaps in homeostatic thinking. Hence, the literature on physiological regulation has become sprinkled with terms such as predictive homeostasis (Moore-Ede, 1986), reactive homeostasis (Moore-Ede, 1986), homeorhesis (Bauman, 2000; Bauman & Currie, 1980; Waddington, 1957, 1968), homeorheusis (Nicolaidis, 2011), homeokinetics (Soodak & Iberall, 1978), rheostasis (Mrosovsky, 1990), homeodynamics (Yates, 1982, 1994, 2008), teleophoresis (Chilliard, 1986; Chilliard et al., 2000), poikilostasis (Kuenzel, Beck, & Teruyama, 1999), heterostasis (Selye, 1973), allodynamic regulation (Berntson & Cacioppo, 2000, 2007) and allostasis (Sterling, 2004, 2012; Sterling & Eyer, 1988).
Overview of Allostasis
By far the most influential of these alternative models has been Sterling and Eyer’s (1988) allostasis, a term they defined as achieving stability through change. More specifically, Sterling and Eyer coined the term allostasis to reflect the process whereby in order to be adaptive, organisms must be able to change the defended levels of one or more regulated parameters as needed to adjust to new or changing environments. For example, in an especially stressful environment an individual might maintain, and defend, an elevated level of blood pressure relative to the level maintained in a less-stressful environment. Sterling and Eyer argued that a strict interpretation of homeostasis disallowed an organism’s defending a different level, as it went against the necessity of constancy of the internal environment maintained by an invariant set point. Sterling and Eyer explicitly incorporated learning and anticipatory responding in their description of allostasis, thus incorporating a second major departure from canonical views of homeostasis.
To summarize, the originally stated basic tenets of allostasis are that (1) the most efficient regulation is anticipatory, relying upon experience or learning from past events, (2) rather than regulated variables having invariant set points, the defended level of a regulated value can and should change to optimally cope with the demands presented by environmental changes, and (3) optimal regulation is achieved by a central command center (in the brain) that directs the activation/deactivation of the multiple responses that influence one or more regulated variables in order to arrive at the most cost-beneficial compromises. As discussed below, this might include activating a hormonal stress response that, while facilitating an animal’s overall ability to respond to a challenge to one regulated variable, may also lead to concurrent activation of responses that have opposite and competing effects on a different regulated variable.
A growing cadre of scientists has embraced allostasis, contending that it has important advantages over the canonical view of homeostasis in the way that we conceptualize physiological regulation and in the way that it relates to health and disease (e.g., Ganzel, Morris, & Wethington, 2010). An additional consequence has been that the breadth of allostasis enabled broadening the scope of regulation to include responding to psychosocial/socioeconomic stressors and how regulatory adjustments are made to minimize their impact. For example, McEwen and Wingfield (2010) believe that allostasis provides useful supplemental terminology that clarifies some ambiguities associated with the terms stress and homeostasis. This evolution of the concept of allostasis is reflected by Peters and McEwen’s (2012) description of allostasis as an “active process by which living organisms adapt to potential threats to their survival and changes in their environment (often referred to as ‘stressors’) in order to maintain homeostasis and promote survival” (p. 1). There is no indication that homeostatic principles were originally developed to address the effect of complex psychosocial stressors on physiological processes relevant to health and illness. Because of all of these reasons, allostasis seems poised to make the first serious challenge to the nearly century-long dominance of homeostasis.
Allostasis has quickly grown in popularity, with numerous scholars moving it in directions not included in its original description. Perhaps most importantly, whereas Sterling and Eyer (1988) viewed allostatic regulation as achieving optimal and efficient operation of key systems in the body with minimal energy expenditure, others have redefined allostasis as the process that occurs when maintenance of key variables comes at a considerable underlying cost, and allostasis is consequently associated with pathophysiological regulation by many (Power, 2004). McEwen and Stellar (1993) suggested the term “allostatic load” to account for the cost to the response system for maintaining a regulated variable at a value chronically displaced from its previous level by prolonged activation of compensatory effectors. Day (2005) posed the reasonable question as to whether the cost of regulation is already inherent within the model of homeostasis (i.e., homeostatic load) without requiring allostasis, the point being that while the cost of responding is important, it does not necessarily differentiate between homeostasis and allostasis.
The legitimate question has been raised as to whether the concept of allostasis provides any new value in understanding regulatory physiology (Power, 2004; Woods & Ramsay, 2007). Carpenter (2004) stated that allostasis provides no added value because it is a vaguely characterized cluster of concepts that have always been part of the ordinary conceptual basis of homeostasis. Cabanac (2006) asserted that allostasis actually blurs the understanding of the regulatory process.
A complicating factor in evaluating the potential value of allostasis is the lack of a common understanding of the concept; i.e., there is as yet no general consensus as to what the model actually is other than it goes beyond homeostasis in some specific ways. In the concluding commentary of an edited volume about allostasis, Power (2004) noted that the contributors differed considerably in their conception of allostasis. Berridge (2004) makes the similar point that because allostasis is a relatively new term it is difficult to predict which of its meanings will become most accepted. The point is that there is still considerable variability in how the term is being used, and this may partially explain why the translation of allostatic principles into clinical research and practice is only just beginning (Peters & McEwen, 2012).
Thus, the field is now confronted with two competing models of regulation that are not easily integrated. For example, Sterling (2004) suggested that allostasis should replace homeostasis (see also Le Moal, 2007) while Power and Schulkin (2012) contend that allostasis and homeostasis are compatible and complementary components of physiological regulation. At the same time, new insights based on empirical studies of regulation have challenged key assumptions of both models. Thus, a critical analysis and comparison of homeostasis and allostasis, as well as physiological regulation per se, is warranted.
The task is rendered more challenging because views of homeostasis have evolved considerably since it was first introduced. Likewise, despite its much shorter history, the understanding of allostasis is dynamically changing as the term is used in different ways by different investigators. In an attempt to clarify the strengths and weaknesses of both homeostasis and allostasis, we begin with a brief historical context for homeostasis, followed by discussion of the perceived shortcomings that prompted Sterling and Eyer to propose their new model of allostatic regulation (Sterling, 2004). We also consider contemporary views and novel findings that impact how both models might be conceptualized.
Homeostasis
In the 19th century, Claude Bernard (1870) stressed the necessity of the body’s maintaining a stable internal environment so that vital processes could proceed optimally and independently of environmental fluctuations and perturbations. He viewed the body’s organ systems as having the capacity, and indeed the responsibility, to detect and subsequently to counter, harmful deviations in one or another internal variable critical to maintain life. Hence, Bernard’s milieu intérieur was equipped with a functional network of regulatory systems which enabled the organism to thrive and reproduce despite environmental challenges. As discussed above, Cannon (1929) later coined the term homeostasis to describe the collective activity of bodily systems that monitor and protect parameters vital for life from harmful deviation. Many of these processes are unnoticed at the conscious level and rely upon behavioral, autonomic, endocrine and other responses to thwart everyday challenges to our internal world. The roster of vital parameters was initially quite small and included oxygen and energy supply to tissues (e.g., blood glucose), body temperature, and cardiovascular parameters such as blood volume, pressure and osmolality. Each of these was considered to be regulated homeostatically by several levels of sensors and effectors (e.g., Dworkin, 1993). Additional physiological variables have since been included within the homeostatic umbrella such as body adiposity, and there are others that might be considered such as sleep (or certain categories of sleep) and reward.
Sterling (2004) stated that the premise of proposing allostasis as an alternative model to homeostasis was based on the assumption that homeostasis is inadequate in certain respects, in particular on its reliance on reactive, post-hoc responses and its insistence on defending a fixed set point. We therefore briefly review these assumptions.
Constancy of Regulated Parameters
Although the opposite has often been claimed, neither Bernard nor Cannon actually considered the value of a regulated variable to be maintained at an invariant level or set point. Bernard viewed the stabilityof the internal environment as a requirement for life (see Langley, 1973), using the French word “fixité” as the descriptor. Unfortunately, this term has oftentimes been mistranslated to imply constancy rather than stability, and has led to the misconception that homeostasis exists to attain an unchanging or fixed ideal level of the regulated variable; i.e., that any variable that is homeostatically regulated has an invariant set point (Yates, 1996). For example, a leading textbook of physiology described homeostasis as maintaining a static, or constant, internal environment (Guyton, 1982).
Like Bernard, Cannon (1929) understood that regulated variables are just that, variable, and consequently selected the word homeostasis rather than homostasis to indicate similarity, thereby allowing some variability, rather than sameness. Cannon (1932) viewed homeostatic regulation as creating a relatively stable state or range within which the value of a parameter could vary, as opposed to one that was set and immobile [see a more thorough discussion of this issue by Carpenter (2004)]. As a final point, it is important to realize that stability of the regulated variable occurs because of altered activity (or instability) of the responses influencing the regulated variable. This important realization was made by Richet as early as 1890 and was incorporated by Cannon in his discussion of homeostasis (1929). The point is that whereas Sterling and Eyer (1988) and many others claim that homeostasis implies an invariant set point, it was not in fact included in Cannon’s definition; rather, and as discussed in a later section, the application of engineer control theory into consideration of biological regulation incorporated the existence of a set point, and many biologists adapted it into their own consideration of homeostasis.
Anticipatory control
Until the last few decades, homeostasis was considered by many to be mainly reflexive and post-hoc, reacting to on-going perturbations in vital systems via negative feedback-type responses. Langley (1973) described homeostasis as a self-regulating negative feedback system which maintains constancy of the internal environment. Most contemporary views understand that reliance upon negative feedback systems that do not get activated until a perturbation of the regulated variable occurs is inefficient and fails to encompass much of regulation. The point is that while negative feedback is certainly an essential component of regulation, it has become recognized that a qualitatively different approach to the defense of bodily parameters is often apparent, one that can reduce or even prevent a perturbation; i.e., based on past experience, organisms learn to make a priori responses in anticipation of an impending disturbance, mitigating their impact (e.g., Moore-Ede, 1986).
Neither learning nor anticipation was considered in Cannon’s descriptions of homeostasis. Dworkin (1993) reviewed the origins of this novel insight about homeostasis and stated that not only is learning an integral part of homeostasis, but learning is in fact the mechanism that gives the body its wisdom. The importance of learning processes in homeostasis is beyond dispute and numerous reviews are available (Dworkin, 1993, 2007; Domjan, 2005; Siegel, 2008; Woods, 1991).
Learned responses that anticipate environmental perturbations and activate countering reflexes on an a priori basis are more the rule, and they are not necessarily inconsistent with Bernard’s and Cannon’s basic premise. Adjusting the homeostatic canon to recognize the ability of organisms to take advantage of past experience in order to preempt likely perturbations simply shifts the activity of inevitable corrective responses to an earlier time point, and perhaps allows a broader range of responses to be recruited.
Nonetheless, many remain loyal to the original manifesto and have continued to interpret homeostatic regulation as relying on after-the-fact negative-feedback. Koob and LeMoal (2001) equate homeostasis with negative feedback, and Berridge (2004) contends that anticipatory responses that preemptively defend a regulated variable from becoming deviated are not homeostatic because the responses are not triggered by a perturbation of the regulated variable.
This reluctance to broaden the scope of homeostasis has been pervasive and has led to a certain amount of consternation from those who believe that homeostatic concepts should be allowed to advance with the advent of new knowledge. For example, Carpenter (2004) stated that “negative feedback … was taught as if it were the only kind of biological control system, and this folk-belief eventually invaded tertiary education” (p. 187). Carpenter considers it a travesty that homeostasis has come to be synonymous with direct negative feedback with a fixed set point, and like ourselves, believes that it is overly restrictive to limit homeostasis to achieving regulatory control solely via the action of negative feedback. Because concepts such as negative feedback and anticipatory responding were unknown when Bernard and Cannon were writing, it seems reasonable that the definition of homeostasis should be modifiable in parallel with contemporary knowledge about how regulation is achieved.
As a concept, homeostasis encompasses the idea that the internal milieu operates optimally when a given bodily variable is stabilized within a specified range, and that myriad physiological strategies exist to sense, maintain and defend the regulated variable within that range. Certainly negative feedback plays an important role as regulated variables are perturbed, but this is but one strategy that might be available in a contemporary homeostatic armamentarium. We presume that Cannon would have included these strategies as part of his wisdom of the body (Cannon, 1932). Indeed, Cannon (1945), in discussing Pavlov’s work, was aware that the brain makes what were then called psychic secretions that were not elicited by ingested food and that nonetheless were a part of the regulatory process of digestion.
An important drawback of equating negative feedback with homeostasis is that it can skew how data are interpreted. For example, if blood glucose levels decrease prior to a bout of eating, the decline can be (and has been) viewed as a deviation in a regulated variable that elicits feeding as a corrective response to restore blood glucose levels via negative feedback (Campfield & Smith, 2003; Mayer, 1955). Conversely, the same premeal drop in blood glucose can be viewed as an anticipatory regulatory response that serves to lessen what would otherwise be a larger prandial hyperglycemia. Indeed, premeal changes of glucose are associated with premeal-anticipatory secretion of insulin that serves to blunt the postprandial rise in glucose. When animals know a meal is imminent, they make numerous anticipatory responses that enable them to more efficiently cope with the homeostatic imbalance created when the food is absorbed (Berthoud, Bereiter, Trimble, Siegel, & Jeanrenaud, 1981; Drazen, Vahl, D’Alessio, Seeley, & Woods, 2006; Teff, 2011; Vahl, Drazen, Seeley, D’Alessio, & Woods, 2010; Woods et al., 1977). These so-called cephalic responses are preemptive rather than reactive in a negative feedback sense.
Strategies that may appear to be anticipatory use initial sensory information in one area of the body as an early signal of an event that could potentially perturb a perhaps critical regulated variable elsewhere in the body, and they thereby activate corrective effectors; i.e., this occurs preemptively, before the critical variable is perturbed. For example, when entering into a cold environment, temperature sensors in the skin are activated before there is any change of core temperature (the variable that is presumably regulated homeostatically); and one effect of increased activity from the relevant skin thermosensors is to trigger a thermoeffector reflex loop leading to increased metabolic rate or peripheral vasoconstriction that can stabilize core temperature and thereby buffer it from the potential effects of being in the cold environment (Kanosue, Crawshaw, Nagashima, & Yoda, 2010; Morrison & Nakamura, 2011).
A thermoeffector loop describes a neuronal circuit that is initiated by a receptor that is sensitive to temperature (or else a change in temperature) and which, when activated, sends a neural signal to a specific motor unit in the central nervous system. The motor unit in turn activates an appropriate effector response. For example, a reduction of temperature might activate a cold sensor that is neuronally wired to peripheral blood vessels causing vasoconstriction.
Perhaps the most compelling examples of anticipatory regulatory strategies involve the use of initially arbitrary cues that, by virtue of prior associative experience, become conditioned to elicit corrective responses. We (Ramsay, Seeley, Bolles, & Woods, 1996; Ramsay & Woods, 1997; Strubbe & Woods, 2004; Woods, 1991; Woods & Ramsay, 2000; Woods & Strubbe, 1994), and many others (Dworkin, 1993; Schulkin, 2011; Siegel, 2008; Siegel & Allan, 1998; Somjen, 1992), have argued that learned anticipatory processes play a major role in regulatory physiology.
