

Abstract
Hfq is a post-transcriptional regulator that binds U- and A-rich regions of sRNAs and their target mRNAs to stimulate their annealing in order to effect translation regulation and, often, to alter their stability. The functional importance of Hfq and its RNA-binding properties are relatively well understood in Gram-negative bacteria, whereas less is known about the RNA-binding properties of this riboregulator in Gram-positive species. Here, we describe the structure of Hfq from the Gram-positive pathogen Listeria monocytogenes in its RNA-free form and in complex with a U6 oligoribonucleotide. As expected, the protein takes the canonical hexameric toroidal shape of all other known Hfq structures. The U6 RNA binds on the “proximal face” in a pocket formed by conserved residues Q9, N42, F43, and K58. Additionally residues G5 and Q6 are involved in protein-nucleic and inter-subunit contacts that promote uracil specificity. Unlike Staphylococcus aureus (Sa) Hfq, Lm Hfq requires magnesium to bind U6 with high affinity. In contrast, the longer oligo-uridine, U16, binds Lm Hfq tightly in the presence or absence of magnesium, thereby suggesting the importance of additional residues on the proximal face and possibly the lateral rim in RNA interaction. Intrinsic tryptophan fluorescence quenching (TFQ) studies reveal, surprisingly, that Lm Hfq can bind (GU)3G and U6 on its proximal and distal faces, indicating a less stringent adenine-nucleotide specificity site on the distal face as compared to the Gram-positive Hfq proteins from Sa and Bacillus subtilis and suggesting as yet uncharacterized RNA-binding modes on both faces.
Keywords: Hfq, Listeria monocytogenes, RNA binding, post-transcription regulator, structural biology
INTRODUCTION
Hfq is a pleiotropic post-transcriptional regulator belonging to the Sm/Lsm superfamily and is found in approximately half of all sequenced bacteria (Tsui et al. 1994; Sun et al. 2002; Valentin-Hansen et al. 2004). This family is characterized structurally by two Sm motifs that form a conserved core, which typically contains an N-terminal α helix and a five-stranded, highly twisted antiparallel β sheet (Kambach et al. 1999). Sm protomers typically assemble into functional hexameric or heptameric rings (Collins et al. 2001; Mura et al. 2001; Toro et al. 2001; Walke et al. 2001). Hfq is an RNA chaperone that binds preferentially to A-, AU-, and U-rich sequences of small noncoding RNAs (sRNA) and their target mRNAs to facilitate their annealing, thus influencing translation and RNA stability and degradation (Zhang et al. 1998, 2002; Wassarman et al. 2001; Moller et al. 2002; Storz et al. 2004).
The first high-resolution structure of any Hfq bound to RNA was the Staphylococcus aureus (Sa) Hfq-AU5G complex (Schumacher et al. 2002). The structure revealed a hexameric toroidal assembly with the AU5 bound to the “proximal face” of the protein, with 1 nt bound per subunit and the 3′-G inserted into a central pore of the hexamer. The proximal face is defined here as the side of the hexamer that contains the sole α helix of Sa Hfq. In contrast, the crystal structure of Escherichia coli (Ec) Hfq bound to A15 RNA showed polyadenosine binding to the opposite or “distal face” (Link et al. 2009). Unlike the proximal face, the distal face of Ec Hfq has three distinct nucleobase binding sites per protomer, originally called the A-R-N motif, where A is an adenine-only binding site, R is a purine-nucleotide specificity site with a strong preference for adenine (Robinson et al. 2014), and N is a nondiscriminatory solvent-exposed nucleotide that is the likely distal-face RNA entry and exit location. As a consequence of this motif, a maximum of 18 nt can readily occupy the distal face of Ec Hfq. These structures established two fundamental mechanisms utilized by Hfq to bind U-rich and A-rich RNA tracts. Subsequent structures of Sa Hfq bound to A4 (Horstmann et al. 2012) as well as Bacillus subtilis (Bs) Hfq bound to (AG)3A (Someya et al. 2012) revealed that the distal face of Hfq from Gram-positive species also binds A-rich tracts but, in contrast to Gram-negative Ec Hfq, Sa and Bs Hfq contain only two distinct nucleotide binding sites, which compose the A-L binding motif, where A is specific for adenine binding and L is a seemingly promiscuous linker site. Hence, the distal face of Sa, Bs, and likely all Gram-positive Hfq proteins readily accommodates only a maximum of 12 nt. Indeed, only one distal-face binding site, the originally designated purine-nucleotide specificity site, which forms a deep pocket on the distal face at the interface of two adjacent Hfq subunits, appears to be conserved between Hfq proteins from Gram-negative and Gram-positive bacteria.
The vital role played by Hfq of Gram-negative bacterial species in fitness, response to a wide variety of environmental stresses, and virulence is well documented (Chao and Vogel 2010). Specifically, Hfq has been shown to be an important virulence factor in Vibrio cholera (Vc), Pseudomonas aeruginosa (Pa), Neisseria meningitides (Nm), and Salmonella typhimurium (St) (Sonnleitner et al. 2003; Ding et al. 2004; Sittka et al. 2007; Fantappie et al. 2009). Additionally, Ec and Salmonella enterica Hfq have been shown to confer antibiotic resistance through their respective regulation of the multidrug efflux pumps AcrB and SmvA (Yamada et al. 2010; Hayashi-Nishino et al. 2012). There is, however, far less information to indicate that Hfq plays a similarly important role in Gram-positive bacteria. Regardless, Hfq has been shown to interact with several sRNAs in both Bs and Sa and was recently shown to affect the stability of certain sRNA in B. subtilis (Dambach et al. 2013), but in S. aureus was not essential for the stability of the identified sRNAs or duplex formation with their target mRNAs (Huntzinger et al. 2005; Geisinger et al. 2006; Heidrich et al. 2006, 2007; Boisset et al. 2007; Preis et al. 2009). Moreover, an Hfq null strain of S. aureus exhibited no deleterious phenotypes, leading to the notion that Hfq plays only a small role in gene expression and RNA metabolism or that there are yet unidentified redundant proteins that render Hfq nonessential (Bohn et al. 2007). On the other hand, a more recent study indicates that the role of Hfq in S. aureus is actually strain-dependent, whereby hfq knockouts led to changes in expression profiles in several strains including MRSA strains and resulted in lowered virulence (Liu et al. 2010).
A more prominent role for an Hfq from a low GC-content Gram-positive bacteria is found in the food-borne human pathogen Listeria monocytogenes (Lm), the causative agent of listeriosis, in which Hfq has been directly implicated in stress tolerance and virulence (Christiansen et al. 2004). Further, there have been numerous sRNAs recently identified in Lm, both experimentally and through bioinformatics analyses, that might serve as Hfq binding partners (Christiansen et al. 2006; Mandin et al. 2007; Nielsen et al. 2008; Toledo-Arana et al. 2009). Interestingly, of the 50 sRNA identified in Lm by Toledo-Arana et al., 21 are absent in the avirulent strain Listeria innocua. Three sRNAs, LhrA, LhrB, and LhrC, have been shown to interact directly with Hfq, with the half-life of LhrA increased by ∼15-fold when Hfq is present (Christiansen et al. 2006). LhrA also prevents association of its target mRNA with the 30S ribosomal subunit in an Hfq-dependent manner (Nielsen et al. 2010).
