

Abstract
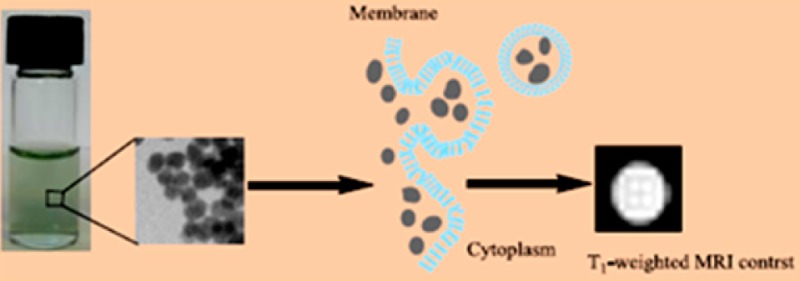
A simple one-step method for preparing biocompatible nanoparticles of gadolinium ferrocyanide coordination polymer KGd(H2O)2[Fe(CN)6]·H2O is reported. The crystal structure of this coordination polymer is determined by X-ray powder diffraction using the bulk materials. The stability, cytotoxicity, cellular uptake, and MR phantom and cellular imaging studies suggest that this coordination-polymer structural platform offers a unique opportunity for developing the next generation of T1-weighted contrast agents with high relaxivity as cellular MR probes for biological receptors or markers. Such high-relaxivity MR probes may hold potential in the study of molecular events and may be used for in vivo MR imaging in biomedical research and clinical applications.
Introduction
Although it is a relatively new modality, magnetic resonance imaging (MRI) has emerged as a prominent noninvasive and nonradioactive imaging tool in diagnostic medicine and biomedical research.1,2 In general, the MRI signal intensity (SI) is a function of the water proton density of tissues under examination and the relative longitudinal and transverse relaxation times, T1 and T2, of the protons in the tissues.3 In some cases, image contrast can be directly produced by the inherent variation of proton density in different tissues by altering the various image acquisition parameters. In other cases, it is necessary to administer a contrast agent (CA) to improve the quality of the image and aid in diagnosis.4 The use of a contrast agent also makes it possible to visualize certain pathological regions externally, such as organs or cells of the human body via target-specific delivery of the agent.5−8 The majority of existing MRI CAs can be divided into two different types based on which relaxation time they alter to a greater extent, T1-weighted and T2-weighted agents.9 The most widely used CAs in clinical imaging are the paramagnetic Gd3+-chelates that can produce T1-weighted or positive contrast enhancement.1−4,9 The principal drawback of such CAs is their limited relaxivity because the observed relaxivity values are usually a few percent of the theoretically attainable values predicted by the Solomon–Bloembergen–Morgan (SBM) theory.10 As the result, rather high tissue concentrations (0.1–0.3 mM) are required for these CAs to be effective when used with MR scanners of low magnetic fields from 0.3 to 3.0 T. The paramagnetic relaxation enhancement has two separate components (i.e., inner-sphere and outer-sphere relaxation contributions). The mechanism of inner-sphere relaxation in small molecular Gd3+-chelates requires that at least one water molecule be directly coordinated to the paramagnetic metal center and be able to undergo a rapid exchange with the water molecules in the outer sphere.1−4 Although increasing the number of coordination water molecules can lead to a significant increase of the inner-sphere relaxivity, the thermodynamic stability and/or kinetic inertness of the Gd3+-chelates will decrease as a result. The consequence is to make the complexes more susceptible to displacement by proteins, biological ligands, or endogenous metal ions, often resulting in the toxicity of the CAs. Therefore, all of the current clinical Gd3+-chelates contain strictly one coordinated water molecule.1−4,9 Furthermore, because of nuclear magnetic relaxation dispersion with increasing Larmor frequency, the relaxivity of such CAs decreases drastically with increasing magnetic field strength to render them less effective on high-field scanners.11 Recently, the use of high-field scanners (4.7 to 9.0 T) has become increasingly prevalent because they provide greater spatial resolution, a shortened data acquisition time, and an improved signal-to-noise ratio.12 Another drawback of these CAs is that as small molecules, the Gd3+-chelates act largely as extracellular agents because they are unable to penetrate cells without attaching to a cell-targeting vector.13 However, high-relaxivity and cell-penetrating superparamagnetic iron oxides (SPIOs) have been developed as effective T2-weighted contrast agents for both clinical diagnosis and cellular imaging.14−16 However, the negative contrast enhancement produced by SPIOs is far less desirable than that produced by the small-molecule T1 agents.17 For example, it is often difficult to differentiate between the darkened spots produced by the accumulation of a T2 CA and signals caused by bleeding, calcification, metal deposits, or other artifacts from the background.18 In addition, the high magnetic susceptibility of the T2 CAs often induces a distortion of the magnetic field on neighboring normal tissues, which complicates the interpretation of MR radiograms.17,18 It should be noted that because of the strong magnetic coupling in SPIOs, water coordination to the metal centers in such CAs is not necessary. Instead, the field inhomogeneity induced by the presence of the tiny magnetic particles can cause a rapid dephasing in the transverse plane, which in turn accelerates the T2 relaxation as well as the T1 relaxation, albeit to a much smaller extent for the latter. The r2/r1 ratio found in most SPIOs is usually larger than 10.19