But are these examples of homeostasis? We contend that homeostasis easily accommodates this anticipatory strategy and still captures the essence of the concept of regulation originally espoused by Bernard and Cannon; i.e., no one doubts the existence or in fact the necessity of anticipatory responding in physiological regulation. Rather, what is in contention is whether the original homeostatic model can be modified to accommodate this new understanding, or whether some new and perhaps qualitatively different concept such as allostasis is needed.
Scope of Homeostasis
Over the last few decades, homeostatic principles have been stretched to accommodate situations never intended by Cannon. A common misuse of the term is to invoke it to describe any activity that facilitates the return of any modifiable parameter in the body to a preperturbation level. The range in the literature for such parameters is wide and expanding; i.e., the negative feedback or reflexive aspect of homeostasis has been applied to the amount of transmitter stored in an axon terminal, or the amount of fat or glycogen stored in some cells; to cellular regulatory systems; and to numerous other physiological parameters. At another level, homeostasis has been applied to responses to the level of stress experienced by individuals or by groups of individuals; to living up to personally held values; and so on. We believe that many of these examples dilute the meaning of the term, often straying so far from the original intent of maintaining parameters whose regulated level is critical for continued existence (at least until reproductive age), and that the stretching of the definition has led to confusion, ambiguity and potential dissatisfaction with homeostasis. It is not that control of these variables is unimportant for normal functioning of cells, organs or individuals, or to their social and psychological well-being, but rather that homeostasis was never intended to explain all control processes in biology, never mind psychology.
Physiological Regulation vs. Physiological Control
In order to understand homeostasis and/or allostasis, it is helpful to clarify how the terms regulation and control have been used (see, Brobeck, 1965; Cabanac, 2006). Control refers to the process of activating (or deactivating) effector responses that regulate or stabilize a regulated variable, either by reversing a perturbation that has already occurred and been detected by a sensor (e.g., effectors activated via negative feedback control) or by preempting and thus helping circumvent or minimize an impending perturbation (e.g., effectors activated by anticipatory control). Indeed, a fundamental premise of any regulatory system is that deviations (or anticipated deviations) of regulated variables are detected and trigger effector responses which counter (or mitigate) the deviation, and it is this property that distinguishes regulation from a passive steady-state (Cabanac, 2006).
For certain vital parameters, this can be conceptually straightforward. Oxygen content of the blood is regulated (Lopez-Barneo, Ortega-Saenz, Pardal, Pascual, & Piruat, 2008) and when available oxygen falls below a threshold value, this deviation is detected by sensors in the carotid body that control sensorimotor reflexes to increase respiration and restore oxygen levels. Oxygen levels are regulated and respiration is considered the controlled effector. During exercise, respiration can be dramatically increased to defend/maintain stability of the oxygenation of the blood and tissues.
Food intake, and its relationship to body adiposity, provides a more complex example. Common lay usage often implies that food intake per se is regulated, and terms such as appetite, hunger and satiation have evolved to describe the ‘need’ to initiate or end meals. In point of fact, body adiposity or a close correlate is more likely the regulated variable. Over 60 years ago, Kennedy (1950) recognized that energy intake is normally well-matched to energy expenditure, resulting in body adiposity being maintained at relatively stable levels over long intervals. Considered in this light, food intake should be considered as a controlled effector whose activity contributes to the regulation/maintenance of body adiposity over long intervals.
Investigating individual bouts of eating (i.e., meals) can be misleading when considering the relationship of food intake with adiposity because parameters of eating can be extremely flexible in order to cope with environmental demands or competing behaviors and yet still help maintain a stable body adiposity. Collier demonstrated this in a clever series of experiments. He found that rats are quite flexible with regard to specifics of the parameters of food taking, readily adjusting their daily patterns of meal timing, size and number to accommodate environmental constraints, yet always achieving a schedule that allows maintenance of a stable body weight (Collier, 1986; Collier, Johnson, Hill, & Kaufman, 1986; Collier, Johnson, & Mitchell, 1999). Collier viewed animals as economists that constantly balance the procurement of food with numerous other behavioral activities, yet still maintaining a stable body weight. In this schema, eating is considered an effector that is activated in the homeostatic maintenance of body fat.
Most instances of food intake are preemptive with regard to meeting energy demands and maintaining body adiposity. Meals are initiated at times dictated by habit, time of day, convenience, opportunity and other factors as opposed to being reactions to deficits of available energy (Strubbe & Woods, 2004; Woods, 1991, 2009). This diversity of times when meals occur allows eating to be adaptive with regard to one’s lifestyle, and the overall pattern makes the linkage of food intake in individual meals to the regulation of body fat seem remote.
Biological influences over food intake could be implemented by when meals are begun and/or by how much is consumed once a meal is underway. As discussed above, the timing of meal onset can be quite plastic as lifestyle circumstances intervene. The preferred strategy to control daily (or weekly) food intake is consequently via changes in the amount of food eaten in individual meals, once a meal begins; i.e., any regulatory (homeostatic or allostatic) effector mechanism involving food intake is most adaptively manifest as changes in meal size. This is a highly efficient mode of control that takes advantage of a fluctuating environment and yet functions to maintain adiposity, albeit with a longer time constant than occurs for many other homeostatically regulated parameters (Ramsay et al., 1996; Seeley, Ramsay, & Woods, 1997; Strubbe & van Dijk, 2002; Strubbe & Woods, 2004; Woods, Decke, & Vasselli, 1974; Woods & Strubbe, 1994).
The same example reveals another important point about regulation. Effector responses, and especially those utilizing overt behaviors such as eating, often impact numerous bodily parameters at the same time. This can lead to situations where the response (e.g., eating) may assist in the regulation of one defended variable (e.g., body adiposity) while at the same time perturbing others (e.g., increasing blood glucose, plasma osmolality and body temperature). These conflicts posed by the ingestion of food were described by Woods (1991) as the “eating paradox.” When conditions are stable and predictable, individuals learn to make premeal responses that allow taking in sufficient calories while minimizing perturbations to other parameters. Analogously, a dehydrated and heat-stressed animal may defend blood osmolality by not using evaporative heat loss mechanisms to counter the heat stress even though this strategy results in an elevated body temperature. The important point is that regulatory effectors, and especially behavioral responses such as eating, often have multiple functions and consequences, and they thus interact in numerous complex ways with diverse regulatory systems.
Homeostasis, Allostasis and the Application of Engineering Control Theory
The adoption of engineering concepts and terminology by the life sciences and their application to physiological regulation (Wiener, 1948) caused changes in prevailing perceptions of homeostasis that have led to unfortunate contemporary misconceptions. Physiological negative feedback loops were soon described from this perspective, and control theory became firmly entrenched in biological explanations (Mrosovsky, 1990). Thus, physiological negative feedback loops were commonly described as including a reference signal or set point against which the value of the regulated variable is compared. When the value of the regulated variable aligns with the set point, effectors are not recruited. Thus, when a discrepancy occurs between the actual and the idealized set value, it is described as creating an error signal. The error signal activates an effector that moves the regulated variable back toward the value of the set point. As the corrective effector responses take effect, countering the effect of the perturbation, the regulated variable begins to return toward set point values and the deviation (error signal) is reduced.
It is usually assumed that the magnitude of corrective effector responses that are elicited is proportional to the magnitude of the error signal (called proportional control). Thus, as effector mechanisms move the regulated variable back toward reference levels, despite the continued presence of the disturbance, the error signal consequently diminishes and the compensatory responses wane in parallel. Accordingly, the regulated variable returns to close to reference levels.
This somewhat mechanical view of negative feedback resulted in the frequent incorporation of a set point as an explicit or implicit component of homeostasis. Thus, a stable set point or reference signal as used in engineering became linked to homeostatic models of physiological regulation, and while some argue that this analogy can be useful, others argued that it can also be misleading (Berridge, 2004; Gordon, 2001; Kanosue et al., 2010; Werner, 2010). For example, the Commission for Thermal Physiology (2001, p. 266–267) of the International Union of Physiological Sciences noted that the use of the term set point for temperature regulation “has evoked much confusion, as it has been used for different phenomena …[including]…. a central reference signal (which obviously does not exist explicitly in the thermoregulatory system).”
In physiology, the term set point is used metaphorically to indicate that a regulatory system operates ‘as if’ there was an engineering type of set point or reference signal (Hardy, 1965); i.e., a set point is a hypothetical construct that is inferred by assessing whether an animal defends a given value of one or another variable using behavioral and/or physiological responses. When the regulated variable is at a value where all effectors are at minimal or basal levels of activity, this would be considered the null point or null zone and would correspond to what is metaphorically considered as the set point.
Furthermore, rather than being ‘set,’ these set points are often adjustable and can be altered by diverse naturalistic factors (e.g. circadian and seasonal changes, maturation, hormonal status, nutrition, stress) (Hammel, 1970; Satinoff, 2005). Common examples of regulation with an adjustable set point include the increased and well-defended level of body temperature (fever) when an individual has an infection and the increase in otherwise well-regulated body fat stores as some species prepare for migration or hibernation. Mrosovsky’s (1990) thoughtful monograph on this topic defines changes in the level of the variable around which regulation occurs as rheostasis. As an aside, it should be noted that 25 years prior to Sterling and Eyer’s (1988) assertion that allostatic regulation was needed because homeostasis is limited to having an invariant set point, others had already modified the model of homeostatic regulation to include an adjustable set point (Hammel, 1965, 1970; Hammel et al., 1963).
Investigation and consideration of thermoregulation has led to the development of several physiologically plausible neuronal models that endow the homeostatic system with the ‘as if’ appearance of a set point (Bligh, 1972, 1998, 2006; Boulant, 1981, 2000, 2006; Hammel, 1965; Hammel, Jackson, Stolwijk, Hardy, & Strømme, 1963; Wyndham & Atkins, 1968). An important dictum of these homeostatic models was that the sensory input from diverse thermal receptors located in the skin and throughout the body converges on an integrated neural circuit in the central nervous system so that elicitation of effector activity can be centrally coordinated. It was argued that if there were no central coordination, opposing effectors could operate concurrently and in competition with one another. For example, Bligh (1998) argued that a general principle of homeostatic regulation is that the activities of two effectors with opposite influences on the regulated variable are not concurrently active.
Thus, these neuronal models were designed so that sensory information indicating a challenge to the regulated variable not only provides an excitatory input that activates corrective effector responses, but also provides inhibitory input to the opposing effectors. Bligh (1998) described this as being analogous to the reciprocal cross-inhibition system described by Sherrington (1906) that prevents opposing flexor and extensor muscles from contracting concurrently. This proposed design feature prevents the uneconomical concurrent activation of opposing effectors while defending thermal stability, and it fits with the observation that opposing effectors are not found to be concurrently active (e.g., shivering to increase heat production while simultaneously sweating or panting to increase heat loss).
These models were also able to account for an adjustable set point (Hammel, 1965, 1970; Hammel et al., 1963) by allowing additional signals from other systems to influence the elicitation and magnitude of corrective effector responses at the thermoregulatory coordinating interface in the central nervous system (CNS). This additional design feature allowed the level of the set point to be adjustable while maintaining the key feature of homeostatic regulation that detects challenges to, or deviations of, regulated variables and triggers appropriate effector responses to stabilize the variable. While much of this work was based on the thermoregulatory system, these principles are considered to be general and relevant to understanding other homeostatically regulated systems as well (Bligh, 1998, 2006; Boulant, 2006; Cabanac, 2006). Further, although succumbing to the lure of control theory terminology, all of these models of homeostasis nonetheless recognized that physiological regulation has movable set points.
A model provided by Gordon (2009) provides an excellent example that illustrates how thermoregulation is viewed within this sort of homeostatic framework. He points out that an interpretational complexity arises in studies of homeostasis because an observed change in a regulated variable can result either from an environmental disturbance that forces the variable away from its defended value (e.g., entering into a warm room) or else from a rheostatic adjustment of the set point value that causes effectors to move the variable toward a new level (e.g., a bacterial infection eliciting fever).
Gordon (1983, 2005, 2009) refers to changes in a regulated variable that reflect a deviation away from a defended level as being ‘forced’ while changes in a variable that are due to a rheostatic adjustment of the defended value are termed ‘regulated.’ Gordon proposed that forced and regulated changes could be distinguished by inference because behavioral and physiological homeostatic effectors are activated to oppose or compensate for a forced change whereas effector mechanisms act in the opposite direction to facilitate a change that is regulated because of a rheostatic set point adjustment. This is consistent with the commonly accepted attribute of homeostasis discussed above that behavioral and autonomic effectors work in concert to defend the set point (e.g., Bligh, 1998; Cabanac, 2006). Gordon’s model (Figure 1) illustrates the coordination of behavioral and autonomic effectors in moving the measured value of the regulated variable toward the target value of an adjustable set point.
Figure 1.

“Summary of behavioral and autonomic responses of a homeotherm when subjected to manipulation of body and set-point temperature. Graphs on the left represent the relationship between the set point (dashed line) and core temperature (solid line).” [This figure and figure legend are reprinted with permission (Gordon, 2009, p. 894).]
Figure 1 depicts a homeostatic thermoregulatory model that employs adjustable set points and coordinated effector activity. The left column illustrates the relationship between the regulated variable of core temperature (solid line) and the hypothetical centrally determined set point (dashed line). The middle column illustrates how the behavioral effector in a thermal gradient (i.e., the animal’s actively moving to a warmer or cooler end of the gradient) defends the set point, and the third column illustrates how autonomic effectors defend the set point [i.e., metabolism (heat production effectors), skin blood flow (heat loss effectors), evaporation (e.g., heat-loss effectors of sweating, panting, saliva spreading), and piloerection (heat loss effectors).]
In the top row, core temperature is depicted as having the variability that would typically be observed in the null zone and when the effectors are all functioning at the most energetically efficient or basal levels to maintain normothermia. Measured changes in core temperature (Rows 2–5) illustrate how behavioral and autonomic effectors act in coordination to defend the set point by correcting the discrepancy between the set point and the actual core temperature. It is important to note that in Gordon’s model, both behavioral and autonomic patterns of responses are either collectively warming or collectively cooling the body, consistent with the requirements of a homeostatic model that uses an adjustable set point. Further, and also consistent with homeostasis, the five patterns of autonomic and behavioral effector activity depicted are the only ones possible; i.e., concurrent activation of opposing effectors is prohibited. If concurrent activation of opposing effectors did occur, it would require a different, non-homeostatic model of regulation.
The necessity of a central integrator for physiological regulation
A key aspect of most models of regulation is the premise that the central nervous system (CNS) contains an integrator/controller that coordinates responses with regard to each regulated variable’s set point, and how best to cope with the magnitude of ongoing or potential perturbations. In 1986, Smullin [as discussed by Bligh (1998)], constructed a functional physical model of thermoregulation to test the theoretical validity of the neuronal implementation of an adjustable set point and central integrator for homeostatic thermoregulation and that prevented concurrent activation of competing effectors. Experiments using this model demonstrated that efficient regulation could be achieved using a physiologically tenable design.
