

Abstract
Antibodies against surface molecules of human tumors are now frequently administered in combination with strong chemotherapy, increasing therapeutic efficacy but making the task of elucidating immunological events more difficult. Experiments on genetically manipulated mice indicate that antibody efficacy is greatest when IgG antibody coating tumor cells is engaged by the Fcγ-receptors of effector cells, chiefly the monocyte/macrophage lineage. Evidence suggests lesser roles for NK cells, neutrophils, receptor-mediated cytotoxicity and complement-mediated cytotoxicity. The classical mode of killing employed by macrophages is phagocytosis, but much has to be learned about optimally activating macrophages for this task, and about any other modes of cytotoxicity used. There is renewed interest in antigenic modulation, which implies removal of therapeutic antibody linked with antigen from target-cell surfaces. It is now apparent that this removal of immune complexes can be achieved either by internalization by the target cell, or by transfer of the complexes to another cell by trogocytosis. In trials, anti-idiotype antibodies surprisingly proved therapeutically more effective than anti-CD20, despite anti-idiotype being more effectively removed from target-cell surfaces by antigenic modulation. This anomalous result might reflect the fact that persistence of anti-CD20 immune complexes in large amounts induces serious effector modulation, which paralyzes macrophage attacks on antibody-coated cells. The case for effector modulation is argued by analogy with the therapeutic suppression of autoimmune inflammation by effector modulation, achieved by infusion either of normal IgG in large amounts, or of anti-red cell IgG in relatively small amounts.
Introduction
The modern era of antibody therapy of cancer started in the 1970s with attacks on selected molecular targets on malignant cells, an approach immeasurably enhanced by the development of monoclonal antibodies (mAb). Initial therapeutic results were modest,1 but improved when the precision of antibody therapy was combined with broadly cytotoxic chemotherapy.2,3 Unfortunately, the chemotherapy makes the task of evaluating immunological events arising from the antibody activity more difficult. A persuasive case has been made for treating some patients with antibody alone,4,5 but this has not been widely followed. Nor can total confidence be placed in conclusions drawn from antibody monotherapy in animals, which is dominated by the use of inbred mice with possible complications arising from activating endogenous retroviruses.6,7
A further cautionary note arises from the variation in biological mechanisms utilized by evolution for a given task. The medieval philosopher William of Ockham coined the dictum that “concepts should not be multiplied unless necessary”, known as Ockham’s razor. This has proved useful in the physical sciences but not in biology, where, quoting Francis Crick,8 “(Ockham’s razor) can be a very dangerous implement. It is thus very rash to use simplicity and elegance as a guide in biological research”. Crick’s words seem particularly apt for antibody therapy.
Three problems associated with antibody therapy will be discussed here: the killing of antibody-coated tumor cells; antigenic modulation; and effector modulation. To provide settings for these problems, we describe two examples of antibody therapy: human B-cell lymphomas treated with mouse monoclonal anti-idiotype (anti-Id); and the same tumor types treated with chimeric anti-CD20. Two examples of trials of these therapies come from the Stanford University Division of Oncology. Both are close to being antibody monotherapies, in that other accompanying anti-tumor agents were absent or of minor severity.
Anti-idiotype therapy
This approach uses as targeted epitopes the variable amino acid sequences which serve the antibody recognition function on surface immunoglobulin (Ig) of B lymphocytes. The totality of these epitopes is the idiotype (Id). Originally,9,10 the Id was described as “confined to the tumor-cell surface”. This soon had to be qualified: in most cases the surface idiotype is displayed on monomeric surface IgM (mol wt ~180,000), while a minute but variable amount of pentameric IgM (mol wt ~950,000) is secreted.11 This small amount can provide an appreciable extracellular idiotypic barrier, consuming anti-Id and often requiring a preliminary plasmapheresis.
The Stanford group overcame formidable logistical problems to provide a series of 45 cases of low-grade B-cell lymphoma treated with 52 courses of custom-made monoclonal anti Id.12–14 Some cases also received α-interferon, IL-2, or chlorambucil. A total of 66% achieved a significant remission (reduction by approx. 50% of measurable disease), including 18% complete remission (CR), and including in turn 13% prolonged CR. Five of the 6 patients in the last group, 3–8 years into their remissions, had blood and marrow samples examined for tumor Id. Very low levels were detected in all patients, but they all subsequently remained in remission and this has been maintained up to the time of writing; a striking example of tumor dormancy.
Anti-Id therapy is now in abeyance due to the logistical difficulties involved in preparing individual antibodies for each patient. However, follicular lymphomas have been found to present an unusual glycan on their variable domains, close to the idiotypic epitopes, so there is a prospect that, for these tumors, an antibody of good affinity aimed at the glycan could be an effective single substitute for multiple anti-Id preparations.15
Anti-CD20 therapy
CD20 is a small cell-surface molecule of mol wt 33078. It is found on the B-cell lineage, from early B cells up to, but excluding, plasma cells. It has 4 trans-membrane strands, cytoplasmic N- and C-terminus, two extracellular loops, and no recognized ligand. Its function is not clear, but it may be involved in B-cell activation and trans-membrane calcium flux.16 Normally it is neither secreted nor shed in significant amounts. The anti-CD20 mAb in widespread use is rituximab, a chimeric molecule in which C-terminus of mouse anti-CD20 VH and Vκ domains are fused genetically to N-terminus of human CH1 and Cκ domains, respectively. The human constant domains are from the IgG1 isotype.
Anti-CD20 antibodies have revealed differences in effector functions independent of the heavy chain isotype.17 Type I antibodies (including rituximab) redistribute CD20 to membrane rafts and activate complement efficiently. Type II antibodies have not been seen to redistribute to rafts, and generally activate complement poorly, but they induce receptor-mediated death much more readily than type I. X ray crystallography of immune complexes containing Type I or Type II antibodies yielded no clear explanation for these differences, but revealed several features which might be contributory.18
A single course of treatment with rituximab generally consists of four intravenous infusions at one week intervals, at a dose of 375 mg/m2 of surface area. In a pioneering trial,1 such doses were given to 37 patients with low-grade B-cell lymphoma who had relapsed after chemotherapy. There was a 46% response rate, including 8% CR. Median time to progression was 10.2 months. It was noted that the results compared favorably with standard chemotherapy, with the antibody having a better safety profile.
Drugs and doses for chemotherapy coincident with rituximab are discussed by Maloney.2 Improvements in survival in follicular lymphoma over four decades, culminating in this combined treatment, have been set out.3 Resurgent tumor appearing after remissions induced by anti-CD20 therapy is sometimes seen to be CD20-negative, probably more often among the more aggressive lymphomas.19,20
Problem 1: killing antibody-coated tumor cells
Three major paths are available for killing tumor cells coated by IgG antibody. 1) Receptor-mediated cytotoxicity (RMC), the receptor being the target antigen and the initiating event being the union with antibody. Multiple forms of RMC have been described, of which receptor-mediated apoptosis21 is the best understood. 2) Antibody-dependent cell-mediated cytotoxicity (ADCC) is initiated by the antibody Fcγ engaging Fcγ-receptors on the effector cells (Figure 1).22–25 3) Complement-mediated cytotoxicity (CMC) can occur when complement is activated by the clustered Fcγ regions of the coating antibody.
Figure 1.