The use of nanoparticulate coordination polymers as contrast agents in different imaging modalities represents an important new direction in the development of next-generation contrast agents.20−30 Of particular relevance to the design of T1-weighted MR CAs is the ability to tailor the porosity, water coordination, and thermodynamic/kinetic stability in such crystalline polymers through the proper selection of the metal centers and ligands. In this article, we describe the synthesis, characterization, and MR imaging studies of biocompatible nanoparticles (NPs) of a coordination polymer, KGd(H2O)2[Fe(CN)6]·H2O. We have demonstrated that these NPs exhibit extremely large T1 relaxivity because of the presence of two water molecules coordinated to the Gd(III) center. Furthermore, such NPs also possesses unprecedentedly high thermodynamic stability and kinetic inertness due to the maximum ligand field stabilization energy (LFSE) found in the [Fe(CN)6]4– ligand, coupled with the considerable lattice energy of the extended 3D solid-state structure.31 We have also shown that such NPs have no cytotoxicity and can be readily internalized by cells to act as effective cellular MR probes.
Methods and Materials
Synthesis of the Bulk KGd(H2O)2[Fe(CN)6]·H2O Material
Equimolar aqueous solutions of Gd(NO3)3 (0.25 mmol, 20 mL) and K4[Fe(CN)6] (0.25 mmol, 20 mL) were mixed at room temperature under vigorous stirring. The solution turned pale yellow and cloudy after continuous stirring at room temperature for ca. 30 min. The reaction product was dialyzed using a regenerated cellulose tubular membrane (MWCO = 12 000–14 000) against distilled water for 8 h, followed by lyophilization. The solid material obtained from this process was redispersed in water by sonication to form a slurry and lyophilized one more time to increase its crystallinity. The bulk material was collected, washed with water and acetone, and dried in vacuum.
Synthesis of PVP-Citrate-Coated KGd(H2O)2[Fe(CN)6]·H2O Nanoparticles
An aqueous solution of Gd(NO3)3 (1 mM, 10 mL) containing 50 mg of sodium citrate was added dropwise to an aqueous mixture of K4[Fe(CN)6] (1 mM, 10 mL) and PVP (111 mg, MW = 8000) under vigorous stirring for ca. 3 h. The reaction product was dialyzed using a regenerated cellulose tubular membrane (MWCO is 12 000–14 000) against distilled water for 2 days. The solid product was collected by lyophilization.
Metal Analysis
Metal analysis was carried out using inductively coupled plasma optical emission spectroscopy (ICP-OES) with a PerkinElmer Optima 3200 system. A sample of 60 mg of KGd(H2O)2[Fe(CN)6]·H2O bulk material was first decomposed at 620 °C for ca. 6 h to obtain an amorphous oxide powder that was then extracted with 5 mL of concentrated HNO3. After dilution in a volumetric flask, the solution was analyzed by ICP-OES for potassium, gadolinium, and iron.
T1 and T2 Relaxivity Measurements
Homogenous aqueous dispersions of NPs with measured concentrations of the Gd3+ ions in the range of 5.0 × 10–3 to 3.5 × 10–2 mM were used for T1 and T2 relaxivity measurements at 25 °C using a 60 MHz Bruker MiniSpec relaxometer and a 7.0 T Bruker Biospec small animal MRI system (Bruker, Inc., Billerica, MA). For relaxivity measurements at 1.4 T, T1 and T2 relaxiviy values were obtained from the MiniSpec relaxometer using conventional inversion recovery (T1) and multiecho spin echo (T2) acquisitions. For relaxivity measurements at 7.0 T, an inversion recovery gradient echo sequence with TE = 4 ms was used for T1 relaxiviy measurements. The inversion time was varied between 30 and 2000 ms. T2 measurements were performed using a spin-echo sequence of TR of 10 000 ms and a TE of 10.6–340 ms. The T1-weighted MR images were acquired using the 7.0 T scanner with a matrix size of 128 × 128, a field of view of 3.0 × 3.0 cm2, a slice thickness of 0.5 mm, a TE of 9.4 ms, and a TR of 13.9–1500 ms. Data analysis was performed by fitting to relaxivity curves with self-written programs.