Models of thermoregulation such as this were designed to resolve concerns arising from the use of engineering principles to explain physiological temperature regulation; i.e., they were designed to function in a way that was compatible with the current understanding of how homeostasis was thought to operate. That said, many concerns about this approach to understanding physiological regulation have been raised (Werner, 2010). Yates (1996) argued that engineered physical models “… illustrate nothing about biology because programmers are ghosts in those machines, and it is the designer’s intentionality, not that of the machine, that is observed” (p. 683). Yates further states that the “failure of models based on machines that do work or process information to explain homeostasis has provoked an intellectual crisis in science” (p. 683). While this position may sound extreme, it emphasizes that demonstrating that a biologically plausible system can be designed to achieve a pre-conceived homeostatic outcome does not provide evidence that such an approach actually exists in physiology. The value of such models is that they provide a well-described hypothesis that is suitable for testing. The important point is that whereas much theorizing can occur about how a physiological system could be designed and constructed to appear as if control theory concepts are applicable, they lack biological validity in and of themselves.
In 1978, Satinoff challenged the widely accepted belief that thermoregulation depends on a central neural integrator. Citing findings by Carlisle and Ingram (1973), she argued that a single central integrator is unlikely to exist because when temperatures in the spinal cord and hypothalamus were manipulated in different directions, opposing effector responses were concurrently elicited. In accord with the standard view of homeostasis, Satinoff (1978) recognized that this was anomalous because “in a normal animal in a hot or cold environment, one would never see such opposing behaviors” (p. 20).
Nonetheless, in light of these experimental findings of concurrently active opposing effector responses, Satinoff concluded that the model of thermoregulation must be modified to include separate integrators for each individual thermosensor-effector loop, rather than having a single overall central integrator. In her model, temperature regulation would be the arithmetic sum of activity in whichever thermoactive sensor-effector loops were turned on at any point of time, whether pushing temperature in the same or different directions. Satinoff’s (1978) analysis began a movement away from the traditional homeostatic conceptualization of thermoregulation as being a unified model with a single central controller to achieve effector coordination. The current view of the thermoregulatory system’s structure and function was well described by Romanovsky (2007b), and it is much different than the traditional, unified homeostatic model outlined above that incorporates engineering control theory.
In point of fact, many contemporary models have abandoned the concept of a coordinated central controller. Mammalian body temperature regulation is thought to have evolved utilizing multiple thermoeffector loops, each with its own independent central control (McAllen, Tanaka, Ootsuka, & McKinley, 2010). From that perspective, the structure and function of complex regulatory systems, including thermoregulation, are considered to have been created through evolutionary ‘tinkering’ (Jacob, 1977). As Jacob eloquently described, “Evolution does not produce novelties from scratch. It works on what already exists, either transforming a system to give it new function or combining several systems to produce a more elaborate one” (p. 1164) …. “… living organisms are historical structures: literally creations of history. They represent, not a perfect product of engineering, but a patchwork of odd sets pieced together when and where opportunities arose” (p. 1166). It follows from this that the thermoregulatory system is not organized in the way that an engineer might ideally design (e.g., McAllen et al., 2010). This view contrasts sharply with the model Bligh (1998) designed that was intended to provide a biologically feasible method for coordinating effectors so as to prevent concurrent opposing effector activation and to provide the functional attributes of an adjustable set point, both of which were believed to be integral to homeostasis. Kanosue and colleagues (2010) present a contemporary homeostatic model of the thermoregulatory system that fits the data and that has no reference signal (or set point), no error signal and no overall coordinator for the thermoeffectors.
As reviewed by others (McAllen et al., 2010; Romanovsky, 2007b), there has been reluctance to broad acceptance of the view that thermoregulation occurs via independent control circuits without central coordination. McAllen and colleagues (2010) described the problem succinctly, suggesting that the reluctance stems from the fact that thermal regulatory mechanisms ‘appear’ to function seamlessly together as a precisely coordinated control system seeking a set point. Despite this appearance, Romanovsky (2007a, 2007b) explains that the apparent coordination of thermoeffectors typically observed does not require or imply a common central coordinator as had been previously proposed, but instead can result from the common influence of the regulated variable (core temperature) acting at the sensor of each independent thermoeffector loop. For example, as a thermoeffector’s activity alters core temperature, the newly altered level of core temperature necessarily influences the activation/cessation of other thermoeffector mechanisms, each according to its own thresholds. While this example is based on thermoregulation, Romanovsky (2007b) makes the important point that the principles are relevant and apply to physiological regulation for other regulated variables.
A regulatory system consisting of diverse sensor-effector circuits that operate independently has significant implications for how set points are understood in homeostatic regulation. Without a unified control system in the CNS to coordinate effector activity in defense of a common set point, regulation instead results from the sum of influences of each independent effector’s action. Each effector loop has its own threshold that determines its individual activity, and thus each effector may be described as having its own ‘set point’ reflecting the sum of the individual thresholds of the activated sensory neurons (Kobayashi, Okazawa, Hori, Matsumura, & Hosokawa, 2006; Romanovsky, 2007b; Werner, 2010). A thermoeffector loop provides an illustrative example (e.g., Kobayashi et al., 2006; Romanovsky, 2007b; Tajino et al., 2011). If a cold temperature exceeds the activation threshold of a receptor located on the sensory neuron of a thermoeffector loop, the sensory neuron changes its basal firing rate in accord with stimulus intensity and sends its signal via a neural circuit to a thermoeffector which will respond if it receives sufficient inputs (Tajino et al., 2011).
Figure 2 depicts a single regulatory sensor-effector loop. This view of a sensor-effector loop allows for integration of afferent information as well as for modulatory influences from other inputs to that circuit. However, individual thermoeffector loops are largely independent from one another, and there is not a central controller that receives all of the relevant afferent signals from the thermoeffectors in order to coordinate the effector activity. Kobayashi and colleagues (2006) suggest describing the sensory neuron as a ‘comparator’ rather than simply a sensor. This is because the decision about whether a neural signal is generated is determined by the activation threshold of the receptors on that sensory neuron.
Figure 2.
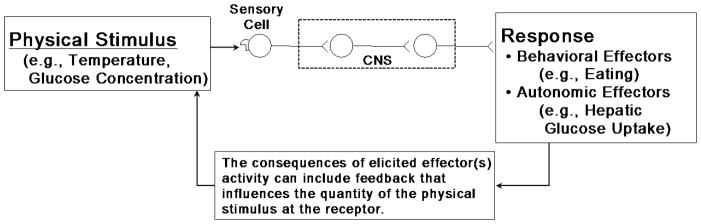
Schematic representation of a single regulatory sensor - effector loop. Although the sensory cell is depicted in the periphery, they also exist within the CNS. The activation threshold of the receptor on the primary sensory neuron is triggered when the quantity of the physical stimulus is sufficient. Note: all such loops involve the CNS and are multi-synaptic.
Note that this narrow definition of a set point is quite different from the commonly held belief that there is an integrated central set point that serves to coordinate the action of all of the thermoregulatory system’s different effectors with the goal of defending a common specified value (Romanovsky, 2007b). Instead, the stabilized level of a regulated variable is better conceived as a ‘balance point’ that reflects the summed action of all of the influences on that variable (Romanovsky, 2007b; Werner, 2010; Kanosue et al, 2010). The point is that whereas in common parlance the term set point refers to a hypothetically optimal level of a parameter that is monitored, maintained and defended (e.g., 37° C for body temperature), in actuality body temperature is the consequence of multiple individual and independent thermoeffector loops, each with its own threshold (or ‘set point’) for activation, whose collective activity results in a value that reflects the current conditions and for which there is no central integrator that coordinates effector activity. Figure 3 depicts a balance point model of homeostasis that yields a pattern of effector activity that appears as if it might be the result of purposeful coordination by a central command center but instead occurs without monitoring the magnitude of a presumed regulated variable, without using a comparator to evaluate the regulated value relative to a set point value in order to generate an error signal, and in fact without any central coordination of effector activity.
Figure 3.

Schematic describing how a homeostatic model explains thermoregulatory effector activity when body temperature (Tcore) is comparably elevated due to being in a high ambient temperature or an infection-induced fever. Panel A. Schematic depicting how the activity of multiple independent sensor-effector loops contributes to the balance point of a regulated variable. Panel B. Two different patterns of effector activity (adjusted for basal activity levels during normothermia) that result from five different thermo-effector loops. When ambient temperature is high, cooling responses are activated; during a fever, warming responses are activated. As in Gordon’s homeostatic model (see Fig. 1), the same pattern of coordinated effector activity occurs to move Tcore in the same direction, but this approach does not measure the value of the regulated variable and compare it to a set-point value.
This view of homeostasis clarifies how to interpret the term ‘regulated variable.’ There is not a unified, integrated control system that is constantly calculating and monitoring the value of an assumed regulated variable. Rather, what is considered to be the regulated variable reflects the scientific community’s current understanding of the functional outcome of a regulatory system (e.g., defense of core temperature, body adiposity or blood glucose). The term provides an organizational construct to categorize the regulatory function of multiple effectors acting at once, and the consequent value that is attained reflects not a set point and rather a balancing or settling point. Therapeutic strategies would not be intended to target a non-existent central representation of a regulated variable, but would instead shift the balance point of a regulated variable via targeted alterations in effector loop activity.
It should be noted that the suggestion that a term such as balance point or settling point be used in the context of homeostatic regulation is not new (Berridge, 2004; Booth, 2008; Hardy, 1965; Romanovsky, 2007b; Wirtshafter & Davis, 1977). Kanosue and colleagues (2010) point out that it is cumbersome for ‘balance point’ theory to describe homeostatic processes, because the notion that core temperature is determined as the balance of active and passive processes lacks a clear delineation of regulation. For example, in the case of fever versus exposure to excessive ambient heat, both situations can result in an elevated balance point for core temperature. Those studying physiological regulation are interested in understanding how the effector responses differ in these situations (see different patterns of effector activity for an elevation in core temperature in Figures 1 and 3). However, simply knowing that the level of the balance point has increased is uninformative about underlying effector activity. Thus, additional clarification is needed to describe the influence of the regulatory effectors on the regulated variable in these different situations.1
Another problem hinders the adoption of terminology that correctly describes the current view of homeostatic regulation. Traditional views of homeostasis based on characteristics of engineered controllers have been so influential that the concepts are deeply entrenched in science and medicine. Thus, the use of terms like reference signal, set point, error signal, and the belief in the existence a single central coordinator for each regulatory system, are commonplace. In the field of thermoregulation, Romanovsky (2007b) states that “… in the minds of biologists, physicians, and students, the term set point is strongly, perhaps inseparably associated with the reference signal of a unified thermoregulatory system” (p. R43). We agree, and believe that the constraints imposed by this commonly held misunderstanding of homeostasis may be an impediment to achieving a deeper understanding of physiological regulation.
As a final point on the evolution of homeostatic concepts, all of the same points can be made about allostasis. Allostasis has its roots set firmly in homeostasis, and the same control theory terms and models are commonly used to explain allostatic regulation. In fact, two of the most fundamental features of allostasis are that it has an adjustable rather than an invariant ‘set point’ and that regulatory responses are coordinated by a ‘central controller.’ Based on contemporary views of regulation, and the discussion above, we believe that a balance-point model should be applied to both homeostasis and allostasis.
Experimental Findings that Are Inconsistent with Homeostasis
Hammel (1965, 1970), who is credited with the concept that an adjustable set point is an important component of homeostasis, nonetheless recognized that commonly-held homeostatic and regulatory concepts need revision (1988). Two of Hammel’s major concerns (1989, 1990) are discussed below as they relate to findings that he believed were inexplicable using a homeostatic model based on negative feedback.
Hammel (1990) was perplexed by the findings of Johansen (1961) who had cold-challenged armadillos and found that step decreases in ambient temperature resulted in persistent and excessive increases in core temperature and oxygen consumption that exceeded the values that had been present before the perturbations. “These studies demonstrate a conspicuous overcompensation to the cold stressing armadillos. In other words, the body temperature is drastically raised at the expense of an increased metabolism.” (Johansen, 1961, p. 133). For example, armadillos exposed to ambient temperatures of −5° C shivered and generated sufficient heat to raise rectal temperature up to 3° C higher than the temperature found in a neutral thermal environment. Hammel was intrigued by this finding, stating “This is an unusual response when compared with the response of most endotherms to cold. The magnitude of the response seems excessive and we may ponder the adaptive value of it. More interesting to me, however, is to inquire about the thermal stimuli which initiate and sustain the response. …. The core temperature of many endotherms decreases when exposed to cold. Removing heat from the core by natural cold exposure or by an internal heat exchanger and, thereby, lowering the core temperature, elicits shivering and/or nonshivering thermogenesis in all conscious, awake endotherms without any known exception.” (Hammel, 1990, p. 175). Because core temperature was assumed to be the homeostatically regulated variable, a deficit in core body heat was believed to be the critical signal driving the shivering response. Thus, Hammel found it odd that shivering and other heat-producing effector responses would be sustained as core temperature increased above control values.
Mercer and Hammel (1989) subsequently found that the substantial increase in metabolic rate and core temperature was not a temporary dynamic response as the animals maintained their elevated metabolic rate and increased core temperature throughout a 3-hour exposure period. Furthermore, they determined that armadillos had core thermosensitivities similar to those of other homeothermic mammals, thus ruling out the possible explanation that armadillos are insensitive to changes in core temperature.
Based on thermoregulatory research with the armadillo and other similar findings, Hammel wondered how a physiological disturbance could trigger effector responses that: 1) continue to be sustained even though the value of the regulated variable has been returned to its pre-perturbation level, and 2) are so large that they result in a persistent overcompensation of the regulated variable (Hammel 1989, 1990; Hammel & Simon, 1994; Mercer & Hammel, 1989). In canonical views of homeostasis, perturbation of a regulated variable causes an error signal that elicits effector responses that counter or compensate for the disturbance of the regulated variable. As compensatory responses take effect, the regulated variable returns toward its homeostatically-defended pre-perturbation level such that the deviation is reduced and the error signal dissipates. Without the error signal, the effector responses should not be sustained and therefore should not result in an overcompensation. Furthermore, elevation in core temperature above the normally defended level should activate effectors to lower core temperature while inhibiting effectors that increase temperature, thereby preventing concurrent competition between opposing effectors.
Mercer and Hammel (1989) offered several possible explanations to account for this observation, one of which included the elevation of an adjustable set point for core temperature. However, as previously explained, regulation appears to result from a balance-point model and not from a set-point model that invokes hypothetical components (e.g., a central coordinator, a monitored regulated variable, an error signal, a set point) whose existence has not been empirically supported. Thus, Mercer and Hammel’s adjustable set point explanation seems unlikely. A contemporary interpretation might describe the armadillo’s core temperature as settling at a new elevated balance point in the cold environment as a result, at least in part, of thermal sensors located near the body surface activating thermoeffectors that elicit heat production and/or reduce heat loss.
Although contemporary views of both homeostasis and allostasis incorporate the idea that learning enables anticipatory activation of effector responses that can diminish or eliminate the perturbation of the regulated variable, this also does not offer a suitable explanation for overcompensation. While learned responses play an important role in regulation by offsetting the effect of the disturbance, these learned responses do not overcompensate for the disturbance (Dworkin, 1993). Learned responses can shift a dependent variable in the direction opposite to that caused by the disturbance, but only if the response is elicited and the disturbance is smaller than anticipated (e.g., conditioned responses elicited by a conditioned stimulus) or if the disturbance diminishes more quickly that the learned responses dissipate (e.g., as occurs in drug withdrawal) (Dworkin, 1993, Siegel, Baptista, Kim, McDonald, & Weise-Kelly, 2000; Ramsay & Woods, 1997). While past experience with cold exposure could have enabled the armadillos to elicit a prompt and enhanced effector response, learned responses would be unlikely to explain such large and prolonged overcompensations of core temperature in the continued presence of cold exposure.
