

Abstract
The oocyte-to-embryo transition (OET) is thought to be mainly driven by post-transcriptional gene regulation. However, expression of both RNAs and proteins during the OET has not been comprehensively assayed. Furthermore, specific molecular mechanisms that regulate gene expression during OET are largely unknown. Here, we quantify and analyze transcriptome-wide, expression of mRNAs and thousands of proteins in Caenorhabditis elegans oocytes, 1-cell, and 2-cell embryos. This represents a first comprehensive gene expression atlas during the OET in animals. We discovered a first wave of degradation in which thousands of mRNAs are cleared shortly after fertilization. Sequence analysis revealed a statistically highly significant presence of a polyC motif in the 3′ untranslated regions of most of these degraded mRNAs. Transgenic reporter assays demonstrated that this polyC motif is required and sufficient for mRNA degradation after fertilization. We show that orthologs of human polyC-binding protein specifically bind this motif. Our data suggest a mechanism in which the polyC motif and binding partners direct degradation of maternal mRNAs. Our data also indicate that endogenous siRNAs but not miRNAs promote mRNA clearance during the OET.
Keywords: Caenorhabditis elegans, embryogenesis, maternal mRNA, oocyte-to-embryo transition, posttranscriptional gene regulation
See also: AT Neff et al (August 2014) and M Stoeckius et al (August 2014)
Introduction
Fertilization of the egg starts a new life cycle. Immediately after fertilization, the zygote reorganizes the entirety of its cellular components to become a mitotically dividing totipotent cell (Stitzel & Seydoux, 2007; Evsikov & Evsikova, 2009; Tadros & Lipshitz, 2009). Although this process is one of the most fundamental events in biology, little is known about the molecular mechanisms that regulate this so-called oocyte-to-embryo transition (OET). Remarkably, mature germ cells are transcriptionally silent, and reprogramming into totipotency during the OET occurs in the absence of transcription in all studied animals (Davidson, 1989; Evsikov et al, 2006). Some of the general post-transcriptional mechanisms that regulate gene expression later in embryogenesis at the ‘maternal-to-zygotic transition’ (MZT) could potentially also control the OET. Examples include translational activation or degradation of mRNAs mediated by sequence elements in 3′ untranslated regions (3′ UTRs) which are recognized and bound by small non-coding RNAs and/or RNA-binding proteins (RBPs) (Vasudevan et al, 2006; Radford et al, 2008; Giraldez, 2010). However, currently, there is no specific 3′ UTR sequence element known that is functionally important during the OET. Finally, even the expression of small non-coding RNAs such as miRNAs which are thought to regulate many developmental processes (Giraldez, 2010; Svoboda & Flemr, 2010) is virtually uncharacterized during the OET in all animals.
The reasons for the lack of understanding of the OET are in part technical—it is difficult to purify precisely staged oocytes before and after fertilization to enable comprehensive biochemical studies. Consequently, the OET has mostly been studied at a single-gene level. A few genome-wide transcriptome studies in early embryogenesis mainly focused on mRNA expression during the MZT (Baugh et al, 2003; Dobson et al, 2004; Hamatani et al, 2004; Wang et al, 2004; Evsikov et al, 2006; Zhang et al, 2009; Peaston et al, 2010; Aanes et al, 2011). Furthermore, mRNA expression during early development is expected to poorly correlate with corresponding protein levels (Grün et al, 2014), and therefore, expression studies should include quantification of protein levels. Although ribosome profiling was recently used as a proxy for translation rates in development and compared to mRNA abundance (Bazzini et al, 2012; Stadler et al, 2012), there is currently no study in any system that quantifies both mRNA and protein levels during early development.
In this study, we used the nematode Caenorhabditis elegans, one of the best-explored models for the OET (Robertson & Lin, 2013), to address some of the outstanding questions. We started by quantifying genome-wide mRNA and protein expression in oocytes and 1-cell-stage embryos. To do so, we purified large populations of stage-specific C. elegans embryos by flow cytometry (‘eFACS’(Stoeckius et al, 2009)). We developed a shotgun proteomics approach (‘in vivo SILAC’ (Grün et al, 2014)), similar to published variants (Fredens et al, 2011; Larance et al, 2011), that allowed us to assess expression changes of thousands of proteins in vivo. To quantify RNA expression changes, we used high-throughput sequencing (‘RNA-seq’) of mRNAs and small RNAs. These data represent, to our knowledge, the first comprehensive atlas of gene expression during the OET in an animal.
We observed that 25% of mRNAs present in the oocyte are degraded at the oocyte-to-embryo transition. We discovered a polyC 3′ UTR motif that was significantly enriched and occurred in over half of all degraded mRNAs. Using transgenic animals expressing 3′ UTR reporter constructs, we show that this polyC motif is required and sufficient to induce clearance of those mRNAs during the OET. We then identified a conserved RBP that binds this motif in vitro. We were unable to identify miRNAs that could explain the observed maternal mRNA clearance. However, we found that degradation of a small but significant subset of maternal mRNAs, mostly those without a polyC motif, is promoted by endogenous siRNAs in the zygote.
Results
To study the composition and dynamics of the transcriptome and thousands of proteins at the OET in C. elegans, we collected roughly one hundred thousand oocytes, 1-cell and 2-cell embryos. These early developmental stages cover a time period of approximately 50 min in vivo (at 20°C) during which two fundamental processes occur: oocyte maturation and fertilization (oocyte to 1-cell embryo, since maturation and fertilization are concomitant processes in C. elegans, we hereafter refer to this transition only as ‘fertilization’) and the first mitosis (1-cell to 2-cell embryo) (Fig1A). While sufficient numbers of precisely staged 1-cell embryos and an enriched 2-cell embryo sample (Materials and Methods) could be automatically collected by a cell-sorting-based method that we established previously (Stoeckius et al, 2009), we manually picked mature oocytes in large numbers after noticing pronounced differences in gene expression of oocytes obtained with commonly used methods (Aroian et al, 1997) (Supplementary Fig S1). High purity of these samples was confirmed by expression analysis of marker genes and non-germ line tissue-specific transcripts (Supplementary Fig S2 and Methods). We extracted protein and RNA from the same samples and averaged expression across at least two biological replicates. Oocyte-to-embryo expression changes were highly reproducible between replicates for transcripts and proteins (Supplementary Fig S3).
Figure 1. Composition and dynamics of transcriptome and proteome at the oocyte-to-embryo transition in Caenorhabditis elegans.

A Schematic representation of the three cell types used for proteome and transcriptome analysis including isolation method, obtained datasets, and the number of independent biological replicates. Oocytes were manually isolated from wild-type hermaphrodites. 1-cell and 2-cell embryos were obtained by eFACS (Stoeckius et al, 2009). The samples cover fertilization (oocyte to 1-cell embryo) and first mitosis (1-cell to 2-cell embryo).
B, C Histograms of mRNA log2-fold changes between oocytes and 1-cell (fertilization, B) and between 1-cell and 2-cell embryos (first mitosis, C). Significant fold changes are highlighted in blue (P < 0.01) (See Materials and Methods for further information).
D RT-qPCRs on equal numbers of independently handpicked 1-cell embryos and oocytes normalized to an exogenous spike were used to measure the fold changes of 50 selected transcripts over several orders of magnitude. These were then used to normalize the fold changes computed from mRNA-seq.
E, F Histograms of protein log2-fold changes between oocytes and 1-cell embryos (E) and between 1-cell and 2-cell embryos (F) computed from quantitative proteomics (in vivoSILAC). Significant fold changes are highlighted in red (P < 0.05) and red (P < 0.01) (See Materials and Methods for further information).
G Log2-fold changes of selected proteins measured between oocytes and 1-cell-stage embryos, some of which are known to have dynamic or constant expression upon fertilization (PIE-1 and MEX-5, OMA-1, MEI-2). Error bars were computed from two independent biological replicate measurements.
H, I Comparison of mRNA and protein log2-fold changes for ˜3,300 genes with confidently quantified expression (Materials and Methods) at fertilization (H) and first mitosis (I). Significant fold changes are highlighted in green (P < 0.05) and blue (P < 0.01) for transcripts and orange (P < 0.05) and red (P < 0.01) for proteins.
We first profiled mRNA expression by sequencing of polyadenylated transcripts (mRNA-seq) and found ˜35% of encoded mRNAs (7,460 genes) expressed (≥ 2 RPKM) in oocytes or embryos (Fig1B and C, Supplementary Fig S2 and Methods). To be able to accurately call up- and down-regulation of transcripts in our data, it is essential to carefully normalize the data, especially because substantial global mRNA expression changes may occur at the OET (Piko & Clegg, 1982; Gilbert et al, 2009; Peaston et al, 2010). We thus performed RT-qPCRs on 50 randomly selected genes in handpicked equal numbers of wild-type (N2) oocytes and living non-fixed 1-cell embryos together with an exogenous spike of RNA (Materials and Methods). These showed good correlation with the sequencing data (Fig1D) and, with the spike, could be used to normalize mRNA expression changes between oocytes and embryos in our RNA-seq data (Materials and Methods). After normalization, the median log2-fold change at the OET was shifted by −0.61, indicating that the overall mRNA content in the embryo is decreased and that normalization to an external control is essential to reveal this behavior.
We used in vivo SILAC to quantitatively measure abundance changes (Grün et al, 2014) of roughly 3,300 proteins at the OET with a false discovery rate of 5% (Fig1E and F). In vivo SILAC has been shown to allow precise and reproducible measurement of protein fold changes in vivo by mass spectrometry (Fredens et al, 2011; Larance et al, 2011; Grün et al, 2014). We normalized the fold changes based on the assumption that a set of 100 structural, metabolic, and translation machinery proteins remain constant during this transition (Materials and Methods). Indeed, using this normalization, the few proteins that are known to be constant, or up- or down-regulated during the OET, followed the expected behavior in our data (Fig1G) (Srayko et al, 2000; Cuenca et al, 2003; Lin, 2003).
Protein and mRNA expression dynamics reflect the biological processes occurring during the OET
While we observed widespread changes in transcript abundance at the OET (Fig1B), few transcripts underwent substantial changes after mitosis (Fig1C). On the protein level, we observed no difference in the overall strength and dynamic range of changes at the OET (Fig1E) compared to the first cell division (Fig1F). Overall, 178 proteins and 2,022 mRNAs were more than twofold down-regulated (P < 0.05) after fertilization, indicating clearance of many maternally supplied components. Notably, the observed expression changes are consistent with different functional requirements in oocytes and zygotes. Based on gene ontology (GO) classifications (Materials and Methods), strongly down-regulated proteins were functionally enriched in genes related to meiotic spindle organization (11 genes, P < 2e-3; Fisher's exact test) such as CYN-4 (Fig1G), indicating the switch form meiotic to mitotic cell cycle in the zygote. Consistently, the meiotic spindle component MEI-2 decreased roughly threefold from 1-cell to 2-cell stage. Moreover, genes involved in eggshell formation, such as CHS-1, were among the most strongly down-regulated proteins in 1-cell embryos (Fig1G). Transcripts down-regulated upon fertilization were also enriched in genes involved in cell-cycle switching such as gld-2 (14 genes, P < 1e-5; Fisher's exact test). Perhaps as expected, maternal sterile (34 genes, P < 0.04, Fisher's exact test) and embryonic lethal (87 genes, P < 0.03, Fisher's exact test) phenotypes were overrepresented among these genes (Materials and Methods).