To understand more fully the determinants of the Lm Hfq–RNA interaction at the atomic level, and Gram-positive Hfq-RNA binding in general, we determined the X-ray crystal structures of ligand-free Lm Hfq and an Lm Hfq-U6 RNA complex. This is the fourth bacterial species from which an Hfq–uridine-rich RNA complex crystal structure has been solved, with S. aureus, E. coli, and S. typhimurium being the others. The structure of the complex, while in many respects similar to previous structures, reveals new, critical contributors to uracil binding and specificity. Fluorescence polarization-based Hfq-RNA binding measurements also indicate that magnesium plays a role in RNA binding to the proximal face of the protein. Intriguingly, RNA-Hfq tryptophan fluorescence quenching assays (Robinson et al. 2014) suggest strongly that Lm Hfq is a more promiscuous RNA-binding protein than Hfq proteins from other Gram-positive bacteria.
RESULTS AND DISCUSSION
Listeria monocytogenes Hfq structure
Lm Hfq, like the Gram-positive Sa Hfq, is composed of 77 aa residues per subunit. Full-length Lm Hfq was crystallized using the hanging drop-vapor diffusion method. The resulting crystals took the orthorhombic space group P21221 with a = 63.6 Å, b = 66.8 Å, c = 106.5 Å (Table 1). The structure was determined by molecular replacement (MR) using an Lm Hfq homology model (PHYRE v2.0) as the search model. The structure was refined to Rwork and Rfree values of 21.3% and 27.1%, respectively, to 2.60 Å resolution. The asymmetric unit (ASU) contains two stacked trimers with the N termini of one trimer forming crystal contacts with the distal face of the other trimer. Due to some disorder in the C terminus, no electron density was observed for the last 2 to 5 residues of a given subunit (Table 1). The biologically relevant Hfq hexamer is generated by a crystallographic twofold symmetry operation. The global structure of the protein exhibits the typical Sm fold that is observed for Hfq from other species, with each protomer consisting of a single α helix (α1, residues 6–19) and a five-stranded, highly twisted antiparallel β sheet with β1 composed of residues 22–27, β2 residues 29–39, β3 residues 44–47, β4 residues 52–57, and β5 residues 62–66 (Fig. 1A). The homohexamer is stabilized primarily by hydrogen bonds between β4 and β5 of adjacent protomers. As seen in other Hfq proteins, β2 is twisted back on itself. Structural alignment of the Lm Hfq hexamer with Sa Hfq, Bs Hfq, and Ec Hfq hexamers results in RMSDs (root mean square deviations) of 0.95 Å, 0.92 Å, and 1.58 Å, respectively, showing expected similarity with Hfq proteins from other Gram-positive bacteria. The larger RMSD between Lm Hfq and Ec Hfq arises primarily from differences in the loops between β-strands β3 and β4 and the β1-loop-β2 region.
TABLE 1.
Selected crystallographic data and refinement statistics
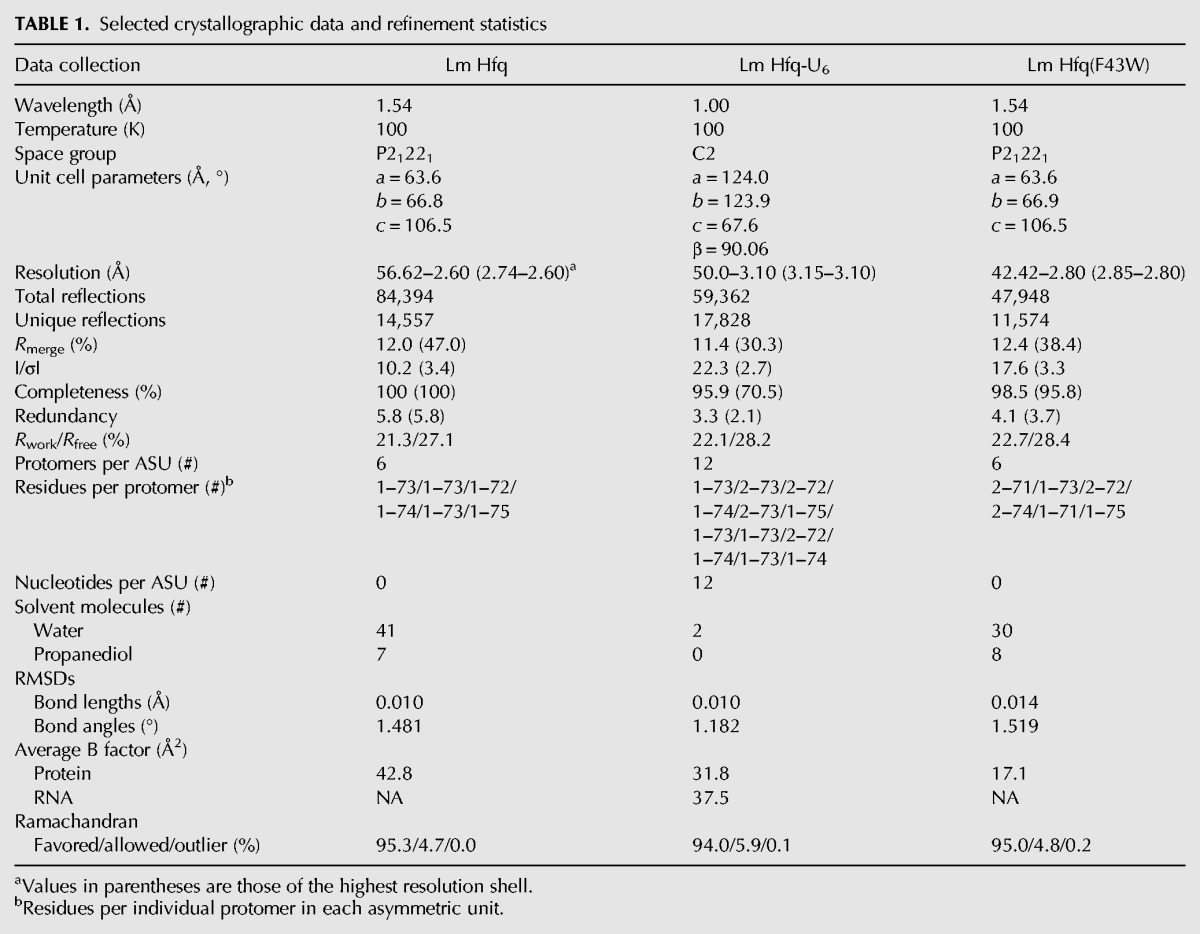
FIGURE 1.

Structures of Lm Hfq and the Lm Hfq-U6 complex. (A) View into the proximal face of the Lm Hfq hexamer. The protein is shown as a cartoon and surface. The N and C termini of the protein are labeled, while the α helices are colored red, and the five-stranded twisted antiparallel β sheet is shown in cyan. The secondary elements of one protomer are labeled. (B) The asymmetric unit of the Lm Hfq–U6 complex structure. Notice the N terminus-distal face packing. The Lm Hfq is shown as a blue cartoon and the bound RNA as an atom-colored cartoon. (C) An Fo – Fc electron density difference map calculated after molecular replacement and one round of rigid body refinement in Phenix and prior to the addition of the U6 RNA. The Cα trace of the protein backbone is shown with each protomer individually colored. The electron density difference map (green mesh) is contoured at 3.5 σ. (D) View into the proximal face of the Lm Hfq–U6 complex structure. The protein is shown as a cartoon and surface colored blue, with the RNA shown as sticks and colored by atom type, with carbon atoms colored green.