Leaching of CN– and Gd3+ Ions from Nanoparticles under Different Conditions
Leaching experiments were performed to verify the stability of NPs under physiologically relevant conditions. About 200 mg NPs were sealed in a membrane dialysis bag (MWCO = 3000). This dialysis bag was gently stirred in 25 mL solutions of distilled water at pH 7 or 1 for 24 h to reach equilibrium. The leaching experiment was also conducted in saline solution or in a solution containing a biologically relevant divalent metal ion (i.e., Zn2+, Ca2+, or Mg2+) at a concentration of 100 ppm for each ion with the pH value adjusted to 7.4. The resulting solutions were analyzed for free CN– ions by a fluorometric method using the cyanide test kit from LaMotte Co. (Chestertown, MD; code 7387-01). The calibration curve was established using the standard KCN solutions with concentrations at the ppm level. The quantitative determination of the leached Gd3+ ions was carried out by the ICP-OES method.
Cell Viability Assay
Cytotoxicity of PVP-coated NPs was evaluated using an MTT viability assay. Hela cells were seeded in a 96-well plate at a density of 1 × 105 cells per well with the DMEM low-glucose medium and incubated for 12 h at 37 °C in an atmosphere of 5% CO2 and 95% air to allow cells to attach to the surface. Cells in each well were then treated with 100 μL of fresh medium containing varying concentrations of the NPs and then incubated for 24 or 48 h. Control wells contained the same medium without NPs. The cells were incubated in media containing 0.1 mg/mL 3-[4,5-dimethylthialzol-2-yl]-2,5-diphenyltetrazolium bromide (i.e., the MTT dye) for 3 h. After the MTT solution was removed, the precipitated violet crystals were dissolved in 200 μL of DMSO. The absorbance was measured at 560 and 630 nm using a microplate reader. The assay was run in triplicate, and the results were presented as percent viable cells.
Conjugation of Fluorescent Dye Molecules to the Surfaces of Nanoparticles
To conjugate the fluorescent dye molecules to the surfaces of NPs, we used a slightly modified synthetic procedure derived from the above-mentioned process for preparing PVP-citrate-coated KGd(H2O)2[Fe(CN)6]·H2O NPs. Specifically, 200 mg of PVP (MW = 40000) was added to an aqueous solution of K4[Fe(CN)6] (1 mM, 50 mL) under stirring, followed by the addition of a 50 mL Gd(NO3)3 solution (1 mM) containing 100 mg of citric acid. The resultant solution was stirred for ca. 3 h before it was dialyzed and lyophilized. The product was resuspended in 2 mL of aqueous PVP solution (100 mg PVP/mL). Ethylenediamine (1.5 equiv) and 1-ethyl-3-(3-(dimethylamino)propyl)-carbodiimide (EDC, 1.2 equiv) reagents were then added to it under vigorous stirring. The resultant mixture was stirred for 24 h before the excess ethylenediamine was removed by dialysis against distilled water for 2 days. Next, 5 mL of carboxyfluorescein (0.35 mg/mL) was allowed to react with 1.2 equiv of EDC (∼1.5 mg) for 24 h. The ethylenediamine-coated NPs were then added to 2 mL of the above reaction mixture and stirred for ca. 24 h. Finally, the product was dialyzed for 2 days to remove unconjugated dye molecules. The fluorescence spectrum of the dye-conjugated NPs clearly showed the presence of dye molecules that are covalently bound to the surfaces of NPs.
Cellular Uptake Studies
Confocal scanning microscopy was used to visualize the cellular uptake of the fluorescence dye-labeled NPs in Hela cells. The cells were first seeded in an 8-well chamber slide at a density of approximately 1.5 × 105 cells per well and incubated at 37 oC for 24 h in a complete medium without antibiotics. The cells were then incubated with the fluorescent dye-labeled NPs for 3 h at the same temperature. After the cells were washed with the PBS buffer solution three times to remove free NPs, the cellular uptake of NPs was directly imaged on the living cells under the confocal microscope with the 488 nm excitation wavelength.
