

Abstract
To persist and cause disease in the host, Mycobacterium tuberculosis must adapt to its environment during infection. Adaptations include changes in nutrient utilization and alterations in growth rate. M. tuberculosis Rv1422 is a conserved gene of unknown function that was found in a genetic screen to interact with the mce4 cholesterol uptake locus. The Rv1422 protein is phosphorylated by the M. tuberculosis Ser/Thr kinases PknA and PknB, which regulate cell growth and cell wall synthesis. Bacillus subtilis strains lacking the Rv1422 homologue yvcK grow poorly on several carbon sources, and yvcK is required for proper localization of peptidoglycan synthesis. Here we show that Mycobacterium smegmatis and M. tuberculosis strains lacking Rv1422 have growth defects in minimal medium containing limiting amounts of several different carbon sources. These strains also have morphological abnormalities, including shortened and bulging cells, suggesting a cell wall defect. In both mycobacterial species, the Rv1422 protein localizes uniquely to the growing cell pole, the site of peptidoglycan synthesis in mycobacteria. An M. tuberculosis ΔRv1422 strain is markedly attenuated for virulence in a mouse infection model, where it elicits decreased inflammation in the lungs and shows impaired bacterial persistence. These findings led us to name this gene cuvA (carbon utilization and virulence protein A) and to suggest a model in which deletion of cuvA leads to changes in nutrient uptake and/or metabolism that affect cell wall structure, morphology, and virulence. Its role in virulence suggests that CuvA may be a useful target for novel inhibitors of M. tuberculosis during infection.
INTRODUCTION
Regulated pathways that allow physiologic adaptations in response to changes in the environment are critical for the growth and survival of all bacteria. In the case of the human pathogen Mycobacterium tuberculosis, where infection can persist for decades and cause several different disease manifestations, the bacterium must adapt to multiple environments during the prolonged course of infection. These environments include intracellular milieus within host phagocytic cells, such as neutrophils, macrophages, and dendritic cells, which may be part of organized granulomas, as well as in extracellular environments such as those occurring in caseating granulomas. In addition to mechanisms to defend against and modulate host antibacterial activities, a critical adaptation of M. tuberculosis during infection is utilization of host lipids as a primary carbon source (1–3). Another key bacterial adaptation during M. tuberculosis infection is regulation of replication, which changes from rapid growth during the first few weeks of infection to markedly reduced bacterial replication following the onset of adaptive immunity (3, 4). Gaining insight into bacterial adaptive mechanisms is critical for understanding tuberculosis pathogenesis and may lead to novel therapeutic approaches that target these bacterial adaptive processes.
M. tuberculosis Rv1422 encodes a protein of unknown function that has homologues in other mycobacteria and across a broad range of bacterial phyla. Our interest in this protein as potentially having an important role in bacterial adaptation during infection was motivated by three observations. One is our previous finding that it is phosphorylated in M. tuberculosis cells by the Ser/Thr protein kinases PknA and PknB. These kinases are essential for growth and regulate cell division, peptidoglycan (PGN) synthesis, and cell morphology (5–9), suggesting that the Rv1422 protein may play a role in one or more of these processes. Second, Rv1422 was identified in a genome-wide transposon screen for genetic interactions with the mce1 and mce4 loci. The mce4 locus encodes a cholesterol uptake system (2, 10), and the mce1 locus has been implicated in the transport of other lipids (11), suggesting that the Rv1422 protein may function in utilization of cholesterol or other lipids. In addition, an M. tuberculosis strain with a transposon insertion in Rv1422 was defective for replication in mouse spleens in an 8-week infection model, suggesting that Rv1422 might also be important in M. tuberculosis virulence (12).
An orthologue of Rv1422, yvcK, has been investigated in the model organism Bacillus subtilis, where a null mutant of this gene was found to have partial growth defects on media containing several different carbon sources, including tricarboxylic acid cycle intermediates (13). Upon transfer to medium containing carbon sources in which this organism grew poorly, bulging and lysis of cells were seen, indicating perturbation of PGN synthesis. Subsequently, YvcK was found to be distributed in a circumferential pattern around the long axis of the cell in B. subtilis, paralleling the distribution of PGN synthesis (14). Overexpression of MreB, a filament-forming protein that is similarly distributed around the circumference of the cell (15), was shown to suppress the carbon source-specific growth and morphology defects of the yvcK null mutant. In addition, the PGN synthesis protein PBP1 was found to be mislocalized in the ΔyvcK strain, in a carbon source-dependent manner (14). Thus, although its mechanism of action is not known, YvcK appears to be required for proper localization of PGN synthesis and production of a normal cell wall in B. subtilis.
These data from M. tuberculosis and B. subtilis led us to ask whether Rv1422 plays a role in carbon source utilization, cell growth, and virulence in mycobacteria. We identified growth defects on multiple carbon sources for deletion mutants of M. tuberculosis Rv1422 and the Mycobacterium smegmatis homologue MSMEG_3080 compared to the wild-type strains. Because of the importance of cholesterol as a carbon source for M. tuberculosis during infection (2, 16, 17), we used cholesterol as a representative carbon source for additional investigation of the function of Rv1422. When the M. smegmatis and M. tuberculosis mutant strains were grown on cholesterol, both had morphological abnormalities, including bulging and shortened cell length, indicating a role for Rv1422 in cell wall synthesis. We also show a striking localization of Rv1422 protein-green fluorescent protein (GFP) fusions exclusively to the growing cell pole, the site of PGN synthesis in mycobacteria (8), and demonstrate that the M. smegmatis deletion strain is hypersusceptible to several β-lactam antibiotics when grown on cholesterol, findings that link the Rv1422 protein to cell growth and cell wall synthesis. Consistent with these in vitro phenotypes, the virulence of the M. tuberculosis mutant was markedly attenuated in a mouse infection model. Based on these results, we have named this gene cuvA (carbon utilization and virulence protein A).
MATERIALS AND METHODS
Strains, media, plasmids, and primers.
M. tuberculosis H37Rv and M. smegmatis mc2155 (18) were used as wild-type strains and were the parental strains in which mutants were made. Escherichia coli TOP10 (Invitrogen) was used for cloning and was grown in LB broth. For routine growth, M. tuberculosis and M. smegmatis were grown at 37°C in Middlebrook 7H9 liquid medium (Difco) supplemented with 0.5% albumin, 0.2% glucose, 0.085% NaCl, 0.2% glycerol, and 0.05% Tween 80 (7H9-ADC-Tw). Kanamycin (20 μg ml−1) or hygromycin (50 μg ml−1) was added to liquid or agar medium when appropriate. Details of primers, plasmids, and strains are shown in Table S1 in the supplemental material.
For growth of M. smegmatis on different carbon sources in liquid medium, cells were grown in minimal medium (MM) (1 g/liter KH2PO4, 2.5 g/liter Na2HPO4, 0.5 g/liter asparagine, 0.5 g/liter MgSO4 · 7H2O, 0.5 mg/liter CaCl2, 0.1 mg/liter ZnSO4, 50 mg/liter ferric ammonium citrate, and 0.05% tyloxapol) (2) containing 0.01% (wt/vol) glucose, glycerol, cholesterol, propionate, gluconate, or citrate. MM plus 0.2% glucose was used as a positive control for maximal growth. For measurements of growth on solid medium, cells grown in Middlebrook 7H9 liquid medium to an optical density at 600 nm (OD600) of 0.5 were washed in phosphate-buffered saline containing 0.05% tyloxapol (PBS-Tx), and 10-fold serial dilutions in PBS were spotted on agar plates containing MM as described above (without tyloxapol) plus a 0.01% concentration of the carbon source. MM plates containing 0.2% glucose were included as a control. Plates were incubated at 37°C and photographed after 1 month for M. tuberculosis and after 2 days for M. smegmatis.
For subcellular localization, CuvA was expressed as a carboxy-terminal GFP fusion. cuvA was PCR amplified from genomic DNA of M. tuberculosis H37RV by use of primers Rv1422AE and Rv1422rPac1. gfp was PCR amplified from pTracerCMV (Invitrogen) by use of primers GFP3PacI and GFP4XK. Overlap PCR was then carried out using primers Rv1422AE and GFP4XK, giving rise to the fusion PCR product Rv1422-gfp. The Rv1422-gfp PCR product was digested with AseI and XbaI (the AseI site in the Rv1422 gene was eliminated by overlap PCR mutagenesis to create a silent mutation) and cloned 3′ of the acetamidase promoter in the integrating vector pMV306AC to obtain pMV306Ac-Rv1422-gfp (pMV306Ac-cuvA-gfp).
For localization of M. smegmatis PBP1 (encoded by MSMEG_6900), a C-terminal MSMEG_6900-red fluorescent protein (RFP) fusion protein was expressed. The MSMEG_6900 gene was amplified by PCR from M. smegmatis genomic DNA by use of primers MSMEG6900-F and MSMEG6900-R. Similarly, rfp was PCR amplified using primers Rfp_F and Rfp_R. Overlap PCR was then carried out using primers MSMEG6900-F and Rfp_R. The resulting fusion PCR product was cloned into the NdeI and XbaI sites of pMV261Ac, 3′ of the acetamidase promoter, to obtain pMV261Ac-MSMEG6900-rfp. All recombinant clones used in this study were sequenced to rule out any mutations.
Generation of M. smegmatis and M. tuberculosis cuvA deletion strains.
To delete cuvA (MSMEG_3080) from M. smegmatis, a 1,008-bp region (L arm) 5′ to MSMEG_3080, including 6 codons of MSMEG_3080, was PCR amplified using primers MSMEG3080-cond-1 and MSMEG3080-cond-2. Similarly, a 1,040-bp region (R arm) 3′ to MSMEG_3080, including 100 bp of the MSMEG_3080 gene, was PCR amplified using primers MSMEG3080-cond-3 and MSMEG3080-cond-4. Overlap PCR was then carried out with primers MSMEG3080-cond-1 and MSMEG3080-cond-4. The PCR product contained a PacI site between the arms and was cloned into pRH1351 (19). A hygromycin resistance cassette was introduced at the PacI site, and the targeting construct thus obtained was electroporated into M. smegmatis mc2155. Candidate deletion strains were obtained using a two-step double-counterselection method as previously described (19, 20). PCR amplification of genomic DNAs from candidate mutant strains, using primers annealing to flanking regions of MSMEG_3080, yielded the expected 1.5-kb PCR product, confirming the replacement of MSMEG_3080 (cuvA) in the genome by the hygromycin cassette (see Fig. S1 in the supplemental material). For complementation of the M. smegmatis cuvA deletion strain, cuvA was PCR amplified using primers MSMEG3080-int-1 and MSMEG3080-int-2 and cloned under the control of the tet promoter in pMind (21). This complemented strain was grown in the presence of 5 ng/ml tetracycline to induce expression of cuvA.