Lm Hfq-U6 RNA complex
Upon inspection of the packing of the apo Lm Hfq crystals, soaking in smaller biologically relevant RNAs seemed possible. Hence, U6 RNA was added to drops containing preformed apo Hfq crystals in an attempt to obtain an Lm Hfq-U6 complex. The addition of the RNA resulted in a pseudoorthorhombic crystal that now takes the monoclinic space group C2 with a = 124.03 Å, b = 123.93 Å, c = 67.60 Å, and β = 90.06° (Table 1). Diffraction of these crystals was reduced to a 3.1 Å-resolution. The structure was determined by MR using the refined apo Lm Hfq structure as the search model. There are 12 subunits (two hexamers) in the asymmetric unit (Fig. 1B). After MR and one round of rigid body refinement, strong Fo – Fc electron density was observed for the RNA on the proximal face (Fig. 1C). RNA was built into the map and subsequently refined to Rwork and Rfree values of 21.9% and 28.0%, respectively (Fig. 1C,D; Table 1). Since the electron density defining the RNA does not indicate definitively the 5′ and 3′ ends of the RNA, we conclude the RNA is statistically disordered, which likely contributes to the slightly enlarged difference between Rwork and Rfree. The Lm Hfq protein does not undergo any dramatic local or global change upon RNA binding (RMSD between Lm Hfq and Lm Hfq-U6 is 0.68 Å), thereby indicating a preformed polyuridine binding pocket.
Lm Hfq recognition of uridine
Lm Hfq binds U6 RNA in the proximal pore of the protein with each interface between subunits creating a binding site that accommodates a single nucleobase. Thus, one U6 RNA molecule wraps around the pore and occupies all six available binding sites per hexamer (Fig. 1C,D). The uracil nucleobase stacks between residues N42 and F43 of one subunit and residue F43′ of an adjacent subunit (Fig. 2A). Lm Hfq recognition of the uracil is effected by a network of hydrogen bonds involving residues G5, Q6, Q9, N42, and K58. Key to uracil specificity are the hydrogen bonds from the Nε and Oε side chain atoms of Q6 to the O4 and N3(H) atoms of the uracil. However, for such a readout to occur, the Nε of residue Q6 is locked into place by a hydrogen bond from the Oδ side chain oxygen of residue N42, which, in turn, is buttressed by a hydrogen bond from its Nδ atom to the carbonyl oxygen of residue G5″ from an adjacent subunit (Fig. 2A). This interaction better positions Q6 to bind these uracils, although it should be noted that there is little difference in the positions of G5 and Q6 in the presence or absence of RNA, again indicating that the uracil binding site is preformed. The O2 position of uracil is engaged in hydrogen bonding with the Nε side chain nitrogen of residue Q9. The Oε side chain oxygen of residue Q9 hydrogen bonds to the Nζ atom of K58, which also hydrogen bonds to the peptide backbone carbonyl oxygen of N42, thus completing the engulfment of the uracil ring (Fig. 2A). These are the predominant interactions in our twelve independent views of the uracil binding pocket. The protein, however, does display some conformational flexibility in this area that allows alternative hydrogen bond networks to form. In one subunit, residue N42 rotates so that its Nδ reads the O4 position of uracil directly while foregoing its bonding with Q6 and G5″ (Supplemental Fig. S1A). In another subunit, the Oε atom of residue Q6 engages in a hydrogen bond to the Nε of residue Q9 while still forming a hydrogen bond to the N3(H) of the uracil. In all subunits, the preference for RNA binding is enhanced directly by a hydrogen bond between the 2′ hydroxyl group of the ribose and the Nδ atom of residue H59 (Supplemental Figs. S1B, S2A). Further, in two of the 12 uridine binding sites, a water molecule is observed. This water is bound by the Nδ of the H59 side chain and the F43 peptide backbone carbonyl oxygen as well as the 2′ hydroxyl group of the ribose and phosphate of the RNA (Supplemental Fig. S1B).
FIGURE 2.

Uridine binding to the proximal face of Hfq. (A) The interface between Lm Hfq subunits creates a binding site for a single uracil base. Key RNA-interacting residues are labeled in each panel. All residues and the nucleotide are shown as sticks whereby the oxygen atoms are colored red, nitrogen atoms blue, phosphorus atoms orange, and carbons either green (RNA) or magenta (protein). Residue F43 stacks with the base while residues Q6 and Q9 engage in hydrogen bonds with the O2, N3, and O4 atoms of the base. Residue N42 hydrogen bonds with residues Q6 and G5″ to position the Q6 side chain for base read out. Residue K58 hydrogen bonds with both the side chain of residue Q9 and the backbone carbonyl of residue N42 and might be able to read the exocyclic O2 position of the base. Hydrogen bonding is indicated by blue dashes. (B–D) The proximal-face uridine-binding pockets of (B) Gram-positive Sa Hfq (PDB: 1KQ2), (C) Gram-negative St Hfq (PDB: 2YLC), and (D) Ec Hfq (PDB: 4HT9) reveal high conservation of key binding residues including Q9, F43, and K58 of Lm Hfq. Residue N42 of Lm Hfq corresponds to Q41 in the Hfq proteins of both Gram-negative species, whereas Sa Hfq has a lysine (K41) at this position. None of these Hfq proteins utilizes a residue corresponding to Lm Hfq Q6 for RNA binding. Interacting residues and nucleotides are shown as sticks and key hydrogen bonds are depicted by dashed blue lines.
The initial Fo – Fc electron density difference maps showed a large region of positive density in the center of the RNA molecule and pore (Fig. 1C). We were unable to identify any reasonable candidates in our crystallization condition that were likely to reside in this position and believe that this is simply an accumulation of spurious electron density along a pseudocrystallographic axis. It is worth noting, however, that in the higher resolution crystal structure of Ec Hfq bound to proximal face-bound AU6A (Wang et al. 2011), a water coordinated metal ion was observed in a similar location and made contacts to the RNA phosphate backbone. Although this might still be the case for the Lm Hfq-U6 complex, placement of a similarly hydrated metal ion is too speculative at the current resolution. We do note, however, that magnesium does have a significant impact on the binding of U6 (see below).
Uracil binding to Hfq from Gram-positive and Gram-negative bacteria
As described above, the proximal-face pore-binding mode of uracil to Lm Hfq is similar but not identical to those utilized by other Hfq proteins. One key common feature of Hfq-uridine tract binding is the use of a conserved aromatic residue, a polar residue, and the same aromatic residue from a neighboring subunit to provide base-stacking interactions (Fig. 2). Further, in common among Hfq proteins is the hydrogen bond made between a conserved glutamine (Lm Hfq residue Q9; Ec [Wang et al. 2011, 2013], St [Sauer and Weichenrieder 2011], and Sa [Schumacher et al. 2002] Hfq residue Q8) and the exocyclic O2 atom of uracil. In all four proteins, the Oε of this glutamine typically hydrogen bonds to the side chain ε amino group of a conserved lysine residue (K58 in Lm Hfq, K57 in Sa Hfq, and K56 in Ec and St Hfq). In Sa Hfq, the Oε atom of this glutamine (Q8) can form a weak hydrogen bond with the N3H atom of the uracil and thereby contribute to discrimination against cytosine (Schumacher et al. 2002). Interestingly, the residue corresponding to N42 of Lm Hfq is a glutamine in the Hfq proteins of Gram-negative species such as Ec (Q41) and St (Q41) and a lysine (K41) in Sa Hfq (Fig. 2; Supplemental Fig. S3). Both Ec Q41 and St Q41 make a hydrogen bond to the O4 oxygen of the uracil base, and this lysine in Sa Hfq also plays a role in uracil binding affinity and specificity, as it can interact favorably only with the O4 oxygen of uracil (Fig. 2B–D). Furthermore, the peptide backbone carbonyl groups of these glutamines in Ec and St Hfq and the lysine residue from Sa Hfq hydrogen bond to the Nζ of the conserved lysine residues, K56, K56, and K57, respectively, thereby buttressing the side chain conformations of residue Q8, their respective uracil O2-binding glutamines. In contrast, Lm Hfq residue N42 is typically not engaged in a similar bond to O4 but rather acts as a bridge between residues Q6 and G5″, thus aiding Q6 to engage in base-specifying hydrogen bonds to the N3 and O4 atoms of uracil. Whereas the use of Lm Hfq residue Q6, which is located at the N terminus of the sole Hfq α helix, in RNA binding is newly described, similar use of the corresponding residues in Hfq proteins from other Gram-positive bacteria is unlikely as they are incapable of engaging in the same type of interactions. More concretely, the corresponding residue in Sa Hfq is a glutamate (E5) and an isoleucine or valine in the Hfq proteins of other bacilli (Supplemental Fig. S3). Although Sa Hfq residue E5 is flanked by asparagines N4 and N6, which, in principle, could fulfill the role of Lm Hfq residue Q6, neither residue is positioned properly nor long enough to interact with the uracil base (Fig. 2B) (Schumacher et al. 2002). Thus, despite the conservation of several key residues, the uracil recognition mechanisms of Hfq proteins differ in several key aspects even among the Gram-positive bacteria, with the most notable being the recruitment of residues G5 and Q6 to aid in base specificity.