We found that 111 proteins were significantly up-regulated more than twofold upon fertilization (P < 0.05, Materials and Methods). The most highly up-regulated proteins were enriched in regulators of gene expression and transcription (18 genes, P < 1.4e-4, Fisher's exact test), such as the essential Tata-associated factor-4 (TAF-4) (Fig1G). Interestingly, we observed many RNA-binding proteins known to be essential for fate specification, such as POS-1, MEX-6, and SPN-4, to be up-regulated more than fourfold (Fig1G). RNAPII is thought to be transcriptionally silent in the early embryo (Seydoux & Fire, 1994; Seydoux & Dunn, 1997; Baugh et al, 2003; Guven-Ozkan et al, 2008). Surprisingly, we found 193 mRNAs significantly up-regulated more than twofold upon fertilization (P < 0.05, Materials and Methods). We investigate the nature of this up-regulation in a parallel study (see Discussion and Stoeckius et al, 2014). The 53 most strongly ‘up-regulated’ transcripts (twofold with P < 0.01, > 4 RPKM in 1-cell embryos and not down-regulated in 2-cell embryos (< 1.4-fold)) were also significantly associated with transcription initiation (six genes, P < 1.6e-5; Fisher's exact test) and, more generally, DNA conformation change (nine genes, P < 1e-4, Fisher's exact test). These findings suggest that expression changes required for the onset of zygotic transcription are already initiated in the 1-cell embryo.
Protein and transcript expression changes are decoupled
Although transcripts and proteins with major expression changes at the OET belong to similar functional categories, transcriptome and proteome changes appeared to be largely decoupled upon fertilization (Spearman's correlation coefficient (ρ) = 0.17; Fig1H). This observation illustrates the broad post-transcriptional gene regulation at the OET affecting protein abundance independent of transcript levels. In stark contrast, the massive changes we observed on mRNA expression during the OET were much reduced when comparing 1- and 2-cell embryos, while protein abundances were changing altogether as before, suggesting that mostly de novo translation and protein turnover are encompassing the first mitosis (Fig1I).
In summary, we measured mRNA expression of roughly 7,500 genes and protein abundance changes for approximately 3,300 proteins. Expression dynamics of proteins and mRNAs are decoupled but reflect biological processes occurring concomitant with the OET.
The majority of down-regulated transcripts contain a polyC motif in their 3′ UTRs
Sequence elements in 3′ UTRs can direct mRNA translational activation, silencing, and decay. To identify molecular mechanisms that could regulate the increase or decrease in protein abundance and the widespread clearance of maternal mRNAs that we observed, we performed a de novo search for sequence motifs specifically enriched or depleted in 3′ UTRs of down-regulated transcripts and up- or down-regulated proteins compared to the entire pool of mRNAs using MEME (Bailey & Elkan, 1994).
We only discovered one extremely significantly enriched motif (MEME E-value < 3e-155), a stretch longer than 8 cytosine nucleotides (hereafter referred to as ‘polyC motif’; Fig2A) in the 3′ UTR of down-regulated transcripts. In many cases, we identified extended stretches of ˜12 cytosine nucleotides (Fig2B). Out of the 6,429 expressed genes at the OET that have an annotated 3′ UTR, ˜1,000 contain a polyC motif (> 90% quantile of motif score distribution across all 3′ UTRs; see Materials and Methods). These genes are on average 2.1-fold more highly expressed in oocytes compared to genes without a motif (P < 1e-40; Wilcoxon rank-sum test, Supplementary Fig S4A) suggesting a stabilization of genes that contain a polyC motif in the oocytes. At the OET, polyC-containing genes are strongly down-regulated (P < 1e-232, Wilcoxon rank-sum test; Fig2C), and this further progresses in the 2-cell stage. The probability to find the motif in the 3′ UTR of a gene decreases sharply for less strongly down-regulated genes. Moreover, genes that are expressed in oocytes and harbor a motif have a 94% probability to be down-regulated at the 1-cell stage.
Figure 2. A polyC motif directs transcript degradation upon fertilization.
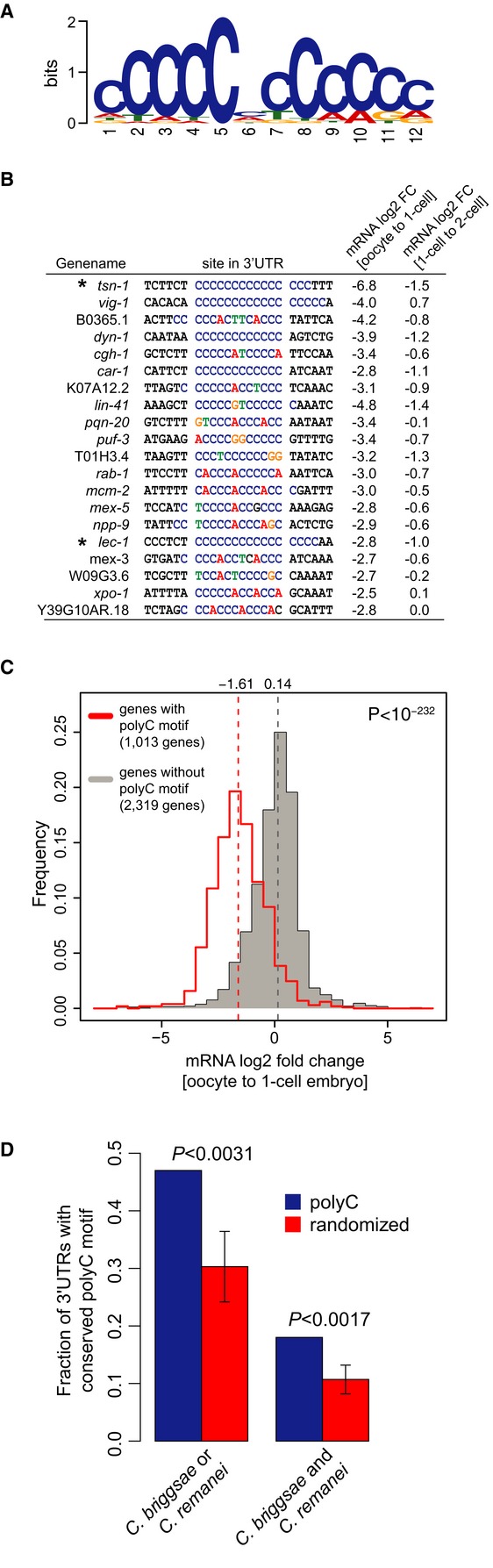
- Highly significant sequence motif identified in the 3′ UTR of 36% of down-regulated genes at the OET determined by MEME (P = 0, Fisher's exact test).
- Examples of 20 down-regulated mRNAs containing a polyC motif including the measured mRNA log2-fold changes between oocytes and 1-cell embryos, and between 1-cell and 2-cell embryos. Asterisks indicate the genes for which 3′ UTR reporter strains were generated.
- Histograms of mRNA fold changes between oocytes and 1-cell embryos for genes with (red) and without (gray) a polyC motif. The median (dotted vertical lines) is significantly shifted between the two distributions (P < 10−232, Wilcoxon rank-sum test).
- Fraction of genes with conserved occurrence of polyC (blue bars) and random motifs (red bars) in the 3′ UTR of C. briggsae and/or C. remanei. Error bars and P-values were deduced by comparing to conservation of 50 randomized motifs.
Taken together, we observe a polyC motif in the majority of transcripts that are down-regulated at the OET, and these genes are among the most highly expressed genes in oocytes.
Function and conservation of the polyC motif and the genes harboring a motif
To investigate the functional significance of the polyC motif-containing genes, we performed GO term analysis. Diverse biological functions were overrepresented among genes with high motif scores (> 90% quantile of the score distribution) in comparison with genes with low motif scores (< 50% quantile of the score distribution). For instance, significantly enriched GO terms were associated with signaling (P < 4.9e-29), cytoskeleton organization (P < 5.6e-21), and organelle organization (P < 1.4e-18) and, more specifically, comprised gamete generation (P < 9.7e-13) and vulval development (P < 1.9e-12). We observed a significant enrichment of maternal sterile (P < 4e-5) and embryonic lethal phenotypes (P < 1.6e-9).
To further investigate the functional significance of the polyC motif, we measured conservation in other nematode species, C. briggsae and C. remanei, separated from C. elegans by more than 30 million years of evolution (Cutter, 2008). PolyC motifs were not significantly conserved when analyzing sequence conservation in alignments of orthologous 3′ UTR across these species. However, this approach is likely compromised by the difficulty to infer correct alignments for relatively lowly conserved 3′ UTR sequence. To circumvent this problem, we defined conservation simply by the presence of a polyC motif somewhere in a set of orthologous 3′ UTRs. With this strategy, we observed that conservation was significant (P < 3.1e-3; Fig2D). Interestingly, a similar motif has been suggested to regulate mRNA stability (Makeyev & Liebhaber, 2002) and alternative polyadenylation (Ji et al, 2013) in cells, but has not been implicated in the OET or early development of any organism.
The polyC motif is sufficient and necessary to direct mRNA decay at the OET
To test functionality of the polyC motif in vivo, we cloned the 3′ UTR of two candidates (tsn-1 and lec-1) with a wild-type motif and a mutated version of the motif into GFP reporter strains (Fig3A and B). The candidates had one obvious polyC motif in their 3′ UTR but differed in down-regulation strength upon fertilization (asterisks in Fig2B). All transgenic lines were produced by single copy insertion into the same position into the genome (Frøkjaer-Jensen et al, 2008a,b). The reporter transcripts with wild-type 3′ UTR decreased at comparable levels to the endogenous transcript upon fertilization. Mutation of the polyC motif led to a complete abolishment of regulation in the case of lec-1 (Fig3A) and an over 64-fold decreased regulation in case of tsn-1 (Fig3B), indicating that mRNA clearance of the two candidates is controlled by the polyC motif in their 3′ UTR.
Figure 3. PolyC motif is sufficient and necessary to induce mRNA degradation upon fertilization.