The cuvA (Rv1422) gene was deleted from the M. tuberculosis H37Rv genome by use of similar methods. One-kilobase regions 5′ and 3′ to Rv1422 were PCR amplified and ligated to the 5′ and 3′ ends, respectively, of a hygromycin-chloramphenicol cassette. The ligated DNA fragment was introduced into a temperature-sensitive mycobacterial suicide vector harboring the sacB and xylE genes. The targeting vector was transformed into wild-type M. tuberculosis H37Rv, and candidate deletion strains were obtained by counterselection on sucrose-containing plates at 38°C. Deletion of cuvA was confirmed by PCR amplification of genomic DNA, using primers annealing to flanking regions of the gene. The expected size of 2.1 kb, resulting from the replacement of cuvA by the hygromycin-chloramphenicol cassette, confirmed the deletion of the gene (see Fig. S1 in the supplemental material). The complementing construct was obtained by cloning cuvA downstream of the hsp70 promoter in the integrating vector pJEB402 (22).
Microplate alamarBlue assay (MABA) of growth.
M. smegmatis strains were shaken at 37°C in 7H9-ADC-Tw (without glycerol) until they reached an OD600 of ∼0.5. The cells were spun down, washed twice in PBS-Tx (PBS plus 0.05% tyloxapol), and diluted to a calculated OD600 of 0.001 in MM alone or with added 0.2% glucose or one of the following added to 0.01%: glucose, cholesterol, glycerol, gluconate, citrate, or propionate. Tetracycline (5 ng/ml) was present in all cultures. Two hundred microliters of each culture was placed in a 96-well clear-bottom plate, and 20 μl filtered alamarBlue (Life Technologies) was added. Fluorescence (excitation/emission wavelengths = 550 nm/590 nm) was measured every hour from the bottom of the well in a plate reader (Tecan), with incubation at 37°C. The exponential-phase portions of the growth curves were analyzed by nonlinear curve fitting to an exponential growth model, and the rate constants were compared by an extra-sum-of-squares F test, using GraphPad Prism 5.0 software.
MABA susceptibility determination.
M. smegmatis cultures were prepared as described above for the MABA growth curves, except that they were diluted to a calculated OD600 of 0.005 in MM containing 0.01% cholesterol. Glucose was added to half of the cultures to obtain cultures with 0.01% cholesterol plus 0.2% glucose; the other cultures contained 0.01% cholesterol only. To half of the cholesterol-only cultures and half of the cholesterol-plus-glucose cultures, clavulanate was added to a final concentration of 2.5 μg/ml. For each antibiotic tested, 2-fold dilutions were made, and M. smegmatis cultures (190 μl) and antibiotics (10 μl) were mixed in clear 96-well plates. After overnight (for glucose plus cholesterol) or 2-day (for cholesterol only) incubation, 50 μl of 0.5× alamarBlue reagent diluted in 10% Tween 80 was added. After overnight incubation, fluorescence (excitation/emission wavelengths = 550 nm/590 nm) was measured. Growth inhibition was calculated using the following formula: % inhibition = {1 − [(F − Fmin)/(Fmax − Fmin)]} × 100, where Fmin is the fluorescence from wells without growth and Fmax is the fluorescence from control wells containing no antibiotic. The MIC90 was defined as the concentration of antibiotic that caused 90% growth inhibition (23).
M. tuberculosis H37Rv was tested for antibiotic susceptibility in a similar way, except that cultures were preincubated in PBS-Tx for 2 days and then diluted to an OD600 of 0.05, and the clavulanate concentration was 7.5 μg/ml. Plates were incubated for 1 week before alamarBlue was added and analyzed as described above. The final concentration ranges of the antibiotics tested were as follows: rifampin (M. tuberculosis), 0.003 to 0.64 μg/ml; rifampin (M. smegmatis), 0.3 to 80 μg/ml; levofloxacin, 0.01 to 2.56 μg/ml; ampicillin, 4 to 1,000 μg/ml; cefotaxime, 4 to 1,000 μg/ml; cephalothin, 2 to 500 μg/ml; and meropenem, 0.2 to 50 μg/ml. For both M. smegmatis and M. tuberculosis, each antibiotic was tested in at least three biological replicates, with technical duplicates for every experiment. MICs between strains were compared using the t test in Microsoft Excel.
Microscopy and fluorescent vancomycin staining.
For cellular localization of CuvA, the recombinant clone pMV306-pacet-Rv1422-gfp was transformed into M. smegmatis strain mc2155 and into an M. tuberculosis leucine-pantothenate double-auxotrophic strain (18, 24). The pMV306-pacet-gfp plasmid, expressing GFP alone, was transformed into M. smegmatis as a control. The M. smegmatis strains were grown to an OD600 of 0.1 to 0.2 in 7H9 broth, induced with 0.2% acetamide for 6 to 8 h, and visualized by microscopy. The M. tuberculosis strains were grown to an OD600 of 0.5 and inoculated onto a layer of nutrient agarose medium on microscope slides (agarose pads) made with Middlebrook 7H9 medium containing glucose and 0.2% acetamide. Cells on these agarose pads were observed 24 h after growth.
For staining of sites of peptidoglycan synthesis, vancomycin-Alexa 568 (Van-Alexa 568; Molecular Probes) was prepared and used as previously described (8). Van-Alexa 568 was added to cultures at an OD600 of 0.1 to 0.2 to achieve a final concentration of 5 μg/ml, and growth was continued for another 2.5 h in the dark. The cells were washed, transferred onto a glass slide, and air dried, and a coverslip was mounted using Prolong Gold antifade reagent (Invitrogen). Cells were observed using a Zeiss AxioImager.Z2 microscope with a 63× differential interference contrast (DIC) oil-immersion objective. For fluorescence, green and red fluorescence filters were used. Images were captured by use of a CoolSNAP HQ2 camera (Photometrics), acquired with AxiVision 4.8 software, and processed by Adobe Photoshop CS5.
For examination of cell morphology, cells were grown in the indicated medium, pelleted, and fixed in 4% paraformaldehyde prepared in PBS for 2 h. Ammonium chloride was then added to a final concentration of 50 mM, and cells were pelleted and then resuspended in PBS. Five microliters of the cell suspension was spotted on a glass slide, which was air dried, mounted with a coverslip, and observed using either a 63× or 100× differential interference contrast oil-immersion objective. Images were processed using Adobe Photoshop CS5. Measurements of cell length were performed using ImageJ software, and statistical analysis of cell length was performed with one-way analysis of variance (ANOVA) and the nonparametric Kruskal-Wallis test, using GraphPad Prism software. For examination of live M. smegmatis, cells were grown overnight on agarose pads on glass microscope slides. The cells were then directly observed on the agarose pads.
Transposon mutagenesis.
To identify mutations that suppress the cholesterol growth defect of the ΔcuvA strain of M. tuberculosis, we performed transposon mutagenesis using the φMycoMarT7 phagemid (25, 26). Preparation of the phage stock, its titration, and subsequent transduction of the cuvA deletion strain were carried out as described previously (27). Approximately 120,000 transductants were plated on cholesterol-agar plates containing 20 μg/ml kanamycin and incubated at 37°C. After 5 weeks, 60 colonies were streaked on cholesterol-agar plates, and 20 showed substantial growth. Genomic DNA was isolated, and three restriction enzymes (BamHI, SphI, and SacII) were used individually to completely digest the genomic DNA. The recognition sites of these enzymes are absent in the transposon. The fragmented DNAs were purified and treated with T4 DNA ligase to circularize the fragments. PCR amplification was then carried out using outward primers annealing to the ends of the transposon. The PCR products were analyzed by agarose gel electrophoresis, and strong-intensity bands were extracted and sequenced to identify the site of transposon insertion.
Mouse infection experiments.
Mouse infection experiments were performed by the NIAID-sponsored Tuberculosis Animal Research and Gene Evaluation Task Force (TARGET). Mouse infection protocols were approved by the Animal Care and Use Committee at Johns Hopkins School of Medicine, Baltimore, MD, and all experiments involving mice were carried out according to these protocols.
To determine the effect of cuvA deletion on growth of M. tuberculosis in vivo, wild-type H37Rv, the cuvA deletion strain (RH478), and the complemented ΔcuvA strain (RH480) were used to perform aerosol infection of BALB/c mice. Prior to infection, all M. tuberculosis stains were confirmed to produce phthiocerol dimycolate and to have positive neutral red staining (data not shown). For time-to-death experiments, strains were grown to mid-log phase and diluted to an OD600 of 0.2 in 10 ml of 7H9 Middlebrook liquid medium supplemented with oleic acid-albumin-dextrose-catalase (OADC). Sixteen BALB/c mice per strain were infected for 30 min in a Glas-col aerosol machine. At 1 day postinfection, 3 mice per strain were sacrificed. Lungs from each mouse were homogenized, and dilutions of lung homogenate were plated on 7H11-antibiotic selective plates. Numbers of CFU were recorded after 3 to 4 weeks of incubation at 37°C. The remaining 12 mice infected with each strain were monitored to record the number of days to death or severe illness for each mouse. Statistical analysis of survival was performed with the log rank (Mantel-Cox) test, using GraphPad Prism 5.0 software.
To determine the numbers of CFU of each strain in the lungs and spleen following infection, the three strains were grown to an OD600 of 0.05 to 0.1 in Middlebrook 7H9 liquid medium supplemented with OADC. Twenty-four BALB/c mice per strain were infected as described above. Four mice per strain were sacrificed on days 1, 14, 28, 56, 84, and 112 following infection. The lungs and spleen of each mouse were weighed, and gross pathology pictures were taken. Three-fourths of each lung and spleen was homogenized, and dilutions were plated on 7H11 selective plates. The numbers of CFU were counted after 3 to 4 weeks at 37°C. The remaining 1/4 of each lung and spleen was stained with hematoxylin and eosin for histopathology. The number of CFU in the lungs, lung weight, and spleen weight for each strain were compared at the indicated time points by one-way ANOVA with Tukey's posttest, using GraphPad Prism 5.0 software.
RESULTS
CuvA is required for optimal growth and normal cell morphology on several carbon sources.