These thermoregulatory findings with the armadillo foreshadow several issues that pose problems for homeostatic interpretations and which we believe call attention to aspects of regulation that are better incorporated with allostasis. First, a fundamental tenet of homeostasis is that effectors operate in an efficient and apparently coordinated manner. Accordingly, when confronted with a cold challenge, the armadillo should respond in an energetically efficient manner that maintains core temperature without producing more heat than is necessary to offset the cold challenge. Yet, the armadillo persistently overcorrects its core temperature at the expense of an increased metabolism. As we discuss later, an emerging view of allostasis includes overactive effector responses that can shift the balance point of a regulated variable in the direction opposite to that caused by the ongoing disturbance.
Second, the armadillo’s prolonged elevation of metabolic heat production is metabolically costly beyond what is required to defend core temperature against the cold ambient temperature. While such metabolic inefficiency is not compatible with a homeostatic view, excessive costs associated with driving effector responses sufficiently to shift the balance point of core temperature is a concept that has been linked with the allostatic perspective (i.e., as allostatic load).
Third, concurrent activation of opposing effectors could be occurring. It is possible that cold ambient temperatures activate peripheral sensors of thermoeffector loops that elevate core temperature. In turn, the elevated core temperature activates opposing thermoeffector loops located in the core and together these opposing influences balance at the observed core temperature. While inefficient concurrent activation of opposing effectors is inconsistent with homeostatic principles, a balance point model of allostasis can accommodate it and recognize that this inefficiency contributes to allostatic load on effector systems.
The following sections expand upon how these and other examples of apparently non-homeostatic forms of regulation are being included under an allostatic model.
How Does the Concept of Allostasis Move Us Forward in Accounting for Findings that Are Inconsistent with Homeostasis?
The original use of the term allostasis was in the context of the regulatory adjustments that occur when humans and other animals must contend with perceived or anticipated chronic social demands and stresses (Sterling and Eyer, 1988), and this emphasis remains a major focus (Schulkin, 2011). The idea was that psychosocial stressors contribute to adverse health outcomes via chronic actions exerted through, and upon, regulatory systems. Stress has in fact been described as a process that initiates and maintains allostasis. Indeed, in the psychobiological literature the concept of allostasis is heavily entangled with the concept of stress and how the body copes with stressful events. For example, Ganzel and colleagues (2010) describe the prototypical stress hormones, cortisol and epinephrine, as being principal physiological mediators of allostasis. While the problems inherent in bundling stress and allostasis have been well-aired elsewhere (Day, 2005), it is nonetheless worth discussing some of the implications as they pertain to comparisons of homeostasis and allostasis.
An observed change of the level of a regulated variable has been described by some as the sine qua non of allostasis; e.g., “Regulation around an altered apparent steady-state is the essence of allostasis” (Goldstein & Kopin, 2007, p. 116), “An allostatic state can be defined as a state of chronic deviation of the regulatory system from its normal (homeostatic) operating level” (Koob, 2010, p. 10). Psychosocial stress has been suggested as one of the major causes for a change in the level of a regulated variable. For example, if blood pressure becomes chronically elevated by continued psychosocial stress, the change could be interpreted as an appropriate allostatic adjustment to meet the situational demands, but it may also have undesirable health consequences, especially if the hypertension becomes severe and/or chronic. Allostatic regulation was suggested to employ an adjustable set point and so a chronic elevation of blood pressure would be interpreted as an adaptation to an increasingly stressful environment. Alternatively, an adjustable set-point model of homeostasis would view this chronic elevation of blood pressure as either a stress-induced ‘forced’ hypertension or a stress-induced ‘regulated’ hypertension (e.g., see Figure 1). Because contemporary models of regulation do not include central controllers and/or set points, chronic hypertension would be interpreted as a new balance point that reflects the influence of stress on effector responses. As explained later, distinguishing between allostasis and homeostasis will depend on how effector responses interact to influence the hypertensive balance point.
In a thoughtful review, Boulos and Rosenwasser (2004) suggested limiting what could potentially be an overly inclusive definition of allostasis to those persistent changes in the defended level of regulated variables that are believed to result from psychosocial stress or the chronic disruption of circadian timing (e.g., shift work). Their recommendation to restrict allostasis to changes in defended levels that result from psychosocial stressors was based on the recognition that there are so many diverse examples of changes in the defended value of regulated parameters that allowing such a broad inclusion criterion for allostasis might diminish its potential usefulness.
Physiological research on homeostasis has typically focused on delivering well-defined disturbances to regulated variables in order to study the corrective responses that are elicited and how these responses can occur in anticipation of a potential disturbance. Having a functional homeostatic regulatory system has obvious relevance to health because unopposed challenges to critical regulated variables such as body temperature, blood osmolality, blood volume, blood oxygen, or blood glucose threaten viability. Thus, the focus of homeostasis has been primarily on life-maintaining regulatory physiology, and there is no indication that homeostatic principles were originally developed to address the effect of complex psychosocial stressors on physiological processes relevant to health and illness.
In contrast, consider the nature of low SES as a psychosocial stressor. McEwen and Gianaros (2010) describe how low socioeconomic status (SES) can be considered a psychosocial stressor that is linked to poor health. They contend that the stress of having a low SES, with its myriad of correlates, has physiological consequences that include responses of the autonomic nervous system, the hypothalamo-pituitary-adrenal (HPA) axis, and the cardiovascular, metabolic and immune systems. The responses made by these diverse systems represent allostatic adaptations to the stress of low SES that have both beneficial and adverse health consequences. Just as homeostatic functioning is relevant to health, the allostatic adaptations caused by low SES also have health consequences that are important to understand. Further, these stress-induced responses are typically considered to be relatively non-selective and to involve multiple systems (e.g., Day, 2005), although responses to stress can be more specific than is commonly appreciated (Goldstein, 2013). Unlike stress responses, homeostatic regulatory responses are more selective and targeted at correcting the eliciting or impending perturbation. Thus, we agree with Boulos and Rosenwasser (2004) that allostasis offers a useful new perspective when considering the effects of complex psychosocial stress that goes beyond typical homeostatic approaches.
As discussed earlier, physiological descriptions of homeostatic regulation based on engineered control systems are being discarded and replaced, in large part due to basic research in the field of thermoregulation. Nonetheless, it is still commonplace for homeostasis to be described using concepts and terms that came from an analogy to engineering (e.g., set points, reference signals, error signals, and centrally coordinated homeostats/comparators), which, unfortunately, are misleading. Romanovsky (2007b) bemoaned the use of this kind of terminology, stating it is now likely inseparably linked in the minds of many in the scientific community with the older view that homeostasis functions via a centrally unified and coordinated regulatory system.
This also appears to be the case for much of the theorizing about allostasis (e.g., Ahmed & Koob, 2005; Berntson & Cacioppo, 2007; Goldstein, 2004, 2008, 2012; Le Moal & Koob, 2007). Because allostasis is a derivative of homeostasis, the adoption of homeostatic concepts and terminology should come as no surprise. To describe current perspectives of allostasis, it is consequently necessary to use many of the same terms, although we will attempt to clarify them in terms of contemporary understanding. We are sensitive to the concern that using outmoded homeostatic concepts to describe allostasis may hinder its progress as a useful theoretical construct.
The influential work of Koob, Le Moal and their colleagues illustrates how apparent changes in set level can be interpreted using an allostatic model of drug addiction. Their model is based on the reasonable premise that reward and hedonia are physiologically regulated and that sensor-effector loops exist that influence the value of these variables (Koob, 2004, 2009; Koob & Le Moal, 1997, 2008; Solomon, 1980; Solomon & Corbit, 1974). The rewarding pharmacological effects of addictive drugs are thus considered to reflexively activate anti-reward effectors that limit excessive activity in the reward system. However, according to Koob and LeMoal, brain anti-reward effectors can become overactivated with continued drug taking, and create an overcompensated state of the regulated variable characterized by a chronic deviation of the reward set point (2008).
The overcorrection caused by the brain anti-reward systems results in an aversive state that motivates increased drug taking behavior to obtain drug effects that oppose it (Koob & LeMoal, 2005, 2008). Taking additional drug only temporarily ameliorates the aversive reward deficit state because it also causes further overactivation of anti-reward effectors causing further allostatic dysregulation. Koob and LeMoal describe this as a cycle of spiraling dysregulation of brain reward/antireward mechanisms, and that as it progressively increases, it results in compulsive drug use. Stated another way, as drug taking continues, the balance point for the level of reward/hedonia keeps shifting farther and farther from its initial value due primarily to the progressive overactivation of brain antireward effectors which interact with behavioral effectors seeking drug reward. As discussed below, this model of addiction provides excellent examples of how allostasis can be differentiated from homeostasis. In this model, allostatic dysregulation results from the overactivation of regulatory effector responses that lead to a negative reward state. Koob and Le Moal (2004) state that when the drug-induced disturbance of the reward system is appropriately balanced by a counter-regulatory response, homeostasis is maintained and allostasis does not develop. However, when the counter-adaptive opponent process becomes excessive and overcompensates for the drug’s rewarding effects, allostasis develops as reflected by the concomitant shift in the reward balance point into an aversive (negative) state.
From a homeostatic perspective, it is inexplicable why a regulatory response that is intended to defend hedonic homeostasis would cause a persistent overcorrection that places the individual in a negative reward state. However, in contrast to what would be expected to occur based on traditional homeostatic explanations, this shift of the balance point into a negative reward state is thought to reflect the influence of responses that do not readily shut off once engaged (Koob, 2010), and/or responses that exhibit “residual hysteresis,” reflecting an allostatic aspect of addiction (Koob & Le Moal, 2004, p. 158). The observation of overly intense and/or persistent responses is commonly attributed to allostasis (e.g., Ganzel et al., 2010; Karatsoreos & McEwen, 2011; McEwen & Gianaros, 2010; Peters & McEwen, 2012).
Anticipatory activation of effector responses (e.g., autonomic, neuroendocrine, stress responses) has been suggested as a possible explanation for these dysregulatory characteristics of allostasis (Koob, 2010). Because learned effector responses can be activated prior to the occurrence of an anticipated disturbance, as occurs with cephalic insulin secretion for example, it follows they can cause dysregulatory effects of their own if the anticipated disturbance does not occur (Lê, Poulos, & Cappell, 1979; Mansfield & Cunningham, 1980; Woods, 1991). However, this type of error of is consistent with and in fact predicted by principles of homeostatic regulation in contemporary usage, and it is different from the dysregulation that results from allostatic overcorrections because the latter occur in the continued presence of the disturbance.
An important insight in this interpretation of how allostasis differs from homeostasis is that motivational consequences arise that encourage drug-taking behavior. The overactive anti-reward effectors are hypothesized to shift the hedonic equilibrium into an aversive reward-deficit state that can be countered by self-medicating with the drug, both to alleviate the reward-deficit state and perhaps to obtain the drug’s rewarding effects as well. Thus, in this conceptualization, two opposing effectors are operating simultaneously. The overactive anti-reward effectors decrease reward and shift the balance point in the anhedonic direction, whereas the behavioral effector increases drug intake and reward, which reduces the aversive state thereby strengthening these behavioral drug-taking responses via negative reinforcement (Koob, 2009). Concurrent competition between these two opposing effector systems creates an allostatic vicious cycle model of addiction (Edwards & Koob, 2010; Koob & Le Moal, 2001, 2008).
As discussed above, concurrent activation of competing effector systems was purposefully excluded from most homeostatic models of physiological regulation. Gordon’s (2009) description of homeostatic thermoregulation (see Figure 1) illustrates how behavioral and autonomic effects work together rather than in competition. In contemporary homeostatic models, the null zone refers to the balance point (or zone) of a regulated variable when its controlling effectors are at minimal or basal activity. Of course, when perturbation of a homeostatically regulated variable occurs, effectors are activated that require energy and there is undoubtedly some associated cost (homeostatic load). Yet, homeostasis is believed to minimize these costs by not having opposing effectors in concurrent competition. An allostatic model of regulation that allows concurrent activation of competing effectors will also settle at a balance point but it does so inefficiently and at greater cost (allostatic load) (Figure 4). This view of allostasis was described by Goldstein (2004, p. 108) using the analogy of an overheated apartment during the change in seasons from winter to spring; “Continued use of the furnace and air conditioner in opposition to one another, an example of an inefficient ‘allostatic state,’ consumes fuel and contributes to wear and tear on both pieces of equipment.”
Figure 4.
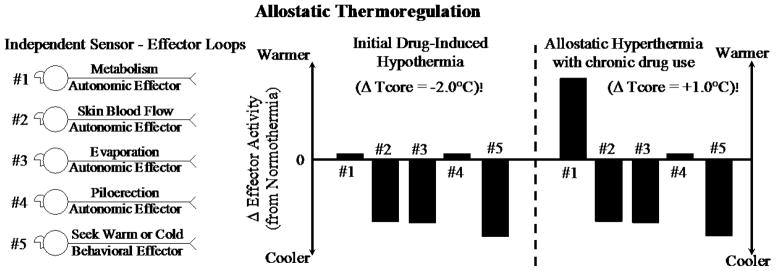
Schematic describing how an allostatic model explains thermoregulatory effector activity when body temperature (Tcore) drops during an initial drug administration but then reverses and eventually becomes an allostatic hyperthermia (sign reversal) over repeated drug administrations. The schematic in Figure 3 (Panel A) depicts how the activity of multiple independent sensor-effector loops contributes to the balance point of a regulated variable. The effector activity (adjusted for basal activity levels during normothermia) that results from five different thermo-effector loops changes with chronic drug use. When the drug is initially administered, cooling responses are activated which cause hypothermia. However, with repeated drug use, Tcore becomes hyperthermic which is due to the increase in metabolic heat production that overcompensates for the concurrent effectors that favor cooling.
Based on this analysis, the concept of allostasis as suggested by Koob and Le Moal (2001, 2004, 2005, 2008) is better able than homeostasis to account for dysregulations that result from overactive or persistent effector responses that overcorrect a regulated variable (i.e., shift the balance point) in the direction opposite to the disturbance. Excessive effector activity can cause overcorrections during the presence of the disturbance or, if the effectors do not shut off properly when the disturbance ceases, continued effector activity can cause a persistent shift in the balance point of the regulated variable in the absence of the disturbance. The allostatic vicious cycle model of addiction (Edwards & Koob, 2010; Koob & Le Moal, 2001, 2008) allows overactive anti-reward effectors to concurrently compete with behavioral drug-taking effectors that increase reward, which is incompatible with a homeostatic interpretation. As described in the next section, others (Colpaert, Deseure, Stinus, & Adriaensen, 2006; Ossipov et al., 2004) have also suggested that concurrent competition between opposing effectors may be a mechanism underlying drug addition. We believe that a strong rationale for the adoption of allostasis as an alternative regulatory model can be made if it is able to explain findings that are incompatible with homeostatic interpretations.
Sign-Reversals: Are they Allostatic or Homeostatic?
