

Summary
Heterogeneity is a hallmark of stem cell populations, in part due to the molecular differences between cells undergoing self-renewal and those poised to differentiate. We examined phenotypic and molecular heterogeneity in pluripotent stem cell populations, using public gene expression data sets. A high degree of concordance was observed between global gene expression variability and the reported heterogeneity of different human pluripotent lines. Network analysis demonstrated that low-variability genes were the most highly connected, suggesting that these are the most stable elements of the gene regulatory network and are under the highest regulatory constraints. Known drivers of pluripotency were among these, with lowest expression variability of POU5F1 in cells with the highest capacity for self-renewal. Variability of gene expression provides a reliable measure of phenotypic and molecular heterogeneity and predicts those genes with the highest degree of regulatory constraint within the pluripotency network.
Highlights
-
•
Gene expression variability is highly concordant with population heterogeneity
-
•
Genes within the pluripotency network have distinct variability profiles
-
•
Expression variability is a network property important for pluripotency
Gene expression variability is a simple measure of heterogeneity in stem cell populations and is informative about the behavior of pluripotency genes. Network analysis demonstrates that genes within the pluripotency network have distinct and characteristic variability profiles, such that genes highly connected in the network have the lowest variability. Wells and colleagues propose that this identifies genes that may be under the highest degree of regulatory constraint.
Introduction
Pluripotency can only be propagated in the context of phenotypic heterogeneity: cells flux between states of self-renewal and competency-to-differentiate, but the origin and importance of molecular heterogeneity in these processes remains controversial. Some argue that stem cell heterogeneity is largely a consequence of culture conditions rather than a necessary or inherent property (Smith, 2013), but there is clear evidence that heterogeneity at the molecular level, exemplified by cyclic expression of differentiation-inducing transcription factors, describes critical features of the pluripotent phenotype (Singh et al., 2013). Mouse embryonic stem cells (mESCs) under standard culture conditions exhibit highly variable Nanog expression, permitting the breadth of pluripotency phenotypes to manifest in the stem cell population (Chambers et al., 2007; Hayashi et al., 2008). Low Nanog enhances the competency of mESCs to respond to extrinsic signals required for differentiation, whereas high levels are associated with self-renewal. Mice hemizygous for Pou5f1 express half the wild-type level of Pou5f1 transcript, resulting in the stabilization of Nanog expression and propagation of a ground state of self-renewal (Karwacki-Neisius et al., 2013). Although the ability to grow mESCs in a “ground state” has generated much debate about the physiological significance of stem cell heterogeneity (Karwacki-Neisius et al., 2013; Smith, 2013), it unequivocally demonstrates that variability in the expression of key members of the pluripotency network will drive phenotypic heterogeneity.
Studies of early embryogenesis in other model organisms provide further evidence that expression variability is an essential driver of phenotypic outcome. For example, wild-type Caenorhabditis elegans have a highly predictable genetic network specifying intestinal cell fate that has been well characterized, where the 20 cells that make up the gut descend from a single progenitor (Raj et al., 2010). Expression variability is an intrinsic characteristic of genes composing this developmental network and underlies cell-cell differences in endodermal differentiation outcomes. Mutations in the key transcription factor skn-1 resulted in significant variability in the expression of downstream targets end-1, end-3, and elt-2, even between cells from isogenic individuals (Raj et al., 2010). However, some expression variability of end-1, end-3, and elt-2 was tolerated, providing a level of robustness to the differentiation outcomes driven by these genes, and the level of expression variability was concordant with the deleted gene’s connectivity in the regulatory network. Similarly, the propagation of gene expression variability at different stages of the sea urchin Strongylocentrotus purpuratus development was identified as an important driver of phenotypic diversity (Garfield et al., 2013). These in vivo studies demonstrate the utility of expression variability as a parameter that is directly related to the range of phenotypic outcomes that could be derived from a single well-specified gene regulatory network.
Single-cell expression profiling has allowed researchers to test the idea that gene expression variability reflects true biological variation in cellular mRNA levels. For example, in an analysis of individual pancreatic islet cells, the transcripts of insulin genes Ins1 and Ins2 were highly correlated with each other (Pearson R 0.9), but not other genes (Bengtsson et al., 2005). This supports a model where insulin genes are coexpressed at a high level in some cells and a low level in other cells to produce a spectrum of insulin-producing states within the larger tissue compartment, rather than the generation of two distinct cell populations that display uniquely high or low transcriptional activity. The concordance of any two transcripts in a single cell must be dependent not just on the transcriptional activity of the parent genes, but also on the stability of each mRNA. As a result, single-cell analyses will necessary reveal the stochastic nature of the molecular process of transcription, whereas bulk measures of mRNA across populations of cells will report the average mRNA level. An outstanding question for the field is therefore how to measure, and interpret, the variation of gene expression across a population of cells.
We have previously shown that the coefficient of variation (CoV) identifies variability in repeated measures of the same population (Mar et al., 2011c) to provide a snapshot of each gene across a population of cells and allow these to be classified as either stable (low CoV) or changing (high CoV). The stable genes in a network may represent the elements that help to define key features of phenotype common to all cells in the population. Conversely, highly variable genes are expressed in some individuals in the population but absent in others. In a pluripotency network, these genes may represent elements which fluctuate as an asynchronous stem cell population moves between the transient states of self-renewal and competency-to-differentiate. The propagation of gene expression variability across a pluripotency network may therefore be essential to the regulation of a pluripotent phenotype.
Results
Gene Expression Variability Reflects Population Heterogeneity
Experience tells us that the averages derived from a pool of cells are relatively insensitive to fluctuations of individuals within the pool. We modeled this in Figure S1A (available online) to demonstrate that the CoV was an order of magnitude more sensitive than the mean to fluctuations of even 5% of the cells in a series of pooled measures, and confirmed that the CoV was not intensity dependent. To demonstrate that phenotypic variability within a population was concordant with global gene expression variability in real-world data sets, we examined three independently generated human stem cell microarray experiments. Each experimental series contained subpopulations defined by differing levels of cell-surface markers, which reportedly corresponded with different efficiencies of self-renewal or lineage priming. Figure 1 ordered these from lowest to highest pluripotency based on the published phenotypes. We predicted that populations with low CoV (a ratio of absolute and variable expression) would be less heterogeneous than populations with high CoV, and this holds for the three experimental series examined here. The populations with mixed phenotypes demonstrated the highest overall expression variability. For example partially reprogrammed induced pluripotent stem cell (iPSC) (“iPSC-low,” Vitale) showed the highest gene expression variability. These cells were described as a mixed population of progenitor cells not able to produce all germ layers in a teratoma (Vitale et al., 2012). In contrast, the human embryonic stem cell (hESs) that had been fluorescence-activated cell sorting (FACS) sorted prior to profiling using two pluripotency surface markers (Figure 1C, all P fractions, Hough) had low variability of gene expression (Hough et al., 2009). This population was further fractionated (Figure 1D), P7 cells selected on the highest combined surface expression of GCTM2 and CD9 were reported to have the highest self-renewal capacity and had the lowest CoV, whereas cells in the P4 fraction isolated from the other end of the FACS spectrum had the highest CoV of this series. The increased gene expression variability in the P4 fraction is consistent with a mixed cell population with transitioning phenotypes, where higher numbers of cells were either transiently primed toward a lineage, or committed to differentiation.