Lm Hfq binding affinity for uridine-rich tracts is length- and magnesium-dependent
Despite the large number of interactions between multiple side chains of Lm Hfq and the uridine nucleoside, we found initially that the measured binding affinity of Lm Hfq for U6 is significantly lower than that observed for Hfq proteins from other species (Sauer and Weichenrieder 2011; Horstmann et al. 2012; Robinson et al. 2014) with the Kd of U6-Lm Hfq interaction ∼3.4 μM (Supplemental Fig. S4A). This is in sharp contrast to the 70 nM affinity of U6 for Sa Hfq (Horstmann et al. 2012) and is yet sevenfold higher than U6 binding to Ec Hfq (Table 2). This lower affinity is surprising given the similarity of the uridine binding modes between these Hfq proteins and the large number of Lm Hfq-base contacts. Inspection of the electrostatic surface potentials (Baker et al. 2001) suggests that, while the proximal face of the four proximally bound Hfq proteins is largely electropositive, the distribution of this charge is variable (Fig. 3). Indeed, the interior of the Lm pore is electrostatically less positive than the Sa, St, and Ec Hfq proteins (Fig. 3). This difference in electrostatic potential might be less accommodating of the phosphate groups of the oligoribonucleotide, which sit in the pore and are not contacted directly by any residues (Fig. 1D) and, hence, may hinder smaller pore-bound RNAs from binding with the higher affinities observed for other U-tract RNA-Hfq complexes (Sauer and Weichenrieder 2011; Horstmann et al. 2012). Another contributor to the low affinity of Lm Hfq for U6 could be the need for additional flanking RNA to effect higher affinity binding. Previously, we measured the dissociation constant for Ec Hfq with both U6 and U16 RNA. The measured Kd values in those experiments were 766 nM and 6.6 nM, respectively, indicating that longer U-tracts, indeed, lead to higher affinity (Table 2; Robinson et al. 2014). In accord, a binding study using a longer U-tract RNA, U18, and Ec Hfq at half the salt concentration used in our experiments (100 mM versus 200 mM) resulted in a Kd of 120 pM (Olejniczak 2011). These binding data are consistent with the ability of the longer U-tract to bind in the pore of Lm Hfq, thus providing selectivity, while the remaining uridines interact with other residues on the proximal face and potentially the lateral rim and result in high-affinity binding (Murina et al. 2013; Panja et al. 2013). In support of this hypothesis, we determined the Kd of the Lm Hfq-U16 complex (Supplemental Fig. S4B). The equilibrium dissociation constant is 10.2 nM, 330 times higher affinity than that of Lm Hfq-U6 binding (Table 2).
TABLE 2.
Equilibrium dissociation constants (Kd) for selected Hfq–RNA complexes

FIGURE 3.

Surface electrostatic potentials of selected Hfq proteins. Views of the surface electrostatic potentials of the proximal faces of (A) Lm Hfq, (B) Sa Hfq (PDB: 1KQ1), (C) St Hfq (PDB:2YLB), and (D) Ec Hfq (PDB:1HK9), contoured at ±3 kT/e (T = 310 K). Calculations were made using the APBS plug-in in PyMol (Baker et al. 2001). N.B.: The pore of Lm Hfq, the U6 RNA binding location, is less positive than the other three Hfq proteins. The electrostatic surface of the proximal pore of each protein likely has a strong influence on the binding affinity of each protein for U6 RNA.
The highest reported binding affinity of U6 for any Hfq protein was observed for St Hfq, where the Kd was 13 nM (Sauer and Weichenrieder 2011). This value was obtained in the presence of 10 mM magnesium. As our previous Lm Hfq-RNA binding experiments were devoid of magnesium, we tested the effect of this divalent cation on U6 and U16 binding to Lm Hfq and U6 binding to Ec Hfq. The latter protein was reassessed because we previously found an inexplicably poor affinity of Ec Hfq for U6 (Robinson et al. 2014). However, this binding experiment was conducted in the absence of magnesium. For both Hfq proteins, the binding affinity for U6 was greatly increased in the presence of 10 mM magnesium, with the Kd for Lm Hfq dropping 343-fold to 9.9 nM and the Kd for Ec Hfq dropping 232-fold to 3.3 nM (Supplemental Fig. S4C; Table 2). Interestingly, a smaller though significant increase in binding affinity was measured for U16, with a calculated Kd of 2.7 nM in the presence of 10 mM magnesium versus 10.2 nM in the absence of magnesium. Hence, magnesium makes a significant difference for Hfq-U6 affinity and is a likely important contributor to in vivo binding of smaller RNA tracts. In contrast, longer tracts such as U16 appear to be less dependent upon magnesium for high-affinity binding, a result that likely arises from additional interactions with proximal face and lateral rim residues of Hfq. Regardless, magnesium binding to the sugar-phosphate backbone of U6 and U16 would be expected to neutralize its strong negative charge and thereby stabilize RNA binding in the nonpolar proximal pore of Lm Hfq. The beneficial electrostatic effect of this divalent cation on St and Ec Hfq binding to U6 is less clear, but this ion could play a key role in stabilizing a higher-affinity binding conformation of the RNA.
As an additional interrogation of the role of magnesium in Lm Hfq-RNA binding affinity and to ensure that residue Q6 is, indeed, important for U6 binding and not simply an artefact of crystal packing, we created a glutamine-to-alanine mutation at position 6 and measured the affinity of the purified Lm Hfq Q6A protein to U6 and U16 RNA in the presence of magnesium. This substitution, which results in the loss of the carboxamide side chain, abolished binding to U6 and reduced the affinity for U16 by ∼50-fold (Table 2), thereby demonstrating its importance in uracil binding to the proximal face.