In considering the effects of repeated drug administrations and the development of drug tolerance, Dworkin (1993) described the widely held belief that even with very many administrations, drug effects sometimes diminish to zero but do not invert to the opposite direction. While we initially also espoused this view (Ramsay & Woods, 1997), we now recognize that there are instances where an underlying effector response persistently overcompensates for the effect of the disturbance in the continued presence of the drug. Chronically tolerant individuals can in fact exhibit persistent sign reversals (inversions) of the regulated variable in the opposite direction to the initial drug effect despite the presence of maximal drug concentrations, indicating that these reversals are not rebound effects or instances of drug-withdrawal. Similar sign-reversal phenomena also occur with non-drug disturbances. Along with persistent activation of effectors and concurrent activation of opposing effectors, such phenomena challenge many of the assumptions underlying homeostatic regulation and have important implications for understanding allostasis.
To the best of our knowledge, this phenomenon was first termed a ‘sign-reversal’ in an investigation of the analgesic effects of μ-opioid receptor agonists (Bruins Slot, Pauwels, & Colpaert, 2002). Colpaert and colleagues (Bruins Slot & Colpaert, 1999; Bruins Slot et al., 2002; Colpaert, 1996; Colpaert & Fregnac, 2001) had developed a formal mathematical model that made the surprising and provocative prediction that under some parametric conditions, the effects caused by an input may reverse even as the input is maintained at a constant magnitude. They tested this prediction and observed that the analgesic effect of continuously administered fentanyl does in fact reverse during the course of a prolonged, continuous administration as shown in Figure 5 (Bruins Slot et al., 2002, p. 177). In other words, the rats became hyperalgesic despite the continued administration of the opiate to maintain drug concentrations. The animals did not become fully tolerant to the analgesia as indicated by their return to near baseline (or control) levels as would be predicted by homeostatic models. Instead, they became excessively tolerant by exhibiting a persistent hyperalgesic overcorrection.2
Figure 5.
“Nociceptive threshold in rats at different times (hr, days) following the implantation of an osmotic pump releasing fentanyl. On day 0 of the experiment, rats were implanted subcutaneously with an osmotic pump that released 0.31 mg rat−1day−1 of fentanyl (n = 14); control animals (n = 13) were implanted with a pump delivering 0.12 ml day−1 of saline. Nociceptive thresholds were measured by the paw-pressure vocalization test before and at different time intervals after pump implantation. A 750 g cut-off value was used to prevent tissue damage. Results are expressed as the mean (± s.e.m.) paw pressure (in g) inducing vocalization. Differences between fentanyl-treated (closed circle) and saline-treated (open circle) (control) groups were determined with the Student’s t-test subsequent to two-way ANOVA; *p<0.05; **p<0.01; ***p<0.001.” [This figure and figure legend are reprinted with permission (Bruins Slot et al., 2002, p. 177)].
The authors emphasized that the hyperalgesic state was not a withdrawal effect resulting from discontinuation of the opioid or from a precipitated rebound effect as would occur with the administration of an opioid antagonist. This issue is critical to the current discussion because withdrawal and precipitated rebound effects are explicable within a homeostatic model of drug tolerance (e.g., Ramsay & Woods, 1997).
This sign-reversal phenomenon with opioids is often referred to as opioid-induced hyperalgesia and numerous reviews are available (Angst & Clark, 2006; Chu, Angst, & Clark, 2008; Lee, Silverman, Hansen, Patel, & Manchikanti, 2011; Ossipov et al., 2004; Ossipov, Lai, King, Vanderah, & Porreca, 2005; Ossipov, Lai, Vanderah, & Porreca, 2003). An overactive hyperalgesic response is believed to mediate the sign-reversal state of opioid-induced hyperalgesia (Célèrier, Laulin, Corcuff, Le Moal, & Simonnet, 2001). Mao (2002) and Vanderah and colleagues (2001) emphasize that the hyperalgesic overcompensation cannot be explained as a withdrawal effect (or ‘mini-withdrawal’) because the opioid concentrations are held constant during the continuous drug administration.
While clinical research on individuals with a history of opioid drug abuse has inherent limitations (e.g., non-random assignment to drug use history), this corpus of work nonetheless illustrates how these sign reversals are not easily explained by common homeostatic views of adaptation (Compton, 1994; Compton, Canamar, Hillhouse, & Ling, 2012; Compton, Charuvastra, & Ling, 2001; Ren, Shi, Epstein, Wang, & Lu, 2009). For example, Compton found that cold-pressor pain tolerance counter-intuitively decreases as methadone dose increases (1994). Chronic methadone users report maximal pain sensitivity at the time that they have the peak concentrations of the analgesic opioid (i.e., the increased pain sensitivity cannot be explained as a withdrawal effect from a waning drug concentration), and this is thought to be indicative of an over-activated corrective response. As another example, opiate addicts continue to make hyperalgesic responses, despite being abstinent from opioids for at least 5 months, suggesting that these responses are persistent and not easily deactivated even though the initial eliciting perturbation is no longer present (Ren et al., 2009).
A long-standing belief about drug addiction is that drug-dependent individuals who stop taking their drug become increasingly motivated to re-administer the drug to prevent or ameliorate drug withdrawal (e.g., Baker, Piper, McCarthy, Majeskie, & Fiore, 2004). In an allostasis model, aversive sign-reversal states that exist in the presence of maintained drug concentrations can motivate increased drug consumption, and this argument has been made by others to explain drug taking in human addicts (Compton, 1994; White, 2004). While tolerance represents a homeostatic balance between drug effect and effector adaptations that is motivationally neutral (until drug concentrations wane and withdrawal begins), allostasis reflects a dysregulated imbalance between drug effects and excessive effector activity that causes a sign-reversal.
Analogous themes permeate several current models of addiction that are similar in this regard to the allostatic model of drug addiction proposed by Koob and Le Moal (2001). Colpaert and colleagues (Colpaert, Deseure, Stinus, & Adriaensen, 2006) hypothesized that drug-opposite, sign-reversal states have motivational properties that encourage further drug administration. Ossipov and colleagues (Ossipov et al., 2004) proposed a vicious circle-of-addiction hypothesis where overactive effector responses shift the balance point of the regulated variable into an aversive sign-reversal state which motivates drug taking. Pharmacologically increasing the drug effect via increased drug-taking behavior leads to the further overactivation of effector responses, and the restoration of the sign-reversal state which drives (motivates) further increases in drug taking.
Sign-reversals have also been reported in the osmoregulatory system (e.g., Na, Morris, Johnson, Beltz, & Johnson, 2007). The amount of sodium in the blood is related to several regulated parameters including blood volume and blood pressure. When the drug, furosemide, is administered, it reduces the kidney’s ability to retain sodium in the body, resulting in sodium loss to the urine and consequent hyponatremia. Rats respond to furosemide by increasing their intake of an otherwise not consumed hypertonic sodium solution to reverse the deficit. After several repetitions of this furosemide regimen, many rats persistently over-consume sodium salts, and this is indicative of an effector response that is excessive and does not turn itself off even though the challenges that reduced blood sodium concentration no longer occur; i.e., these rats persistently consume excess quantities of sodium throughout their lives.
This finding is confusing from a homeostatic perspective because the compensatory behavioral response (sodium consumption) is greater than what is necessary to correct the perturbation, and because this dysregulated response persists over time even when the conditions that initially elicited it are no longer present. Further, there appears to be a motivational component to the persistent behavior (Bernstein, 2003). This non-homeostatic finding might be better understood using an allostatic model.
Suggestions of sign-reversals have also been reported for the temperature-altering effects of drugs. Gordon and colleagues (Gordon, Rowsey, & Yang, 2002) found that rats exhibit hypothermia during an initial nicotine administration and that chronic tolerance develops with repeated administrations. However, by the fifth administration, the rats develop a significant sign-reversal hyperthermia that the authors described as paradoxical. As another example, rats become hypothermic when initially administered nitrous oxide (Kaiyala & Ramsay, 2005; Ramsay, Kaiyala, Leroux, & Woods, 2005). However, over repeated administrations, rats develop chronic tolerance to this effect and eventually exhibit a hyperthermic sign-reversal (Kaiyala, Chan, & Ramsay, 2012).
The overcompensation of effector responses that mediate sign-reversal phenomena are probably what attracted Hammel’s attention to Johansen’s (1961) research with the armadillo, where step decreases in ambient temperature caused increases in core temperature (i.e., a change in the regulated variable opposite in direction to the effects caused by the disturbance). While Hammel (1989, 1990) wondered why effector responses were sustained after the effect of the disturbance on the regulated variable was corrected and why the effector responses were of sufficient magnitude to cause an overcorrection, we now recognize that these are two characteristics commonly ascribed by some to allostatic regulation. Since that initial demonstration, several others have reported cold-induced elevations of core temperature caused by over-compensation of autonomic thermoregulatory effectors (Katz, Frank, McGwin Jr., Finch, & Gordon, 2012; Székely, Szelényi, Pétervári, & Balaskó, 2001; Yang & Gordon, 2005).
There is a paucity of research designed to identify and investigate sign-reversal phenomena. When these phenomena have been observed, the investigators usually describe their findings with terms like paradoxical, inexplicable, surprising, puzzling, and counter-intuitive. We believe this reflects the inability of sign-reversals to be explained within the dominant homeostatic model, and that the absence of a theoretical model to make sense of these kinds of observations has hindered research.
Thus, the allostatic model may provide a better framework for investigating these phenomena; i.e., the persistent maintenance of a sign-reversed response requires continued energy expenditure and likely places a load on those effectors, as well as on reflexes that oppose the sign reversal (i.e., concurrent activation of competing effectors). All of these nicely fit into McEwen’s concept of allostatic load and its potential adverse health effects. Stress responses may also play an important role in sign-reversal phenomena that are characteristic of allostasis (e.g., Edwards et al., 2012; Koob, 2010). Stress responses that are slow to resolve or do not shut off even after the disturbance has ended have been suggested to contribute to allostasis (e.g., Gourley, Swanson, & Koleske, 2013; Herman, 2013; Olausson, Kiraly, Gourley, & Taylor, 2013).
To summarize, while sign-reversal phenomena cannot be explained by principles of homeostasis, these observations are compatible with an allostatic model of regulation. Sign-reversals are energetically inefficient in that they involve discoordinated effector responses (e.g., concurrent activation of competing effectors, or responses that are excessive in magnitude or duration relative to the eliciting disturbance). The inefficiency of sign-reversals nicely supports the concept of the costs associated with allostatic load.
Why Allostasis? - Robustness Versus Fragility in Physiological Regulation
If physiological regulation does function in different ways, described as homeostatically and allostatically, it is worthwhile to speculate as to how and why this may have occurred. It is important to recognize that regulatory mechanisms evolved to counter the myriad of common as well as uncommon challenges that confront a species in its historic environmental niche. While homeostasis may appear elegantly simple in an abstract context, it is in actuality quite complex in terms of the processes that are employed to stabilize multiple vital, and often interacting, parameters simultaneously.
As discussed above, regulatory systems are complex because they have been shaped by evolutionary tinkering. Biological regulatory mechanisms evolved to be robust for reasons of survival and reproduction. Biological robustness is the property of active maintenance of a specific bodily function despite external and internal perturbations (Kitano et al., 2004). Csete and Doyle (2002) contend that whereas improvements in robustness are relatively easy to achieve in an evolutionary sense by adding to regulatory complexity, the increased robustness comes with a cost. They have demonstrated mathematically that while highly complex regulatory systems can be quite robust to cope with naturalistic disturbances, this increase in robustness comes at the cost of an equal increase in fragility (i.e., susceptibility to failure) of the system. Kitano and colleagues explain that “… systems that evolved to be robust against various but uncommon perturbations exhibit extreme fragility against unusual, unanticipated perturbations ….. In this context, many diseases can be viewed as exposed fragilities in normal biologic design, with disease progression mediated by acquisition of established and emergent robustness mechanisms” (Kitano et al., 2004, p. S6–S7).
We believe that many instances of allostatic regulation may be the result of exposed fragilities in homeostatic regulation. Fragilities in homeostasis are manifest as dysregulation, which explains why these phenomena do not seem to be homeostatic. Thus, these phenomena require a different explanatory model to understand dysregulation, and many believe that allostasis is assuming that role. Health problems such as drug addiction and obesity (as well as other disorders of modern society) may result from homeostatic fragility exposed by evolutionarily unanticipated perturbations. Many aspects of contemporary society have challenged our highly robust, yet fragile, homeostatic regulatory system with evolutionarily unanticipated perturbations. For example, the electric light has “confounded predictions based on the evolutionary experience of several million years” (Moore-Ede, 1986, p. R744).
Morris and colleagues (Morris, Na, & Johnson, 2008) provide a good example from the study of sodium appetite and the adverse health consequences that result from excess salt consumption, noting that disturbed sodium homeostasis likely plays a role in promoting excessive sodium intake. Because large quantities of salt have become available in the human diet over the past approximately 5000 years, this dysregulation could be the result of an exposed fragility in homeostasis (e.g., contributing to allostasis and allostatic load on the cardiovascular system).
There are numerous other examples of novel challenges to our regulatory system. Consumption of purified and highly potent drugs represent another one of these novel perturbations, as does the easy access to highly palatable and calorically dense foods as well as the use of artificial sweeteners. The case has also been made that the stressors of everyday living in modern life comprise a psychosocial stress that perturbs many regulated systems in ways that did not previously exist, perhaps predisposing to the rising incidence of depression, obesity and other chronic conditions (e.g., Karatsoreos & McEwen, 2011).
Cabanac (2006), Mrosovsky (1990) and others have made the similar point that findings that are incompatible with homeostatic principles may relate to the oddity of situations that provide challenges out of the context in which the regulatory systems evolved. However, when the homeostatic system is challenged in ways consistent with their evolutionary history, the response to the challenge appears coordinated and synchronous. Although uncommon, naturalistic examples of opposing effector competition have been described.3 The implication is that homeostasis may remain the dominant explanatory framework when regulatory systems are challenged in evolutionarily appropriate or predictable ways. However, novel ways of perturbing the regulatory system may expose fragilities in homeostasis that lead to dysregulatory or allostatic states.
Concluding Thoughts
In broad terms, we suggest that allostasis be considered a dysregulatory (or disordered) form of physiological regulation that involves effector loops that over-respond in magnitude or duration and/or that compete concurrently with other effectors. We believe that a potentially fruitful strategy to distinguish between homeostasis and allostasis will involve analyzing the underlying sensor-effector loops that contribute to the value of a regulated variable as well as the kinds of regulatory errors that occur.
For example, because homeostasis is complex, regulatory errors can occur in everyday situations, such as when an error of prediction happens (e.g., failing to make an anticipatory response when indicated or making an anticipatory response when the expected disturbance does not occur) (Lê et al., 1979; Mansfield & Cunningham, 1980; Siegel, 2001; Siegel, Hinson, Krank, & McCully, 1982). These kinds of errors can be explained using contemporary models of homeostasis. In contrast, we believe there are at least three kinds of effector activity which can occur separately or in combination and that would require an allostatic, rather than a homeostatic, explanation: 1) an effector response that eventually overcorrects an initial perturbation (i.e., causes a sign-reversal) despite the continued presence of the disturbance, 2) an effector response that remains active even though the disturbance that elicited it is no longer present and the continued effector activity is causing a persistent shift in the balance point of a regulated variable, and 3) concurrent effector competition caused by triggering events that occur in a manner or situation not anticipated in an evolutionary sense. We suspect that stress responses are likely to play an important role in determining the activity of effector loops that result in an allostatic state.