Cellular MR Imaging
Approximately 1 × 105 PC3 cells were placed in T25 flasks and incubated for 48 h. The cells were then rinsed with serum-free medium and incubated with varying concentrations of NPs for 6 h. The treated cells were washed with PBS three times and harvested by trypsinisation. Cells were then pelleted in 0.6 mL tubes for imaging studies. MR imaging of cell pellets was performed on a Bruker 9.4-T MRI scanner at 37 °C using a conventional gradient echo acquisition with an inversion recovery preparation. The other acquisition parameters were field of view = 3.0 × 3.0 cm2, matrix = 128 × 128, and slice thickness = 3.0 mm.
Determination of Intracellular Gadolinium and Iron Concentrations
Intracellular Gd and Fe concentrations were determined from the cell lysate using ICP-MS. After incubation with NPs, cells were washed three times with PBS and centrifuged. Cells were then lysed with concentrated nitric acid and diluted to determine the Gd concentration using ICP-MS.
Results and Discussion
Synthesis and Characterization of Bulk and Biocompatible Nanoparticulate Forms of KGd(H2O)2[Fe(CN)6]·H2O
The direct combination of a soluble Gd(III) salt with K4[Fe(CN)6] in aqueous solution resulted in the formation of a pale-yellow precipitate with the metallic ratio K/Gd/Fe from the elemental analysis being close to unity and a singlet IR stretching band at 2070 cm–1, suggesting the formation of a single-phase coordination polymer. However, because of poor crystallinity, the powder X-ray diffraction (XRD) patterns of this product could not be indexed. Refluxing of the reaction in water or an organic solvent (e.g., ethanol and DMF) to increase crystallinity caused the product to decompose into a mixture that invariably contained Prussian blue (PB) and other unidentified components. Although the single-crystal structure of a coordination polymer formed from these two starting materials was reported in 1996,32 there are few synthesis details except for the mention that single crystals were obtained from the diffusion method in a U tube for 6 months given in the structural report. We were able to shorten the crystal-growth time to ∼45 days using 1 M nitric acid as the crystallization medium with a similar diffusion method, but the yield we obtained for the intended compound was less than 1% whereas the major product formed was PB powder. To develop a suitable synthesis method that could later be adopted for preparing NPs of this coordination polymer, we explored a variety of preparative techniques including hydro(solvo)thermal synthesis and mechanical milling. The best method we have found is an unusual low-temperature solid-state crystallization process. This method entails the mixing of an equimolar quantity of aqueous Gd(NO3)3 and K4[Fe(CN)6] solutions at millimolar concentrations, followed by stirring the solution at room temperature for ∼8 h. The product was dialyzed in water to remove potassium and nitrate ions. The solution was then frozen while water was removed under vacuum to afford a light-yellow powder. This product was redispersed in water and lyophilized one more time. The yield was virtually quantitative. This synthesis process resembles the hydrothermal reaction for crystal growth in that the supersaturating condition suitable for crystallization is slowly induced at low temperatures. The advantage of such an approach is that the heating of the reaction is avoided to prevent the reaction product from decomposing into Prussian blue.
The X-ray powder diffraction (XRD) patterns of this bulk sample were well indexed on the basis of an orthorhombic unit cell with the same lattice parameters as those obtained from the single-crystal structure analysis (Supporting Information). Therefore, atomic parameters of the latter structure were used as the starting model in the Rietveld refinement.33 The final refinement in space group Pnma gave a = 12.6098(4) Å, b = 13.6161(4) Å, c = 7.2490(3) Å, V = 1244.63(7) Å3, and ρcalc = 2.4351(1) g/cm3 (Figure S1). The selected interatomic distances are summarized in Table 1.
