

Abstract
Legionella spp. cause the severe pneumonia Legionnaires' disease. The environmental bacteria replicate intracellularly in free-living amoebae and human alveolar macrophages within a distinct, endoplasmic reticulum (ER)-derived compartment termed the Legionella-containing vacuole (LCV). LCV formation requires the bacterial Icm/Dot type IV secretion system (T4SS) that translocates into host cells a plethora of different “effector” proteins, some of which anchor to the pathogen vacuole by binding to phosphoinositide (PI) lipids. Here, we identified by unbiased pulldown assays in Legionella longbeachae lysates a 111-kDa SidC homologue as the major phosphatidylinositol 4-phosphate [PtdIns(4)P]-binding protein. The PI-binding domain was mapped to a 20-kDa P4C [PtdIns(4)P binding of SidC] fragment. Isothermal titration calorimetry revealed that SidC of L. longbeachae (SidCLlo) binds PtdIns(4)P with a Kd (dissociation constant) of 71 nM, which is 3 to 4 times lower than that of the SidC orthologue of Legionella pneumophila (SidCLpn). Upon infection of RAW 264.7 macrophages with L. longbeachae, endogenous SidCLlo or ectopically produced SidCLpn localized in an Icm/Dot-dependent manner to the PtdIns(4)P-positive LCVs. An L. longbeachae ΔsidC deletion mutant was impaired for calnexin recruitment to LCVs in Dictyostelium discoideum amoebae and outcompeted by wild-type bacteria in Acanthamoeba castellanii. Calnexin recruitment was restored by SidCLlo or its orthologues SidCLpn and SdcALpn. Conversely, calnexin recruitment was restored by SidCLlo in L. pneumophila lacking sidC and sdcA. Together, biochemical, genetic, and cell biological data indicate that SidCLlo is an L. longbeachae effector that binds through a P4C domain with high affinity to PtdIns(4)P on LCVs, promotes ER recruitment to the LCV, and thus plays a role in pathogen-host interactions.
INTRODUCTION
Legionella spp. are environmental, amoeba-resistant bacteria that upon inhalation cause a severe pneumonia termed Legionnaires' disease. Legionella longbeachae and Legionella pneumophila are the major causative agents of the respiratory ailment (1–4). Whereas L. longbeachae causes around 30% of the cases in Australia and New Zealand, L. pneumophila is associated with up to 90% of all cases in Europe and the United States. The environmental niches, transmission route, and disease epidemiology of L. longbeachae and L. pneumophila differ considerably (5). L. longbeachae is associated with compost and potting soils (3, 6), whereas L. pneumophila is found ubiquitously in natural and man-made water systems (7, 8). The preferences of the Legionella spp. for distinct environmental niches are reflected in major physiological and genetic differences. L. longbeachae is capsulated, nonflagellated, and, based on transcriptome analysis, does not show a pronounced growth phase-dependent life cycle (9, 10). On the other hand, L. pneumophila is motile and exhibits a growth phase-dependent biphasic life style, which alternates between a replicative, nonvirulent phase and, upon reaching stationary growth phase, a transmissible, virulent form (11).
Legionella spp. replicate intracellularly in free-living amoebae and mammalian macrophages within a distinct compartment, the “Legionella-containing vacuole” (LCV) (7, 12). The mechanism of intracellular replication appears to be evolutionarily conserved, as in protozoan, as well as metazoan phagocytes LCVs avoid fusion with lysosomes yet communicate extensively with the endosomal and secretory vesicle-trafficking pathways and eventually fuse with the endoplasmic reticulum (ER) (13, 14). Accordingly, a number of small GTPases implicated in endosomal and secretory vesicle trafficking localize to LCVs in amoebae (15), as well as in macrophages (16). The formation of LCVs by L. pneumophila has been studied in some detail. In contrast, knowledge about the formation of LCVs by L. longbeachae is very limited, but the process appears to be different (17).
LCV formation and intracellular growth of most, if not all, Legionella spp. is determined by a type IVB secretion system (T4SS) called Icm/Dot (intracellular multiplication/defective organelle trafficking) (18). L. longbeachae (9) or L. pneumophila (19, 20) lacking a functional Icm/Dot T4SS fails to replicate intracellularly and is avirulent. The Icm/Dot T4SS translocates an astonishing total of several hundred so-called effector proteins into host cells, where they presumably subvert host signal transduction and vesicle-trafficking pathways. For L. pneumophila, more than 300 Icm/Dot substrates are known to date (21–23). A number of these effectors have been mechanistically characterized and subvert components of the endocytic, retrograde, or secretory vesicle-trafficking machinery, including small GTPases of the Arf and Rab (21, 24–26), as well as the Ran (27, 28) families; the vacuolar H+-ATPase (29); the autophagy machinery (30); the retromer complex (31); and phosphoinositide (PI) lipids (32, 33).
Phosphoinositides are low-abundance cellular lipids that are spatiotemporally controlled by PI kinases and/or phosphatases and regulate eukaryotic membrane dynamics and signal transduction in concert with small GTPases (34, 35). L. pneumophila and many other intracellular bacteria target and subvert these crucial lipids (36, 37). In particular, the Icm/Dot substrates SidF (38) and SidP (39) have been identified as PI phosphatases due to their CX5R phosphatase motif. SidF is a PI 3-phosphatase that hydrolyzes in vitro endosomal phosphatidylinositol 3,4-bisphosphate [PtdIns(3,4)P2] and also phosphatidylinositol 3,4,5-trisphosphate [PtdIns(3,4,5)P3] yielding phosphatidylinositol 4-phosphate [PtdIns(4)P] directly or indirectly through the activity of the host PI 5-phosphatase OCRL1/Dd5P4 present on LCVs (40). SidP is a PI 3-phosphatase that in vitro hydrolyzes PtdIns(3)P, as well as PtdIns(3,5)P2, thus promoting the evasion of the endocytic pathway by LCVs.
Several other L. pneumophila Icm/Dot substrates anchor to the LCV membrane by binding to PI lipids. The 73-kDa glycosyltransferase SetA (41) and the 131-kDa retromer interactor RidL (31) preferentially bind PtdIns(3)P, a signpost of endosomal trafficking. In contrast, the 106-kDa ER interactor SidC and its paralogue SdcA (42), as well as the 73-kDa Rab1 guanine nucleotide exchange factor (GEF)/adenylyltransferase (AMPylase) SidM (also called DrrA), specifically bind PtdIns(4)P (43), a marker of secretory-vesicle trafficking. SidC anchors to LCVs (44) by binding to PtdIns(4)P via a novel 20-kDa PtdIns(4)P-interacting domain, termed “P4C” [PtdIns(4)P binding of SidC] (42, 45). The P4C domain can be employed as a PtdIns(4)P-selective probe in cell biological and biochemical assays (43, 45, 46), similar to structurally unrelated eukaryotic PtdIns(4)P-binding domains (47, 48). L. pneumophila lacking sidC and sdcA is defective for the transition from tight to spacious LCVs and the recruitment of the ER to the LCV (42, 45, 49). Thus, SidC might represent a bacterial tethering factor. While the function of SidC remains elusive, the effector plays a role in monoubiquitination of Rab1, and the N-terminal domain of SidC (amino acids [aa] 1 to 608) reveals a novel fold (50, 51). SidM, on the other hand, is the major L. pneumophila PtdIns(4)P-interacting effector, which binds the PI lipid through a unique 12-kDa “P4M” (PtdIns(4)P binding of SidM) domain (43) with an affinity in the nanomolar range (52). Thus, the Rab1 modulator SidM links the exploitation of host PI metabolism and the subversion of small GTPases.
L. longbeachae is predicted to produce more than 110 Icm/Dot substrates (9, 23). However, none of these putative effectors has been mechanistically characterized to date. More than 66 L. pneumophila effectors are missing in L. longbeachae, and 50 novel Icm/Dot substrates have been identified, yet only a minority are conserved. For example, L. longbeachae lacks SidM, while SidC is present. In this study, SidC was identified in L. longbeachae lysates as the major PtdIns(4)P-binding Icm/Dot substrate, which binds through a P4C domain with high affinity to PtdIns(4)P on LCVs, promotes calnexin recruitment to LCVs, and is implicated in pathogen-host interactions.
MATERIALS AND METHODS
Growth of cells and bacteria.
Dictyostelium discoideum strain Ax3 harboring the plasmid pCaln-GFP (see Table S1 in the supplemental material) was grown axenically in HL-5 medium containing 20 μg ml−1 Geneticin sulfate at 23°C (42, 53). Acanthamoeba castellanii was grown in PYG medium (54, 55) at 23°C. RAW 264.7 macrophages were grown in RPMI 1640 medium (Gibco) supplemented with 10% fetal calf serum (FCS) and 2 mM l-glutamine at 37°C with 5% CO2.