Figure 1.
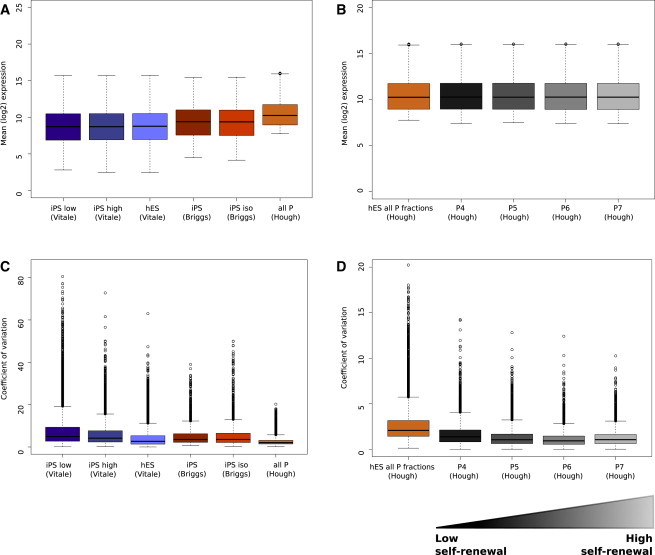
Expression Variability Is Concordant with Population Heterogeneity
(A) and (C) display the average log2 expression levels and the corresponding CoV profiles of each cell phenotype from three independent experimental series.
(B) and (D) illustrate the same metrics between subfractions of a stem cell colony with differing pluripotency phenotypes. The x axes describe the cell phenotype and the experimental series, and the y axes describe the population metric as either coefficient of variation or average log2 gene expression.
Gene Expression Variability Is a Network Feature Persistent in Different Network Types
Given that all the stem cell populations contained some degree of heterogeneity, we exploited this to find highly stable parts of a molecular stem cell network. We postulated that genes with low CoV would identify genes with stable expression across the cell population and highly variable genes may be informative about parts of the network that reflect cell-cell differences within the pluripotent cell population. We tested this hypothesis by extending known pathways (the PluriNet; Müller et al., 2011) and KEGG Extracellular matrix receptor interaction pathway) to construct a pluripotency network that consisted of 1,150 genes (see Experimental Procedures for detail).
We examined the relationships between elements of this network using several approaches: the first was based on the degree of coexpression (Pearson correlation, Figure 2A), which should reflect coordinated patterns of expression across different cell populations. The second and third used both known and predicted protein-protein interactions (PPI network Figure 2C; Figure S2A; STRING, Figure S2B), to ask whether the formation of signaling complexes might also impact on the stability of the network. In all three cases, molecules with a large number of connections (a high degree of connectivity) displayed the most stable expression.
Figure 2.
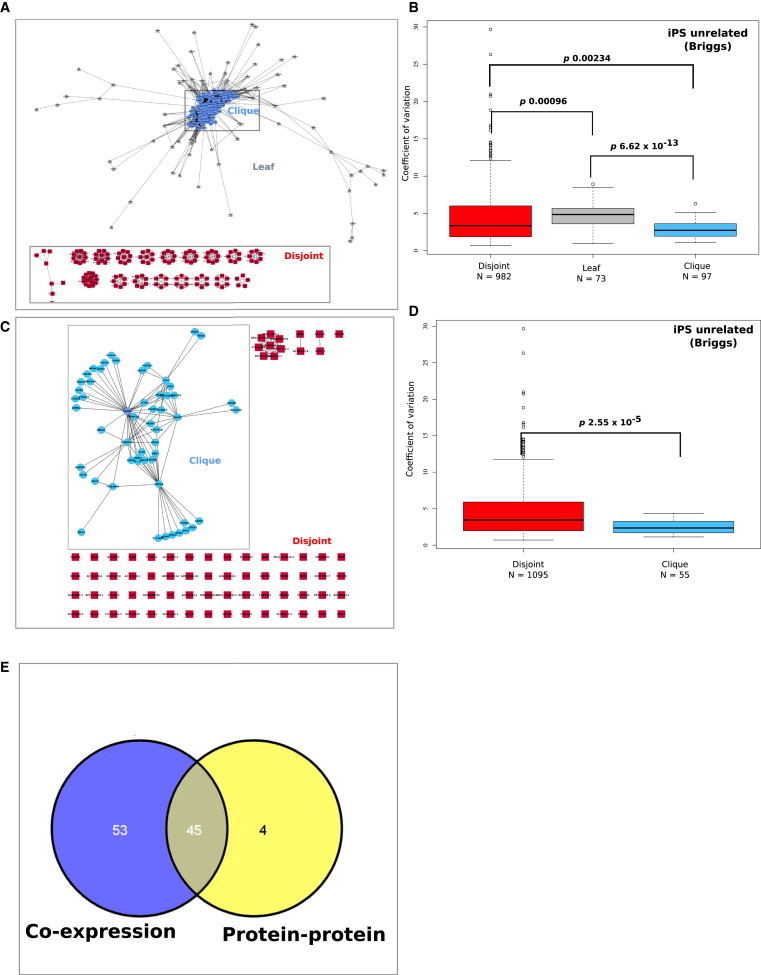
Gene Expression Variability Is Concordant with Network Connectivity
(A) Coexpression network derived from iPS unrelated (Briggs) data set. An edge A→B is drawn whenever there is a correlation in average log2 expression greater than or equal to 0.995. Identifiable substructures were arbitrarily defined as the clique, leaf, and disjoint regions. The color of each node reflects the region to which it has been assigned.
(B) CoV profiles for each region in the coexpression network in the iPS unrelated (Briggs) data set. The x axis describes the network regions and y axis describes the coefficient of variation. The p values assess significant differences in gene expression variability between each network region (p, 0.05, Wilcoxon rank sum).
(C) Protein-protein interaction (PPI) network derived from genes in the full coexpression network using the BisoGenet plug-in for Cytoscape. An edge A→B is drawn whenever there is experimental data that validates an interaction between the protein products of each gene. The densely connected region (defined as the clique) clearly separated from the other nodes (defined as disjoint); no leaf nodes were produced in this network.
