

SUMMARY
While conceptual principles governing plant immunity are becoming clear, its systems-level organization and the evolutionary dynamic of the host-pathogen interface are still obscure. We generated a systematic protein-protein interaction network of virulence effectors from the ascomycete pathogen Golovinomyces orontii and Arabidopsis thaliana host proteins. We combined this dataset with corresponding data for the eubacterial pathogen Pseudomonas syringae and the oomycete pathogen Hyaloperonospora arabidopsidis. The resulting network identifies host proteins onto which intraspecies and interspecies pathogen effectors converge. Phenotyping of 124 Arabidopsis effector-interactor mutants revealed a correlation between intra- and interspecies convergence and several altered immune response phenotypes. The effectors and most heavily targeted host protein co-localized in sub-nuclear foci. Products of adaptively selected Arabidopsis genes are enriched for interactions with effector targets. Our data suggest the existence of a molecular host-pathogen interface that is conserved across Arabidopsis accessions, while evolutionary adaptation occurs in the immediate network neighborhood of effector targets.
INTRODUCTION
The spread of pathogens is predicted to change in the wake of global warming, generating emerging epidemics threatening human welfare and security. Plants evolved a sophisticated two-layered defense system to detect and defend against the majority of potential pathogens (Chisholm et al., 2006; Dodds and Rathjen, 2010; Jones and Dangl, 2006). Activation of extracellular plant pattern-recognition receptor kinases by highly conserved microbe-associated molecular patterns (MAMPs) triggers a complex cellular response termed MAMP-triggered immunity (MTI) that can stop microbial proliferation. Host-adapted pathogens are equipped with diverse suites of virulence effectors, which are delivered into the plant cell by various and mostly poorly understood means. Effector proteins interact with host proteins to undermine MTI and to modify host physiology, thus enhancing pathogen proliferation (Feng and Zhou, 2012; Raffaele and Kamoun, 2012; Win et al., 2012). Plants evolved highly polymorphic intracellular nucleotide-binding site, leucine-rich repeat (NLR) proteins to recognize intracellular effectors. Activation of NLRs is also poorly understood in all but a few cases, but can proceed by either direct effector-NLR interaction or upon effector modification of an NLR-associated host target protein. NLR activation results essentially in a more rapid and higher amplitude MTI output known as effector-triggered immunity (ETI) (Chisholm et al., 2006; Dodds and Rathjen, 2010; Jones and Dangl, 2006).
While the conceptual principles of the plant immune system have been elucidated, knowledge of its systems-level organization and the evolutionary dynamic of the molecular host-pathogen interface are rudimentary. Filling this gap represents an important goal in the quest for targeted crop improvement (Dangl et al., 2013; Pardey et al., 2013). The reference eudicot Arabidopsis thaliana (hereafter Arabidopsis) is host to a range of evolutionary diverse pathogens including bacteria, oomycetes and fungi. We defined an effector-host network featuring protein interactions between effectors from the oomycete Hyaloperonospora arabidopsidis (Hpa) and the eubacterium Pseudomonas syringae (Psy) and 8,000 Arabidopsis host proteins and integrated this with a first generation Arabidopsis interactome map (Consortium, 2011; Mukhtar et al., 2011). Our results suggested that effectors from these evolutionarily diverse pathogens converge onto common host proteins, which were characterized by a high interaction degree and a central position in the host protein network. Immune function was demonstrated for 15/17 proteins that are effector targets shared by both pathogens (Mukhtar et al., 2011). It remained to be determined whether the effector convergence onto common targets extended to Arabidopsis pathogens from other kingdoms from the microbial tree of life, how effector convergence related broadly to phenotypic relevance and how the central network position of many targeted host proteins accommodated the selective pressure imposed by pathogens.
While the effector set of facultative phytopathogenic bacteria like Psy is confined to 10–40 effectors per strain (Baltrus et al., 2011), the genomes of eukaryote obligate biotrophic plant-pathogens encode extensive apparent effector arsenals (Baxter et al., 2010; Raffaele and Kamoun, 2012; Win et al., 2012). Only a minute fraction of these are functionally characterized, largely because genetic screens are challenging or as yet impossible in most of these organisms. The increased availability of genome sequences and improved bioinformatic prediction pipelines facilitate identification of proteins carrying signatures of virulence effectors. Recent additions include the powdery mildew fungi, an economically important class of plant pathogens with annotated candidate effector repertoires in several species, strains, and formae speciales (Hacquard et al., 2013; Pedersen et al., 2012; Wicker et al., 2013). The obligate biotrophic ascomycete Golovinomyces orontii (Gor) causes powdery mildew on Arabidopsis (Micali et al., 2008). The available genome resources make Gor an excellent pathogen to identify effectors and their interacting host proteins. Additionally, this ascomycete belongs to a different taxonomic kingdom than the eubacterium Psy and the stramenopile Hpa (Figure 1A), providing an evolutionary contrast from which to challenge the principles we previously proposed regarding the structure of a plant-pathogen interactome network (Mukhtar et al., 2011).
Figure 1. Gor effector identification and interactome mapping.
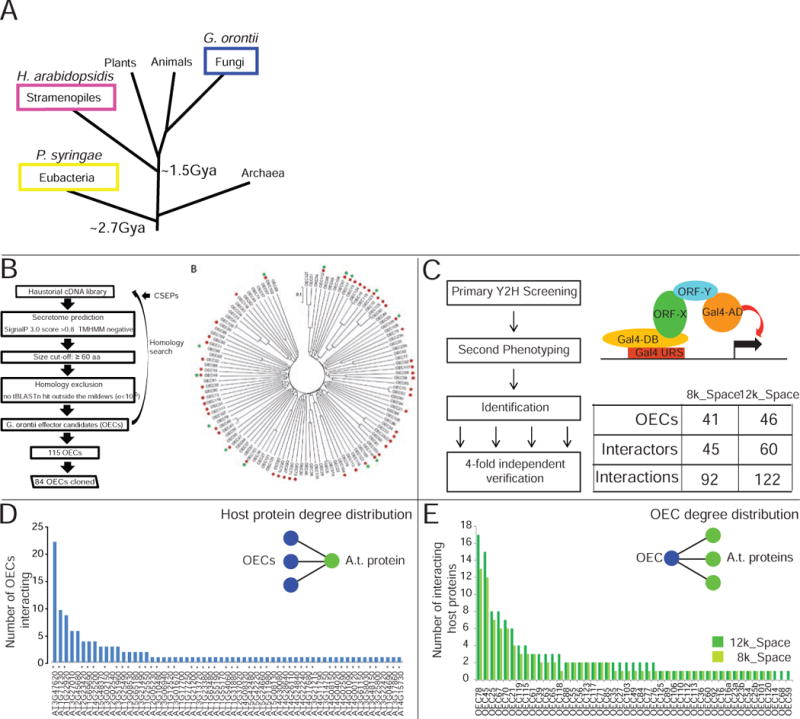
A. Golovinomyces orontii is a pathogenic ascomycete that diverged approximately 2.7 and 1.5 billion years ago (Gya), respectively, from the other Arabidopsis pathogens (Kemen and Jones, 2012; Markow, 2005). B. Effector identification pipeline and family relationships of identified and cloned Gor effector candidates (OEC) and the presence of homologs in the powdery mildews Blumeria graminis f. sp. hordei (green dots) and Erysiphe pisi (red dots). C. Our Y2H pipeline consists of three interrogation steps: screening, phenotyping and four-fold verification, resulting in the indicated number of interactions. D. Degree distribution of Arabidopsis proteins interacting with OECs. Asterisks indicate 8k_space proteins. E. Degree distribution of OECs interacting with Arabidopsis proteins in the 8k_space (light green) and 12K_space (dark green). See also Figure S1, Table S1.