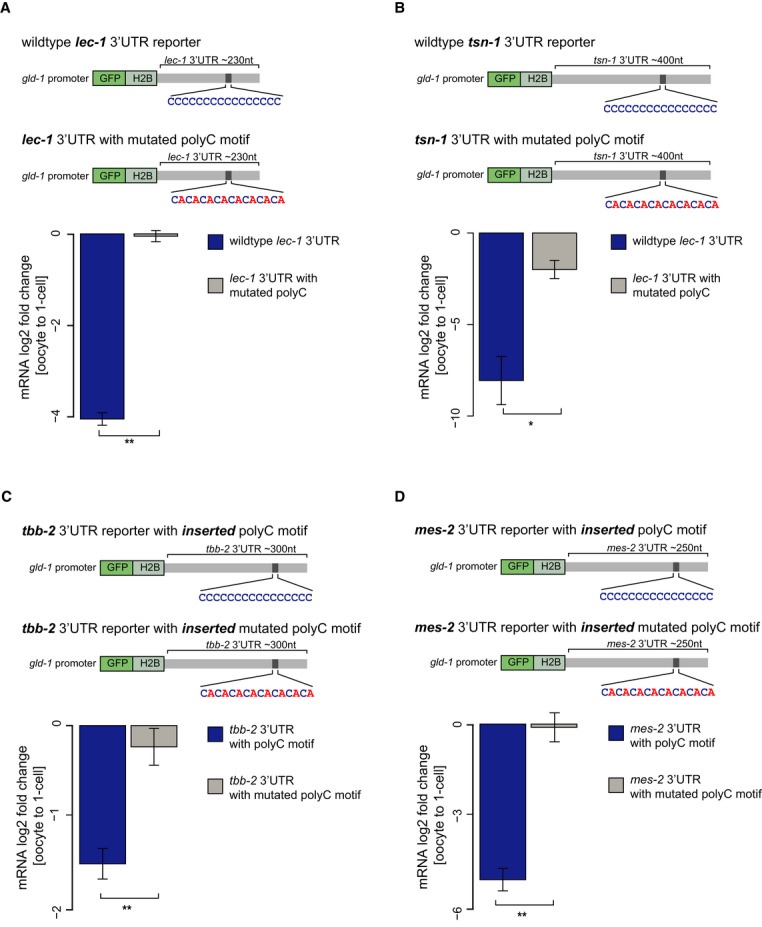
A, B Schematic representation of reporter constructs used for testing whether the polyC motif is necessary to induce mRNA decay at the OET in vivo. Reporters were generated that contained a gld-1 promoter fused to GFP-histone 2B and the 3′ UTRs of lec-1 (A) and tsn-1 (B) with unaltered or mutated versions (CACACA…) of the polyC motif. Transgenic animals expressing a single copy construct at a fixed location in the genome were generated by MosSCI (Frøkjaer-Jensen et al, 2008a,b). Expression changes of the reporters in handpicked ooyctes and 1-cell-stage embryos were then profiled by qRT-PCRs. Mutation of the polyC motif leads to inhibition of down-regulation of the lec-1 reporter and a roughly 64-fold decreased down-regulation of the tsn-1 reporter mRNA upon fertilization as determined by RT-qPCRs on handpicked oocytes and 1-cell embryos.
C, D Schematic representation of reporter constructs used for testing sufficiency of the polyC motif to induce mRNA decay at the OET in vivo. The polyC motif and a mutated version of the motif were inserted into the 3′ UTRs of tbb-2 (C) and mes-2 (D) that do not contain the motif. Expression changes of the reporters in handpicked ooyctes and 1-cell-stage embryos were then profiled by qRT-PCRs. Insertion of the motif but not the mutated version leads to a threefold down-regulation of the tbb-2 reporter (C) and an over 16-fold down-regulation of the mes-2 reporter (D) upon fertilization.
To test whether the polyC motif is not only required but even sufficient to induce degradation during the OET, we inserted the consensus polyC motif and separately a mutated control motif into the tbb-2 and mes-2 3′ UTRs (Fig3C and D) that do not contain the motif and are not post-transcriptionally regulated in the germ line (Merritt et al, 2008). The insertion of the polyC motif resulted in a threefold reduction of tbb-2 reporter and over 16-fold reduction of mes-2 reporter expression upon fertilization (Fig3C and D). This down-regulation is lost for mes-2 and reduced for the tbb-2 reporter, when only a mutated version of the polyC motif is inserted into the 3′ UTR (Fig3C and D). Moreover, we do not observe a trend toward higher or lower reporter expression in oocytes when inserting the motif compared to reporters with a mutated variant of the motif (Supplementary Fig S5C).
Together, the reporter assays strongly suggest that polyC motifs in 3′ UTRs are required and sufficient to confer down-regulation of transcripts at the OET.
A set of conserved RNA-binding proteins bind the polyC motif
We next explored which RNA-binding protein binds to the polyC motif. In human, the polyC motif is bound by polyC-binding proteins 1 and 2 (PCPB1, PCBP2) (Makeyev & Liebhaber, 2002). By reciprocal blast, we found three potential PCBP homologues in the C. elegans genome (Fig4A). We performed pull-down assays from worm lysates using RNA oligonucleotides containing the polyC motif or a mutated motif that was also used in the reporter assays (Materials and Methods). Proteins binding to the bait-RNAs were subsequently identified by mass spectrometry and quantified by label-free quantification. Only two proteins were highly significantly and reproducibly bound to the polyC motif compared to the mutated control (Fig4B). These two proteins (PES-4 and F26B1.2) turned out to be the two closest predicted homologues of the human PCBP1/2 (Fig4A). The third predicted homologue (Y59A8B.10) was only significantly enriched in one of the two independent biological replicates of our biotinylated RNA pull-down experiments (Supplementary Fig S4D).
Figure 4. PolyC motif is bound by conserved RNA-binding protein in vitro.
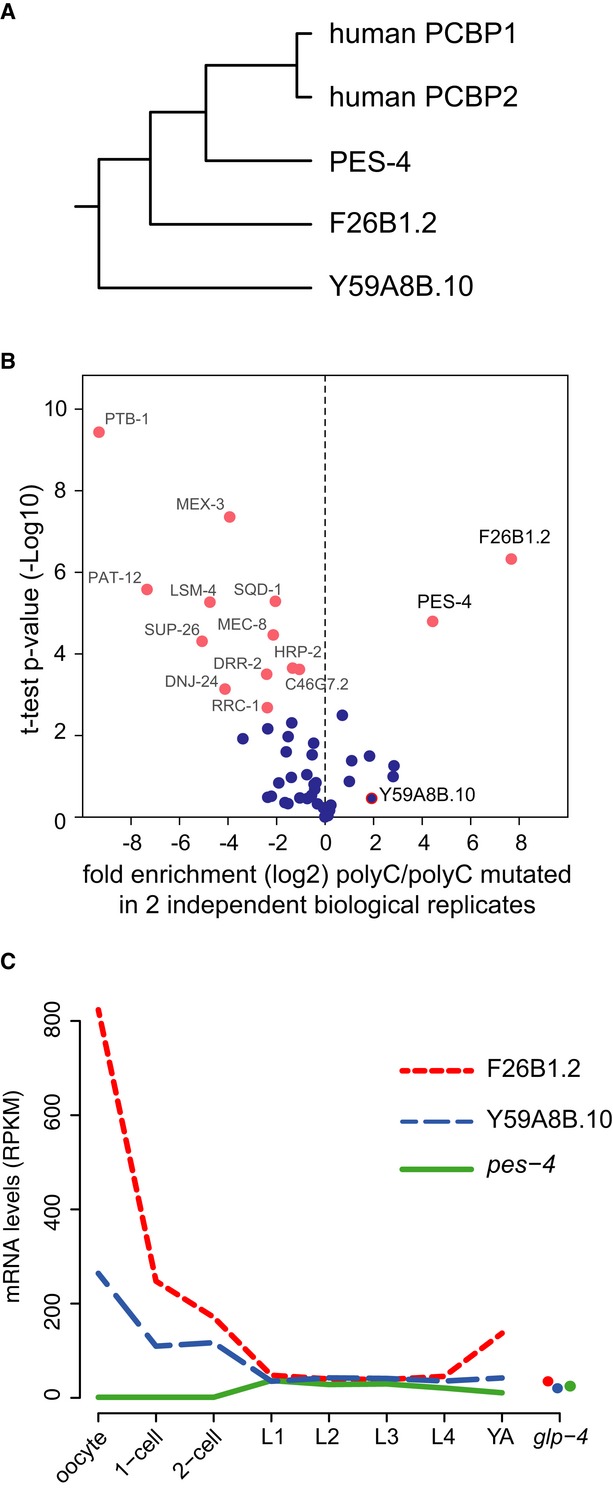
- Phylogenetic tree of three predicted Caenorhabditis elegans homologues of human polyC-binding protein-1 (PCBP1). For each gene, the amino acid sequence of the longest isoform was extracted, and sequences were aligned using ClustalW with default parameters.
- Biotinylated RNA pull-down assay coupled to mass spectrometry to identify polyC-binding proteins in adult worm lysates. Two independent biological replicates were measured in triplicates by mass spectrometry and quantified. Observed enrichment (log2-fold change) binding to polyC motif versus the mutated motif (CACACA…) is plotted against the negative logarithmic P-value of the t-test resulting in a volcano plot. Proteins with a high fold change and low P-value are significant interactors.
- mRNA expression depicted in RPKM of predicted C. elegans homologues of PCBP1 in oocytes, 1-cell and 2-cell-stage embryos and throughout larval development and in germline-less adult worms (glp-4). Expression data for larval stages and young adults were recently published from our laboratory (Grün et al, 2014).
We next investigated the expression pattern of these genes in our data and throughout larval and adult development using our previously published mRNA and protein datasets (Grün et al, 2014). We did not measure protein levels for these genes in our OET datasets. While pes-4 mRNA is almost not expressed (< 1 RPKM) in our samples, we observed high expression of F26B1.2 (˜820 RPKM) and Y59A8B.10 (˜264 RPKM) in oocytes with a sharp expression decrease in embryos and relatively low expression (˜40 RPKM) in larval stages and adults (Fig4C), suggesting a specific function of these two proteins in oocytes or at the OET.
Together, we found that the polyC motif is bound by close homologues of human PCBP1/2 in vitro. Expression of these indicates that F26B1.2 and Y59A8B.10 act during oogenesis and/or the OET and may thus have a conserved function in regulation of mRNA stability of polyC motif-containing genes as it has been suggested in human cells (Makeyev & Liebhaber, 2002).
Endogenous siRNAs but not miRNAs contribute to maternal mRNA clearance
Small non-coding RNAs have also been implicated in post-transcriptional regulation that drives mRNA turnover during development (Giraldez, 2010; Rouget et al, 2010; Svoboda & Flemr, 2010). To investigate whether small non-coding RNAs are involved in mRNA clearance at the OET in C. elegans, we mined our small RNA-sequencing data generated from a parallel study (Stoeckius et al, 2014) for small RNA expression. We first asked whether expressed miRNAs can explain transcript or protein changes of putative conserved miRNA target genes predicted by PicTar (Lall et al, 2006). Neither looking directly for targets of differentially expressed miRNAs nor taking an unbiased approach and asking for an enrichment of miRNA targets among differentially expressed genes provided compelling evidence for miRNA-directed regulation.
We then studied endogenous siRNAs in the zygote. We separated the prominent class of 22G-RNAs into two distinct groups based on 26G-RNA evidence for the same locus, which at least in part allowed us to distinguish silencing from non-silencing endo-siRNAs (Materials and Methods). Interestingly, genes that give rise to silencing 22G-RNAs in the 1-cell embryo are overall mildly but significantly down-regulated at the OET (P < 6.3e-59, Wilcoxon rank-sum test; Fig5A) and are enriched in genes associated with oogenesis (P < 6.3e-8; Fisher's exact test) and female germ cell development (Reinke et al, 2004) (P < 1e-16; Fisher's exact test). In summary, while we did not find indications for miRNA-directed mRNA turnover at the OET, we provide evidence that down-regulation of a subset of maternal mRNAs (˜10%), many of which are lacking a polyC motif, is likely due to endogenous siRNAs.
