

Abstract
High-density lipoproteins (HDLs) are athero-protective, primarily because of their ability to promote cholesterol flux from peripheral tissues to the liver by reverse cholesterol transport (RCT). The delivery of HDL-cholesteryl esters (CE) into cells is mediated by the HDL receptor, scavenger receptor class B type I (SR-BI), a promising target for enhancing whole body cholesterol disposal and preventing cardiovascular disease. A detailed understanding of the structural determinants underlying proper SR-BI/HDL alignment that supports the selective uptake of HDL-CE into cells remains lacking. To this end, we exploited CD36, a class B scavenger receptor with a predicted topology similar to that of SR-BI that binds HDL but is unable to mediate efficient selective uptake of HDL-CE. We generated a series of SR-BI/CD36 chimeric receptors that span the extracellular (EC) domain of SR-BI to delineate regions that are essential for SR-BI’s cholesterol transport functions. All 16 SR-BI/CD36 chimeras were transiently expressed in COS-7 cells, and their plasma membrane localization was confirmed. The majority of SR-BI/CD36 chimeric receptors displayed significant reductions in their ability to (i) bind HDL, (ii) deliver HDL-CE to cells, (iii) mediate efflux of free cholesterol (FC) to HDL, and (iv) redistribute plasma membrane domains of FC. We also demonstrated that changes in SR-BI function were independent of receptor oligomerization. Altogether, we have identified discrete subdomains, particularly in the N-terminal and C-terminal regions of the EC domain of SR-BI, that are critical for productive receptor–ligand interactions and the various cholesterol transport functions of SR-BI.
For decades, epidemiological studies have reported an inverse relationship between the risk for developing coronary heart disease and plasma concentrations of high-density lipoproteins (HDLs).1,2 Indeed, HDL protects against the development and progression of atherosclerosis by several mechanisms, including reductions in oxidative damage, endothelial dysfunction, and inflammation (reviewed in ref (3)). A growing body of evidence now strongly suggests that the “function” of HDL is a better indicator of cardiovascular risk and has ignited an ongoing debate about whether higher HDL-cholesterol (HDL-C) levels are truly athero-protective.4−6 This is especially important in light of a recent Mendelian randomization study that revealed a lack of association between a lowered risk of myocardial infarction and genetic mechanisms that increase plasma HDL-C levels.7
Relevant to the studies described herein, HDL is also known to be athero-protective primarily by virtue of its role in reverse cholesterol transport (RCT) whereby HDL transports cholesterol from peripheral tissues to the liver for excretion via bile formation.8,9 In the final steps of RCT, scavenger receptor class B type I (SR-BI), the most physiologically relevant HDL receptor,10 facilitates the selective uptake of cholesteryl ester (CE) from HDL into hepatocytes for disposal.11,12 The selective uptake process involves two steps in which (i) HDL must bind to the extracellular (EC) domain of SR-BI and (ii) lipid alone is transferred from HDL to the plasma membrane, without holoparticle uptake.13−15 The selective uptake activity of SR-BI is not dependent on other proteins,16 and our recent evidence of the existence of SR-BI oligomers17,18 supports the notion that HDL-CE uptake occurs via a nonaqueous pathway, possibly involving the formation of a “hydrophobic channel”.19
The anti-atherogenic properties of SR-BI and its ability to promote RCT are firmly established by studies in mice and humans. Hepatic overexpression of SR-BI in mice8,20,21 markedly lowered HDL-C levels, enhanced cholesterol catabolism and excretion, and slowed atherosclerosis.22−24 On the other hand, a 50% reduction in the level of SR-BI expression25 or full disruption of the SR-BI gene26,27 in mice significantly increased plasma HDL-C levels yet dramatically accelerated atherosclerosis.27−29 More recently, mutations of SR-BI, identified in patients with high HDL-C levels,30,31 were associated with impaired cholesterol transport functions.31,32 The correlation of elevated HDL levels with high levels of plasma cholesterol is due to the dramatic defect in RCT resulting from SR-BI deficiency33 and is a perfect example of how the flux of cholesterol is a more important determinant of cardiovascular risk than steady-state levels of HDL-C.
One approach to improving cholesterol flux out of the body, and ultimately HDL “function”, is to enhance the removal of HDL-CE via SR-BI. A critical first step in developing new approaches for promoting the selective uptake of HDL-CE requires a clear understanding of the structural determinants underlying proper SR-BI/HDL alignment via “productive complex” formation.34 SR-BI is a glycosylated cell surface receptor35,36 that consists of a large EC domain anchored by two transmembrane domains and two cytoplasmic domains.37 CD36, a class B scavenger receptor with a predicted topology similar to SR-BI, binds HDL but is unable to mediate efficient HDL-CE selective uptake.13,14 This feature of CD36 was exploited to generate SR-BI/CD36 chimeric receptors.13,14 These “domain swap” chimeras were generated by exchanging the cytoplasmic domains, the transmembrane domains, and/or the EC domain of SR-BI with those of CD36. In these studies,13,14 only chimeras containing the EC domain of SR-BI were able to mediate selective uptake of HDL-CE regardless of the origin of the transmembrane domains and/or cytoplasmic domains, thus establishing that the EC domain of SR-BI is the sole requirement for the selective uptake of HDL-CE. In fact, a spectrum of rather distinct SR-BI-mediated activities appears to be an inherent property of the EC domain of SR-BI, including the bidirectional flux of free cholesterol (FC), increased cellular FC mass, and enhanced sensitivity of FC to exogenous cholesterol oxidase.38,39
To further define and characterize extracellular subdomains within SR-BI that are required for cholesterol transport, we constructed a series of 16 SR-BI/CD36 chimeric receptors that span the entire EC domain of SR-BI to delineate regions that are essential for HDL binding and HDL-CE selective uptake. Because CD36 binds HDL but cannot mediate efficient selective uptake, we hypothesized that our entire panel of SR-BI/CD36 chimeras would bind HDL at levels similar to that of wild-type SR-BI, but only some chimeras would be unable to mediate selective uptake of HDL-CE. We also anticipated that these chimeras would provide insight into which subdomains are required for efflux of FC to HDL and distribution of FC in the plasma membrane.
Experimental Procedures
Materials
The following antibodies were used: anti-SR-BI antibody directed against either the C-terminal cytoplasmic domain or the EC domain (Novus Biologicals, Inc., Littleton, CO), anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (Millipore, Billerica, MA), peroxidase-conjugated goat anti-rabbit secondary IgG, and peroxidase-conjugated sheep anti-mouse secondary IgG (GE Healthcare), fluorescein isothiocyanate-conjugated goat anti-rabbit secondary IgG (BD Biosciences, San Jose, CA), and Alexa633-conjugated goat anti-rabbit secondary IgG (Invitrogen). Human HDL (1.063–1.21 g/mL) was purchased from Biomedical Technologies, Inc. [125I]Sodium iodide and [3H]cholesterol were purchased from PerkinElmer, and [3H]cholesteryl oleyl ether (COE) was purchased from American Radiolabeled Chemicals, Inc. (St. Louis, MO). EZ-Link sulfo-NHS-LC-biotin and streptavidin agarose resin were obtained from Thermo Fisher Scientific (Rockford, IL). Perfluorooctanoic acid (PFO), cholesterol oxidase (Streptomyces), acyl:CoA cholesterol acyltransferase (ACAT) inhibitor (i.e., Sandoz 58-035), and cholesterol, 4-cholesten-3-one, and cholesteryl oleate standards were obtained from Sigma-Aldrich (St. Louis, MO). All other reagents were of analytical grade.