(D) CoV profiles for each region in the PPI network in the iPS unrelated (Briggs) data set. The p values assess significant differences in gene expression variability between each network region (p, 0.05, Wilcoxon rank sum).
(E) Venn diagram illustrates the overlap between genes in the coexpression and protein-protein interaction networks for the clique region.
For the coexpression network, we assessed the connectivity (degree) of genes that were coexpressed in iPSC from the Briggs iPSC cell data set (n = 18; Briggs et al., 2013) and defined three network regions, the clique, leaf, and disjoint regions (Figure 2A; Table S1). The dense central network region (clique) represented genes that were coexpressed with a large number of other genes. Nanog was not included in any of the network regions, as the canonical transcript was not present on the HT12-V3-Illumina chips (see Experimental Procedures for further detail). However many other known pluripotency regulators including POU5F1, DNMT3b, SOX2, DPPA4, LIN28, CLDN7, FGFR4, and ZFP42 (REX1) were represented in the clique region, as well OVOL2, USP44, and SRFP2, which have emerging roles in pluripotency (Fuchs et al., 2012; Mirotsou et al., 2007; Zhang et al., 2013). The unifying feature of this region of the coexpression network was enrichment for genes with low expression variability (Figure 2B, p, 0.00234 Wilcoxon rank sum) rather than common amplitude of expression. For example, POU5F1 and SOX2 were highly expressed, DNA damage repair factor C1orf86 was expressed at a low level, and the mesodermal specification marker HEY2 was intermediate.
The majority (85%) of genes in the coexpression network formed small, disjointed subnetworks, such that any gene in this region was coexpressed with a relatively small number of partners (Figure 2A). Among genes in this region were a number of G-coupled protein receptors (e.g., GPR124, GPR137), ribosomal proteins (e.g., RPL24, RPS2), and small nucleolar RNAs (e.g., SNORA10, SNORD109A). This region of the network was enriched for the most variable genes, suggesting that they may be expressed in some cells, but not in others. These showed functional enrichment for mitotic and cell cycle biological processes (Bonferroni-adjusted p value < 0.05; Figure S2C), which is consistent with an asynchronously dividing cell population.
The concordance between gene expression variability and network connectivity was also evident when we examined other types of relationships between the genes in our pluripotency network. For example, we built edges between the genes based on known protein-protein interactions (Figures 2C, 2D, and S2A). The network regions with fewer physical (PPI) relationships were highly enriched for the most variable genes, and genes with many protein partners were less variable (p, 2.5 × 10−5 Wilcoxon rank sum). The gene overlap between the clique regions of the coexpression network and PPI network was substantial (Figures 2E and S3), indicating that genes whose expression is correlated with a large number of partners, are also likely to interact with a large number of partners at the protein level. We predict that as the cells transition out of a pluripotency phenotype, the network structure (coexpression or protein partnerships) would change. This led us to investigate whether differences in expression variability of the network members might also reflect phenotypic differences between pluripotent and nonpluripotent cell populations.
Differences in Gene Expression Variability and Network Connectivity Reflect Changes in Stem Cell Phenotypes
The Hough ESC data set provided an opportunity to examine changes in the expression of genes in a series of stem cell fractions with varying potential for differentiation and self-renewal (Hough et al., 2009). We used the existing coexpression network, and analyzed the changes in the pattern of expression variability for each human ESC (hESC) fraction. The overall pattern of variability in each network region was high in P4 and low in P7 (Figure 3A–3C), consistent with our observations concerning global gene expression variability in these populations (Figure 1D).
Figure 3.
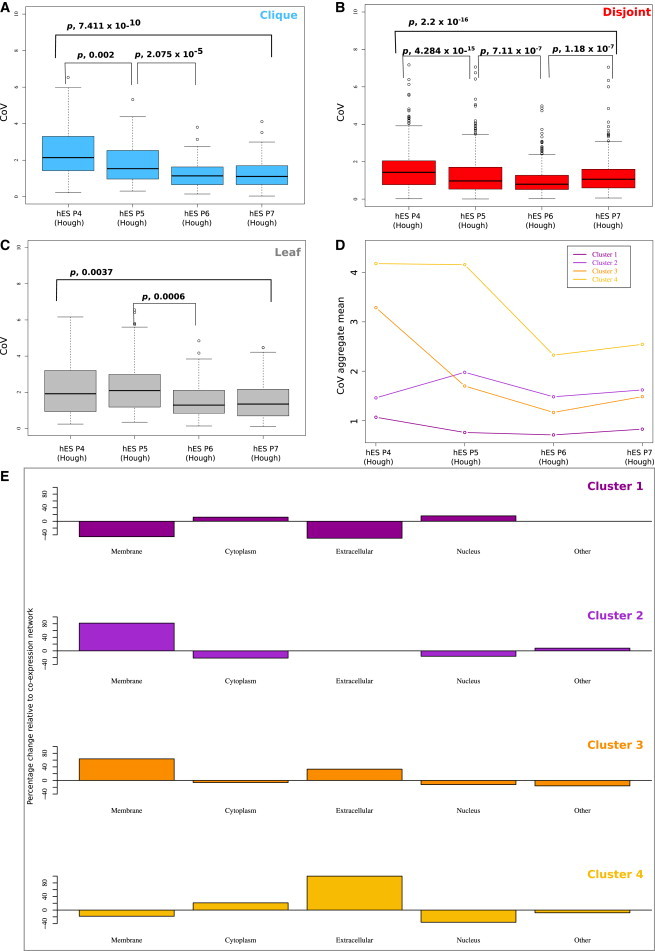
Differences in Gene Expression Variability and Network Connectivity Reflect Changes in Stem Cell Phenotypes
(A–C) Box plots illustrating gene expression variability in each coexpression network region as a stem cell moves from a self-renewing (P7) to differentiating (P4) phenotype. The x axes describe the subfraction (P4-P7) from the Hough data set, and the y axes describe the coefficient of variation. The p values assess significant differences between stem cell fractions within each region (p, 0.05, Wilcoxon rank sum).
(D) Four clusters of genes within the coexpression network displaying distinct CoV patterns between subfractions (K-means cluster analysis). Cluster 1, n = 566; cluster 2, n = 211; cluster 3, n = 188; cluster 4, n = 65. The x axes describe the subfraction (P4-P7) from the Hough data set, and the y axes describe the aggregate mean for CoV.
(E) Percentage change in the proportion of gene products predicted to be located in the plasma membrane, cytoplasm, nucleus or extracellular matrix for each cluster relative to the network. The total proportions of each location in clusters 1 (p, 0.02, chi-square test) and 2 (p, 0.0006, chi-square test) are significantly different from that of the coexpression network.