L. longbeachae NSW150, L. pneumophila JR32, and derivatives of these strains (see Table S1 in the supplemental material) were cultured on charcoal yeast extract agar (CYE) plates for 3 days (56). Overnight cultures were incubated for 18 to 21 h in ACES [N-(2-acetamido)-2-aminoethanesulfonic acid] yeast extract medium (AYE) (56) at 37°C on a turning wheel. To maintain plasmids, chloramphenicol (Cam) was used at 10 μg ml−1 (L. longbeachae, solid media), 2.5 µg ml−1 (L. longbeachae, liquid media), or 5 μg ml−1 (L. pneumophila). Escherichia coli strains were grown on LB (Luria-Bertani) plates and LB medium (Invitrogen) with the appropriate antibiotics. To induce protein production, isopropyl-β-d-thiogalactopyranoside (IPTG) was added at a concentration of 0.5 mM to liquid cultures.
Molecular cloning.
The plasmid pSD01 (see Table S1 in the supplemental material) expressing His6-sidCLlo under the control of a T7 promoter was constructed by amplifying sidCLlo by PCR with the oligonucleotides oSD07 and oSD08 (see Table S2 in the supplemental material), using genomic DNA of L. longbeachae NSW150 as template. The PCR product was digested with NdeI and NheI and ligated into pET28a(+) cut with the same enzymes. This plasmid was used to construct plasmid pSD02, where silent mutations were introduced with the QuikChange kit (Agilent Technologies) using oSD22, oSD23, oSD28, and oSD29 in order to eliminate the BamHI and SalI restriction sites in the sidCLlo gene.
Plasmid pSD03, encoding glutathione S-transferase (GST)-SidCLlo_609-782 (an N-terminal fusion of GST with a fragment of the SidCLlo protein comprising amino acids 609 to 782), was generated by PCR amplification of sidCLlo_609-782 using pSD02 as the template and oSD18 and oSD19, cut with BamHI and SalI, and ligated into pGEX4T-1. The same procedure was used to construct pSD04 (GST-SidCLlo_1-340; oSD16/oSD17), pSD05 (GST-SidCLlo_341-608; oSD24/oSD25), and pSD06 (GST-SidCLlo_783-969; oSD26/oSD27). To generate pSD07, sidCLlo was cut with BamHI/SalI from pSD02 and ligated into pGEX4T-1. The plasmids pSD13 and pSD14 were constructed by liberating sidCLlo from pSD02 by BamHI/SalI restriction and ligation into pMMB207-C-M45 (42), yielding pSD13, or into pCR77 (31), yielding pSD14. To construct pIH47, the sdcA gene was cut from pCR78 (45) using BamHI/SalI and ligated into pCR80 (31) cut with the same enzymes.
To construct plasmid pIH60 encoding GST-SidCLpn_1-608-Llo_609-969, the fragments sidCLpn_1-608 and sidCLlo_609-969 were amplified by PCR using the templates pCR02 and pSD14 and the oligonucleotides oCR1/oIH39 or oIH40/oSD27, respectively. Silent mutations that led to the formation of PstI sites were introduced using oIH39 and oIH40. Subsequently, the DNA was digested with the appropriate enzymes (pGEX-4T-1, BamHI/SalI; SidCLpn1-608, BamHI/PstI; SidCLlo609-969, PstI/SalI), and a three-fragment ligation was conducted. The plasmid pMH01 encoding GST-SidCLlo_1-608-Lpn_609-917 was generated by a one-step sequence- and ligation-independent cloning strategy (57), using PCR-amplified sidCLlo_1-608, sidCLpn_609-917, and the linearized pGEX4T-1 vector as templates. The following templates and primers were used: pGEX4T-1 (pGEX4T-1; oMH01/oMH02), sidCLlo_1-608 (pSD14; oMH03/oMH04), and sidCLpn_609-917 (pCR02; oMH05/oMH06). Thus, sidCLlo_1-608 or sidCLpn_609-917 generated overlapping ends with pGEX4T-1 and sidCLpn_609-917 or pGEX4T-1 and sidCLlo_1-608, respectively.
For chromosomal deletion of sidCLlo (llo3098), the suicide plasmid pIH33 was generated as follows: the up- and downstream flanking regions of sidCLlo were amplified using oIH019/oIH020 (upstream flanking region; ∼950 bp) or oIH021/oIH022 (downstream flanking region; ∼850 bp), cut with BamHI/XbaI, and ligated together with a kanamycin (Kan) resistance cassette (cut from pUC4K with BamHI) into pLAW344 (cut with XbaI). All PCR-based constructs were sequenced.
Construction of L. longbeachae ΔsidC.
For the chromosomal deletion of sidCLlo (llo3098), L. longbeachae was grown overnight in AYE to exponential growth phase. The overnight culture was used to set up a fresh culture (1:5) in AYE, which was incubated on a shaker for 5 to 6 h until it reached an optical density at 600 nm (OD600) of 0.5 to 0.7. An overnight culture of E. coli ST18 with the deletion plasmid pIH33 was grown in LB medium to a final OD600 of 0.5 to 0.7 in the presence of 50 μg ml−1 aminolevulinic acid. The cells were harvested and mixed at a 1:1 ratio, spotted on a CYE plate containing 50 μg ml−1 aminolevulinic acid, and incubated for 24 h at 37°C. The lawn was harvested in sterile phosphate-buffered saline (PBS), and dilutions were plated on CYE plates with 50 μg ml−1 Kan. Resistant clones were grown in AYE broth containing 50 μg ml−1 Kan overnight and spotted onto CYE plates containing 50 μg ml−1 Kan or 50 μg ml−1 Kan and 2% sucrose. Sucrose-resistant clones were grown in AYE containing 50 μg ml−1 Kan and spotted onto CYE plates or CYE plates with 50 μg ml−1 Kan or 10 μg ml−1 Cam. Kan-resistant and Cam-sensitive clones were analyzed by PCR (amplification of the complete region with oIH23/oIH24), and successful deletion was verified by sequencing.
Transformation of L. longbeachae.
For transformation, L. longbeachae was grown overnight in AYE to late exponential growth phase, diluted 1:4 in fresh medium, and incubated on a shaker until it reached an OD600 of 0.4 to 0.5. Three milliliters of culture per transformation reaction was harvested and washed three times with 10% ice-cold sterile glycerol. Finally, the pellet of electrocompetent cells was resuspended in 50 μl ice-cold 10% glycerol freshly mixed with ∼3 μg of DNA and electroporated (2.5 kV; 200 Ω; 0.25 μF). The cultures were cultivated for 3.5 h at 37°C on a turning wheel and plated out on CYE plates with the appropriate antibiotic. Colonies appeared after 3 days.
PI bead pulldown assays.
The PI bead pulldown was done with L. longbeachae and L. pneumophila lysates and agarose beads covalently bound to PIs as described previously (43). The washed beads were boiled with SDS sample buffer and, after SDS-PAGE, visualized by staining with Coomassie brilliant blue or silver. The Coomassie-stained gel bands were excised and subjected to matrix-assisted laser desorption ionization–time of flight mass spectrometry (MALDI-TOF MS) analysis.
Protein lipid overlays with PIP strips and PIP arrays.
Commercially available PIP strips and PIP arrays (Echelon Biosciences Inc., USA) were used to visualize the different binding affinities of GST-tagged proteins. The GST fusion proteins were purified, and the experiment was performed as previously described (42, 43, 45, 58), but more rigorous washing conditions were applied. As primary and secondary antibodies, a mouse monoclonal anti-GST antibody (Sigma) and a goat anti-mouse peroxidase-labeled antibody were used, respectively, followed by enhanced chemiluminescence (ECL) detection (GE Healthcare).
Isothermal titration calorimetry.
Interaction studies by isothermal titration calorimetry (ITC) were performed using a VP-ITC microcalorimeter (MicroCal Inc., USA). Di-octyl-PtdIns(4)P [di-C8-PtdIns(4)P] (Echelon Biosciences Inc., USA) was dissolved to 250 μM in running buffer and titrated into the cell containing His6-SidCLlo or His6-SidCLpn at 25 μM. Measurements were carried out in 20 mM HEPES buffer, pH 7.5, 50 mM NaCl, and 1 mM Tris(2-carboxyethyl)phosphine (TCEP) at 25°C. Data were analyzed using the ITC software provided by the manufacturer (MicroCal Inc.).