Table 1. Selected Interatomic Distances.
| atom 1 | atom 2 | distance, Å | atom 1 | atom 2 | distance, Å |
|---|---|---|---|---|---|
| Gd | N1 × 2 | 2.40112 | Fe | C1 × 2 | 1.92208 |
| N2 × 2 | 2.42436 | C2 × 2 | 1.89469 | ||
| N3 × 2 | 2.46226 | C3 × 2 | 1.90203 | ||
| O1 × 1 | 2.55421 | K/O3 | O1 | 2.85365 | |
| O2 × 1 | 2.60647 | O2 | 2.99319 | ||
| N3 | 2.99319 |
The structure of KGd(H2O)2[Fe(CN)6]·H2O consists of Fe2+ octahedra and Gd3+ biface-capped trigonal prisms joined in a 3D framework by CN– groups (Figure 1). The Fe atom is coordinated by six C atoms of the CN– groups whereas the Gd atom is coordinated by six N atoms and additionally by two O atoms of water molecules O1 and O2 (Table 1). The cavities in the framework are filled up by the K+ ion and water of crystallization (O3), showing site-occupancy disorder in the same cavity. Both K and O3 are slightly shifted from each other by 0.4 Å so that K distances to nearby O and N atoms are in the range of 2.85 to 2.99 Å, whereas the O3 water molecule forms two H bonds with O1 and O2 (Table 1). Thermal gravimetric analysis (TGA) on the bulk sample showed a two-step loss of water (i.e., the first step of weight loss was found in the range from room temperature to about 100 oC, corresponding to the loss of one water molecule, and the second step of weight loss was found in the range from 100 to 160 oC, corresponding to the loss of two water molecules (Figure S2)). This observation is consistent with the existence of one zeolitic and two coordinative water molecules per formula as revealed by the powder X-ray structure determination. Furthermore, the Fourier transform infrared (FT-IR) spectra of the bulk sample of KGd(H2O)2[Fe(CN)6]·H2O showed a strong characteristic C≡N stretching vibration at 2070 cm–1, attributable to the Fe(II)-C≡N–Gd(III) bonding mode in the structure (vide infra).
Figure 1.
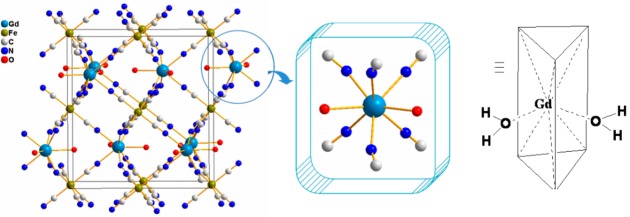
Unit-cell packing diagram of KGd(H2O)2[Fe(CN)6]·H2O (left) with the K+ ion and zeolitic water molecule omitted for clarity. The coordination environment of the Gd3+ ion (middle and right) showing two water molecules directly bound to the metal center.
The reaction of Gd(NO3)3 in a sodium citrate solution with K4[Fe(CN)6] in an aqueous solution containing polyvinylpyrrolidone (PVP) under similar conditions to the above preparation led to the formation of stable PVP-citrate-coated KGd(H2O)2[Fe(CN)6]·H2O NPs (PVP-C-KGdFeCN NPs). We found that the concurrent use of sodium citrate as a capping agent and PVP as a coating polymer is necessary for controlling the size and stabilizing the NPs in aqueous solution and in the solid state. When either sodium citrate or PVP was used alone, the NPs would aggregate and separate from the solution after the aqueous dispersion was left to stand at room temperature for a few days. The FT-IR spectra of the NPs after 24 h of dialysis to remove unbound citrate and PVP contained the characteristic IR vibrations attributable to the citrate anion and PVP in addition to the C≡N stretching vibration at 2070 cm–1, confirming the presence of both the capping agent and the coating polymer on the surface (Figure S3). The elemental analysis of the sample digested with nitric acid showed the expected K/Gd/Fe to be 1:1:1. Furthermore, the XRD patterns of the PVP-C-KGdFeCN NPs match well with those of the bulk sample (Figure S4).
The morphology and the size of the NPs were examined by transmission electronic microscopy (TEM). As shown in Figure 2, the TEM image of PVP-C-KGdFeCN NPs reveals irregularly shaped NPs with a relatively broad size distribution. The average size of the NPs is approximately 25 ± 10 nm. Energy-dispersive X-ray analysis (EDXA) showed that the nanomaterial contains gadolinium, potassium, and iron (Figure S5 of the SI).
Figure 2.
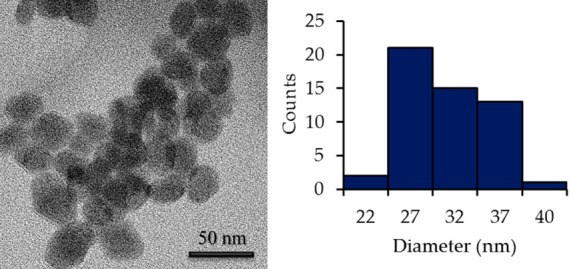
TEM image of PVP-C-KGdFeCN NPs.