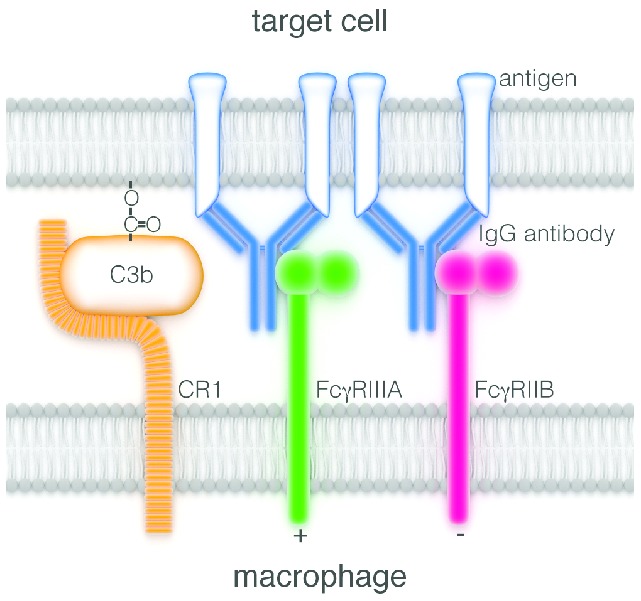
One of the multiple contacts22 forming a synapse between a macrophage and a target cell coated with IgG antibody. Initially antigen molecules have been cross-linked by the bivalent antibody, causing under some circumstances receptor-mediated cytotoxicity. But death is more likely to result from subsequent engagement of antibody Fcγ regions with activating FcγR (here of the class FcγRIIIA) on the macrophage, thus stimulating ADCC activity by the macrophage. This activity is modulated or repressed if inhibitory FcγRIIB receptors are simultaneously engaged. NK cells and granulocytes23 are also capable of killing targets coated with IgG antibody, such encounters being most likely in the bloodstream. NK cells lack FcγRIIB, so any NK cytotoxicity lacks the modulation transmitted by these receptors. The complement system is also activated by the clustered Fcγ regions of coating antibody. Some of the resulting complement products, notably C3b, link covalently to the target-cell surface and might promote opsonization via their affinity for one or other of the macrophage complement receptors (here the receptor CR1 is arbitrarily depicted adjacent to the FcγR). Acting thus complement is a powerful opsonizer for phagocytosis of microorganisms by macrophages and neutrophils, but it has not been consistently shown to assist in attacking antibody-coated tumor cells. It has actually been seen to impede ADCC by NK cells,24 which lack complement receptors. Finally C3b has been reported to have an enigmatic role in promoting antigenic modulation.25
Receptor-mediated cytotoxicity
Receptor-mediated cytotoxicity (RMC) evoked by antibody is reported frequently in vitro but infrequently in vivo, where cell survival is less precarious and any RMC is difficult to observe. Below are a variety of reports suggesting contributory roles for RMC, but a lack of proof for major roles.
Receptors regularly mediating apoptosis, Fas and TNF receptors (TNFR-I and TNFR-II), are widely distributed throughout many tissues and, therefore, the cause of too much toxicity to be therapeutically useful. A humanized mAb to CD44 has been reported26 to have a specific pro-apoptotic effect in vitro, without cross-linking, against those human chronic lymphocytic leukemia (CLL) cells that express the protein kinase ZAP-70. This antibody also cleared the leukemic cells completely from immunodeficient engrafted mice.
The ability of anti-Id mAbs to stimulate tyrosine phosphorylation within tumor samples correlated with their ability to induce tumor regression,14 suggesting that this metabolic response at least contributed to tumor cell death. The cross-linking of coating anti-CD20 mAb by anti-mouse Ig antibody led to apoptosis of lymphoma cell lines in vitro,27 but such a manipulation has not been reported in vivo.
The apparent RMC caused by type II anti-CD20 in vitro has been demonstrated using lymphoma cell lines and primary CLL cells as targets. It has been associated with agglutination, relocalization of actin, and dispersal of lysosomal contents.28 The events are complex but add to the promise of Type II antibodies as therapeutic agents.
There has been a proliferation of terms to describe forms of RMC which appear to be neither apoptotic nor necrotic (and to which the term “programmed cell death” is often applied in the absence of any clear program). One particular conundrum involves autophagy, long regarded as a normal housekeeping process but perhaps an important accelerator of cell death in the presence of certain stresses.29 Much work is being undertaken on these miscellaneous forms of cell death, and precise descriptions are likely to emerge. A plea has also arisen for greater precision and consistency in describing the progress of apoptosis itself.30
Antibody-dependent cell-mediated cytotoxicity: the major killing mechanism?
An initial note is needed about the term antibody-dependent cell-mediated cytotoxicity (ADCC). It was introduced to describe killing by NK cells of targets coated by IgG antibody. This is extracellular killing in which perforin-generated pores allow entry of granzymes into the cytoplasm of the targeted cell. However, taken literally, the term could refer to any method used by an effector cell to kill an antibody-coated target, including phagocytosis, NK-type killing, and obscure mechanisms. Others31 have found it useful to use the term in this broad literal sense, and the same practice will be followed here.
In 2000, Clynes et al.,31 expanding on earlier work,32 reported the use of mAb to treat syngeneic and xenografted tumors in mice bearing genetically manipulated Fcγ-receptors (FcγR). Results showed clearly that the engagement of both activating and inhibitory FcγR on effector cells was a dominant component in killing the antibody-coated cells in vivo.
When the antibody TA99 was used to treat the B16 mouse melanoma, partial suppression of the tumor, seen in mice with normal FcγR, was lost completely in γ−/− mice, which lack the activating γ chains required in mice for FcγR I, III and IV.33
A similar result was achieved when comparing γ+/+ mice (with normal FcγR) and γ−/− mice, both bearing tumor xenografts treated with systemic injections of mAb. However, here the loss of therapy in γ−/− mice was incomplete. The xenografts were either of human breast carcinoma (treated with trastuzumab) or of human B-cell lymphoma (treated with rituximab). The carcinoma was reduced by 96% in the γ+/+ mice, and by 23% in the γ−/− mice. The lymphoma was reduced by more than 99% in the γ+/+ mice, and by 29% in the γ−/−mice. Thus, in both these cases, the disabling of FcγR unmasked the presence of some far less efficient killing, which could have been RMC, CMC, blocked function of the target antigen, or an unrecognized mechanism.
Another arm of this study involved the use of mice with intact activating FcγR, but with the inhibitory FcγRIIB genetically deleted. These animals revealed a dramatic improvement in the response of the B16 melanoma to antibody therapy: wild-type mice responded to antibody therapy by a 3-fold reduction in tumor load, while the FcγRIIB−/− mice responded with a 100-fold reduction. The breast carcinoma xenograft in mice with normal FcγR responded to sub-therapeutic doses of trastuzumab with slowed but relentless growth, whereas in FcγRIIB−/− mice these doses of antibody yielded complete inhibition of growth.
The relevance to human clinical practice of the involvement of the activating FcγR in antibody therapy has been supported by studies of the treatment of follicular lymphoma by rituximab. FcγRIIIA, a major activating FcγR on macrophages and NK cells, displays a 158V allotype with a higher affinity for human Fcγ, and mediating ADCC better, than the alternative 158F. Higher clinical responses were seen in V/V subjects than in V/F or F/F, consistent with a major therapeutic role for ADCC.34,35 Similar results were obtained on examining overall survival in a series of trials of anti-CD20 plus chemotherapy, and in a trial of chemotherapy alone.36 With the combined therapy, having at least one V allele conferred improved survival versus the homozygous F/F. No such dependence on genotype was seen in the trial of chemotherapy alone. Studies relating responses to a polymorphism displayed by another activating receptor, FcγRIIA, have yielded disparate results.35,36
The correlation of the V allele with a favorable response to anti-CD20 therapy has not been seen with chronic lymphocytic leukemia.37,38 This suggests that one or more mechanisms other than ADCC are dominant in the antibody therapy of this disease.
Macrophages: the major effector cells?