Figure 5. Endogenous siRNAs contribute to mRNA turnover upon fertilization.
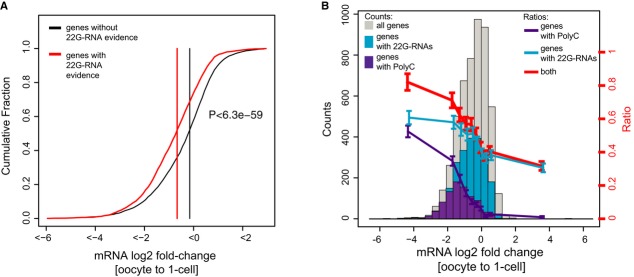
- Cumulative fraction of mRNA log2-fold changes upon fertilization. Genes that are targeted by silencing 22G-RNAs are significantly shifted toward negative fold changes compared to genes that are not targeted by silencing 22G-RNAs (P < 6.4e-59; Wilcoxon rank-sum test).
- Histograms of mRNA fold changes between oocytes and 1-cell embryos. Shown are fold changes for all genes with log2 RPKM > 1 in either embryos or oocytes (gray), for genes with a polyC motif (purple) and for genes that are predicted targets of silencing siRNAs (blue). Lines represent the fraction of genes carrying a polyC motif (purple) or being a predicted target of silencing siRNAs (blue) and by both (red) as a function of the log2-fold change at the OET.
Together, up to 60% of the most strongly down-regulated genes upon fertilization can be explained by endogenous siRNAs and/or the polyC-directed mRNA down-regulation (Fig5B).
Discussion
This study represents the first comprehensive gene expression atlas during the OET in any animal. We performed extensive analyses to quantify around 7,500 expressed mRNAs, 3,300 proteins and all known classes of small non-coding RNAs in oocytes, 1-cell and 2-cell embryos and to investigate expression dynamics between these stages. Previous studies interrogating this transition on a single-gene level could not detect, but only suggest, the clearly orchestrated and highly dynamic changes between transcriptome and proteome that we describe here in detail. Our study illustrates broad post-transcriptional gene regulation at the OET in C. elegans and maps the dynamic mRNA changes as well as protein changes that occur at this transition. All of these data are easily accessible on our developmental transcript and protein expression database at http://elegans.mdc-berlin.de.
Perhaps surprisingly, our data indicated that some mRNAs confidently increased more than twofold upon fertilization. It is believed that RNAPII is inactive in oocytes and embryos until the 3- to 4-cell embryo in C. elegans (Seydoux & Fire, 1994; Seydoux & Dunn, 1997; Guven-Ozkan et al, 2008). These studies relied on in situ hybridization of single transcripts (Seydoux & Fire, 1994) or immunohistochemistry visualizing C-terminal domain phosphorylation of RNAPII (Seydoux & Dunn, 1997; Guven-Ozkan et al, 2008). Neither of these techniques is as sensitive as RNA sequencing used in this study, and therefore, the possibility that mRNA transcription begins even earlier remains. We thus note that the detected subtle up-regulation of mRNAs can in principle be attributed to transcription in the 1-cell embryo. Another explanation is that some of the observed up-regulation could be caused by readenylation, which would result in an increased probability of transcript capture by polyA selection-based techniques used for mRNA-seq. An increase of mRNA abundance at the OET can also be due to transcripts transferred by sperm during fertilization. This has recently been suggested in various organisms (Krawetz, 2005), and we investigated paternal contributions in C. elegans elsewhere (Stoeckius et al, 2014). In short, the data suggest that over 80% of the strongly up-regulated mRNAs can be explained by paternally contributed transcripts (Stoeckius et al, 2014).
We note that, in C. elegans, oocytes are produced in an assembly line-like fashion, undergoing oocyte growth, maturation, and fertilization in quick succession (McCarter et al, 1999). We thus caution that some of the mRNA and protein changes that we observe could have been potentially occurring in a continuum during oocyte development or initiated shortly before fertilization.
A polyC motif and endogenous siRNAs coordinate maternal mRNA clearance at the OET
Zygotic genome activation and maternal mRNA clearance are a hallmark of the MZT in all studied organisms. In C. elegans, zygotic RNAPII transcription is initiated in the 3- to 4-cell embryo (Seydoux & Fire, 1994; Seydoux & Dunn, 1997; Baugh et al, 2003; Guven-Ozkan et al, 2008), which is accompanied by a degradation of a subset of maternal mRNAs (Baugh et al, 2003). Our data show that around 30% of the ˜7,500 expressed mRNAs in the C. elegans oocyte are already targeted for degradation at the OET. This suggests a strictly maternal program of mRNA clearance at the OET in C. elegans prior to zygotic genome activation that has also been suggested in zebrafish (Aanes et al, 2011) and flies (Tadros et al, 2007). While the mechanism of this first wave of mRNA clearance in zebrafish remains unknown, in flies, it has been shown that de novo translation of the RBP SMAUG during egg activation is essential for maternal mRNA destabilization (Tadros et al, 2007). Although many miRNAs are dynamically expressed at the OET (Stoeckius et al, 2014), we surprisingly did not find indications for miRNA-directed regulation of mRNA degradation, as it has been described in other species at the MZT (Giraldez et al, 2006; Bushati et al, 2008). However, our data provide ample evidence for endogenous siRNA-directed clearance of transcripts. This is in line with the observation that only very few miRNAs have embryonic lethal phenotypes in C. elegans (Miska et al, 2007) and that siRNAs but not miRNAs seem essential for very early embryogenesis in mice (Tam et al, 2008; Watanabe et al, 2008; Suh et al, 2010).
We discovered a cis-regulatory motif (‘polyC’) which had a much more pronounced effect on mRNA degradation than siRNAs at the OET. This motif is clearly present in at least one-third of the thousands of transcripts that are degraded upon fertilization and mostly absent in transcripts that are not degraded. We remark that more degenerate versions of this motif may explain even more degraded transcripts. Our functional data from in vivo reporter assays strongly argue that the polyC motif is both required and sufficient to mark the corresponding mRNAs for degradation after fertilization. Together, our data show that the presence or absence of the polyC motif largely determines whether an mRNA is degraded or not immediately after fertilization. Interestingly, although the motif is significantly conserved in 3′ UTRs across nematodes, the overall amount of conservation was moderate, suggesting that either the set of transcripts degraded upon fertilization is evolving rapidly or that different mechanisms of transcript degradation evolved in the other nematodes. Nonetheless, we note that a similar 3′ UTR motif in vertebrates is bound by a polyC-binding protein (PCBP). PCBPs belong to the hnRNP K homology (KH) domain-containing RBPs that have been shown to be involved in multiple post-transcriptional pathways including mRNA stability and translation regulation (Makeyev & Liebhaber, 2002). Indeed, by RNA pull-down assays coupled to mass spectrometry, we could confirm that the polyC motif in C. elegans is bound by close homologues of the human PCBP in vitro.
How can the mRNA clearance at the OET be regulated by the polyC motif? The two simplest scenarios include a) de novo translation or activation of a negative trans-acting RBP (or small RNA) comparable to observations in early development of flies (Tadros et al, 2007) and zebrafish (Giraldez et al, 2006) that induces degradation of target mRNAs, or b) degradation and/or deactivation of a stabilizing trans-acting RBP, which in turn leads to destabilization of its bound mRNAs. We believe that the latter is more likely because we observe decay of the polyC-binding protein homologues at the OET. Moreover, the polyC motif has previously been described as a stabilizing element (Makeyev & Liebhaber, 2002). Our data thus suggest degradation of a stabilizing RBP that results in clearance of its target mRNAs at the OET, which may serve as an entry point for future studies elucidating the functional details of the motif and its trans-acting factors.
To pinpoint the function of the polyC-binding proteins on polyC motif-containing genes, we performed knockdown of the predicted homologues. Single RNAi of any candidate did not result in an observable phenotype. Although double knockdown of two PCBP homologues (F26B1.2 and Y59A8B.10) caused reproducible embryonic lethality after roughly 5 embryonic cleavages, we observed only very subtle expression changes of mRNAs containing the polyC motif. One likely reason for this could be that we were not able to achieve sufficient double or triple knockdown of the candidates or miss more distantly related homologues in our predictions. In fact, using less stringent reciprocal best blast, we could identify three more distantly related homologues of the human polyC-binding protein totaling up to six potential homologues in the C. elegans genome (Supplementary Fig S4E). However, knockdown of these also did not yield conclusive results potentially because we were not able to achieve sufficient triple or quadruple knockdown of the candidates. In D. melanogaster, SMAUG is a newly translated trans-acting factor that induces mRNA destabilization together with unknown factors upon egg activation (Tadros et al, 2007). It is thus also likely that we failed to identify a co-factor involved in the regulation of polyC motif-containing gene expression. It is also possible that the polyC motif influences mRNA stability indirectly at this transition by regulating mRNA translation. We observe that proteins of genes containing the motif are also more strongly down-regulated at the OET (Supplementary Fig S5A), and we do not observe this effect for genes targeted by 22G-RNAs (Supplementary Fig S5B).
Altogether, we provide the first genome-wide characterization of joint changes in RNA and protein expression at the oocyte-to-embryo transition and begin to uncover molecular mechanisms that drive these changes in the nematode C. elegans. We believe that our analyses lead to a better understanding of this fundamental developmental transition in animal development.
Materials and Methods
Caenorhabditis elegans maintenance
Strains were maintained using previously described methods (Brenner, 1974; Stiernagle, 2006) on OP50-seeded NGM plates at permissive temperatures. For all experiments, strains were cultivated at 24°C. Unless otherwise noted, the wild-type strain used was the Bristol N2.
Isolation of 1-cell and 2-cell embryos by eFACS
1-cell- and 2-cell-stage embryos were obtained by fluorescence-activated cell sorting (eFACS) as described previously (Stoeckius et al, 2009) in an FACSAriaIII flow cytometer (BD Biosciences, USA). Microscopic examination of the sorted embryos indicated that the 1-cell embryo sample was virtually pure (> 98% 1-cell-stage embryos), while the 2-cell-stage embryo sample was a mixture of 1-cell-stage (35%), 2-cell-stage (60%), and older (< 5%) embryos. Moreover, purity of the stages was further validated by checking for marker gene expression (see below) (Supplementary Fig S2). Roughly 100,000 embryos for each independent biological replicate were used for RNA and protein extraction.
Isolation of wild-type oocytes
We isolated oocytes from wild-type worms (see Supplementary Fig S1). Adult wild-type hermaphrodites (N2) were cut using a razor blade in PBS containing 0.5% BSA and 0.02% Tween. Oocytes were picked by mouth pipetting under a stereo microscope (Leica), washed thoroughly in PBS, and lyzed in TriZol LS (Invitrogen, USA). Only preparations containing > 98% pure oocytes were used. A fraction of the isolated oocytes was checked for endomitosis by fluorescence microscopy (Zeiss, Germany) with a nuclear dye. Moreover, purity of oocytes was further validated by checking for marker gene expression (see below) (Supplementary Fig S2). Roughly 100,000 oocytes for each independent biological replicate were used for RNA and protein extraction.