Plasmids
The murine SR-BI (GenBank accession number Q61009) and rat CD36 (GenBank accession number AAH72543) coding regions were cloned into the pSG5 vector (Invitrogen) to produce pSG5[mSR-BI]13and pSG5[rCD36]40 (herein termed SR-BI and CD36, respectively). The following SR-BI/CD36 chimeric receptors were generated by replacing the designated SR-BI sequence with the corresponding region of CD36: SC(51–56), SC(63–71), SC(75–79), SC(85–89), SC(92–98), SC(140–148), SC(153–165), SC(184–192), SC(219–223), SC(232–237), SC(271–277), SC(282–287), SC(297–302), SC(370–375), SC(385–394), and SC(423–434). Cloning, mutagenesis, and sequencing were performed by Top Gene Technologies (Pointe-Claire, QC).
Cell Maintenance and Transfection
COS-7 cells were maintained in DMEM (Invitrogen), 10% calf serum (Invitrogen), 2 mM L-glutamine, 50 units/mL penicillin, 50 μg/mL streptomycin, and 1 mM sodium pyruvate. Cells were transiently transfected with FuGENE 6 (Fisher Scientific) according to the manufacturer’s protocol and as previously described.13 Cells were assayed 48 h post-transfection, with the exception of free cholesterol efflux assays, in which cells were assayed 72 h post-transfection.
Cell Lysis
COS-7 cells transiently expressing wild-type SR-BI or SR-BI/CD36 chimeric receptors were washed twice with cold PBS (pH 7.4) and lysed with 1% NP-40 cell lysis buffer13 containing protease inhibitors (1 μg/mL pepstatin, 0.2 mM phenylmethanesulfonyl fluoride, 1 μg/mL leupeptin, and 10 μg/mL aprotinin). Protein concentrations were determined by the Lowry method as previously described.41
Cell Surface Receptor Expression
Two methods were used to assess receptor expression at the cell surface. Cell surface biotinylation of SR-BI and SR-BI/CD36 chimeric receptors in transiently transfected COS-7 cells was performed as previously described.42 In separate experiments, cell surface expression of SR-BI and SR-BI/CD36 chimeric receptors was verified by flow cytometry as described using the Accuri 6 cytometer (BD Biosciences; Blood Center of Wisconsin) or FACS Calibur (Flow Cytometry Core, Medical College of Wisconsin).43
Receptor Oligomerization
COS-7 cells transiently expressing wild-type SR-BI or SR-BI/CD36 chimeric receptors were lysed in PBS containing protease inhibitors. Receptor oligomerization was assessed by 6% PFO–PAGE as previously described.42
HDL Labeling, Cell Association of [125I]HDL, and Uptake of [3H]HDL-COE
Human HDL was doubly radiolabeled with nonhydrolyzable [3H]COE and [125I]dilactitol tyramine as described previously.13 Preparations of radiolabeled HDL had an average 3H specific activity of 242.25 dpm/ng of protein and an average 125I specific activity of 279.11 dpm/ng of protein. COS-7 cells transiently transfected with cDNA encoding empty pSG5 vector, wild-type SR-BI, or SR-BI/CD36 chimeric receptors were assayed for cell association of [125I]HDL and selective uptake of nonhydrolyzable [3H]COE as previously described.13 Statistical analysis was conducted by one-way analysis of variance (ANOVA).
Free Cholesterol Efflux and Cholesterol Oxidase Sensitivity
COS-7 cells expressing the empty pSG5 vector, wild-type SR-BI, or SR-BI/CD36 chimeric receptors were prelabeled with [3H]cholesterol and assayed for the release of free cholesterol from cells to HDL or for sensitivity to exogenous cholesterol oxidase 72 or 48 h post-transfection, respectively, as previously described.39 Statistical analysis was conducted by one-way ANOVA.
Results
Design of SR-BI/CD36 Chimeras
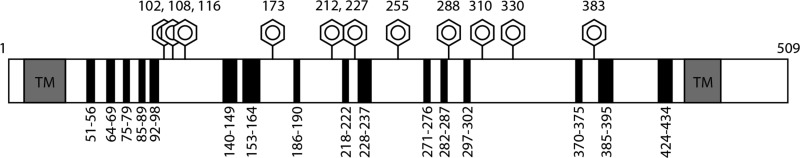
A detailed schematic outlining the location of each chimeric region within the EC domain of SR-BI is shown in Figure 1A. Murine SR-BI and rat CD36 primary sequences were aligned using ClustalW [72% similar sequences (http://www.ebi.ac.uk/Tools/msa/clustalw2/)], and protein secondary structure was predicted using Jpred analysis [http://www.compbio.dundee.ac.uk/www-jpred/ (Figure 1B)]. To generate a panel of chimeric receptors, we chose regions of similar secondary structure, taking care to avoid sites of N-linked glycosylation,35,36 as well as regions containing cysteine residues and disulfide bonds.43 We identified 16 small regions of interest that were suitable for swapping and that spanned the length of SR-BI’s EC domain, with each region ranging in length from 5 to 13 amino acids. Chimeras were named on the basis of the amino acids in the SR-BI sequence that were swapped for the corresponding regions in CD36 as follows: SC(51–56), SC(63–71), SC(75–79), SC(85–89), SC(92–98), SC(140–148), SC(153–165), SC(184–192), SC(219–223), SC(232–237), SC(271–277), SC(282–287), SC(297–302), SC(370–375), SC(385–394), and SC(423–434).
Figure 1.

Schematic and design of SR-BI/CD36 chimeric receptors. (A) Panel of SR-BI/CD36 chimeras created by replacement of small regions of SR-BI sequence with the corresponding CD36 sequence with a similar secondary structure (avoiding sites of glycosylation and disulfide bonds). Shown is a linear representation of SR-BI, with boxed gray regions representing the N- and C-terminal transmembrane domains (TM), black boxes representing all swapped regions (with residue ranges), and hexagons marking residues that harbor N-linked glycosylations. (B) SR-BI (GenBank accession number Q61009) and CD36 (GenBank accession number AAH72543) sequences were aligned using ClustalW, and secondary structure prediction was conducted using Jpred. Boxed regions represent sequences with similar secondary structures. The symbols represent the following: asterisks for fully conserved residues, colons for highly similar residues, periods for weakly similar residues, dollar signs for Cys residues, number signs for Asn residues of the N-linked glycosylation consensus sequence, E for extended (β sheet), H for α helix, and dashes for random coils.