RESULTS
Definition and cloning of candidate Golovinomyces orontii virulence effectors
We identified Gor effector candidates (OECs) iteratively using a bioinformatic pipeline (Figure 1B). Using several criteria including a size cut-off, presence of an N-terminal secretion signal and, due to the rapidly evolving nature of effectors, a lack of homology outside the powdery mildew fungi, we predicted 103 OECs de novo from a sequenced haustorial Gor cDNA library (Weßling et al., 2012). This set of OECs, and candidate effector sequences from the barley powdery mildew Blumeria graminis f. sp. hordei (Bgh), were then used as templates for iterative homology searches in the same haustorial cDNA library (Pedersen et al., 2012; Weßling et al., 2012). These iterations identified two and ten additional OECs, respectively, yielding a total of 115 OECs. Sequence relatedness among Gor effectors were usually low (Figure 1B). Sequences with similarity to OECs were rare in the Bgh genome, but frequent in the genome of the pea powdery mildew Erysiphe pisi (Figure 1B). This pattern is consistent with the different evolutionary distances between Gor and the two other powdery mildew species and suggests that conserved effectors may constitute elements of a putative powdery mildew core effector set. We subsequently cloned the Open Reading Frames (ORFs) of mature OECs without signal peptides into Gateway Entry vectors and verified the identity of 84 full-length OECs clones (73% success rate) (Table S1).
Systematic host-protein interactome mapping using the G. orontii effector candidates
We defined protein-protein interactions between 69 OECs and 12,000 Arabidopsis proteins encoded by sequence-verified ORFs (12k_space) (15 of the 84 OECs auto-activated and were not screened). A subset of 8,000 ORFs (8k_space) was previously used to generate the Arabidopsis Interactome 1 (AI-1) and the Plant-Pathogen Immune Network 1 (PPIN-1) (Consortium, 2011; Mukhtar et al., 2011). We used a stringent Y2H mapping pipeline, which yields high-quality data with a low false-discovery rate (Braun, 2012; Dreze et al., 2010) (Figure 1C), to assemble a Gor effector-host interactome network (Gor_EHIn12k) (Figure S1, Table S1). The subset of interactions of OECs ith host-proteins within the previously screened 8k_space is denoted Gor_EHIn8k (Figure S1). In Gor_EHIn12k we found on average 2.3 interaction partners for positive OECs (Figure 1E); 38 OECs yielded no interactor. Conversely, 16 host proteins interacted with multiple, typically phylogenetically unrelated, OECs (Figure 1D, Figure S1C). This new dataset allowed us to consider whether the host interactors of OECs included previously observed interactors of oomycete and bacterial effectors, and whether the convergence previously observed for Psy and Hpa extended to the Gor effector host network.
Integrated network map reveals interspecies effector convergence onto shared host proteins
The interactions of effectors from all three pathogens (Gor, Hpa and Psy) with host proteins were integrated with interactions among host proteins from AI-1, PPIN-1 and the literature, to yield a comprehensive Plant-Pathogen Immune Network 2 (PPIN-2) (Figure S2, Table S1). To generate a network produced with identical experimental parameters, effector interactions with the Arabidopsis 8k_space proteins were extracted and integrated with their mutual interactions from the systematic AI-1MAIN dataset to yield PPIN-28k_sys (Figure 2A, Table S1). PPIN-28k_sys consists of 178 Arabidopsis host proteins and 123 effectors connected by 421 effector host-protein interactions and 162 interactions among host proteins (Figure S1B). PPIN-28k_sys was used for all subsequent statistical analyses unless otherwise noted. An overview describing the different datasets is provided in Figure S1.
Figure 2. Network integration.
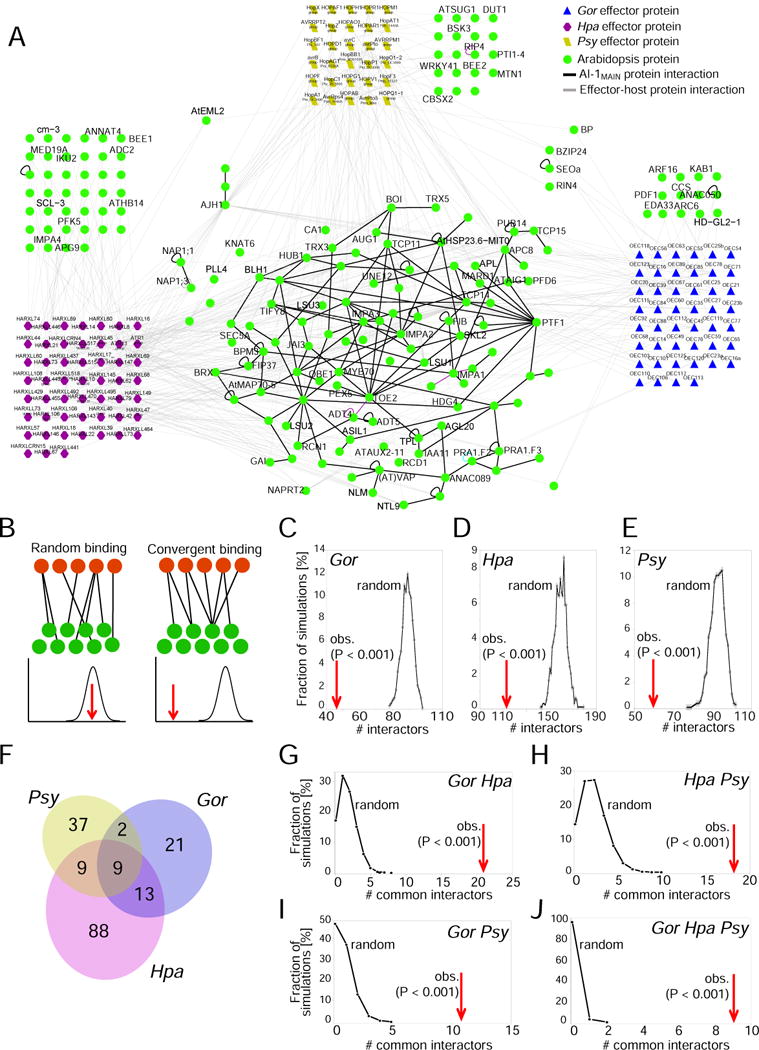
A. The integrated PPIN-28k_sys network of host proteins interacting with Gor, Hpa, and Psy effectors and physical interactions among host proteins derived from AI-1MAIN in the 8k_space. B. Random and convergent interaction of Arabidopsis proteins (green) with effectors (red) can be distinguished by degree-preserving random network rewiring and simulation. C. Random interactors observed in degree-preserving network rewiring simulations of Gor effector-host protein interactions vs. observed value. D. As in C, but for Hpa effector-target interactions. E. As C for Psy effector-host protein interactions. F. Venn diagram showing observed overlap between effector-interactors from the three pathogens. G. Simulated random and observed overlap between Gor and Hpa effector-interactors. H. As in G but between Hpa and Psy effector-interactors. I. As in G but between Gor and Psy effector-interactors. J. As in G but between Gor, Hpa, and Psy effector-interactors. In C–E and G–J, random simulations are shown by black lines and observed values are highlighted by red arrows. See also Figure S2 and Table S1.
One large cluster was apparent within PPIN-28k_sys in which 88 of the 178 effector-interactors (49%) are connected to each other by AI-1MAIN interactions (Figure 2A). Eighty-six effector-interacting host proteins did not interact with any other effector-interactor in AI-1MAIN, although some were connected by interactions from other datasets, e.g. PPIN-1 or literature data (Figure S2A). Degree-preserving random network rewiring revealed that the effector-interactors were less connected to each other in AI-1MAIN than expected by chance (exp. p < 0.001, Figure S2B). This finding may indicate that effectors collectively target different parts of the overall network rather than a functionally coherent subnetwork.
Gene Ontology (GO) enrichment analysis on TAIR10 annotations of effector targets returned mostly high level categories of regulatory processes (Table S2), including defense signaling, potentially due to our previous data being incorporated into the TAIR annotation. We focused on specific, and hence more informative, GO-terms that annotate less than 100 genes in AI-1MAIN and found terms related to development (e.g. floral organ development), auxin and salicylic acid mediated signaling and others (p-value 0.005; Table S2). Both of these phytohormones play important roles in plant pathogen interactions (Robert-Seilaniantz et al., 2011). The functional categories are consistent with analysis of specific bacterial effectors and their targets (Deslandes and Rivas, 2012; Win et al., 2012).