If expression variability is an important network descriptor, then genes that change from highly variable in the P4 fraction to highly constrained in the P7 fraction, or vice versa, might identify changes in the pluripotency network that permit cells in the population to transition between these states. We sought to identify coordinated patterns of change in expression variability across the fractions using K-means clustering and expected the majority of genes to display the same trend. Four distinct clusters were identified (Figure 3D; Table S3). Expression variability was highest in the transitioning population (P4) and lowest in the self-renewing fraction (P7) in 2 clusters (clusters 3 and 4), but, surprisingly, these clusters were very small and together composed approximately 24% of the total coexpression network. Clusters 1 and 2 (76% of the network) displayed little change in expression variability across the hESC fractions, potentially representing parts of the network that are coordinately regulated across the transitioning cell phenotypes. Gene families featured in cluster 1 included those coding for zinc finger proteins, ribosomal proteins, proteasome subunits, and ATP synthases.
We next examined the molecular processes common to genes that showed highly variable patterns of CoV across the hESC fractions. We first assessed whether genes in the entire coexpression network were predicted to be located in the plasma membrane, cytoplasm, nucleus, extracellular matrix, or unknown (other). We then addressed whether each cluster represented the expected proportion of each subcellular category, shown as a percentage of the network baseline in Figure 3E. A chi-square analysis revealed skewed distributions of these subcellular categories in clusters 1 (p, 0.02, chi-square test) and 2 (p, 0.0006, chi-square test), with 50% reduction of plasma membrane components in the largest cluster (cluster 1, Figure 3E). That is, the cell-cell interaction molecules had different levels of expression variability in the different P-fractions: EPCAM (cluster 3, plasma membrane) and CLDN7 (cluster 4, plasma membrane) showed highest variability in the P4 group, and lowest variability in the highly self-renewing P7 fraction. These elements have been previously identified as upregulated in human and mouse pluripotent cell types (Nagaoka et al., 2010; Xu et al., 2010) and are known to directly interact with key pluripotency regulators OCT4, SOX2, and NANOG, but the mechanism by which they maintain pluripotency is unknown. Clusters 3 and 4 were also highly enriched for plasma membrane and extracellular components respectively, but the small cluster size makes this difficult to functionally evaluate.
It is possible that changes in the pattern of expression variation between hESC fractions was a consequence of the underlying coexpression network, which we constructed using an iPSC data set. We therefore assessed changes in CoV across the Hough data set using the PluriNet, which is enriched in hESC sorted using the CD9-GCTM2 strategy (Kolle et al., 2011). Although genes belonging to the PluriNet were used to construct our coexpression network the PluriNet itself represents a highly curated PPI network, and is therefore not subject to the same assumptions about regulatory constraint or network connectivity as our coexpression network. Consistent with our previous findings, the P4 fraction displayed the lowest degree of coexpression, and the P7 fraction displaying the highest (Figure 4A). The PluriNet genes were expressed in all of the Hough stem cell fractions, and the pathway showed significant differential expression (attract ANOVA, p < 0.01) across the fractions (P7-P4). The attract analysis identified two groups of genes, which showed strikingly graduated expression across the stem cell fractions (Figure 4B) with the majority expressed at the highest level in the P7 fraction, and lowest level in the P4 cells. In contrast, clustering the PluriNet genes by CoV generated three subsets (Figure 4C; Table S4): The CoV changes across every cluster are suggestive of differences in regulatory constraints on the PluriNet were different for each fraction, and possibly most critical in the transitioning fractions. For example, key pluripotency regulators POU5F1 and DNMT3b belonged to cluster 2, which together with cluster 1 was most variable I the P4 fraction, with variability lowest in the transitioning fractions.
Figure 4.
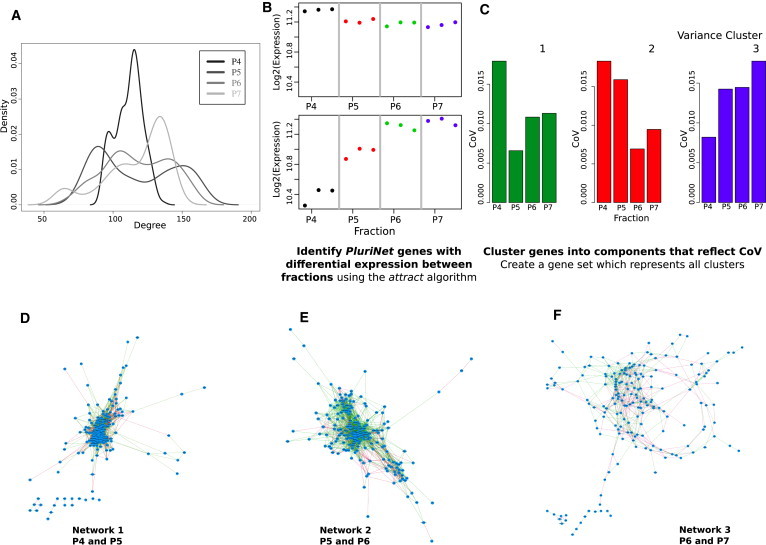
Differences in Coexpression Describe Differences in Regulatory Constraint
(A) The degree of coexpression within each colony subfraction (Figure 4A). The x axis represents the degree of coexpression for all genes in the PluriNet, and y axis represents the density.
(B) Probe sets driving phenotypic differences between stem cell fractions for the PluriNet. Log2 (expression) on the y axis and sample phenotypes are listed across the x axis. Each point represents the average expression for each cell type.
(C) Three clusters of genes within the PluriNet with distinct CoV patterns between subfractions (K-means cluster analysis).
(D–F) Coexpression networks illustrate the degree of coexpression between colony subfractions P4-P5 (D), P5-P6 (E), and P6-P7 (F).
Because coexpression between network elements is suggestive of a regulatory relationship (Allocco et al., 2004), high levels of regulatory constraint should manifest as high levels of coexpression between PluriNet elements (and vice versa). We tested this hypothesis by constructing three coexpression networks (Figures 4D–4F; Table S5): each representing coexpression between the 196 genes represented in the three PluriNet clusters, as cell populations transition between adjacent fractions. Figures 4D–4F illustrate an increase in coexpression as cells move from pluripotency to lineage commitment. We observe limited coexpression between P7 and P6 fractions (Figure 4E, Network 3), likely to be driven by divergence between the fractions, rather than differences within either fraction (Figure 4A). This may reflect a phenotypic transition point that disrupts constraint on the network, resulting in limited coexpression between PluriNet elements. As cells in the population become primed toward a lineage, the degree and the range of coexpression increased (Figure 4D). For example, the cell-signaling molecule LCK displayed a steady increase in connectivity (degree) from 7 in Network 3 (pluripotent) to 22 in Network 1 (differentiating). This profile is consistent with increased constraint on lineage specific markers and a reduction in the possible number of lineages a cell can commit to as the population becomes more sensitive to differentiation signals. Such structural differences in the network are likely to describe regulatory changes that a stem cell undergoes during transition from a plastic (pluripotent), to a more constrained (differentiating) phenotype.