Circular dichroism.
Far-UV circular-dichroism (CD) experiments were carried out on a J-715 spectropolarimeter (Jasco Inc., Japan) in the wavelength range of 190 to 260 nm at 25°C using a quartz cuvette with a path length of 1 mm and a scan speed of 20 nm/min. Proteins were diluted to 0.05 mg ml−1 in distilled H2O (dH2O) for the measurements. All spectra were accumulated 12 times and analyzed with the analysis program CDSSTR (59).
Western blot analysis.
For Western blot analysis, affinity-purified polyclonal rabbit antibodies were used as primary antibodies: anti-SidCLlo (this study) (Sequence Laboratories Goettingen GmbH) or anti-SidCLpn (42). Goat anti-rabbit peroxidase-labeled antibody (GE Healthcare) was used as a secondary antibody.
Phagocyte infection.
Infection of phagocytes was performed as described previously (42, 43, 45). Briefly, phagocytes (amoebae or macrophages) were seeded out in the appropriate medium 1 day prior to the experiment. An AYE culture was inoculated at an OD600 of 0.1 18 to 21 h before infection. The bacteria were added at a specific multiplicity of infection (MOI), spun down to synchronize the infection, and incubated at 25°C (D. discoideum), 30°C (A. castellanii), or 37°C and 5% CO2 (macrophages).
Immunofluorescence microscopy.
For immunofluorescence microscopy, RAW 264.7 macrophages infected with L. longbeachae were fixed, permeabilized, and treated with polyclonal rabbit anti-SidCLlo (this study) (Sequence Laboratories Goettingen GmbH), anti-SidCLpn (42), or monoclonal mouse anti-GST (Sigma) antibodies as described previously (42, 45). As secondary antibodies, Cy5-coupled goat anti-rabbit (Invitrogen) or goat anti-mouse (Jackson) antibodies were used at a 1:200 dilution. Alternatively, D. discoideum amoebae producing calnexin-green fluorescent protein (GFP) or P4CLpn-GFP were infected at an MOI of 20 for 2 h or 1 h, respectively, with red fluorescent L. longbeachae, and the recruitment of the GFP fusion proteins to LCVs was quantified in live cells. Images were acquired with a Leica SP5 TCS confocal laser scanning fluorescence microscope.
PtdIns(4)P on LCVs was detected in homogenates of Legionella-infected amoebae by purified recombinant GST fusion probes as described previously (42, 45). Briefly, D. discoideum Ax3 producing calnexin-GFP was infected with red fluorescent L. pneumophila JR32 (MOI, 100; 1 h) at 25°C, washed with Sörensen phosphate buffer (SorC) (2 mM Na2HPO4, 15 mM KH2PO4, 50 μM CaCl2 · 2 H2O, pH 6.0), and homogenized. The samples were spun onto coverslips, fixed with 4% paraformaldehyde, washed with SorC, and incubated with the GST-tagged proteins for 15 min. Immunofluorescence staining was performed with a monoclonal mouse anti-GST antibody (Sigma) and a Cy5-coupled anti-mouse antibody (Jackson).
Intracellular replication assay.
A. castellanii amoebae or RAW 264.7 macrophages were infected with Legionella cultures as described previously (42, 45, 60, 61). A. castellanii was infected in Ac buffer [4 mM MgSO4 · 7 H2O, 0.4 mM CaCl2, 3.4 mM sodium citrate, 0.05 mM Fe(NH4)2(SO4)2 · 6 H2O, 2.5 mM Na2HPO4 · 7 H2O, 2.5 mM KH2PO4, pH 6.5]. The bacteria were spun down on the cells for 5 min and incubated for 20 min at 37°C (t0). RAW 264.7 macrophages were infected in RPMI medium supplemented with l-glutamine and FCS at 37°C and 5% CO2. The infected cells were centrifuged for 10 min and incubated for another 10 min at 37°C and 5% CO2 (t0). The supernatant of the infected cells was plated out on CYE plates at different time points. The CFU were counted after 3 days of incubation at 30°C (amoebae) or 37°C and 5% CO2 (macrophages).
Amoeba competition assay.
The amoeba competition assays were performed as described previously (61). Briefly, A. castellanii amoebae were seeded into a 96-well plate 1 h before the experiment and incubated at 30°C. After attachment of the cells, the medium was exchanged for Ac buffer. The stationary Legionella AYE cultures were diluted in Ac buffer and added to the A. castellanii-containing wells. The infected cells were centrifuged (450 × g; 10 min) and incubated for 1 h at 37°C, followed by exchanging the Ac buffer. Bacterial input controls were plated on CYE plates with and without antibiotics. After 3 days the supernatant of the infected amoebae was collected, the cells were lysed with 0.8% saponin for 10 min, and the homogenate was added to the supernatant. Fifty microliters of a 1:1,000 dilution was used to infect a freshly seeded layer of A. castellanii amoebae. The collected samples were diluted and plated out in triplicate on CYE plates with and without antibiotics.
RESULTS
Identification of L. longbeachae SidC in a screen for PI-binding proteins.
The PtdIns(4)P-binding Icm/Dot substrate SidM from L. pneumophila was identified by unbiased pulldown experiments using agarose beads coupled to PI lipids (43). We adopted this approach to screen in an unbiased manner for PI-binding proteins from L. longbeachae. To this end, L. longbeachae lysates were incubated with beads coupled to PtdIns or different PI lipids, the beads were washed, and bound proteins were separated by SDS-PAGE. Silver staining revealed a single dominant protein with an apparent molecular mass of approximately 110 kDa eluting from beads coupled to PtdIns(4)P (Fig. 1A). The protein band was excised from a Coomassie brilliant blue-stained gel run in parallel (Fig. 1C), and the corresponding protein was identified by MALDI-TOF MS as the L. longbeachae SidC orthologue (SidCLlo). SidCLlo shares 40% overall identity with SidC from L. pneumophila (SidCLpn), while the paralogue of SidCLpn, SdcALpn (72% identity with SidCLpn [see Fig. S1 in the supplemental material]), is not present in L. longbeachae. As a control, PtdIns(4)P- or PtdIns-coupled beads were incubated with L. pneumophila lysates (Fig. 1B and D), and the ∼75-kDa protein specifically retained by the PI was identified as the major PtdIns(4)P-binding L. pneumophila effector SidM (43), which is lacking in L. longbeachae (9). Thus, SidCLlo represents the major PtdIns(4)P-binding protein of L. longbeachae.
FIG 1.

Identification of L. longbeachae SidC in a screen for PI-binding proteins. Shown are pulldowns of lysates from L. longbeachae wild-type (A and C) or L. pneumophila wild-type (B and D) strains using agarose beads coated with different PIs or PtdIns. Bacterial proteins retained by washed beads were separated by SDS-PAGE and visualized by staining with silver (A and B) or Coomassie brilliant blue (C and D). The dominant proteins (*) eluting from beads coated with PtdIns(4)P with an apparent molecular mass of ∼110 kDa (A and C) or ∼75 kDa (B and D) were identified by mass spectrometry as L. longbeachae SidC or L. pneumophila SidM, respectively. The proteins retained by PtdIns(4,5)P2-coated beads were heat shock protein 90 or elongation factor Tu. The data are representative of two independent experiments.
Binding of SidCLlo or SidCLlo-SidCLpn chimeras to PtdIns(4)P in vitro and identification of the PI-binding domain.
The PI-binding specificity of SidC was tested in vitro using commercially available PIP strips and PIP arrays, where PIs and other lipids are immobilized on nitrocellulose membranes. To this end, we heterologously produced GST fusion proteins of SidCLlo and fragments thereof, affinity purified the fusion proteins, and assayed binding by protein-lipid overlay assays using an anti-GST antibody. GST-SidCLlo specifically bound to PtdIns(4)P, but not to other PIs or lipids spotted onto a PIP strip (Fig. 2A). To map the PtdIns(4)P-binding domain of SidCLlo, we constructed N-terminal fusions of GST with fragments of the protein comprising amino acids 1 to 340, 341 to 608, 609 to 782, or 783 to 969, respectively (see Fig. S1 in the supplemental material). Among these fragments, only SidCLlo_609-782 bound to PtdIns(4)P (Fig. 2A). This fragment corresponds to the PtdIns(4)P-binding P4C domain of SidCLpn, which comprises the amino acids 609 to 776 (45). Therefore, the corresponding fragment of L. longbeachae SidC was termed SidCLlo_P4C. SidCLlo_P4C and SidCLpn_P4C are 45% identical on an amino acid level (see Fig. S1 in the supplemental material).
FIG 2.