The results of Clynes et al.31 indicate that the predominant cells engaged in killing antibody-coated tumor possess both activating and inhibitory FcγR. Cells fitting such a description are monocytes/macrophages, dendritic cells, and neutrophils.39 NK cells, the cells used routinely to assess ADCC function in candidate anti-tumor antibodies, have not been shown to express the inhibitory FcγRIIB (apart from a subpopulation of mouse NK cells comprising <1% of the total40). These facts led to the conclusion that macrophages are the major effector cells involved.31
Supporting evidence for macrophages as the major effector cells arose from observations on depletion of normal B lymphocytes in mice injected with mouse anti-CD20 mAb.41,42 Mice genetically deficient in NK function or complement showed unimpaired depleting activity, whereas macrophage removal by clodronate grossly impaired it.41
Macrophages, when including site-specialized descendants such as osteoclasts, Kuppfer cells, Langerhans cells, microglia and others, are ubiquitous throughout the body. A histological survey of seven of the most common human solid malignancies revealed tumor-infiltrating cells accumulating preferentially in the stromal bands between tumor cells. In all tumor types, the macrophage lineage accounted for most of the infiltrating cells.43 Next in frequency were Tαb+ lymphocytes, while hardly any NK cells were identified. Large numbers of the macrophages stained positively for FcγRIII. Macrophages thus seem ideally placed and equipped for dealing with antibody-coated tumor.
Neutrophils as effector cells?
Albenesi et al.23 have reported a series of experiments in which neutrophils appeared to be capable of reducing antibody-coated tumors in mice. Intravenous antibody injections starting on Days -1 to +2 were effective against small subcutaneous inocula (given on Day 0) of syngeneic melanoma or xenogeneic breast carcinoma. Evidence from genetically modified or macrophage-depleted mice implicated neutrophils, acting through their FcγR, as the tumor-reducing effectors.
Neutrophils have also shown a surprising ability to phagocytose human leukemic B lymphoblasts coated with obinutuzumab,44 an anti-CD20 mAb with an enhanced Fcγ docking site for FcγRIII.
Neutrophils, unlike macrophages, are not regularly seen to infiltrate healthy tissue or tumors. They must, therefore, be attracted to a target as part of an inflammatory process. To attract inflammatory infiltrates to large dispersed tumors, for example by anaphylatoxins from widespread activation of complement, could risk a dangerous systemic inflammatory response. However, the report from Albanesi et al.23 does suggest that neutrophils could usefully eradicate antibody-coated small tumor residues in certain accessible sites.
What is the cytotoxic mechanism destroying antibody-coated cells?
The conclusion that destruction of antibody-coated tumor is carried out principally by macrophages has been inferred from the sites, prevalences and characteristics of the candidate lineages, and from genetic manipulations of the FcγR that they carry. But much remains to be learned about macrophage adaptation and killing activity45 when faced with the challenge of large numbers of antibody-coated tumor cells. Whether the classical method by which macrophages enact ADCC (phagocytosis by the zipper mechanism46,47) is available on the scale required remains to be determined. An observation of NK-type extracellular killing (utilizing granzymes and perforin) performed by a subset of rat macrophages has been recorded48 but no similar reports have been found.
Hubert et al.22 have described synapses formed in mice, during tumor rejection, between antibody-coated tumor cells and macrophages or neutrophils. These synapses consisted of multifocal contacts, each displaying accumulated actin, FcγR and phosphotyrosines. It is plausible that such engagements have a good chance of killing the tumor cell, but deaths could not be demonstrated in vitro so the cellular and subcellular events remain obscure. Unlike NK cells, which upon separation from blood are sufficiently activated to kill antibody-coated targets in vitro, monocytes/macrophages from blood need to be cultured in the presence of activators like GM-CSF and IFN-γ.49 Then they have been seen to kill, in vitro, CLL cells coated with rituximab, with inhibition in the presence of cytochalasin D, suggesting that killing required at least the ability to phagocytose.49 It might be noted that eosinophils have been observed to kill by repeatedly engulfing and regurgitating targets50 so that killing variations on the theme of phagocytosis are at least feasible. Non-activated mouse peritoneal macrophages, when presented with lymphocytic targets bearing capped antigen-antibody on their uropods, and therefore without a circumferential zipper, attach to the uropods and remove the immune-complex caps (by the process now known as trogocytosis51) without killing the target cell.47 The same macrophages, presented with lymphocytes with a complete IgG antibody coating, are shown to be capable of ingesting as many as 6 of these targets, which are then rapidly degraded within the phagocytic vacuoles.47 In contrast to the failure of macrophages to kill capped cells, capping was seen to encourage killing of target lymphocytes by NK-mediated ADCC.52
It is clear that ADCC of antibody-coated targets in vitro by NK cells from normal volunteers is a poor surrogate for killing the same targets by autologous macrophages in vivo. NK cells have a limited distribution. They lack the inhibitory FcγRIIB and they display their own inhibitory systems to deter attacks on autologous cells,53 systems which will be variably activated by the non-autologous targets used in routine laboratory assessments of NK ADCC.
Complement-mediated cytotoxicity
As a consequence of activation of complement at an antibody-coated cell surface, the death of that cell by CMC can occur either by the lytic action of complement’s membrane-attack complex, or by attachment of myeloid effectors to C3b and other complement breakdown products bound to the target-cell surface. Against these possibilities, mammalian cells have available a formidable array of defences, both in extracellular fluids and on cell surfaces.54 Before the primacy of cellular effectors in disposing of antibody-coated cells had been established, a number of publications over 25 years had reported that complement had no detectable role in antibody treatment of tumor.41,55–57 Non-complement-activating antibodies appeared to reduce tumor at least as well as good complement activators; and animals with severe complement deficiencies appeared to respond at least as well to antibody therapy as did normal animals.
However, there have been reports in which complement has been shown to contribute to tumor cell death.58,59 This would be favored by circumstances in which ADCC under-performs, as is probable in rituximab-treated CLL.37,38
In an attempt to assess whether appreciable CMC is occurring in rituximab-treated follicular lymphoma, a correlation was sought between tumor response and expression by the cells of three complement inhibitors: CD46 (membrane co-factor protein), CD55 (decay accelerating factor) and CD59. No such correlation was found,60 suggesting no significant involvement of complement.
Appreciable complement activation, even if not directly involved in cell death, could provide a systemic boost for macrophage ADCC. Thus the anaphylatoxin C5a has been shown to increase the ratio of activating to inhibitory FcγR on macrophages.61 Contrasting with this is a warning about a possible negative local effect of complement activation. There is evidence that C3b deposition on rituximab-coated target cells inhibits the interaction between the rituximab Fcγ and the FcγRIIIA on NK cells.24 NK cell activation and ADCC are thereby diminished. Whether a similar effect occurs with macrophage ADCC needs to be determined, bearing in mind that macrophages, unlike NK cells, display CD35 (CR1) and sometimes CRIg (complement receptor of the Ig superfamily). CR1 and CRIg are the principal receptors for binding C3b.
One approach to enhancing antibody cytotoxicity
The demonstration that linkage of coating antibodies to FcγR of effector cells is a dominant component of effective antibody therapy in several animal models,31,32 and is apparently important in antibody therapy of follicular lymphoma in man,34–36 has led to engineering of therapeutic antibodies in order to favor their linkage to activating FcγR.
All human FcγR, activating and inhibitory, share a binding site at the N-terminus of Fcγ between the two Cγ2 domains and reaching the hinge, with the glycans attached to each domain appearing to form a partial floor to the site.62 There are differences in the detailed binding of each FcγR, but all the bindings appear similar and only one receptor at a time can be accommodated in the site. The activating receptor FcγRIIIA was chosen as the template to which the Fcγ would be remoulded. One criterion of success would be an improvement in the ratio of (KA for union with FcγRIIIA) to (KA for union with FcγRIIB). Three approaches have been tried.
The Fcγ is remoulded by mutagenesis, leading to increases in the KA ratio of 9- and 10-fold.63,64
Human IgG is synthesized with its Cγ2 glycans non-fucosylated, producing increases in the KA ratio approaching 50-fold.65
A bispecific FabIgG construct is synthesized by attaching an Fab’γ module, with antibody specificity for FcγRIIIA, to the rituximab hinge. The attachment process deliberately and severely disables the docking of FcγRIIB at its adjoining site,62,66 giving a KA ratio or more than 1000.