Assessment of sample purity
To validate purity of our 1-cell, 2-cell embryos, and oocytes, we checked our datasets for expression of known zygotic and older embryo marker genes, as well as non-germ line tissue-specific transcripts, in our sequencing data. While early embryo and oocyte marker genes are highly expressed (e.g. oma-1; Supplementary Fig S2A and B), we did not observe zygotic transcripts that have been described to be expressed in four-to-eight cell embryos (e.g. med-1, end-3, end-1; Supplementary Fig S2A and B) and did not detect transcripts which are highly expressed in muscle (myo-3, myo-2, hlh-1), neurons (unc-8, unc-25), gut (elt-2, ges-1), and sperm (msp-10, msp-81, msp-56, spe-9; Supplementary Fig S2A). In conclusion, our samples obtained contain 1-cell and 2-cell embryos, and oocytes, respectively, with high purity.
Stable isotope labeling by amino acids in Caenorhabditis elegans (in vivo SILAC)
Worms were metabolically labeled with 15N213C6-Lysine (Cambridge Isotope Laboratories, hereafter referred to as ‘heavy’ lysine) by feeding them with the metabolically labeled lysine auxotroph Escherichia coli strain AT713. Worms were cultivated for one generation on peptone-free NGM plates supplemented with antibiotic–antimycotic (Invitrogen, USA) seeded with an excess of labeled bacteria. Oocytes isolated from ‘heavy’ labeled worms from a spe-9 (hc88TS) mutant background were directly compared to ‘light’ wild-type oocytes and ‘light’ 1-cell-stage and ‘light’ 2-cell-stage embryos. Protein fold changes between wild-type oocytes, 1-cell and 2-cell embryos were then calculated by computing the ratios of proteins.
RNA extraction
For all samples, RNA was isolated by two rounds of freeze–thaw lysis in Trizol LS reagent (Invitrogen, USA) according to the manufacturer's protocol. RNA was co-precipitated with Glycoblue (Ambion, USA) for 30 min at −80°C. Subsequently, RNA was DNAse treated (RQ1 DNAse, Promega) and was re-extracted with Acid Phenol Chloroform (Ambion, USA). RNA concentration was measured by means of absorption spectrometry at a wavelength of 260 nm in a NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies, USA). RNA integrity was determined by capillary gel electrophoresis on a Bioanalyzer (Agilent, USA).
Protein extraction
Proteins were isolated from exactly the same samples in parallel with RNA using the Trizol LS reagent (Invitrogen, USA) with some modifications from the manufacturer's protocol. In short, proteins were precipitated in acetone (85% final concentration) over night at −20°C. The protein pellet was washed three times in 3 M guanidine-HCl in ethanol with a final wash in 100% ethanol. Pellet was solubilized by boiling and sonication in NuPAGE LDS loading buffer (Invitrogen, USA).
PolyA RNA isolation
PolyA mRNA was purified from 1 μg of total RNA using the Dynalbeads mRNA Purification Kit (Invitrogen, USA) according to the manufacturer's protocol. Depletion of rRNAs was validated by capillary gel electrophoresis on a Bioanalyzer (Agilent, USA). PolyA RNA was subsequently processed for sequencing (see below).
Constructing sequencing libraries for transcriptome analysis
PolyA-selected RNA was fragmented into approximately 250 nt fragments by chemical fragmentation (200 mM Tris acetate pH 8.2, 500 mM potassium acetate, 150 mM magnesium acetate) at 94°C for exactly 3.5 min in a thermocycler. Fragmented RNAs were isolated with RNA Clean beads (Beckman Coulter, USA) according to manufacturer's instructions. Fractionation was checked by capillary electophoresis in a RNA Pico 6000 chip using the Bioanalyzer (Agilent Technologies, USA). First-strand cDNA synthesis was accomplished using Superscript III Reverse Transcriptase and random primers (Invitrogen, USA), followed by second-strand synthesis using DNA Polymerase I and RNaseH (Invitrogen USA). Double-stranded DNA was purified with Agencourt AMPure beads XP (Beckman Coulter, USA), and quality was checked by capillary gel electrophoresis on the Bioanalyzer with the Agilent DNA 1000 kit (Agilent Technologies, USA). dsDNA libraries subsequently processed for sequencing using the Genomic DNA Sample Prep Kit (Illumina, USA) according to the manufacturer's protocol.
Cluster generation and sequencing
Cluster generation as well as sequencing of the prepared libraries was performed on the Illumina cluster station (Illumina, USA) and HiSeq2000 or GAIIx (Illumina, USA) according to the manufacturer's protocols (Illumina, USA).
Gene models
We used revised and extended modENCODE gene models (Gerstein et al, 2010), comprising 64,826 transcripts which correspond to 21,774 different genes.
Processing of mRNA-sequencing output
All libraries were sequenced by paired-end sequencing (2 × 101 nt reads) yielding around 60–80 million reads per experiment if performed on GAIIx and more than 100 million reads if performed on HiSeq2000. The paired-end reads of all samples were mapped to the transcriptome sequences using the read alignment software BWA (Li & Durbin, 2010). Prior to read mapping, we removed consecutive strings of basecalls with lowest Phred quality score from the 3′ end of the reads and kept only those with a minimum remaining length of 30 bases after trimming. We ran BWA with a minimum seed length of 30 and default parameters otherwise. The fraction of reads mapping to the transcriptome ranged from ˜60% to ˜85%. To quantify expression of a given gene locus, we aggregated reads across all isoforms derived from this locus. Reads mapping to multiple loci were distributed uniformly among these loci. Expression was quantified in reads per kilobase of transcript sequence per million mapped reads (RPKM) (Pepke et al, 2009), normalizing by the total length of exonic sequence obtained after merging all isoforms. The mean expression μi of gene i was computed as the average across biological replicates, and expression variability was estimated by the standard deviation σi.
We assigned a confidence value to the expression quantification of gene i, given by max(0,1 − μi/σi). Similarly, we calculated mean and standard deviation of the log2-fold changes between two samples by Gaussian error propagation. We assume a Gaussian distribution of log2-fold changes at each transition and consider the mean as an estimator for the expected log2-fold change. This allows us to infer a z-score and hence a P-value for each log2-fold change.
The reproducibility of transcript fold changes across independent biological replicates was good (Spearman's correlation coefficient > 0.87).
We then applied a threshold of 2 RPKM to filter out transcripts expressed at background level. We were left with roughly 7,500 expressed genes in either oocytes or embryos (Supplementary Table S1).
Normalization of mRNA fold changes
Inappropriate normalization when computing expression fold changes can result in artificial increases of transcript abundance during this period of transcriptional silence (Peaston et al, 2010). These problems are mainly caused by two circumstances. First, fertilization involves the fusion of two cells and merging both their transcript and protein contents, and second, it can trigger vast degradation of RNAs, as it has been shown in mice (Piko & Clegg, 1982). It is unknown how the proteome changes upon fertilization; however, it is likely that similar problems also affect protein expression changes at the OET.
For our transcriptome data, we thus performed RT-qPCRs for 50 genes on an equal number of independently handpicked oocytes and 1-cell-stage embryos into which a fixed amount of human total RNA was spiked. Normalization of the RT-qPCR to human GPDH and human actin permitted the quantification of absolute expression changes between oocytes and embryos. After normalization, the sequencing and RT-qPCR-derived fold changes were in good agreement (Fig1D, Spearman's correlation coefficient ˜0.73). Reverse transcription performed with random- or oligo-d(T)12-16 primers leads to comparable results (data not shown). After normalization, the median log2-fold change at the OET was shifted by -0.61. All primer sequences can be found in Supplementary Table S3.
Normalization of protein fold changes
In vivo SILAC allowed us to quantitatively measure fold changes of ˜3,300 proteins between oocytes and embryos at a false discovery rate of 5% (on peptide identifications), with good correlation between independent biological replicates (Supplementary Fig S3; Spearman's correlation coefficient (ρ) ˜0.6). We have previously shown that fold changes determined by in vivo SILAC accurately represent changes in protein abundance in vivo (Grün et al, 2014). Lacking the ability for comparable sensitive spike in techniques as we used for our RT-qPCRs, we normalized our data based on the assumption that abundance of a set of roughly 100 structural, metabolic, and translation machinery proteins is not changing at this transition and could be used for normalization (Supplementary Table S2).
Mapping and processing of small RNA data
Reads from all small RNA-sequencing libraries were mapped to the C. elegans genome (WS190). Prior to read mapping, we removed consecutive strings of basecalls with lowest Phred quality score from the 3′ end of the reads and kept only those with a minimum remaining length of 15 bases after trimming. We ran BWA with a minimum seed length of 15 and default parameters otherwise. We assigned a functional annotation to the locus of each read using a hierarchy based on expression of different classes of non-coding RNA. Overlap with annotations of coding and non-coding RNAs in sense and antisense direction was tested in the following order: mature miRNA, miRNA precursor, rRNA, tRNA, snRNA, snoRNA, 21U-RNA, coding exon, coding intron, repeat sequence. If an overlap of at least 5 bases was observed, the read was assigned to the respective class of non-coding RNA, and deeper levels of the hierarchy were not tested.
For normalization of small RNA expression, we assumed constant abundance of total microRNA in oocytes and embryos. This assumption was validated by RT-qPCR-based expression quantification of 10 different microRNAs for equal numbers of oocytes and embryos normalized to equal concentration of spiked-in external RNA. Within each class of small RNA, we converted expression into reads per one million of microRNA reads. For the inference of differentially regulated small RNAs within each class, we eliminated global changes by computing a linear regression and eliminating the intercept.
Specific classes of small non-coding RNAs antisense to protein coding genes were extracted based on the length of the mapped reads and the 5′-most nucleotide. The most ubiquitous class, 22G-endo-siRNAs, contains small RNAs of length 22 starting with a G. We divided this prominent class into two distinct groups: 22G-RNAs for which we have indication of upstream 26G-RNAs (antisense reads of length 26 starting with a G) were annotated as silencing 22G-RNAs (Vasale et al, 2010). 22G-RNAs without 26G-RNA evidence most likely belong to a class of CSR-1 small RNAs that are not silencing but rather involved in chromosome segregation (Claycomb et al, 2009; Van Wolfswinkel et al, 2009). These were here classified as ‘other siRNAs’.
Hand-picking embryos and oocytes for validations
Adult wild-type hermaphrodites (N2) were cut using a razor blade in PBS containing 0.5% BSA and 0.02% Tween. A total of 300 1-cell embryos and 300 oocytes were collected from cut gravid wild-type (N2) hermaphrodites by mouth pipetting under a stereomicroscope (Leica, Germany). Oocytes and embryos were washed thoroughly in PBS containing 0.5% BSA and 0.02% Tween and lyzed in Trizol LS Reagent (Invitrogen, USA). 0.5 μg of HeLa total RNA was added to each sample to normalize the subsequent RT-qPCRs on an exogenous spike.