Proton Relaxivity Measurements and Phantom Imaging Studies of PVP-C-KGdFeCN NPs
The efficiency of a CA is measured by its ability to enhance the proton relaxation of water and is commonly expressed as proton relaxivity.34 The latter is defined as the longitudinal or transverse relaxation rate increase of the water protons by unity concentration of the agent in mM. To evaluate the efficacy of our NPs as MRI contrast agents, we performed a series of proton T1 and T2 relaxation measurements in order to determine their longitudinal and transverse relaxivity values, r1 and r2, at both low (1.4 T) and high (7.0 T) magnetic field strengths. The 1.4 T relaxometry results were obtained on a Bruker Minispec 60 MHz relaxometer, and the 7.0 T results were obtained on a Bruker Biospec 7.0-T MRI scanner. Experimentally, the change in relaxation rate with increasing concentration of CA was measured, and the numeric value of the relaxivity, r1 or r2, was then extracted from the plot of 1/T1 (or 1/T2) vs the concentration of Gd3+ ions in NPs using the following equation
where 1/Ti,d (i = 1, 2) is the diamagnetic contribution to the relaxation rate and [Gd] is the concentration of Gd3+ ions in NPs. The relaxation rate of pure water is taken as the diamagnetic contribution in all of the experiments. All of the data are reported on a per Gd3+ ion basis at two different magnetic fields in Table 2 and compared to commercial contrast agent Magnevist (i.e., Gd-DTPA) and several known nanoparticle-based Gd(III) MR contrast agents. As shown in Figure 3, the resulting values for these NPs are r1 = 17 mM–1 s–1 and r2 = 24 mM–1 s–1 at 7.0 T and r1 = 35 mM–1 s–1 and r2 = 38 mM–1 s–1 at 1.4 T, respectively. For comparison, the relaxivity values of Gd-DTPA are r1 = 3.4 mM–1 s–1 and r2 = 3.7 mM–1 s–1 at 1.4 T (Table 2).
Table 2. Comparison of Relaxivity Data of Several Selected Nanoparticulate Gd3+-Based Contrast Agents.
| nanoparticulate Gd-based CA | r1 (mM–1 s–1) | r2 (mM–1 s–1) | r2/r1 ratio | field (T) | ref |
|---|---|---|---|---|---|
| KGd(H2O)2[Fe(CN)6]·H2O | 16.8 | 23.9 | 1.4 | 7.0 | this work |
| KGd(H2O)2[Fe(CN)6]·H2O | 35.2 | 38.4 | 1.1 | 1.4 | this work |
| Gd(DTPA) | 3.4 | 3.7 | 1.1 | 1.4 | this work |
| Gd(BDC)1.5(H2O)2a | 35.8 | 55.6 | 1.6 | 3.0 | (15)(a) |
| Gd(H2O)2[Fe(CN)6]·2H2O | 13.3 | 20.1 | 1.5 | 4.7 | (16)(d) |
| GdPO4 | 13.9 | 15.0 | 1.1 | 0.47 | (14) |
| Gd2O3 | 4.8 | 16.9 | 3.5 | 7.0 | (23) |
BDC = terephthalic acid.
Figure 3.
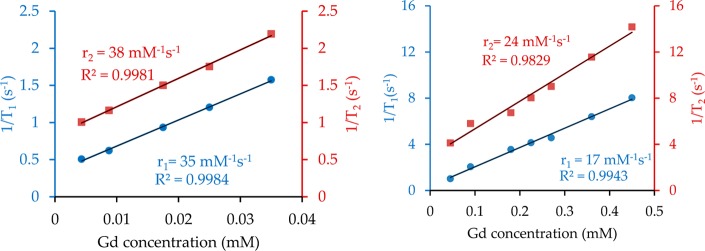
Plots of 1/Ti (i = 1, 2) vs the Gd3+ concentration at magnetic field strengths of 1.4 T (left) and 7.0 T (right) for PVP-C-KGdFeCN NPs.