All three of the above constructs spare the two additional docking sites (for FcRn and C1q) carried by the Fcγ. All give enhanced ADCC in vitro with NK cells as effectors; but they cannot readily be compared by ADCC in vitro with macrophages as effectors, because for this no standardized procedure exists.
An example of approach (2) in the above list is provided by the type II non-fucosylated mAb obinutuzumab, recently granted approval for use, in combination with chlorambucil, in previously untreated CLL.67 This follows clinical trials which revealed improved progression-free survival for the combination versus chlorambucil alone, and then for the combination versus rituximab plus chlorambucil. The source of the superiority of obinutuzumab over rituximab is not clear: it could arise from superior RMC and/or superior ADCC performance, or there could be other factors.
Problem 2: antigenic modulation
Antigenic modulation may be defined as an antibody-induced redistribution of a cell-surface antigen, which confers cellular resistance to subsequent attacks on the same antigen by antibody plus cytotoxic effectors. Studies of anti-Id and anti-CD20 activities have strongly suggested that ‘redistribution’ in this definition can imply two completely different types of movement, internalization by the target cell and trogocytosis by effector cells, an ambiguity dealt with later.
Old and Boyse68 first described modulation in 1963 for mouse TL (thymus leukemia) antigens. Leukemic cells bearing TL antigens survived passaging in mice which had been immunized to produce anti-TL antibodies. The recovered cells were TL-negative as judged by resistance to anti-TL plus complement, but regained positivity on being re-passaged in antibody-negative hosts. An explanation for these phenomena was provided by Stackpole et al.69 They showed that an initial exposure of TL-positive cells to anti-TL serum in vitro led to movement of antigen-antibody complexes over the surface of the cell to form patches and then a cap, from both of which endocytosis occurred to leave the cell surface almost entirely cleared of that antigen. The cells were then found to be resistant to cytotoxic attack by anti-TL plus syngeneic complement.
Binding of C3b to the target cells, via either the classical or alternative pathway of complement activation, was sometimes needed for the successful internalization of the immune complexes.25 The reason for this remains unexplained. Modulation was enhanced upon increasing the cross-linking of immune complexes by adding an anti-Ig reactive with the coating antibody. A similar enhancement was observed upon exposing antibody-coated cells to monocytes, apparently using the monocytic membrane with its FcγR for cross-linking.70
Antigenic modulation of cell-surface IgM
The commonest class of Ig to be found on the surface of both normal and neoplastic B lymphocytes is IgM in its monomeric form (mol wt ~180 000). Surface IgM was found to exhibit rapid antigenic modulation in response to either antigen or polyclonal anti-Ig preparations.71 Immune complexes patched, migrated towards one pole of the cell to form a cap and then a trailing uropod, and were continually internalized. Modulation conferring resistance to attack by antibody and syngeneic complement was apparent by the time the complexes were minimally aggregated: capping and extensive endocytosis were not necessary.72 Post-modulation sparseness of surface IgM was aggravated by a reduced surface delivery of newly synthesized molecules.73
Modulation in vitro induced by individual monoclonal anti-Id antibodies was found to be much slower than occurred with the polyclonal antibodies used in early work.74,75 Nevertheless most anti-Id antibodies, and antibody directed at Ig constant regions (e.g. anti-μ), were among the fastest modulators (judged by internalization) in a group of antibodies aimed at a variety of lymphocytic surface proteins.74–77 Slowness of modulation due to some anti-Id antibodies was attributed to ‘monogamous’ binding, in which the two antibody sites of the anti-Id molecule attached to the two Fv regions on a single cell-surface IgM molecule.74
How important is antigenic modulation in protecting a tumor cell against an attack by anti-Id therapeutic antibody? In treating a guinea-pig lymphoblastic B-cell leukemia with polyclonal anti-Id preparations, a comparison was made between a bivalent reagent (IgG) and a univalent reagent (Fab/c) prepared by removing one Fab arm of the IgG.78 In vitro the IgG induces rapid modulation upon cross-linking the cell-surface IgM, the Fab/c no modulation at all. In the added presence of macrophages expressing FcγR, Fab/c does cause modulation but at a slower rate than the IgG. In vivo the univalent Fab/c gave a notably greater therapeutic effect than did the IgG, strongly suggesting that antigenic modulation significantly impedes antibody therapy of tumor.
Antigenic modulation of CD20
After studies of antigenic modulation in vitro revealed little or no internalization induced by anti-CD20 mAbs,76,77 it was hoped that antigenic modulation of CD20 would not be a problem. Nor was it evident in nodal biopsies from cases of B-cell lymphoma treated with rituximab.79 So it caused some surprise when in 2003 Jilani et al.80 produced clear evidence of antigenic modulation (absence of detectable rituximab and CD20 on target cells) in blood samples from the majority of 65 patients with CLL who had been treated with rituximab-containing protocols. Mixing of CLL or Raji cells with rituximab in vitro left detectable rituximab bound to CD20 on the cell surfaces. However, these immune complexes became undetectable upon the addition of plasma, a finding that suggests the C3-dependence of internalization reported for modulation of TL antigens.25 The antigenic modulation of CD20 was accompanied by a downmodulation of CD20 mRNA, consistent with the observed reduction in delivery to the plasma membrane of IgM after modulation of that antigen had taken place.73
Beers et al.81 reported in 2010 that antigenic modulation reduced the efficacy of type I but not of type II anti-CD20 in eliminating in vivo mouse lymphocytes which had been transduced with human CD20. The modulation involved internalization by the target cells of the CD20-antibody complexes. Normal human B cells, and cells from most cases of CLL and mantle cell lymphoma, showed similar CD20 internalization, while cells from most cases of FL and diffuse large B-cell lymphoma were more resistant.
Antigenic modulation of CD20: the role of FcγRIIB
In 1994 Vervoordeldonk et al.82 reported that the FcγRII on malignant B cells, now known to be FcγRIIB, promoted the antigenic modulation induced by an anti-CD19 mAb. They were surprised to observe that the FcγR, as judged by an unchanged surface density, did not co-modulate with the anti-CD19 mAb, and put forward as one possible explanation a recycling of the endocytosed FcγR to the surface. Such recycling of human FcγRIIB, either free of immune complexes,83 or both with and without them,84 has now been reported.
CD20 (when targeted by type I antibodies) and CD38 have now been added to the B-cell antigens whose modulation is promoted by FcγRIIB, with a greater number of antigens failing to show promotion.85 At sufficient cell densities, promotion of modulation of compliant antigens by FcγRIIB can be attained in either cis or trans fashion.85
A possible source of confusion might be noted. Occasional B-cell lymphomas exhibit deregulation of FcγRIIB and tumor progression, associated with chromosomal 1q21–23 rearrangements.86,87
Antigenic modulation of CD20: the role of trogocytosis
The speed of modulation of CD20 during rituximab treatment of CLL, supported by observations in vitro on CLL cells and Burkitt and mantle-cell lymphoma lines, led to the proposition that modulation was occurring by trogocytosis with the leukemic cell as donor.88,89 This implied that an effector cell such as a monocyte/macrophage became attached by its FcγR to the immune complexes on the leukemic cell surface, prompting the effector cell to nibble off and endocytose the immune complexes together with the attached FcγR, bits of adjacent membrane and possibly significant numbers of bystander receptors.