Binding of SidCLlo or SidCLlo-SidCLpn chimeras to PtdIns(4)P in vitro and identification of the PI-binding domain. Shown is a protein-lipid overlay assay of 100 pmol/spot (A and C) or serial 2-fold dilutions (1.56 to 100 pmol/spot) (B and D) of the lipids indicated. The binding of affinity-purified GST-SidCLlo, GST-SidCLpn, and GST-SidCLlo-SidCLpn chimeras or GST-SidC fragments to lipids immobilized on nitrocellulose membranes was analyzed using an anti-GST antibody. LPA, lysophosphatidic acid; LPC, lysophosphocholine; PtdIns, phosphatidylinositol; PtdIns(x)P, phosphatidylinositol x-phosphate (phosphoinositide); PE, phosphatidylethanolamine; PC, phosphatidylcholine; S1P, sphingosine-1-phosphate; PA, phosphatidic acid; PS, phosphatidylserine. The data shown are representative of three (A and B) or two (C and D) independent experiments.
Next, we compared the binding of SidCLlo, SidCLlo_P4C, SidCLpn, and SidCLpn_P4C to PIs or PtdIns using PIP arrays, onto which the lipids were spotted in 2-fold dilution series. Using these PIP arrays, SidCLlo and SidCLlo_P4C were found to bind with apparently similar affinities almost exclusively to PtdIns(4)P and only very weakly to PtdIns(3)P (Fig. 2B). In contrast, under the same conditions, full-length SidCLpn did not bind to any PI, whereas SidCLpn_P4C specifically bound to PtdIns(4)P with an affinity similar to that of SidCLlo or SidCLlo_P4C, respectively. Compared to previously published results showing that SidCLpn binds to PtdIns(4)P (42, 45), we employed harsher washing conditions in the current experiments to remove background signal. The seemingly higher affinity of SidCLlo than SidCLpn for PtdIns(4)P might provide an explanation as to why, in bacterial lysates, only SidCLlo and not SidCLpn was precipitated by PtdIns(4)P-coupled agarose beads (Fig. 1) (43). Taking the data together, SidCLlo represents the major PtdIns(4)P-binding protein of L. longbeachae, which strongly interacts with the PI lipid through a P4C domain located near the C terminus.
To further explore the contribution of the N terminus of SidC to PtdIns(4)P binding, we constructed chimeric proteins comprising the N-terminal fragment of SidCLlo or SidCLpn (amino acids 1 to 608) and the C-terminal fragments of SidCLpn (aa 609 to 917) or SidCLlo (aa 609 to 969), respectively. The chimeric proteins SidCLlo_1-608-Lpn_609-917 and SidCLpn_1-608-Llo_609-969 were found to selectively bind PtdIns(4)P with an apparent affinity similar to that of SidCLlo using PIP strips (Fig. 2C) or PIP arrays (Fig. 2D). In summary, these results confirm that the C terminus of SidC, including the P4C domain, represents the major PtdIns(4)P-binding determinant, and the findings are in agreement with the notion that the N terminus does not significantly affect the binding affinity to the PI lipid.
Isothermal titration calorimetry and circular dichroism of SidCLlo and SidCLpn.
Full-length SidCLlo appears to bind PtdIns(4)P with high affinity and more strongly than SidCLpn (Fig. 2B). To quantify the binding affinities of the two proteins, we performed ITC using purified N-terminally His6-tagged proteins and synthetic di-octyl-PtdIns(4)P (Fig. 3A). The ITC studies revealed that SidCLlo binds di-octyl-PtdIns(4)P with a dissociation constant (Kd) of 71 nM compared to SidCLpn, binding the PI with a Kd of 243 nM. The 3.4-fold-lower Kd of SidCLlo for binding to di-octyl-PtdIns(4)P matches its higher affinity for di-hexadecanoyl-PtdIns(4)P observed in the protein-lipid overlay assay.
FIG 3.

Isothermal titration calorimetry and circular dichroism of SidCLlo and SidCLpn. (A) Isothermal titration calorimetry of 25 μM His6-SidCLlo or His6-SidCLpn in the presence of 250 μM di-C8-PtdIns(4)P. (B) Far-UV circular dichroism spectra of full-length SidCLlo (red) and SidCLpn (black). The signal unit is converted into mean residue weight ellipticity (MRW). The helical structure is evidenced by strong negative ellipticities at around 208 and 225 nm.
To investigate whether a different secondary structure could be the reason for these substantial differences in the PI-binding affinity, CD assays were performed (Fig. 3B). The CD measurements indicated similar secondary-structure elements for SidCLlo and SidCLpn, namely, α-helices (SidCLlo = 0.63; SidCLpn = 0.74), β-sheets (SidCLlo = 0.06; SidCLpn = 0.07), and loops (SidCLlo = 0.12; SidCLpn = 0.08). Hence, the overall composition and extent of secondary-structure elements does not account for the different binding affinities of the SidC homologues to PtdIns(4)P. In summary, SidCLlo shows an approximately 4-fold-higher affinity toward PtdIns(4)P than SidCLpn, yet the two proteins share similar overall secondary structures.
Icm/Dot-dependent localization of SidCLlo and SidCLpn on L. longbeachae LCVs.
SidCLlo and SidCLpn are 40% identical; however, a polyclonal antiserum against SidCLpn (42) does not recognize SidCLlo in Western blots, and conversely, a polyclonal antiserum against SidCLlo does not show any cross-reactivity against SidCLpn (Fig. 4A). Using the specific antibodies, we found that L. longbeachae heterologously produces SidCLpn and L. pneumophila produces SidCLlo (Fig. 4A).
FIG 4.
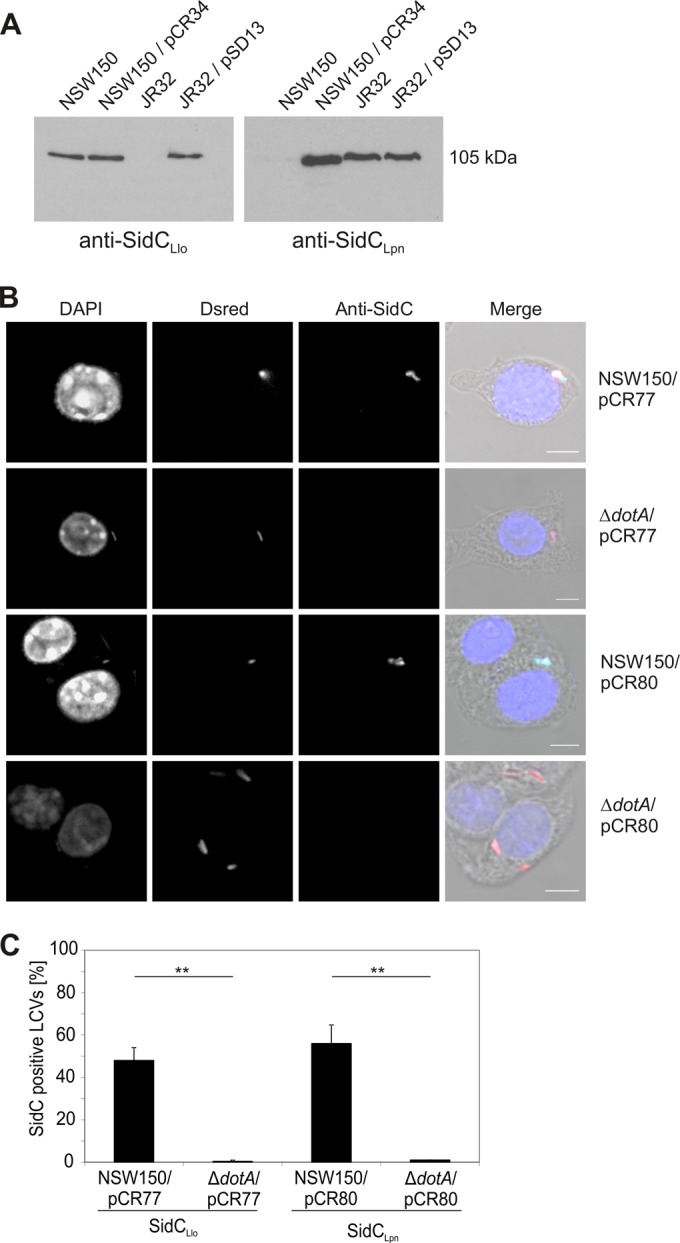
Icm/Dot-dependent localization of SidCLlo and SidCLpn on L. longbeachae LCVs. (A) Production of SidCLlo and SidCLpn in L. longbeachae or L. pneumophila was analyzed in L. longbeachae NSW150, L. pneumophila JR32, or the wild-type strains harboring pCR34 (M45-SidCLpn) or pSD13 (M45-SidCLlo). Overnight cultures were boiled, the proteins were separated by SDS-PAGE, and SidCLlo or SidCLpn was visualized by Western blotting using specific rabbit polyclonal anti-SidCLlo or anti-SidCLpn antibodies, which do not cross-react. The data are representative of three different experiments. (B) Confocal laser scanning micrographs of RAW 264.7 macrophages infected (MOI, 50) for 1 h with L. longbeachae wild-type NSW150 or a dotA mutant strain producing the red fluorescent protein Dsred (pCR77) or Dsred and SidCLpn (pCR80), fixed, and immunostained for SidC with specific anti-SidC antibodies (light blue). The DNA was stained with DAPI (4′,6-diamidino-2-phenylindole) (dark blue). Bars, 5 μm. (C) Percentages of LCVs decorated with either SidCLlo or SidCLpn. Means and standard deviations of three independent experiments scoring 50 LCVs each are shown (**, P < 0.01).