Discussion
The role of cellular heterogeneity in stem cell biology is controversial, perhaps in part because the field is driven by the need to obtain “purer” populations of stem cells with predictable growth and differentiation properties. mESCs can be manipulated into a “ground state” of self-renewal using MEK/ERK and GSK3 inhibitors (Wray et al., 2010), a state that can be recapitulated by genetic manipulation of the levels of Pou5F1 and stabilization of the expression of Nanog (Karwacki-Neisius et al., 2013). Although this raises questions about the stability of stem cell phenotypes in culture (Karwacki-Neisius et al., 2013; Smith, 2013), it provides evidence that variability in the expression of members of the pluripotency network is a key driver of phenotypic variability in stem cell populations. Understanding the functional heterogeneity of stem cells requires laborious phenotyping, expression profiling is a commonly adopted phenotyping method. However, bioinformatics workflows focus on average population measures, and rarely consider how representative these measures are for individual cell behaviors. Although our population-based CoV approach does not trace the variability of individual cells, it does estimate the variability across the entire population. Our analyses suggest that profiling experiments used to benchmark new stem cell cultures should consider both relative expression, and expression variability of the pluripotency network.
We have shown that expression variability is associated with network structure in a surprisingly generalizable manner. In three independently constructed networks we observed that gene expression variability was greatest in network regions with fewer connections. Conversely, highly connected network regions also exhibited the most stable, least variable pattern of expression. These observations were reproduced across different types of networks, as well as independently generated stem cell data sets (iPS and hES), and this suggests that gene expression variability is an intrinsic network property.
There are a few caveats that should be considered in the interpretation of our findings. In the first instance, we chose to use quantile normalization, a method that is commonly applied to microarray data sets, and this may impact on the stability and distribution of variance across the data sets that we used. The use of background correction may amplify variability in very low-expressed probes, and we removed these by intensity thresholding the data prior to analysis. The strength of the correlations that we observed across numerous data sets gives us some confidence that CoV patterns reflect an underlying biology, and not the normalization process. We have not attempted to assess data sets subjected to a large number of amplification rounds, as this is known to compress the linear range of gene expression measurements, and we predict this would also impact on reliable variance measures. Others have shown patterns of expression variability in single-cell measures of stem cell populations using a variety of means: gene dosage and protein fluctuation (Karwacki-Neisius et al., 2013); mRNA levels that are cell cycle dependent (Singh et al., 2013). We conclude that an assessment of expression variability will become an important aspect of single-cell profiling experiments, as well as array-style studies that have sufficient depth of repeated measurement.
Gene Expression Variability Is an Essential Feature of Human Pluripotent Cell Populations
Given the repeated observations that stem cells are intrinsically heterogeneous under a range of culture conditions, we asked whether heterogeneity was a key feature of different human stem cell populations and the networks that govern them. We identified low gene expression variability in strongly pluripotent iPS and hES populations with high capacity for self-renewal and high variability in heterogeneous populations with low pluripotent capacity. This illustrates that the general trend is for increased gene expression variability in human stem cell populations with a transitioning phenotype, where lower levels of pluripotency are associated with higher number of cells transiently primed or already committed to differentiation.
Phenotypic variation in stem cell populations may also arise from culture conditions, iPSC derivation methods and FACS sorting protocols prior to nucleic acid isolation. However, it would be a mistake to dismiss all heterogeneity as a culture artifact: within a single hESC colony, key pluripotency regulators (POU5F1, DNMT3b, SOX2, DPPA4, LIN28, CLDN7, FGFR4, and ZFP42) displayed low variability in the strongly self-renewing fraction, and high variability in the differentiating fraction. Although a population-based CoV approach does not itself identify mechanisms leading to variability between individual cells in a population, it provides a snapshot of the level of stability a gene displays within a population, allowing us to make more targeted inferences regarding the contribution a gene makes to phenotype. The identification of genes with high variability in the population lends support to the idea that distinct subpopulations exist within the larger stem cell compartment. For example, changes in patterns of variability between self-renewing (P7) and differentiating (P4) phenotypes are likely to indicate changes in the level of regulatory constraint imposed on members of the pluripotency network, and we postulate this is a major factor in defining the different phenotypes. Very recently expression heterogeneity in some human ESC populations was shown to be regulated by cell-cycle-related expression variability in transcription factors that drive lineage commitment (Singh et al., 2013), demonstrating that molecular heterogeneity can describe critical features of the pluripotent phenotype, providing a mechanism for cells to flux between self-renewal and differentiation.
Gene Expression Variability Reflects the Level of Regulatory Constraint on Network Members
As stem cell populations differentiate, alterations in regulatory control are observable via changes in expression variability in the network (Huang et al., 2007, 2009; Swiers et al., 2006). Small fluctuating differences are unlikely to influence average measures but may signify departures from, or altered occupancy of discrete cellular states that have regulatory consequences, and lead to significant changes in expression variance across the stem cell population. We observed that transition from self-renewal to lineage commitment was accompanied by changes in the underlying network structure, such that elements became increasingly coregulated as the population became more sensitive to differentiation signals. In the Hough data set, variability of the pluripotency network increased as cells transitioned from highly pluripotent and self-renewing (P7) to the more heterogeneous P4 fraction. However, different members of the pluripotency network exhibited unique variance profiles that could be clustered across subfractions of a hESC colony. This highlights a critical difference in average versus variability analysis approaches: Highly correlated changes on average reflect large changes in the population phenotype, but these may not be coordinately regulated within a population. For example, the increased connectivity and variability of POU5F1 in the transitioning networks implies that the rate at which regulators silence expression of pluripotency genes during lineage commitment differs between members of the population. This type of profile is likely to drive differences in competency between the fractions to produce all germ layers in a teratoma (Hough et al., 2009) and captures the elements of stochasticity inherent to lineage commitment. Such differences in variability could indicate differences in constraints associated with RNA biogenesis, and possibly RNA stability, but without lab-based validation it is difficult to determine which aspect is the major contributor to the variability profiles that we have observed. It might be reasonable to assume that different genes will be stabilized by multiple convergent regulatory processes, including chromatin state, microRNA networks, and translational efficiency. Rather than speculating on individual processes, we propose that gene expression variability reflects the totality of regulatory mechanisms that constrain or diversify the phenotypic output.