Lm Hfq does not appear to recognize specifically the 3′ end of poly-U RNA
Previous comparison of the RNA backbone conformations of the Sa Hfq–AU5G and St Hfq–U6 complex crystal structures showed that, when bound to St Hfq, the RNA backbone adopted a constricted conformation, whereas in Sa Hfq a dilated conformation was observed (Sauer and Weichenrieder 2011). The difference in the conformation stems from the positioning of the 3′ end of the RNA. In St Hfq, both the 2′ and 3′ hydroxyls of the RNA reside proximal to residue H57, whereas this is not the case in Sa Hfq, as the dilated conformation of the phosphates has pushed the hydroxyls too far from H58, thereby preventing hydrogen bonding. Comparison of Lm Hfq-bound RNA with the U-rich tracts of the St, Sa, and Ec Hfq-RNA complexes (Supplemental Fig. S2) reveals that U6 from Lm Hfq is most similar to the constricted conformation of poly-U RNA binding observed in the St Hfq-U6 complex. However, it is less likely that the 3′ end of the Lm Hfq-bound U6 is specifically recognized, as the 2′ hydroxyl of all twelve uridines interact with H59 with nearly all engaged in a strong hydrogen bond, while all but two 3′ hydroxyl groups are in poor position to hydrogen bond strongly (Supplemental Figs. S1B, S2A). To ensure that this was not the result of the statistical disorder of the U6, we attempted to force a geometrically proper hydrogen bond between the 3′ OH and residue H59. However, none of these bonds withstood refinement and ended with only the 2′ OH-H59 interaction. To test biochemically if 3′ hydroxyl recognition is important for specific RNA binding by Lm Hfq, we utilized a 5′-fluorescein-labeled oligonucleotide, U15dUp, in which the 3′-terminal uridine was modified to remove the 2′ hydroxyl group (dU) and its 3′ hydroxyl modified with a phosphate (dUp). Addition of the phosphate makes this more similar to an internal nucleotide, while removal of the 2′ hydroxyl eliminates further hydrogen bonding capabilities of the 3′ end. The binding affinity of this oligonucleotide for Lm Hfq was essentially identical to the affinity of the Lm Hfq for U16 in the presence or absence of magnesium (Table 2; Supplemental Fig. S4) suggesting that Lm Hfq, unlike St Hfq, does not specifically recognize the 3′ end of the RNA via the free 3′ hydroxyl alone or at the least that Lm Hfq does not bind this terminal nucleotide with higher affinity than internal uridines. Alternatively, it is possible that a long U-tract, such as U16, competes with recognition of the 3′ end and is able to displace this terminal nucleotide. Regardless our structural and binding data suggest that Lm Hfq binds U-rich tracts differently from St Hfq and perhaps highlight a functional difference between Gram-negative and Gram-positive Hfq proteins.
Tryptophan fluorescence quenching: novel (GU3)G and U6 binding modes
In order to understand more fully the RNA sequence preferences and potential RNA binding loci of Lm Hfq, a series of tryptophan fluorescence quenching (TFQ) experiments were carried out using a variety of RNA sequences. This approach takes advantage of the intrinsic fluorescence quenching of any tryptophan by nucleobases within the proximity of the indole ring (5 Å or less) and has been used recently to probe the Hfq-RNA interfaces of Ec and Sa Hfq (Supplemental Fig. S5; Robinson et al. 2014). Wild-type Lm Hfq has no tryptophans and three single tryptophan-containing Lm Hfq proteins were newly created, expressed, and purified for these TFQ experiments (Supplemental Fig. S6A,B). These mutants are as follows: R17W (on the lateral rim), F26W (a distal face A-site component), and L33W (a site proximal to the distal face A-site). The TFQ data from a F43W mutant, which probes proximal-face RNA binding (Fig. 2A) and has been characterized previously (Robinson et al. 2014), are presented again to provide a fuller contextual understanding of the different RNA binding modes of Lm Hfq. RNA sequences assayed in the TFQ experiments include the “control” sequences A15 and U6, which were expected to bind preferentially the distal and proximal faces, respectively, G7, and the alternating purine-pyrimidine tracts, AUAUAUA (denoted [AU]3A), ACACACA (denoted [AC]3A), and GUGUGUG (denoted [GU]3G).
As expected, A15 strongly quenches the F26W mutant without exhibiting any significant interaction with the other mutant sites (Fig. 4), although the relatively close distal-face residue, L33W, shows minor quenching at the higher 4-μM RNA concentration (Supplemental Fig. S6D). In sum, these data underscore the tight binding of this adenine tract to the distal-face purine nucleotide binding site (Fig. 4). In contrast, U6 unexpectedly quenched mutants on both the distal and proximal faces. Quenching of the F43W proximal face mutant was expected based on previous studies on Sa Hfq (Schumacher et al. 2002) and Ec Hfq (Wang et al. 2011, 2013; Robinson et al. 2014). However, quenching of the A-site F26W mutant represents the first observed interaction of a polyuridine tract with the distal face (Fig. 4A,B). Even though Lm Hfq is able to accommodate U-rich RNA tracts on both its RNA-binding faces, significantly greater quenching of the F43W mutant at both tested RNA concentrations indicates a far stronger preference for the proximal side. In contrast, U16 RNA does not quench F26W but essentially fully quenches F43W and lateral rim mutants (discussed in more detail below), indicating that poly-U sequences in the context of longer RNA prefer to bind to the proximal face (Fig. 4). Smaller uridine-containing tracts are also observed to bind to the distal face when intermingled with purine nucleobases. Thus, as observed for Sa Hfq, (AU)3A was able to bind to both the distal and proximal faces of Lm Hfq (Fig. 4A,B; Robinson et al. 2014). Substitution of the uridines by cytidines appears to address these smaller oligoribonucleotides to the distal face only, as (AC)3A quenches only F26W, which was also observed for Sa Hfq (Fig. 4A). Intriguingly, both F26W and F43W are quenched by (GU)3G, whereas no quenching of either residue was observed for G7, indicative of no RNA binding (Fig. 4A,B). We should note that any potential complications of secondary structure formation of these oligoribonucleotides are minimal as the experiments are conducted at 25°C and the calculated Tm values for an (AU)3 duplex is 12°C and for a (GU)3 duplex is 8.5°C. Further, thermal denaturation studies on G7, which under some specific conditions might form a G-quartet, revealed no discernable hyperchromic shift in its UV absorption from 20°C to 80°C, strongly suggesting no ordered base-stacked structure is present at 25°C.
FIGURE 4.

Tryptophan fluorescence quenching (TFQ) of Lm Hfq mutants by a series of RNAs. (A) TFQ of Gram-positive A-site pocket residue F26W. (B) TFQ of the proximal face-uracil stacking residue F43W (taken from Robinson et al. 2014). (C) TFQ of lateral rim residue R17W. (D) TFQ of lateral rim residue S39W. The abscissa shows the RNA sequence used and the ordinate the percent quenched. Each RNA and its associated quenching (shown as a bar) are colored the same with A15 in red, U6 in blue, U16 in cyan, (GU)3G in green, (AU)3A in lavender, (AC)3A in burgundy, and G7 in gold. The solid bar indicates the percent quenched at 1-μM RNA concentration and the striped bar at 4-μM concentration.
The quenching of F43W by (GU)3G was reported earlier but is unexplained, as the U6-bound Lm structure suggests that the exocyclic N2 nitrogen of guanine would clash with residue Q9 (Robinson et al. 2014). In order to analyze the consequence of this Phe-to-Trp change further, we determined the crystal structure of the Lm Hfq F43W mutant (Table 1). Superposition of the mutant and WT Lm Hfq structures reveals that the introduced mutation does not alter the shape of the preformed binding pocket, suggesting that any bound guanosine could not be positioned in the pocket identically to that of a bound uracil (Supplemental Fig. S6C). However, without relevant structures of Lm Hfq-(GU)3G complexes, we can only speculate on its binding mode. It is possible that the guanine ring binds but does not penetrate as deeply into this binding pocket as the uracil base or that this purine base forces the protein to adopt a locally altered conformation. Alternatively, the uracils of the (GU)3G might bind as observed in the Lm Hfq-U6 complex structure but the guanines flip out into the pore in order to avoid steric clash. Although less likely, the quenching of F43W might arise as a consequence of a guanosine binding in the open pore as seen in the Sa Hfq-AU5G complex (Schumacher et al. 2002), thereby positioning one or more of the other bases of the (GU)3G tract close to the W43 residue to elicit tryptophan fluorescence quenching.