It is important to emphasize that distinguishing homeostasis and allostasis cannot be accomplished by evaluating the balance level of the regulated variable and instead requires measurement of the underlying effectors. Of course, we realize that these suggestions are speculative and likely to be revised. Our goal is to provide a more rigorous and better-defined model of allostasis that can be distinguished from homeostasis and thereby encourage basic research on behavioral and autonomic effector loops as well as translational research pertinent to the pathogenesis and treatment of human health concerns. Table 1 provides an overview of primary attributes of the two models that we believe are important to compare and contrast in order to help clarify the distinction between them, along with related issues that we consider important for understanding regulation more generally.
Table 1.
Key Attributes of Homeostatic and Allostatic Models of Physiological Regulation
| Homeostasis | Allostasis | Comments on Pertinent Issues | |
|---|---|---|---|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
The case could be made that the understanding of physiological regulation will eventually become sufficiently sophisticated that the organizing value of models like homeostasis and allostasis will no longer be useful. If regulation reflects a balance of passive external (e.g., ambient temperature) and active regulatory processes (e.g., controlled thermo-effector activity), then it may eventually be possible to measure the contributions of all of the individual components that together determine the balance point of the regulated variable. This level of understanding would obviate the need for explanatory models like homeostasis or allostasis. The technical feasibility of measuring these determinants is more advanced for some regulatory systems (e.g., temperature) than for others (e.g., pain, reward), and this likely explains why research on thermoregulation has played such a significant role in elucidating physiological regulation more generally. However, for the foreseeable future, models like homeostasis and allostasis will continue to have value in generating testable hypotheses about the relationships of sensor-effector loops and how they contribute to the regulated variable.
To conclude, regulatory phenomena have been recognized that cannot be accommodated by homeostasis; e.g., the persistent sign reversal of regulated variables when stimulated in certain ways and disturbances that elicit concurrent effector competition. These behaviors come with a cost that fits well with the concept of allostatic load. The argument can be made that certain aspects of modern society present situations that never existed historically in an evolutionary sense, and for which homeostatic systems are not equipped to handle. These situations reveal the fragility (i.e., allostatic) side of our finely-tuned and otherwise robust regulatory (i.e., homeostatic) systems, and lead to responses that may represent compromised balances among multiple and sometimes concurrently competing dysregulated effectors, and that are associated with (often severe) adverse health effects such as drug addiction, depression and obesity.
Acknowledgments
This work was supported by NIH grants: DA023484 & DA033453 (DSR) and DK017844 (SCW).
The authors gratefully acknowledge Karl Kaiyala’s contributions to our understanding of physiological regulation. We also thank the Helen Riaboff Whiteley Center, located at the University of Washington’s Friday Harbor Laboratories, for providing the ideal environment for us to write this manuscript.
Footnotes
An analogous situation exists in the study of drug tolerance. Ramsay & Woods (1997) explained how the simple description of an observed change in a measured variable following a drug administration does not distinguish the contributions of regulatory responses, direct drug action, and other influences on the variable.
The same research group was able to demonstrate the analogous ‘mirror opposite’ of the μ-opioid sign-reversal effect by giving a continuous infusion of a high-efficacy 5-hydroxytryptamine 1A agonist. This agonist caused an initial hyperalgesia followed by a sign reversal to analgesia despite the continued presence of the drug (Bruins Slot, Koek, Tarayre, & Colpaert, 2003). Interestingly, the sign-reversal analgesia persists for a long duration even after administration of the agonist has ceased (Colpaert et al., 2004).
Refinetti (2010) contends that the oscillatory circadian system’s influence on core body temperature mediated via heat production and heat loss thermoeffectors is opposed by the homeostatic thermoregulatory system’s behavioral effectors, with a net result of a dampened circadian pattern of core temperature that reflects “the integrated output of the two systems” (p. 574). Refinetti (2010, p. 575) further suggests that this “violation of homeothermy” can be explained as a conflict between the evolutionary histories of the homeostatic and circadian systems and thus is not simply a “curious deviation from an ideal pattern.” Although the concurrent activation of competing effectors elicited by the circadian and homeostatic systems fits an allostatic pattern of dysregulation, it is a naturalistic example that is neither provoked by a disturbance nor the result of an artificial experimental condition. Thus, we would not consider this concurrent competition of effectors as an example of allostasis but rather that it simply represents how contemporary homeotherms have evolved.
References
- Ahmed SH, Koob GF. Transition to drug addiction: a negative reinforcement model based on an allostatic decrease in reward function. Psychopharmacology (Berl) 2005;180(3):473–490. doi: 10.1007/s00213-005-2180-z. [DOI] [PubMed] [Google Scholar]
- Angst MS, Clark JD. Opioid-induced hyperalgesia: a qualitative systematic review. Anesthesiology. 2006;104(3):570–587. doi: 10.1097/00000542-200603000-00025. [DOI] [PubMed] [Google Scholar]
- Baker TB, Piper ME, McCarthy DE, Majeskie MR, Fiore MC. Addiction motivation reformulated: An affective processing model of negative reinforcement. Psychological Review. 2004;111(1):33–51. doi: 10.1037/0033-295x.111.1.33. [DOI] [PubMed] [Google Scholar]
- Bauman DE. Regulation of nutrient partitioning during lactation: Homeostasis and homeorhesis revisited. In: Cronjé P, Boomker EA, editors. Ruminant physiology: digestion, metabolism, growth, and reproduction. Wallingford, Oxon, UK; New York, NY, USA: CABI Pub; 2000. pp. 311–328. [Google Scholar]
- Bauman DE, Currie WB. Partitioning of nutrients during pregnancy and lactation: a review of mechanisms involving homeostasis and homeorhesis. Journal of Dairy Science. 1980;63(9):1514–1529. doi: 10.3168/jds.s0022-0302(80)83111-0. [DOI] [PubMed] [Google Scholar]
- Bernard C. Lessons on the phenomena of life common to animals and vegetables. Second lecture, the three forms of life. In: Langley LL, editor. Homeostasis: Origins of the concept 1973. Stroudsberg, PA, USA: Dowden, Hutchinson & Ross, Inc; 1870. pp. 129–151. [Google Scholar]
- Bernstein IL. Interaction between natural motivational systems and those which respond to drugs of abuse. Appetite. 2003;41(3):333–334. doi: 10.1016/j.appet.2003.06.001. [DOI] [PubMed] [Google Scholar]
- Berntson GG, Cacioppo JT. From homeostasis to allodynamic regulation. In: Cacioppo JT, Tassinary LG, Berntson GG, editors. Handbook of psychophysiology. 2. Cambridge, UK; New York, NY, USA: Cambridge University Press; 2000. pp. 459–481. [Google Scholar]
- Berntson GG, Cacioppo JT. Integrative physiology: Homeostasis, allostasis, and the orchestration of systemic physiology. In: Cacioppo JT, Tassinary LG, Berntson GG, editors. Handbook of Psychophysiology. 3. Cambridge England; New York: Cambridge University Press; 2007. pp. 433–452. [Google Scholar]
- Berridge KC. Motivation concepts in behavioral neuroscience. Physiology & Behavior. 2004;81(2):179–209. doi: 10.1016/j.physbeh.2004.02.004. [DOI] [PubMed] [Google Scholar]
- Berthoud HR, Bereiter DA, Trimble ER, Siegel EG, Jeanrenaud B. Cephalic Phase, Reflex Insulin-Secretion - Neuroanatomical and Physiological Characterization. Diabetologia. 1981;20:393–401. doi: 10.1007/Bf00254508. [DOI] [PubMed] [Google Scholar]
- Bligh J. Neuronal models of mammalian temperature regulation. In: Bligh J, Moore RE, editors. Essays on temperature regulation. Amsterdam: North-Holland Pub. Co; 1972. pp. 105–120. [Google Scholar]
- Bligh J. Mammalian homeothermy: An integrative thesis. Journal of Thermal Biology. 1998;23(3–4):143–258. doi: 10.1016/S0306-4565(98)00014-X. [DOI] [Google Scholar]
- Bligh J. A theoretical consideration of the means whereby the mammalian core temperature is defended at a null zone. Journal of Applied Physiology. 2006;100(4):1332–1337. doi: 10.1152/Jappalphysiol.01068.2005. [DOI] [PubMed] [Google Scholar]
- Booth DA. Physiological regulation through learnt control of appetites by contingencies among signals from external and internal environments. Appetite. 2008;51(3):433–441. doi: 10.1016/j.appet.2008.06.008. [DOI] [PubMed] [Google Scholar]
- Boulant JA. Hypothalamic mechanisms in thermoregulation. Federation Proceedings. 1981;40(14):2843–2850. [PubMed] [Google Scholar]
- Boulant JA. Role of the preoptic-anterior hypothalamus in thermoregulation and fever. Clinical Infectious Diseases. 2000;31(Suppl 5):S157–161. doi: 10.1086/317521. [DOI] [PubMed] [Google Scholar]
- Boulant JA. Neuronal basis of Hammel’s model for set-point thermoregulation. Journal of Applied Physiology. 2006;100(4):1347–1354. doi: 10.1152/japplphysiol.01064.2005. [DOI] [PubMed] [Google Scholar]
- Boulos Z, Rosenwasser AM. A chronobiological perspective on allostasis and its application to shift work. In: Schulkin J, editor. Allostasis, homeostasis and the costs of physiological adaptation. New York: Cambridge University Press; 2004. pp. 228–301. [Google Scholar]
- Brobeck JR. Exchange, control, and regulation. In: Yamamoto WS, Brobeck JR, editors. Physiological controls and regulations. Philadelphia: Saunders; 1965. pp. 1–14. [Google Scholar]
- Bruins Slot LA, Colpaert FC. Experimental realization of a signal transduction algorithm. Journal of Theoretical Biology. 1999;200(1):39–48. doi: 10.1006/jtbi.1999.0973. [DOI] [PubMed] [Google Scholar]
- Bruins Slot LA, Koek W, Tarayre JP, Colpaert FC. Tolerance and inverse tolerance to the hyperalgesic and analgesic actions, respectively, of the novel analgesic, F 13640. European Journal of Pharmacology. 2003;466(3):271–279. doi: 10.1016/s0014-2999(03)01566-8. [DOI] [PubMed] [Google Scholar]
- Bruins Slot LA, Pauwels PJ, Colpaert FC. Sign-reversal during persistent activation in mu-opioid signal transduction. Journal of Theoretical Biology. 2002;215(2):169–182. doi: 10.1006/jtbi.2001.2509. [DOI] [PubMed] [Google Scholar]
- Cabanac M. Adjustable set point: to honor Harold T. Hammel. Journal of Applied Physiology. 2006;100(4):1338–1346. doi: 10.1152/japplphysiol.01021.2005. [DOI] [PubMed] [Google Scholar]
- Campfield LA, Smith FJ. Blood glucose dynamics and control of meal initiation: a pattern detection and recognition theory. Physiological Reviews. 2003;83(1):25–58. doi: 10.1152/physrev.00019.2002. [DOI] [PubMed] [Google Scholar]
- Cannon WB. Organization for physiological homeostasis. Physiological Reviews. 1929;9(3):399–431. [Google Scholar]
- Cannon WB. The wisdom of the body. New York: W. W. Norton & Company; 1932. [Google Scholar]
- Cannon WB. The way of an investigator: A scientist’s experiences in medical research. 1965. New York: Hafner Publishing Company; 1945. [Google Scholar]
- Carlisle HJ, Ingram DL. The effects of heating and cooling the spinal cord and hypothalamus on thermoregulatory behaviour in the pig. Journal of Physiology. 1973;231(2):353–364. doi: 10.1113/jphysiol.1973.sp010237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter RH. Homeostasis: a plea for a unified approach. Advances in Physiology Education. 2004;28(1–4):180–187. doi: 10.1152/advan.00012.2004. [DOI] [PubMed] [Google Scholar]
- Célèrier E, Laulin JP, Corcuff JB, Le Moal M, Simonnet G. Progressive enhancement of delayed hyperalgesia induced by repeated heroin administration: a sensitization process. Journal of Neuroscience. 2001;21(11):4074–4080. doi: 10.1523/JNEUROSCI.21-11-04074.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chilliard Y. Bibliographic review: Quantitative variations and metabolism of lipids in adipose tissue and the liver during the gestation-lactation cycle. 1. In the rat. Reproduction Nutrition Development. 1986;26(5A):1057–1103. [PubMed] [Google Scholar]
- Chilliard Y, Ferlay A, Faulconnier Y, Bonnet M, Rouel J, Bocquier F. Adipose tissue metabolism and its role in adaptations to undernutrition in ruminants. Proceedings of the Nutrition Society. 2000;59(1):127–134. doi: 10.1017/s002966510000015x. [DOI] [PubMed] [Google Scholar]
- Chu LF, Angst MS, Clark D. Opioid-induced hyperalgesia in humans: molecular mechanisms and clinical considerations. Clinical Journal of Pain. 2008;24(6):479–496. doi: 10.1097/AJP.0b013e31816b2f43. [DOI] [PubMed] [Google Scholar]
- Collier GH. The dialogue between the House Economist and the Resident Physiologist. Nutrition & Behavior. 1986;3(1):9–26. [Google Scholar]
- Collier GH, Johnson DF, Hill WL, Kaufman LW. The economics of the law of effect. Journal of the Experimental Analysis of Behavior. 1986;46(2):113–136. doi: 10.1901/Jeab.1986.46-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collier G, Johnson DF, Mitchell C. The relation between meal size and the time between meals: Effects of cage complexity and food cost. Physiology & Behavior. 1999;67(3):339–346. doi: 10.1016/S0031-9384(99)00086-4. [DOI] [PubMed] [Google Scholar]
- Colpaert FC. System theory of pain and of opiate analgesia: no tolerance to opiates. Pharmacological Reviews. 1996;48(3):355–402. [PubMed] [Google Scholar]
- Colpaert FC, Deseure K, Stinus L, Adriaensen H. High-efficacy 5-hydroxytryptamine 1A receptor activation counteracts opioid hyperallodynia and affective conditioning. Journal of Pharmacology and Experimental Therapeutics. 2006;316(2):892–899. doi: 10.1124/jpet.105.095109. [DOI] [PubMed] [Google Scholar]