To evaluate whether the apparent OEC convergence onto common host proteins (Figure 1D) was significant, we simulated OECs randomly interacting with AI-1MAIN proteins. The frequency distribution of the randomly observed values obtained in these simulations was used to calculate an experimental P value for OEC interactions with host proteins (Figure 2B). We performed 10,000 simulations with all proteins in the AI-1MAIN network represented according to their degree in AI-1MAIN. The mean random expectation of more than 80 OEC interacting proteins is significantly higher than the experimentally observed value of 45 Arabidopsis proteins interacting with OECs in EHIn8k_sys (Figure 2C; exp. P value < 0.0001). Thus, OEC effectors converge onto a small set of host proteins. We refer to the convergence of effectors from a single pathogen species onto common host proteins as “intraspecies convergence”. Applying this novel analysis to Hpa and Psy effectors revealed the same striking and significant intraspecies convergence as observed for OECs (Figure 2D,E; exp. P value < 0.0001). Thus, effectors of pathogens from diverse kingdoms exhibit intraspecies convergence onto host proteins.
We previously observed convergence of the combined Psy and Hpa effector sets onto common host proteins (“interspecies convergence”), several of which were hubs (highly interconnected host proteins) (Mukhtar et al., 2011). The Gor_EHIn data enabled us to extend the finding of convergence to a divergent fungal pathogen. Effectors from the three pathogens exhibited remarkable overlap with regard to shared host interacting proteins within EHIn8k_sys; this included 24 host proteins interacting with effectors from two pathogens and nine host proteins interacting with effectors from all three (Figure 2F). We performed simulations for all pairwise, and the three-fold, combinations of the three pathogens. In each case the experimentally observed overlap was significantly higher than expected by chance (Figure 2 G–H; exp. P value < 0.0001).
Thus, we observed significant intra- and interspecies convergence of effectors from three evolutionarily highly diverse (hemi-)biotrophic pathogens. This strongly suggests that the convergence is the product of natural selection, and that the respective host proteins are functionally relevant to the pathogen.
Genetic support for effector-interactors: altered infection phenotypes
We tested the functional relevance of 124 effector-interacting host proteins in PPIN-28k_sys using available T-DNA insertion mutants (Alonso and Ecker, 2006). We focused on exon insertions early in genes and tested independent alleles when available (Table S3). We confirmed by PCR both T-DNA insertion into the gene of interest and homozygosity for a total of 179 T-DNA lines. We did not confirm each line as an mRNA null leaving the formal possibility of phenotypic false negatives. These validated mutants were phenotyped using the Gor isolate MPIPZ (Spanu et al., 2010), which is virulent on the Col-0 genetic background of the mutants (Micali et al., 2008; Weßling et al., 2012); Psy strain Pto DC3000, also virulent on Col-0; and three Hpa isolates: Emwa1, Emoy2, and Noco2. The Hpa isolates Emwa1 and Emoy2 are avirulent on Col-0 due to RPP4-mediated recognition whereas Hpa Noco2 is virulent on Col-0 (van der Biezen et al., 2002). The Hpa isolates were selected to detect both enhanced disease susceptibility (eds) and enhanced disease resistance (edr) phenotypes.
At least one altered infection phenotype was detected for 63 of the 124 tested effector-interactors (51% validation rate) (Figure 3A,B; Figure S3, Table S3). The 63 interactors with infection phenotypes will be referred to as effector targets. Of these, 25 (40%) demonstrated exclusively eds phenotypes upon pathogen challenge, indicating that the corresponding wild type proteins function in immune responses. Mutants for 21 targets (33%) exhibited exclusively edr phenotypes upon pathogen challenge, most pronounced in response to the virulent Hpa isolate Noco2. These host proteins may facilitate pathogen sustenance in the host. Alternatively, they may repress an activator of immune signaling, a function possibly stabilized by the interacting virulence effectors; thus, in the absence of the putative negative regulator, effector action is neutralized and host resistance increases. We noted divergent disease phenotypes for 17 (27%) effector targets, i.e. mutants exhibited an edr phenotype with one pathogen or Hpa isolate, and an eds phenotype with a second pathogen or a different Hpa isolate, as described (Pieterse et al., 2012). Their existence suggests that these host proteins may have disease-specific functions. The TCP transcription factors TCP13, TCP14, and TCP19, which exhibited eds phenotypes with the biotrophic pathogens Hpa and Gor, but edr phenotypes with the hemibiotrophic Psy, are particularly interesting in this context.
Figure 3. Phenotypic characterization of effector-interactor mutants.
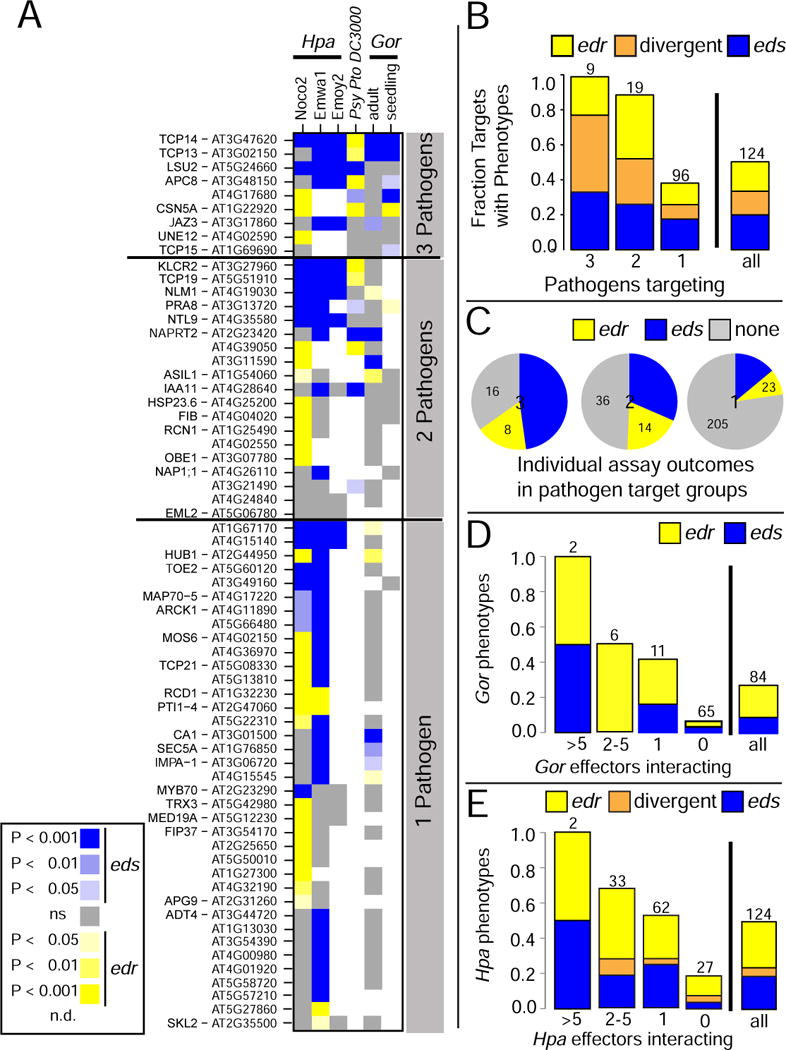
A. Heat-map summarizing the outcome of phenotypic analyses of mutants in genes encoding the indicated effector-interactors in infection assays with the noted pathogens and developmental stages. Host proteins are sorted by the number of pathogens interacting with them, then by number of observed phenotypes and performed assays. Mutant lines for 59 proteins interacting with effectors from a single pathogen did not show any disease phenotype and are not shown. Refer to Table S3 for raw data for all phenotyped loci and Figure S3 for complete results for all tested lines. B. Fraction of mutant lines for proteins interacting with effectors from the indicated number of pathogens that exhibited an edr, eds or divergent phenotypes across the assays. C. Pie chart representation of the phenotype density; the number of observed phenotypes relative to individual assays performed for that group. Each pie displays data for proteins that interacted with effectors from the number of pathogens given in the center. D. Fraction of mutant lines for proteins targeted by the indicated number of Gor effectors for which edr or eds phenotypes were observed. Numbers above bars indicate the number of targets in that class. E. As in D, but for proteins targeted by the indicated number of Hpa effectors for which edr or eds phenotypes were observed. Numbers above bars indicate the number of effector-interactors in the class.
Effector convergence correlates with altered infection phenotypes
The nonrandom nature of effector-host protein connectivity suggested that the network topology of the plant immune system is the product of natural selection, and consequently that the convergence we observed is biologically meaningful. We explored whether a relationship exists between intra- and interspecies effector convergence and altered pathogen infection phenotypes. A host protein was considered a point of intraspecies convergence when at least two effectors from the same pathogen interacted with it, and an object of interspecies convergence when effectors from different pathogens interacted with it.