We then investigated whether L. longbeachae translocates SidCLlo and SidCLpn in an Icm/Dot-dependent manner and where the proteins localize in the host cell. To this end, we infected RAW 264.7 macrophages with red fluorescent L. longbeachae wild-type NSW150 or a ΔdotA mutant strain or with the corresponding strains producing SidCLpn. Immunofluorescence analysis of the infected macrophages revealed that endogenous SidCLlo, as well as heterologously produced SidCLpn, localizes to the LCV membrane in an Icm/Dot-dependent manner (Fig. 4B). Approximately 50 to 55% of the LCVs harboring wild-type L. longbeachae were decorated with either SidCLlo or SidCLpn, whereas less than 1% of the pathogen vacuoles containing ΔdotA mutant bacteria stained positive for Icm/Dot substrates (Fig. 4C). Together, these results indicate that SidCLlo is an Icm/Dot substrate localizing to the LCV membrane [likely to the cytoplasmic side, as PtdIns(4)P faces the host cell cytoplasm], and the L. longbeachae Icm/Dot T4SS translocates SidCLlo with an efficiency similar to that of SidCLpn.
L. longbeachae ΔsidC and L. pneumophila ΔsidC-sdcA are outcompeted by wild-type strains in amoebae.
To characterize the Icm/Dot substrate SidCLlo genetically, we constructed by allelic exchange a defined chromosomal deletion mutant of L. longbeachae strain NSW150 lacking the sidC gene (ΔsidC; strain IH02). To this end, the protocol routinely employed for L. pneumophila had to be adapted as outlined in Materials and Methods, since L. longbeachae produces a capsule (9), which greatly reduces the transformation efficiency.
Upon infection of A. castellanii amoebae (Fig. 5A) or RAW 264.7 macrophages (Fig. 5B) for 3 days, 3 or 2 orders of magnitude more CFU of L. longbeachae wild-type strain NSW150 were recovered, respectively. Whereas, the L. longbeachae ΔsidC mutant grew at the same rate as the wild-type strain, a ΔdotA mutant did not grow at all. However, upon coinfection of A. castellanii with L. longbeachae ΔsidC and NSW150, the mutant was outcompeted by the wild-type strain and was no longer detectable within 24 days of infection (Fig. 5C). Similarly, upon coinfection of A. castellanii with L. pneumophila ΔsidC-sdcA and the wild-type strain JR32, the mutant was outcompeted by the wild-type strain (Fig. 5D). The competition defect of the L. longbeachae ΔsidC mutant strain appeared to be weaker than the defect of the corresponding L. pneumophila ΔsidC-sdcA mutant, as the latter was no longer detectable after as little as 12 days. In summary, L. longbeachae or L. pneumophila lacking individual or multiple sidC paralogues grows at a wild-type rate in amoebae or macrophages but was outcompeted by the respective wild-type strains upon coinfection of A. castellanii.
FIG 5.

Intracellular growth and competition of L. longbeachae ΔsidC or L. pneumophila ΔsidC-sdcA. (A and B) For intracellular growth assays A. castellanii amoebae (A) or RAW 264.7 macrophages (B) were infected (MOI, 0.1) with single strains of L. longbeachae wild-type NSW150, ΔsidC, or ΔdotA, and CFU were determined at the time points indicated. (C and D) For competition assays, A. castellanii amoebae were coinfected (MOI, 0.01 each) with L. longbeachae ΔsidC and NSW150 (C) or L. pneumophila ΔsidC-sdcA and JR32 (D). The mutant strains were outcompeted by the respective wild-type strains. The data shown are representative of three independent experiments (A and B), or means and standard deviations of three independent experiments are shown (C and D).
LCVs harboring L. longbeachae ΔsidC are impaired for calnexin recruitment.
Pathogen vacuoles containing L. longbeachae acquire the ER, as substantiated by colocalization with the ER marker KDEL using fluorescence microscopy and by electron microscopy (17). D. discoideum amoebae producing the ER marker calnexin-GFP have been instrumental in analyzing the recruitment of the ER to pathogen vacuoles containing L. pneumophila and the role of the SidC paralogues in this process (42, 45, 49, 62).
To analyze a role of SidC in the formation of L. longbeachae-containing vacuoles, D. discoideum producing calnexin-GFP was infected for 2 h with red fluorescent L. longbeachae strains, and the recruitment of calnexin was observed in live amoebae (Fig. 6A). Whereas approximately 80% of the LCVs harboring wild-type L. longbeachae NSW150 acquired calnexin, the ER marker was absent from vacuoles containing ΔdotA mutant bacteria (Fig. 6B), indicating that the T4SS system is crucial for the formation of an ER-derived, replicative L. longbeachae compartment. In amoebae infected with L. longbeachae ΔsidC, only about 20% of LCVs acquired calnexin. Thus, SidCLlo promotes the recruitment of the ER to LCVs harboring L. longbeachae, similar to its function in L. pneumophila. Calnexin recruitment to LCVs was entirely restored by complementing the L. longbeachae ΔsidC mutant with different sidC paralogues, i.e., either SidCLlo, SidCLpn, or SdcALpn produced from plasmid-borne genes (Fig. 6B).
FIG 6.

LCVs harboring L. longbeachae ΔsidC are impaired for calnexin recruitment. (A and C) Confocal micrographs of D. discoideum amoebae producing calnexin (Caln.)-GFP and infected (MOI, 20) with L. longbeachae wild-type NSW150, ΔdotA, or ΔsidC (A) or L. pneumophila wild-type JR32, ΔicmT, or ΔsidC-sdcA (C) harboring pCR77 (DsRed), pSD14 (DsRed; SidCLlo), pCR80 (DsRed; SidCLpn), or pIH047 (DsRed; SdcALpn). Bars, 5 μm. (B and D) Percentages of calnexin-GFP-positive LCVs in live amoebae. Means and standard deviations of two independent experiments scoring 100 LCVs each are shown (*, P < 0.05).
We also analyzed whether the defect in calnexin acquisition of LCVs harboring L. pneumophila ΔsidC-sdcA mutant bacteria is complemented by SidCLlo. To this end, D. discoideum producing calnexin-GFP was infected for 1 h with red fluorescent L. pneumophila strains, and the recruitment of calnexin was tested in live amoebae (Fig. 6C). As previously observed, upon infection of the amoebae with wild-type L. pneumophila strain JR32, approximately 75% of LCVs stained calnexin positive in an Icm/Dot-dependent manner (45). In the absence of the two sidC paralogues, only less than 30% of LCVs acquired calnexin, and the recruitment defect was fully restored by producing not only SidCLpn, but also SidCLlo from plasmid-borne genes (Fig. 6D). In summary, these results indicate that L. longbeachae SidC promotes the recruitment of calnexin to LCVs, and SidCLlo, SidCLpn, or SdcALpn is functionally redundant.
The PtdIns(4)P probes P4CLlo and P4CLpn decorate LCVs.
To investigate whether the PtdIns(4)P-binding domain of SidCLlo, P4CLlo, decorates LCVs and might be useful as a PI-binding probe, we affinity purified the corresponding GST fusion protein and employed the probe to stain LCVs in lysates of D. discoideum producing calnexin-GFP infected with red fluorescent L. pneumophila (Fig. 7A). LCV binding of GST-P4CLlo was also compared to that of GST-P4CLpn (45) or GST-PHFAPP1 (63). Purified GST-P4CLlo stained more than 80% of the LCVs in homogenates in L. pneumophila-infected amoebae, which is similar to the percentage of LCVs stained by GST-P4CLpn or GST-PHFAPP1, used as positive controls (Fig. 7B). Purified GST was employed as a negative control and did not bind to any LCVs.
