

Abstract
The charge-transporting activity of the Na+,K+-ATPase depends on its surrounding electric field. To isolate which steps of the enzyme’s reaction cycle involve charge movement, we have investigated the response of the voltage-sensitive fluorescent probe RH421 to interaction of the protein with BTEA (benzyltriethylammonium), which binds from the extracellular medium to the Na+,K+-ATPase’s transport sites in competition with Na+ and K+, but is not occluded within the protein. We find that only the occludable ions Na+, K+, Rb+, and Cs+ cause a drop in RH421 fluorescence. We conclude that RH421 detects intramembrane electric field strength changes arising from charge transport associated with conformational changes occluding the transported ions within the protein, not the electric fields of the bound ions themselves. This appears at first to conflict with electrophysiological studies suggesting extracellular Na+ or K+ binding in a high field access channel is a major electrogenic reaction of the Na+,K+-ATPase. All results can be explained consistently if ion occlusion involves local deformations in the lipid membrane surrounding the protein occurring simultaneously with conformational changes necessary for ion occlusion. The most likely origin of the RH421 fluorescence response is a change in membrane dipole potential caused by membrane deformation.
Introduction
In muscle and nerve cells, rapid changes in transmembrane electrical potential (Vm) occur via voltage-dependent Na+- and K+-channels. Following an action potential, the Na+,K+-ATPase reestablishes the resting Na+ and K+ concentration gradients across the membrane. Therefore, for a complete understanding of nerve and muscle function, it is crucial to identify this enzyme’s transmembrane voltage-dependent steps.
Transmembrane voltage dependence of the overall Na+,K+-pump cycle turnover has been observed in some cells (1–8). The most popular explanation for this is that the transported ions pass a narrow access channel to enter and leave their binding sites on the protein’s extracellular face (9–12). As the ions migrate through the channel-like structure to reach their binding sites, they would pass through some of the electric field created across the membrane by the electrical transmembrane potential, and hence, binding would be voltage-dependent.
After the Na+,K+-ATPase binds K+ ions from the extracellular medium, the ions are occluded within the protein. The term “occlusion” represents an entrapment of the ion within the protein matrix so that access to either the cytoplasm or the extracellular medium is limited. Occlusion is an effective way of preventing simultaneous access of the transported ions to both the cytoplasm and the extracellular medium, which would seriously diminish the effectiveness of ion pumping.
Although most if not all of available electrophysiological data on intact cells can be explained within the framework of transmembrane voltage-dependent binding of ions within an extracellular access channel, some data on purified open membrane fragments containing the Na+,K+-ATPase point toward charge movement associated with occlusion reactions. For example, Ganea et al. (13) found that the kinetics of the conformational change E1P(Na+)3 → E2PNa+3 (whereby Na+ ions are deoccluded to the extracellular face of the protein) are significantly slowed by the presence of an intramembrane electric field produced by bound perchlorate ions. These results suggest that extracellular Na+ deocclusion is associated with movement of charge. More recently, Myers et al. (14) found that the membrane-bound voltage-sensitive fluorescent probe RH421 (closely related to di-8-ANEPPS) responds to K+ occlusion by the E2P form of the enzyme with a fluorescence change, thus suggesting that K+ occlusion from the extracellular medium involves charge movement. Thus, measurements on the native enzyme in intact cells and purified enzyme in open membrane fragments seem to be yielding different information concerning the reaction steps in which charge movement is occurring.
In 2004, Peluffo et al. (15) suggested a new means of discriminating between binding and occlusion as the origin of transmembrane voltage-dependence. They proposed that, by studying the voltage-dependence of inhibition of the Na+,K+-ATPase by quaternary organic ammonium ions, the voltage-dependent step could be isolated. It was already known that certain ions of this type inhibit the Na+,K+-ATPase competitively with extracellular K+ ions (16). Forbush (17) found that benzyl-substituted quaternary ammonium ions, e.g., BTEA (benzyltriethylammonium), were particularly effective in inhibiting the kinetics of release of occluded Rb+ ions, and the study’s results were consistent with the ammonium ions being able to bind to the extracellular transport sites but without undergoing occlusion. Based on the evidence that BTEA cannot be occluded, Peluffo et al. (15) reasoned that if its inhibition of the Na+,K+-ATPase was voltage-dependent, it would be clear evidence for voltage-dependent ion binding. This, in fact, was the result they found (15,18,19).
To investigate the possibility of charge movement associated with occlusion, here we use the same tool as used by Peluffo et al. (15), the BTEA ion. However, instead of using isolated cells (Peluffo et al. (15) used cardiac myocytes), we investigate the interaction with purified Na+,K+-ATPase-containing open membrane fragments, i.e., the system from which all evidence for charge movement associated with occlusion has come. To detect any changes in electric field caused by BTEA binding, we use the voltage-sensitive fluorescent membrane probe RH421. The fluorescence of this probe is known to be sensitive to changes in local electric field in the membrane caused by movement of charged or dipolar species (20,21). Styrylpyridinium dyes of this type were first used to follow the kinetics of partial reactions of the Na+,K+-ATPase by Klodos and Forbush (22,23), and have been widely used since that time (13,24–29). If ion binding is a voltage-dependent reaction and causes a change in local electric-field strength in the adjacent membrane, one would expect binding of BTEA to the Na+,K+-ATPase to cause a drop in RH421 fluorescence, as has been observed after addition of K+ (14,30,31). In fact, as we will show here, this is not what is observed; only occludable ions (K+, Rb+, or Cs+) cause a drop in fluorescence. At first glance, this seems to suggest ion binding is not a voltage-dependent reaction, and that only occlusion is—which is in contradiction to the results of Peluffo et al. (15,18) and Peluffo and Berlin (19). However, all of the results can in fact be reconciled if one makes a distinction regarding the origin of the electric field influencing the kinetics of different reaction steps of the Na+,K+-ATPase, i.e., whether the electric field influencing a particular reaction comes from the transmembrane electrical potential difference, or it originates from intramembrane electrical charges.
Materials and Methods
Enzyme and reagents
Na+,K+-ATPase-containing open membrane fragments from pig kidney were purified as described by Klodos et al. (32). The specific ATPase activity at 37°C and pH 7.4 was measured according to Ottolenghi (33) to be 2020 μM ATP hydrolyzed h−1 (mg of protein)−1 at saturating substrate concentrations. The protein concentration was 4.98 mg mL−1 according to the Peterson modification (34) of the Lowry method (35) using bovine serum albumin as a standard.
RH421 (N-(4-Sulfobutyl)-4-(4-(p-(dipentylamino)phenyl)butadienyl)-pyridinium salt) was obtained from Molecular Probes (Eugene, OR) and used without further purification. RH421 was added to Na+,K+-ATPase-containing membrane fragments from an ethanolic stock solution. The dye spontaneously partitions into membranes.
DMPC (dimyristoylphosphatidylcholine) was obtained from Auspep (Parkville, Australia). Unilamellar vesicles were prepared by the ethanol injection method described in Zouni et al. (36,37). Dialysis tubing was purchased from Spectrum Laboratories (Rancho Dominguez, CA). The DMPC concentration of the vesicle suspension was 3 mM, determined by a Phospholipid-C test kit (Novachem, Collingwood, Australia). All fluorescence measurements carried out using vesicles were performed in buffer containing 30 mM Tris (tris[hydroxymethyl]aminomethane), 1 mM EDTA, and 150 mM NaCl. The pH was adjusted to 7.2 with HCl.
The origins of the reagents used were: BTEA chloride (99%, Sigma, Castle Hill, Australia); imidazole (≥99%; Sigma); Tris (minimum 99.9%, Sigma); ouabain octahydrate (≥95%, Sigma); NaCl (suprapure grade, Merck, Kilsyth, Australia); KCl (analytical grade, Merck); RbCl (AnalaR BDH, Merck); CsCl (99.9%, Alfa Aesar, Ward Hill, MA); MgCl2·6H2O (analytical grade, Merck); EDTA (99%, Sigma); ATP disodium·3H2O (special quality, Roche, Castle Hill, Australia); NaOH (analytical grade, Merck); and HCl (0.1 N Titrisol solution, Merck).
Proteolytic cleavage, gel electrophoresis, and immunoblotting
Extensive proteolysis was performed in a reaction mixture containing 100 μg of purified Na+,K+-ATPase membrane fragments in 30 mM histidine, pH 7.1, and 1 mM EDTA, in the presence of either 20 mM KCl or BTEA. Membranes were preincubated for 5 min at 24°C before addition of 20 μg of trypsin, and the mixtures were incubated at 37°C for increasing time intervals, after which proteolysis was terminated with SDS (sodium dodecyl sulfate) sample buffer (140 mM Tris, 20% glycerol, 2% SDS, and 0.01% Coomassie Blue R250, pH 7.2) containing 1% trichloroacetic acid to irreversibly inhibit the protease. Posttryptic membranes were analyzed by SDS-polyacrylamide gel electrophoresis (PAGE) overnight at 150 V and 12 mA per gel. A quantity of 10 μg of protein was loaded onto 8% SDS-PAGE, and protein fragments on the gel were transferred to PVDF membranes and visualized by Western blotting using a C-terminal α-subunit antibody (peptide 1002-1016), as described in Cornelius et al. (38).