Generation of transgenic reporter strains
3′ UTRs of tsn-1 and lec-1 (either the wild-type or mutant version of the poly C motif) were cloned into a vector-containing gld-1 promoter, to drive expression in the germ line, GFP::H2B, and the candidate 3′ UTR and recombined with the MosSCI Destination vector, pCFJ151, using LR clonase and were injected into EG4322 for integration onto chromosome II according to the MosSCI direct insertion protocol (Frøkjaer-Jensen et al, 2008a,b). We integrated the polyC motif into a region of the 3′ UTRs of mes-2 and tbb-2 that did not alter the predicted (RNAfold) secondary structure of the 3′ UTR. All clones were sequence verified by Sanger sequencing. Stable integrated lines were maintained on NGM plates seeded with OP50 at 25°C. Adult worms were then checked for GFP expression on a confocal microscope (Leica, Germany). The reporters were expressed throughout the germ line. We then handpicked oocytes and 1-cell- to 2-cell-stage embryos from at least two independent stable integrated lines and measured the mRNA expression change of the reporter, the endogenous gene, and controls by RT-qPCRs.
GO term analysis
GO term analysis was performed in R using the GOstats package. Overrepresented GO terms were computed against the background of all genes expressed (> 2 RPKM) in oocytes or embryos.
RNAi phenotype enrichment analysis
For the enrichment analysis of RNAi phenotypes, data for all available RNAi experiments (WS190) were downloaded from Wormbase.org. All genes with support from less than two experiments were discarded. Phenotypes were considered if observed in at least 50% of all experiments performed on the gene of interest.
Motif identification with MEME
We screened for overrepresented motifs in defined gene sets using MEME (Bailey & Elkan, 1994). To identify specifically enriched motifs, we compared to background sequences that were similar in expression and sequence length. To define theses background sequences, we computed for each gene in the set the relative length and expression difference to all other genes not contained in the set. We then selected the nearest neighbors in the space of relative expression and length differences among all background genes. In case of small gene sets, we constructed larger groups of background sequenced by including next nearest neighbors. We ran MEME against this background set in order to construct position-specific priors (Bailey et al, 2010) with parameters -mod zoops -nmotifs 6 -minw 6 -maxw 12 -minsites 5 -maxsites 20000.
After running MEME on a set of candidate genes, all genes were scored with the MEME derived position weight matrix (PWM) and the hit with the maximum score was recorded. Genes with motif hits exceeding the 90% quantile of the motif score distribution were considered bona fide polyC motifs. Genes with a motif score less than the 50% quantile of the score distribution were assumed to not contain a functional polyC motif.
Sequence conservation analysis
To analyze motif conservation, we generated randomized controls by shuffling the PWM. We first shuffled columns, and in a second step, rows were shuffled. This strategy preserves the polarity of the matrix and makes the score distribution of the randomized motif approximately comparable to the real motif (assuming uniform base composition). The same strategy as for the real motif was applied to infer best matching positions for the control motif. We used three-way alignments of C. elegans, C. briggsae and C. remanei to measure motif conservation. We retrieved genome-wide alignments from the UCSC genome browser (Dreszer et al, 2012) and extracted 3′ UTR alignments based on our C. elegans 3′ UTR annotation (Mangone et al, 2010).
Biotinylated RNA pull-down assay
Biotinylated RNA-protein pull-down assay was performed as described previously (Lee & Schedl, 2001). Worm lysate was incubated with 1 μg of biotinylated RNA oligo containing the polyC motif (5′-CCCCCCCCCCCC), a mutated polyC motif (5′-CACACACACACA) or no RNA as control. After four washes, proteins bound to the beads were eluted by applying two times 100 μl elution buffer (6 M urea/2 M thiourea in 10 mM HEPES (pH 8.0), 100 U Benzonase (Merck)) and 1 time elution buffer B (100 mM glycine pH 2.5/100 U Benzonase (Merck)). The collected protein eluates were combined in a fresh 2-ml Eppendorf tube, and ethanol precipitated overnight at 4°C and centrifuged with 20,000 g at 4°C for 60 min. The supernatant was removed; the protein pellet was air-dried and processed for mass spectrometry.
SILAC protein quantification by mass spectrometry
After determining protein concentration by amido black (Dieckmann-Schuppert & Schnittler, 1997) ‘heavy’ labeled sample was mixed with each of the ‘light’ samples at 1:1 ratio. Total protein for every sample was separated under reducing conditions by SDS-PAGE on a 4–12% NuPAGE gradient gel (Invitrogen, USA) according to the manufacturer's instructions. Proteins were fixed in 50% methanol, 10% acetic acid and stained by Colloidal Coomassie Blue (Invitrogen, USA). Gel lanes were cut into 16 slices, and samples were processed following standard in-gel digest protocol (Shevchenko et al, 2000) using Lysyl endopeptidase (LysC) (Wako, Japan). Stop and go extraction (STAGE) tips containing C18 empore disks (3M, USA) were used to purify and store peptide extracts as described previously (Rappsilber et al, 2003).
Online LC-MS/MS (liquid chromatography coupled with tandem mass spectrometry) analysis was performed by separating peptide mixtures by reversed-phase chromatography using the Eksigent NanoLC—1D Plus system (Eksigent, USA) on in-house manufactured 10-cm fritless silica microcolumns with an inner diameter of 75 μm as described previously (Selbach et al, 2008). Columns were packed with ReproSil-Pur C18-AQ 1.9 μm resin (Dr. Maisch GmbH, Germany). Separation was performed using a 10–60% acetonitrile gradient (240 min or 360 min) with 0.5% acetic acid at a flow rate of 200 nl/min. Eluting peptides were directly ionized by electrospray ionization and transferred into the orifice of a LTQ-Orbitrap hybrid mass spectrometer (classic, XL or Velos instruments, Thermo Fisher, USA). Mass spectrometry was performed in the data-dependent mode with one full scan in the Orbitrap (m/z = 300–1,700; R = 60,000; full scan target value = 1 × 106) and fragmentation in the LTQ using collision-induced dissociation. Dynamic exclusion for selected precursor ions was 60 s. Two independent biological replicates were measured for each stage.
The MaxQuant software package (version 1.0.13.13) was used to identify and quantify proteins (Cox & Mann, 2007; Cox et al, 2009). SILAC duplets were extracted from isotope patterns, re-calibrated, and quantified by the Quant module (Settings: heavy label Lys-8; maximum of four labeled amino acids per peptide; polymer detection enabled; top 6 MS/MS peaks per 100 Da). Peak lists were searched on a MASCOT search engine (version 2.2, MatrixScience, USA) against an in-house curated database for C. elegans and E. coli (MG1655) plus common contaminants (e.g. BSA). All protein sequences were also reversed to generate a target-decoy database to determine false discovery rates (Elias & Gygi, 2007). Carbamidomethylation of cysteine was selected as fixed modification, and oxidation of methionine and acetylation of the protein N-terminus were used as variable modifications. LysC was selected as protease (full specificity) with a maximum of 3 missed cleavages. A mass tolerance of 0.5 Da was selected for fragment ions. A minimum of six amino acids per identified peptide and at least one peptide per protein group were required. False discovery rate was set to 5% at the peptide and protein levels. Protein ratios were calculated from the median of all normalized peptide ratios using only unique peptides or peptides assigned to the protein group with the highest number of peptides (‘Occam's razor’ peptides). Protein log2-fold changes were averaged across independent biological replicates, and variability was estimated by the standard deviation. The SignificanceB measure calculated by the MaxQuant software was used as an estimate for the P-value of a log2-fold change. These P-values are based on the assumption of a Gaussian distribution as a noise model for protein fold changes. To arrive at P-values for averaged fold changes, we computed the quantiles corresponding to the P-value of each replicate, averaged these quantiles and transformed the average back into an average P-value. A SILAC ratio with a ratio count higher than two was assigned a maximum confidence value of one, and zero confidence was assigned to the remaining SILAC ratios. Confidence values were averaged across replicates to obtain a confidence value for the average fold change.
Biotinylated RNA pull-down protein sample processing
Precipitated proteins from pull-down experiments were solubilized in 6 M urea/2 M thiourea (10 mM HEPES (pH 8.0)), reduced with 1 mM DTT, alkylated 1 mM chloroacetamide and in-solution digested with Lysyl endopeptidase (LysC) (protein:enzyme ratio 50:1; Wako, Osaka, Japan) for 3 h at room temperature. After dilution of the samples 4 times with digestion buffer (50 mM ammonium bicarbonate in water (pH 8.0)), sequence-grade-modified trypsin (Promega, Madison, WI, USA) was added (protein:enzyme ratio 50:1) and digested over night. Trypsin and LysC activity was quenched by acidification of the sample with TFA to pH ˜2. Peptides were extracted and desalted using the Stage tips (Rappsilber et al, 2003). Prior to MS, peptides were separated by reversed-phase liquid chromatography (EASY-nLC system, Thermo Scientific) on in-house manufactured 20-cm fritless silica microcolumns with an inner diameter of 75 μm, packed with ReproSil-Pur C18-AQ 3 μm resin (Dr. Maisch GmbH, Germany), with a gradient of 8–60% acetonitrile over 240 min with a constant flow rate of 200 nl/min (hydrophilic solvent: 0.5% acetic acid), and eluting peptides were directly ionized by electrospray ionization and transferred into a Q Exactive mass spectrometer (Thermo Scientific). Mass spectrometry was performed in the data-dependent positive mode with one full scan (m/z range = 300–1,700; R = 70,000; target value: 3 × 106; maximum injection time = 120 ms). The 10 most intense ions with a charge state greater than one were selected (R = 35,000, target value = 5 × 105; isolation window = 4 m/z; maximum injection time = 120 ms). Dynamic exclusion for selected precursor ions was set to 30 s.
MS/MS data were analyzed by MaxQuant software v1.2.2.5. The internal Andromeda search engine was used to search MS/MS spectra against an in-house curated decoy database for C. elegans and E. coli (MG1655). The search included variable modifications of methionine oxidation and N-terminal acetylation and fixed modification of carbamidomethyl cysteine. Minimal peptide length was set to six amino acids, and a maximum of two missed cleavages was allowed. The false discovery rate (FDR) was set to 1% for peptide and protein identifications. If the identified peptide sequence set of one protein was equal to or contained another protein's peptide set, these two proteins were grouped together and the proteins were not counted as independent hits. Two biological replicates of polyC, mutated polyC, and control (no RNA) samples were measured three times each, and protein abundance was determined by label-free quantification (LFQ) as described (Hubner et al, 2010). Unique and razor peptides were considered for quantification with a minimum ratio count of 1. Retention times were recalibrated based on the built-in nonlinear time-rescaling algorithm. MS/MS identifications were transferred between LC-MS/MS runs with the ‘Match between runs’ option in which the maximal retention time window was set to 2 min. For every peptide, corresponding total signals from multiple runs were compared to determine peptide ratios. Median values of all peptide ratios of one protein then represent a robust estimate of the protein ratio. Analysis of label-free data was performed using the Perseus tool of MaxQuant. LFQ intensity values were logarithmized, and missing values were imputed with random numbers from a normal distribution, whose mean and standard deviation were chosen to best simulate low abundance values below the noise level (width = 0.3; down shift = 1.8). PolyC, mutated polyC, and control samples were selected as individual groups of 6 replicates (3 technical replicates from two individual experiments). Proteins changing significantly between samples were extracted by a volcano plot-based strategy which combines t-test P-values and log ratios as previously described (Hubner et al, 2010). We used a permutation-based FDR of 1% as cutoff. To define polyC-specific interaction partners, we first selected all proteins significantly enriched in the pulldowns with polyC or the mutated motif relative to the control. In this subset, we selected proteins significantly enriched in the polyC versus the mutated motif pulldown as polyC-specific binders.