To probe if the citrate-coating layer on the surfaces of the NPs would impede water exchange between the bulk and coordinated H2O molecules, we intentionally prepared the NPs under the same synthesis conditions with PVP as the only coating layer of the NPs. As shown in Figure S6, there is a slight increase in the r1 value from 35 to 37 mM–1 s–1 for the PVP-coated NPs, whereas the simultaneous change in the r2 value for such NPs from 38 to 47 mM–1 s–1 appears to be more significant. These results suggest that the presence of the citrate coating layer on the surfaces of the NPs may block the access of the bulk water to the sites of the coordinated H2O because the slight increase in the r1 relaxivity may well be due to the increased outer-sphere contribution as a result of a higher r2 value in the PVP-coated NPs. However, the opposite may be said about the tight, covalently bound citrate coating layer; that is, its presence on the surfaces of the NPs can act as a magnetic shield to reduce to the effectiveness of the localized tiny magnetic fields generated by the NPs, which in turn would induce the r2 relaxation of the bulk water protons. It is conceivable that the Gd(III) ions on the surface of the NPs have a higher ability to induce the relaxation of the bulk water protons than the contribution to Gd(III) inside the NPs. We therefore prepared PVP-C-KGdFeCN NPs with four different sizes by controlling the nucleation rate and coating. The relaxivity measurements showed a clear size dependence (Figure 4). It is interesting that both r1 and r2 relaxivities decrease with increasing particle size at almost the same rate.
Figure 4.
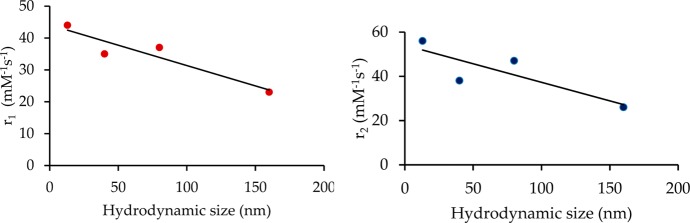
Plots of r1 (left) and r2 (right) vs the hydrodynamic particle size for PVP-C-KGdFeCN NPs.
These observed values of r1 relaxivity would place the current NPs among the best nanoparticle-based T1-weighted CAs that contain the Gd3+ ion.19,20,27,36,37 Besides the extremely high r1 relaxivity, the small r2/r1 ratios of ca. 1.4 at 7.0 T and 1.1 at 1.4 T indicate that these NPs are suitable as a T1-weighted agent to enhance the longitudinal relaxation of the water protons. In contrast, metal oxide-based NPs, particularly iron oxide NPs, have a tendency to exhibit superparamagnetic behavior with an exceedingly large r2/r1 ratio (i.e., r2/r1 > 10).19 As a result, iron and other metal oxide NPs typically act as highly effective T2-weighted CAs.35
According to SBM theory, the mechanism of inner-sphere relaxation in small molecular Gd3+-chelates can be understood from the following equation10
where q is the number of water molecules directly coordinated to the Gd3+ center, [C] is the molar concentration of the contrast agent in mM, T1M is the longitudinal relaxation time of the bound water, and τM is the mean residence lifetime of the coordinated water molecule. If T1M and τM are fixed, then increasing the number of water molecules bound to the Gd3+ center from one to two will result in a doubling of the inner-sphere T1 relaxivity. However, the decreased thermodynamic stability and/or kinetic inertness will render the complex highly susceptible to displacement by proteins, biological ligands, or endogenous Zn2+, Ca2+, and Mg2+ ions.1−4 It is tempting to conjecture that the presence of two water molecules directly coordinated to the Gd(III) center as well as the zeolitic water in the structural cavities plays an important role in contributing to the inner-sphere longitudinal relaxation of the water protons. Furthermore, the structural rigidity resulting from the 3D extended polymeric network and the reduced tumbling rate of such particulate CA also favor high relaxivity. Recently, Perrier and coworkers reported the synthesis and NMR relaxivity studies of NPs based on a similar coordination polymer with the formula Gd(H2O)2[Fe(CN)6]·H2O, where Gd3+ contains two coordinated water molecules as well, although the cytotoxicity and cellular penetrating ability of these NPs remained to be seen.27 As expected, the PEGylated NPs of the coordination polymer also exhibit high T1-weighted relaxivity with a low r2/r1 ratio (Table S1). However, the [Fe(CN)6]3– anion in Gd(H2O)2[Fe(CN)6]·H2O contains the low-spin Fe(III) center with S = 1/2, which may be magnetically coupled with the Gd(III) center with S = = 7/2 to give rise to a significant contribution of the outer-sphere relaxation to the overall T1-weighted relaxivity, thus making the distinction of the coordinated water molecules to the inner-sphere longitudinal relaxation from the outer-sphere relaxation impossible. However, because the [Fe(CN)6]4– anion in our KGd(H2O)2[Fe(CN)6]·H2O contains the low-spin Fe(II) center with S = 0, the Gd(III) ions can be viewed as isolated paramagnetic centers, thus rendering the description of their T1-weighted relaxivity using the SBM model meaningful.