Fluorescence measurements
Fluorescence measurements were carried out with a model No. RF-5301 PC spectrofluorophotometer (Shimadzu, Kyoto, Japan) using quartz semimicro cuvettes. The temperature was maintained at 30°C via a circulating water bath. The value λex was 577 nm (bandwidth 5 nm) with an OG530 filter (Schott Advanced Optics, Duryea, PA) in the excitation path. λem was 670 nm (bandwidth 5 nm) with an RG645 filter (Schott Advanced Optics) in front of the photomultiplier.
The relative fluorescence change, ΔF/F0, of membrane-bound RH421 associated with interaction of either K+ or BTEA with the phosphorylated form of the Na+,K+-ATPase was determined by measuring the change in fluorescence, ΔF, of a suspension of open Na+,K+-ATPase-containing membrane fragments noncovalently labeled with RH421 and preequilibrated with 2 mM Na2ATP after K+ or BTEA addition and then dividing ΔF by the fluorescence before K+ or BTEA addition, F0. All measurements were performed in buffer containing 30 mM imidazole, 5 mM MgCl2, 1 mM EDTA, and 150 mM NaCl. The buffer pH was adjusted to 7.4 with HCl, i.e., >4 pH units above the apparent pKa of membrane-bound RH421 of 3.1 (39). The final ΔF/F0 values were corrected for dilution. The RH421 concentration used was always <1 μM to avoid dye-induced inhibition of Na+,K+-ATPase function (40).
In the absence of K+ ions, the enzyme cycle is inhibited at the dephosphorylation step (41); hence, in the buffer solution described above, the enzyme accumulates in a phosphorylated state. The NaCl, MgCl2, and ATP concentrations of the buffer were chosen so that enzyme phosphorylation proceeds at its maximum rate, thus ensuring the maximum degree of phosphorylation of enzyme before addition of either K+ or BTEA (26,42). The NaCl concentration of 150 mM is, however, far below the half-saturating Na+ concentration for extracellular binding of ∼400 mM (43), so that the majority of the enzyme is expected to be in the E2P state before K+ or BTEA addition. Simulations based on a recently published kinetic model (44) indicate that under the buffer conditions used here, 84% of the enzyme would be in an E2P state (40% E2P, 30% E2PNa+, 6% E2PNa+2, and 8% E2PNa+3), 15% in the E1P(Na+)3 state, and <1% in unphosphorylated states.
Electrostatic calculation of intramembrane electric field strength
To calculate the extent of change in electric-field strength due to ion binding, the Poisson-Boltzmann (PB) equation was solved using the PBEQ utility of CHARMM (45). Two structures of the Na+,K+-ATPase, PDB:2ZXE with two bound K+ ions (46) and PDB:3WGU with three bound Na+ ions (47), were obtained from the Orientations of Proteins in Membranes (OPM) database (http://opm.phar.umich.edu/) (48), positioned and aligned relative to a membrane, and the change in electrostatic potential around the protein due to ion binding was calculated.
The membrane was modeled as a 25 Å-thick low-dielectric (εm = 2) slab, mimicking a typical membrane bilayer hydrocarbon region, with the interfacial region modeled with the same high dielectric as bulk water (εi = 80), as suggested by studies of a heterogeneous lipid bilayer (49). Bulk solvent was represented by a high dielectric (εw = 80) with a 150 mM 1:1 ionic solution. A range of experimental and computational studies have yielded estimates for the dielectric constant of protein ranging from 2–3 to 10–30 (near the protein surface) (50–53). For these calculations, we chose εp = 2 and 10, to provide a range of extents of shielding of the electric fields due to ion binding. The solvent-excluded protein region was determined with a water-sized reentrant probe (54), which was assigned a dielectric constant εp. A map of the assignments of dielectric constants can be seen in the left columns of Fig. S1 and Fig. S2 in the Supporting Material.
All atomic charges, except those on the ions of interest, were removed to isolate their contribution to the electrostatic potential. The linear PB equation was solved for all systems on a grid with 0.5 Å spacing. B-spline interpolation was used to interpolate charge on the grid. Periodic boundary conditions were used, imposing a rectangular box with x and y dimensions (i.e., in the plane of the membrane) of 100 × 100 Å to simulate the known density of protein (55) present in the membrane fragments used for the experimental measurements. For each protein crystal structure used, the height of the box (z dimension, perpendicular to the membrane surface) was chosen to cover the whole protein plus an extra 10 Å. The definitions of axes and coordinates used were those provided in the structures by the OPM database. The full three-dimensional potential was calculated, although for visualization, specific projections have been chosen. Maps of the electrostatic potential and electric-field strength were produced for y = 0, and are representative of other projections. For ease of comparison with experimental data, the electric field within the membrane was computed by averaging over the grid outside the protein boundary, represented as a function of z in Fig. S3.
Results
Effect of BTEA on tryptic cleavage of the Na+,K+-ATPase
Extensive trypsin cleavage in the presence of K+ completely removes cytoplasmic protein domains of the Na+,K+-ATPase, but leaves intact membrane-embedded trans-membrane protein domain pairs. These are referred to as 19-kDa fragments, of which the C-terminal fragment comprising TM7-TM10 is the largest intact part of the α-subunit after proteolysis (56). Occluded K+ is essential to obtain an intact 19-kDa C-terminal fragment. Incubation of enzyme with trypsin in the presence of ions that are not occluded results in complete cleavage of the 19-kDa fragment. Therefore, an analysis of tryptic cleavage patterns in the presence of BTEA allows one to determine whether that ion is occluded by the Na+,K+-ATPase.
Na+,K+-ATPase-containing membrane fragments were equilibrated with either K+ or BTEA, and tryptic cleavage was carried out as described under Materials and Methods. As shown in Fig. 1, although BTEA (20 mM) produced a cleavage pattern similar to K+ during the initial incubation period (5–10 min), it failed to protect the 19-kDa C-terminal fragment (i.e., the α-subunit of the Na+,K+-ATPase was completely cleaved at times >20 min). In contrast, in the presence of the same concentration of K+, the 19-kDa fragment is resistant against cleavage even after 90 min. Occluded K+ tethers the transmembrane domains and protects the C-terminal third of the α-subunit. An ion that binds at the binding site but is not occluded will not protect the membrane-embedded fragments. Hence, although BTEA initially binds in the ion-binding path, it fails to occlude in a K+-like manner and is not able to protect membrane-embedded domains. This supports the results of Forbush (17) and Peluffo et al. (18), based on kinetic data, that although BTEA competes with K+ for binding to the same sites, it does not allow occlusion.
Figure 1.
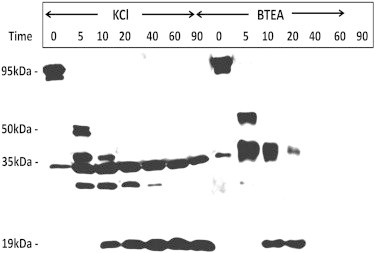
Immunoblot using C-terminal antibody against the α-subunit. Proteolytic cleavage, SDS-PAGE, and immunoblotting were performed as described under Materials and Methods. Incubation with trypsin was allowed to proceed for the indicated time intervals (minutes) before quenching with SDS sample buffer with TCA. Zero time indicates control samples where SDS sample buffer/TCA was added before addition of trypsin.
Interaction of BTEA with lipid vesicles and RH421
Before testing the interaction of BTEA with the Na+,K+-ATPase, we tested for any direct interaction of BTEA with RH421 and with lipid membranes. BTEA itself was found to exhibit no observable fluorescence when excited with the same wavelength used for RH421 (577 nm). The addition of BTEA up to a concentration of 10 mM to DMPC vesicles noncovalently labeled with 0.5 μM of RH421 was found to cause no observable change in the fluorescence of RH421 when excited at 577 nm and the emission was detected at 670 nm (the same wavelengths used for subsequent measurements using Na+,K+-ATPase-containing membrane fragments). Therefore, either BTEA does not bind to the lipid phase of membranes or, if it does, it does not interfere with the fluorescence of RH421. This is in contrast to the situation in aqueous solution. Addition of BTEA (up to 10 mM) to 0.5 μM RH421 in imidazole buffer was found to cause an increase in fluorescence intensity (up to ∼30%) of RH421 across its entire fluorescence emission spectrum.