Effector-interactors were binned by whether they interacted with effectors from three, two or one pathogen(s) (Figure 2F). The rate of altered infection-related phenotypes was evaluated for each bin and compared to the overall rate (“all” column) (Figure 3B). We noted a positive correlation between the degree of interspecies convergence and the probability of observing an infection phenotype in that bin (Figure 3B). To exclude that our observation was due to deeper phenotypic interrogation of the most highly targeted proteins, we also calculated the phenotype density for proteins interacting with effectors from three, two, or one pathogen as the fraction of assays (individual squares in Figure 3B) in which an edr or eds phenotype was observed divided by the total number of assays performed in this group. This analysis confirmed the correlation between convergence and phenotypic relevance of the targeted host protein (Figure 3C).
We then evaluated the phenotypic relevance of genes encoding host proteins that are objects of intraspecies convergence (Figure 2C–E). We binned host proteins according to the number of effectors from each pathogen interacting with them and evaluated how often an altered immunity phenotype could be observed with the respective pathogen. We found edr or eds phenotypes for all mutants in genes encoding the two proteins targeted by more than five Gor or Hpa effectors (Figure 3D,E). The fraction of phenotypically validated host targets decreased proportional to the degree at which effectors are connected to the respective plant proteins (Figure 3D,E). Thus, the extent of intraspecies effector convergence onto host targets is also directly correlated to the functional relevance of the targeted proteins.
We wondered whether the host proteins that interact with effectors from multiple pathogens were also targeted repeatedly by the suite of effectors from any individual pathogen. All nine Arabidopsis proteins targeted by effectors from all three pathogens are also intraspecies convergence points for at least one pathogen (Figure S3). Furthermore, 16 of 23 proteins targeted by effectors from two pathogens are also points of intraspecies convergence. Thus, the most commonly targeted Arabidopsis proteins are objects of both intra- and interspecies effector convergence (Fisher’s exact test, P < 0.001).
Effector convergence is exemplified by TCP14, a member of a large family of transcriptional regulators typically recognized to function in plant development (Martín-Trillo and Cubas, 2010). TCP14 was the most targeted host protein, interacting with 23 distinct OECs, 25 Hpa effectors and four Psy effectors, and exhibiting disease phenotypes in all assays (Figure 3A). The related family members TCP13, TCP15 and TCP19 were also targeted multiple times by effectors from at least two pathogens and exhibited altered infection phenotypes. These findings suggest an important and possibly universal role of this class of TFs during infection, consistent with their emerging role as targets of phytoplasma effectors (Sugio et al., 2011) and the recent demonstration of their importance in plant immunity (Kim et al., 2014).
Effectors co-localize with TCP14 in planta
To independently validate the convergence concept using cell biological methods, we tested TCP14 for co-localization with 11 of the 25 interacting Hpa effectors, focusing on those effectors demonstrated to localize to the nucleus (Caillaud et al., 2012), 19 of the 23 Gor effectors not previously localized, and three of four Psy effectors (Table S4).
TCP14 localized to sub-nuclear foci in transgenic Arabidopsis plants expressing a functional TCP14-YFP fusion (Figure S4). We used this knowledge to develop a transient over-expression assay platform in Nicotiana benthamiana where TCP14-RFP was also localized to sub-nuclear foci (Figure 4A). Under these conditions, TCP14-RFP did not co-localize with a wide set of controls with which it did not interact in Y2H: free YFP (Figure 4B), representative effectors (HopBC1, HaRxL62 and OEC56), an unrelated TF (bZIP5), or an unrelated, sub-nuclear body-localized protein (PhyB) (Chen et al., 2005) (Figure S4). We then demonstrated that TCP14 can re-localize into sub-nuclear foci 64% and 74% of the tested Hpa and Gor effectors, respectively, and one of three Psy effectors (Figure 4C, Figure S4). Thus, the majority (67%) of tested interactions between effectors and TCP14 were validated by re-localization in vivo (Table S4). We confirmed these findings via co-immunoprecipitation for single effectors from each pathogen (Figure 4D). We thus validated in planta the majority of effector interactions with the most heavily targeted host protein, TCP14, consistent with our claim that the observed convergence onto the plant protein is not an artefact.
Figure 4. TCP14 re-localizes effectors to sub-nuclear foci.
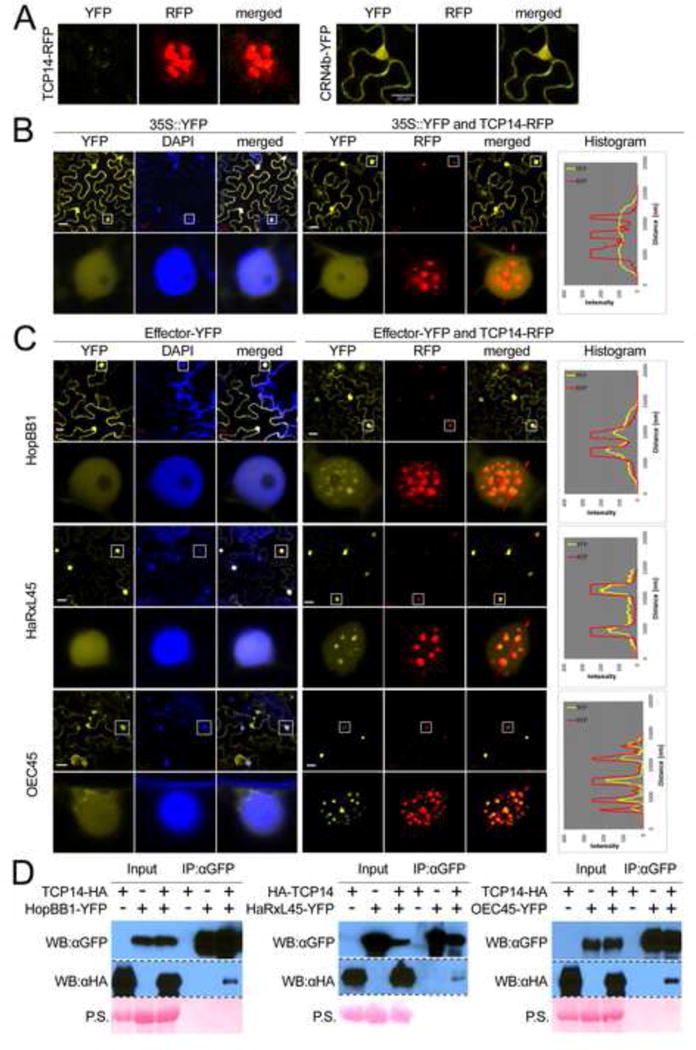
A. Technical control demonstrating that YFP- and RFP channels do not leak into each other. The images show localization of TCP14-RFP and CRN4b-YFP; image data for YFP and RFP channels were collected for both. The same settings were then applied to all assays below. Note that TCP14-YFP forms sub-nuclear foci. B and C. The lower panel exhibits an enlarged view of a representative nucleus boxed in the upper panel. The histogram illustrates the intensity of fluorescent signal across the path indicated by the red arrow. All confocal pictures were taken 40–48 h after infiltration of Agrobacterium strains expressing the different fluorophore–tagged proteins. B. Negative control: TCP14 does not re-localize YFP. C. TCP14 re-localizes effectors from Psy (HopBB1), Hpa (HaRxL45) and Gor (OEC45) to sub-nuclear foci. D. TCP14 is co-immunoprecipitated by HopBB1, HaRxL45, and OEC45. All proteins were expressed from the CaMV 35S promoter in N. benthamiana leaves. ‘P.S’ in D denotes Ponceau S staining. See also Figure S4 and Table S4.
Proteins interacting with common effector targets are likely under positive selection
We sought evidence for the evolutionary relevance of our effector targets from population genomics. We used the complete genomes of 80 accessions sequenced in the context of the 1001 Genomes project and mapped on the Col-0 reference genome (Cao et al., 2011). These were collected in eight regions distributed over Europe and Asia, where Arabidopsis naturally occurs and thus provide a large spatial and phylogenetic sample of genotypes adapted to different environments (http://1001genomes.org/data/). For all proteins in AI-1MAIN we calculated Tajima’s D (DT) and Watterson’s estimator θ (θW) to assess the allele frequency deviation from neutrality and scaled mutation rate, respectively (Tajima, 1989; Watterson, 1975) (Table S5). As the two statistics lead to different ordered gene rankings, we also built a consensus ranking based on the relative positions of each gene in the two ranked lists (Dθ-ranking). In addition, we constructed consensus protein sequences from 81 Arabidopsis accessions (80 plus the Col-0 reference) by majority voting (Altmann et al., in preparation) and used this resource to identify single nucleotide polymorphisms (SNPs) that give rise to altered amino acid sequences (amino acid polymorphisms, AAPs) in the 2,653 AI-1MAIN proteins (Table S5).