Acknowledgments
We are grateful to Rueyling Lin for providing us with the TX189[P(oma-1)::oma-1::GFP] strain. All other strains used in this project were provided by the Caenorhabditis Genetics Center, which is funded by the National Center for Research Resources. We thank all members of the Rajewsky laboratory for discussions and support, particularly Lena von Oertzen for technical assistance. We thank Hans-Peter Rahn for help with FACS. We thank J. Solana and S. Grosswendt for performing PAT assays. We acknowledge Claudia Langnick and Mirjam Feldkamp from the Wei Chen laboratory (MDC) for the sequencing runs, and Wei Chen for helpful discussions concerning next generation sequencing technologies. M.S. thanks Michelle Kudron in the lab of Valerie Reinke and the lab of Antonio Giraldez where some revision experiments could be performed. M.S. thanks BIMSB/NYU International PhD program for funding. N.R. thanks the NYU Department of Biology for funding stays at NYU where he carried out part of the work. NR also thanks FP and Kris Gunsalus for many fruitful interactions. D.G. received funding from the European Community's Seventh Framework Programme (FP7/2007-2013) under grant agreement HEALTH-F4-2010-241504 (EURATRANS). F.P. acknowledges funding from the National Institutes of Health Grant R01HD046236.
Author contributions
NR and MSt conceived this study. MSt designed, performed, and lead most of the experiments. DG contributed the entire computational analyses. MK performed the mass spectrometry measurements, supervised by MSe. SA prepared sequencing libraries. MH performed C. elegans injections. FT contributed polyC reporter cloning. FP supervised MSt in early stages of the project. NR supervised MSt, DG, and the project. MSt, DG, and NR interpreted the data and wrote the paper. Data analyzed herein have been deposited in the NCBI Gene Expression Omnibus under accession number GSE58141.
Conflict of interest
The authors declare that they have no conflict of interest.
Supplementary information for this article is available online: http://emboj.embopress.org
References
- Aanes H, Winata C, Mathavan S. Zebrafish mRNA sequencing deciphers novelties in transcriptome dynamics during maternal to zygotic transition. Genome Res. 2011;21:1328–1338. doi: 10.1101/gr.116012.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aroian RV, Field C, Pruliere G, Kenyon C, Alberts BM. Isolation of actin-associated proteins from Caenorhabditis elegans oocytes and their localization in the early embryo. EMBO J. 1997;16:1541–1549. doi: 10.1093/emboj/16.7.1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL, Elkan C. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc Int Conf Intell Syst Mol Biol. 1994;2:8–36. [PubMed] [Google Scholar]
- Bailey TL, Bodén M, Whitington T, Machanick P. The value of position-specific priors in motif discovery using MEME. BMC Bioinformatics. 2010;11:179. doi: 10.1186/1471-2105-11-179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baugh LR, Hill AA, Slonim DK, Brown EL, Hunter CP. Composition and dynamics of the Caenorhabditis elegans early embryonic transcriptome. Development. 2003;130:889–900. doi: 10.1242/dev.00302. [DOI] [PubMed] [Google Scholar]
- Bazzini AA, Lee MT, Giraldez AJ. Ribosome profiling shows that miR-430 reduces translation before causing mRNA decay in zebrafish. Science 80- 2012;336:233–237. doi: 10.1126/science.1215704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushati N, Stark A, Brennecke J, Cohen SM. Temporal reciprocity of miRNAs and their targets during the maternal-to-zygotic transition in Drosophila. Curr Biol. 2008;18:501–506. doi: 10.1016/j.cub.2008.02.081. [DOI] [PubMed] [Google Scholar]
- Claycomb JM, Batista PJ, Pang K, Gu W, Vasale J, van Wolfswinkel J, Chaves DA, Shirayama M, Mitani S, Ketting RF, Conte D, Mello CC. The Argonaute CSR-1 and its 22G-RNA cofactors are required for holocentric chromosome segregation. Cell. 2009;139:123–134. doi: 10.1016/j.cell.2009.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J, Mann M. Is proteomics the new genomics? Cell. 2007;130:395–398. doi: 10.1016/j.cell.2007.07.032. [DOI] [PubMed] [Google Scholar]
- Cox J, Matic I, Hilger M, Nagaraj N, Selbach M, Olsen JV, Mann M. A practical guide to the MaxQuant computational platform for SILAC-based quantitative proteomics. Nat Protoc. 2009;4:698–705. doi: 10.1038/nprot.2009.36. [DOI] [PubMed] [Google Scholar]
- Cuenca AA, Schetter A, Aceto D, Kemphues K, Seydoux G. Polarization of the C. elegans zygote proceeds via distinct establishment and maintenance phases. Development. 2003;130:1255–1265. doi: 10.1242/dev.00284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cutter AD. Divergence times in Caenorhabditis and Drosophila inferred from direct estimates of the neutral mutation rate. Mol Biol Evol. 2008;25:778–786. doi: 10.1093/molbev/msn024. [DOI] [PubMed] [Google Scholar]
- Davidson EH. Gene Activity in Early Development. 3 edn. New York, NY: Academic Press; 1989. [Google Scholar]
- Dieckmann-Schuppert A, Schnittler HJ. A simple assay for quantification of protein in tissue sections, cell cultures, and cell homogenates, and of protein immobilized on solid surfaces. Cell Tissue Res. 1997;288:119–126. doi: 10.1007/s004410050799. [DOI] [PubMed] [Google Scholar]
- Dobson AT, Raja R, Abeyta MJ, Taylor T, Shen S, Haqq C, Pera RAR. The unique transcriptome through day 3 of human preimplantation development. Hum Mol Genet. 2004;13:1461–1470. doi: 10.1093/hmg/ddh157. [DOI] [PubMed] [Google Scholar]
- Dreszer TR, Karolchik D, Zweig AS, Hinrichs AS, Raney BJ, Kuhn RM, Meyer LR, Wong M, Sloan CA, Rosenbloom KR, Roe G, Rhead B, Pohl A, Malladi VS, Li CH, Learned K, Kirkup V, Hsu F, Harte RA, Guruvadoo L, et al. The UCSC Genome Browser database: extensions and updates 2011. Nucleic Acids Res. 2012;40:D918–D923. doi: 10.1093/nar/gkr1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias JE, Gygi SP. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat Methods. 2007;4:207–214. doi: 10.1038/nmeth1019. [DOI] [PubMed] [Google Scholar]
- Evsikov AV, Graber JH, Brockman JM, Hampl A, Holbrook AE, Singh P, Eppig JJ, Solter D, Knowles BB. Cracking the egg: molecular dynamics and evolutionary aspects of the transition from the fully grown oocyte to embryo. Genes Dev. 2006;20:2713–2727. doi: 10.1101/gad.1471006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evsikov AV, Evsikova CM. Gene expression during the oocyte-to-embryo transition in mammals. Mol Reprod Dev. 2009;76:805–818. doi: 10.1002/mrd.21038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredens J, Engholm-Keller K, Giessing A, Pultz D, Larsen MR, Højrup P, Møller-Jensen J, Færgeman NJ. Quantitative proteomics by amino acid labeling in C. elegans. Nat Methods. 2011;8:845–847. doi: 10.1038/nmeth.1675. [DOI] [PubMed] [Google Scholar]
- Frøkjaer-Jensen C, Davis MW, Hopkins CE, Newman BJ, Thummel JM, Olesen S-P, Grunnet M, Jorgensen EM. Single-copy insertion of transgenes in Caenorhabditis elegans. Nat Genet. 2008a;40:1375–1383. doi: 10.1038/ng.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frøkjær-Jensen C, Davis WM, Hopkins CE, Jorgensen EM. Single-copy insertion of transgenes in Caenorhabditis elegans. Nat Genet. 2008b;40:1375–1383. doi: 10.1038/ng.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamberi C, Gottlieb E. Internally controlled poly(A) tail assay to study gene regulation. Biotechniques. 2002;33:476–478. doi: 10.2144/02333bm02. 480. [DOI] [PubMed] [Google Scholar]
- Gerstein MB, Lu ZJ, Van Nostrand EL, Cheng C, Arshinoff BI, Liu T, Yip KY, Robilotto R, Rechtsteiner A, Ikegami K, Alves P, Chateigner A, Perry M, Morris M, Auerbach RK, Feng X, Leng J, Vielle A, Niu W, Rhrissorrakrai K, et al. Integrative analysis of the Caenorhabditis elegans genome by the modENCODE project. Science. 2010;330:1775–1787. doi: 10.1126/science.1196914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert I, Scantland S, Sylvestre E-L, Gravel C, Laflamme I, Sirard M-A, Robert C. The dynamics of gene products fluctuation during bovine pre-hatching development. Mol Reprod Dev. 2009;76:762–772. doi: 10.1002/mrd.21030. [DOI] [PubMed] [Google Scholar]
- Giraldez AJ, Mishima Y, Rihel J, Grocock RJ, Van DS, Inoue K, Enright AJ, Schier AF. Zebrafish MiR-430 promotes deadenylation and clearance of maternal mRNAs. Science. 2006;312:75–79. doi: 10.1126/science.1122689. [DOI] [PubMed] [Google Scholar]
- Giraldez AJ. microRNAs, the cell's Nepenthe: clearing the past during the maternal-to-zygotic transition and cellular reprogramming. Curr Opin Genet Dev. 2010;20:369–375. doi: 10.1016/j.gde.2010.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grün D, Kirchner M, Thierfelder N, Stoeckius M, Selbach M, Rajewsky N. Conservation of mRNA and Protein Expression during Development of C. elegans. Cell Rep. 2014;6:565–577. doi: 10.1016/j.celrep.2014.01.001. [DOI] [PubMed] [Google Scholar]
- Guven-Ozkan T, Nishi Y, Robertson SM, Lin R. Global transcriptional repression in C. elegans germline precursors by regulated sequestration of TAF-4. Cell. 2008;135:149–160. doi: 10.1016/j.cell.2008.07.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamatani T, Carter MG, Sharov AA, Ko MSH. Dynamics of Global Gene Expression Changes during Mouse Preimplantation Development. Dev Cell. 2004;6:117–131. doi: 10.1016/s1534-5807(03)00373-3. [DOI] [PubMed] [Google Scholar]
- Hubner NC, Bird AW, Cox J, Splettstoesser B, Bandilla P, Poser I, Hyman A, Mann M. Quantitative proteomics combined with BAC TransgeneOmics reveals in vivo protein interactions. J Cell Biol. 2010;189:739–754. doi: 10.1083/jcb.200911091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji X, Wan J, Vishnu M, Xing Y, Liebhaber S. αCP Poly(C) binding proteins act as global regulators of alternative polyadenylation. Mol Cell Biol. 2013;33:2560–2573. doi: 10.1128/MCB.01380-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krawetz SA. Paternal contribution: new insights and future challenges. Nat Rev Genet. 2005;6:633–642. doi: 10.1038/nrg1654. [DOI] [PubMed] [Google Scholar]