It is interesting to recall an experiment described in 1976 in one of the major papers47 on the zipper hypothesis of phagocytosis. Normal mouse mesenteric B cells, while capping and internalizing surface immune complexes formed by adding anti-IgM antibody at 37°C, were transferred to cold monolayers of peritoneal macrophages. When the mixed cell suspension was warmed, macrophages that had attached to lymphocytic caps proceeded to piecemeal phagocytosis (that is, trogocytosis) of the cap plasma membranes. The process is recorded in striking electron micrographs44 that show membrane-derived vacuoles, identified by labeled immune complexes, entering the macrophage cytoplasm. The B cells survived. Control B cells from the same experiment, diffusely coated with anti-IgM, were ingested and killed by the macrophages. These results suggest that trogocytosis requires: (1) a target membrane with a sufficient density of Fcγ-displaying immune complexes; (2) an incomplete encircling of the cell by the immune complexes, to avoid closing the phagocytic zipper.
Trogocytosis is not easily observed in vivo but is strongly suggested if donor cells are seen to have lost from their surfaces the targeted antigen plus significant amounts of bystander antigens. Such evidence has been presented by Rossi et al.90,91 Patients receiving the mAb epratuzumab (humanized anti-CD22), as immunotherapy for SLE, showed major reductions of CD22 on blood B lymphocytes, plus significant reductions of CD19 and CD21. B lymphocytes were not markedly reduced in number but may have been sufficiently disabled by trogocytosis to account for clinical remissions.
Problem 3: effector modulation
As previously pointed out15 it is instructive, when viewing the Stanford trials of anti-Id and anti-CD20,1,13 to compare times from first antibody treatment to first response. Counting from the beginning of antibody infusions (Day 0), the delays before tumor responses were noted: in the anti-Id trial, 8–16 days; in the anti-CD20 trial, 28–133 (median 71) days. Two questions arise about the strikingly delayed anti-CD20 response. What caused the delay? Was the eventual response due to the intended anti-CD20 attack on the tumor or to some other mechanism?
The second question is more easily answered. It has been suggested that the delayed response reflects a slowly developing anti-idiotypic ‘vaccinal’ attack on the tumor, but evidence for such a mechanism has not been substantiated. Instead, pharmacokinetic data from a large multicenter trial92 of rituximab monotherapy reveal statistically significant correlations between median plasma antibody levels and tumor responses at all time points after the first 3 of the 4 weekly infusions. For example, at 105 days after beginning the first infusion, median levels were: in responders (62 patients) 25.4 μg/mL; in non-responders (42 patients) 5.9 μg/mL. This is persuasive evidence for antibody attack being an essential component of the successful therapy.
A tentative answer to the first question is that the immune system has responded to sudden exposure to a large number of Fcγ-displaying immune complexes by entering a state of effector modulation. This may be defined15 as a failure of immunological effectors to kill antibody-coated targets at antibody levels predicted to lie within the cytotoxic range. It implies a much more prolonged failure than a transient overwhelming of immunological capacity. The postulate derives from an analogy with suppression of autoimmune inflammation brought about by one of two measures. (1) Infusion of normal human IgG up to a remarkable 2 g/kg. The infusion is rendered immunosuppressive by the presence of aggregates,93,94 and possibly has some inherent immunosuppressive activity.95 (2) Infusion of human IgG antibody to the rhesus D antigen, into D-positive subjects, at up to 75 μg/kg/day. Either of these measures, applied originally96 to the autoimmune disease acute idiopathic thrombocytopenic purpura, typically curtails the phagocytic destruction of platelets for a period of weeks without affecting immune complexes on the platelet surface. This represents a clear analogy with therapeutic antibody coating the surface of a lymphoma cell for weeks without causing its destruction. The analogy is described in more detail in an earlier review.15
Studies of suppression of autoimmune inflammation have revealed interesting pathways with interesting overlaps in function.93–100 It is hoped that such work will illuminate delayed responses to antibody therapy of tumor. In the meantime, it is interesting to note again the faster and better therapeutic responses to anti-Id than to anti-CD20, despite antigenic modulation in vitro of anti-Id being much the more impressive. Antigenic modulation of anti-Id might here, in balance, be favoring therapy by rapidly reducing immune complexes threatening effector modulation.
Recently developed protein kinase inhibitors and pro-apoptotic drugs might be incorporated into formidable combinations with antibodies, and might in some situations supplant them. But antibodies will retain the ability to strike at multiple targets on the surfaces of cancer cells, often with exquisite specificity and remarkable freedom from serious toxicity. We do, however, need to have a better understanding of their modes of action.
Acknowledgments
I am indebted to Dr Doris Taylor for drawing (and amending) Figure 1.
Footnotes
Authorship and Disclosures
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Maloney DG, Grillo-López AJ, White CA, Bodkin D, Schilder RJ, Neidhart JA, et al. IDEC-C2B8 (Rituximab) anti-CD20 monoclonal antibody therapy in patients with relapsed low-grade non-Hodgkin’s lymphoma. Blood. 1997;90(6):2188–95 [PubMed] [Google Scholar]
- 2.Maloney DG. Anti-CD20 antibody therapy for B-cell lymphomas. N Engl J Med. 2012;366(21):2008–16 [DOI] [PubMed] [Google Scholar]
- 3.Tan D, Horning SJ, Hoppe RT, Levy R, Rosenberg SA, Sigal BM, et al. Improvements in observed and relative survival in follicular grade 1–2 lymphoma during 4 decades: the Stanford University experience. Blood. 2013;122(6):961–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ghielmini M. Single agent rituximab is a valid option for first-line treatment in follicular lymphoma. Ann Oncol. 2011;22(Suppl 4):iv81 [Google Scholar]
- 5.Martinelli G, Schmitz SF, Utiger U, Cerny T, Hess U, Bassi S, et al. Long-term follow-up of patients with follicular lymphoma receiving single-agent rituximab at two different schedules in trial SAKK 35/98. J Clin Oncol. 2010;28(29):4480–4 [DOI] [PubMed] [Google Scholar]
- 6.Barré-Sinoussi F, Montagnier L, Lidereau R, Sisman J, Wood J, Chermann JC. Enhancement of retrovirus production by anti-interferon serum. Ann Microbiol (Paris). 1979;130B(3):349–62 [PubMed] [Google Scholar]
- 7.Young GR, Eksmond U, Salcedo R, Alexopoulou L, Stoye JP, Kassiotis G. Resurrection of endogenous retroviruses in antibody-deficient mice. Nature. 2012;491(7426):774–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Crick FH. In What Mad Pursuit: A Personal View of Scientific Discovery. Basic Books; New York, New York. 1988; p.146 [Google Scholar]