A likely cause is interaction between the benzene ring of BTEA and the chromophore of RH421. In aqueous solution, RH421 aggregates strongly, quenching its fluorescence (37). Interaction between RH421 and BTEA would, thus, increase the separation between RH421 molecules and would be expected to decrease the degree of quenching. A major reason for the high fluorescence of RH421 when bound to lipid membranes is dye binding in monomeric form to the membrane coupled to dye disaggregation in the aqueous phase (37). For comparison, addition of vesicles (250 μM lipid) to RH421 under the same conditions was found to cause a 680% increase in fluorescence (i.e., a 23-fold-greater increase than that caused by BTEA). The magnitude of the large increase in fluorescence due to vesicle addition is comparable with previous data using different excitation and emission wavelengths (36). Thus, lipid is much more effective than BTEA in decreasing fluorescence quenching of the dye.
Interaction of BTEA with phosphorylated Na+,K+-ATPase
Addition of BTEA to Na+,K+-ATPase-containing membrane fragments labeled with RH421 and phosphorylated with ATP causes a small but statistically significant (P < 0.01, Wilcoxon signed rank test) increase in the fluorescence level (see Fig. 2). Fitting a single-site binding model to the BTEA data according to
| (1) |
yields an apparent dissociation constant, Kd, of 0.63 (±0.10) mM and a maximum relative fluorescence change at saturating BTEA concentrations, (ΔF/F0)max, of 0.055 (±0.002). To determine whether the fluorescence increase caused by BTEA is related to an interaction with the Na+,K+-ATPase we carried out a control experiment in which 1.5 mM ouabain was added to the enzyme before BTEA addition. Ouabain is known to bind preferentially to the E2P form of the enzyme, and inhibit extracellular K+ binding (38). In the presence of ouabain, the fluorescence increase caused by BTEA was almost completely abolished.
Figure 2.
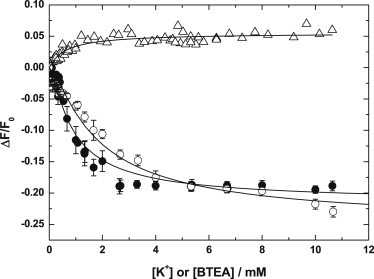
Concentration dependencies of relative fluorescence changes, ΔF/F0, accompanying the addition of either BTEA chloride or KCl to Na+,K+-ATPase-containing membrane fragments (33 μg/mL or 0.23 μM) noncovalently labeled with RH421, 50 nM. In measurements in which either BTEA or K+ were added alone, F0 refers to the fluorescence level before the addition of either BTEA or K+ ions. In measurements in which K+ was added at a constant [BTEA], F0 refers to the fluorescence level before the addition of K+. The three titrations shown are: BTEA alone (Δ), K+ alone (●), and K+ in the presence of 4 mM BTEA (○). (Solid lines) Nonlinear least-squares fits to the data of Eq. 1, in the case of the BTEA titration, or Eq. 2 in the case of both K+ titrations. Each measurement for both K+ titrations was performed in triplicate; (plotted points) average value. Each measurement for the BTEA titration was performed once, but a much larger number of BTEA concentrations were used to allow for a statistical test on the significance of the small fluorescence increase observed.
The small increase in fluorescence caused by BTEA is in stark contrast to the behavior observed when Na+ or K+ interacts with the protein. The release of Na+ ions from the phosphorylated form of the protein causes a significant increase in the fluorescence of RH421 (24,26,29). The interaction of K+ ions with the phosphorylated form of the protein causes a significant drop in the fluorescence of RH421 (14,30,31). Therefore, if binding alone of Na+ or K+ to ion transport sites on the E2P state of the protein (i.e., before occlusion) were causing a change in intramembrane electric-field strength detectable by RH421, one would expect also binding of BTEA to the protein to cause a drop in fluorescence, not an increase.
Because BTEA is bound but not occluded, the logical conclusion is, thus, that the drop in RH421 fluorescence caused by interaction of Na+ or K+ with the phosphorylated protein is in fact due to ion occlusion, i.e., the formation of E1P(Na+)3 in the case of Na+ and the formation of E2P(K+)2 in the case of K+, not ion binding per se.
A remaining question is the origin of the small increase in RH421 fluorescence caused by BTEA addition. However, with ion occlusion as the cause of the decrease in fluorescence arising from Na+ or K+ interaction with the protein, this can easily be explained. In the absence of K+ ions, a series of sequential coupled equilibria would exist linking the states E1P(Na+)3, E2PNa+3, E2PNa+2, E2PNa+, and E2P due to the presence of Na+ needed to support phosphorylation by ATP. According to Le Châtelier’s principle, binding of BTEA to the E2P state would lead to a shift away from the E1P(Na+)3 state toward E2P, and hence further deocclusion of Na+. The drop in the amount of enzyme in an occluded state would be expected to lead to a slight increase in fluorescence. As described under Materials and Methods, before BTEA addition computer simulations of the Na+,K+-ATPase cycle (44) indicate that ∼15% of the enzyme is expected to occupy the E1P(Na+)3 state. Thus, the increase in RH421 fluorescence caused by BTEA addition and a consequent drop in Na+ occlusion is entirely consistent with the decrease in fluorescence cause by K+ addition and the resulting increase in K+ occlusion.
Finally, a further conclusion from the experimental results is that, because RH421 detects local electric-field-strength changes, ion occlusion by the Na+,K+-ATPase must involve a movement of charge, as previously proposed by Ganea et al. (13). Occlusion must involve a conformational change of the protein, which moves charge either away from or toward RH421. However, the exact path of the charge movement, parallel or perpendicular to the membrane face, will be absolutely critical for the fluorescence response, and the path followed does not necessarily have to be across the membrane (i.e., perpendicular to the surface), as will be discussed later.
Interaction of K+, Rb+, and Cs+ with phosphorylated Na+,K+-ATPase
The effect of K+ addition to phosphorylated Na+,K+-ATPase on the level of RH421 fluorescence is shown in Fig. 2. There it can be seen that at saturating K+ concentrations the drop in fluorescence caused is 4–5 times greater than the increase in fluorescence caused by BTEA. The fluorescence drop caused by K+ is completely abolished if the enzyme is preincubated with 1.5 mM ouabain, indicating that it is due to interaction with the Na+,K+-ATPase.
The concentration dependence of the relative fluorescence change, ΔF/F0, could be fitted to the equation
| (2) |
where Kd is the apparent microscopic dissociation constant of the K+ ions. This equation is based on a model in which a fluorescence change is assumed to occur only after two K+ ions have already bound to the E2P state of the enzyme. This is consistent with recent stopped-flow results of Myers et al. (14), in which only a single kinetic phase of K+ occlusion was observed although it is known that two K+ ions are occluded. An attempt was also made to fit two identical independent binding-site models to the experimental data, but the goodness-of-fit was much improved by use of Eq. 2. For example, the χ-squared value was reduced to ∼40% of that obtained by the two independent identical binding-site models (mathematically described by an expression analogous to Eq. 1). This result, therefore, gives further support to the conclusion that K+ occlusion is the cause of the RH421 fluorescence change and that for the pig kidney enzyme used here, occlusion only occurs once two K+ ions have bound. The apparent K+ microscopic dissociation constant determined from the fitting was 0.42 (±0.03) mM, and the value of (ΔF/F0)max was found to be −0.215 (±0.005). It must be noted, however, that the value of Kd is an apparent value because we have not taken into account competition from Na+ or Mg2+ ions, which are present in the solution to allow phosphorylation by ATP, and the K+ binding equilibria would also be perturbed by the subsequent occlusion and dephosphorylation reactions in the Na+,K+-ATPase cycle. A true estimation of the affinity of the ion binding sites for K+ can only be obtained from kinetic studies.
The fit obtained to the data shown in Fig. 2 is still not perfect. Particularly in the region 1–4 mM K+, there appears to be a systematic deviation between the experimental data points and the theoretical curve. This could be due to positive cooperativity in K+ binding. However, because the dissociation constants derived from the fitting are in any case only apparent values (as explained above), adding further complexity to the model to obtain a better fit will not yield further information of any significant value.
We also tested the effect of addition of Rb+ and Cs+ ions to phosphorylated Na+,K+-ATPase. At a concentration of 20 mM, RbCl and CsCl both caused decreases in fluorescence. The values of (ΔF/F0) were −0.165 (±0.008) and −0.156 (±0.009) for Rb+ and Cs+, respectively. For comparison, the value for K+ at the same concentration was −0.193 (±0.006). Therefore, although all three ions cause drops in RH421 fluorescence, the effectiveness appears to be K+ > Rb+ ≈ Cs+.