Mutant strains containing cuvA deletions were constructed by allelic exchange in both the pathogen M. tuberculosis and the rapid-growing nonpathogen M. smegmatis (see Fig. S1 in the supplemental material). A complemented M. tuberculosis ΔcuvA strain was constructed by expressing cuvA under the control of the hsp70 promoter in the vector pJEB402 (22) to allow stable expression during prolonged incubation in vitro and during infection experiments. The same construct was used to complement the M. smegmatis mutant with M. tuberculosis cuvA. To complement the M. smegmatis mutant with the M. smegmatis cuvA homologue MSMEG_3080, this gene was expressed under the control of a Tet repressor-regulated promoter to allow inducible expression of this gene (21).
When grown in Middlebrook 7H9 liquid medium containing 0.2% glucose, the M. tuberculosis ΔcuvA (Fig. 1A) and M. smegmatis ΔcuvA (see Fig. S4A in the supplemental material) strains grew at similar rates and to the same final densities as the wild-type parental strains. Based on genetic data linking the Rv1422 protein to the Mce4 cholesterol transport system (28), we initially examined growth of the deletion strains in minimal medium (MM) with cholesterol added as a carbon source. In contrast to the results obtained with glucose-containing 7H9 medium, we observed altered growth phenotypes for both the M. smegmatis and M. tuberculosis ΔcuvA strains when they were grown on MM plus cholesterol, though these growth defects were not identical in the two species.
FIG 1.
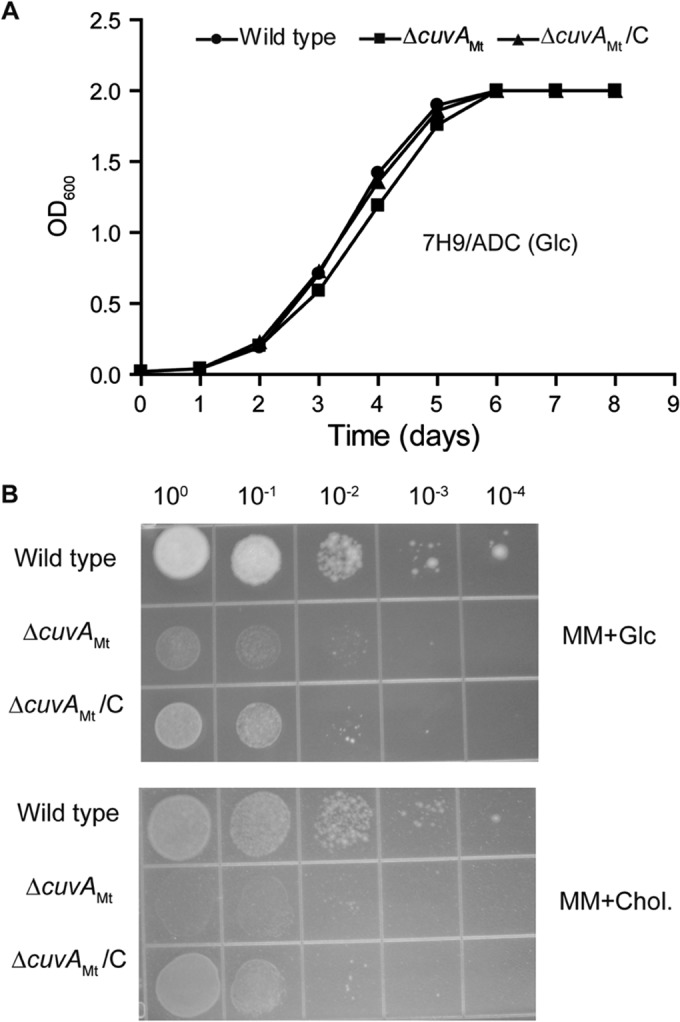
Growth of M. tuberculosis strains in liquid and solid media. (A) Wild-type M. tuberculosis H37Rv, ΔcuvAMt, and complemented ΔcuvA (ΔcuvAMt/C) strains were grown in 7H9-ADC-Tw liquid medium with 0.2% glucose, and the OD600 was measured daily. (B) Growth of the same strains on agar plates containing MM plus 0.2% glucose or MM plus 0.01% cholesterol. Serial 10-fold dilutions were spotted and incubated at 37°C, and photographs were taken after 1 month of incubation. In both panels, data shown are from one experiment that was repeated at least twice with similar results. Glc, 0.2% glucose; Chol., 0.01% cholesterol.
For M. tuberculosis, our attempts to compare growth by A600 measurements of wild-type, ΔcuvA, and complemented strains in liquid MM containing 0.01% cholesterol were limited by marked clumping of all strains and by the medium being cloudy and forming precipitates. We therefore used an alternative measure of growth, the microplate alamarBlue assay (MABA), which measures dye reduction in metabolically active growing cells. Data from this assay did not show a clear growth defect of the M. tuberculosis ΔcuvA strain in liquid medium containing cholesterol, although all M. tuberculosis strains grew poorly and required a high inoculum to grow in this medium, limiting interpretation of this result (see Fig. S2 in the supplemental material). On solid medium, however, the M. tuberculosis ΔcuvA strain had a clear growth defect compared to the wild type (Fig. 1B). When grown in Middlebrook 7H9 medium, washed, and spotted onto agar plates, the wild-type strain grew and formed colonies on MM agar with either 0.01% cholesterol or 0.2% glucose, although the growth on MM plus cholesterol was slower. The ΔcuvA strain grew more slowly than the wild type on MM plus 0.2% glucose and showed minimal growth on cholesterol, even after prolonged incubation. These growth phenotypes were complemented by expression of cuvA in the M. tuberculosis mutant.
The observation that the B. subtilis yvcK deletion strain had growth defects on multiple carbon sources led us to compare the growth of wild-type M. tuberculosis to that of the cuvA deletion strain on solid MM containing several other carbon sources. Each compound was added to a final concentration of 0.01%, the concentration of cholesterol that has been used in mycobacterial growth media because of its low aqueous solubility (2, 17). As shown in Fig. 2, the M. tuberculosis cuvA mutant strain grew much more slowly than the wild type on each of these media, as indicated by less-dense growth and an absence of visible growth at lower dilutions. In each case, the complemented strain showed growth similar to, though slightly less than, that of the wild type. These data indicate that the growth defect of the ΔcuvA strain is not specific but rather is a general defect in utilization of these several different carbon sources.
FIG 2.
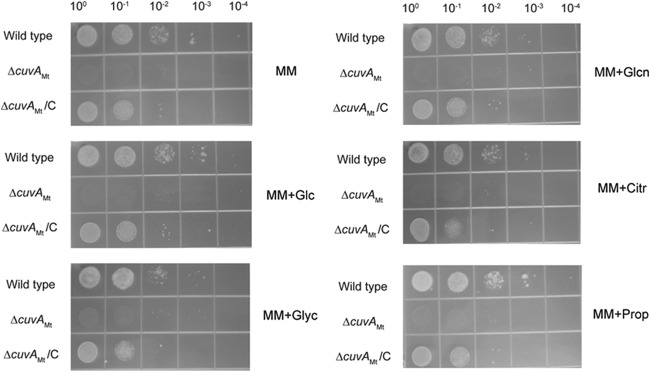
Growth of M. tuberculosis strains on MM agar containing added carbon sources. Serial 10-fold dilutions of wild-type M. tuberculosis H37Rv, ΔcuvAMt, and complemented ΔcuvA (ΔcuvAMt/C) strains were spotted on MM agar alone or containing the indicated compounds, added to a concentration of 0.01%, and then incubated at 37°C, and photographs were taken after 1 month of incubation. Data shown are from one experiment that was repeated at least twice with similar results. Glc, glucose; Glyc, glycerol; Glcn, gluconate; Citr, citrate; Prop, propionate.
Because CuvA was shown to be phosphorylated in M. tuberculosis (5, 29), to determine whether phosphorylation of CuvA is important for its function in nutrient utilization, complementing constructs encoding CuvA proteins in which the phosphoacceptor Thr residue was replaced with either a nonphosphorylatable (Thr325Ala) or a phospho-mimetic (Thr325Glu) amino acid were tested. Both of these mutant alleles also fully complemented the cholesterol growth defect (see Fig. S3 in the supplemental material), suggesting that phosphorylation of Thr325 is not required for CuvA function in cholesterol utilization under the conditions examined in these experiments.
We then examined growth of the M. smegmatis ΔcuvA strain. This strain had substantial, statistically significant growth defects in the MABA when grown in liquid MM alone or with several carbon sources present at 0.01%, though growth was similar to that of the wild type in MM plus glucose or gluconate (Fig. 3). The mutant growth defects were well complemented in each case by MSMEG_3080 expressed in trans. Addition of 0.2% glucose both increased the growth rate and eliminated the difference between the wild-type and mutant strains grown on cholesterol or propionate (data not shown), indicating that the growth defect is not likely due to toxic effects of defective cholesterol catabolism as has been observed in other mutants that cannot utilize cholesterol (16). When grown on solid medium, the ΔcuvA strain grew less than the wild type on MM alone or with added cholesterol, but it grew as well as the wild type on 0.2 or 0.01% glucose (see Fig. S4B in the supplemental material). These data show that the growth defects of the M. smegmatis ΔcuvA strain, like those of M. tuberculosis, are not specific.
FIG 3.

(A to H) Growth of M. smegmatis strains on different carbon sources. M. smegmatis mc2155 (wild type), ΔcuvAMs, and complemented ΔcuvA (ΔcuvAMs/C) strains were diluted to an OD600 of 0.001, grown in liquid MM alone or with added carbon sources, and analyzed using the MABA performed with technical duplicates. Except for hiGlc medium, which contained 0.2% glucose, all media contained compounds added to a concentration of 0.01%. The exponential growth phase of the ΔcuvAMs strain was significantly slower than that of the wild-type or ΔcuvAMs/C strain in MM alone (P < 0.0001), MM plus cholesterol (P < 0.0001), MM plus glycerol (P < 0.0001), MM plus citrate (P < 0.005), and MM plus propionate (P < 0.0001). The growth rates were not significantly different (P > 0.05) when cells were grown on 0.2% glucose or 0.01% glucose or gluconate. Glc, glucose; Glyc, glycerol; Citr, citrate; Chol, cholesterol; Glcn, gluconate; Prop, propionate. Error bars show 1 standard deviation (SD).