- Colpaert FC, Fregnac Y. Paradoxical signal transduction in neurobiological systems. Molecular Neurobiology. 2001;24(1–3):145–168. doi: 10.1385/MN:24:1-3:145. [DOI] [PubMed] [Google Scholar]
- Colpaert FC, Wu WP, Hao JX, Royer I, Sautel F, Wiesenfeld-Hallin Z, Xu XJ. High-efficacy 5-HT1A receptor activation causes a curative-like action on allodynia in rats with spinal cord injury. European Journal of Pharmacology. 2004;497(1):29–33. doi: 10.1016/j.ejphar.2004.06.026. [DOI] [PubMed] [Google Scholar]
- Compton MA. Cold-pressor pain tolerance in opiate and cocaine abusers: correlates of drug type and use status. Journal of Pain and Symptom Management. 1994;9(7):462–473. doi: 10.1016/0885-3924(94)90203-8. [DOI] [PubMed] [Google Scholar]
- Compton P, Canamar CP, Hillhouse M, Ling W. Hyperalgesia in heroin dependent patients and the effects of opioid substitution therapy. Journal of Pain. 2012;13(4):401–409. doi: 10.1016/j.jpain.2012.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Compton P, Charuvastra VC, Ling W. Pain intolerance in opioid-maintained former opiate addicts: effect of long-acting maintenance agent. Drug and Alcohol Dependence. 2001;63(2):139–146. doi: 10.1016/s0376-8716(00)00200-3. [DOI] [PubMed] [Google Scholar]
- Csete ME, Doyle JC. Reverse engineering of biological complexity. Science. 2002;295(5560):1664–1669. doi: 10.1126/Science.1069981. [DOI] [PubMed] [Google Scholar]
- Day TA. Defining stress as a prelude to mapping its neurocircuitry: no help from allostasis. Progress in Neuropsychopharmacology & Biological Psychiatry. 2005;29(8):1195–1200. doi: 10.1016/j.pnpbp.2005.08.005. [DOI] [PubMed] [Google Scholar]
- Domjan M. Pavlovian conditioning: a functional perspective. Annual Review of Psychology. 2005;56:179–206. doi: 10.1146/annurev.psych.55.090902.141409. [DOI] [PubMed] [Google Scholar]
- Drazen DL, Vahl TP, D’Alessio DA, Seeley RJ, Woods SC. Effects of a fixed meal pattern on ghrelin secretion: Evidence for a learned response independent of nutrient status. Endocrinology. 2006;147(1):23–30. doi: 10.1210/En.2005-0973. [DOI] [PubMed] [Google Scholar]
- Dworkin BR. Learning and physiological regulation. Chicago: University of Chicago Press; 1993. [Google Scholar]
- Dworkin BR. Interoception. In: Cacioppo JT, Tassinary LG, Berntson GG, editors. Handbook of Psychophysiology. 3. Cambridge England; New York: Cambridge University Press; 2007. pp. 482–506. [Google Scholar]
- Edwards S, Koob GF. Neurobiology of dysregulated motivational systems in drug addiction. Future Neurology. 2010;5(3):393–401. doi: 10.2217/fnl.10.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards S, Vendruscolo LF, Schlosburg JE, Misra KK, Wee S, Park PE, Koob GF. Development of mechanical hypersensitivity in rats during heroin and ethanol dependence: Alleviation by CRF1 receptor antagonism. Neuropharmacology. 2012;62(2):1142–1151. doi: 10.1016/J.Neuropharm.2011.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganzel BL, Morris PA, Wethington E. Allostasis and the human brain: Integrating models of stress from the social and life sciences. Psychological Review. 2010;117(1):134–174. doi: 10.1037/a0017773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein DS. Merging of the homeostat theory with the concept of allostatic load. In: Schulkin J, editor. Allostasis, homeostasis and the costs of physiological adaptation. New York: Cambridge University Press; 2004. pp. 99–112. [Google Scholar]
- Goldstein DS. Computer Models of Stress, Allostasis, and Acute and Chronic Diseases. Stress, Neurotransmitters, and Hormones: Neuroendocrine and Genetic Mechanisms. 2008;1148:223–231. doi: 10.1196/Annals.1410.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein DS. Stress, allostatic load, catecholamines, and other neurotransmitters in neurodegenerative diseases. Cellular and Molecular Neurobiology. 2012;32(5):661–666. doi: 10.1007/s10571-011-9780-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein DS. Differential responses of components of the autonomic nervous system. Handbook of Clinical Neurology. 2013;117:13–22. doi: 10.1016/B978-0-444-53491-0.00002-X. [DOI] [PubMed] [Google Scholar]
- Goldstein DS, Kopin IJ. Evolution of concepts of stress. Stress. 2007;10(2):109–120. doi: 10.1080/10253890701288935. [DOI] [PubMed] [Google Scholar]
- Gourley SL, Swanson AM, Koleske AJ. Corticosteroid-induced neural remodeling predicts behavioral vulnerability and resilience. Journal of Neuroscience. 2013;33(7):3107–3112. doi: 10.1523/JNEUROSCI.2138-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon CJ. A review of terms for regulated vs. forced, neurochemical-induced changes in body temperature. Life Sciences. 1983;32(12):1285–1295. doi: 10.1016/0024-3205(83)90802-0. [DOI] [PubMed] [Google Scholar]
- Gordon CJ. The therapeutic potential of regulated hypothermia. Emergency Medicine Journal. 2001;18(2):81–89. doi: 10.1136/emj.18.2.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon CJ. Temperature and toxicology: an integrative, comparative, and environmental approach. Boca Raton, FL: Taylor & Francis; 2005. [Google Scholar]
- Gordon CJ. Autonomic nervous system: Central thermoregulatory control. In: Squire LR, editor. Encyclopedia of Neuroscience. Vol. 1. Oxford: Academic Press; 2009. pp. 891–898. [Google Scholar]
- Gordon CJ, Rowsey PJ, Yang YL. Effect of repeated nicotine exposure on core temperature and motor activity in male and female rats. Journal of Thermal Biology. 2002;27(6):485–492. Pii S0306-4565(02)00021-9. [Google Scholar]
- Guyton AC. Human physiology and mechanisms of disease. 3. Philadelphia: Saunders; 1982. [Google Scholar]
- Hammel HT. Neurons and temperature regulation. In: Yamamoto WS, Brobeck JR, editors. Physiological controls and regulations. Philadelphia: Saunders; 1965. pp. 71–97. [Google Scholar]
- Hammel HT. Concept of the adjustable set temperature. In: Hardy JD, Gagge AP, Stolwijk JAJ, editors. Physiological and behavioral temperature regulation. Springfield, Ill: Charles C. Thomas; 1970. pp. 676–683. [Google Scholar]
- Hammel HT. Anesthetics and Body-Temperature Regulation. Anesthesiology. 1988;68(6):833–835. [PubMed] [Google Scholar]
- Hammel HT. Homeostasis: Embracing negative feedback enhanced and sustained by positive feedback. In: Paganelli CV, Farhi LE, editors. Physiological function in special environments. New York: Springer-Verlag; 1989. pp. 191–202. [Google Scholar]
- Hammel HT. Negative plus positive feedback. In: Bligh J, Voigt K, editors. Thermoreception and temperature regulation. Berlin; New York: Springer-Verlag; 1990. pp. 174–182. [Google Scholar]
- Hammel HT, Jackson DC, Stolwijk JA, Hardy JD, Strømme SB. Temperature Regulation by Hypothalamic Proportional Control with an Adjustable Set Point. Journal of Applied Physiology. 1963;18:1146–1154. doi: 10.1152/jappl.1963.18.6.1146. [DOI] [PubMed] [Google Scholar]
- Hammel HT, Simon E. In: Pleschka K, Gerstberger Rd, editors. Salt gland excretion enhanced during cross circulation of blood between two Pekin ducks: Evidence for positive feedback; Integrative and cellular aspects of autonomic functions: temperature and osmoregulation: proceedings of the International Symposium on Integrative and Cellular Aspects of Autonomic Functions held in Bad Nauheim; Germany. 29–30 July 1993; Montrouge, France: J. Libbey Eurotext; 1994. pp. 497–508. [Google Scholar]
- Hardy JD. The “set-point” concept in physiological temperature regulation. In: Yamamoto WS, Brobeck JR, editors. Physiological controls and regulations. Philadelphia: Saunders; 1965. pp. 98–116. [Google Scholar]
- Herman JP. Neural control of chronic stress adaptation. Frontiers in Behavioral Neuroscience. 2013;7(61) doi: 10.3389/fnbeh.2013.00061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob F. Evolution and tinkering. Science. 1977;196(4295):1161–1166. doi: 10.1126/science.860134. [DOI] [PubMed] [Google Scholar]
- Johansen K. Temperature regulation in the nine-banded armadillo (Dasypus novemcinctus mexicanus) Physiological Zoölogy. 1961;34:126–144. [Google Scholar]
- Kaiyala KJ, Chan B, Ramsay DS. Robust thermoregulatory overcompensation, rather than tolerance, develops with serial administrations of 70% nitrous oxide to rats. Journal of Thermal Biology. 2012;37:30–40. doi: 10.106/j.jtherbio.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiyala KJ, Ramsay DS. Assessment of heat production, heat loss, and core temperature during nitrous oxide exposure: A new paradigm for studying drug effects and opponent responses. American Journal of Physiology: Regulatory, Integrative, and Comparative Physiology. 2005;288:692–701. doi: 10.1152/ajpregu.00412.2004. [DOI] [PubMed] [Google Scholar]
- Kanosue K, Crawshaw LI, Nagashima K, Yoda T. Concepts to utilize in describing thermoregulation and neurophysiological evidence for how the system works. European Journal of Applied Physiology. 2010;109(1):5–11. doi: 10.1007/s00421-009-1256-6. [DOI] [PubMed] [Google Scholar]
- Karatsoreos IN, McEwen BS. Psychobiological allostasis: resistance, resilience and vulnerability. Trends in Cognitive Sciences. 2011;15(12):576–584. doi: 10.1016/J.Tics.2011.10.005. [DOI] [PubMed] [Google Scholar]
- Katz LM, Frank JE, McGwin G, Jr, Finch A, Gordon CJ. Induction of a prolonged hypothermic state by drug-induced reduction in the thermoregulatory set-point. Therapeutic Hypothermia and Temperature Management. 2012;2(2):61–66. doi: 10.1089/ther.2012.0011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy GC. The Hypothalamic Control of Food Intake in Rats. Proceedings of the Royal Society B-Biological Sciences. 1950;137(889):535–549. doi: 10.1098/Rspb.1950.0065. [DOI] [PubMed] [Google Scholar]
- Kitano H, Oda K, Kimura T, Matsuoka Y, Csete M, Doyle J, Muramatsu M. Metabolic syndrome and robustness tradeoffs. Diabetes. 2004;53:S6–S15. doi: 10.2337/Diabetes.53.Suppl_3.S6. [DOI] [PubMed] [Google Scholar]
- Kobayashi S, Okazawa M, Hori A, Matsumura K, Hosokawa H. Paradigm shift in sensory system - Animals do not have sensors. Journal of Thermal Biology. 2006;31(1–2):19–23. doi: 10.1016/J.Jtherbio.2005.11.011. [DOI] [Google Scholar]
- Koob GF. Allostatic view of motivation: implications for psychopathology. Nebraska Symposium on Motivation. 2004;50:1–18. [PubMed] [Google Scholar]
- Koob GF. Dynamics of neuronal circuits in addiction: reward, antireward, and emotional memory. Pharmacopsychiatry. 2009;42(Suppl 1):S32–41. doi: 10.1055/s-0029-1216356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF. The role of CRF and CRF-related peptides in the dark side of addiction. Brain Research. 2010;1314:3–14. doi: 10.1016/j.brainres.2009.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF, Le Moal M. Drug abuse: hedonic homeostatic dysregulation. Science. 1997;278(5335):52–58. doi: 10.1126/science.278.5335.52. [DOI] [PubMed] [Google Scholar]
- Koob GF, Le Moal M. Drug addiction, dysregulation of reward, and allostasis. Neuropsychopharmacology. 2001;24(2):97–129. doi: 10.1016/S0893-133X(00)00195-0. [DOI] [PubMed] [Google Scholar]
- Koob GF, Le Moal M. Drug addiction and allostasis. In: Schulkin J, editor. Allostasis, homeostasis and the costs of physiological adaptation. NewYork: Cambridge University Press; 2004. pp. 150–163. [Google Scholar]
- Koob GF, Le Moal M. Plasticity of reward neurocircuitry and the ‘dark side’ of drug addiction. Nature Neuroscience. 2005;8(11):1442–1444. doi: 10.1038/nn1105-1442. [DOI] [PubMed] [Google Scholar]
- Koob GF, Le Moal M. Addiction and the brain antireward system. Annual Review of Psychology. 2008;59:29–53. doi: 10.1146/annurev.psych.59.103006.093548. [DOI] [PubMed] [Google Scholar]
- Kuenzel WJ, Beck MM, Teruyama R. Neural sites and pathways regulating food intake in birds: a comparative analysis to mammalian systems. Journal of Experimental Zoology. 1999;283(4–5):348–364. doi: 10.1002/(sici)1097-010x(19990301/01)283:4/5<348::aid-jez5>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- Langley LL. Homeostasis: origins of the concept. Stroudsburg, Pa: Dowden; 1973. [Google Scholar]
- Lê AD, Poulos CX, Cappell H. Conditioned Tolerance to the Hypothermic Effect of Ethyl-Alcohol. Science. 1979;206(4422):1109–1110. doi: 10.1126/Science.493999. [DOI] [PubMed] [Google Scholar]
- Le Moal M. Historical approach and evolution of the stress concept: a personal account. Psychoneuroendocrinology. 2007;32(Suppl 1):S3–9. doi: 10.1016/j.psyneuen.2007.03.019. [DOI] [PubMed] [Google Scholar]
- Le Moal M, Koob GF. Drug addiction: pathways to the disease and pathophysiological perspectives. European Neuropsychopharmacology. 2007;17(6–7):377–393. doi: 10.1016/j.euroneuro.2006.10.006. [DOI] [PubMed] [Google Scholar]
- Lee M, Silverman SM, Hansen H, Patel VB, Manchikanti L. A comprehensive review of opioid-induced hyperalgesia. Pain Physician. 2011;14(2):145–161. [PubMed] [Google Scholar]
- Lopez-Barneo J, Ortega-Saenz P, Pardal R, Pascual A, Piruat JI. Carotid body oxygen sensing. European Respiratory Journal. 2008;32(5):1386–1398. doi: 10.1183/09031936.00056408. [DOI] [PubMed] [Google Scholar]
- Mansfield JG, Cunningham CL. Conditioning and Extinction of Tolerance to the Hypothermic Effect of Ethanol in Rats. Journal of Comparative and Physiological Psychology. 1980;94(5):962–969. doi: 10.1037/H0077824. [DOI] [PubMed] [Google Scholar]
- Mao J. Opioid-induced abnormal pain sensitivity: implications in clinical opioid therapy. Pain. 2002;100(3):213–217. doi: 10.1016/S0304-3959(02)00422-0. [DOI] [PubMed] [Google Scholar]
- Mayer J. Regulation of Energy Intake and the Body Weight - the Glucostatic Theory and the Lipostatic Hypothesis. Annals of the New York Academy of Sciences. 1955;63(1):15–43. doi: 10.1111/J.1749-6632.1955.Tb36543.X. [DOI] [PubMed] [Google Scholar]
- McAllen RM, Tanaka M, Ootsuka Y, McKinley MJ. Multiple thermoregulatory effectors with independent central controls. European Journal of Applied Physiology. 2010;109(1):27–33. doi: 10.1007/s00421-009-1295-z. [DOI] [PubMed] [Google Scholar]
- McEwen BS, Gianaros PJ. Central role of the brain in stress and adaptation: links to socioeconomic status, health, and disease. Annals of the New York Academy of Sciences. 2010;1186:190–222. doi: 10.1111/j.1749-6632.2009.05331.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEwen BS, Stellar E. Stress and the individual. Mechanisms leading to disease. Archives of Internal Medicine. 1993;153(18):2093–2101. [PubMed] [Google Scholar]