SR-BI/CD36 Chimeric Receptors Are Expressed at the Cell Surface
cDNA encoding wild-type SR-BI and SR-BI/CD36 chimeric receptors was transiently transfected into COS-7 cells and examined for cell surface expression using two different methods. First, we used receptor biotinylation to assess the level of expression of chimeric receptors at the cell surface (Figure 2A). Although all chimeric receptors localize to the cell surface, five chimeras [SC(51–56), SC(85–89), SC(153–165), SC(297–302), and SC(370–375)] exhibited levels of expression lower than that of wild-type SR-BI. With the exception of SC(51–56) and SC(297–302), a lower level of expression for these chimeric receptors was confirmed by flow cytometry analyses using antibodies directed against the EC domain of SR-BI (Figure 2B). Interestingly, flow cytometry analyses also revealed significantly increased levels of expression of SC(92–98), SC(219–223), and SC(282–287), which was not mirrored by our biotinylation experiments.
Figure 2.

SR-BI/CD36 chimeric receptors are expressed at the cell surface. (A) COS-7 cells transiently expressing wild-type SR-BI or SR-BI/CD36 chimeric receptors were assessed for cell surface expression by sulfo-NHS-LC biotinylation. Immunoblot analyses of biotinylated SR-BI at the cell surface (from 150 μg of total lysate) (top panel) and in 20 μg of total cell lysate (middle panel) are shown using an antibody directed against the C-terminal cytoplasmic domain of SR-BI. GAPDH was detected as a loading control (bottom panel). Data are representative of three independent experiments. (B) Surface expression of wild-type SR-BI or SR-BICD36 chimeras in COS-7 cells assessed by flow cytometry using an antibody directed against the extracellular domain of SR-BI. Data are expressed as a percentage of SR-BI expression following subtraction of empty vector values. Data are the average of at least nine independent transfections. As determined by one-way ANOVA, *p = 0.01–0.05, **p < 0.005, and ***p < 0.001.
Chimeric Receptors Display Variable Ability To Bind HDL and Mediate Selective Uptake of HDL-COE
To determine whether swapping regions of the SR-BI EC domain with a corresponding region of CD36 altered SR-BI function, wild-type and chimeric SR-BI/CD36 receptors were tested for their ability to bind HDL and mediate selective uptake of HDL-COE following transient transfection in COS-7 cells. Our analysis revealed that 11 of the 16 chimeric receptors [SC(63–71), SC(75–79), SC(92–98), SC(140–148), SC(153–165), SC(219–223), SC(282–287), SC(297–302), SC(370–375), SC(385–394), and SC(423–434)] displayed a markedly reduced ability to bind HDL [ranging from 6 to 55% of that of wild-type SR-BI (Figure 3A)]. These decreases in the level of binding were accompanied by similar decreases in the rate of selective uptake of HDL-CE (11–46% of that of wild-type SR-BI) (Figure 3B). The SC(271–277) chimeric receptor was also defective in HDL binding and mediating the uptake of HDL-CE; however, the decreases, though statistically significant, were not as drastic. While SC(85–89) bound HDL at levels similar to that of wild-type SR-BI, its ability to mediate the selective uptake of HDL-CE was decreased by 30%. On the other hand, while SC(75–79) and SC(297–302) exhibited statistically significant decreases in their ability to bind HDL, their ability to mediate the selective uptake of HDL-CE was reduced, though not statistically significant.
Figure 3.

HDL binding and selective uptake of HDL-COE in cells expressing wild-type SR-BI and SR-BI/CD36 chimeric receptors. COS-7 cells transiently expressing wild-type SR-BI or SR-BI/CD36 chimeric receptors were incubated at 37 °C for 1.5 h with [125I]DLT/[3H]COE-labeled HDL (10 μg of HDL protein/mL), after which cells were processed. In parallel, cells were plated in separate wells to assess cell surface expression by flow cytometry. Cell-associated HDL (A) and selective uptake of HDL-COE (B), both normalized to cell surface expression, are shown. Data represent means ± SD of at least three independent experiments, each performed in triplicate. As determined by one-way ANOVA, *p = 0.01–0.05, **p < 0.005, and ***p < 0.001.
Several Chimeric Receptors Display a Reduced Ability To Stimulate the Efflux of Free Cholesterol to HDL
Apart from its role in facilitating HDL-CE uptake, SR-BI also has the ability to stimulate the release of free cholesterol from cells.44−46 Therefore, COS-7 cells transiently expressing either wild-type SR-BI or SR-BI/CD36 chimeric receptors were assessed for their ability to stimulate the efflux of FC to HDL acceptors. Analysis of our panel of chimeric receptors revealed that seven chimeras [SC(63–71), SC(92–98), SC(140–148), SC(153–165), SC(219–223), SC(385–394), and SC(423–434)] displayed a marked reduction in their ability to stimulate the efflux of FC to HDL [45–79% decreases in efflux compared to that of wild-type SR-BI (Figure 4)]. As noted above, these same receptors also had an impaired ability to bind HDL and mediate selective uptake of HDL-CE. Interestingly, despite the inability to bind HDL, SC(282–287), SC(297–302), and SC(370–375) showed no differences in their capacity to release FC from cells to HDL as compared to that of wild-type SR-BI.
Figure 4.

Efflux of [3H]cholesterol to HDL by wild-type SR-BI and SR-BI/CD36 chimeric receptors. COS-7 cells transiently expressing empty pSG5 vector, wild-type SR-BI, or SR-BI/CD36 chimeric receptors were prelabeled with [3H]cholesterol and incubated with 50 μg/mL HDL for 4 h at 37 °C to measure the efflux of [3H]cholesterol to the HDL acceptor. Values represent means ± SD of eight independent experiments, each performed in quadruplicate. As determined by one-way ANOVA, *p = 0.01–0.05 and **p < 0.001.
Several Chimeric Receptors Display a Weakened Ability To Redistribute Plasma Membrane Pools of Free Cholesterol
Upon addition of exogenous cholesterol oxidase, a higher membrane content of cholestenone reflects the ability of SR-BI to increase the size of the pool of plasma membrane FC available for oxidation.47 COS-7 cells transiently expressing either wild-type SR-BI or SR-BI/CD36 chimeric receptors were assessed for their ability to redistribute plasma membrane pools of FC upon treatment of cells with exogenous cholesterol oxidase 48 h post-transfection. Analysis of our panel of chimeric receptors revealed that five chimeras [SC(63–71), SC(92–98), SC(140–148), SC(153–165), and SC(219–223)] displayed significant decreases in their levels of cholestenone production (Figure 5), suggesting that these chimeras were defective in their ability to redistribute FC in the plasma membrane. It is interesting to note that receptors harboring chimeric regions in the C-terminal half of the EC domain of SR-BI displayed wild-type levels of cholestenone production, despite having impaired abilities to mediate HDL-CE uptake and/or efflux of FC to HDL. These observations reinforce the notion that the cholesterol transport functions of SR-BI are separable,39 and that individual (possibly overlapping) regions of the EC domain are responsible for each task of SR-BI.
Figure 5.

Sensitivity of cells expressing wild-type SR-BI and SR-BI/CD36 chimeric receptors to cholesterol oxidase. COS-7 cells transiently expressing wild-type SR-BI or SR-BI/CD36 chimeric receptors were prelabeled with [3H]cholesterol and incubated with 0.5 unit of exogenous cholesterol oxidase for 4 h to determine cholesterol oxidase sensitivity. Values represent means ± SD of three independent experiments, each performed in quadruplicate. As determined by one-way ANOVA, *p = 0.01–0.05, **p < 0.005, and ***p < 0.001.