- 9.Stevenson GT, Stevenson FK. Antibody to a molecularly-defined antigen confined to a tumour cell surface. Nature. 1975;254(5502):714–6 [DOI] [PubMed] [Google Scholar]
- 10.Hough DW, Eady RP, Hamblin TJ, Stevenson FK, Stevenson GT. Anti-idiotype sera raised against surface immunoglobulin of human neoplastic lymphocytes. J Exp Med. 1976;144(4):960–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stevenson FK, Hamblin TJ, Stevenson GT, Tutt AL. Extracellular idiotypic immunoglobulin arising from human leukemic B lymphocytes. J Exp Med. 1980;152(6):1484–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miller RA, Maloney DG, Warnke RA, Levy R. Treatment of B-cell lymphoma with monoclonal anti-idiotype antibody. N Engl J Med. 1982;306(9):517–22 [DOI] [PubMed] [Google Scholar]
- 13.Meeker TC, Lowder J, Maloney DG, Miller RA, Thielemans K, Warnke R, et al. A clinical trial of anti-idiotype therapy for B cell malignancy. Blood. 1985;65(6):1349–63 [PubMed] [Google Scholar]
- 14.Davis TA, Maloney DG, Czerwinski DK, Lilies TM, Levy R. Anti-idiotype antibodies can induce long-term complete remissions in non-Hodgkin’s lymphoma without eradicating the malignant clone. Blood. 1998;92(4):1184–90 [PubMed] [Google Scholar]
- 15.Stevenson FK, Stevenson GT. Follicular lymphoma and the immune system: from pathogenesis to antibody therapy. Blood. 2012;119(16):3659–67 [DOI] [PubMed] [Google Scholar]
- 16.Riley JK, Sliwkowski MX. CD20: a gene in search of a function. Semin Oncol. 2000;27(6, Suppl 12):17–24 [PubMed] [Google Scholar]
- 17.Beers SA, Chan CH, James S, French RR, Attfield KE, Brennan CM, et al. Type II (tositumomab) anti-CD20 monoclonal antibody out performs type I (rituximab-like) reagents in B-cell depletion regardless of complement activation. Blood. 2008;112(10):4170–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Klein C, Lammens A, Schäfer W, Georges G, Schwaiger M, Mössner E, et al. Epitope interactions of monoclonal antibodies targeting CD20 and their relationship to functional properties. Mabs. 2013;5(1):22–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Davis TA, Czerwinski DK, Levy R. Therapy of B-cell lymphoma with anti-CD20 antibodies can result in the loss of CD20 antigen expression. Clin Cancer Res. 1999;5(3):611–5 [PubMed] [Google Scholar]
- 20.Kennedy GA, Tey SK, Cobcroft R, Marlton P, Cull G, Grimmett K, et al. Incidence and nature of CD20-negative relapses following rituximab therapy in aggressive B-cell non-Hodgkin’s lymphoma: a retrospective review. Br J Haematol. 2002;119(2):412–6 [DOI] [PubMed] [Google Scholar]
- 21.Marsden VS, Strasser A. Control of apoptosis in the immune system: Bcl-2, BH3-only proteins and more. Annu Rev Immunol. 2003;21:71–105 [DOI] [PubMed] [Google Scholar]
- 22.Hubert P, Heitzmann A, Viel S, Nicolas A, Sastre-Garau X, Oppezzo P, et al. Antibody-dependent cell cytotoxicity synapses form in mice during tumor-specific antibody immunotherapy. Cancer Res. 2011;71(15):5134–43 [DOI] [PubMed] [Google Scholar]
- 23.Albanesi M, Mancardi DA, Jönsson F, Iannascoli B, Fiette L, Di Santo JP, et al. Neutrophils mediate antibody-induced anti-tumor effects in mice. Blood. 2013;122(18):3160–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang SY, Racila E, Taylor RP, Weiner GJ. NK-cell activation and antibody-dependent cellular cytotoxicity induced by rituximab-coated target cells is inhibited by the C3b component of complement. Blood. 2008;111(3):1456–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stackpole CW, Jacobson JB, Galuska S. Antigenic modulation in vitro. II. Modulation of thymus leukemia (TL) antigenicity requires complement component C3. J Immunol. 1978;120(1):188–97 [PubMed] [Google Scholar]
- 26.Zhang S, Wu CC, Fecteau JF, Cui B, Chen L, Zhang L, et al. Targeting chronic lymphocytic leukemia cells with a humanized monoclonal antibody specific for CD44. Proc Natl Acad Sci USA. 2013;110(15):6127–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shan D, Ledbetter JA, Press OW. Apoptosis of malignant human B cells by ligation of CD20 with monoclonal antibodies. Blood. 1998;91(5):1644–52 [PubMed] [Google Scholar]
- 28.Ivanov A, Beers SA, Walshe CA, Honeychurch J, Alduaij W, Cox KL. Monoclonal antibodies directed to CD20 and HLA-DR can elicit homotypic adhesion followed by lysosome-mediated cell death in human lymphoma and leukemia cells. J Clin Invest. 2009;119(8):2143–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Scarlatti F, Granata R, Meijer AJ, Codogno P. Does autophagy have a licence to kill mammalian cells? Cell Death Differ. 2009;16(1):12–20 [DOI] [PubMed] [Google Scholar]
- 30.Bucur O, Stancu AL, Khosravi-Far R, Almasan A. Analysis of apoptosis methods recently used in Cancer Research and Cell Death & Disease publications. Cell Death Dis. 2012;3:e263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Clynes RA, Towers TL, Presta LG, Ravetch JV. Inhibitory Fc receptors modulate in vivo cytotoxicity against tumor targets. Nat Med. 2000;6(4):443–6 [DOI] [PubMed] [Google Scholar]
- 32.Clynes R, Takechi Y, Moroi Y, Houghton A, Ravetch JV. Fc receptors are required in passive and active immunity to melanoma. Proc Natl Acad Sci USA. 1998;95(2):652–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nimmerjahn F, Bruhns P, Horiuchi K, Ravetch JV. FcγRIV: a novel FcR with distinct IgG subclass specificity. Immunity. 2005;23(1):41–51 [DOI] [PubMed] [Google Scholar]
- 34.Cartron G, Dacheux L, Salles G, Solal-Celigny P, Bardos P, Colombat P, et al. Therapeutic activity of humanized anti-CD20 monoclonal antibody and polymorphism in IgG Fc receptor FcγRIIIa gene. Blood. 2002;99(3):754–58 [DOI] [PubMed] [Google Scholar]
- 35.Weng WK, Levy R. Two immunoglobulin G fragment C receptor polymorphisms independently predict response to rituximab in patients with follicular lymphoma. J Clin Oncol. 2003;21(21):3940–7 [DOI] [PubMed] [Google Scholar]
- 36.Persky DO, Dornan D, Goldman BH, Braziel RM, Fisher RI, LeBlanc M, et al. Fc gamma receptor 3a genotype predicts overall survival in follicular lymphoma patients treated on SWOG trials with combined monoclonal antibody plus chemoptherapy but not chemotherapy alone. Haematologica. 2012;97(6):937–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Farag SS, Flinn IW, Modali R, Lehman TA, Young D, Byrd JC. FcγRIIIa and FcγRIIa polymorphisms do not predict response to rituximab in B-cell chronic lymphocytic leukemia. Blood. 2004;103(4):1472–4 [DOI] [PubMed] [Google Scholar]
- 38.Dornan D, Spleiss O, Yeh R-F, Duchateau-Nguyen G, Dufour A, Zhi J, et al. Effect of FCGR2A and FCGR3A variants on CLL outcome. Blood. 2010;116(20):4212–22 [DOI] [PubMed] [Google Scholar]
- 39.Desjarlais JR, Lazar GA, Zhukovsky EA, Chu SY. Optimizing engagement of the immune system by anti-tumor antibodies: an engineer’s perspective. Drug Discov Today. 2007;12(21–22):898–910 [DOI] [PubMed] [Google Scholar]