FIG 7.

The PtdIns(4)P probes P4CLlo and P4CLpn decorate LCVs. (A) D. discoideum amoebae producing calnexin-GFP were infected (MOI, 100; 1 h) with L. pneumophila producing DsRed, homogenized, fixed, incubated with the purified GST-tagged proteins indicated, and treated with an anti-GST antibody. Bars, 1 μm. (B) Percentages of GST-positive LCVs. Means and standard deviations of three independent experiments scoring 100 LCVs each are indicated (**, P < 0.01; ***, P < 0.001). (C) D. discoideum amoebae producing P4CLpn-GFP were infected (MOI, 20; 1 h) with L. longbeachae wild-type NSW150, ΔdotA, or ΔsidC harboring pCR77 (DsRed) or pSD14 (DsRed; SidCLlo), and acquisition of the PtdIns(4)P probe was analyzed in live amoebae. Bars, 10 μm. (D) Percentages of P4CLpn-positive LCVs. Means and standard deviations of three independent experiments scoring at least 70 LCVs each are indicated (***, P < 0.001).
Finally, to test whether L. longbeachae LCVs accumulate the PI lipid PtdIns(4)P, D. discoideum producing P4CLpn-GFP was infected with red fluorescent L. longbeachae wild-type NSW150, ΔdotA, or ΔsidC, and the recruitment of the PtdIns(4)P probe was assayed in live amoebae (Fig. 7C). Under these conditions, approximately 70% of the LCVs harboring wild-type L. longbeachae stained positive for the PtdIns(4)P marker, while only less than 20% of the vacuoles harboring the ΔdotA mutant strain accumulated the probe (Fig. 7D). The deletion of sidC or the overproduction of SidCLlo in the ΔsidC mutant did not affect the percentage of PtdIns(4)P-positive LCVs. Taken together, dependent on the Icm/Dot T4SS, L. longbeachae LCVs accumulate the PI lipid PtdIns(4)P, which is likely used as a membrane anchor for Icm/Dot-translocated effectors, such as SidC.
DISCUSSION
L. longbeachae and L. pneumophila cause clinically indistinguishable respiratory diseases, yet the ecological niches, modes of transmission, physiologies, and effector protein sets of the two Legionella species are significantly different. Here, we analyze the first effector protein of L. longbeachae by biochemical and genetic approaches. Using an unbiased pulldown assay, a homologue of the Icm/Dot substrate SidC was identified as the major PtdIns(4)P-binding L. longbeachae protein. SidCLlo binds PtdIns(4)P with high affinity through its P4C domain, localizes in an Icm/Dot-dependent manner to the PtdIns(4)P-positive LCV membrane, and promotes the acquisition of the ER in the pathogen vacuole.
SidCLlo was the only L. longbeachae effector protein candidate that bound to a PI lipid in pulldown assays using bacterial lysates. However, other L. longbeachae effector proteins might also interact with distinct PI lipids with lower affinity. L. pneumophila, on the other hand, produces several Icm/Dot substrates that bind to PtdIns(4)P or PtdIns(3)P, including the Rab1 GEF/AMPylase SidM (43), the ER interactor SidC and its paralogue SdcA (42), the glycosyltransferase SetA (41), and the retromer interactor RidL (31).
In pulldown experiments using L. pneumophila lysates, SidM was the only protein that bound to PI-coupled agarose beads (43). Even in the absence of SidM, i.e., in lysates from an L. pneumophila ΔsidM mutant strain, SidCLpn or other proteins were not precipitated by the beads. The binding affinity of SidCLpn to PtdIns(4)P is 3.4-fold lower than that of SidCLlo (Fig. 3). The lower binding affinity of SidCLpn likely accounts for the fact that the protein does not bind PtdIns(4)P-coupled beads in bacterial lysates, but at this point, we cannot rule out the possibility that another L. pneumophila protein masks the PtdIns(4)P-binding site of SidCLpn.
SidCLlo and SidCLpn show overall similar secondary-structure compositions, indicating that the differences in PtdIns(4)P-binding affinity are not due to large structural variances. The P4C domains of the two effectors share 45% identity (see Fig. S1 in the supplemental material), possibly accounting for the observed differences in PtdIns(4)P binding. However, in contrast to the full-length SidC proteins, the P4C domains bound PtdIns(4)P with similar apparent affinities (Fig. 2B). The affinities of the P4C domains for PtdIns(4)P could not be further quantified, due to the insolubility of the proteins at the high concentrations required for ITC measurements (data not shown). Chimeric SidC proteins comprising the N terminus of SidCLlo or SidCLpn and the reciprocal C terminus bound to PtdIns(4)P similarly to the corresponding P4C domains alone (Fig. 2D). These results indicate that the P4C domain represents the major, if not the only, PtdIns(4)P-binding determinant of SidC. Furthermore, assuming that the N termini of the SidC chimeras are correctly folded, the findings also suggest that the N terminus does not significantly affect the PI-binding affinity of the full-length SidC othologues. Thus, the amino acid exchanges in the P4C domains of SidCLlo and SidCLpn might preferentially account for the observed differences in binding to PtdIns(4)P.
Whereas the affinities of SidCLlo and SidCLpn for PtdIns(4)P differ in vitro, similar amounts of the endogenously or heterologously produced effectors bind to the L. longbeachae pathogen vacuole in infected macrophages (Fig. 4). Under the conditions used, L. longbeachae produced comparable amounts of endogenous SidCLlo and heterologous SidCLpn (Fig. 4A). These findings are in agreement with the notion that, in addition to PtdIns(4)P, the SidC paralogues might bind a (protein) coreceptor on the LCV membrane, which contributes to anchoring the effector to the LCV.
L. longbeachae lacking sidC was not defective for intracellular growth but was outcompeted by the corresponding wild-type strain, NSW150, in an amoeba competition assay (Fig. 5). Similarly, L. pneumophila ΔsidC-sdcA showed no intracellular growth phenotype (44, 45) but was outcompeted by the parental wild-type strain, JR32 (Fig. 5). Thus, the L. longbeachae ΔsidC mutant strain lacking a single Icm/Dot substrate grows intracellularly in the same robust manner as L. pneumophila strains lacking individual or whole families of paralogous effectors. Notably, L. longbeachae lacking SidC was outcompeted by wild-type bacteria more slowly than L. pneumophila lacking two SidC paralogues. The competition assay comprising dilution and reinfection steps is rather complex (see Materials and Methods) and probably selects for intracellular growth of the bacteria, as well as for persistence of the bacteria in the medium after their release from amoebae. It is conceivable that the two L. pneumophila SidC paralogues fulfill more pleiotropic functions during infection than the single L. longbeachae SidC effector, and accordingly, in a complex assay, the L. pneumophila deletion mutant would have a stronger phenotype.
LCVs harboring L. longbeachae ΔsidC are severely impaired for ER recruitment (Fig. 6). Interestingly, the phenotype is fully complemented, not only by providing the sidCLlo gene in trans, but also by sidCLpn or sdcALpn. The reciprocal complementation of the L. pneumophila ΔsidC-sdcA mutant by sidCLlo was equally efficient. Therefore, even though the orthologous SidC effectors share only 40% identity, they seem to be functionally redundant. In summary, the work documented in this study describes the biochemical and cell biological characterization of an L. longbeachae T4SS-translocated effector protein. Moreover, a protocol was established for the generation of defined L. longbeachae deletion mutant strains, which should be useful for a more comprehensive genetic and cellular analysis of L. longbeachae-phagocyte interactions.
Supplementary Material
ACKNOWLEDGMENTS
We thank Gudrun Pfaffinger for excellent technical assistance and Carmen Buchrieser (Institut Pasteur) for providing the L. longbeachae wild-type NSW150 and ΔdotA mutant strains.
The work of the H.H. group was funded by the Max von Pettenkofer Institute, Ludwig-Maximilians University Munich, the German Research Foundation (DFG) (HI 1511/1-1 and SPP1580), and the Swiss National Science Foundation (SNF) (31003A-125369). A.I., M.H., and A.C. acknowledge the DFG for generous financial support.
Footnotes
Published ahead of print 14 July 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.01685-14.