Impaired Function of SR-BI/CD36 Chimeras Is Independent of Oligomer Formation
We17,18 and others48−51 have shown that SR-BI is able to form dimers and higher-order oligomers in cells and tissues. It is hypothesized that oligomerization forms a “hydrophobic channel” that facilitates the delivery of cholesterol from HDL to the plasma membrane.19,48−50 While the putative oligomerization domains are thought to localize to the transmembrane domains,18,51 it is certainly possible that regions within the EC domain of SR-BI may also contribute to SR-BI oligomerization. To determine whether exchanging small EC regions of SR-BI for corresponding areas in CD36 altered the ability of SR-BI to form multimeric complexes and facilitate its normal cholesterol transport functions, we performed PFO–PAGE, a variation of native polyacrylamide gel electrophoresis performed in the presence of perfluorooctanoic acid, that is often used to evaluate the molecular mass of homomultimeric membrane protein complexes.32,52−55 As shown in Figure 6, all chimeric receptors displayed wild-type patterns of oligomerization with bands appearing at ∼75, 150, and >330 kDa, representing the monomer, dimer, and higher-order oligomer species, respectively. While the dimeric and oligomeric bands do not appear clearly for the SC(153–165) chimeric receptor, other blots (not shown) do show formation of these complexes. These data suggest that the amino acids within the swapped extracellular regions are not required for the formation of multimeric SR-BI complexes.
Figure 6.
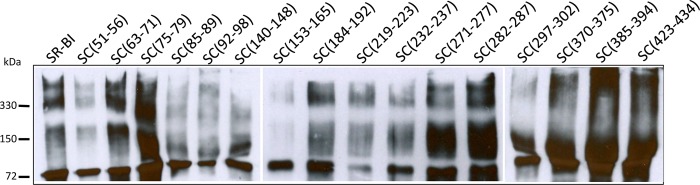
SR-BI/CD36 chimeric receptors display wild-type oligomerization patterns. COS-7 cells transiently expressing wild-type SR-BI or SR-BI/CD36 chimeric receptors were lysed and subjected to 6% PFO–PAGE. Protein bands were detected using an antibody directed against the C-terminal tail of SR-BI.
Discussion
The goal of this study was to design chimeric SR-BI/CD36 receptors that would define the boundaries of small extracellular regions of SR-BI that are responsible for its various cholesterol transport functions. As CD36 binds HDL but is unable to mediate efficient selective uptake of HDL-CE,13,14 we hypothesized that all of the chimeric receptors would bind HDL, but only some would be unable to mediate selective uptake of HDL-COE. Surprisingly, a majority of the 16 chimeric receptors were unable to bind HDL and mediate the selective uptake of HDL-CE. Further, many of these SR-BI/CD36 chimeras were also unable to stimulate the efflux of FC to HDL or enhance the accessibility of membrane FC to exogenous cholesterol oxidase. Together, our findings move the field forward by identifying discrete subdomains of the extracellular domain of SR-BI that are critical for this receptor’s various cholesterol transport functions.
Constructing chimeras between functionally distinct yet structurally homologous receptors is a unique approach to mapping the functional determinants of proteins.56−59 For our studies, the use of SR-BI/CD36 chimeras was ideal as the two proteins belong to the class B family of scavenger receptors and have EC domains sequences that are 72% similar. We used flow cytometry and cell surface biotinylation analyses to demonstrate that all SR-BI/CD36 chimeras were present at the cell surface. The only differences noted were for SC(51–56) and SC(297–302). This discrepancy may be explained by variations in transient expression of the mutant receptor in separate experiments.
Our analyses revealed that the chimeras could be separated into four different groups in terms of HDL binding and selective uptake of HDL-CE. In the first group, SC(51–56), SC(184–192), and SC(232–237) chimeric receptors had no effect on any of the SR-BI-mediated functions that were tested. Interestingly, replacement of residues 85–89 exhibited only 30% less CE uptake, despite 50% less expression of this chimeric receptor at the cell surface. In the second group, as would be expected, defective functions correlated with a decreased level of cell surface expression, but for only SC(153–165) and SC(370–375), suggesting some level of impairment in receptor trafficking. In the third group, impaired abilities to bind HDL and mediate the selective uptake of HDL-CE were observed for SC(63–71), SC(140–148), SC(271–277), SC(297–302), SC(385–394), and SC(423–434), despite normal levels of expression at the cell surface. In the final group, SC(92–98), SC(219–223), and SC(282–287) displayed extremely low levels of HDL binding and selective uptake of HDL-CE despite their high levels of cell surface expression (41–59% higher than that of wild-type SR-BI).
In a previous study, we designed nonconservative and conservative mutations at V67, L140/L142, V164, and V221 to demonstrate that the hydrophobicity of this region of the N-terminal half of the EC domain was critical for SR-BI’s cholesterol transport functions.42 Many of the chimeras tested in this study harbored swapped residues that overlapped with these regions. Kyte–Doolittle analysis60 of these swapped segments confirmed that the hydrophobicity of these regions was indeed altered (data not shown), corroborating our findings that changes in hydrophobicity in this region of the N-terminal half of the EC domain of SR-BI have an impact on receptor function. Recently, Neculai et al. published the crystal structure for the EC domain of LIMP-2,61 a member of the class B scavenger receptor family to which SR-BI and CD36 also belong.62,63 By homology modeling, a structure of the EC domain of SR-BI was inferred and suggested the presence of a three-helix bundle that projects hydrophobic side chains that may be required for HDL binding. Two of these α-helices are fully replaced by our chimeras, SC(140–148) and SC(153–165), and display defective HDL binding. Given that LIMP-2 shares 34% sequence identity and 56% sequence homology with human SR-BI,61 the presence of a helix bundle in this area of SR-BI deserves extensive investigation.
The C-terminal half of the EC domain of SR-BI contains cysteine (Cys) residues that are involved in intramolecular disulfide bonds43,64 that most likely maintain the EC domain of SR-BI in a specific conformation that supports its cholesterol transport functions.43 Although we took care to avoid Cys residues in the design of our chimeric receptors, many SR-BI/CD36 swapped regions are located between Cys251 and Cys384, the first and last extracellular Cys residues, respectively, in the linear sequence of SR-BI. As such, we can speculate that the impaired HDL binding and CE uptake abilities observed with the chimeras within this region may be due to the fact that CD36 residues could alter the conformation of this extracellular region in a manner that prevents proper formation of intramolecular disulfide bonds. Recently, mutation of a highly conserved proline residue at position 297 in SR-BI was discovered in patients with very high HDL-C levels.31 In our studies, SC(297–302) displays an inability to bind HDL and a reduced ability to mediate uptake of HDL-CE, which supports the findings of Vergeer et al.31 The only other known extracellular mutations of human SR-BI are located at Ser112 and Thr17530,32 and do not reside in any of the chimeric receptors generated for this study.