- 40.Dutertre CA, Bonnin-Gélizé E, Pulford K, Bourel D, Fridman WH, Teillaud JL. A novel subset of NK cells expressing high levels of inhibitory FcγRIIB modulating antibody-dependent function. J Leukoc Biol. 2008;84(6):1511–20 [DOI] [PubMed] [Google Scholar]
- 41.Uchida JC, Hamaguchi Y, Oliver JA, Ravetch JV, Poe JC, Haas KM, Tedder TF. The innate mononuclear phagocyte network depletes B lymphocytes through Fc receptor-dependent mechanisms during anti-CD20 antibody immunotherapy. J Exp Med. 2004;199(12):1659–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hamaguchi Y, Xiu Y, Komura K, Nimmerjahn F, Tedder TF. Antibody isotype-specific engagement of Fcγ receptors regulates B lymphocyte depletion during CD20 immunotherapy. J Exp Med. 2006;203(3):743–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Van Ravenswaay Claasen HH, Kluin PM, Fleuren GJ. Tumor infiltrating cells in human cancer. On the possible role of CD16+ macrophages in antitumor cytotoxicity. Lab Invest. 1992;67(2):166–74 [PubMed] [Google Scholar]
- 44.Golay J, Da Roit F, Bologna L, Ferrara C, Leusen JH, Rambalda A, et al. Glycoengineered CD20 antibody obinutuzumab activates neutrophils and mediates phagocytosis through CD16B more efficiently than rituximab. Blood. 2013;122(20):3482–91 [DOI] [PubMed] [Google Scholar]
- 45.Locati M, Mantovani A, Sica A. Macrophage activation and polarization as an adaptive component of innate immunity. Adv Immunol. 2013;120:163–84 [DOI] [PubMed] [Google Scholar]
- 46.Griffin FM, Griffin JA, Leider JE, Silverstein SC. Studies on the mechanism of phagocytosis. I. Requirements for circumferential attachment of particle-bound ligands to specific receptors on the macrophage plasma membrane. J Exp Med. 1975;142(5):1263–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Griffin FM, Griffin JA, Silverstein SC. Studies on the mechanism of phagocytosis. II. The interaction of macrophages with anti-immunoglobulin IgG-coated bone-marrow derived lymphocytes. J Exp Med. 1976;144(3):788–809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Baba T, Iwasaki S, Maruoka T, Suzuki A, Tomaru U, Ikeda H. Rat CD4+CD8+ macrophages kill tumor cells through an NKG2D- and granzyme/perforin-dependent mechanism. J Immunol. 2008;180(5):2999–3006 [DOI] [PubMed] [Google Scholar]
- 49.Lefebvre ML, Krause SW, Salcedo M, Nardin A. Ex vivo-activated human macrophages kill chronic lymphocytic leukemia cells in the presence of rituximab: mechanism of antibody-dependent cellular cytotoxicity and impact of human serum. J Immunother. 2006;29(4):388–97 [DOI] [PubMed] [Google Scholar]
- 50.Sanderson CJ, Thomas JA. A comparison of the cytotoxic activity of eosinophils and other cells by 51chromium release and time lapse microcinematography. Immunology. 1978;34(4):771–80 [PMC free article] [PubMed] [Google Scholar]
- 51.Joli E, Hudrisier D. What is trogocytosis and what is its purpose? Nat Immunol. 2003;4(9):815. [DOI] [PubMed] [Google Scholar]
- 52.Rudnicka D, Oszmiana A, Finch DK, Strickland I, Schofield DJ, Lowe DC, et al. Rituximab causes a polarization of B cells that augments its therapeutic function in NK-cell-mediated antibody-dependent cellular cytotoxicity. Blood. 2013;121(23):4694–702 [DOI] [PubMed] [Google Scholar]
- 53.Vilches C, Parham P. KIR: rapidly evolving receptors of innate and adaptive immunity. Ann Rev Immunol. 2002;20:217–51 [DOI] [PubMed] [Google Scholar]
- 54.Law SK, Reid KB. Control of complement. In Complement, 2nd ed IRL Press, Oxford, 1995; pp22–27 [Google Scholar]
- 55.Hersey P. New look at antiserum therapy of leukaemia. Nature New Biol. 1973;244(131):22–4 [DOI] [PubMed] [Google Scholar]
- 56.Lanier LL, Babcock GF, Raybourne RB, Arnold LW, Warner NL, Haughton G. Mechanism of B cell lymphoma therapy with passive xenogeneic anti-idiotype serum. J Immunol. 1980;125(4):1730–6 [PubMed] [Google Scholar]
- 57.Stevenson GT, Glennie MJ. Surface immunoglobulin of B-lymphocytic tumours as a therapeutic target. Cancer Surv. 1985;4(1):214–44 [PubMed] [Google Scholar]
- 58.Bernstein ID, Tam MR, Nowinski RC. Mouse leukemia: therapy with monoclonal antibodies against a thymus differentiation antigen. Science. 1980;207(4426):1980. [DOI] [PubMed] [Google Scholar]
- 59.Bologna L, Gotti E, Manganini M, Rambaldi A, Intermesoli T, Introna M, et al. Mechanism of action of type II, glycoengineered, anti-CD20 monoclonal antibody GA101 in B-chronic lymphocytic leukemia whole blood assays in comparison with rituximab and alemtuzumab. J Immunol. 2011;186(6):3762–9 [DOI] [PubMed] [Google Scholar]
- 60.Weng WK, Levy R. Expression of complement inhibitors CD46, CD55 and CD59 on tumor cells does not predict the clinical outcome after rituximab treatment in follicular non-Hodgkin lymphoma. Blood. 2001;98(5):1352–7 [DOI] [PubMed] [Google Scholar]
- 61.Shushakova N, Skokowa J, Schulman J, Baumann U, Zwirner J, Schmidt RE, et al. C5a anaphylatoxin is a major regulator of activating versus inhibitory FcγRs in immune complex-induced lung disease. J Clin Invest. 2002;110(12):1823–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sondermann P, Huber R, Oosthuizen V, Jacob U. The 3.2-Å crystal structure of the human IgG1 Fc fragmentBFcγRIII complex. Nature. 2000;406(6793):267–73 [DOI] [PubMed] [Google Scholar]
- 63.Lazar GA, Dang W, Karki S, Vafa O, Peng JS, Hyun L, et al. Engineered antibody Fc variants with enhanced effector function. Proc Natl Acad Sci USA. 2006;103(11):4005–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Stavenhagen JB, Gorlatov S, Tuaillon N, Rankin CT, Li H, Burke S, et al. Fc optimization of therapeutic antibodies enhances their ability to kill tumor cells in vitro and controls tumor expansion in vivo via low-affinity activating Fcγ receptors. Cancer Res. 2007;67(18):8882–90 [DOI] [PubMed] [Google Scholar]
- 65.Shields RL, Lai J, Keck R, O’Connell LY, Hong K, Meng YG, et al. Lack of fucose on human IgG1 N-linked oligosaccharide improves binding to human FcγRIII and antibody-dependent cellular toxicity. J Biol Chem. 2002;277(30):26733–40 [DOI] [PubMed] [Google Scholar]
- 66.Leong WS, Thomas KA, Chan CH, Stevenson GT. A standardized conversion of IgG antibody to bispecific form with inversely altered affinities for Fcγ-receptors II and III. Mol Immunol. 2011;48(5):760–8 [DOI] [PubMed] [Google Scholar]
- 67.Ratner M. Genentech’s glyco-engineered antibody to succeed rituxan. Nat Biotechnol. 2014;32(1):6–7 [DOI] [PubMed] [Google Scholar]
- 68.Old LJ, Boyse EA. Antigenic properties of experimental leukemias. I. Serological studies in vitro with spontaneous and radiation-induced leukemias. J Natl Cancer Inst. 1963;31:977–95 [PubMed] [Google Scholar]
- 69.Stackpole CW, Jacobson JB, Lardis MP. Antigenic modulation in vitro. I. Fate of thymus-leukemia (TL) antigen-antibody complexes following modulation of TL antigenicity from the surfaces of mouse leukemia cells and thymocytes. J Exp Med. 1974;140(4):939–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Schroff RW, Farrell MM, Klein RA, Stevenson HC, Warner NL. Induction and enhancement by monocytes of antibody-induced modulation of a variety of human lymphoid cell surface antigens. Blood. 1985;66(3):620–6 [PubMed] [Google Scholar]