We asked whether the direct effector-interactors exhibit evidence for balancing selection, as indicated by positive DT values (Figure S5A). No significant deviation from random expectation could be detected for any of four effector-interactor groups: i) all effector-interactors, ii) interactors of effectors from two or three pathogens, iii) interactors of effectors from three pathogens, iv) phenotypically supported effector-interactors (Figure S5A). For three of the four groups the mean of DT for effector-interactors is lower than that of random controls, whereas for the group targeted by three pathogens the mean is slightly higher.
The lack of a strong signal can likely be explained by our previous observation that many effector targets are central proteins in the network, which likely cannot tolerate much variation without adverse effects on protein function. We therefore asked whether instead there might be evidence for the selective pressure imposed by pathogens in the network neighborhood of the effector-interactors. To this end, we explored whether the AI-1MAIN interaction partners of effector-interactors are subject to balancing selection, but no such evidence could be detected for any of the effector-interactor groups (P 0.51 – 0.94) (Figure S5B). It is possible that a majority of interacting proteins mediating non-immune functions may mask any potential signal from the few interacting proteins involved in immune functions. We therefore adopted an inverse approach and investigated whether effector-interactors are preferential interaction partners of proteins encoded by genes under balancing selection. For each cut-off of the top ranking genes in the combined Dθ-ranked list we counted the cumulative number of interacting effector-interactors in AI-1MAIN (red dots) separately for each of the four effector groups noted above (Figure 5). To estimate the specificity of the observations in the context of the experimentally derived network structure, we performed rewiring controls of AI-1MAIN and counted randomly interacting effector-interactors for the same top Dθ-ranking proteins (Figure 5A). The number of interacting effector-interactors in the real AI-1MAIN network is always significantly higher than random across a range of cut-offs, demonstrating a preferential interaction of proteins encoded by genes under balancing selection with our effector-interactors. These findings are supported by similar results obtained with an AAP based ranking, although with slightly different top-ranking proteins. These polymorphic proteins show the greatest signal with effector-interactors targeted by effectors from three pathogens. The protein with the greatest number of AAPs is an intracellular TIR-NLR type immune receptor (AT1G31540), which interacts with TCP14 and is characterized by the third highest ranking θ value (0.021) among all AI-1MAIN proteins. Previously, TIR-NLR was identified as the dominant RAC1 gene, mediating resistance to Albugo candida in the Ksk-1 accession (Borhan et al., 2004).
Figure 5. Proteins with high natural genetic variation interact with effector-interactors.
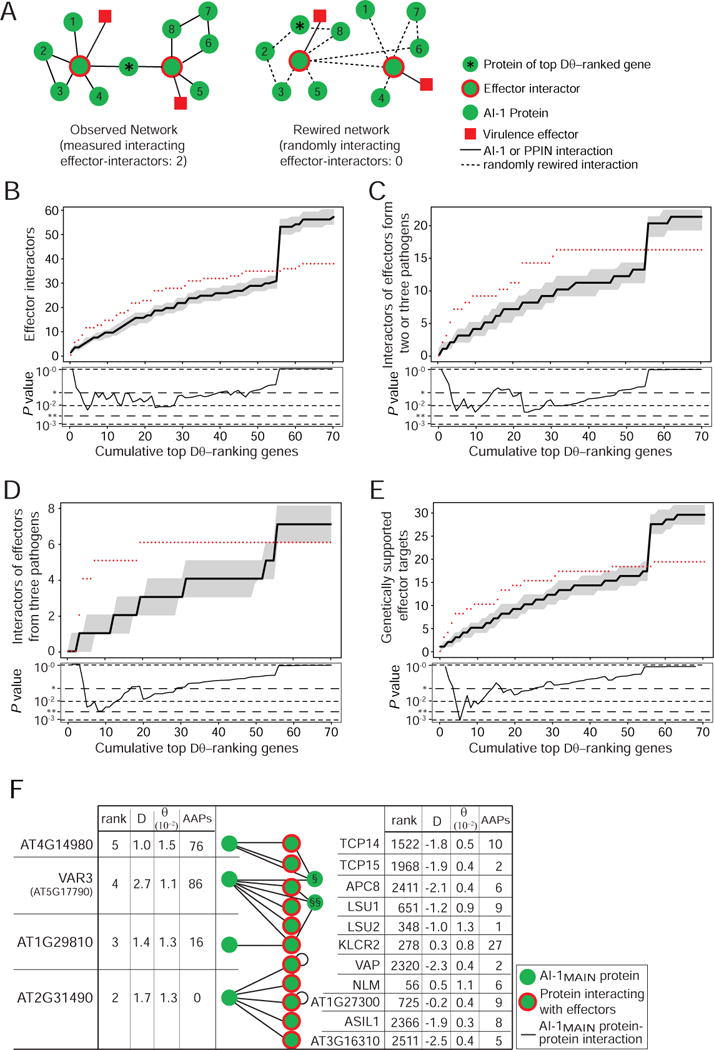
A. Schematic illustration of the analysis in B–E: in the AI-1MAIN network (left) the effector-interactors directly interacting with top Dθ-ranking gene products are counted and compared to the distribution of counts observed in 1,000 randomly rewired networks (single example shown). Effectors are shown for illustration only and not included in the analysis. B. Analysis as described in A. Plotted along the Y-axis are cumulative counts of effector-interactors interacting with proteins encoded by the top Dθ-ranking × genes. Data from AI-1MAIN are shown as red dots, the black line shows the median of 1,000 randomly rewired networks, grey shaded areas show the 25th and 75th percentiles of values from rewiring controls. The lower panel shows the corresponding experimental P values (* 0.05; ** 0.005). The steep rise in the simulations at x = 56 is caused by a high-degree protein (NLM1, AT4G19030) at that position; the many rewired interactions for this protein increase the count of random interactors in all categories. C. As in B but counting proteins interacting with effectors from two or three pathogens. D. As in B but counting proteins interacting with effectors from three pathogens. E. As in B but counting proteins whose mutation caused altered immune phenotypes. F. Among the 13 interaction partners of the five most selected proteins are eleven effector-interactors, including the five most targeted proteins. The tables show for all top Dθ-ranking proteins and effector-interactors the relative combined rank, DT, θW and the count of AAPs. The non-effector-interactor interactors of the variable proteins are § AT1G51580 and §§ ZPF7. See also Figure S5 and Table S5.
The network of four of the top Dθ-ranking five proteins further shows that even the two interaction partners that are not effector-interactors are members of a common subnetwork by virtue of multiple interactions with variable proteins and effector-interactors (Figure 5F). The underlying biological reasons of how the increased genetic variation is beneficial in the evolutionary battle remain to be elucidated. A Gene Ontology enrichment analysis of the top Dθ-ranking genes did not yield conclusive results, though, partly because many of these genes have not been characterized, yet.
The evidence for preferential interaction of proteins encoded by genes under balancing selection with effector-interactors contrasts with the conservation of effector-interactors themselves, which show signs of purifying selection (Figure 5F). Together these data demonstrate that at least a subset of proteins targeted by multiple evolutionary distant pathogens are under purifying selection and in such instances variation at the level of neighbors in the protein interaction network becomes a substrate for balancing selection.
DISCUSSION
We identified interactions between candidate virulence effector proteins from the obligate biotrophic powdery mildew fungus Golovinomyces orontii and proteins from its host, Arabidopsis. We added these to previously defined interactions between the same set of host proteins and effector suites from two pathogens derived from different kingdoms. Analysis of the combined data allowed us to significantly extend our previously defined principles of how plant pathogens have independently evolved effectors to converge onto a limited, shared set of host proteins. Our main conclusions will propel future hypothesis testing, ultimately resulting in the definition of key plant machinery modulated by diverse pathogens to increase their fitness during infection.