The findings from this study are also consistent with other reports that ascribe the various lipid transport functions of SR-BI to discrete functional extracellular subdomains of the receptor.19,39,42,43,65 For example, three SR-BI/CD36 chimeras [SC(282–287), SC(297–302), and SC(370–375)] exhibited normal levels of efflux compared to that of wild-type SR-BI, despite being unable to bind HDL. The differences in these activities were independent of SR-BI expression at the cell surface at 48 and 72 h, the times post-transfection at which HDL binding and FC efflux were measured, respectively (data not shown). The swapped segments are all located in the same extracellular region that harbors the Cys residues and intramolecular disulfide bonds. The normal levels of efflux parallel our previous findings that suggest this region of the C-terminal half of the EC domain of SR-BI may not be responsible for efflux functions, even with possible disruptions in conformation of this portion of the protein.43 Our findings are also consistent with previous observations that the efflux function of SR-BI is not dependent on HDL binding.43,44 Similarly, all chimeric receptors with swapped regions in the C-terminal half of the EC domain of SR-BI were able to reorganize plasma membrane pools of free cholesterol to an extent similar to that of wild-type SR-BI, as judged by the membrane content of cholestenone upon addition of exogenous cholesterol oxidase. Again, these findings are in agreement with previous studies demonstrating that mutations in this region of SR-BI do not affect the receptor’s ability to maintain accessibility to exogenous cholesterol oxidase42,43 and support the notion that HDL binding is not required for this function of SR-BI.43,66
It has been hypothesized that the delivery of HDL-C to the plasma membrane is facilitated by the formation of SR-BI oligomers.19 We have previously reported that self-association of SR-BI is likely mediated by regions within the C-terminal half of SR-BI.18 Specifically, this region possesses a “GXXXG” dimerization motif, as well as a putative leucine zipper, both of which are known to mediate protein dimerization.67,68 Indeed, SC(423–434) disrupts both of these domains, and most cholesterol transport functions were impaired. However, PFO–PAGE analysis suggests that SC(423–434) and all other chimeras maintain their ability to form oligomers. On the basis of these data, we cannot exclude the possibility that mutation of these motifs may not have been disruptive enough to prevent oligomerization or that these motifs work in concert with other, as yet unidentified, regions of the receptor that are responsible for SR-BI oligomerization. Alternatively, the HDL binding site may differ between the two proteins (i.e., it is located in two noninterchangeable regions of CD36 and SR-BI). On the other hand, if the entire HDL binding site in CD36 was indeed swapped into SR-BI, it is possible that the secondary structure of CD36 differs from that of SR-BI, and this portion of the receptor must interact or fold with other regions of the CD36 EC domain (that are not present within the EC domain of SR-BI) to function properly.
Taken together, our findings confirm that the N-terminal region is required for efficient efflux of FC to HDL and the ability to redistribute FC in the plasma membrane, supporting our previous findings42 and suggesting that the hydrophobicity of this region may be critical for efficient cholesterol transport. Further, our data support the notion that while a specific fold or conformation of the C-terminal half of the EC domain of SR-BI is required for mediating proper binding to HDL and selective uptake of HDL-CE, disruption of this region does not significantly impact the ability of this receptor to facilitate the release of free cholesterol from cells to HDL. Creation of smaller swapped regions between the two receptors or investigation of the role of specific amino acids by site-directed mutagenesis may help further define the discrete boundaries of the subdomains in the EC domain of SR-BI that are responsible for its various cholesterol transport functions.
Acknowledgments
We thank Jeff Woodliff for assisting with the flow cytometry experiments. We are grateful to Dr. Victor Drover for helpful discussions and design of the SR-BI/CD36 chimeras. We thank Dr. Roy Silverstein for critical review of the manuscript.
Glossary
Abbreviations
- CD36
cluster of differentiation 36
- CE
cholesteryl ester
- COE
cholesteryl oleyl ether
- EC
extracellular
- FC
free cholesterol
- HDL
high-density lipoprotein
- HDL-C
high-density lipoprotein-cholesterol
- PAGE
polyacrylamide gel electrophoresis
- PFO
perfluorooctanoic acid
- RCT
reverse cholesterol transport
- SD
standard deviation
- SR-BI
scavenger receptor class B type I.
Author Present Address
⊥ G.A.K.: Blood Research Institute, Blood Center of Wisconsin, Milwaukee, WI, 53226.
This work was supported by National Institutes of Health Grant R01HL058012 (D.S.) and American Heart Association Predoctoral Fellowship 10PRE2790023 (G.A.K.). Generous support was also provided by the Raymond & Bernice Eschenberg Fund of the Greater Milwaukee Foundation (D.S.).
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Gordon T.; Castelli W. P.; Hjortland M. C.; Kannel W. B.; Dawber T. R. (1977) High density lipoprotein as a protective factor against coronary heart disease. Am. J. Med. 62, 707–714. [DOI] [PubMed] [Google Scholar]
- Miller G. J.; Miller N. E. (1975) Plasma high density lipoprotein concentration and the development of ischaemic heart disease. Lancet 1, 16–19. [DOI] [PubMed] [Google Scholar]
- Rye K. A.; Barter P. J. (2014) Cardioprotective functions of HDLs. J. Lipid Res. 55, 168–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Athyros V. G.; Katsiki N.; Karagiannis A.; Mikhailidis D. P. (2012) Should raising high-density lipoprotein cholesterol be a matter of debate?. J. Cardiovasc. Med. (London, U.K.) 13, 254–259. [DOI] [PubMed] [Google Scholar]
- Joy T.; Hegele R. A. (2008) Is raising HDL a futile strategy for atheroprotection?. Nat. Rev. Drug Discovery 7, 143–155. [DOI] [PubMed] [Google Scholar]