To further investigate the growth defects of the cuvA mutant, we undertook a series of additional experiments. We first examined cell shape and observed that the growth defect of the M. smegmatis ΔcuvA strain grown in cholesterol-containing liquid MM was accompanied by altered cell morphology, with cells that were shorter than those of the wild type and appeared to have thickened, wider cell poles (Fig. 4A). Measurement of the cholesterol-grown cells demonstrated that the difference in cell length was highly significant (Fig. 4B). These cell length and morphology defects were fully complemented by the wild-type cuvAMs allele. When the M. smegmatis ΔcuvA strain was grown on solid MM plus cholesterol, cell morphology was also affected, with many cells showing asymmetric bulging (Fig. 4C). This phenotype is very similar to the ΔyvcK morphology phenotype of B. subtilis strains grown on gluconate, where the bifunctional PGN synthesis enzyme PBP1 is mislocalized (14). This M. smegmatis ΔcuvA phenotype is also very similar to the morphological abnormalities seen in a PBP1 depletion strain of M. smegmatis, where PGN synthesis is directly affected (30).
FIG 4.
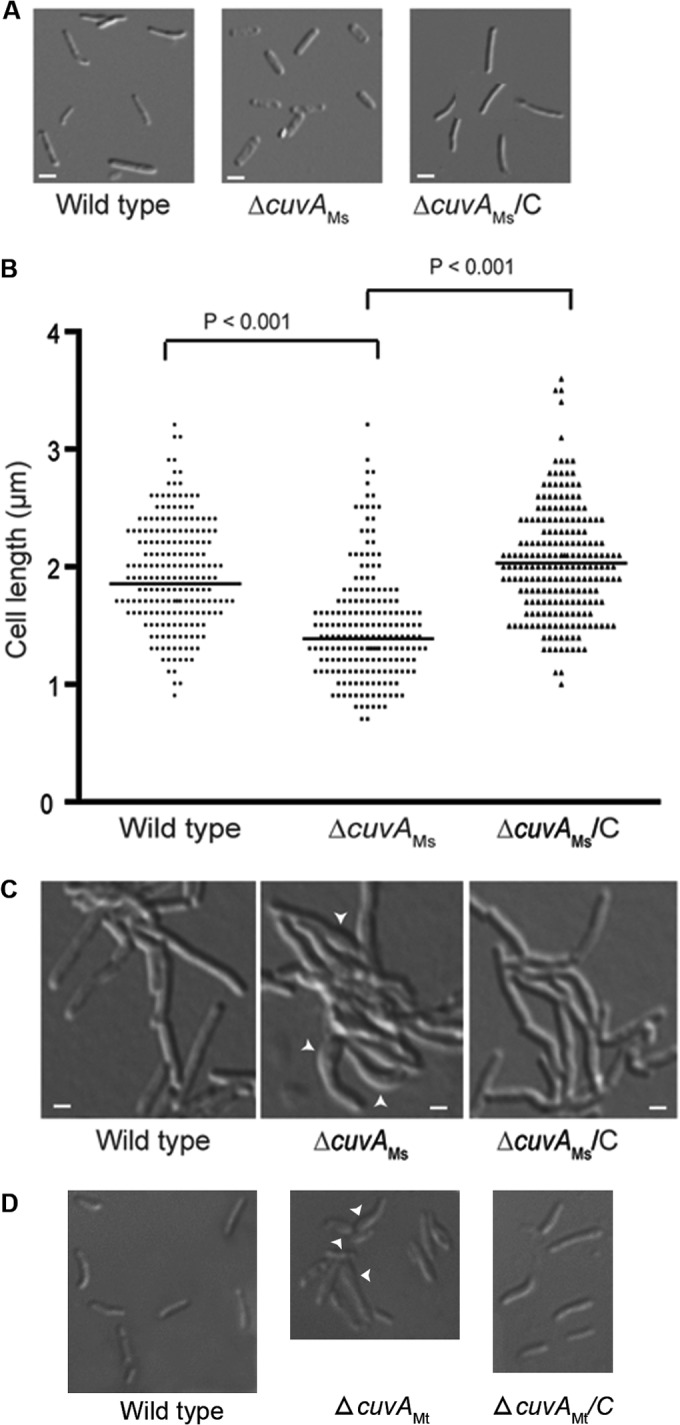
Morphology of M. smegmatis and M. tuberculosis cells. (A) M. smegmatis mc2155 (wild type), ΔcuvAMs, and complemented ΔcuvA (ΔcuvAMs/C) strains were grown in MM plus 0.01% cholesterol, harvested after 24 h, and examined by light microscopy. Bars = 1 μm. (B) Length distribution of M. smegmatis cells obtained after 24 h of growth in MM plus 0.01% cholesterol, determined by measuring over 200 cells of each strain and analyzed using ImageJ software. The horizontal lines indicate mean cell lengths. (C) Cells of each M. smegmatis strain were grown overnight on agarose pads made with MM plus 0.01% cholesterol and then photographed. (D) Wild-type M. tuberculosis H37Rv, ΔcuvAMt, and complemented ΔcuvA (ΔcuvAMt/C) strains were grown to late stationary phase in MM liquid medium plus 0.01% cholesterol and then examined by light microscopy using a 100× oil-immersion objective. Arrowheads in panels C and D indicate areas of abnormal cell morphology.
Because of the minimal growth of the M. tuberculosis ΔcuvA strain on solid MM containing cholesterol, there were too few cells to identify a morphological phenotype of the ΔcuvA strain on this medium. When ΔcuvA cells were grown to late stationary phase in liquid MM with 0.01% cholesterol, however, we observed a bulging phenotype similar to that observed for M. smegmatis (Fig. 4D). This phenotype was seen in 42% of ΔcuvA cells versus 11% of wild-type cells (P < 0.0001, with over 200 cells analyzed per strain) and 12% of complemented cells. As seen for M. smegmatis, this M. tuberculosis phenotype was not seen for cells grown on MM plus glucose. These morphological phenotypes strongly suggest that there is a defect in PGN synthesis or structure in the ΔcuvA deletion strains.
CuvA localizes asymmetrically to the growing cell pole in mycobacteria.
Peptidoglycan synthesis in mycobacteria and other actinomycetes is targeted to the septum and asymmetrically to the growing cell pole (8, 31). The growth and morphology defects we observed in the ΔcuvA strains, together with the data from B. subtilis indicating that YvcK plays a role in regulating cell wall synthesis (14), led us to investigate the subcellular localization of CuvA. We first determined that M. tuberculosis cuvA is a functional orthologue of the M. smegmatis gene by showing that M. tuberculosis cuvA fully complemented the cholesterol growth defect of the M. smegmatis ΔcuvA strain (see Fig. S5 in the supplemental material). We then made a cuvAMt-gfp fusion and confirmed that this construct also complemented the growth defect of the ΔcuvAMs strain in MM containing 0.01% cholesterol (see Fig. S5).
Using the cuvAMt-gfp fusion, we found that CuvA-GFP strongly localized to one pole of most cells (Fig. 5), similar to the pattern we observed previously for Wag31 (DivIVA), an essential protein that we showed to be required for polar localization of PGN synthesis in mycobacteria (8). To determine the timing of CuvA localization and whether this protein is targeted to the old or new pole, we performed time-lapse live-cell microscopy of M. smegmatis expressing CuvA-GFP and growing on nutrient agarose containing MM with 0.01% cholesterol, overlaid on microscope slides. As shown in Movie S1 in the supplemental material, we found that CuvA-GFP localized to the old pole in most cells. We also observed that cell growth occurred predominantly at one pole of the mycobacterial cell, as we and others have shown previously (8, 32), and that CuvA consistently localized to this growing pole. This localization of CuvA to the growing cell pole occurred when cells were grown on medium containing either glucose or cholesterol as the carbon source. Similar to these observations in M. smegmatis, CuvA also localized to one pole in M. tuberculosis (Fig. 6). Though we were not able to perform time-lapse microscopy with M. tuberculosis, observation of cells that appeared to have recently undergone cell division indicated that CuvA also localizes to the old, actively growing cell pole in this species. A nonphosphorylatable form of CuvA in which Thr325 was replaced with Ala also localized to the cell pole in M. tuberculosis (not shown).
FIG 5.

Colocalization of CuvA and nascent peptidoglycan synthesis in M. smegmatis. Wild-type M. smegmatis mc2155 cells expressing GFP alone or GFP fused to CuvAMt were grown in MM-ADC-TW containing 0.2% glucose or MM containing 0.01% cholesterol. Vancomycin labeled with Alexa 568 was used to bind to newly synthesized peptidoglycan. Fluorescence and DIC microscopy images were obtained to determine the localization of CuvAMt-GFP and nascent peptidoglycan synthesis. Bars = 1 μm.
FIG 6.
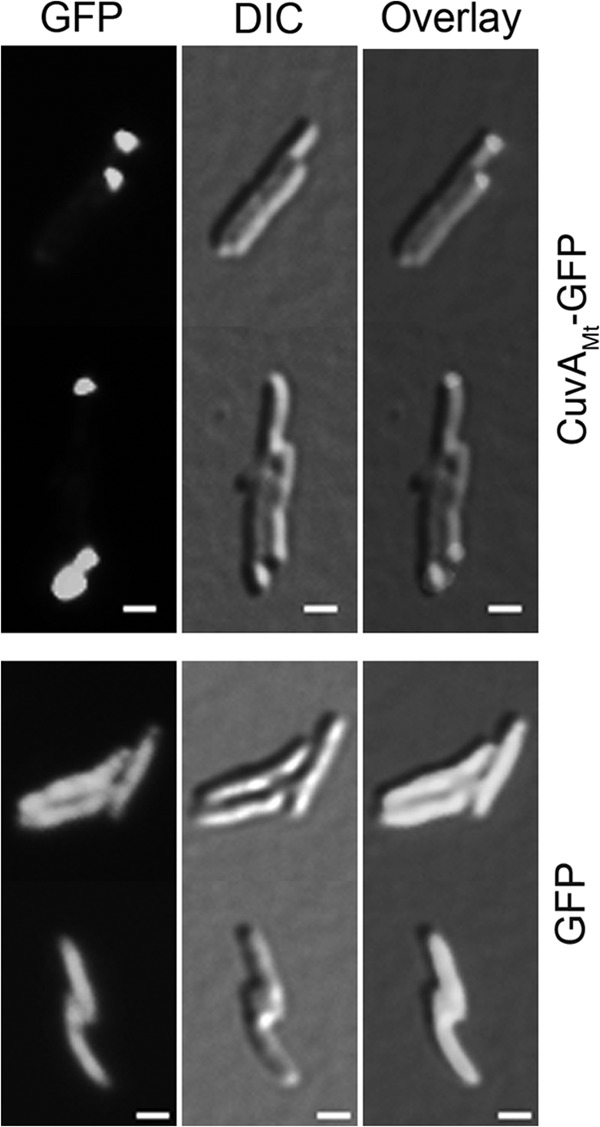
Localization of CuvA in M. tuberculosis. An M. tuberculosis leucine-pantothenate auxotroph strain (24) expressing CuvAMt-GFP or unfused GFP was grown on agarose pads containing MM-ADC with 0.2% glucose and acetamide to induce expression of cuvAMt-gfp. Fluorescence and DIC microscopy images were obtained as described in Materials and Methods after 24 h of induction to determine the localization of CuvAMt-GFP. Bars = 1 μm.