The differences in the effects of the interaction of K+, Rb+, and Cs+ with the Na+,K+-ATPase in comparison with the interaction of BTEA with the protein are most likely due to different sizes of the ions. K+, Rb+, and Cs+ have ionic diameters of 2.76, 3.04, and 3.34 Å, respectively (57). BTEA, on the other hand, is nonspherical and its longest linear dimension is ∼9.0 Å, roughly three times the size of the metal ions. Its larger size probably prevents the protein conformational change required for ion occlusion, as suggested by Forbush (17).
Interaction of K+ with phosphorylated Na+,K+-ATPase in the presence of BTEA
To obtain further information on the interaction of BTEA with the phosphorylated protein, we carried out titrations of the Na+,K+-ATPase with K+ in the presence of BTEA (see Fig. 2). As the concentration of BTEA is increased, the affinity of the enzyme for K+ decreases. At a BTEA concentration of 4 mM, the apparent K+ dissociation constant of the protein from fitting to Eq. 2 was found to be 0.86 (±0.07) mM, i.e., twice the value found from the titration with K+ in the absence of BTEA.
This result is consistent with BTEA binding to the Na+,K+-ATPase in competition with K+, as previously suggested by others (15–19). Competition does not by itself imply that BTEA binds to the same sites as K+. Competitive binding would also be observed if BTEA acted as a plug, blocking the access channel and preventing K+ from reaching transport sites situated more deeply in the protein interior. However, the finding of Peluffo et al. (15,18) that inhibition of the Na+,K+-ATPase by BTEA is voltage-dependent and dissipates a similar fraction of the transmembrane electric field as extracellular K+ binding reactions is a strong indication that BTEA does itself penetrate deeply into the extracellular access channel. Thus, all of the data are in accord with the conclusion that BTEA binds to transport sites from the extracellular face of the protein but that the electric field caused by its binding is not sensed by RH421 in the adjacent lipid phase of the membrane.
The value of (ΔF/F0)max found from the fitting was −0.250 (±0.007). The magnitude of the maximum fluorescence drop caused by addition of K+ is thus 0.035 greater in the presence of 4 mM BTEA than in its absence. This greater fluorescence drop can be explained by K+ ions not only being occluded but also driving BTEA out of the binding sites. The results of the BTEA titration in Fig. 2 show that BTEA itself causes an increase in fluorescence. Therefore, the K+ titration in the presence of BTEA starts at a higher initial fluorescence level than in the absence of BTEA. Because of this, although the final fluorescence level is the same at saturating K+ concentrations for both titrations, the maximum relative fluorescence change in the presence of BTEA is greater.
Electrostatic calculation of intramembrane field-strength changes induced by extracellular ion binding
If, as the results presented above suggest, RH421 does not detect the electric-field-strength change induced by extracellular ion binding, the question arises why this is so. Bühler et al. (58) previously estimated the magnitude of the intramembrane electric-field-strength change produced by the release of Na+ ions from Na+,K+-ATPase-containing membrane fragments at the extracellular face. Considering the entire membrane (protein and surrounding lipid) as a homogeneous dielectric medium, they calculated the electric-field-strength change, ΔE, in a plane within the membrane at the depth where the ions are located when 3 Na+ ions are released from the E2P state to the extracellular medium using
| (3) |
where np is the number of elementary charges per unit area located at a distance r from the cytoplasmic interface of the membrane, e0 is the elementary charge, ε0 is the permittivity of vacuum, ε is the dielectric constant of the membrane, and d is the membrane thickness. The factor (1 – r/d) represents the fractional distance across the membrane that the Na+ ions must pass from their binding sites to the extracellular medium. Depending on the depth at which the ions are located, Bühler et al. (58) estimated a value of ΔE in the range 5 × 107–3 × 108 V m−1. This range was based on an upper limit of the surface density of Na+,K+-ATPase molecules in the membrane of 1 × 1016 m−2 (55), and hence a value of np for 3 bound Na+ ions of 3 × 1016 m−2 and a membrane dielectric constant of 2.
At the time Bühler et al. (58) performed their calculations, no structural data at atomic resolution was available for the Na+,K+-ATPase. There are other major limitations of the model just described. In fact, the membrane is far from being a homogeneous dielectric and the K+ ions are not uniformly distributed across a plane within the membrane. The membrane fragments consist of individual protein molecules embedded within a lipid bilayer membrane, and the K+ ions are only bound to the protein. Furthermore, comparison of the spectral properties of RH421 in homogenous solvents to those bound to lipid vesicles have shown (39) that the membrane-bound dye experiences a dielectric constant in the range 70–80, i.e., close to water and far higher than that assumed in the above calculation. Therefore, a more accurate calculation of the electric field strength produced in the lipid phase of the membrane by protein-bound K+ ions would have to consider these facts.
We now explore this question using continuum electrostatics calculations of the fully atomistic protein structures for the Na+,K+-ATPase to determine the electric-field strengths generated by the binding of extracellular ions. Calculations were done for two conformational states: E2∙Pi∙2K+ (PDB:2ZXE) with the binding of 2 K+ ions (46), and E1∼P∙ADP∙3Na+ (PDB:3WGU) with the binding of 3 Na+ ions (47). The value of the electric-field strength perpendicular to the membrane bilayer was calculated from the electrostatic potential obtained from the solution to the PB equation, with results shown in Fig. 3 a (PDB:2ZXE) and Fig. 3 b (PDB:3WGU), for protein dielectric constant, εp = 10. Results for the scalar potential and electric field strength for both dielectric constants, εp = 2 and 10, are shown in Fig. S1 and Fig. S2. Although the electric-field strength can be as large as 108 V m−1 at the protein-membrane boundary (for the 3 Na+ ion binding case, but much lower for the 2 K+ ion case), it decays by an order of magnitude within 20 Å laterally from the protein-membrane boundary. Importantly, the electric field decays rapidly in the high dielectric membrane interfacial region, being an order-of-magnitude lower, and suggesting little interaction with field-sensitive RH421 molecules.
Figure 3.
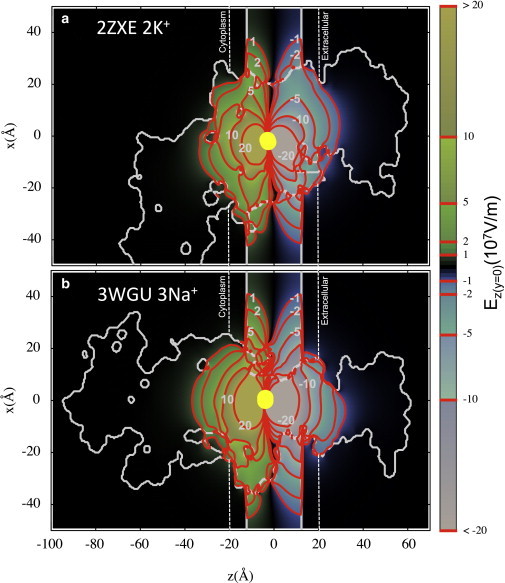
Electric field calculated for PDB:2ZXE due to binding of 2 K+ ions (a), and for PDB:3WGU due to binding of 3 Na+ ions (b), in units of 107 V/m (truncated at 20 × 107 V/m). (Solid vertical lines) Boundaries for the implicit membrane (dielectric constant εm = 2), with interfacial region (with εi = 80) indicated (between the dashed and solid lines). The protein was assigned a dielectric constant εp = 10 (with alternative εp = 2, shown in Fig. S1 and Fig. S2 in the Supporting Material). (Solid curves) Protein boundary. (Yellow circles) Bound ions. Additional calculations are provided in Fig. S1, Fig. S2, and Fig. S3. To see this figure in color, go online.
To better relate to our experiments, the total electric-field strength and its average components perpendicular to the membrane are shown in Fig. S3. Periodic boundary conditions that assume an experimental density of protein in membrane (55) are taken into account, corresponding to one protein every 100 × 100 Å. The electric field averaged outside the protein in slabs parallel to the membrane is ∼107 V m−1 in the membrane core, decaying in the interfacial region to 106 V m−1. If this field strength were across the entire membrane thickness of ∼4 nm, it would correspond to a transmembrane electrical potential of 4 mV. From measurements on intact cells, it is known that typical relative fluorescence changes for dyes of this class are in the range 5–20%/100 mV (59). Taking 20%/100 mV as an upper limit, then for a 4 mV change in Vm, one would expect a relative fluorescence change of 0.8%.