- Stylianou I. M.; Bauer R. C.; Reilly M. P.; Rader D. J. (2012) Genetic basis of atherosclerosis: Insights from mice and humans. Circ. Res. 110, 337–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voight B. F.; Peloso G. M.; Orho-Melander M.; Frikke-Schmidt R.; Barbalic M.; Jensen M. K.; Hindy G.; Holm H.; Ding E. L.; Johnson T.; Schunkert H.; Samani N. J.; Clarke R.; Hopewell J. C.; Thompson J. F.; Li M.; Thorleifsson G.; Newton-Cheh C.; Musunuru K.; Pirruccello J. P.; Saleheen D.; Chen L.; Stewart A.; Schillert A.; Thorsteinsdottir U.; Thorgeirsson G.; Anand S.; Engert J. C.; Morgan T.; Spertus J.; Stoll M.; Berger K.; Martinelli N.; Girelli D.; McKeown P. P.; Patterson C. C.; Epstein S. E.; Devaney J.; Burnett M. S.; Mooser V.; Ripatti S.; Surakka I.; Nieminen M. S.; Sinisalo J.; Lokki M. L.; Perola M.; Havulinna A.; de Faire U.; Gigante B.; Ingelsson E.; Zeller T.; Wild P.; de Bakker P. I.; Klungel O. H.; Maitland-van der Zee A. H.; Peters B. J.; de Boer A.; Grobbee D. E.; Kamphuisen P. W.; Deneer V. H.; Elbers C. C.; Onland-Moret N. C.; Hofker M. H.; Wijmenga C.; Verschuren W. M.; Boer J. M.; van der Schouw Y. T.; Rasheed A.; Frossard P.; Demissie S.; Willer C.; Do R.; Ordovas J. M.; Abecasis G. R.; Boehnke M.; Mohlke K. L.; Daly M. J.; Guiducci C.; Burtt N. P.; Surti A.; Gonzalez E.; Purcell S.; Gabriel S.; Marrugat J.; Peden J.; Erdmann J.; Diemert P.; Willenborg C.; Konig I. R.; Fischer M.; Hengstenberg C.; Ziegler A.; Buysschaert I.; Lambrechts D.; Van de Werf F.; Fox K. A.; El Mokhtari N. E.; Rubin D.; Schrezenmeir J.; Schreiber S.; Schafer A.; Danesh J.; Blankenberg S.; Roberts R.; McPherson R.; Watkins H.; Hall A. S.; Overvad K.; Rimm E.; Boerwinkle E.; Tybjaerg-Hansen A.; Cupples L. A.; Reilly M. P.; Melander O.; Mannucci P. M.; Ardissino D.; Siscovick D.; Elosua R.; Stefansson K.; O’Donnell C. J.; Salomaa V.; Rader D. J.; Peltonen L.; Schwartz S. M.; Altshuler D.; Kathiresan S. (2012) Plasma HDL cholesterol and risk of myocardial infarction: A Mendelian randomisation study. Lancet 380, 572–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji Y.; Wang N.; Ramakrishnan R.; Sehayek E.; Huszar D.; Breslow J. L.; Tall A. R. (1999) Hepatic scavenger receptor BI promotes rapid clearance of high density lipoprotein free cholesterol and its transport into bile. J. Biol. Chem. 274, 33398–33402. [DOI] [PubMed] [Google Scholar]
- Kozarsky K. F.; Donahee M. H.; Rigotti A.; Iqbal S. N.; Edelman E. R.; Krieger M. (1997) Overexpression of the HDL receptor SR-BI alters plasma HDL and bile cholesterol levels. Nature 387, 414–417. [DOI] [PubMed] [Google Scholar]
- Acton S.; Rigotti A.; Landschulz K. T.; Xu S.; Hobbs H. H.; Krieger M. (1996) Identification of scavenger receptor SR-BI as a high density lipoprotein receptor. Science 271, 518–520. [DOI] [PubMed] [Google Scholar]
- Brundert M.; Ewert A.; Heeren J.; Behrendt B.; Ramakrishnan R.; Greten H.; Merkel M.; Rinninger F. (2005) Scavenger receptor class B type I mediates the selective uptake of high-density lipoprotein-associated cholesteryl ester by the liver in mice. Arterioscler., Thromb., Vasc. Biol. 25, 143–148. [DOI] [PubMed] [Google Scholar]
- Out R.; Hoekstra M.; Spijkers J. A.; Kruijt J. K.; van Eck M.; Bos I. S.; Twisk J.; Van Berkel T. J. (2004) Scavenger receptor class B type I is solely responsible for the selective uptake of cholesteryl esters from HDL by the liver and the adrenals in mice. J. Lipid Res. 45, 2088–2095. [DOI] [PubMed] [Google Scholar]
- Connelly M. A.; Klein S. M.; Azhar S.; Abumrad N. A.; Williams D. L. (1999) Comparison of class B scavenger receptors, CD36 and SR-BI, shows that both receptors mediate HDL-cholesteryl ester selective uptake but SR-BI exhibits a unique enhancement of cholesteryl ester uptake. J. Biol. Chem. 274, 41–47. [DOI] [PubMed] [Google Scholar]
- Gu X.; Trigatti B.; Xu S.; Acton S.; Babitt J.; Krieger M. (1998) The efficient cellular uptake of high density lipoprotein lipids via scavenger receptor class B type I requires not only receptor-mediated surface binding but also receptor-specific lipid transfer mediated by its extracellular domain. J. Biol. Chem. 273, 26338–26348. [DOI] [PubMed] [Google Scholar]
- Pittman R. C.; Knecht T. P.; Rosenbaum M. S.; Taylor C. A. Jr. (1987) A nonendocytotic mechanism for the selective uptake of high density lipoprotein-associated cholesterol esters. J. Biol. Chem. 262, 2443–2450. [PubMed] [Google Scholar]
- Liu B.; Krieger M. (2002) Highly purified scavenger receptor class B, type I reconstituted into phosphatidylcholine/cholesterol liposomes mediates high affinity high density lipoprotein binding and selective lipid uptake. J. Biol. Chem. 277, 34125–34135. [DOI] [PubMed] [Google Scholar]
- Sahoo D.; Darlington Y. F.; Pop D.; Williams D. L.; Connelly M. A. (2007) Scavenger receptor class B type I (SR-BI) assembles into detergent-sensitive dimers and tetramers. Biochim. Biophys. Acta 1771, 807–817. [DOI] [PubMed] [Google Scholar]
- Sahoo D.; Peng Y.; Smith J. R.; Darlington Y. F.; Connelly M. A. (2007) Scavenger receptor class B, type I (SR-BI) homo-dimerizes via its C-terminal region: Fluorescence resonance energy transfer analysis. Biochim. Biophys. Acta 1771, 818–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigueza W. V.; Thuahnai S. T.; Temel R. E.; Lund-Katz S.; Phillips M. C.; Williams D. L. (1999) Mechanism of scavenger receptor class B type I-mediated selective uptake of cholesteryl esters from high density lipoprotein to adrenal cells. J. Biol. Chem. 274, 20344–20350. [DOI] [PubMed] [Google Scholar]
- Ueda Y.; Gong E.; Royer L.; Cooper P.; Francone O.; Rubin E. M. (2000) Relationship between Expression Levels and Atherogenesis in Scavenger Receptor Class B, Type I Transgenics. J. Biol. Chem. 275, 20368–20373. [DOI] [PubMed] [Google Scholar]
- Wang N.; Arai T.; Ji Y.; Rinninger F.; Tall A. R. (1998) Liver-specific overexpression of scavenger receptor BI decreases levels of very low density lipoprotein apoB, low density lipoprotein apoB, and high density lipoprotein in transgenic mice. J. Biol. Chem. 273, 32920–32926. [DOI] [PubMed] [Google Scholar]