Gene Expression Variance Patterns across a Network Reflect Features of Robustness
Cells as complex systems have the tendency to produce coherent rather than chaotic behaviors in the face of environmental changes and perturbations. A key feature of this coherence is what Kitano (2004) defines as robustness. Robustness is observable in the context of gene regulatory networks, where loss of a key regulator rarely results in catastrophic loss of function, and is not necessarily reflected in phenotypic changes (Raj et al., 2010). In this regard, the stochastic behavior of individual molecules in a network, which are representative of the entire cell population, may be buffered such that essential events are highly predictable, but a more relaxed state of entropy may exist in the absence of a biological imperative. In a recent review, MacArthur and Lemischka (2013) addressed this idea in more detail, postulating that molecular and cellular heterogeneity can be explored in terms of entropy behaviors, where a system that allows both highly regulated, and highly stochastic events will also permit the full complement of phenotypes arising from a population, even despite perturbation of key regulators in individual cells. Although such effects become more apparent at the level of single molecules, transcripts, and cells, population-based analyses echo the behavior of individual cells in the population. Our analysis is consistent with these ideas, and proposes that the CoV describes the stability of a gene across a cell population, and in doing so, is a surrogate estimate of genes under different entropy constraints. We have demonstrated that genes with different CoV have variable input into a network, suggesting that genes with different variability in expression make different contributions to phenotype. In order to confer a canalized phenotype, a network should possess structural elements which improve robustness against perturbation while contributing to highly conserved core processes that are shared by all members of the population (Kitano, 2004). This provides the network with features of stability and flux (or adaptability), which we suggest is reflected in genes displaying low and high variability in expression respectively.
Elements in the disjoint region of the network with high expression variability and low connectivity contribute to the phenotypic heterogeneity we observe in pluripotent stem cell populations and are likely to be independently regulated. Genes in the largest variability cluster (cluster 1) primarily (91%) belong to the disjoint region of the coexpression network, and gene expression variability remains unchanged during lineage commitment. This profile suggests these elements are unlikely to contribute to key differences between pluripotent and differentiated cell types, but rather, are involved in a number of independently regulated cellular functions. The diversity of regulation, combined with reduced connectivity and increased variability is likely to confer the ability to widen the range of phenotypes available to the population.
Elements in the network clique display low variability and high connectivity, supporting the hypothesis that these are the most stable elements of the pluripotency network and are under the highest regulatory constraints. We propose low variability and high connectivity provide stability to the network, contributing to highly conserved core processes common to all members of the pluripotent cell population. Clique elements displayed this profile in both coexpression and PPI networks, with a very high degree of membership overlap. Known (EPCAM, ZSCAN10, OCT4, DPPA4, DNMT3b, CLDN6) and emerging (OVOLD2 [Zhang et al., 2013], USP44 [Fuchs et al., 2012], SRFP2 [Mirotsou et al., 2007]) regulators of pluripotency are located in the clique, consistent with previous findings that expression level of a gene correlates with the number of interactions and essentiality of a gene product in PPI networks (Jeong et al., 2001; Lehner, 2008; Pál et al., 2003). Furthermore, the coexpression network clique captured membrane specific and secreted factors (CDH3, EPHA1, MARVELD3) previously identified as concordant with self-renewal (Eiges et al., 2001; Fuchs et al., 2012; Kolle et al., 2009; Patel and Simon, 2010; Zhang et al., 2013). Changes in network integrity accompanied phenotypic divergence during a possible switch point in differentiation (P7-P6), such that expression of these elements became less coordinated and predictable. We conclude that high connectivity and low variability classifies those stable elements in the pluripotency network under the highest degree of regulatory constraint. Changes in constraint during transition are likely to identify the critical phenotypic regulators of different cell states. We therefore propose that the combination of genes with high connectivity and low variability and low connectivity and high variability confer features of robustness to the pluripotency phenotype, providing the pluripotent cell population with the ability to flux between self-renewal, the competency to respond to differentiation signals, and lineage priming.
Conclusions
The global constraints on the availability of mRNA can be inferred from the variability of gene expression, and this, in turn, impacts on cell phenotype. Reduced gene expression variability in highly connected network regions may be informative of the level of regulation placed on a network element. Thus, an opportunity exists to understand how densely interacting elements of the pluripotency network reduce variability across the pluripotent population, and whether regions of high variability provide an indicator of genes which are permissive of phenotypic plasticity. Such a metric enables us to make useful and more targeted predictions about what regulates a cell phenotype and may provide insight into changes in the levels of regulation of network elements driving cell-fate transitions.
Experimental Procedures
Microarray Data sets
Public microarray data sets (accessions from GEO: GSE13201, GSE42956, ArrayExpress: ID E-MTAB-1040) were derived on the Illumina HT-12v3 microarray platform. Raw data were summarized using Bead Studio (Illumina). Background correction (affy) and quantile normalization was performed using R statistical software Bioconductor package lumi (Du et al., 2008). We tested the distribution of variability in each phenotype and found no significant differences (Figure S1C). Full details on data set selection and normalization procedures are provided in Supplemental Experimental Procedures.
Simulating Gene Expression Changes in the Cell Population
We used python programming language to model a matrix of 107 cells, reflecting the size of a typical cell population in culture. A 1D array fitting a normal distribution was simulated using the range of expression values typically seen in the linear range of a microarray experiment (5,000–50,000 fu). The mean, median, SD, and covariance were calculated, and normality was tested based on D’Agostino’s K-squared test. Randomized “pooled” samples (representing a summary of 106 entries, or one “pool”) were taken from the original array and the mean and CoV of these pooled samples were exported to a table (n = 100 pools). Increasing percentages (we selected 1%, 5%, 10%, and 20%) of entries in the original array were perturbed, and the degree of perturbation was also scaled (we selected 5%−50% in increasing increments of 5%), prior to resampling randomized pooled samples for each perturbation, as described above. The proportional deviation from the original population values were recorded and were visualized in a line graph where n = 100 for either the CoV or the mean at each point.
Population Variance
The coefficient of variation (CoV), computed for each gene by dividing the SD of its expression measures across a sample population by its average expression. A Wilcoxon rank sum test assessed whether the differences between the distributions were statistically significant.
Network Construction
KEGG (Kanehisa, 2002) and PluriNet (Müller et al., 2011) pathways were assessed using the attract algorithm (Mar et al., 2011a, 2011b). Correlated partners of the synexpression groups were computed at a Pearson coefficient cutoff of +0.9. A single list of genes was generated that comprised members of the ECMR interaction and PluriNet pathways, and their correlated partners of expression. Those gene pairs with a Pearson R value equal to or above +0.995 and below −0.995 were selected as network nodes. The network was visualized using a force directed spring embedded layout in Cytoscape, where the correlation coefficient between the pair of genes represents an edge weight (Shannon et al., 2003). Associated with an edge was either positive (Pearson R ≥ 0.995; green) or negative (Pearson R ≤ −0.995; red) correlation in gene expression. Cytoscape plug-ins for BisoGenet (Martin et al., 2010) and STRING.db (Francheschini et al., 2012) were used for protein-protein and literature-based networks, respectively.