The finding that (AU)3A, (GU)3G, and (AC)3A quench the F26W mutant supports an R-L binding motif for the distal face of Lm Hfq in which the R site accommodates purine bases and a nondiscriminating linker site (L site) stacks with the intervening pyrimidine. This is in sharp contrast to Sa Hfq, which has been shown to exhibit a more stringent (A-L)n motif with the R site, in fact, an adenine-only binding pocket (Horstmann et al. 2012; Robinson et al. 2014). Regardless, Lm Hfq prefers to bind the (GU)3G sequence on its proximal face, as the quenching is far more robust at all tested concentrations and approaches nearly 60% of the proximal-face quenching by U6 (Fig. 4A,B).
The lack of quenching of the R17W mutant was expected for all the RNA sequences tested, as we would expect only longer RNA sequences to interact with and quench this residue. Previous data revealed that Ec hfq mRNA will quench R17W in E. coli (Robinson et al. 2014). Indeed, recent studies show that arginines at positions 16, 17, and 19 in Ec Hfq are critical for function but do not affect the ability of Hfq to bind short RNA sequences such as those used in our study (Panja et al. 2013). It should be noted that Sa Hfq contains lysine, alanine, and glutamine residues at positions equivalent to 16, 17, and 19, whereas Lm Hfq is more similar to Ec Hfq, with an arginine-lysine-lysine triplet (Supplemental Fig. S3). To test the ability of the lateral rim site to bind longer RNA, we performed TFQ with R17W and U16 RNA and were surprised to find no quenching. It became apparent though that this mutation disrupts binding to the lateral rim site, as the affinity of the R17W protein for U16 was significantly lower (Kd = 2.8 μM) (Table 2). However, as a key component of our TFQ approach to account for any mutation-induced loss of affinity and the resulting loss of quenching, we introduce tryptophans at additional sites proximal to our site of primary interest. Hence, we created and tested the Lm Hfq S39W mutant, which has the closest approach to R17 of ∼4.5 Å. The Lm Hfq S39W protein shows wild-type binding to U16 (Kd = 2.7 nM) (Table 2). As anticipated, the S39W mutant was quenched significantly by U16 but not U6 RNA (Fig. 4D), thus supporting the notion that S39 and R17 form part of the proximal face sRNA binding path.
CONCLUSIONS AND PERSPECTIVE
The crystal structure of the Lm Hfq-U6 complex reveals a new interaction utilized in uracil recognition and binding in which the side chain of residue Q6, restricted stereochemically by a G5-N42-Q6 interaction network, reads the N3 and O4 positions of the uracil base eliciting specificity. Intriguingly, our TFQ studies indicate that U6 and (GU)3G RNA bind to both faces of Lm Hfq, although both U-containing sequences prefer to bind to the proximal face. To the best of our knowledge, this is the first instance of uridine tract binding to the distal-face “A-site.” Combined, these results support the contention that the RNA binding pockets of Hfq proteins have the potential to bind a wider variety of RNA sequences than previously envisioned. Clearly, a fuller understanding of the Lm Hfq-U6 distal-face binding, Lm Hfq-(GU)3G proximal-face binding, and Lm Hfq binding to longer and more structured RNA sequences will require additional structural and biochemical studies. The resulting data will likely require the development of a new, more comprehensive RNA-binding site nomenclature by the Hfq community. More globally, our data demonstrate that, while Hfq proteins from various species of both Gram-negative and Gram-positive bacteria have highly conserved structures, the atomic details of their binding to the same RNA sequences can vary significantly.
MATERIALS AND METHODS
Protein expression, purification, and site-directed mutagenesis
Listeria monocytogenes Hfq was overexpressed in E. coli strain ER2566 Δhfq using the pTYB11 vector. The cells were grown in Luria Broth (LB) containing 50 µg/mL ampicillin at 37°C to an OD600 between 0.4 and 0.6. Expression was induced with 0.5 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) for 20 h at 15°C. Cells were harvested at 4°C and stored at −80°C or lysed immediately using a microfluidizer. To remove contaminating RNA and DNA, 10 µg/mL DNase and 10 µg/mL RNase were added to the crude lysate and stirred at 4°C for 2 h before clarification by centrifugation at 33,000g for 30 min at 4°C. Hfq was purified using the IMPACT-CN system as described (Moller et al. 2002). Mutations in the WT Lm hfq gene were generated using standard protocols. Lm Hfq is less stable than other Hfq proteins, and any attempt to reduce the salt and DTT concentrations in the final storage buffer as well as maintain the protein at high concentration resulted in aggregation and precipitation. Hence, the protein was stored in 20 mM (4-[2-hydroxyethyl]-1-piperazineethanesulfonic acid) (HEPES)-Na, pH 8.0, 500 mM NaCl, 0.5 mM EDTA, 2 mM DTT at concentrations <6 mg/mL.
Crystallization and data collection
Crystallization trials were performed using the hanging drop-vapor diffusion method at room temperature (22°C) by mixing equal volumes of Lm Hfq at 5.5 mg/mL and the crystallization reservoir solution. A solution containing 100 mM HEPES-Na, pH 7.5 and 40% 1,2-propanediol yielded crystals within 1 to 3 wk. The crystals were dipped briefly into a 100 mM HEPES-Na, pH 7.5 and 45% 1,2-propanediol solution and flash frozen in a nitrogen stream at 100K. X-ray diffraction intensity data were collected to a resolution of 2.6 Å using a Rigaku FR-E+ Superbright rotating copper anode with an HTC imaging plate detector. Data reduction was done using iMOSFLM and SCALA in the CCP4 suite (Collaborative Computational Project 1994; Battye et al. 2011). The crystals take the space group P21221 with the cell dimensions a = 63.6 Å, b = 66.8 Å, c = 106.5 Å.
To obtain crystals of the Hfq-U6 complex, a reservoir solution of 100 mM HEPES-Na, pH 7.5 and 45% 1,2-propanediol was mixed 1:1 with 100 μM U6 RNA dissolved in 10 mM sodium cacodylate, pH 6.5. Apo crystals were then transferred to the hanging drop, the well was sealed, and the crystals were soaked for 5 d and then frozen directly in liquid nitrogen. X-ray diffraction intensity data were collected to a resolution of 3.1 Å at the Advanced Photon Source (APS) at Argonne National Laboratory on beamline 22-ID-D. Data reduction was done using HKL2000 (Otwinowski and Minor 1997). The crystals take the monoclinic space group C2 with the cell dimensions a = 124.0, b = 123.9, c = 67.6, and β = 90.1°. Analysis of both the apo Lm Hfq and Lm Hfq-U6 complex intensity data using phenix.xtriage revealed no twinning.
Structure determination and refinement
The apo Lm Hfq structure was solved by molecular replacement using Phaser (McCoy 2007) with an Hfq protomer, which was generated through the Protein Homology/Analogy Recognition Engine (PHYRE) v2.0 server, serving as the search model. The asymmetric unit contains two trimers. The Hfq-U6 structure was solved by molecular replacement using the contents of the asymmetric unit from the apo structure as a search model. The asymmetric unit contains two hexamers. Both models were subjected to an initial round of rigid body refinement in PHENIX.Refine, and noncrystallographic symmetry restraints were employed (Adams et al. 2010). The RNA and residues that were not part of the search model were built iteratively using COOT (Emsley and Cowtan 2004), and additional refinement was carried out using PHENIX. Due to the symmetry of the space group, the RNA backbone is statistically disordered whereby each phosphate group has an occupancy of 0.83 and an occupancy of 0.17 as the 5′ and 3′ hydroxyl groups. During refinement, however, a single, arbitrary chosen 5′ and 3′ end were assigned and all backbone occupancies were held at 1.0. As a consequence, the B factors of the phosphate and 5′ and 3′ hydroxyl groups reflected the lowered occupancies. We compared this refinement strategy to one in which a phosphate was added to connect the free 5′ and 3′ ends, and all phosphate occupancies were set to 0.83. We observed essentially no change in the position and conformation of the RNA backbone, indicating that our refinement strategy did not impact the structure or its interpretation.