- Lall S, Grün D, Krek A, Chen K, Wang Y-L, Dewey CN, Sood P, Colombo T, Bray N, Macmenamin P, Kao H-L, Gunsalus KC, Pachter L, Piano F, Rajewsky N. A genome-wide map of conserved microRNA targets in C. elegans. Curr Biol. 2006;16:460–471. doi: 10.1016/j.cub.2006.01.050. [DOI] [PubMed] [Google Scholar]
- Larance M, Bailly AP, Pourkarimi E, Hay RT, Buchanan G, Coulthurst S, Xirodimas DP, Gartner A, Lamond AI. Stable-isotope labeling with amino acids in nematodes. Nat Methods. 2011;8:849–851. doi: 10.1038/nmeth.1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MH, Schedl T. Identification of in vivo mRNA targets of GLD-1, a maxi-KH motif containing protein required for C. elegans germ cell development. Genes Dev. 2001;15:2408–2420. doi: 10.1101/gad.915901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26:589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin R. A gain-of-function mutation in oma-1, a C. elegans gene required for oocyte maturation, results in delayed degradation of maternal proteins and embryonic lethality. Dev Biol. 2003;258:226–239. doi: 10.1016/s0012-1606(03)00119-2. [DOI] [PubMed] [Google Scholar]
- Makeyev AV, Liebhaber SA. The poly(C)-binding proteins: a multiplicity of functions and a search for mechanisms. RNA. 2002;8:265–278. doi: 10.1017/s1355838202024627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangone M, Manoharan AP, Thierry-Mieg D, Thierry-Mieg J, Han T, Mackowiak SD, Mis E, Zegar C, Gutwein MR, Khivansara V, Attie O, Chen K, Salehi-Ashtiani K, Vidal M, Harkins TT, Bouffard P, Suzuki Y, Sugano S, Kohara Y, Rajewsky N, et al. The landscape of C. elegans 3′UTRs. Science. 2010;329:432–435. doi: 10.1126/science.1191244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarter J, Bartlett B, Dang T, Schedl T. On the control of oocyte meiotic maturation and ovulation in Caenorhabditis elegans. Dev Biol. 1999;205:111–128. doi: 10.1006/dbio.1998.9109. [DOI] [PubMed] [Google Scholar]
- Merritt C, Rasoloson D, Ko D, Seydoux G. 3′ UTRs are the primary regulators of gene expression in the C. elegans germline. Curr Biol. 2008;18:1476–1482. doi: 10.1016/j.cub.2008.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miska EA, Alvarez-Saavedra E, Abbott AL, Lau NC, Hellman AB, McGonagle SM, Bartel DP, Ambros VR, Horvitz HR. Most Caenorhabditis elegans microRNAs are individually not essential for development or viability. PLoS Genet. 2007;3:e215. doi: 10.1371/journal.pgen.0030215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peaston AE, Graber JH, Knowles BB, de Vries WN. Interrogating the transcriptome of oocytes and preimplantation embryos. Methods Enzymol. 2010;477:481–510. doi: 10.1016/S0076-6879(10)77024-5. [DOI] [PubMed] [Google Scholar]
- Pepke S, Wold B, Mortazavi A. Computation for ChIP-seq and RNA-seq studies. Nat Methods. 2009;6:S22–S32. doi: 10.1038/nmeth.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piko L, Clegg KB. Quantitative changes in total RNA, total poly(A), and ribosomes in early mouse embryos. Dev Biol. 1982;89:362–378. doi: 10.1016/0012-1606(82)90325-6. [DOI] [PubMed] [Google Scholar]
- Radford HE, Meijer HA, de Moor CH. Translational control by cytoplasmic polyadenylation in Xenopus oocytes. Biochim Biophys Acta. 2008;1779:217–229. doi: 10.1016/j.bbagrm.2008.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappsilber J, Ishihama Y, Mann M. Stop and go extraction tips for matrix-assisted laser desorption/ionization, nanoelectrospray, and LC/MS sample pretreatment in proteomics. Anal Chem. 2003;75:663–670. doi: 10.1021/ac026117i. [DOI] [PubMed] [Google Scholar]
- Reinke V, Gil IS, Ward S, Kazmer K. Genome-wide germline-enriched and sex-biased expression profiles in Caenorhabditis elegans. Development. 2004;131:311–323. doi: 10.1242/dev.00914. [DOI] [PubMed] [Google Scholar]
- Robertson S, Lin R. The oocyte-to-embryo transition. Adv Exp Med Biol. 2013;757:351–372. doi: 10.1007/978-1-4614-4015-4_12. [DOI] [PubMed] [Google Scholar]
- Rouget C, Boureux A, Franco B, Papin C, Simonelig M. Maternal mRNA deadenylation and decay by the piRNA pathway in the early Drosophila embryo. Nature. 2010;467:1128–1132. doi: 10.1038/nature09465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selbach M, Schwanhäusser B, Thierfelder N, Fang Z, Khanin R, Rajewsky N. Widespread changes in protein synthesis induced by microRNAs. Nature. 2008;455:58–63. doi: 10.1038/nature07228. [DOI] [PubMed] [Google Scholar]
- Seydoux G, Fire A. Soma-germline asymmetry in the distributions of embryonic RNAs in Caenorhabditis elegans. Development. 1994;120:2823–2834. doi: 10.1242/dev.120.10.2823. [DOI] [PubMed] [Google Scholar]
- Seydoux G, Dunn MA. Transcriptionally repressed germ cells lack a subpopulation of phosphorylated RNA polymerase II in early embryos of Caenorhabditis elegans and Drosophila melanogaster. Development. 1997;124:2191–2201. doi: 10.1242/dev.124.11.2191. [DOI] [PubMed] [Google Scholar]
- Shevchenko A, Loboda A, Ens W, Standing KG. MALDI quadrupole time-of-flight mass spectrometry: a powerful tool for proteomic research. Anal Chem. 2000;72:2132–2141. doi: 10.1021/ac9913659. [DOI] [PubMed] [Google Scholar]
- Srayko M, Buster DW, Bazirgan OA, McNally FJ, Mains PE. MEI-1/MEI-2 katanin-like microtubule severing activity is required for Caenorhabditis elegans meiosis. Genes Dev. 2000;14:1072–1084. [PMC free article] [PubMed] [Google Scholar]
- Stadler M, Artiles K, Pak J, Fire A. Contributions of mRNA abundance, ribosome loading, and post- or peri-translational effects to temporal repression of C. elegans heterochronic miRNA targets. Genome Res. 2012;22:2418–2426. doi: 10.1101/gr.136515.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiernagle T. 2006. pp. 1–11. WormBook Maintenance of C. elegans. [DOI] [PMC free article] [PubMed]
- Stitzel ML, Seydoux G. Regulation of the oocyte-to-zygote transition. Science. 2007;316:407–408. doi: 10.1126/science.1138236. [DOI] [PubMed] [Google Scholar]
- Stoeckius M, Maaskola J, Colombo T, Rahn H-P, Friedländer MR, Li N, Chen W, Piano F, Rajewsky N. Large-scale sorting of C. elegans embryos reveals the dynamics of small RNA expression. Nat Methods. 2009;6:745–751. doi: 10.1038/nmeth.1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoeckius M, Grün D, Rajewsky N. Paternal RNA contributions in the Caenorhabditis elegans zygote. EMBO J. 2014;33:1740–1750. doi: 10.15252/embj.201488117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh N, Baehner L, Moltzahn F, Melton C, Shenoy A, Chen J, Blelloch R. MicroRNA function is globally suppressed in mouse oocytes and early embryos. Curr Biol. 2010;20:271–277. doi: 10.1016/j.cub.2009.12.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svoboda P, Flemr M. The role of miRNAs and endogenous siRNAs in maternal-to-zygotic reprogramming and the establishment of pluripotency. EMBO Rep. 2010;11:590–597. doi: 10.1038/embor.2010.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tadros W, Goldman AL, Babak T, Menzies F, Vardy L, Orr-Weaver T, Hughes TR, Westwood JT, Smibert CA, Lipshitz HD. SMAUG is a major regulator of maternal mRNA destabilization in Drosophila and its translation is activated by the PAN GU kinase. Dev Cell. 2007;12:143–155. doi: 10.1016/j.devcel.2006.10.005. [DOI] [PubMed] [Google Scholar]
- Tadros W, Lipshitz HD. The maternal-to-zygotic transition: a play in two acts. Development. 2009;136:3033–3042. doi: 10.1242/dev.033183. [DOI] [PubMed] [Google Scholar]
- Tam OH, Aravin AA, Stein P, Girard A, Murchison EP, Cheloufi S, Hodges E, Anger M, Sachidanandam R, Schultz RM, Hannon GJ. Pseudogene-derived small interfering RNAs regulate gene expression in mouse oocytes. Nature. 2008;453:534–538. doi: 10.1038/nature06904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Wolfswinkel J, Claycomb JM, Batista PJ, Mello CC, Berezikov E, Ketting RF. CDE-1 affects chromosome segregation through uridylation of CSR-1-bound siRNAs. Cell. 2009;139:135–148. doi: 10.1016/j.cell.2009.09.012. [DOI] [PubMed] [Google Scholar]
- Vasale JJ, Gu W, Thivierge C, Batista PJ, Claycomb JM, Youngman EM, Duchaine TF, Mello CC, Conte D. Sequential rounds of RNA-dependent RNA transcription drive endogenous small-RNA biogenesis in the ERGO-1/Argonaute pathway. Proc Natl Acad Sci USA. 2010;107:3582–3587. doi: 10.1073/pnas.0911908107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasudevan S, Seli E, Steitz JA. Metazoan oocyte and early embryo development program: a progression through translation regulatory cascades. Genes Dev. 2006;20:138–146. doi: 10.1101/gad.1398906. [DOI] [PubMed] [Google Scholar]
- Wang QT, Piotrowska K, Ciemerych MA, Milenkovic L, Scott MP, Davis RW, Zernicka-Goetz M. A Genome-Wide Study of Gene Activity Reveals Developmental Signaling Pathways in the Preimplantation Mouse Embryo. Dev Cell. 2004;6:133–144. doi: 10.1016/s1534-5807(03)00404-0. [DOI] [PubMed] [Google Scholar]
- Watanabe T, Totoki Y, Toyoda A, Kaneda M, Kuramochi-Miyagawa S, Obata Y, Chiba H, Kohara Y, Kono T, Nakano T, Surani MA, Sakaki Y, Sasaki H. Endogenous siRNAs from naturally formed dsRNAs regulate transcripts in mouse oocytes. Nature. 2008;453:539–543. doi: 10.1038/nature06908. [DOI] [PubMed] [Google Scholar]
- Zhang P, Zucchelli M, Bruce S, Hambiliki F, Stavreus-Evers A, Levkov L, Skottman H, Kerkelä E, Kere J, Hovatta O. Transcriptome profiling of human pre-implantation development. PLoS ONE. 2009;4:e7844. doi: 10.1371/journal.pone.0007844. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.