Experimentally, measured relative fluorescence changes on Na+,K+-ATPase-containing membrane fragments sometimes exceed 100% (24,42,58). Thus, the measured fluorescence responses vastly exceed what would be expected from a transmembrane electric field strength change. This demonstrates that RH421 is unlikely to detect the electric fields resulting from the binding of extracellular ions. This theoretical result explains the experimental finding that ion binding to the extracellular transport sites of the Na+,K+-ATPase does not cause a drop in RH421 fluorescence per se. However, this does not necessarily mean that Na+ or K+ binding to the E2P state is not an important charge-transporting reaction step of the enzyme’s reaction cycle.
Discussion
The results presented here show that interaction of the occludable ion K+ with the Na+,K+-ATPase in the E2P state causes a significant drop in fluorescence of the voltage-sensitive membrane probe RH421, in confirmation of previous results (14,30,31). Schvartz et al. (28) reported that the occludable Rb+ ion also stimulates a drop in RH421 fluorescence when it interacts with the E2P state of the enzyme. This agrees with our own findings reported here. Furthermore, we have found that the Cs+ ion, which is also known to substitute for extracellular K+, becomes occluded and undergoes transport into the cell cytoplasm (60,61), and stimulates a drop in RH421 fluorescence. The nonoccludable ion BTEA, on the other hand, does not cause a drop in RH421 fluorescence. Nevertheless, BTEA decreases the binding affinity of K+ with the E2P state, indicating that although it is not occluded, it competes with K+ for binding to the same sites on the protein. This is in agreement with previous studies using other techniques (15–19). The results obtained here, therefore, indicate that RH421 responds to ion occlusion, not to ion binding.
This finding has important structural implications for the functioning of the Na+,K+-ATPase. RH421 is a voltage-sensitive dye that binds to lipid membranes and responds to its local electric field strength. It is known to be particularly sensitive to the amount of positive charge within the membrane at the level of the glycerol backbone of the lipid bilayer (20,21,58). For this reason, it, and related styrylpyridinium dyes, such as di-8-ANEPPS, have widely been used as probes of the membrane dipole potential, which has a value in the range 200–400 mV, is positive in the membrane interior, and whose value depends on lipid structure, e.g., headgroup and degree of chain saturation (20,21,62–64). The fact that occlusion of K+ by the E2P state of the Na+,K+-ATPase causes a drop in fluorescence of RH421 indicates that this reaction must be associated with a redistribution of charge within the membrane, and more precisely with an increase in the amount of positive charge in the vicinity of the dye at the level of the phospholipids’ glycerol backbone.
Although consistent with the finding of Ganea et al. (13) that the conformational transition E1P → E2P, which deoccludes Na+ from the protein, is an important charge-transporting step of the enzyme’s reaction cycle, this result would appear, at first glance, to be inconsistent with other studies indicating that the enzyme’s conformational transitions do not make major contributions to charge translocation across the membrane (65,66). However, to fully understand the origin of an electric-field effect of ion occlusion on RH421, we believe that the direction of charge movement needs to be considered. During binding of extracellular K+, Na+ and K+ are exchanged through an extracellular ion pathway between the transmembrane helices (67). Thus, the major component of the K+ and Na+ ions’ translational motion is perpendicular to the membrane surface. Hence, their movement will be most significantly affected by electric fields whose field lines are also perpendicular to the membrane surface, as in the case of the transmembrane electrical potential. On the other hand, if an enzyme reaction step causes a movement of charge parallel to the membrane surface, then the charges do not pass any part of the transmembrane electrical potential gradient and no dependence on the transmembrane potential difference would be observed.
It is known that RH421 binds to the lipid membrane adjacent to the protein with its conjugated chromophore undergoing rotational diffusion within a cone angle of 20–30° relative to the membrane normal (68,69). Therefore, movement of charge parallel to the surface of the membrane could strongly influence the electrical potential drop along the length of the dye’s chromophore and cause a significant fluorescence response, particularly if the charge movement occurs at one end of its chromophore (see Fig. 4). Parallel movement of charge away from or toward one end of the chromophore would cause a redistribution of electrons along the length of the dye molecule; this would change the energy of its ground electronic state, causing an electrochromic shift of its absorbance or fluorescence excitation spectrum, and hence a change in its fluorescence intensity.
Figure 4.
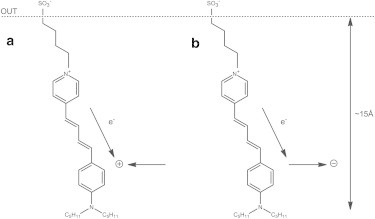
(Schematic diagram) Membrane-bound RH421. Two possible scenarios are shown that could both lead to a decrease in the fluorescence of RH421, as observed when either Na+ or K+ ions are occluded by the phosphorylated enzyme. The movement of either positive charge toward the amino end of the molecule (scenario a) or negative charge away from the amino end (scenario b) would both lead to redistribution of electrons within the ground-state RH421 chromophore toward the amino nitrogen and a shift in its fluorescence excitation spectrum to shorter wavelengths, i.e., higher energy.
Similarly, lyotropic ions, such as perchlorate, bind to the lipid membrane adjacent to the protein and their electric field could strongly influence the kinetics of reactions in which charge is moved parallel to the membrane surface. Of course, these charges would not be the Na+ or K+ ions, but charges or dipoles on amino-acid side chains of the protein or neighboring lipid molecules. Thus, all of the results can be explained by ion occlusion reactions being associated with the movement of charged amino-acid side chains of the protein or dipolar groups associated with the surrounding lipid membrane (e.g., carbonyl dipoles of phospholipid ester linkages, the dipole of the cholesterol molecule, or by hydrating water dipoles) parallel to the membrane surface and with either no component or a very small component perpendicular to the membrane surface.
It is interesting now to consider the location of charged residues on the protein or its surrounding lipid, which may move on occlusion. Any charged amino-acid residues on the α-subunit of the protein located near the ion binding sites are likely to be shielded from dye by the intervening protein mass, as are the transported ions themselves. Thus, even if some do move laterally, which crystal structural data indicate is the case (46,67), this is unlikely to be the cause of the fluorescence change detected by RH421. The movement of charged residues on the β-subunit is also an unlikely cause of the RH421 response because fluorescence changes of styrylpyridinium dyes of similar structure to RH421 associated with the activity of the sarcoplasmic reticulum Ca2+-ATPase, which lacks a β-subunit, have also been reported (70).
Now we turn our attention to the protein’s lipid surroundings. In 2011, by a combination of low resolution x-ray crystallography and molecular-dynamics simulations, Sonntag et al. (71) found evidence that conformational changes of the sarcoplasmic reticulum Ca2+-ATPase between its E2 and E2P states (occluded and nonoccluded states, respectively) are accompanied by local deformations in the surrounding lipid bilayer, so as to minimize any hydrophobic/hydrophilic mismatch between the protein and the membrane. Because the Ca2+-ATPase is closely related to the Na+,K+-ATPase, it is highly probable that such mutual adaptations of the protein and the membrane are also occurring when the Na+,K+-ATPase occludes ions.
If the deformations of the lipid bilayer result in the movement of lipid dipoles or dipoles of associated water molecules or a change in the density of packing of these dipoles, this could easily produce a change in electric field strength, which might be detected by RH421. Because RH421 and related styrylpyridinium dyes are known to bind to the lipid phase of membranes and their fluorescence excitation spectra are known to be sensitive to the membrane dipole potential, whose magnitude is determined in part by the lipid packing density (20,72,73), lipid membrane deformation accompanying the conformational changes of the enzyme necessary for occlusion of the transported ions is probably the most likely cause of the fluorescence response of RH421. In fact, a reorganization of the lipids surrounding the Na+,K+-ATPase caused by a conformational change of the protein was already suggested as the origin of the RH421 fluorescence response by Frank et al. (40).
This mechanism is explained diagrammatically in Fig. 5. In Fig. 5 A, two membranes are shown; on the left, one with a low membrane dipole potential, ψd; and on the right, one with a high dipole potential. In the membrane on the left, the hydrocarbon chain tails of the lipids are disordered. In this disordered state, each chain occupies a relatively large amount of space. This forces the lipid headgroups apart and leads to a relatively low lipid packing density in the membrane. Each lipid headgroup has associated with it a dipole, positive in the interior of the membrane, which originates from a combination of the dipoles of the carbonyl groups of the ester linkages among the headgroups of the lipids and the hydrocarbon chains, water dipoles, and charges on the headgroups themselves. A low packing density of these dipoles causes a low value of the membrane dipole potential. In the membrane shown on the right of Fig. 5 A, the chains are more ordered. This increases the lipid packing density and also the dipole potential. At the same time, the greater extension of the ordered chains increases the bilayer thickness.
Figure 5.