REFERENCES
- 1. Newton HJ, Ang DK, van Driel IR, Hartland EL. 2010. Molecular pathogenesis of infections caused by Legionella pneumophila. Clin. Microbiol. Rev. 23:274–298. 10.1128/CMR.00052-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gomez-Valero L, Rusniok C, Cazalet C, Buchrieser C. 2011. Comparative and functional genomics of Legionella identified eukaryotic like proteins as key players in host-pathogen interactions. Front. Microbiol. 2:208. 10.3389/fmicb.2011.00208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Whiley H, Bentham R. 2011. Legionella longbeachae and legionellosis. Emerg. Infect. Dis. 17:579–583. 10.3201/eid1704.100446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hilbi H, Hoffmann C, Harrison CF. 2011. Legionella spp. outdoors: colonization, communication and persistence. Environ. Microbiol. Rep. 3:286–296. 10.1111/j.1758-2229.2011.00247.x [DOI] [PubMed] [Google Scholar]
- 5. Hilbi H, Jarraud S, Hartland E, Buchrieser C. 2010. Update on Legionnaires' disease: pathogenesis, epidemiology, detection and control. Mol. Microbiol. 76:1–11. 10.1111/j.1365-2958.2010.07086.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Steele TW, Moore CV, Sangster N. 1990. Distribution of Legionella longbeachae serogroup 1 and other legionellae in potting soils in Australia. Appl. Environ. Microbiol. 56:2984–2988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rowbotham TJ. 1980. Preliminary report on the pathogenicity of Legionella pneumophila for freshwater and soil amoebae. J. Clin. Pathol. 33:1179–1183. 10.1136/jcp.33.12.1179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hoffmann C, Harrison CF, Hilbi H. 2014. The natural alternative: protozoa as cellular models for Legionella infection. Cell Microbiol. 16:15–26. 10.1111/cmi.12235 [DOI] [PubMed] [Google Scholar]
- 9. Cazalet C, Gomez-Valero L, Rusniok C, Lomma M, Dervins-Ravault D, Newton HJ, Sansom FM, Jarraud S, Zidane N, Ma L, Bouchier C, Etienne J, Hartland EL, Buchrieser C. 2010. Analysis of the Legionella longbeachae genome and transcriptome uncovers unique strategies to cause Legionnaires' disease. PLoS Genet. 6:e1000851. 10.1371/journal.pgen.1000851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kozak NA, Buss M, Lucas CE, Frace M, Govil D, Travis T, Olsen-Rasmussen M, Benson RF, Fields BS. 2010. Virulence factors encoded by Legionella longbeachae identified on the basis of the genome sequence analysis of clinical isolate D-4968. J. Bacteriol. 192:1030–1044. 10.1128/JB.01272-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Molofsky AB, Swanson MS. 2004. Differentiate to thrive: lessons from the Legionella pneumophila life cycle. Mol. Microbiol. 53:29–40. 10.1111/j.1365-2958.2004.04129.x [DOI] [PubMed] [Google Scholar]
- 12. Horwitz MA. 1983. Formation of a novel phagosome by the Legionnaires' disease bacterium (Legionella pneumophila) in human monocytes. J. Exp. Med. 158:1319–1331. 10.1084/jem.158.4.1319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Isberg RR, O'Connor TJ, Heidtman M. 2009. The Legionella pneumophila replication vacuole: making a cosy niche inside host cells. Nat. Rev. Microbiol. 7:13–24. 10.1038/nrmicro1967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hilbi H, Haas A. 2012. Secretive bacterial pathogens and the secretory pathway. Traffic 13:1187–1197. 10.1111/j.1600-0854.2012.01344.x [DOI] [PubMed] [Google Scholar]
- 15. Urwyler S, Nyfeler Y, Ragaz C, Lee H, Mueller LN, Aebersold R, Hilbi H. 2009. Proteome analysis of Legionella vacuoles purified by magnetic immunoseparation reveals secretory and endosomal GTPases. Traffic 10:76–87. 10.1111/j.1600-0854.2008.00851.x [DOI] [PubMed] [Google Scholar]
- 16. Hoffmann C, Finsel I, Otto A, Pfaffinger G, Rothmeier E, Hecker M, Becher D, Hilbi H. 2014. Functional analysis of novel Rab GTPases identified in the proteome of purified Legionella-containing vacuoles from macrophages. Cell Microbiol. 16:1034–1052. 10.1111/cmi.12256 [DOI] [PubMed] [Google Scholar]
- 17. Asare R, Abu Kwaik Y. 2007. Early trafficking and intracellular replication of Legionella longbeachae within an ER-derived late endosome-like phagosome. Cell Microbiol. 9:1571–1587. 10.1111/j.1462-5822.2007.00894.x [DOI] [PubMed] [Google Scholar]
- 18. Cazalet C, Jarraud S, Ghavi-Helm Y, Kunst F, Glaser P, Etienne J, Buchrieser C. 2008. Multigenome analysis identifies a worldwide distributed epidemic Legionella pneumophila clone that emerged within a highly diverse species. Genome Res. 18:431–441. 10.1101/gr.7229808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Segal G, Purcell M, Shuman HA. 1998. Host cell killing and bacterial conjugation require overlapping sets of genes within a 22-kb region of the Legionella pneumophila genome. Proc. Natl. Acad. Sci. U. S. A. 95:1669–1674. 10.1073/pnas.95.4.1669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Vogel JP, Andrews HL, Wong SK, Isberg RR. 1998. Conjugative transfer by the virulence system of Legionella pneumophila. Science 279:873–876. 10.1126/science.279.5352.873 [DOI] [PubMed] [Google Scholar]
- 21. Hubber A, Roy CR. 2010. Modulation of host cell function by Legionella pneumophila type IV effectors. Annu. Rev. Cell Dev. Biol. 26:261–283. 10.1146/annurev-cellbio-100109-104034 [DOI] [PubMed] [Google Scholar]
- 22. Zhu W, Banga S, Tan Y, Zheng C, Stephenson R, Gately J, Luo ZQ. 2011. Comprehensive identification of protein substrates of the Dot/Icm type IV transporter of Legionella pneumophila. PLoS One 6:e17638. 10.1371/journal.pone.0017638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lifshitz Z, Burstein D, Peeri M, Zusman T, Schwartz K, Shuman HA, Pupko T, Segal G. 2013. Computational modeling and experimental validation of the Legionella and Coxiella virulence-related type-IVB secretion signal. Proc. Natl. Acad. Sci. U. S. A. 110:E707–E715. 10.1073/pnas.1215278110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Itzen A, Goody RS. 2011. Covalent coercion by Legionella pneumophila. Cell Host Microbe 10:89–91. 10.1016/j.chom.2011.08.002 [DOI] [PubMed] [Google Scholar]
- 25. Xu L, Luo ZQ. 2013. Cell biology of infection by Legionella pneumophila. Microbes Infect. 15:157–167. 10.1016/j.micinf.2012.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sherwood RK, Roy CR. 2013. A Rab-centric perspective of bacterial pathogen-occupied vacuoles. Cell Host Microbe 14:256–268. 10.1016/j.chom.2013.08.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rothmeier E, Pfaffinger G, Hoffmann C, Harrison CF, Grabmayr H, Repnik U, Hannemann M, Wölke S, Bausch A, Griffiths G, Müller-Taubenberger A, Itzen A, Hilbi H. 2013. Activation of Ran GTPase by a Legionella effector promotes microtubule polymerization, pathogen vacuole motility and infection. PLoS Pathog. 9:e1003598. 10.1371/journal.ppat.1003598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Simon S, Wagner MA, Rothmeier E, Müller-Taubenberger A, Hilbi H. 2014. Icm/Dot-dependent inhibition of phagocyte migration by Legionella is antagonized by a translocated Ran GTPase activator. Cell Microbiol. 16:977–992. 10.1111/cmi.12258 [DOI] [PubMed] [Google Scholar]
- 29. Xu L, Shen X, Bryan A, Banga S, Swanson MS, Luo ZQ. 2010. Inhibition of host vacuolar H+-ATPase activity by a Legionella pneumophila effector. PLoS Pathog. 6:e1000822. 10.1371/journal.ppat.1000822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Choy A, Dancourt J, Mugo B, O'Connor TJ, Isberg RR, Melia TJ, Roy CR. 2012. The Legionella effector RavZ inhibits host autophagy through irreversible Atg8 deconjugation. Science 338:1072–1076. 10.1126/science.1227026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Finsel I, Ragaz C, Hoffmann C, Harrison CF, Weber S, van Rahden VA, Johannes L, Hilbi H. 2013. The Legionella effector RidL inhibits retrograde trafficking to promote intracellular replication. Cell Host Microbe 14:38–50. 10.1016/j.chom.2013.06.001 [DOI] [PubMed] [Google Scholar]