- Arai T.; Rinninger F.; Varban L.; Fairchild-Huntress V.; Liang C. P.; Chen W.; Seo T.; Deckelbaum R.; Huszar D.; Tall A. R. (1999) Decreased selective uptake of high density lipoprotein cholesteryl esters in apolipoprotein E knock-out mice. Proc. Natl. Acad. Sci. U.S.A. 96, 12050–12055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozarsky K. F.; Donahee M. H.; Glick J. M.; Krieger M.; Rader D. J. (2000) Gene transfer and hepatic overexpression of the HDL receptor SR-BI reduces atherosclerosis in the cholesterol-fed LDL receptor-deficient mouse. Arterioscler., Thromb., Vasc. Biol. 20, 721–727. [DOI] [PubMed] [Google Scholar]
- Ueda Y.; Gong E.; Royer L.; Cooper P. N.; Francone O. L.; Rubin E. M. (2000) Relationship between expression levels and atherogenesis in scavenger receptor class B, type I transgenics. J. Biol. Chem. 275, 20368–20373. [DOI] [PubMed] [Google Scholar]
- Varban M. L.; Rinninger F.; Wang N.; Fairchild-Huntress V.; Dunmore J. H.; Fang Q.; Gosselin M. L.; Dixon K. L.; Deeds J. D.; Acton S. L.; Tall A. R.; Huszar D. (1998) Targeted mutation reveals a central role of SR-BI in hepatic selective uptake of high density lipoprotein cholesterol. Proc. Natl. Acad. Sci. U.S.A. 95, 4619–4624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigotti A.; Trigatti B. L.; Penman M.; Rayburn H.; Herz J.; Krieger M. (1997) A targeted mutation in the murine gene encoding the high density lipoprotein (HDL) receptor scavenger receptor class B type I reveals its key role in HDL metabolism. Proc. Natl. Acad. Sci. U.S.A. 94, 12610–12615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trigatti B.; Rayburn H.; Vinals M.; Braun A.; Miettinen H. (1999) Influence of the high density lipoprotein receptor SR-BI on reproductive and cardiovascular pathophysiology. Proc. Natl. Acad. Sci. U.S.A. 96, 9322–9327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun A.; Trigatti B. L.; Post M. J.; Sato K.; Simons M.; Edelberg J. M.; Rosenberg R. D.; Schrenzel M.; Krieger M. (2002) Loss of SR-BI expression leads to the early onset of occlusive atherosclerotic coronary artery disease, spontaneous myocardial infarctions, severe cardiac dysfunction, and premature death in apolipoprotein E-deficient mice. Circ. Res. 90, 270–276. [DOI] [PubMed] [Google Scholar]
- Covey S. D.; Krieger M.; Wang W.; Penman M.; Trigatti B. L. (2003) Scavenger receptor class B type I-mediated protection against atherosclerosis in LDL receptor-negative mice involves its expression in bone marrow-derived cells. Arterioscler., Thromb., Vasc. Biol. 23, 1589–1594. [DOI] [PubMed] [Google Scholar]
- Brunham L. R.; Tietjen I.; Bochem A. E.; Singaraja R. R.; Franchini P. L.; Radomski C.; Mattice M.; Legendre A.; Hovingh G. K.; Kastelein J. J.; Hayden M. R. (2011) Novel mutations in scavenger receptor BI associated with high HDL cholesterol in humans. Clin. Genet. 79, 575–581. [DOI] [PubMed] [Google Scholar]
- Vergeer M.; Korporaal S. J.; Franssen R.; Meurs I.; Out R.; Hovingh G. K.; Hoekstra M.; Sierts J. A.; Dallinga-Thie G. M.; Motazacker M. M.; Holleboom A. G.; Van Berkel T. J.; Kastelein J. J.; Van Eck M.; Kuivenhoven J. A. (2011) Genetic variant of the scavenger receptor BI in humans. N. Engl. J. Med. 364, 136–145. [DOI] [PubMed] [Google Scholar]
- Chadwick A. C.; Sahoo D. (2012) Functional Characterization of Newly-Discovered Mutations in Human SR-BI. PLoS One 7, e45660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.; Da Silva J. R.; Reilly M.; Billheimer J. T.; Rothblat G. H.; Rader D. J. (2005) Hepatic expression of scavenger receptor class B type I (SR-BI) is a positive regulator of macrophage reverse cholesterol transport in vivo. J. Clin. Invest. 115, 2870–2874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T.; Krieger M.; Kan H. Y.; Zannis V. I. (2002) The effects of mutations in helices 4 and 6 of apoA-I on SR-BI-mediated cholesterol efflux suggest that formation of a productive complex between reconstituted HDL and SR-BI is required for efficient lipid transport. J. Biol. Chem. 277, 21578–21584. [DOI] [PubMed] [Google Scholar]
- Babitt J.; Trigatti B.; Rigotti A.; Smart E.; Anderson R. G. W.; Xu S.; Krieger M. (1997) Murine SR-BI, a high density lipoprotein receptor that mediates selective lipid uptake, is N-glycosylated and fatty acylated and colocalizes with plasma membrane caveolae. J. Biol. Chem. 272, 13242–13249. [DOI] [PubMed] [Google Scholar]
- Vinals M.; Xu S.; Vasile E.; Krieger M. (2003) Identification of the N-Linked Glycosylation Sites on the High Density Lipoprotein (HDL) Receptor SR-BI and Assessment of Their Effects on HDL Binding and Selective Lipid Uptake. J. Biol. Chem. 278, 5325–5332. [DOI] [PubMed] [Google Scholar]
- Krieger M. (1999) Charting the fate of the “good cholesterol”: Identification and characterization of the high-density lipoprotein receptor SR-BI. Annu. Rev. Biochem. 68, 523–558. [DOI] [PubMed] [Google Scholar]
- Connelly M. A.; de la Llera-Moya M.; Monzo P.; Yancey P.; Drazul D.; Stoudt G.; Fournier N.; Klein S. M.; Rothblat G. H.; Williams D. L. (2001) Analysis of chimeric receptors shows that multiple distinct functional activities of scavenger receptor, class B, type I (SR-BI) are localized to the extracellular receptor domain. Biochemistry 40, 5249–5259. [DOI] [PubMed] [Google Scholar]
- Connelly M. A.; de la Llera-Moya M.; Peng Y.; Drazul-Schrader D.; Rothblat G. H.; Williams D. L. (2003) Separation of lipid transport functions by mutations in the extracellular domain of scavenger receptor class B, type I (SR-BI). J. Biol. Chem. 278, 25773–25782. [DOI] [PubMed] [Google Scholar]
- Ibrahimi A.; Sfeir Z.; Magharaie H.; Amri E. Z.; Grimaldi P.; Abumrad N. A. (1996) Expression of the CD36 homolog (FAT) in fibroblast cells: Effects on fatty acid transport. Proc. Natl. Acad. Sci. U.S.A. 93, 2646–2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowry O. H.; Rosebrough N. J.; Farr A. L.; Randall R. J. (1951) Protein measurement with the folin phenol reagent. J. Biol. Chem. 193, 265–275. [PubMed] [Google Scholar]
- Papale G. A.; Nicholson K.; Hanson P. J.; Pavlovic M.; Drover V. A.; Sahoo D. (2010) Extracellular hydrophobic regions in scavenger receptor BI play a key role in mediating HDL-cholesterol transport. Arch. Biochem. Biophys. 496, 132–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papale G. A.; Hanson P. J.; Sahoo D. (2011) Extracellular disulfide bonds support scavenger receptor class B type I-mediated cholesterol transport. Biochemistry 50, 6245–6254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Llera-Moya M.; Rothblat G. H.; Connelly M. A.; Kellner-Weibel G.; Sakar S. W.; Phillips M. C.; Williams D. L. (1999) Scavenger receptor BI (SR-BI) mediates free cholesterol flux independently of HDL tethering to the cell surface. J. Lipid Res. 40, 575–580. [PubMed] [Google Scholar]