Our identification and analysis of Gor candidate effectors serves two functions. First, it paves the way for mechanistic studies of single effectors and elucidation of the infection strategies of fungal pathogens in general. Incidentally several OECs that interact with functionally relevant targets, such as the highly connected OEC78 or the less connected OEC49 and OEC101, have homologues in Bgh and Erysiphe pisi and may be part of a putative powdery mildew core effector set (Figure 1B). Unfortunately, it was not possible to clearly differentiate host proteins as being targeted by core or non-core proteins, which prevented a deeper characterization.
Second, our systematic and unbiased identification of Gor effector interactors enabled combined analyses of host-pathogen interaction networks when integrated with our previous data for Hpa and Psy (Mukhtar et al., 2011). The result is a map of Arabidopsis proteins interacting with effectors from three destructive pathogen lineages: bacteria, oomycetes, and fungi (Figure 2A, Table S1). We demonstrate that most of the host interactors are genuine targets, in the sense that loss-of-function results in altered host immune system function (Figure 3A,B; Figure S3, Table S3). In the combined PPIN-2 network, we also significantly expand our previous evidence for interspecies effector convergence (Mukhtar et al., 2011).
We discovered evidence for combined intraspecies convergence by effectors of each of the three pathogens (Figure 2F). Our data indicate that effector-target convergence evolved independently in all kingdoms of life. Thus, convergence per se may be an important, if not necessary, feature of host-pathogen interactions. The mechanistic and evolutionary rationale for this convergence is speculation, but may suggest that successful biotrophic pathogens need to manipulate a largely shared set of physiological host networks and proteins. They may achieve this, in the face of receptor-based host immune surveillance, by homology-independent functional redundancy that uses different effectors to modulate different nodes in a common set of sub-networks, as suggested by our findings (Figure S1C). Additional pathogenicity strategies evolved by other biotrophic or non-biotrophic pathogens may drive the evolution of idiosyncratic pathogen species-specific host targets, or of completely new host machinery that is required to support lifestyles beyond those of the pathogens whose effector suites we have surveyed.
Genome sequencing of pathogenic bacterial strains revealed that effector complements are only marginally overlapping, even between strains that otherwise exhibit very high genomic sequence identity (97%) (Baltrus et al., 2011). Likewise, genome sequencing in a variety of oomycete lineages reveals diverse expansion and contraction in effector families (Pais et al., 2013; Stergiopoulos et al., 2012). Thus, the observed intraspecies convergence of effectors supports our suggestion of functional redundancy mediated by different effectors to maintain host protein targeting. The effector complement would thus be buffered against loss or rapid selection against specific effectors due to host recognition. Importantly, the plant immune system can blunt effector evolution by detecting effector-dependent host target modifications. It is much more efficient to guard a limited number of important host targets than to evolve receptors for each effector (Jones and Dangl, 2006), especially in cases where both the effector and the host receptor are under frequency-dependent balancing selection (Van der Hoorn et al., 2002). Our observations that in our systematic network (1) effectors converge onto limited number of targets, and (2) a large fraction of targets, in turn, interact with highly polymorphic proteins that are under balancing selection across the Arabidopsis population support this notion. Alternative explanations for effector convergence include the sequential delivery of effectors targeting the same host-protein, but at different time-points in the course of host colonization, and/or cooperativity of effectors that might act together to modify host protein functions. Intriguingly, most Hpa effectors conferred enhanced virulence on only a subset of accessions after DC3000 delivery and pathogenicity assays, indicating variation between accessions in the susceptibility to effector manipulation (Fabro et al., 2011).
Integrating these network concepts with our extensive reverse genetic data, we demonstrate that effector convergences strongly correlate with mutant infection phenotypes (Figure 3B–E). Our genetic data convincingly reinforce our interpretation that network convergence is due to selection of effector interactions with host proteins. Our mutant phenotyping results also show that most host-targets of multiple pathogens have previously unknown plant immune system functions. This conclusion is reinforced by the fact that effector-interactors are also preferential interaction partners of intracellular NLR receptors, likely reflecting guarding of these virulence targets by the plant immune system (Mukhtar et al., 2011).
To demonstrate that interactions between effectors and highly targeted host-proteins can occur in planta, we investigated TCP14, the host protein most commonly targeted by effectors. These experiments demonstrated that the majority of tested effectors that interact with TCP14 in Y2H are re-localized in the nucleus to characteristic TCP14 sub-nuclear foci (Figure 4C, Figure S4, Table S4). The function of the TCP14 foci and the function(s) of specific effectors within them are now the object of active investigation. Together with extended genetic support for TCP14 function in response to infection, our findings strengthen the hypothesis that the majority of effector-TCP14 interactions reflect genuine protein-protein interactions that function during infections by diverse pathogens.
Plants and microbial pathogens are engaged in a constant evolutionary battle in which pathogens can expand their suites of effectors via horizontal gene transfer, or evolution of new alleles of existing effectors, driving selection in plants to respond accordingly. Each successful evolutionary step in the plant can be overcome by subsequent modification of pathogen effector deployment. Yet both pathogen virulence and plant immune function have fitness costs that can drive the arms race into balanced trench warfare (Holub, 2001). The current shape of the plant immune system is a consequence of these counteracting forces. The network we define here is uniquely well supported by broad mutant phenotype analysis and cell biological investigation.
EXPERIMENTAL PROCEDURES
Effector prediction and cloning
Secreted proteins were identified in a cDNA library from isolated haustoria by employing SignalP3.0 at a HMM threshold of 0.8 and the TMHMM algorithm (http://www.cbs.dtu.dk/services/TMHMM (Bendtsen et al., 2004). A size cut-off of ≥60 amino acids was applied. The absence of homologs in unrelated species was confirmed by BLAST. In a second prediction round, 491 Bgh Candidates for Secreted Effector Proteins (Pedersen et al., 2012) and effector candidates from the first round were used as templates for tBLASTn and BLASTp analyses of the haustorial cDNA library. Putative homologs of predicted effectors were then subjected to SignalP3.0 and TMHMM analysis and the absence of homologs in unrelated species was confirmed to prevent false positives. Finally, the conservation of OECs in the genomes of Bgh and E. pisi was queried by tBLASTn. A multiple sequence alignment of the OECs was generated using ClustalW (http://www.ebi.ac.uk/Tools/msa/clustalw2/) and imported into MEGA5 (Tamura et al., 2011). The phylogeny was computed with default parameters using the Neighbor-joining algorithm and a p-distance model for AA substitutions. For OEC cloning, primers spanning the mature protein (without signal peptide) were generated and used for PCR from a mix of cDNA constructed from plant material at one, three, five and ten dpi with G. orontii using a proof-reading polymerase.
Y2H analysis
A detailed description of the Y2H pipeline can be found in the Supplemental Information.
Phenotypic assays
We used Arabidopsis thaliana Columbia (Col-0) unless mentioned otherwise. Single mutants for eds1–2 (Bartsch et al., 2006), sid2-2 (Dewdney et al., 2000), and mlo2–6 (Consonni et al., 2006) in the Col-0 genetic background were used as controls for the relevant infection assays. The set of homozygous T-DNA insertion lines is described in Table S3. Pathogen infection assays were performed as described in the Supplemental methods.
Statistical Analysis
Details of the statistical analyses and controls can be found in the Supplemental Information.
Re-localization of effectors by TCP14
Vector constructs
Coding sequences of Psy effectors and coding sequences of Hpa and Gor effectors lacking the signal peptides were amplified by PCR, and then Gateway-cloned into the destination vectors pGWB41, pGWB660, pGWB642, pGWB644, pGWB645, and a modified pMDC7 respectively (Akimoto-Tomiyama et al., 2012; Nakagawa et al., 2007) (Table S4). The plasmids were transformed into the Agrobacterium tumefaciens strain GV3101 for transient expression assays in Nicotiana benthamiana (see Supplemental methods).