Coordinates and structure factors
The coordinates and structure factors of the apo Lm-Hfq, Lm Hfq–U6 complex, and Lm F43W structures, have been deposited in the PDB under the PDB ID codes 4NL2, 4NL3, and 4NOY, respectively.
Tryptophan fluorescence quenching
Tryptophan fluorescence quenching measurements were made using an RF-5301PC spectrofluorophotometer (Shimadzu) at 298 K and followed the protocol of Robinson et al. (2014). Each sample was excited at 298 nm and its fluorescence emission scanned from 320 to 400 nm. A 1-mL sample containing 1 µM Hfq protein in binding buffer (20 mM HEPES-Na, pH 8.0, 200 mM NaCl, 0.5 mM EDTA) was first scanned to determine the maximum fluorescence emission wavelength, which is 343 nm. TFQ was measured after the addition of 1 μM and 4 μM oligoribonucleotides. Each titration was done at least three times. Data were analyzed using Microsoft Excel. Quenching was determined using the arbitrary fluorescence maximal height for each Trp mutant in the absence of any oligoribonucleotide. The quenching percentage was calculated using the equation:
where FR is the fluorescence value after addition of RNA to the Hfq solution, F0 is the initial fluorescence value of the Hfq solution without RNA, and FB is the fluorescence of buffer without RNA or Hfq.
Fluorescence polarization
The RNA binding affinities of Hfq were determined by a fluorescence polarization-based assay. The polarization measurements were done with a PanVera Beacon 2000 instrument (Invitrogen) at 295 K. Hfq was serially aliquoted into 100 µL of binding buffer, which consisted of 20 mM HEPES-Na, pH 8.0, 0.5 mM EDTA, 200 mM NaCl and contained either 1 nM or 0.5 nM 5′-fluorescein-labeled U6 or U16 RNA (IDT DNA Technologies) depending upon the affinity. Samples were excited at 490 nm and emission was detected at 530 nm. Data were analyzed assuming a 1:1 binding stoichiometry between one Hfq hexamer and one molecule of RNA. The data were plotted using KaleidoGraph (Synergy Software), and the generated curves were fit using nonlinear least squared analysis, assuming a bimolecular model such that the Kd values represent the protein concentration at half maximal RNA binding. The binding isotherms were fit to the equation, P = {(Pbound − Pfree) [protein]/(Kd + [protein])} + Pfree, with Pbound being the maximum polarization at saturation, P is the polarization at a given protein concentration, Pfree is the polarization of free fluorescein-labeled RNA, and Kd is the equilibrium dissociation constant for one hexamer binding to the oligoribonucleotide. The measured millipolarization (mP) is equal to P × 10−3. All values were independently determined in triplicate.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
X-ray diffraction data were collected remotely at the Southeast Regional Collaborative Access Team 22-ID-D beamline at the Advanced Photon Source, Argonne National Laboratory, which is supported by the US Department of Energy, Office of Science and the Office of Basic Energy Sciences under Contract No. W-31-109-Eng-38. This work was supported by the Structural Biology and Biophysics (SBB) Training Program (NIH/NIGMS 5T32 GM008487) and Duke University School of Medicine.
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.044032.113.
Freely available online through the RNA Open Access option.
REFERENCES
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. 2010. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66: 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA 2001. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci 98: 10037–10041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battye TG, Kontogiannis L, Johnson O, Powell HR, Leslie AG 2011. iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr D Biol Crystallogr 67: 271–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohn C, Rigoulay C, Bouloc P 2007. No detectable effect of RNA-binding protein Hfq absence in Staphylococcus aureus. BMC Microbiol 7: 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boisset S, Geissmann T, Huntzinger E, Fechter P, Bendridi N, Possedko M, Chevalier C, Helfer AC, Benito Y, Jacquier A, et al. 2007. Staphylococcus aureus RNAIII coordinately represses the synthesis of virulence factors and the transcription regulator Rot by an antisense mechanism. Genes Dev 21: 1353–1366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao Y, Vogel J 2010. The role of Hfq in bacterial pathogens. Curr Opin Microbiol 13: 24–33 [DOI] [PubMed] [Google Scholar]
- Christiansen JK, Larsen MH, Ingmer H, Sogaard-Andersen L, Kallipolitis BH 2004. The RNA-binding protein Hfq of Listeria monocytogenes: role in stress tolerance and virulence. J Bacteriol 186: 3355–3362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christiansen JK, Nielsen JS, Ebersbach T, Valentin-Hansen P, Sogaard-Andersen L, Kallipolitis BH 2006. Identification of small Hfq-binding RNAs in Listeria monocytogenes. RNA 12: 1383–1396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collaborative Computational Project, Number 4. 1994. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr 50: 760–763 [DOI] [PubMed] [Google Scholar]
- Collins BM, Harrop SJ, Kornfeld GD, Dawes IW, Curmi PM, Mabbutt BC 2001. Crystal structure of a heptameric Sm-like protein complex from archaea: implications for the structure and evolution of snRNPs. J Mol Biol 309: 915–923 [DOI] [PubMed] [Google Scholar]
- Dambach M, Irnov I, Winkler WC 2013. Association of RNAs with Bacillus subtilis Hfq. PLoS One 8: e55156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Y, Davis BM, Waldor MK 2004. Hfq is essential for Vibrio cholerae virulence and downregulates σ expression. Mol Microbiol 53: 345–354 [DOI] [PubMed] [Google Scholar]
- Emsley P, Cowtan K 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60: 2126–2132 [DOI] [PubMed] [Google Scholar]
- Fantappie L, Metruccio MM, Seib KL, Oriente F, Cartocci E, Ferlicca F, Giuliani MM, Scarlato V, Delany I 2009. The RNA chaperone Hfq is involved in stress response and virulence in Neisseria meningitidis and is a pleiotropic regulator of protein expression. Infect Immun 77: 1842–1853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geisinger E, Adhikari RP, Jin R, Ross HF, Novick RP 2006. Inhibition of rot translation by RNAIII, a key feature of agr function. Mol Microbiol 61: 1038–1048 [DOI] [PubMed] [Google Scholar]
- Hayashi-Nishino M, Fukushima A, Nishino K 2012. Impact of hfq on the intrinsic drug resistance of Salmonella enterica serovar Typhimurium. Front Microbiol 3: 205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidrich N, Chinali A, Gerth U, Brantl S 2006. The small untranslated RNA SR1 from the Bacillus subtilis genome is involved in the regulation of arginine catabolism. Mol Microbiol 62: 520–536 [DOI] [PubMed] [Google Scholar]
- Heidrich N, Moll I, Brantl S 2007. In vitro analysis of the interaction between the small RNA SR1 and its primary target ahrC mRNA. Nucleic Acids Res 35: 4331–4346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horstmann N, Orans J, Valentin-Hansen P, Shelburne SA III, Brennan RG 2012. Structural mechanism of Staphylococcus aureus Hfq binding to an RNA A-tract. Nucleic Acids Res 40: 11023–11035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huntzinger E, Boisset S, Saveanu C, Benito Y, Geissmann T, Namane A, Lina G, Etienne J, Ehresmann B, Ehresmann C, et al. 2005. Staphylococcus aureus RNAIII and the endoribonuclease III coordinately regulate spa gene expression. EMBO J 24: 824–835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kambach C, Walke S, Young R, Avis JM, de la Fortelle E, Raker VA, Luhrmann R, Li J, Nagai K 1999. Crystal structures of two Sm protein complexes and their implications for the assembly of the spliceosomal snRNPs. Cell 96: 375–387 [DOI] [PubMed] [Google Scholar]