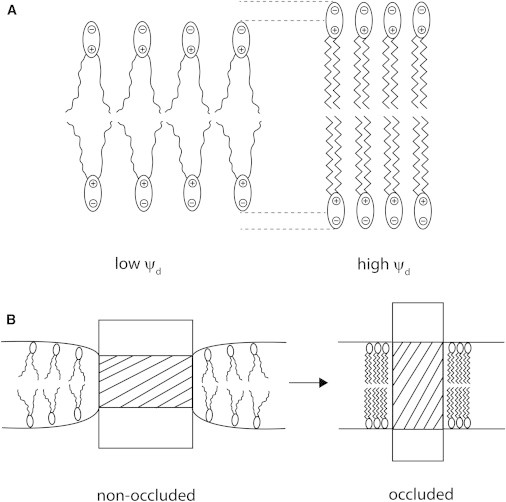
Relation between membrane thickness and dipole potential, ψd. (A) (Left) A membrane is shown with disordered hydrocarbon chains, a relatively small width, low lipid dipolar packing density, and consequently a low ψd. (Right) The membrane has more ordered chains, a greater width, a higher dipole packing density, and consequently a higher ψd. Note: The dipoles shown in the lipid headgroup region represent the net dipole of all contributions to ψd, e.g., water dipoles, carbonyl dipoles, and charges on the headgroups themselves. They are not intended to represent the phosphocholine dipole alone. (B) Protein/membrane structural changes accompanying ion occlusion. (Left) The protein nonoccluded state is shown with a narrow hydrophobic thickness. This necessitates a deformation of surrounding lipid so that the lipid hydrocarbon chains are disordered and the membrane is thinned. The relatively low lipid packing density this produces is responsible for a low ψd. (Right) When the protein occludes ions, its hydrophobic thickness increases. This stretches the hydrocarbon chains of the lipids, increases the membrane thickness, and increases the lipid packing density; hence, ψd increases.
In Fig. 5 B, conformational changes of the Na+,K+-ATPase accompanying ion occlusion are shown schematically. For the purposes of clearly demonstrating the principle, the magnitude of the structural changes the protein undergoes is vastly exaggerated and no molecular detail is shown. The important proposal is that if ion occlusion involves an increase in hydrophobic thickness of the protein (shaded area in the diagram), to avoid energetically unfavorable interaction between hydrophobic portions of the protein and hydrophilic headgroups of the lipid, the membrane must simultaneously increase its hydrophobic thickness. This must involve a straightening and ordering of the hydrocarbon chains of the lipid in the immediate vicinity of the protein, which also has as a consequence an increase in lipid packing density and an increase in membrane dipole potential. Molecules of RH421 situated in the membrane surrounding the protein would thus be exposed to a higher electric-field strength and undergo a shift in their fluorescence excitation spectrum.
The mechanism shown in Fig. 5 B involves a diagonal movement of dipoles on each side of the membrane relative to the center of the bilayer, i.e., there are both vertical and horizontal components to the dipole motion. However, what determines RH421 fluorescence changes is the change in electric-field strength surrounding the dye. Therefore, one must take the dye itself as the frame of reference, not the membrane. If the hydrophobic thickness of the membrane and the lipid packing density increase, the dye must move vertically in parallel with the lipid headgroups so as to avoid its hydrophilic sulfonate group being exposed to the hydrophobic interior of the membrane. Thus, from the point of view of a dye molecule, there is no net vertical movement of the lipid dipoles, only a horizontal displacement (i.e., the change in lipid packing density alone is the decisive factor).
As well as the Na+,K+-ATPase inducing changes in membrane structure that can be detected as electric-field-strength changes by RH421, it seems highly likely that the electrical properties of the surrounding lipid would influence the ability of the Na+,K+-ATPase to occlude ions. Thus, if charged species, either present in the membrane or added to it, compromised the ability of the lipid to readjust to conformational changes of the protein, this could inhibit the kinetics of the ion-occluding reactions of the protein. This could explain some of effects of lyotropic anions on the kinetics of the Na+,K+-ATPase’s partial reactions (13). Changes in membrane rigidity or order are also likely to affect the relative rates of ion occlusion and deocclusion. According to the model presented in Fig. 5 B, ion occlusion would be favored by an ordered membrane with extended lipid hydrocarbon chains. This is indeed what is experimentally observed. The stability of the complex of the Na+,K+-ATPase with occluded Rb+ is dramatically enhanced at low temperatures (74), where one would naturally expect an increase in membrane order.
An important general conclusion of potential relevance for many ion-transporting membrane proteins one can reach from this study is that the term “voltage dependence” is, in fact, too imprecise. It is important to distinguish between the transmembrane voltage dependence of reactions that a membrane protein undergoes and their intramembrane voltage dependence. As discussed here, both can strongly affect different reaction steps of a protein, depending on the direction of charge movement relative to the membrane surface. The membrane-parallel charge movement that we propose could be associated with the occlusion reactions of the Na+,K+-ATPase, and which we suggest is most likely due to movement of charge within the lipid membrane surroundings of the protein, may explain the strong dependence of the kinetics of this enzyme on its lipid environment (75–79).
Acknowledgments
The authors acknowledge helpful correspondence with Prof. Hans-Jürgen Apell, University of Konstanz, Germany, and thank Anne Woods for preparation of figures.
H.H.R. acknowledges, with gratitude, financial support (project grant No. 633252) from the National Health and Medical Research Council (Australia). R.J.C., B.L., and T.W.A. received financial support from the Australian Research Council (Discovery grant No. DP-121003548); J.R.B. received financial support from the National Institutes of Health (grant No. R01-GM057253); and F.C. received financial support from the Danish Research Council.
Supporting Material
References
- 1.Lafaire A.V., Schwarz W. Voltage dependence of the rheogenic Na+/K+ ATPase in the membrane of oocytes of Xenopus laevis. J. Membr. Biol. 1986;91:43–51. doi: 10.1007/BF01870213. [DOI] [PubMed] [Google Scholar]
- 2.Nakao M., Gadsby D.C. [Na] and [K] dependence of the Na/K pump current-voltage relationship in guinea pig ventricular myocytes. J. Gen. Physiol. 1989;94:539–565. doi: 10.1085/jgp.94.3.539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rakowski R.F., Vasilets L.A., Schwarz W. A negative slope in the current-voltage relationship of the Na+/K+ pump in Xenopus oocytes produced by reduction of external [K+] J. Membr. Biol. 1991;121:177–187. doi: 10.1007/BF01870531. [DOI] [PubMed] [Google Scholar]
- 4.Bielen F.V., Glitsch H.G., Verdonck F. Dependence of Na+ pump current on external monovalent cations and membrane potential in rabbit cardiac Purkinje cells. J. Physiol. 1991;442:169–189. doi: 10.1113/jphysiol.1991.sp018788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sagar A., Rakowski R.F. Access channel model for the voltage dependence of the forward-running Na+/K+ pump. J. Gen. Physiol. 1994;103:869–894. doi: 10.1085/jgp.103.5.869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Berlin J.R., Peluffo R.D. Mechanism of electrogenic reaction steps during K+ transport by the NA,K-ATPase. Ann. N. Y. Acad. Sci. 1997;834:251–259. doi: 10.1111/j.1749-6632.1997.tb52256.x. [DOI] [PubMed] [Google Scholar]
- 7.Peluffo R.D., Argüello J.M., Berlin J.R. The role of Na,K-ATPase α subunit serine 775 and glutamate 779 in determining the extracellular K+ and membrane potential-dependent properties of the Na,K-pump. J. Gen. Physiol. 2000;116:47–59. doi: 10.1085/jgp.116.1.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hansen P.S., Buhagiar K.A., Rasmussen H.H. Dependence of Na+-K+ pump current-voltage relationship on intracellular Na+, K+, and Cs+ in rabbit cardiac myocytes. Am. J. Physiol. Cell Physiol. 2002;283:C1511–C1521. doi: 10.1152/ajpcell.01343.2000. [DOI] [PubMed] [Google Scholar]
- 9.Läuger P. Sinauer Associates; Sunderland, MA: 1991. Electrogenic Ion Pumps; pp. 221–224. [Google Scholar]