Immunoprecipitation
HA-tagged TCP14 and eYFP-tagged effectors were expressed in N. benthamiana under control of the 35S constitutive promoter. The binary vectors used for expression were pGWB614 (for TCP14-HA), pGWB715 (for HA-TCP14), pGWB641 (for HaRxL45-YFP and HopBB1-YFP) and pGWB41 (for OEC45-YFP). Agrobacteria carrying each construct (OD=0.2) were infiltrated into N. benthamiana leaves 24 hours prior to harvesting. Proteins were extracted from 0.5 g of fresh tissue using 2 ml extraction buffer (50 mM Hepes pH 7.5; 50 mM NaCl; 10 mM EDTA, pH 8.0; 0.5% Triton-X100; 5 mM DTT and 1× Plant protease inhibitor cocktail from Sigma-Aldrich). Magnetic labeling and separation of tagged proteins was performed using μMACS Epitope Tag Protein Isolation Kit (Miltenyi Biotec). Protein samples were separated by 12% SDS-PAGE. Immunoblots were performed with a 1:1000 dilution of α-HA (Roche) and 1:1000 dilution of α-GFP (Roche). Blots were detected by ECL prime (GE Healthcare).
Supplementary Material
Acknowledgments
We thank Axel Künstner for helpful discussions about the population genetic analysis. This work was funded by grants to JLD from the National Science Foundation (IOS-1257373), the National Institutes of Health (1RO1 GM107444), the Gordon and Betty Moore Foundation (GBMF3030), and the HHMI. RW was supported by PhD fellowship from the International Max-Planck Research School (IMPRS). JLD is an Investigator of the Howard Hughes Medical Institute. LY was funded in part by the Gordon and Betty Moore Foundation through Grant GBMF 2550.02 to the Life Sciences Research Foundation. Y.H. was supported by a Distinguished Guest Professorship, Eberhard-Karls-Universität, Tübingen, Germany to J.L.D. SA and PB are supported by the Deutsche Forschungsgemeinschaft (DFG) grant SFB924. RP and PSL were supported by grants from the Max-Planck society. We thank Dr. Meng Chen, Duke University for 35S:phyB-CFP and useful discussions regarding sub-nuclear foci.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS: OEC identification and cloning: RW, AES, EVLT, RP; OEC-At interactome mapping: RW, LG, JRE, MV, PB; Data Integration and convergence statistics: SA, CG, KFXM, JLD, PB; Immune assays (Hpa, Psy): PE, KW, NM, MSM, JLD; Immune assays (Gor): RW, SH, PSL, RP; Phenotype statistics: SA, PE, RW, EVLT, PB; Co-localization analysis: YH, LY, JLD; Immunoprecipitations: LY, YH, JLD; Provision of several Hpa effectors: JDGJ; Natural variation analysis: SA, SRH, KCB, KFXM, DW, JLD, PB; Manuscript writing: PB, PE, SA, RW, JLD; Critical manuscript reading and editing: PSL, RP; Conception and design: PE, RP, PB, JLD.
Supplemental information includes 5 figures, 7 tables, a glossary, details of the statistical tests and experimental methods and can be found with this article online at [link].
References
- Akimoto-Tomiyama C, Furutani A, Tsuge S, Washington EJ, Nishizawa Y, Minami E, Ochiai H. XopR, a type III effector secreted by Xanthomonas oryzae pv. oryzae, suppresses microbe-associated molecular pattern-triggered immunity in Arabidopsis thaliana. Mol Plant Microbe Interact. 2012;25:505–514. doi: 10.1094/MPMI-06-11-0167. [DOI] [PubMed] [Google Scholar]
- Alonso JM, Ecker JR. Moving forward in reverse: genetic technologies to enable genome-wide phenomic screens in Arabidopsis. Nat Rev Genet. 2006;7:524–536. doi: 10.1038/nrg1893. [DOI] [PubMed] [Google Scholar]
- Baltrus DA, Nishimura MT, Romanchuk A, Chang JH, Mukhtar MS, Cherkis K, Roach J, Grant SR, Jones CD, Dangl JL. Dynamic evolution of pathogenicity revealed by sequencing and comparative genomics of 19 Pseudomonas syringae isolates. PLoS Pathog. 2011;7:e1002132. doi: 10.1371/journal.ppat.1002132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartsch M, Gobbato E, Bednarek P, Debey S, Schultze JL, Bautor J, Parker JE. Salicylic acid-independent ENHANCED DISEASE SUSCEPTIBILITY1 signaling in Arabidopsis immunity and cell death is regulated by the monooxygenase FMO1 and the Nudix hydrolase NUDT7. Plant Cell. 2006;18:1038–1051. doi: 10.1105/tpc.105.039982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baxter L, Tripathy S, Ishaque N, Boot N, Cabral A, Kemen E, Thines M, Ah-Fong A, Anderson R, Badejoko W, et al. Signatures of adaptation to obligate biotrophy in the Hyaloperonospora arabidopsidis genome. Science. 2010;330:1549–1551. doi: 10.1126/science.1195203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendtsen JD, Nielsen H, von Heijne G, Brunak S. Improved prediction of signal peptides: SignalP 3.0. J Mol Biol. 2004;340:783–795. doi: 10.1016/j.jmb.2004.05.028. [DOI] [PubMed] [Google Scholar]
- Borhan MH, Holub EB, Beynon JL, Rozwadowski K, Rimmer SR. The arabidopsis TIR-NB-LRR gene RAC1 confers resistance to Albugo candida (white rust) and is dependent on EDS1 but not PAD4. Mol Plant Microbe Interact. 2004;17:711–719. doi: 10.1094/MPMI.2004.17.7.711. [DOI] [PubMed] [Google Scholar]
- Braun P. Interactome mapping for analysis of complex phenotypes: insights from benchmarking binary interaction assays. Proteomics. 2012;12:1499–1518. doi: 10.1002/pmic.201100598. [DOI] [PubMed] [Google Scholar]
- Caillaud MC, Piquerez SJ, Fabro G, Steinbrenner J, Ishaque N, Beynon J, Jones JD. Subcellular localization of the Hpa RxLR effector repertoire identifies a tonoplast-associated protein HaRxL17 that confers enhanced plant susceptibility. Plant J. 2012;69:252–265. doi: 10.1111/j.1365-313X.2011.04787.x. [DOI] [PubMed] [Google Scholar]
- Cao J, Schneeberger K, Ossowski S, Gunther T, Bender S, Fitz J, Koenig D, Lanz C, Stegle O, Lippert C, et al. Whole-genome sequencing of multiple Arabidopsis thaliana populations. Nat Genet. 2011;43:956–963. doi: 10.1038/ng.911. [DOI] [PubMed] [Google Scholar]
- Chen M, Tao Y, Lim J, Shaw A, Chory J. Regulation of phytochrome B nuclear localization through light-dependent unmasking of nuclear-localization signals. Curr Biol. 2005;15:637–642. doi: 10.1016/j.cub.2005.02.028. [DOI] [PubMed] [Google Scholar]
- Chisholm ST, Coaker G, Day B, Staskawicz BJ. Host-microbe interactions: shaping the evolution of the plant immune response. Cell. 2006;124:803–814. doi: 10.1016/j.cell.2006.02.008. [DOI] [PubMed] [Google Scholar]
- Consonni C, Humphry ME, Hartmann HA, Livaja M, Durner J, Westphal L, Vogel J, Lipka V, Kemmerling B, Schulze-Lefert P, et al. Conserved requirement for a plant host cell protein in powdery mildew pathogenesis. Nature Genetics. 2006;38:716–720. doi: 10.1038/ng1806. [DOI] [PubMed] [Google Scholar]
- Consortium AIM. Evidence for network evolution in an Arabidopsis interactome map. Science. 2011;333:601–607. doi: 10.1126/science.1203877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dangl JL, Horvath DM, Staskawicz BJ. Pivoting the plant immune system from dissection to deployment. Science. 2013;341:746–751. doi: 10.1126/science.1236011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deslandes L, Rivas S. Catch me if you can: bacterial effectors and plant targets. Trends Plant Sci. 2012;17:644–655. doi: 10.1016/j.tplants.2012.06.011. [DOI] [PubMed] [Google Scholar]
- Dewdney J, Reuber TL, Wildermuth MC, Devoto A, Cui J, Stutius LM, Drummond EP, Ausubel FM. Three unique mutants of Arabidopsis identify eds loci required for limiting growth of a biotrophic fungal pathogen. Plant J. 2000;24:205–218. doi: 10.1046/j.1365-313x.2000.00870.x. [DOI] [PubMed] [Google Scholar]