Network Analyses
Network Architecture
The larger network was divided into three regions based with different connectivity.
-
1.
Clique: nodes that form part of the densely connected network core. Characterized by blue circles.
-
2.
Leaf: nodes peripherally connected to the main network hub. Characterized by gray triangles.
-
3.
Disjoint: nodes that were disconnected from the main network. Characterized by red squares.
Figure S2A contains gene lists for each region.
Constructing Networks that Represent Pluripotent and Transitioning Cell Populations
The PluriNet pathway was identified as significant in the attract analysis and was decomposed into distinct modes of expression variability. We used agglomerative hierarchical clustering with average linkage to cluster the log2-transformed CoV data and used the Gap statistic with 1,000 bootstrap samples to determine the number of appropriate variance clusters. A unique list of probes with a 1:1 mapping to official gene symbol represents all genes in these variance clusters, and there are 60, 97, and 39 genes associated with each cluster respectively, totaling 196 unique genes (Figure S3).
The subfractions were grouped as follows: network 1, P4 and P5 microarray data; network 2, P5 and P6 microarray data; network 3, P6 and P7 microarray data.
Pairwise Pearson correlation was used to assess the full list of 196 probes. The gene pairs with a Pearson R value equal to or above +0.9 and below −0.9 were selected as network nodes, with the correlation between them representing an edge. Associated with an edge was either positive (Pearson R ≥ 0.9; green) or negative (Pearson R ≤ −0.9; red) correlation in gene expression, corresponding to the Pearson R coefficient.
Author Contributions
E.A.M.: conception and design, data analysis and interpretation, manuscript writing. J.C.M.: conception, data analysis and interpretation, manuscript editing and approval. A.L.L.: provision of study material, manuscript editing and approval. M.F.P.: provision of study material, manuscript editing and approval. J.Q.: conception, manuscript editing and approval. E.W.: data interpretation and manuscript editing and approval. C.A.W.: conception and design, data interpretation, manuscript editing and approval, financial support.
Acknowledgments
E.A.M. is supported by an Australian Postgraduate Award scholarship and receives an AIBN student stipend. C.A.W. is supported by a QLD government Smart Futures Fellowship. This work was supported by an ARC special research initiative to Stem Cells Australia (C.A.W., E.W., A.L.L., and M.F.P.) and a grant from the National Heart, Lung, and Blood Institute of the Unites States NIH (1R01HL111759; J.Q.). The authors wish to thank Mr. Othmar Korn and Mr. Rowland Mosbergen for their programming advice and assistance with data processing within the Stemformatics environment.
Footnotes
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/3.0/).
Supplemental Information
References
- Allocco D.J., Kohane I.S., Butte A.J. Quantifying the relationship between co-expression, co-regulation and gene function. BMC Bioinformatics. 2004;5:18. doi: 10.1186/1471-2105-5-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bengtsson M., Ståhlberg A., Rorsman P., Kubista M. Gene expression profiling in single cells from the pancreatic islets of Langerhans reveals lognormal distribution of mRNA levels. Genome Res. 2005;15:1388–1392. doi: 10.1101/gr.3820805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briggs J.A., Sun J., Shepherd J., Ovchinnikov D.A., Chung T.L., Nayler S.P., Kao L.P., Morrow C.A., Thakar N.Y., Soo S.Y. Integration-free induced pluripotent stem cells model genetic and neural developmental features of down syndrome etiology. Stem Cells. 2013;31:467–478. doi: 10.1002/stem.1297. [DOI] [PubMed] [Google Scholar]
- Chambers I., Silva J., Colby D., Nichols J., Nijmeijer B., Robertson M., Vrana J., Jones K., Grotewold L., Smith A. Nanog safeguards pluripotency and mediates germline development. Nature. 2007;450:1230–1234. doi: 10.1038/nature06403. [DOI] [PubMed] [Google Scholar]
- Du P., Kibbe W.A., Lin S.M. lumi: a pipeline for processing Illumina microarray. Bioinformatics. 2008;24:1547–1548. doi: 10.1093/bioinformatics/btn224. [DOI] [PubMed] [Google Scholar]
- Eiges R., Schuldiner M., Drukker M., Yanuka O., Itskovitz-Eldor J., Benvenisty N. Establishment of human embryonic stem cell-transfected clones carrying a marker for undifferentiated cells. Curr. Biol. 2001;11:514–518. doi: 10.1016/s0960-9822(01)00144-0. [DOI] [PubMed] [Google Scholar]
- Francheschini A., Szklarczyk D., Frankild S., Kuhn M., Simonovic M., Roth A., Lin J., Minguez P., Bork P., von Mering C. STRING v9.1: protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2012;41:1–8. doi: 10.1093/nar/gks1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs G., Shema E., Vesterman R., Kotler E., Wolchinsky Z., Wilder S., Golomb L., Pribluda A., Zhang F., Haj-Yahya M. RNF20 and USP44 regulate stem cell differentiation by modulating H2B monoubiquitylation. Mol. Cell. 2012;46:662–673. doi: 10.1016/j.molcel.2012.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garfield D.A., Runcie D.E., Babbitt C.C., Haygood R., Nielsen W.J., Wray G.A. The impact of gene expression variation on the robustness and evolvability of a developmental gene regulatory network. PLoS Biol. 2013;11:e1001696. doi: 10.1371/journal.pbio.1001696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi K., Lopes S.M., Tang F., Surani M.A. Dynamic equilibrium and heterogeneity of mouse pluripotent stem cells with distinct functional and epigenetic states. Cell Stem Cell. 2008;3:391–401. doi: 10.1016/j.stem.2008.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hough S.R., Laslett A.L., Grimmond S.B., Kolle G., Pera M.F. A continuum of cell states spans pluripotency and lineage commitment in human embryonic stem cells. PLoS ONE. 2009;4:e7708. doi: 10.1371/journal.pone.0007708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S., Guo Y.P., May G., Enver T. Bifurcation dynamics in lineage-commitment in bipotent progenitor cells. Dev. Biol. 2007;305:695–713. doi: 10.1016/j.ydbio.2007.02.036. [DOI] [PubMed] [Google Scholar]
- Huang A.C., Hu L., Kauffman S.A., Zhang W., Shmulevich I. Using cell fate attractors to uncover transcriptional regulation of HL60 neutrophil differentiation. BMC Syst. Biol. 2009;3:20. doi: 10.1186/1752-0509-3-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong H., Mason S.P., Barabási A.L., Oltvai Z.N. Lethality and centrality in protein networks. Nature. 2001;411:41–42. doi: 10.1038/35075138. [DOI] [PubMed] [Google Scholar]