- 10.Hilgemann D.W. Channel-like function of the Na,K pump probed at microsecond resolution in giant membrane patches. Science. 1994;263:1429–1432. doi: 10.1126/science.8128223. [DOI] [PubMed] [Google Scholar]
- 11.Lu C.-C., Kabakov A., Hilgemann D.W. Membrane transport mechanisms probed by capacitance measurements with megaHertz voltage clamp. Proc. Natl. Acad. Sci. USA. 1995;92:11220–11224. doi: 10.1073/pnas.92.24.11220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rakowski R.F., Gadsby D.C., De Weer P. Voltage dependence of the Na/K pump. J. Membr. Biol. 1997;155:105–112. doi: 10.1007/s002329900162. [DOI] [PubMed] [Google Scholar]
- 13.Ganea C., Babes A., Clarke R.J. Hofmeister effects of anions on the kinetics of partial reactions of the Na+,K+-ATPase. Biophys. J. 1999;77:267–281. doi: 10.1016/S0006-3495(99)76888-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Myers S.L., Cornelius F., Clarke R.J. Kinetics of K+ occlusion by the phosphoenzyme of the Na+,K+-ATPase. Biophys. J. 2011;100:70–79. doi: 10.1016/j.bpj.2010.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Peluffo R.D., Hara Y., Berlin J.R. Quaternary organic amines inhibit Na,K pump current in a voltage-dependent manner: direct evidence of an extracellular access channel in the Na,K-ATPase. J. Gen. Physiol. 2004;123:249–263. doi: 10.1085/jgp.200308872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kropp D.L., Sachs J.R. Kinetics of the inhibition of the Na-K pump by tetrapropylammonium chloride. J. Physiol. 1977;264:471–487. doi: 10.1113/jphysiol.1977.sp011678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Forbush B., III The interaction of amines with the occluded state of the Na,K-pump. J. Biol. Chem. 1988;263:7979–7988. [PubMed] [Google Scholar]
- 18.Peluffo R.D., González-Lebrero R.M., Berlin J.R. Quaternary benzyltriethylammonium ion binding to the Na,K-ATPase: a tool to investigate extracellular K+ binding reactions. Biochemistry. 2009;48:8105–8119. doi: 10.1021/bi900687u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Peluffo R.D., Berlin J.R. Membrane potential-dependent inhibition of the Na+,K+-ATPase by para-nitrobenzyltriethylammonium bromide. Mol. Pharmacol. 2012;82:1–8. doi: 10.1124/mol.111.077008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Clarke R.J. Effect of lipid structure on the dipole potential of phosphatidylcholine bilayers. Biochim. Biophys. Acta. 1997;1327:269–278. doi: 10.1016/s0005-2736(97)00075-8. [DOI] [PubMed] [Google Scholar]
- 21.Clarke R.J., Lüpfert C. Influence of anions and cations on the dipole potential of phosphatidylcholine vesicles: A basis for the Hofmeister effect. Biophys. J. 1999;76:2614–2624. doi: 10.1016/S0006-3495(99)77414-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Klodos I., Forbush B., III Rapid conformational changes of the Na/K pump revealed by a fluorescent dye. J. Gen. Physiol. 1988;92:46a. [Google Scholar]
- 23.Forbush B., III, Klodos I. Rate-limiting steps in Na translocation by the Na/K pump. In: Kaplan J.H., De Weer P., editors. The Sodium Pump: Structure, Mechanism, and Regulation. Rockefeller University Press; New York: 1991. pp. 211–225. [Google Scholar]
- 24.Stürmer W., Bühler R., Läuger P. Charge translocation by the Na,K-pump: II. Ion binding and release at the extracellular face. J. Membr. Biol. 1991;121:163–176. doi: 10.1007/BF01870530. [DOI] [PubMed] [Google Scholar]
- 25.Pratap P.R., Robinson J.D. Rapid kinetic analyses of the Na+/K+-ATPase distinguish among different criteria for conformational change. Biochim. Biophys. Acta. 1993;1151:89–98. doi: 10.1016/0005-2736(93)90075-b. [DOI] [PubMed] [Google Scholar]
- 26.Kane D.J., Fendler K., Clarke R.J. Stopped-flow kinetic investigations of conformational changes of pig kidney Na+,K+-ATPase. Biochemistry. 1997;36:13406–13420. doi: 10.1021/bi970598w. [DOI] [PubMed] [Google Scholar]
- 27.Cornelius F. Rate determination in phosphorylation of shark rectal Na,K-ATPase by ATP: temperature sensitivity and effects of ADP. Biophys. J. 1999;77:934–942. doi: 10.1016/S0006-3495(99)76944-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schvartz P.G., Monti J.L.E., Rossi R.C. Kinetic characterization of intermediates during Na-ATPase and Na,K-ATPase activity using RH421 and Rb+ occlusion. J. Gen. Physiol. 2005;126:30a. [Google Scholar]
- 29.Amoroso S., Agon V.V., Clarke R.J. Photochemical behavior and Na+,K+-ATPase sensitivity of voltage-sensitive styrylpyridinium fluorescent membrane probes. Photochem. Photobiol. 2006;82:495–502. doi: 10.1562/2005-06-08-RA-569. [DOI] [PubMed] [Google Scholar]
- 30.Bühler R., Apell H.-J. Sequential potassium binding at the extracellular side of the Na,K-pump. J. Membr. Biol. 1995;145:165–173. doi: 10.1007/BF00237374. [DOI] [PubMed] [Google Scholar]
- 31.Khalid M., Fouassier G., Clarke R.J. Interaction of ATP with the phosphoenzyme of the Na+,K+-ATPase. Biochemistry. 2010;49:1248–1258. doi: 10.1021/bi9019548. [DOI] [PubMed] [Google Scholar]
- 32.Klodos I., Esmann M., Post R.L. Large-scale preparation of sodium-potassium ATPase from kidney outer medulla. Kidney Int. 2002;62:2097–2100. doi: 10.1046/j.1523-1755.2002.00654.x. [DOI] [PubMed] [Google Scholar]
- 33.Ottolenghi P. The reversible delipidation of a solubilized sodium-plus-potassium ion-dependent adenosine triphosphatase from the salt gland of the spiny dogfish. Biochem. J. 1975;151:61–66. doi: 10.1042/bj1510061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Peterson G.L. A simplification of the protein assay method of Lowry et al. which is more generally applicable. Anal. Biochem. 1977;83:346–356. doi: 10.1016/0003-2697(77)90043-4. [DOI] [PubMed] [Google Scholar]
- 35.Lowry O.H., Rosebrough N.J., Randall R.J. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 36.Zouni A., Clarke R.J., Holzwarth J.F. Static and dynamic studies of the potential-sensitive membrane probe RH421 in dimyristoylphosphatidylcholine vesicles. Biochim. Biophys. Acta. 1993;1153:203–212. doi: 10.1016/0005-2736(93)90406-p. [DOI] [PubMed] [Google Scholar]
- 37.Zouni A., Clarke R.J., Holzwarth J.F. Kinetics of solubilization of styryl dye aggregates by lipid vesicles. J. Phys. Chem. 1994;98:1732–1738. [Google Scholar]
- 38.Cornelius F., Mahmmoud Y.A., Toyoshima C. Metal fluoride complexes of Na,K-ATPase: Characterization of fluoride-stabilized phosphoenzyme analogues and their interaction with cardiotonic steroids. J. Biol. Chem. 2011;286:29882–29892. doi: 10.1074/jbc.M111.259663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Clarke R.J., Zouni A., Holzwarth J.F. Voltage sensitivity of the fluorescent probe RH421 in a model membrane system. Biophys. J. 1995;68:1406–1415. doi: 10.1016/S0006-3495(95)80313-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Frank J., Zouni A., Clarke R.J. Interaction of the fluorescent probe RH421 with ribulose-1,5-bisphosphate carboxylase/oxygenase and with Na+,K+-ATPase membrane fragments. Biochim. Biophys. Acta. 1996;1280:51–64. doi: 10.1016/0005-2736(95)00277-4. [DOI] [PubMed] [Google Scholar]
- 41.Cornelius F. Functional reconstitution of the sodium pump. Kinetics of exchange reactions performed by reconstituted Na/K-ATPase. Biochim. Biophys. Acta. 1991;1071:19–66. doi: 10.1016/0304-4157(91)90011-k. [DOI] [PubMed] [Google Scholar]
- 42.Pilotelle-Bunner A., Cornelius F., Clarke R.J. Mechanism of Mg2+ binding in the Na+,K+-ATPase. Biophys. J. 2009;96:3753–3761. doi: 10.1016/j.bpj.2009.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Babes A., Fendler K. Na+ transport and the E1P-E2P conformational transition of the Na+/K+-ATPase. Biophys. J. 2000;79:2557–2571. doi: 10.1016/S0006-3495(00)76496-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Clarke R.J., Catauro M., Apell H.-J. Quantitative calculation of the role of the Na+,K+-ATPase in thermogenesis. Biochim. Biophys. Acta. Bioenerg. 2013;1827:1205–1212. doi: 10.1016/j.bbabio.2013.06.010. [DOI] [PubMed] [Google Scholar]