- Dodds PN, Rathjen JP. Plant immunity: towards an integrated view of plant-pathogen interactions. Nature Reviews Genetics. 2010;11:539–548. doi: 10.1038/nrg2812. [DOI] [PubMed] [Google Scholar]
- Dreze M, Monachello D, Lurin C, Cusick ME, Hill DE, Vidal M, Braun P. High-quality binary interactome mapping. Methods Enzymol. 2010;470:281–315. doi: 10.1016/S0076-6879(10)70012-4. [DOI] [PubMed] [Google Scholar]
- Fabro G, Steinbrenner J, Coates M, Ishaque N, Baxter L, Studholme DJ, Korner E, Allen RL, Piquerez SJ, Rougon-Cardoso A, et al. Multiple candidate effectors from the oomycete pathogen Hyaloperonospora arabidopsidis suppress host plant immunity. PLoS pathogens. 2011;7:e1002348. doi: 10.1371/journal.ppat.1002348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng F, Zhou JM. Plant-bacterial pathogen interactions mediated by type III effectors. Current Opinion in Plant Biology. 2012;15:469–476. doi: 10.1016/j.pbi.2012.03.004. [DOI] [PubMed] [Google Scholar]
- Hacquard S, Kracher B, Maekawa T, Vernaldi S, Schulze-Lefert P, Ver Loren van Themaat E. Mosaic genome structure of the barley powdery mildew pathogen and conservation of transcriptional programs in divergent hosts. Proc Natl Acad Sci U S A. 2013;110:E2219–2228. doi: 10.1073/pnas.1306807110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holub EB. The arms race is ancient history in Arabidopsis, the wildflower. Nature Reviews Genetics. 2001;2:516–527. doi: 10.1038/35080508. [DOI] [PubMed] [Google Scholar]
- Jones JD, Dangl JL. The plant immune system. Nature. 2006;444:323–329. doi: 10.1038/nature05286. [DOI] [PubMed] [Google Scholar]
- Kemen E, Jones JDG. Obligate biotroph parasitism: can we link genomes to lifestyles? Trends Plant Sci. 2012;17:448–457. doi: 10.1016/j.tplants.2012.04.005. [DOI] [PubMed] [Google Scholar]
- Kim SH, Son GH, Bhattacharjee S, Kim HJ, Nam JC, Nguyen PD, Hong JC, Gassmann W. The Arabidopsis Immune Adaptor SRFR1 Interacts with TCP Transcription Factors that Redundantly Contribute to Effector-Triggered Immunity. Plant J. 2014 doi: 10.1111/tpj.12527. [DOI] [PubMed] [Google Scholar]
- Markow AV. On the origin of the eukaryotic cell. Paleontological Journal. 2005;39:109–116. [Google Scholar]
- Martín-Trillo M, Cubas P. TCP genes: a family snapshot ten years later. Trends Plant Sci. 2010;15:31–39. doi: 10.1016/j.tplants.2009.11.003. [DOI] [PubMed] [Google Scholar]
- Micali C, Gollner K, Humphry M, Consonni C, Panstruga R. The powdery mildew disease of Arabidopsis: A paradigm for the interaction between plants and biotrophic fungi. Arabidopsis Book. 2008;6:e0115. doi: 10.1199/tab.0115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhtar MS, Carvunis AR, Dreze M, Epple P, Steinbrenner J, Moore J, Tasan M, Galli M, Hao T, Nishimura MT, et al. Independently evolved virulence effectors converge onto hubs in a plant immune system network. Science. 2011;333:596–601. doi: 10.1126/science.1203659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa T, Kurose T, Hino T, Tanaka K, Kawamukai M, Niwa Y, Toyooka K, Matsuoka K, Jinbo T, Kimura T. Development of series of gateway binary vectors, pGWBs, for realizing efficient construction of fusion genes for plant transformation. J Biosci Bioeng. 2007;104:34–41. doi: 10.1263/jbb.104.34. [DOI] [PubMed] [Google Scholar]
- Pais M, Win J, Yoshida K, Etherington GJ, Cano LM, Raffaele S, Banfield MJ, Jones A, Kamoun S, Go Saunders D. From pathogen genomes to host plant processes: the power of plant parasitic oomycetes. Genome Biology. 2013;14:211. doi: 10.1186/gb-2013-14-6-211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardey PG, Beddow JM, Kriticos DJ, Hurley TM, Park RF, Duveiller E, Sutherst RW, Burdon JJ, Hodson D. Agriculture. Right-sizing stem-rust research. Science. 2013;340:147–148. doi: 10.1126/science.122970. [DOI] [PubMed] [Google Scholar]
- Pedersen C, Ver Loren van Themaat E, McGuffin LJ, Abbott JC, Burgis TA, Barton G, Bindschedler LV, Lu X, Maekawa T, Wessling R, et al. Structure and evolution of barley powdery mildew effector candidates. Bmc Genomics. 2012;13:694. doi: 10.1186/1471-2164-13-694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pieterse CM, Van der Does D, Zamioudis C, Leon-Reyes A, Van Wees SC. Hormonal modulation of plant immunity. Annual Review of Cell and Developmental Biology. 2012;28:489–521. doi: 10.1146/annurev-cellbio-092910-154055. [DOI] [PubMed] [Google Scholar]
- Raffaele S, Kamoun S. Genome evolution in filamentous plant pathogens: why bigger can be better. Nat Rev Microbiol. 2012;10:417–430. doi: 10.1038/nrmicro2790. [DOI] [PubMed] [Google Scholar]
- Robert-Seilaniantz A, Grant M, Jones JD. Hormone crosstalk in plant disease and defense: more than just jasmonate-salicylate antagonism. Annual Review of Phytopathology. 2011;49:317–343. doi: 10.1146/annurev-phyto-073009-114447. [DOI] [PubMed] [Google Scholar]
- Stergiopoulos I, Kourmpetis YA, Slot JC, Bakker FT, De Wit PJ, Rokas A. In silico characterization and molecular evolutionary analysis of a novel superfamily of fungal effector proteins. Mol Biol Evol. 2012;29:3371–3384. doi: 10.1093/molbev/mss143. [DOI] [PubMed] [Google Scholar]
- Sugio A, Kingdom HN, MacLean AM, Grieve VM, Hogenhout SA. Phytoplasma protein effector SAP11 enhances insect vector reproduction by manipulating plant development and defense hormone biosynthesis. Proc Natl Acad Sci U S A. 2011;108:E1254–1263. doi: 10.1073/pnas.1105664108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123:585–595. doi: 10.1093/genetics/123.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Biezen EA, Freddie CT, Kahn K, Parker JE, Jones JD. Arabidopsis RPP4 is a member of the RPP5 multigene family of TIR-NB-LRR genes and confers downy mildew resistance through multiple signalling components. Plant J. 2002;29:439–451. doi: 10.1046/j.0960-7412.2001.01229.x. [DOI] [PubMed] [Google Scholar]
- Van der Hoorn RA, De Wit PJ, Joosten MH. Balancing selection favors guarding resistance proteins. Trends Plant Sci. 2002;7:67–71. doi: 10.1016/s1360-1385(01)02188-4. [DOI] [PubMed] [Google Scholar]
- Watterson GA. On the number of segregating sites in genetical models without recombination. Theoretical population biology. 1975;7:256–276. doi: 10.1016/0040-5809(75)90020-9. [DOI] [PubMed] [Google Scholar]
- Weßling R, Schmidt SM, Micali CO, Knaust F, Reinhardt R, Neumann U, Ver Loren van Themaat E, Panstruga R. Transcriptome analysis of enriched Golovinomyces orontii haustoria by deep 454 pyrosequencing. Fungal Genet Biol. 2012;49:470–482. doi: 10.1016/j.fgb.2012.04.001. [DOI] [PubMed] [Google Scholar]
- Wicker T, Oberhaensli S, Parlange F, Buchmann JP, Shatalina M, Roffler S, Ben-David R, Dolezel J, Simkova H, Schulze-Lefert P, et al. The wheat powdery mildew genome shows the unique evolution of an obligate biotroph. Nat Genet. 2013;45:1092–1096. doi: 10.1038/ng.2704. [DOI] [PubMed] [Google Scholar]
- Win J, Chaparro-Garcia A, Belhaj K, Saunders DG, Yoshida K, Dong S, Schornack S, Zipfel C, Robatzek S, Hogenhout SA, et al. Effector biology of plant-associated organisms: concepts and perspectives. Cold Spring Harb Symp Quant Biol. 2012;77:235–247. doi: 10.1101/sqb.2012.77.015933. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.