- 71.Schreiner GF, Unanue ER. Membrane and cytoplasmic changes in B lymphocytes induced by ligand-surface immunoglobulin interaction. Adv Immunol. 1976;24:37–165 [DOI] [PubMed] [Google Scholar]
- 72.Gordon J, Stevenson GT. Antigenic modulation of lymphocytic surface immunoglobulin yielding resistance to complement-mediated lysis. II. Relation to redistribution of the antigen. Immunology. 1981;42(1):13–7 [PMC free article] [PubMed] [Google Scholar]
- 73.Glennie M, Stevenson FK, Stevenson GT, Virji M. Cross-linking of surface immunoglobulin inhibits its production via a cyclic nucleotide mechanism. Nature. 1979;281(5729):305–7 [DOI] [PubMed] [Google Scholar]
- 74.Elliott TJ, Glennie MJ, McBride HM, Stevenson GT. Analysis of the interaction of antibodies with immunoglobulin idiotypes on neoplastic B lymphocytes: implications for immunotherapy. J Immunol. 1987;138(3):981–8 [PubMed] [Google Scholar]
- 75.Lane AC, Foroozan S, Glennie MJ, Kowalski-Saunders P, Stevenson GT. Enhanced modulation of antibodies coating guinea pig leukemic cells in vitro and in vivo. The role of FcγR expressing cells. J Immunol. 1991;146(7):2461–8 [PubMed] [Google Scholar]
- 76.Press OW, Howell-Clark J, Anderson S, Bernstein I. Retention of B-cell-specific monoclonal antibodies by human lymphoma cells. Blood. 1994;83(5):1390–7 [PubMed] [Google Scholar]
- 77.Press OW, Farr AG, Borroz I, Anderson SK, Martin PJ. Endocytosis and degradation of monoclonal antibodies targeting B-cell malignancies. Cancer Res. 1989;49(9):4906–12 [PubMed] [Google Scholar]
- 78.Glennie MJ, Stevenson GT. Univalent antibodies kill tumor cells in vitro and in vivo. Nature. 1982;295(5851):712–4 [DOI] [PubMed] [Google Scholar]
- 79.Maloney DG, Liles TM, Czerwinski DK, Waldichuk C, Rosenberg J, Grillo-Lopez A, et al. Phase I clinical trial using escalating single-dose infusion of chimeric anti-CD20 monoclonal antibody in patients with recurrent B-cell lymphoma. Blood. 1994;84(8)2457–66 [PubMed] [Google Scholar]
- 80.Jilani I, O’ Brien S, Manshuri T, Thomas DA, Thomazy VA, Imam M, et al. Transient down-modulation of CD20 by rituximab in patients with chronic lymphocytic leukemia. Blood. 2003;102(10):3514–20 [DOI] [PubMed] [Google Scholar]
- 81.Beers SA, French RR, Claude Chan HT, Lim SH, Jarrett TC, Vidal RM, et al. Antigenic modulation limits the efficacy of anti-CD20 antibodies: implications for antibody selection. Blood. 2010;115(25):5191–201 [DOI] [PubMed] [Google Scholar]
- 82.Vervoordeldonk SF, Merle PA, van Leeuwen EF, van der Schoot CE, von dem Borne AE, Slaper-Cortenbach IC. Fcγ receptor II (CD32) on malignant B cells influences modulation induced by anti-CD19 monoclonal antibody. Blood. 1994;83(6):1632–9 [PubMed] [Google Scholar]
- 83.Zhang CY, Booth JW. Divergent intracellular sorting of FcγRIIA and FcγRIIB2. J Biol Chem. 2010;285(44):34250–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Mousavi SA, Sporstøl M, Fladeby C, Kjeken R, Barois N, Berg T. Receptor-mediated endocytosis of immune complexes in rat liver sinusoidal endothelial cells is mediated by FcγRIIB2. Hepatology. 2007;46(3):871–84 [DOI] [PubMed] [Google Scholar]
- 85.Vaughan AT, Iriyama C, Beers SA, Chan CH, Lim SH, Williams EL, et al. Inhibitory FcγRIIb (CD32b) becomes activated by therapeutic mAb in both cis and trans and drives internalization according to antibody specificity. Blood. 2014;123(5):669–77 [DOI] [PubMed] [Google Scholar]
- 86.Callanan MB, le Baccon P, Mossuz P, Duley S, Bastard C, Hamoudi R, et al. The IgG Fc receptor, FcγRIIb, is a target for deregulation by chromosomal translocation in malignant lymphoma. Proc Natl Acad Sci USA. 2000;97(1):309–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Chen W, Palanisamy N, Schmidt H, Teruya-Feldstein J, Jhanwar SC, Zelenetz AD, et al. Deregulation of FCGR2B expression by 1q21 rearrangements in follicular lymphomas. Oncogene. 2001;20(52):7686–93 [DOI] [PubMed] [Google Scholar]
- 88.Kennedy AD, Beum PV, Solga MD, DiLillo DJ, Lindorfer MA, Hess CE, et al. Rituximab infusion promotes rapid complement depletion and acute CD20 loss in chronic lymphocytic leukemia. J Immunol. 2004;172(5):3280–8 [DOI] [PubMed] [Google Scholar]
- 89.Beum PV, Kennedy AD, Williams ME, Lindorfer MA, Taylor RP. The shaving reaction: rituximab/CD20 complexes are removed from mantle cell lymphoma and chronic lymphocytic leukemia cells by THP–1 monocytes. J Immunol. 2006;176(4):2600–9 [DOI] [PubMed] [Google Scholar]
- 90.Rossi EA, Goldenberg DM, Michel R, Rossi DL, Wallace DJ, Chang CH. Trogocytosis of multiple B-cell surface markers by CD22 targeting with epratuzumab. Blood. 2013;122(17):3020–9 [DOI] [PubMed] [Google Scholar]
- 91.Taylor RP. Gnawing at Metchnikoff’s paradigm. Blood. 2013;122(17):2922–4 [DOI] [PubMed] [Google Scholar]
- 92.Berinstein NL, Grillo-López AJ, White CA, Bence-Bruckler I, Maloney D, Czuczman M, et al. Association of serum rituximab (IDEC-C2B8) concentration and anti-tumor response in the treatment of recurrent low-grade or follicular non-Hodgkin’s lymphoma. Ann Oncol. 1998;9(9):995–1001 [DOI] [PubMed] [Google Scholar]
- 93.Teeling JL, Jansen-Hendriks T, Kuijpers TW, de Haas M, van de Winkel JG, Hack CE, et al. Therapeutic efficacy of intravenous immunoglobulin preparations depends on the immunoglobulin G dimers: studies in immune thrombocytopenia. Blood. 2001;98(4):1095–9 [DOI] [PubMed] [Google Scholar]
- 94.Clynes R. Protective mechanisms of IVIG. Curr Opin Immunol. 2007;19(6):646–51 [DOI] [PubMed] [Google Scholar]
- 95.Yu X, Vasilijevic S, Mitchell DA, Crispin M, Scanlan CN. Dissecting the molecular mechanism of IVIg therapy: the interaction between serum IgG and DC-SIGN is independent of antibody glycoform or Fc domain. J Mol Biol. 2013;425(8):1253–8 [DOI] [PubMed] [Google Scholar]
- 96.Imbach P, Morell A. Idiopathic thrombocytopenic purpura (ITP): immunomodulation by intravenous immunoglobulin (IVIg). Int Rev Immunol. 1989;5:181–8 [DOI] [PubMed] [Google Scholar]
- 97.Samuelsson A, Towers TL, Ravetch JV. Anti-inflammatory activity of IVIG mediated through the inhibitory Fc receptor. Science. 2001;291(5503):484–6 [DOI] [PubMed] [Google Scholar]
- 98.Song S, Crow AR, Siragam V, Freedman J, Lazarus AH. Intravenous antibodies that mimic the action of anti-D in the amelioration of murine ITP act by a mechanism distinct from that of IVIg. Blood. 2005;105(4):1546–8 [DOI] [PubMed] [Google Scholar]
- 99.Siragam V, Crow AR, Brinc D, Song S, Freedman J, Lazarus AH. Intravenous immunoglobulin ameliorates ITP via activating Fcγ receptors on dendritic cells. Nat Med. 2006;12(6):688–92 [DOI] [PubMed] [Google Scholar]
- 100.Park-Min KH, Serbina NV, Yang W, Ma X, Krystal G, Neel BG, et al. FcγRIII-dependent inhibition of interferon-γ responses mediates suppressive effects of intravenous immune globulin. Immunity. 2007;26(1):67–78 [DOI] [PubMed] [Google Scholar]