- Kanehisa M. The KEGG database. Novartis Found. Symp. 2002;247:91–101. discussion 101–103, 119–128, 244–252. [PubMed] [Google Scholar]
- Karwacki-Neisius V., Göke J., Osorno R., Halbritter F., Ng J.H., Weiße A.Y., Wong F.C., Gagliardi A., Mullin N.P., Festuccia N. Reduced Oct4 expression directs a robust pluripotent state with distinct signaling activity and increased enhancer occupancy by Oct4 and Nanog. Cell Stem Cell. 2013;12:531–545. doi: 10.1016/j.stem.2013.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitano H. Biological robustness. Nat. Rev. Genet. 2004;5:826–837. doi: 10.1038/nrg1471. [DOI] [PubMed] [Google Scholar]
- Kolle G., Ho M., Zhou Q., Chy H.S., Krishnan K., Cloonan N., Bertoncello I., Laslett A.L., Grimmond S.M. Identification of human embryonic stem cell surface markers by combined membrane-polysome translation state array analysis and immunotranscriptional profiling. Stem Cells. 2009;27:2446–2456. doi: 10.1002/stem.182. [DOI] [PubMed] [Google Scholar]
- Kolle G., Shepherd J.L., Gardiner B., Kassahn K.S., Cloonan N., Wood D.L.A., Nourbakhsh E., Taylor D.F., Wani S., Chy H.S. Deep-transcriptome and ribonome sequencing redefines the molecular networks of pluripotency and the extracellular space in human embryonic stem cells. Genome Res. 2011;21:2014–2025. doi: 10.1101/gr.119321.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehner B. Selection to minimise noise in living systems and its implications for the evolution of gene expression. Mol. Syst. Biol. 2008;4:170. doi: 10.1038/msb.2008.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacArthur B.D., Lemischka I.R. Statistical mechanics of pluripotency. Cell. 2013;154:484–489. doi: 10.1016/j.cell.2013.07.024. [DOI] [PubMed] [Google Scholar]
- Mar J.C., Matigian N.A., Quackenbush J., Wells C.A. attract: a method for identifying core pathways that define cellular phenotypes. PLoS ONE. 2011;6:e25445. doi: 10.1371/journal.pone.0025445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mar J.C., Wells C.A., Quackenbush J. Defining an informativeness metric for clustering gene expression data. Bioinformatics. 2011;27:1094–1100. doi: 10.1093/bioinformatics/btr074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mar J.C., Matigian N.A., Mackay-Sim A., Mellick G.D., Sue C.M., Silburn P.A., McGrath J.J., Quackenbush J., Wells C.A. Variance of gene expression identifies altered network constraints in neurological disease. PLoS Genet. 2011;7:e1002207. doi: 10.1371/journal.pgen.1002207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin A., Ochagavia M., Rabasa L., Miranda J., Fernandez-de-Cossio J., Bringas R. BisoGenet: a new tool for gene network building, visualisation and analysis. BMC Bioinformatics. 2010;11:1–9. doi: 10.1186/1471-2105-11-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirotsou M., Zhang Z., Deb A., Zhang L., Gnecchi M., Noiseux N., Mu H., Pachori A., Dzau V. Secreted frizzled related protein 2 (Sfrp2) is the key Akt-mesenchymal stem cell-released paracrine factor mediating myocardial survival and repair. Proc. Natl. Acad. Sci. USA. 2007;104:1643–1648. doi: 10.1073/pnas.0610024104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller F.-J., Schuldt B.M., Williams R., Mason D., Altun G., Papapetrou E.P., Danner S., Goldmann J.E., Herbst A., Schmidt N.O. A bioinformatic assay for pluripotency in human cells. Nat. Methods. 2011;8:315–317. doi: 10.1038/nmeth.1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagaoka M., Si-Tayeb K., Akaike T., Duncan S.A. Culture of human pluripotent stem cells using completely defined conditions on a recombinant E-cadherin substratum. BMC Dev. Biol. 2010;10:60. doi: 10.1186/1471-213X-10-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pál C., Papp B., Hurst L.D. Genomic function: rate of evolution and gene dispensability. Nature. 2003;421:496–497. doi: 10.1038/421496b. discussion 497–498. [DOI] [PubMed] [Google Scholar]
- Patel S.A., Simon M.C. Functional analysis of the Cdk7.cyclin H.Mat1 complex in mouse embryonic stem cells and embryos. J. Biol. Chem. 2010;285:15587–15598. doi: 10.1074/jbc.M109.081687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj A., Rifkin S.A., Andersen E., van Oudenaarden A. Variability in gene expression underlies incomplete penetrance. Nature. 2010;463:913–918. doi: 10.1038/nature08781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P., Markiel A., Ozier O., Baliga N.S., Wang J.T., Ramage D., Amin N., Schwikowski B., Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh A.M., Chappell J., Trost R., Lin L., Wang T., Tang J., Matlock B.K., Weller K.P., Wu H., Zhao S. Cell-cycle control of developmentally regulated transcription factors accounts for heterogeneity in human pluripotent cells. Stem Cell Rev. 2013;1:532–544. doi: 10.1016/j.stemcr.2013.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith A. Nanog heterogeneity: tilting at windmills? Cell Stem Cell. 2013;13:6–7. doi: 10.1016/j.stem.2013.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swiers G., Patient R., Loose M. Genetic regulatory networks programming hematopoietic stem cells and erythroid lineage specification. Dev. Biol. 2006;294:525–540. doi: 10.1016/j.ydbio.2006.02.051. [DOI] [PubMed] [Google Scholar]
- Vitale A.M., Matigian N.A., Ravishankar S., Bellette B., Wood S.A., Wolvetang E.J., Mackay-Sim A. Variability in the generation of induced pluripotent stem cells: importance for disease modeling. Stem Cells Transl. Med. 2012;1:641–650. doi: 10.5966/sctm.2012-0043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wray J., Kalkan T., Smith A.G. The ground state of pluripotency. Biochem. Soc. Trans. 2010;38:1027–1032. doi: 10.1042/BST0381027. [DOI] [PubMed] [Google Scholar]
- Xu Y., Zhu X., Hahm H.S., Wei W., Hao E., Hayek A., Ding S. Revealing a core signaling regulatory mechanism for pluripotent stem cell survival and self-renewal by small molecules. Proc. Natl. Acad. Sci. USA. 2010;107:8129–8134. doi: 10.1073/pnas.1002024107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T., Zhu Q., Xie Z., Chen Y., Qiao Y., Li L., Jing N. The zinc finger transcription factor Ovol2 acts downstream of the bone morphogenetic protein pathway to regulate the cell fate decision between neuroectoderm and mesendoderm. J. Biol. Chem. 2013;288:6166–6177. doi: 10.1074/jbc.M112.418376. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.