Our finding that CuvA localizes to the growing cell pole led us to predict that CuvA and new PGN synthesis would colocalize. Fluorescent vancomycin (Van-Alexa 568), which binds to the terminal d-Ala-d-Ala of newly synthesized peptidoglycan precursors, can be used to identify sites of active PGN synthesis in bacteria (31). Using Van-Alexa 568, we observed that most M. smegmatis cells grown in glucose-containing medium were stained predominantly at one pole, consistent with our previous observations (8), though some cells had a weaker signal from the other pole and a few had approximately equal staining of both poles (Fig. 5). CuvA-GFP consistently localized to the pole that showed strong Van-Alexa 568 staining, indicating that CuvA is targeted to sites of active PGN synthesis at the elongating cell pole. Surprisingly, however, whereas asymmetry of PGN synthesis was seen in nearly all cells grown in glucose, in cells grown on cholesterol, strong bipolar staining with fluorescent vancomycin was seen in the majority of cells (Fig. 5). Despite this bipolar vancomycin staining, CuvA consistently localized to one pole in cholesterol-grown cells, indicating that CuvA is not absolutely required for localized PGN synthesis at the cell pole but is consistently targeted to the old cell pole when grown on either glucose or cholesterol.
Based on the finding in B. subtilis that the penicillin binding protein PBP1 is delocalized in a yvcK null mutant (14), we examined the localization of PBP1 in wild-type M. smegmatis and in the ΔcuvA strain. As previously reported (30), we found that a PBP1-RFP fusion protein localized to both cell poles and midcell in wild-type M. smegmatis (see Fig. S6 in the supplemental material). In contrast to the findings in B. subtilis, however, no differences were seen in PBP1-RFP localization in the M. smegmatis ΔcuvA strain compared to the wild type when cells were grown on medium containing either glucose or cholesterol as the carbon source. Based on suppressor mutation results (see below) and its identification as a probable substrate of PknB (6), we also examined the localization of PbpA. As observed with PBP1, the localization of PbpA was the same in the wild type and the ΔcuvA strain (data not shown).
The M. smegmatis cuvA deletion strain is hypersusceptible to β-lactam antibiotics and rifampin.
Previous work had shown that M. smegmatis lacking the bifunctional penicillin binding protein PBP1 is markedly more susceptible to several β-lactam antibiotics, including ampicillin and cephalothin (6- to >20-fold lower MIC), but shows little change in susceptibility to other classes of antibiotics (33). In another study, a screen for M. smegmatis and M. tuberculosis transposon mutants that were hypersusceptible to β-lactams identified several genes involved in cell wall synthesis and cell division, including ponA2 (encoding PBP2), dapB, and homologues of DivIVA and DivIVC genes (34). Based on our data suggesting a role for CuvA in cell wall synthesis or structure, we tested the susceptibility of the M. tuberculosis and M. smegmatis deletion strains to several antibiotics by using MABA. No significant differences in susceptibility of the M. tuberculosis deletion strain compared to the wild type were observed. Because of the high inoculum required for M. tuberculosis to grow in MM, however, there may be small differences in susceptibility between strains that we could not detect with this assay (see Table S1 in the supplemental material).
As shown in Table 1, however, we observed a strikingly increased susceptibility of the M. smegmatis ΔcuvA strain to all four β-lactams tested when the strain was grown on cholesterol. For ampicillin and the first-generation cephalosporin cephalothin, the addition of the β-lactamase inhibitor clavulanate was required to observe this increase in susceptibility, whereas for the third-generation cephalosporin cefotaxime and the carbapenem meropenem, which are less susceptible to hydrolysis by β-lactamases, the increased susceptibility was evident without addition of clavulanate (Table 1). This increased-susceptibility phenotype was reversed in the complemented strain. This hypersusceptibility was also reversed in each case by the addition of 0.2% glucose to the culture medium. The M. smegmatis mutant was not significantly more susceptible to levofloxacin, isoniazid, or gentamicin, none of which act on PGN synthesis. The ΔcuvA strain was, however, significantly more susceptible to the RNA polymerase inhibitor rifampin. While this finding raises the possibility of an effect of cuvA on RNA polymerase, an indirect effect related to an altered cell wall in the mutant, such as increased uptake or decreased efflux of rifampin, may account for this difference.
TABLE 1.
Antibiotic susceptibility of M. smegmatis strainsa
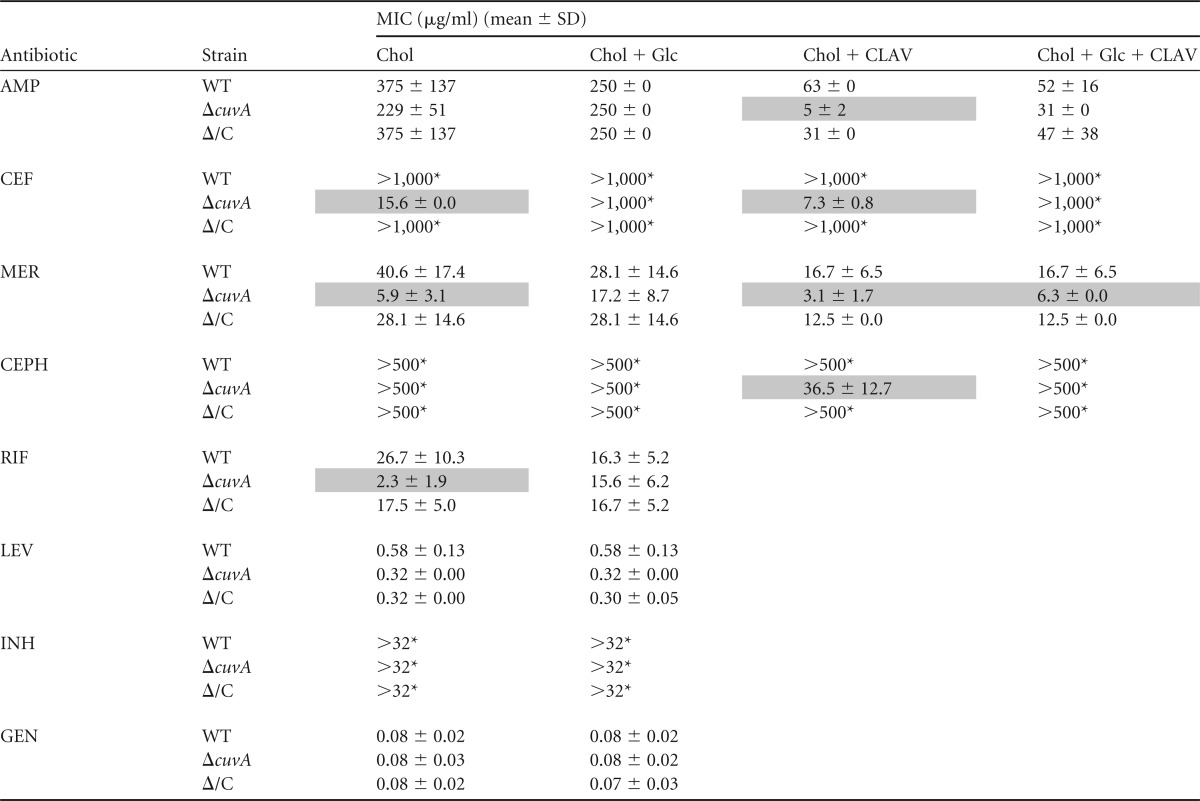
AMP, ampicillin; CEF, cefotaxime; MER, meropenem; CEPH, cephalothin; RIF, rifampin; LEV, levofloxacin; INH, isoniazid; GEN, gentamicin; CLAV, clavulanic acid; Chol, 0.01% cholesterol; Glc, 0.2% glucose; WT, wild type; ΔcuvA, ΔcuvA strain; Δ/C, cuvA-complemented ΔcuvA strain. Shaded boxes indicate results for the ΔcuvA strain that are significantly different from those for the wild type (P < 0.05 and >2-fold difference). *, highest concentration tested.
Suppressors of the CuvA cholesterol growth phenotype map to loci involved in cell wall synthesis and transmembrane transport.
Our phenotypic and localization data suggested that CuvA functions in utilization or uptake of several carbon sources and is required for normal cell wall structure under nutrient-limited conditions. As another approach to gaining insight into the pathways in which CuvA functions, we undertook a saturating transposon mutagenesis experiment to identify suppressors of the M. tuberculosis ΔcuvA cholesterol growth defect. Genes in which two or more insertions were obtained are shown in Table 2. A striking finding was the isolation of several transductants with insertions in pbpA and rodA. These genes are involved in PGN synthesis and cell shape control and are part of the operon containing pknA and pknB, genes encoding essential Ser/Thr kinases that regulate these processes (5, 6, 8, 9, 35). These suppressors thus indicate a link between the ΔcuvA growth phenotype and cell wall synthesis and morphology and are reminiscent of the suppression of the growth and morphology defects of the B. subtilis yvcK mutant by deletion of ponA, the gene encoding PBP1 (14). We also obtained three independent insertions in the forward orientation at the beginning of Rv2683 and one in the 5′ end of the Rv2683 coding sequence, in the same orientation. The location and orientation of these insertions suggest that increased expression of this operon via readthrough expression from the transposon may suppress the ΔcuvA cholesterol growth phenotype. Rv2683 is the first gene in a three-gene operon that encodes a transporter that was recently shown to be essential for M. tuberculosis growth on cholesterol (36). This result suggests that increased expression of this transporter may suppress the ΔcuvA growth defect. Two additional insertions disrupt the response regulator of an uncharacterized two-component system.
TABLE 2.
Transposon insertions that suppress the cholesterol growth defect of the M. tuberculosis ΔcuvA straina
| Gene disrupted | Insertion sites (no. and orientation of insertions) | Probable function of gene/operon |
|---|---|---|
| rodA | +755 (2r), +1291 (1r) | Cell shape/peptidoglycan synthesis, Ser/Thr phosphorylation |
| pbpA | +142 (1r), +232 (1r), +250 (1f), +270 (1f), +541 (1f) | Peptidoglycan synthesis/cell shape, Ser/Thr phosphorylation |
| Rv2683 | −1 (3f), +191 (1f) | Conserved hypothetical regulatory protein/transmembrane transport |
| trcR | +681 (1f), +732 (1r) | Response regulator/TrcRS two-component system |
Genes in which two or more independent insertions were obtained are included in the table. f and r, forward and reverse orientations of the kanamycin marker in the transposon, respectively, with respect to the direction of transcription of the gene disrupted. The number preceding “f” or “r” indicates the number of independent transposon insertions obtained at each site.