- 32. Hilbi H, Weber S, Finsel I. 2011. Anchors for effectors: subversion of phosphoinositide lipids by Legionella. Front. Microbiol. 2:91. 10.3389/fmicb.2011.00091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Haneburger I, Hilbi H. 2013. Phosphoinositide lipids and the Legionella pathogen vacuole. Curr. Top. Microbiol. Immunol. 376:155–173. 10.1007/82_2013_341 [DOI] [PubMed] [Google Scholar]
- 34. Di Paolo G, De Camilli P. 2006. Phosphoinositides in cell regulation and membrane dynamics. Nature 443:651–657. 10.1038/nature05185 [DOI] [PubMed] [Google Scholar]
- 35. Michell RH. 2008. Inositol derivatives: evolution and functions. Nat. Rev. Mol. Cell. Biol. 9:151–161. 10.1038/nrm2334 [DOI] [PubMed] [Google Scholar]
- 36. Pizarro-Cerda J, Cossart P. 2004. Subversion of phosphoinositide metabolism by intracellular bacterial pathogens. Nat. Cell Biol. 6:1026–1033. 10.1038/ncb1104-1026 [DOI] [PubMed] [Google Scholar]
- 37. Weber SS, Ragaz C, Hilbi H. 2009. Pathogen trafficking pathways and host phosphoinositide metabolism. Mol. Microbiol. 71:1341–1352. 10.1111/j.1365-2958.2009.06608.x [DOI] [PubMed] [Google Scholar]
- 38. Hsu F, Zhu W, Brennan L, Tao L, Luo ZQ, Mao Y. 2012. Structural basis for substrate recognition by a unique Legionella phosphoinositide phosphatase. Proc. Natl. Acad. Sci. U. S. A. 109:13567–13572. 10.1073/pnas.1207903109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Toulabi L, Wu X, Cheng Y, Mao Y. 2013. Identification and structural characterization of a Legionella phosphoinositide phosphatase. J. Biol. Chem. 288:24518–24527. 10.1074/jbc.M113.474239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Weber SS, Ragaz C, Hilbi H. 2009. The inositol polyphosphate 5-phosphatase OCRL1 restricts intracellular growth of Legionella, localizes to the replicative vacuole and binds to the bacterial effector LpnE. Cell Microbiol. 11:442–460. 10.1111/j.1462-5822.2008.01266.x [DOI] [PubMed] [Google Scholar]
- 41. Jank T, Bohmer KE, Tzivelekidis T, Schwan C, Belyi Y, Aktories K. 2012. Domain organization of Legionella effector SetA. Cell Microbiol. 14:852–868. 10.1111/j.1462-5822.2012.01761.x [DOI] [PubMed] [Google Scholar]
- 42. Weber SS, Ragaz C, Reus K, Nyfeler Y, Hilbi H. 2006. Legionella pneumophila exploits PI(4)P to anchor secreted effector proteins to the replicative vacuole. PLoS Pathog. 2:e46. 10.1371/journal.ppat.0020046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Brombacher E, Urwyler S, Ragaz C, Weber SS, Kami K, Overduin M, Hilbi H. 2009. Rab1 guanine nucleotide exchange factor SidM is a major phosphatidylinositol 4-phosphate-binding effector protein of Legionella pneumophila. J. Biol. Chem. 284:4846–4856. 10.1074/jbc.M807505200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Luo ZQ, Isberg RR. 2004. Multiple substrates of the Legionella pneumophila Dot/Icm system identified by interbacterial protein transfer. Proc. Natl. Acad. Sci. U. S. A. 101:841–846. 10.1073/pnas.0304916101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ragaz C, Pietsch H, Urwyler S, Tiaden A, Weber SS, Hilbi H. 2008. The Legionella pneumophila phosphatidylinositol-4 phosphate-binding type IV substrate SidC recruits endoplasmic reticulum vesicles to a replication-permissive vacuole. Cell Microbiol. 10:2416–2433. 10.1111/j.1462-5822.2008.01219.x [DOI] [PubMed] [Google Scholar]
- 46. Weber S, Dolinsky S, Hilbi H. 2013. Interactions of Legionella effector proteins with host phosphoinositide lipids. Methods Mol. Biol. 954:367–380. 10.1007/978-1-62703-161-5_23 [DOI] [PubMed] [Google Scholar]
- 47. Lemmon MA. 2008. Membrane recognition by phospholipid-binding domains. Nat. Rev. Mol. Cell. Biol. 9:99–111. 10.1038/nrm2328 [DOI] [PubMed] [Google Scholar]
- 48. Varnai P, Balla T. 2006. Live cell imaging of phosphoinositide dynamics with fluorescent protein domains. Biochim. Biophys. Acta 1761:957–967. 10.1016/j.bbalip.2006.03.019 [DOI] [PubMed] [Google Scholar]
- 49. Weber S, Wagner M, Hilbi H. 2013. Live cell imaging of phosphoinositide dynamics and membrane architecture during Legionella infection. mBio 5:e00839-13. 10.1128/mBio.00839-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Gazdag EM, Schobel S, Shkumatov AV, Goody RS, Itzen A. 2014. The structure of the N-terminal domain of the Legionella protein SidC. J. Struct. Biol. 186:188–194. 10.1016/j.jsb.2014.02.003 [DOI] [PubMed] [Google Scholar]
- 51. Horenkamp FA, Mukherjee S, Alix E, Schauder CM, Hubber AM, Roy CR, Reinisch KM. 2014. Legionella pneumophila subversion of host vesicular transport by SidC effector proteins. Traffic 15:488–499. 10.1111/tra.12158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Schoebel S, Blankenfeldt W, Goody RS, Itzen A. 2010. High-affinity binding of phosphatidylinositol 4-phosphate by Legionella pneumophila DrrA. EMBO Rep. 11:598–604. 10.1038/embor.2010.97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Otto GP, Wu MY, Clarke M, Lu H, Anderson OR, Hilbi H, Shuman HA, Kessin RH. 2004. Macroautophagy is dispensable for intracellular replication of Legionella pneumophila in Dictyostelium discoideum. Mol. Microbiol. 51:63–72. 10.1128/mBio.00839-13 [DOI] [PubMed] [Google Scholar]
- 54. Moffat JF, Tompkins LS. 1992. A quantitative model of intracellular growth of Legionella pneumophila in Acanthamoeba castellanii. Infect. Immun. 60:296–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Segal G, Shuman HA. 1999. Legionella pneumophila utilizes the same genes to multiply within Acanthamoeba castellanii and human macrophages. Infect. Immun. 67:2117–2124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Feeley JC, Gibson RJ, Gorman GW, Langford NC, Rasheed JK, Mackel DC, Baine WB. 1979. Charcoal-yeast extract agar: primary isolation medium for Legionella pneumophila. J. Clin. Microbiol. 10:437–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Jeong JY, Yim HS, Ryu JY, Lee HS, Lee JH, Seen DS, Kang SG. 2012. One-step sequence- and ligation-independent cloning as a rapid and versatile cloning method for functional genomics studies. Appl. Environ. Microbiol. 78:5440–5443. 10.1128/AEM.00844-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Dowler S, Kular G, Alessi DR. 2002. Protein lipid overlay assay. Sci. STKE 2002:pl6. 10.1126/stke.2002.129.pl6 [DOI] [PubMed] [Google Scholar]
- 59. Johnson WC. 1999. Analyzing protein circular dichroism spectra for accurate secondary structures. Proteins 35:307–312 [PubMed] [Google Scholar]
- 60. Tiaden A, Spirig T, Weber SS, Brüggemann H, Bosshard R, Buchrieser C, Hilbi H. 2007. The Legionella pneumophila response regulator LqsR promotes host cell interactions as an element of the virulence regulatory network controlled by RpoS and LetA. Cell Microbiol. 9:2903–2920. 10.1111/j.1462-5822.2007.01005.x [DOI] [PubMed] [Google Scholar]
- 61. Kessler A, Schell U, Sahr T, Tiaden A, Harrison C, Buchrieser C, Hilbi H. 2013. The Legionella pneumophila orphan sensor kinase LqsT regulates competence and pathogen-host interactions as a component of the LAI-1 circuit. Environ. Microbiol. 15:646–662. 10.1111/j.1462-2920.2012.02889.x [DOI] [PubMed] [Google Scholar]
- 62. Lu H, Clarke M. 2005. Dynamic properties of Legionella-containing phagosomes in Dictyostelium amoebae. Cell Microbiol. 7:995–1007. 10.1111/j.1462-5822.2005.00528.x [DOI] [PubMed] [Google Scholar]
- 63. De Matteis MA, Godi A. 2004. PI-loting membrane traffic. Nat. Cell Biol. 6:487–492. 10.1038/ncb0604-487 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.