- McEwen BS, Wingfield JC. What is in a name? Integrating homeostasis, allostasis and stress. Hormones and Behavior. 2010;57(2):105–111. doi: 10.1016/j.yhbeh.2009.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer JB, Hammel HT. Total calorimetry and temperature regulation in the nine-banded armadillo. Acta Physiologica Scandinavica. 1989;135(4):579–589. doi: 10.1111/j.1748-1716.1989.tb08620.x. [DOI] [PubMed] [Google Scholar]
- Moore-Ede MC. Physiology of the circadian timing system: predictive versus reactive homeostasis. American Journal of Physiology. 1986;250(5 Pt 2):R737–752. doi: 10.1152/ajpregu.1986.250.5.R737. [DOI] [PubMed] [Google Scholar]
- Morris MJ, Na ES, Johnson AK. Salt craving: the psychobiology of pathogenic sodium intake. Physiology & Behavior. 2008;94(5):709–721. doi: 10.1016/j.physbeh.2008.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison SF, Nakamura K. Central neural pathways for thermoregulation. Frontiers in Bioscience. 2011;16:74–104. doi: 10.2741/3677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mrosovsky N. Rheostasis: the physiology of change. New York: Oxford University Press; 1990. [Google Scholar]
- Na ES, Morris MJ, Johnson RF, Beltz TG, Johnson AK. The neural substrates of enhanced salt appetite after repeated sodium depletions. Brain Research. 2007;1171:104–110. doi: 10.1016/j.brainres.2007.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolaidis S. Metabolic and humoral mechanisms of feeding and genesis of the ATP/ADP/AMP concept. Physiology & Behavior. 2011;104(1):8–14. doi: 10.1016/j.physbeh.2011.04.058. [DOI] [PubMed] [Google Scholar]
- Olausson P, Kiraly DD, Gourley SL, Taylor JR. Persistent effects of prior chronic exposure to corticosterone on reward-related learning and motivation in rodents. Psychopharmacology. 2013;225(3):569–577. doi: 10.1007/s00213-012-2844-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ossipov MH, Lai J, King T, Vanderah TW, Malan TP, Jr, Hruby VJ, Porreca F. Antinociceptive and nociceptive actions of opioids. Journal of Neurobiology. 2004;61(1):126–148. doi: 10.1002/neu.20091. [DOI] [PubMed] [Google Scholar]
- Ossipov MH, Lai J, King T, Vanderah TW, Porreca F. Underlying mechanisms of pronociceptive consequences of prolonged morphine exposure. Biopolymers. 2005;80(2–3):319–324. doi: 10.1002/bip.20254. [DOI] [PubMed] [Google Scholar]
- Ossipov MH, Lai J, Vanderah TW, Porreca F. Induction of pain facilitation by sustained opioid exposure: relationship to opioid antinociceptive tolerance. Life Sciences. 2003;73(6):783–800. doi: 10.1016/s0024-3205(03)00410-7. [DOI] [PubMed] [Google Scholar]
- Peters A, McEwen BS. Introduction for the allostatic load special issue. Physiology & Behavior. 2012;106(1):1–4. doi: 10.1016/j.physbeh.2011.12.019. [DOI] [PubMed] [Google Scholar]
- Power ML. Viability as opposed to stability: An evolutionary perspective on physiological regulation. In: Schulkin J, editor. Allostasis, homeostasis and the costs of physiological adaptation. New York: Cambridge University Press; 2004. pp. 343–364. [Google Scholar]
- Power ML, Schulkin J. Maternal obesity, metabolic disease, and allostatic load. Physiology & Behavior. 2012;106(1):22–28. doi: 10.1016/j.physbeh.2011.09.011. [DOI] [PubMed] [Google Scholar]
- Ramsay DS, Kaiyala KJ, Leroux BG, Woods SC. Individual differences in initial sensitivity and acute tolerance predict patterns of chronic drug tolerance to nitrous-oxide-induced hypothermia in rats. Psychopharmacology. 2005;181:48–59. doi: 10.1007/s00213-005-2219-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsay DS, Seeley RJ, Bolles RC, Woods SC. Ingestive homeostasis: The primacy of learning. In: Capaldi ED, editor. Why we eat what we eat: the psychology of eating. 1. Washington, DC: American Psychological Association; 1996. pp. 11–27. [Google Scholar]
- Ramsay DS, Woods SC. Biological consequences of drug administration: implications for acute and chronic tolerance. Psychological Review. 1997;104(1):170–193. doi: 10.1037/0033-295x.104.1.170. [DOI] [PubMed] [Google Scholar]
- Refinetti R. The circadian rhythm of body temperature. Frontiers in Bioscience-Landmark. 2010;15:564–594. doi: 10.2741/3634. [DOI] [PubMed] [Google Scholar]
- Ren ZY, Shi J, Epstein DH, Wang J, Lu L. Abnormal pain response in pain-sensitive opiate addicts after prolonged abstinence predicts increased drug craving. Psychopharmacology (Berl) 2009;204(3):423–429. doi: 10.1007/s00213-009-1472-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romanovsky AA. Temperature regulation. In: Petersen OH, editor. Lecture notes. Human physiology. 5. Malden, Mass: Blackwell Pub; 2007a. pp. 603–615. [Google Scholar]
- Romanovsky AA. Thermoregulation: some concepts have changed. Functional architecture of the thermoregulatory system. American Journal of Physiology -Regulatory Integrative and Comparative Physiology. 2007b;292(1):R37–46. doi: 10.1152/ajpregu.00668.2006. [DOI] [PubMed] [Google Scholar]
- Satinoff E. Neural organization and evolution of thermal regulation in mammals. Science. 1978;201(4350):16–22. doi: 10.1126/science.351802. [DOI] [PubMed] [Google Scholar]
- Satinoff E. Thermoregulation. In: Whishaw IQ, Kolb B, editors. The behavior of the laboratory rat: a handbook with tests. Oxford; New York: Oxford University Press; 2005. pp. 226–235. [Google Scholar]
- Schulkin J. Social allostasis: anticipatory regulation of the internal milieu. Frontiers in Evolutionary Neuroscience. 2011;2:111. doi: 10.3389/fnevo.2010.00111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Commission for Thermal Physiology of the International Union of Physiological Sciences. Glossary of terms for thermal physiology. Third Edition. Revised by The Commission for Thermal Physiology of the International Union of Physiological Sciences. The Japanese Journal of Physiology. 2001;51(2):245–280. [PubMed] [Google Scholar]
- Seeley RJ, Ramsay DS, Woods SC. Regulation of food intake: Interactions between learning and physiology. In: Bouton ME, Fanselow MS, editors. Learning, motivation, and cognition: the functional behaviorism of Robert C. Bolles. Washington, DC: American Psychological Association; 1997. pp. 99–115. [Google Scholar]
- Selye H. Homeostasis and heterostasis. Perspectives in Biology and Medicine. 1973;16(3):441–445. doi: 10.1353/pbm.1973.0056. [DOI] [PubMed] [Google Scholar]
- Sherrington CS. The integrative action of the nervous system. New York: C. Scribner’s sons; 1906. [Google Scholar]
- Siegel S. Pavlovian conditioning and drug overdose: When tolerance fails. Addiction Research & Theory. 2001;9(5):503–513. doi: 10.3109/16066350109141767. [DOI] [Google Scholar]
- Siegel S. Learning and the wisdom of the body. Learning & Behavior. 2008;36(3):242–252. doi: 10.3758/lb.36.3.242. [DOI] [PubMed] [Google Scholar]
- Siegel S, Allan LG. Learning and homeostasis: drug addiction and the McCollough effect. Psychological Bulletin. 1998;124(2):230–239. doi: 10.1037/0033-2909.124.2.230. [DOI] [PubMed] [Google Scholar]
- Siegel S, Baptista MA, Kim JA, McDonald RV, Weise-Kelly L. Pavlovian psychopharmacology: the associative basis of tolerance. Experimental and Clinical Psychopharmacology. 2000;8(3):276–293. doi: 10.1037//1064-1297.8.3.276. [DOI] [PubMed] [Google Scholar]
- Siegel S, Hinson RE, Krank MD, McCully J. Heroin “overdose” death: contribution of drug-associated environmental cues. Science. 1982;216(4544):436–437. doi: 10.1126/science.7200260. [DOI] [PubMed] [Google Scholar]
- Solomon RL. The opponent-process theory of acquired motivation: the costs of pleasure and the benefits of pain. Ameican Psychologist. 1980;35(8):691–712. doi: 10.1037//0003-066x.35.8.691. [DOI] [PubMed] [Google Scholar]
- Solomon RL, Corbit JD. An opponent-process theory of motivation. I. Temporal dynamics of affect. Psychological Review. 1974;81(2):119–145. doi: 10.1037/h0036128. [DOI] [PubMed] [Google Scholar]
- Somjen GG. The Missing Error Signal - Regulation Beyond Negative Feedback. News in Physiological Sciences. 1992;7:184–185. [Google Scholar]
- Soodak H, Iberall A. Homeokinetics: a physical science for complex systems. Science. 1978;201(4356):579–582. doi: 10.1126/science.201.4356.579. [DOI] [PubMed] [Google Scholar]
- Sterling P. Principles of Allostasis: Optimal Design, Predictive Regulation, Pathophysiology, and Rational Therapeutics. In: Schulkin J, editor. Allostasis, homeostasis and the costs of physiological adaptation. New York: Cambridge University Press; 2004. pp. 17–64. [Google Scholar]
- Sterling P. Allostasis: a model of predictive regulation. Physiology & Behavior. 2012;106(1):5–15. doi: 10.1016/j.physbeh.2011.06.004. [DOI] [PubMed] [Google Scholar]
- Sterling P, Eyer J. Allostasis: A new paradigm to explain arousal pathology. In: Fisher S, Reason JT, editors. Handbook of life stress, cognition, and health. Chichester; New York: Wiley; 1988. pp. 629–649. [Google Scholar]
- Strubbe JH, van Dijk G. The temporal organization of ingestive behaviour and its interaction with regulation of energy balance. Neuroscience & Biobehavioral Reviews. 2002;26(4):485–498. doi: 10.1016/s0149-7634(02)00016-7. [DOI] [PubMed] [Google Scholar]
- Strubbe JH, Woods SC. The timing of meals. Psychological Review. 2004;111(1):128–141. doi: 10.1037/0033-295X.111.1.128. [DOI] [PubMed] [Google Scholar]
- Székely M, Szelényi Z, Pétervári E, Balaskó M. Thermoregulatory “overshoot” reactions in cold-adapted rats. Journal of Thermal Biology. 2001;26(4–5):491–497. doi: 10.1016/S0306-4565(01)00066-3. [DOI] [Google Scholar]
- Tajino K, Hosokawa H, Maegawa S, Matsumura K, Dhaka A, Kobayashi S. Cooling-Sensitive TRPM8 Is Thermostat of Skin Temperature against Cooling. Plos One. 2011;6(3):e17504. doi: 10.1371/journal.pone.0017504. ARTN. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teff KL. How neural mediation of anticipatory and compensatory insulin release helps us tolerate food. Physiology & Behavior. 2011;103(1):44–50. doi: 10.1016/J.Physbeh.2011.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vahl TP, Drazen DL, Seeley RJ, D’Alessio DA, Woods SC. Meal-Anticipatory Glucagon-Like Peptide-1 Secretion in Rats. Endocrinology. 2010;151(2):569–575. doi: 10.1210/En.2009-1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderah TW, Suenaga NM, Ossipov MH, Malan TP, Jr, Lai J, Porreca F. Tonic descending facilitation from the rostral ventromedial medulla mediates opioid-induced abnormal pain and antinociceptive tolerance. Journal of Neuroscience. 2001;21(1):279–286. doi: 10.1523/JNEUROSCI.21-01-00279.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waddington CH. The strategy of the genes; a discussion of some aspects of theoretical biology. London: Allen & Unwin; 1957. [Google Scholar]
- Waddington CH. Towards a theoretical biology; an IUBS symposium (International Union of Biological Sciences) Vol. 1. Edinburgh: Edinburgh University Press; 1968. (prolegomena) [Google Scholar]
- Werner J. System properties, feedback control and effector coordination of human temperature regulation. European Journal of Applied Physiology. 2010;109(1):13–25. doi: 10.1007/s00421-009-1216-1. [DOI] [PubMed] [Google Scholar]
- White JM. Pleasure into pain: the consequences of long-term opioid use. Addictive Behaviors. 2004;29(7):1311–1324. doi: 10.1016/j.addbeh.2004.06.007. [DOI] [PubMed] [Google Scholar]
- Wiener N. Cybernetics. New York: J. Wiley; 1948. [Google Scholar]
- Wirtshafter D, Davis JD. Set points, settling points, and the control of body weight. Physiology & Behavior. 1977;19(1):75–78. doi: 10.1016/0031-9384(77)90162-7. [DOI] [PubMed] [Google Scholar]
- Woods SC. The eating paradox: how we tolerate food. Psychological Review. 1991;98(4):488–505. doi: 10.1037/0033-295x.98.4.488. [DOI] [PubMed] [Google Scholar]
- Woods SC. The control of food intake: behavioral versus molecular perspectives. Cell Metabolism. 2009;9(6):489–498. doi: 10.1016/j.cmet.2009.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods SC, Decke E, Vasselli JR. Metabolic Hormones and Regulation of BodyWeight. Psychological Review. 1974;81(1):26–43. doi: 10.1037/H0035927. [DOI] [PubMed] [Google Scholar]
- Woods SC, Ramsay DS. Pavlovian influences over food and drug intake. Behavioural Brain Research. 2000;110(1–2):175–182. doi: 10.1016/s0166-4328(99)00194-1. [DOI] [PubMed] [Google Scholar]
- Woods SC, Ramsay DS. Homeostasis: beyond Curt Richter. Appetite. 2007;49(2):388–398. doi: 10.1016/j.appet.2006.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods SC, Strubbe JH. The Psychobiology of Meals. Psychonomic Bulletin & Review. 1994;1(2):141–155. doi: 10.3758/Bf03200770. [DOI] [PubMed] [Google Scholar]
- Woods SC, Vasselli JR, Kaestner E, Szakmary GA, Milburn P, Vitiello MV. Conditioned Insulin-Secretion and Meal Feeding in Rats. Journal of Comparative and Physiological Psychology. 1977;91(1):128–133. doi: 10.1037/H0077307. [DOI] [PubMed] [Google Scholar]
- Wyndham CH, Atkins AR. A physiological scheme and mathematical model of temperature regulation in man. Pflügers Arch. 1968;303(1):14–30. doi: 10.1007/BF00586824. [DOI] [PubMed] [Google Scholar]
- Yang Y, Gordon CJ. Ambient temperature limits and stability of temperature regulation in telemetered male and female rats. Journal of Thermal Biology. 1996;21(5–6):353–363. doi: 10.1016/S0306-4565(96)00021-6. [DOI] [Google Scholar]
- Yates FE. The 10th J. A. F. Stevenson memorial lecture. Outline of a physical theory of physiological systems. Canadian Journal of Physiology and Pharmacology. 1982;60(3):217–248. doi: 10.1139/y82-035. [DOI] [PubMed] [Google Scholar]
- Yates FE. Order and Complexity in Dynamical-Systems - Homeodynamics as a Generalized Mechanics for Biology. Mathematical and Computer Modelling. 1994;19(6–8):49–74. doi: 10.1016/0895-7177(94)90189-9. [DOI] [Google Scholar]
- Yates FE. Homeostasis. In: Birren JE, editor. Encyclopedia of gerontology: age, aging, and the aged. Vol. 1. San Diego: Academic Press; 1996. pp. 679–686. [Google Scholar]
- Yates FE. Homeokinetics/homeodynamics: A physical heuristic for life and complexity. Ecological Psychology. 2008;20(2):148–179. doi: 10.1080/10407410801977546. [DOI] [Google Scholar]