CuvA is required for virulence and persistence in a mouse infection model.
In the context of data indicating that M. tuberculosis strains lacking the Mce4 cholesterol transport system are attenuated during infection and a transposon mutagenesis study suggesting that Rv1422 is required for bacterial replication in mice following intravenous infection (12, 37), the in vitro growth defects of the ΔcuvA strains led us to compare the virulence of the M. tuberculosis ΔcuvA strain and the parental wild-type strain H37Rv in mouse infection experiments. BALB/c mice were infected by the aerosol route with wild-type H37Rv, ΔcuvA, and cuvA-complemented strains. Examination of numbers of CFU present in lungs at serial time points demonstrated that the wild-type and ΔcuvA strains replicated at similar rates until days 14 to 28, after which the number of mutant bacteria declined (Fig. 7A). In contrast, the wild type persisted at a stable level until day 84, after which a small decline was seen. At days 56, 84, and 112, the differences between the wild type and the cuvA strain were highly significant (P < 0.0001 to P < 0.005). By the final time point, 112 days, there was a 200-fold difference in numbers of lung CFU between these strains. The complemented strain showed an intermediate phenotype, with significantly more CFU in the lung than the case with the ΔcuvA strain (P < 0.0001 to P < 0.01) but fewer CFU than the case with the wild type (P < 0.0001 to P < 0.05). This persistence defect of the M. tuberculosis cuvA mutant is similar to, but more pronounced than, the phenotype observed in an mce4 deletion strain, where decreased numbers of CFU were seen at later stages of infection (28). In the spleen, the number of CFU increased for all 3 strains until day 28, after which the wild type increased compared to the mutant, though this phenotype was not well complemented (see Fig. S7 in the supplemental material).
FIG 7.
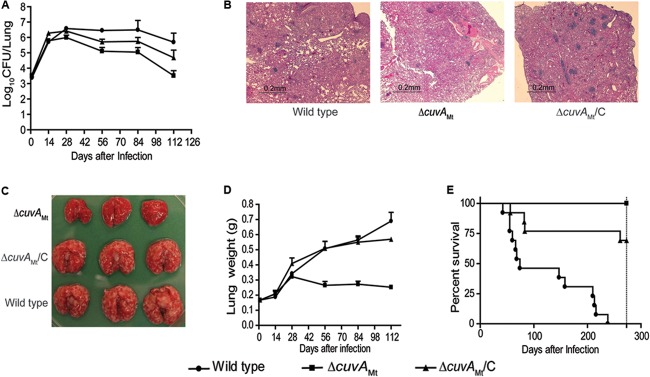
Mouse infection experiments. BALB/c mice were infected by aerosol with wild-type M. tuberculosis H37Rv, the ΔcuvAMt strain, or the complemented ΔcuvA strain (cuvAMt/C). In separate experiments, mice were monitored for morbidity/mortality or sacrificed at serial time points for pathological analysis and measurement of bacterial burden. (A) Numbers of CFU in lungs from day 0 to day 112. Data are means for four mice at each time point. Lungs from mice infected with the wild type had significantly more CFU than those from mice infected with the ΔcuvA strain at days 56, 84, and 112 (P < 0.0001 to P < 0.005). Mice infected with the complemented strain had significantly more CFU in the lung than those infected with the ΔcuvA strain at these time points (P < 0.0001 to P < 0.01) but fewer CFU than those infected with the wild type (P < 0.0001 to P < 0.05). Error bars show 1 SD. (B) Microscopic pathology of sections stained with hematoxylin and eosin on day 84. (C) Gross pathology of lungs on day 84. (D) Lung weights from day 0 to day 112. Data are means for four mice at each time point. Lungs of mice infected with the wild-type or complemented strain were significantly heavier than those of mice infected with the ΔcuvA strain (P < 0.0001 to P < 0.005 at days 54 to 112). Error bars show 1 SD. (E) Survival curves for mice infected with the three strains of M. tuberculosis. Mortality was significantly lower in mice infected with the ΔcuvA strain than in those infected with the wild-type (P < 0.0001) or complemented (P < 0.05) strain. Mortality in the mice infected with the complemented strain was also significantly lower than that in mice infected with the wild type (P < 0.0001).
Gross and microscopic pathology showed striking differences in the extents of disease in the lungs from mice infected with the wild-type and complemented strains and those from the ΔcuvA strain-infected mice. The lungs of wild-type and cuvA-complemented strain-infected mice showed much larger and more numerous tubercles, were significantly heavier (P < 0.0001 to P < 0.005 at days 54 to 112 for the wild type versus the cuvA-complemented strain), and showed much more extensive cellular infiltrates on histopathology (Fig. 7B to D). These phenotypes were well complemented visually and, in the case of lung weight, statistically (P > 0.05 at days 54 to 112 for the wild type versus the complemented strain). The extent of disease in the spleen was also greater in wild-type H37Rv-infected mice than in ΔcuvA strain-infected mice, though the difference was less striking (see Fig. S7 in the supplemental material).
In separate time-to-death/severe morbidity experiments, mice were infected by aerosol, four were sacrificed at day 1 to measure the inoculum (see Fig. S7D in the supplemental material), and the remaining mice were monitored over time. This experiment also showed marked attenuation of the ΔcuvA strain. Fifty percent mortality for wild-type H37Rv was reached at day 68, whereas by day 273, when the experiment was ended, none of the mice infected with the ΔcuvA strain had died (P < 0.0001) (Fig. 7E). There was partial complementation with cuvA, with 25% mortality of mice infected with the complemented strain at day 84 (P = 0.03 for the ΔcuvA versus complemented strain). This incomplete complementation may have resulted from constitutively high-level cuvA expression in the complemented strain, which we found to be >15-fold higher than expression in the wild type. We did not observe polar effects on the expression of genes adjacent to Rv1422 in the cuvA mutant (see Fig. S8).
DISCUSSION
In this work, we have provided an initial characterization of an M. tuberculosis gene of unknown function, Rv1422, and its M. smegmatis orthologue, MSMEG_3080, which we have named cuvA based on altered carbon source utilization in M. smegmatis and M. tuberculosis cuvA deletion strains and decreased virulence phenotypes of the M. tuberculosis deletion mutant. The CuvA protein has homologues across a broad range of diverse bacterial phyla, and its amino acid sequence suggests that CuvA is distantly related to the UPF0052 unidentified protein family (38). The one characterized member of this family is CofD (2-phospho-l-lactate transferase [LPPG]), a protein present in archaea and some bacteria that has been shown to be involved in coenzyme F420 biosynthesis in Methanocaldococcus jannaschii (39, 40). At the level of the primary amino acid sequence, however, CuvA is not significantly similar to CofD. Another M. tuberculosis protein, FbiA (Rv3261), is highly similar to CofD and has been shown to be required for F420 biosynthesis in Mycobacterium bovis BCG (41), indicating that FbiA is the functional orthologue of CofD and that CuvA likely has a different activity.
If it does not function in F420 biosynthesis, then what is the role of CuvA? A B. subtilis strain with a deletion in the cuvA orthologue yvcK was shown to have growth defects on several carbon sources and to be defective in localization of PBP1 and cell wall synthesis. In this work, we have identified several phenotypes of M. tuberculosis and M. smegmatis cuvA deletion mutants that suggest that the CuvA protein is required for utilization of nutrients and for normal cell wall synthesis. In both species, the cuvA mutants show defects in utilization of several carbon sources, including cholesterol, which is important for M. tuberculosis during infection in vivo. In the case of M. smegmatis, growth defects were apparent when cells were grown in liquid MM or on agar MM. The M. smegmatis ΔcuvA strain had growth defects on solid medium similar to those seen in M. tuberculosis, with the exception that the M. smegmatis deletion strain grew well on glucose but the M. tuberculosis strain did not. The basis of this difference in growth on glucose will require further investigation but might result from enhanced metabolic capacity encoded in the much larger genome of the environmental organism M. smegmatis or from the much more efficient uptake of glucose, mediated via the MspA porin, in M. smegmatis than in M. tuberculosis, which lacks this porin (42, 43). Growth defects of the M. tuberculosis ΔcuvA strain were not apparent in liquid medium. The very slow growth of M. tuberculosis in MM broth and the requirement for a high inoculum for M. tuberculosis to grow in this medium may have limited our ability to detect small differences. The mutant strains of both species had similar morphological abnormalities, with asymmetric bulging suggestive of a peptidoglycan defect. Also, in both species, CuvA localized to the growing cell pole, the site of peptidoglycan synthesis required for cell elongation in actinomycetes, including mycobacteria (8, 31).
A notable difference between the M. tuberculosis and M. smegmatis cuvA mutants was the markedly increased susceptibility of the M. smegmatis strain, but not the M. tuberculosis strain, to several cell wall-active antibiotics and to rifampin. Though differences in susceptibility of the M. tuberculosis ΔcuvA strain and the wild type to antibiotics may have been obscured by the poor growth of M. tuberculosis in MM and the high inoculum needed to achieve growth, the magnitude of differences seen in M. smegmatis, particularly for the cephalosporins, suggests that the differences in this phenotype result from differences in the mycobacterial cell envelope in these species. Taken together, the similarities of most phenotypes suggest that the function of CuvA is conserved across these mycobacterial species. This interpretation is also supported by the complementation of both the growth (see Fig. S5 in the supplemental material) and antibiotic susceptibility (not shown) phenotypes of the M. smegmatis ΔcuvA strain by the cuvA gene of M. tuberculosis.