- Ji Y.; Jian B.; Wang N.; Sun Y.; de la Llera Moya M.; Phillips M. C.; Rothblat G. H.; Swaney J. B.; Tall A. R. (1997) Scavenger receptor BI promotes high density lipoprotein-mediated cellular cholesterol efflux. J. Biol. Chem. 272, 20982–20985. [DOI] [PubMed] [Google Scholar]
- Jian B.; de la Llera-Moya M.; Ji Y.; Wang N.; Phillips M. C.; Swaney J. B.; Tall A. R.; Rothblat G. H. (1998) Scavenger receptor class B type I as a mediator of cellular cholesterol efflux to lipoproteins and phospholipid acceptors. J. Biol. Chem. 273, 5599–5606. [DOI] [PubMed] [Google Scholar]
- Kellner-Weibel G.; de la Llera-Moya M.; Connelly M. A.; Stoudt G.; Christian A. E.; Haynes M. P.; Williams D. L.; Rothblat G. H. (2000) Expression of scavenger receptor BI in COS-7 cells alters cholesterol content and distribution. Biochemistry 39, 221–229. [DOI] [PubMed] [Google Scholar]
- Azhar S.; Nomoto A.; Reaven E. (2002) Hormonal regulation of adrenal microvillar channel formation. J. Lipid Res. 43, 861–871. [PubMed] [Google Scholar]
- Landschulz K. T.; Pathak R. K.; Rigotti A.; Krieger M.; Hobbs H. H. (1996) Regulation of scavenger receptor, class B, type I, a high density lipoprotein receptor, in liver and steroidogenic tissues of the rat. J. Clin. Invest. 98, 984–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reaven E.; Cortez Y.; Leers-Sucheta S.; Nomoto A.; Azhar S. (2004) Dimerization of the scavenger receptor class B type I (SR-BI): Formation, function and localization in diverse cells and tissues. J. Lipid Res. 45, 513–528. [DOI] [PubMed] [Google Scholar]
- Gaidukov L.; Nager A. R.; Xu S.; Penman M.; Krieger M. (2011) Glycine dimerization motif in the N-terminal transmembrane domain of the high density lipoprotein receptor SR-BI required for normal receptor oligomerization and lipid transport. J. Biol. Chem. 286, 18452–18464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dragoni I.; Guida E.; McIntyre P. (2006) The cold and menthol receptor TRPM8 contains a functionally important double cysteine motif. J. Biol. Chem. 281, 37353–37360. [DOI] [PubMed] [Google Scholar]
- Kedei N.; Szabo T.; Lile J. D.; Treanor J. J.; Olah Z.; Iadarola M. J.; Blumberg P. M. (2001) Analysis of the native quaternary structure of vanilloid receptor 1. J. Biol. Chem. 276, 28613–28619. [DOI] [PubMed] [Google Scholar]
- Ramjeesingh M.; Huan L. J.; Garami E.; Bear C. E. (1999) Novel method for evaluation of the oligomeric structure of membrane proteins. Biochem. J. 342(Part 1), 119–123. [PMC free article] [PubMed] [Google Scholar]
- Xu J.; Liu Y.; Yang Y.; Bates S.; Zhang J. T. (2004) Characterization of oligomeric human half-ABC transporter ATP-binding cassette G2. J. Biol. Chem. 279, 19781–19789. [DOI] [PubMed] [Google Scholar]
- Schumacher R.; Soos M. A.; Schlessinger J.; Brandenburg D.; Siddle K.; Ullrich A. (1993) Signaling-competent receptor chimeras allow mapping of major insulin receptor binding domain determinants. J. Biol. Chem. 268, 1087–1094. [PubMed] [Google Scholar]
- Allsopp R. C.; Lalo U.; Evans R. J. (2010) Lipid raft association and cholesterol sensitivity of P2X1-4 receptors for ATP: Chimeras and point mutants identify intracellular amino-terminal residues involved in lipid regulation of P2X1 receptors. J. Biol. Chem. 285, 32770–32777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez A.; Wellbrock C.; Gutbrod H.; Dimitrijevic N.; Schartl M. (2001) Ligand-independent dimerization and activation of the oncogenic Xmrk receptor by two mutations in the extracellular domain. J. Biol. Chem. 276, 3333–3340. [DOI] [PubMed] [Google Scholar]
- Hawtin S. R.; Wesley V. J.; Parslow R. A.; Patel S.; Wheatley M. (2000) Critical role of a subdomain of the N-terminus of the V1a vasopressin receptor for binding agonists but not antagonists; functional rescue by the oxytocin receptor N-terminus. Biochemistry 39, 13524–13533. [DOI] [PubMed] [Google Scholar]
- Kyte J.; Doolittle R. F. (1982) A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 157, 105–132. [DOI] [PubMed] [Google Scholar]
- Neculai D.; Schwake M.; Ravichandran M.; Zunke F.; Collins R. F.; Peters J.; Neculai M.; Plumb J.; Loppnau P.; Pizarro J. C.; Seitova A.; Trimble W. S.; Saftig P.; Grinstein S.; Dhe-Paganon S. (2013) Structure of LIMP-2 provides functional insights with implications for SR-BI and CD36. Nature 504, 172–176. [DOI] [PubMed] [Google Scholar]
- Calvo D.; Dopazo J.; Vega M. A. (1995) The CD36, CLA-1 (CD36L1), and LIMPII (CD36L2) gene family: Cellular distribution, chromosomal location, and genetic evolution. Genomics 25, 100–106. [DOI] [PubMed] [Google Scholar]
- Calvo D.; Vega M. A. (1993) Identification, primary structure, and distribution of CLA-1, a novel member of the CD36/LIMPII gene family. J. Biol. Chem. 268, 18929–18935. [PubMed] [Google Scholar]
- Yu M.; Lau T. Y.; Carr S. A.; Krieger M. (2012) Contributions of a disulfide bond and a reduced cysteine side chain to the intrinsic activity of the high-density lipoprotein receptor SR-BI. Biochemistry 51, 10044–10055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temel R. E.; Trigatti B.; DeMattos R. B.; Azhar S.; Krieger M.; Williams D. L. (1997) Scavenger receptor B, type I (SR-BI) is the major route for the delivery of high density lipoprotein cholesterol to the steroidogenic pathway in cultured mouse adrenocortical cells. Proc. Natl. Acad. Sci. U.S.A. 94, 13600–13605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu X.; Kozarsky K.; Krieger M. (2000) Scavenger receptor, class B, type I-mediated [3H]cholesterol efflux to high density and low density lipoproteins is dependent on lipoprotein binding to the receptor. J. Biol. Chem. 275, 29993–30001. [DOI] [PubMed] [Google Scholar]
- Russ W. P.; Engelman D. M. (2000) The GxxxG motif: A framework for transmembrane helix-helix association. J. Mol. Biol. 296, 911–919. [DOI] [PubMed] [Google Scholar]
- Fink A.; Sal-Man N.; Gerber D.; Shai Y. (2012) Transmembrane domains interactions within the membrane milieu: Principles, advances and challenges. Biochim. Biophys. Acta 1818, 974–983. [DOI] [PubMed] [Google Scholar]