- 45.Brooks B.R., Bruccoleri R.E., Karplus M. CHARMM: A program for macromolecular energy minimization and dynamics calculations. J. Comput. Chem. 1983;4:187–217. [Google Scholar]
- 46.Shinoda T., Ogawa H., Toyoshima C. Crystal structure of the sodium-potassium pump at 2.4 Å resolution. Nature. 2009;459:446–450. doi: 10.1038/nature07939. [DOI] [PubMed] [Google Scholar]
- 47.Kanai R., Ogawa H., Toyoshima C. Crystal structure of a Na+-bound Na+,K+-ATPase preceding the E1P state. Nature. 2013;502:201–206. doi: 10.1038/nature12578. [DOI] [PubMed] [Google Scholar]
- 48.Lomize M.A., Lomize A.L., Mosberg H.I. OPM: Orientations of proteins in membranes database. Bioinformatics. 2006;22:623–625. doi: 10.1093/bioinformatics/btk023. [DOI] [PubMed] [Google Scholar]
- 49.Stern H.A., Feller S.E. Calculation of the dielectric permittivity profile for a nonuniform system: Application to a lipid bilayer simulation. J. Chem. Phys. 2003;118:3401–3412. [Google Scholar]
- 50.Gilson M.K., Honig B.H. The dielectric constant of a folded protein. Biopolymers. 1986;25:2097–2119. doi: 10.1002/bip.360251106. [DOI] [PubMed] [Google Scholar]
- 51.Song X. An inhomogeneous model of protein dielectric properties: Intrinsic polarizabilities of amino acids. J. Chem. Phys. 2002;116:9359–9363. [Google Scholar]
- 52.Li L., Li C., Alexov E. On the dielectric “constant” of proteins: Smooth dielectric function for macromolecular modeling and its implementation in DELPHI. J. Chem. Theory Comput. 2013;9:2126–2136. doi: 10.1021/ct400065j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kukic P., Farrell D., Nielsen J.E. Protein dielectric constants determined from NMR chemical shift perturbations. J. Am. Chem. Soc. 2013;135:16968–16976. doi: 10.1021/ja406995j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Allen T.W., Andersen O.S., Roux B. On the importance of atomic fluctuations, protein flexibility, and solvent in ion permeation. J. Gen. Physiol. 2004;124:679–690. doi: 10.1085/jgp.200409111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Deguchi N., Jørgensen P.L., Maunsbach A.B. Ultrastructure of the sodium pump. Comparison of thin sectioning, negative staining, and freeze-fracture of purified, membrane-bound (Na+,K+)-ATPase. J. Cell Biol. 1977;75:619–634. doi: 10.1083/jcb.75.3.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Karlish S.J.D., Goldshleger R., Stein W.D. A 19-kDa C-terminal tryptic fragment of the α-chain of Na/K-ATPase is essential for occlusion and transport of cations. Proc. Natl. Acad. Sci. USA. 1990;87:4566–4570. doi: 10.1073/pnas.87.12.4566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Aylward G., Findlay T. 4th Ed. Wiley; Milton, Australia: 1994. SI Chemical Data. [Google Scholar]
- 58.Bühler R., Stürmer W., Läuger P. Charge translocation by the Na,K-pump: I. Kinetics of local field changes studied by time-resolved fluorescence measurements. J. Membr. Biol. 1991;121:141–161. doi: 10.1007/BF01870529. [DOI] [PubMed] [Google Scholar]
- 59.Loew L.M. How to choose a potentiometric membrane probe. In: Loew L.M., editor. Vol. II. CRC Press; Boca Raton, FL: 1988. pp. 139–151. (Spectroscopic Membrane Probes). [Google Scholar]
- 60.Sachs J.R., Welt L.G. The concentration dependence of active potassium transport in the human red blood cell. J. Clin. Invest. 1967;46:65–76. doi: 10.1172/JCI105512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Forbush B., III Rapid release of 42K or 86Rb from two distinct transport sites on the Na,K-pump in the presence of Pi or vanadate. J. Biol. Chem. 1987;262:11116–11127. [PubMed] [Google Scholar]
- 62.Gross E., Bedlack R.S., Jr., Loew L.M. Dual-wavelength ratiometric fluorescence measurement of the membrane dipole potential. Biophys. J. 1994;67:208–216. doi: 10.1016/S0006-3495(94)80471-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cladera J., O’Shea P. Intramembrane molecular dipoles affect the membrane insertion and folding of a model amphiphilic peptide. Biophys. J. 1998;74:2434–2442. doi: 10.1016/S0006-3495(98)77951-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Starke-Peterkovic T., Clarke R.J. Effect of headgroup on the dipole potential of phospholipid vesicles. Eur. Biophys. J. 2009;39:103–110. doi: 10.1007/s00249-008-0392-y. [DOI] [PubMed] [Google Scholar]
- 65.Wuddel I., Apell H.-J. Electrogenicity of the sodium transport pathway in the Na,K-ATPase probed by charge-pulse experiments. Biophys. J. 1995;69:909–921. doi: 10.1016/S0006-3495(95)79965-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gadsby D.C., Rakowski R.F., De Weer P. Extracellular access to the Na,K pump: pathway similar to ion channel. Science. 1993;260:100–103. doi: 10.1126/science.7682009. [DOI] [PubMed] [Google Scholar]
- 67.Ogawa H., Shinoda T., Toyoshima C. Crystal structure of the sodium-potassium pump (Na+,K+-ATPase) with bound potassium and ouabain. Proc. Natl. Acad. Sci. USA. 2009;106:13742–13747. doi: 10.1073/pnas.0907054106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Visser N.V., van Hoek A., Clarke R.J. Time-resolved fluorescence investigations of the interaction of the voltage-sensitive probe RH421 with lipid membranes and proteins. Biochemistry. 1995;34:11777–11784. doi: 10.1021/bi00037a015. [DOI] [PubMed] [Google Scholar]
- 69.Lambacher A., Fromherz P. Orientation of hemicyanine dye in lipid membrane measured by fluorescence interferometry on a silicon chip. J. Phys. Chem. B. 2001;105:343–346. [Google Scholar]
- 70.Butscher C., Roudna M., Apell H. Electrogenic partial reactions of the SR-Ca-ATPase investigated by a fluorescence method. J. Membr. Biol. 1999;168:169–181. doi: 10.1007/s002329900507. [DOI] [PubMed] [Google Scholar]
- 71.Sonntag Y., Musgaard M., Thøgersen L. Mutual adaptation of a membrane protein and its lipid bilayer during conformational changes. Nat. Commun. 2011;2:304. doi: 10.1038/ncomms1307. [DOI] [PubMed] [Google Scholar]
- 72.Peterson U., Mannock D.A., Pohl E.E. Origin of membrane dipole potential: contribution of the phospholipid fatty acid chains. Chem. Phys. Lipids. 2002;117:19–27. doi: 10.1016/s0009-3084(02)00013-0. [DOI] [PubMed] [Google Scholar]
- 73.Warshaviak D.T., Muellner M.J., Chachisvilis M. Effect of membrane tension on the electric field and dipole potential of lipid bilayer membrane. Biochim. Biophys. Acta. 2011;1808:2608–2617. doi: 10.1016/j.bbamem.2011.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Shani M., Goldschleger R., Karlish S.J.D. Rb+ occlusion in renal (Na+ + K+)-ATPase characterized with a simple manual assay. Biochim. Biophys. Acta. 1987;904:13–21. doi: 10.1016/0005-2736(87)90081-2. [DOI] [PubMed] [Google Scholar]
- 75.Starke-Peterkovic T., Turner N., Clarke R.J. Electric field strength of membrane lipids from vertebrate species: membrane lipid composition and Na+-K+-ATPase molecular activity. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005;288:R663–R670. doi: 10.1152/ajpregu.00434.2004. [DOI] [PubMed] [Google Scholar]
- 76.Yeagle P.L., Young J., Rice D. Effects of cholesterol on (Na+,K+)-ATPase ATP hydrolyzing activity in bovine kidney. Biochemistry. 1988;27:6449–6452. doi: 10.1021/bi00417a037. [DOI] [PubMed] [Google Scholar]
- 77.Klodos I., Fedosova N.U., Plesner L. Influence of intramembrane electric charge on Na,K-ATPase. J. Biol. Chem. 1995;270:4244–4254. doi: 10.1074/jbc.270.9.4244. [DOI] [PubMed] [Google Scholar]
- 78.Sotomayor C.P., Aguilar L.F., Jameson D.M. Modulation of pig kidney Na+/K+-ATPase activity by cholesterol: role of hydration. Biochemistry. 2000;39:10928–10935. doi: 10.1021/bi000717z. [DOI] [PubMed] [Google Scholar]
- 79.Cornelius F. Modulation of Na,K-ATPase and Na-ATPase activity by phospholipids and cholesterol. I. Steady-state kinetics. Biochemistry. 2001;40:8842–8851. doi: 10.1021/bi010541g. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.