The growth defect of the cuvA deletion strains was not specific to a single carbon source. Growth rates of the cuvA mutants on each of the carbon sources tested, with the exception of propionate, were comparable to or higher than those on MM alone, in which asparagine is the primary available energy source. Growth of the M. smegmatis ΔcuvA strain on MM plus propionate, however, was slower than that on MM alone. Mycobacterial strains with defects in either the glyoxylate cycle (icl1 icl2 double null mutant) or the methylcitrate cycle (prpC prpD double null mutant) are unable to grow on cholesterol or propionate (44, 45). Though there is some similarity in the phenotypes of the ΔcuvA strains and the strains disrupted in glyoxylate or methylcitrate pathways, there are important differences that suggest that CuvA does not participate directly in these pathways. First, the M. tuberculosis strains with defects in the glyoxylate or methylcitrate pathways show minimal growth on cholesterol or propionate in liquid MM (17, 45, 46). In contrast, the M. tuberculosis ΔcuvA strain did not have a significant defect in cholesterol-containing liquid medium, and the M. smegmatis ΔcuvA strain grew more slowly but did achieve sustained growth in MM plus cholesterol and MM plus propionate. Second, the M. smegmatis ΔcuvA cholesterol growth defect was fully reversed by the addition of glucose, whereas the growth defects of the icl1 icl2 and prpC prpD M. tuberculosis strains were not reversed by glycerol. In addition, the growth-inhibitory effect of propionate on the M. smegmatis cuvA mutant was not reversed by addition of vitamin B12, in contrast to the case with the prpC prpD and icl1 icl2 mutants, in which B12 addition allows cells to grow on propionate via utilization of the B12-dependent methylmalonyl-coenzyme A (CoA) pathway (17, 45). Thus, cuvA does not appear to be required for the glyoxylate or methylcitrate pathways.
As noted above, our data indicate that the cuvA deletion mutant growth defect is not specific and that CuvA is not an enzyme required for cholesterol catabolism. This inference is consistent with a recent study using genetic approaches and metabolic profiling to map the pathway of cholesterol catabolism in M. tuberculosis, in which the Rv1422 protein was not identified as being required for any step in cholesterol degradation (17). These data, together with rescue of the growth defect by addition of glucose, also suggest that the growth defect is not the result of accumulation of a toxic metabolite from catabolism of the several carbon sources on which the ΔcuvA strain grew less well than the wild type. Though a metabolic defect may explain the cuvA growth phenotypes, these data, together with the genetic interaction of CuvA with both the mce4 cholesterol transport locus and the mce1 locus, which appears to be involved in turnover of mycolic acids (11), suggest a possible alternative mechanism, i.e., that CuvA is required for nutrient uptake into the mycobacterial cell. One mechanism for a broad effect on uptake of many different nutrients would be alterations in the cell envelopes of the ΔcuvA strains that affect cell permeability or transporter functions.
Several of our results indicate that the mutant strains do have an altered cell envelope and suggest that CuvA has a role in the structure and/or function of the mycobacterial cell wall. First, in addition to decreased growth of the M. tuberculosis and M. smegmatis ΔcuvA mutants on MM containing several carbon sources, these cells have an abnormal morphology, showing shortened, bulging cells. This phenotype is strongly suggestive of an effect on cell wall integrity. This altered morphology is highly similar to the previously described phenotype of an M. smegmatis ponA depletion mutant and similar to, though less severe than, the phenotype of a wag31 depletion mutant (8, 30); in both of these mutants, PGN synthesis is disrupted. The phenotype is also very similar to the morphology of the B. subtilis yvcK deletion strain grown on gluconate, in which the PGN biosynthetic enzyme PBP1 is mislocalized (8, 14, 30). The marked increase in susceptibility of the M. smegmatis ΔcuvA strain to several β-lactam antibiotics, which parallels the phenotype observed for a ponA depletion strain, also suggests that the cell wall is altered in this strain. The localization of CuvA to the growing cell pole, the site of new PGN biosynthesis in mycobacteria, is also consistent with a role for this gene in PGN synthesis, though our data show that CuvA is not essential for this function.
Suppressor mutagenesis can be a valuable means to gain insight into the pathway in which a gene of unknown function is active. The suppressor mutagenesis results we obtained provide further support for a role of CuvA in cell wall synthesis and/or transport of nutrients. The inactivating insertions present in pbpA and rodA that restore growth of the M. tuberculosis ΔcuvA strain on cholesterol likely affect the structure of the cell wall. This alteration may allow uptake of cholesterol (and other nutrients) in the ΔcuvA strain, in which cholesterol uptake is otherwise impaired. In addition, for the insertions in Rv2683, their location at the 5′ end of the operon and the orientation of all insertions in the direction of transcription of the operon suggest that these may be gain-of-function mutations resulting from increased expression of genes in the operon from the transposon promoter. Rv2683 is the first gene in a three-gene operon that encodes a transporter that was recently shown to be essential for M. tuberculosis growth on cholesterol (36), suggesting that these insertions may enhance uptake of cholesterol and other compounds into the mycobacterial cell.
The markedly decreased virulence of the cuvA deletion mutant in a mouse model of tuberculosis suggests a critical role for this protein during infection. This attenuation was especially pronounced in the lung, where both gross tubercle formation and microscopic inflammation were markedly decreased with the ΔcuvA strain, and where the number of CFU of the mutant decreased significantly during the course of infection. Though M. tuberculosis appears to utilize cholesterol throughout infection in mice (16), the small difference in lung CFU early during infection and the progressive decline in CFU of the ΔcuvA strain that we observed during the chronic phase parallel the persistence defect seen in an M. tuberculosis strain lacking the Mce4 cholesterol transport system (2, 28). A second prior mouse infection study also showed an mce4 deletion strain to be attenuated (37). In this study, very small differences in numbers of lung CFU were seen, along with decreased lung pathology and significantly increased survival of mice infected with the mce4 strain. Compared to these studies, the decrease in lung CFU of the cuvA mutant in our infection experiment was more pronounced, and the survival of the cuvA mutant was more prolonged, even though we used a more susceptible mouse strain (BALB/c) than that in the previous studies (C57BL/6). In the context of these prior data and the in vitro data presented here, this result suggests that compromised utilization of cholesterol and other nutrients in the ΔcuvA strain during infection likely contributes to the attenuation of the mutant. This interpretation is also consistent with the genetic interaction study that first linked cuvA (Rv1422) to mce4 (28), though other effects of the cuvA deletion, such as a defective cell wall, may also contribute to decreased virulence.
The presence of CuvA homologues encoded in the genomes of a broad range of bacteria suggests a highly conserved function for this protein. Though CuvA is required for optimal nutrient utilization by mycobacteria, as for B. subtilis YvcK, the exact function of M. tuberculosis CuvA remains to be defined at the molecular level. Our data, taken together with the B. subtilis YvcK results, however, indicate a role for CuvA that links cell wall synthesis and nutrient utilization. For M. tuberculosis, the severe attenuation of M. tuberculosis virulence and persistence in the deletion strain suggests that CuvA may be a valuable target for development of novel therapeutics to treat tuberculosis disease and/or latent tuberculosis infection.
Supplementary Material
ACKNOWLEDGMENTS
We thank members of the Husson laboratory for helpful discussions.
This work was supported by NIH grants AI059702 and AI099204 to R.N.H. and by the NIH TARGET consortium contract (N01-AI30036) to William Bishai, Johns Hopkins University. Microscopy resources were provided by the Boston Children's Hospital Intellectual and Developmental Disabilities Research Center (IDDRC) cellular imaging core (NIH grant P30-HD-1865) and the New England Regional Center of Excellence in Biodefense and Emerging Infectious Diseases live-cell imaging core (NIH grant AI0571).
We declare that we have no conflicts of interest.
Footnotes
Published ahead of print 21 July 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.02207-14.
REFERENCES
- 1. McKinney JD, Honer zu Bentrup K, Munoz-Elias EJ, Miczak A, Chen B, Chan WT, Swenson D, Sacchettini JC, Jacobs WR, Jr, Russell DG. 2000. Persistence of Mycobacterium tuberculosis in macrophages and mice requires the glyoxylate shunt enzyme isocitrate lyase. Nature 406:735–738. 10.1038/35021074 [DOI] [PubMed] [Google Scholar]
- 2. Pandey AK, Sassetti CM. 2008. Mycobacterial persistence requires the utilization of host cholesterol. Proc. Natl. Acad. Sci. U. S. A. 105:4376–4380. 10.1073/pnas.0711159105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Russell DG, VanderVen BC, Lee W, Abramovitch RB, Kim MJ, Homolka S, Niemann S, Rohde KH. 2010. Mycobacterium tuberculosis wears what it eats. Cell Host Microbe 8:68–76. 10.1016/j.chom.2010.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. North RJ, Jung Y-J. 2004. Immunity to tuberculosis. Annu. Rev. Immunol. 22:599–623. 10.1146/annurev.immunol.22.012703.104635 [DOI] [PubMed] [Google Scholar]
- 5. Kang CM, Abbott DW, Park ST, Dascher CC, Cantley LC, Husson RN. 2005. The Mycobacterium tuberculosis serine/threonine kinases PknA and PknB: substrate identification and regulation of cell shape. Genes Dev. 19:1692–1704. 10.1101/gad.1311105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dasgupta A, Datta P, Kundu M, Basu J. 2006. The serine/threonine kinase PknB of Mycobacterium tuberculosis phosphorylates PBPA, a penicillin-binding protein required for cell division. Microbiology 152:493–504. 10.1099/mic.0.28630-0 [DOI] [PubMed] [Google Scholar]
- 7. Sureka K, Hossain T, Mukherjee P, Chatterjee P, Datta P, Kundu M, Basu J. 2010. Novel role of phosphorylation-dependent interaction between FtsZ and FipA in mycobacterial cell division. PLoS One 5:e8590. 10.1371/journal.pone.0008590 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 8. Kang CM, Nyayapathy S, Lee JY, Suh JW, Husson RN. 2008. Wag31, a homologue of the cell division protein DivIVA, regulates growth, morphology and polar cell wall synthesis in mycobacteria. Microbiology 154:725–735. 10.1099/mic.0.2007/014076-0 [DOI] [PubMed] [Google Scholar]