- Link TM, Valentin-Hansen P, Brennan RG 2009. Structure of Escherichia coli Hfq bound to polyriboadenylate RNA. Proc Natl Acad Sci 106: 19292–19297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Wu N, Dong J, Gao Y, Zhang X, Mu C, Shao N, Yang G 2010. Hfq is a global regulator that controls the pathogenicity of Staphylococcus aureus. PLoS One 5: e13069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandin P, Repoila F, Vergassola M, Geissmann T, Cossart P 2007. Identification of new noncoding RNAs in Listeria monocytogenes and prediction of mRNA targets. Nucleic Acids Res 35: 962–974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy AJ 2007. Solving structures of protein complexes by molecular replacement with Phaser. Acta Crystallogr D Biol Crystallogr 63: 32–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moller T, Franch T, Hojrup P, Keene DR, Bachinger HP, Brennan RG, Valentin-Hansen P 2002. Hfq: a bacterial Sm-like protein that mediates RNA-RNA interaction. Mol Cell 9: 23–30 [DOI] [PubMed] [Google Scholar]
- Mura C, Cascio D, Sawaya MR, Eisenberg DS 2001. The crystal structure of a heptameric archaeal Sm protein: implications for the eukaryotic snRNP core. Proc Natl Acad Sci 98: 5532–5537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murina V, Lekontseva N, Nikulin A 2013. Hfq binds ribonucleotides in three different RNA-binding sites. Acta Crystallogr D Biol Crystallogr 69: 1504–1513 [DOI] [PubMed] [Google Scholar]
- Nielsen JS, Olsen AS, Bonde M, Valentin-Hansen P, Kallipolitis BH 2008. Identification of a sigma B-dependent small noncoding RNA in Listeria monocytogenes. J Bacteriol 190: 6264–6270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen JS, Lei LK, Ebersbach T, Olsen AS, Klitgaard JK, Valentin-Hansen P, Kallipolitis BH 2010. Defining a role for Hfq in Gram-positive bacteria: evidence for Hfq-dependent antisense regulation in Listeria monocytogenes. Nucleic Acids Res 38: 907–919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olejniczak M 2011. Despite similar binding to the Hfq protein regulatory RNAs widely differ in their competition performance. Biochemistry 50: 4427–4440 [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W 1997. Processing of X-ray diffraction data collected in oscillation mode. In Methods in Enzymology (ed. Carter CW Jr, Sweet RM), Vol. 276: Macromolecular Crystallography, Part A, pp. 307–326 Academic Press, New York: [DOI] [PubMed] [Google Scholar]
- Panja S, Schu DJ, Woodson SA 2013. Conserved arginines on the rim of Hfq catalyze base pair formation and exchange. Nucleic Acids Res 41: 7536–7546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preis H, Eckart RA, Gudipati RK, Heidrich N, Brantl S 2009. CodY activates transcription of a small RNA in Bacillus subtilis. J Bacteriol 191: 5446–5457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson KE, Orans J, Kovach AR, Link TM, Brennan RG 2014. Mapping Hfq-RNA interaction surfaces using tryptophan fluorescence quenching. Nucleic Acids Res 42: 2736–2749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauer E, Weichenrieder O 2011. Structural basis for RNA 3′-end recognition by Hfq. Proc Natl Acad Sci 108: 13065–13070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacher MA, Pearson RF, Moller T, Valentin-Hansen P, Brennan RG 2002. Structures of the pleiotropic translational regulator Hfq and an Hfq-RNA complex: a bacterial Sm-like protein. EMBO J 21: 3546–3556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sittka A, Pfeiffer V, Tedin K, Vogel J 2007. The RNA chaperone Hfq is essential for the virulence of Salmonella typhimurium. Mol Microbiol 63: 193–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Someya T, Baba S, Fujimoto M, Kawai G, Kumasaka T, Nakamura K 2012. Crystal structure of Hfq from Bacillus subtilis in complex with SELEX-derived RNA aptamer: insight into RNA-binding properties of bacterial Hfq. Nucleic Acids Res 40: 1856–1867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnleitner E, Hagens S, Rosenau F, Wilhelm S, Habel A, Jager KE, Blasi U 2003. Reduced virulence of a hfq mutant of Pseudomonas aeruginosa O1. Microb Pathog 35: 217–228 [DOI] [PubMed] [Google Scholar]
- Storz G, Opdyke JA, Zhang A 2004. Controlling mRNA stability and translation with small, noncoding RNAs. Curr Opin Microbiol 7: 140–144 [DOI] [PubMed] [Google Scholar]
- Sun X, Zhulin I, Wartell RM 2002. Predicted structure and phyletic distribution of the RNA-binding protein Hfq. Nucleic Acids Res 30: 3662–3671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toledo-Arana A, Dussurget O, Nikitas G, Sesto N, Guet-Revillet H, Balestrino D, Loh E, Gripenland J, Tiensuu T, Vaitkevicius K, et al. 2009. The Listeria transcriptional landscape from saprophytism to virulence. Nature 459: 950–956 [DOI] [PubMed] [Google Scholar]
- Toro I, Thore S, Mayer C, Basquin J, Seraphin B, Suck D 2001. RNA binding in an Sm core domain: X-ray structure and functional analysis of an archaeal Sm protein complex. EMBO J 20: 2293–2303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsui HC, Leung HC, Winkler ME 1994. Characterization of broadly pleiotropic phenotypes caused by an hfq insertion mutation in Escherichia coli K-12. Mol Microbiol 13: 35–49 [DOI] [PubMed] [Google Scholar]
- Valentin-Hansen P, Eriksen M, Udesen C 2004. The bacterial Sm-like protein Hfq: a key player in RNA transactions. Mol Microbiol 51: 1525–1533 [DOI] [PubMed] [Google Scholar]
- Walke S, Bragado-Nilsson E, Seraphin B, Nagai K 2001. Stoichiometry of the Sm proteins in yeast spliceosomal snRNPs supports the heptamer ring model of the core domain. J Mol Biol 308: 49–58 [DOI] [PubMed] [Google Scholar]
- Wang W, Wang L, Zou Y, Zhang J, Gong Q, Wu J, Shi Y 2011. Cooperation of Escherichia coli Hfq hexamers in DsrA binding. Genes Dev 25: 2106–2117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Wang L, Wu J, Gong Q, Shi Y 2013. Hfq-bridged ternary complex is important for translation activation of rpoS by DsrA. Nucleic Acids Res 41: 5938–5948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wassarman KM, Repoila F, Rosenow C, Storz G, Gottesman S 2001. Identification of novel small RNAs using comparative genomics and microarrays. Genes Dev 15: 1637–1651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada J, Yamasaki S, Hirakawa H, Hayashi-Nishino M, Yamaguchi A, Nishino K 2010. Impact of the RNA chaperone Hfq on multidrug resistance in Escherichia coli. J Antimicrob Chemother 65: 853–858 [DOI] [PubMed] [Google Scholar]
- Zhang A, Altuvia S, Tiwari A, Argaman L, Hengge-Aronis R, Storz G 1998. The OxyS regulatory RNA represses rpoS translation and binds the Hfq (HF-I) protein. EMBO J 17: 6061–6068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang A, Wassarman KM, Ortega J, Steven AC, Storz G 2002. The Sm-like Hfq protein increases OxyS RNA interaction with target mRNAs. Mol Cell 9: 11–22 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.