- 9. Gee CL, Papavinasasundaram KG, Blair SR, Baer CE, Falick AM, King DS, Griffin JE, Venghatakrishnan H, Zukauskas A, Wei JR, Dhiman RK, Crick DC, Rubin EJ, Sassetti CM, Alber T. 2012. A phosphorylated pseudokinase complex controls cell wall synthesis in mycobacteria. Sci. Signal. 5:ra7. 10.1126/scisignal.2002525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mohn WW, van der Geize R, Stewart GR, Okamoto S, Liu J, Dijkhuizen L, Eltis LD. 2008. The actinobacterial mce4 locus encodes a steroid transporter. J. Biol. Chem. 283:35368–35374. 10.1074/jbc.M805496200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Forrellad MA, McNeil M, Santangelo ML, Blanco FC, García E, Klepp LI, Huff J, Niederweis M, Jackson M, Bigi F. 2014. Role of the Mce1 transporter in the lipid homeostasis of Mycobacterium tuberculosis. Tuberculosis (Edinb.) 94:170–177. 10.1016/j.tube.2013.12.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sassetti CM, Rubin EJ. 2003. Genetic requirements for mycobacterial survival during infection. Proc. Natl. Acad. Sci. U. S. A. 100:12989–12994. 10.1073/pnas.2134250100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gorke B, Foulquier E, Galinier A. 2005. YvcK of Bacillus subtilis is required for a normal cell shape and for growth on Krebs cycle intermediates and substrates of the pentose phosphate pathway. Microbiology 151:3777–3791. 10.1099/mic.0.28172-0 [DOI] [PubMed] [Google Scholar]
- 14. Foulquier E, Pompeo F, Bernadac A, Espinosa L, Galinier A. 2011. The YvcK protein is required for morphogenesis via localization of PBP1 under gluconeogenic growth conditions in Bacillus subtilis. Mol. Microbiol. 80:309–318. 10.1111/j.1365-2958.2011.07587.x [DOI] [PubMed] [Google Scholar]
- 15. Garner EC, Bernard R, Wang W, Zhuang X, Rudner DZ, Mitchison T. 2011. Coupled, circumferential motions of the cell wall synthesis machinery and MreB filaments in B. subtilis. Science 333:222–225. 10.1126/science.1203285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chang JC, Miner MD, Pandey AK, Gill WP, Harik NS, Sassetti CM, Sherman DR. 2009. igr genes and Mycobacterium tuberculosis cholesterol metabolism. J. Bacteriol. 191:5232–5239. 10.1128/JB.00452-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Griffin JE, Pandey AK, Gilmore SA, Mizrahi V, McKinney JD, Bertozzi CR, Sassetti CM. 2012. Cholesterol catabolism by Mycobacterium tuberculosis requires transcriptional and metabolic adaptations. Chem. Biol. 19:218–227. 10.1016/j.chembiol.2011.12.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Snapper SB, Melton RE, Mustafa S, Kieser T, Jacobs WR., Jr 1990. Isolation and characterization of efficient plasmid transformation mutants of Mycobacterium smegmatis. Mol. Microbiol. 4:1911–1919. 10.1111/j.1365-2958.1990.tb02040.x [DOI] [PubMed] [Google Scholar]
- 19. Raman S, Hazra R, Dascher CC, Husson RN. 2004. Transcription regulation by the Mycobacterium tuberculosis alternative sigma factor SigD and its role in virulence. J. Bacteriol. 186:6605–6616. 10.1128/JB.186.19.6605-6616.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pelicic V, Jackson M, Reyrat J-M, Jacobs WJ, Gicquel B, Guilhot C. 1997. Efficient allelic exchange and transposon mutagenesis in Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U. S. A. 94:10955–10960. 10.1073/pnas.94.20.10955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Blokpoel MC, Murphy HN, O'Toole R, Wiles S, Runn ES, Stewart GR, Young DB, Robertson BD. 2005. Tetracycline-inducible gene regulation in mycobacteria. Nucleic Acids Res. 33:e22. 10.1093/nar/gni023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Guinn KM, Hickey MJ, Mathur SK, Zakel KL, Grotzke JE, Lewinsohn DM, Smith S, Sherman DR. 2004. Individual RD1-region genes are required for export of ESAT-6/CFP-10 and for virulence of Mycobacterium tuberculosis. Mol. Microbiol. 51:359–370. 10.1046/j.1365-2958.2003.03844.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Collins L, Franzblau SG. 1997. Microplate alamar blue assay versus BACTEC 460 system for high-throughput screening of compounds against Mycobacterium tuberculosis and Mycobacterium avium. Antimicrob. Agents Chemother. 41:1004–1009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sampson SL, Dascher CC, Sambandamurthy VK, Russell RG, Jacobs WR, Bloom BR, Hondalus MK. 2004. Protection elicited by a double leucine and pantothenate auxotroph of Mycobacterium tuberculosis in guinea pigs. Infect. Immun. 72:3031–3037. 10.1128/IAI.72.5.3031-3037.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rubin EJ, Akerley BJ, Novik VN, Lampe DJ, Husson RN, Mekalanos JJ. 1999. In vivo transposition of mariner-based elements in enteric bacteria and mycobacteria. Proc. Natl. Acad. Sci. U. S. A. 96:1645–1650. 10.1073/pnas.96.4.1645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sassetti CM, Boyd DH, Rubin EJ. 2001. Comprehensive identification of conditionally essential genes in mycobacteria. Proc. Natl. Acad. Sci. U. S. A. 98:12712–12717. 10.1073/pnas.231275498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Murry JP, Sassetti CM, Lane JM, Xie Z, Rubin EJ. 2008. Transposon site hybridization in Mycobacterium tuberculosis. Methods Mol. Biol. 416:45–59. 10.1007/978-1-59745-321-9_4 [DOI] [PubMed] [Google Scholar]
- 28. Joshi SM, Pandey AK, Capite N, Fortune SM, Rubin EJ, Sassetti CM. 2006. Characterization of mycobacterial virulence genes through genetic interaction mapping. Proc. Natl. Acad. Sci. U. S. A. 103:11760–11765. 10.1073/pnas.0603179103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Prisic S, Dankwa S, Schwartz D, Chou MF, Locasale JW, Kang CM, Bemis G, Church GM, Steen H, Husson RN. 2010. Extensive phosphorylation with overlapping specificity by Mycobacterium tuberculosis serine/threonine protein kinases. Proc. Natl. Acad. Sci. U. S. A. 107:7521–7526. 10.1073/pnas.0913482107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hett EC, Chao MC, Rubin EJ. 2010. Interaction and modulation of two antagonistic cell wall enzymes of mycobacteria. PLoS Pathog. 6:e1001020. 10.1371/journal.ppat.1001020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Daniel RA, Errington J. 2003. Control of cell morphogenesis in bacteria: two distinct ways to make a rod-shaped cell. Cell 113:767–776. 10.1016/S0092-8674(03)00421-5 [DOI] [PubMed] [Google Scholar]
- 32. Aldridge BB, Fernández-Suárez M, Heller D, Ambravaneswaran V, Irimia D, Toner M, Fortune SM. 2012. Asymmetry and aging of mycobacterial cells lead to variable growth and antibiotic susceptibility. Science 335:100–104. 10.1126/science.1216166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Billman-Jacobe H, Haites RE, Coppel RL. 1999. Characterization of a Mycobacterium smegmatis mutant lacking penicillin binding protein 1. Antimicrob. Agents Chemother. 43:3011–3013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Flores AR, Parsons LM, Pavelka MS. 2005. Characterization of novel Mycobacterium tuberculosis and Mycobacterium smegmatis mutants hypersusceptible to beta-lactam antibiotics. J. Bacteriol. 187:1892–1900. 10.1128/JB.187.6.1892-1900.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Parikh A, Verma SK, Khan S, Prakash B, Nandicoori VK. 2009. PknB-mediated phosphorylation of a novel substrate, N-acetylglucosamine-1-phosphate uridyltransferase, modulates its acetyltransferase activity. J. Mol. Biol. 386:451–464. 10.1016/j.jmb.2008.12.031 [DOI] [PubMed] [Google Scholar]
- 36. Griffin JE, Gawronski JD, DeJesus MA, Ioerger TR, Akerley BJ, Sassetti CM. 2011. High-resolution phenotypic profiling defines genes essential for mycobacterial growth and cholesterol catabolism. PLoS Pathog. 7:e1002251. 10.1371/journal.ppat.1002251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Senaratne RH, Sidders B, Sequeira P, Saunders G, Dunphy K, Marjanovic O, Reader JR, Lima P, Chan S, Kendall S, McFadden J, Riley LW. 2008. Mycobacterium tuberculosis strains disrupted in mce3 and mce4 operons are attenuated in mice. J. Med. Microbiol. 57:164–170. 10.1099/jmm.0.47454-0 [DOI] [PubMed] [Google Scholar]
- 38. Jain E, Bairoch A, Duvaud S, Phan I, Redaschi N, Suzek B, Martin M, McGarvey P, Gasteiger E. 2009. Infrastructure for the life sciences: design and implementation of the UniProt website. BMC Bioinformatics 10:136. 10.1186/1471-2105-10-136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Forouhar F, Abashidze M, Xu H, Grochowski L, Seetharaman J, Hussain M, Kuzin A, Chen Y, Zhou W, Xiao R, Acton T, Montelione G, Galinier A, White R, Tong L. 2008. Molecular insights into the biosynthesis of the F420 coenzyme. J. Biol. Chem. 283:11832–11840. 10.1074/jbc.M710352200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Graupner M, Xu H, White R. 2002. Characterization of the 2-phospho-l-lactate transferase enzyme involved in coenzyme F(420) biosynthesis in Methanococcus jannaschii. Biochemistry 41:3754–3761. 10.1021/bi011937v [DOI] [PubMed] [Google Scholar]
- 41. Choi KP, Bair TB, Bae YM, Daniels L. 2001. Use of transposon Tn5367 mutagenesis and a nitroimidazopyran-based selection system to demonstrate a requirement for fbiA and fbiB in coenzyme F(420) biosynthesis by Mycobacterium bovis BCG. J. Bacteriol. 183:7058–7066. 10.1128/JB.183.24.7058-7066.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Stephan J, Bender J, Wolschendorf F, Hoffmann C, Roth E, Mailänder C, Engelhardt H, Niederweis M. 2005. The growth rate of Mycobacterium smegmatis depends on sufficient porin-mediated influx of nutrients. Mol. Microbiol. 58:714–730. 10.1111/j.1365-2958.2005.04878.x [DOI] [PubMed] [Google Scholar]
- 43. Siroy A, Mailaender C, Harder D, Koerber S, Wolschendorf F, Danilchanka O, Wang Y, Heinz C, Niederweis M. 2008. Rv1698 of Mycobacterium tuberculosis represents a new class of channel-forming outer membrane proteins. J. Biol. Chem. 283:17827–17837. 10.1074/jbc.M800866200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Munoz-Elias EJ, McKinney JD. 2005. Mycobacterium tuberculosis isocitrate lyases 1 and 2 are jointly required for in vivo growth and virulence. Nat. Med. 11:638–644. 10.1038/nm1252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Savvi S, Warner DF, Kana BD, McKinney JD, Mizrahi V, Dawes SS. 2008. Functional characterization of a vitamin B12-dependent methylmalonyl pathway in Mycobacterium tuberculosis: implications for propionate metabolism during growth on fatty acids. J. Bacteriol. 190:3886–3895. 10.1128/JB.01767-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Eoh H, Rhee KY. 2014. Methylcitrate cycle defines the bactericidal essentiality of isocitrate lyase for survival of Mycobacterium tuberculosis on fatty acids. Proc. Natl. Acad. Sci. U. S. A. 111:4976–4981. 10.1073/pnas.1400390111 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.