To further confirm the efficacy and evaluate their performance as effective MR contrast agents, we obtained both T1- and T2-weighted MRI phantom images of PVP-C-KGdFeCN NPs in aqueous solution with various concentrations on a 7.0 T scanner (Figure 5). The T1-weighted images rapidly become brighter with increasing concentration of NPs, and the T2-weighted images respond to the increase in the concentration by slightly darkening the contrast. These results suggest that the current NPs can act as an effective T1-weighted CA at the high magnetic field.
Figure 5.

T1-weighted (left) and T2-weighted (right) MR phantom images of PVP-C-KGdFeCN NPs with various Gd3+ concentrations using a 7.0 T scanner.
Cellular Uptake and Cytotoxicity Studies of PVP-C-KGdFeCN NPs
The ability of NPs to cross the cell membrane is a critical prerequisite for cellular MR imaging applications to be realized. We studied the cellular uptake of our NPs in HeLa cells using the fluorescent confocal microscopic imaging technique. For live cell imaging, cells were incubated with the carboxyfluorescein (CbF) dye-labeled NPs (Figure S7 in the SI), washed with PBS, and directly imaged under a laser scanning confocal microscope without fixation. The advantage of using CbF attached to the NPs as the fluorescent probe is that because of its high anionic negative charge, the CbF dye molecule itself is membrane-impermeable.38 The images of confocal microscopy showed the presence of bright-green fluorescent signals inside the cells that were incubated with the dye-labeled NPs for 3 h. The untreated HeLa cells were used as the negative control. Figure 6 shows the typical confocal fluorescent images of HeLa cells treated with the dye-labeled NPs and the control cells. The uniform fluorescence emission in the perinuclear region of the cell indicates an untargeted cytoplasmic distribution of NPs (i.e., there is no specific binding of NPs to any small organelle in the region). These observations are consistent with the notion that the internalization of such NPs is most likely to occur via endocytosis.
Figure 6.
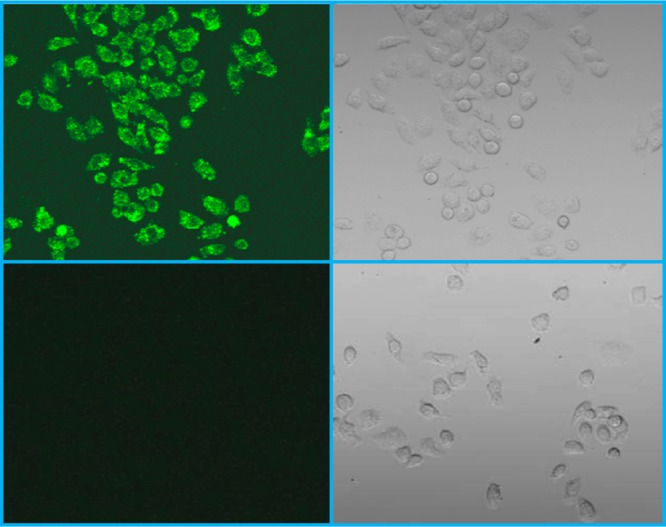
Confocal microscopic fluorescence image of HeLa cells incubated with dye-conjugated NPs for 3 h (top left), the bright-field image of the cells from panel A (top right), the fluorescent image of untreated HeLa cells as the negative control (bottom left), and the bright-field image of the cells from panel C (bottom right).
A quantification of the cellular uptake of KGdFeCN NPs was performed on the HeLa cell lysate using inductively coupled plasma mass spectrometry (ICP-MS). As shown in Figure 7, there is clear correlation between the cellular fluorescence signals and the increase in both intracellular Gd and Fe concentrations. However, the cellular uptake of Gd-DTPA is greatly diminished (i.e., reduced approximately 30-fold) compared to that of KGdFeCN NPs as revealed by the much lower Gd concentration in the cell lysate treated with Gd-DTPA. This is because as a small molecule, Gd-DTPA penetrates the cell membrane mainly by the process of passive diffusion.
Figure 7.
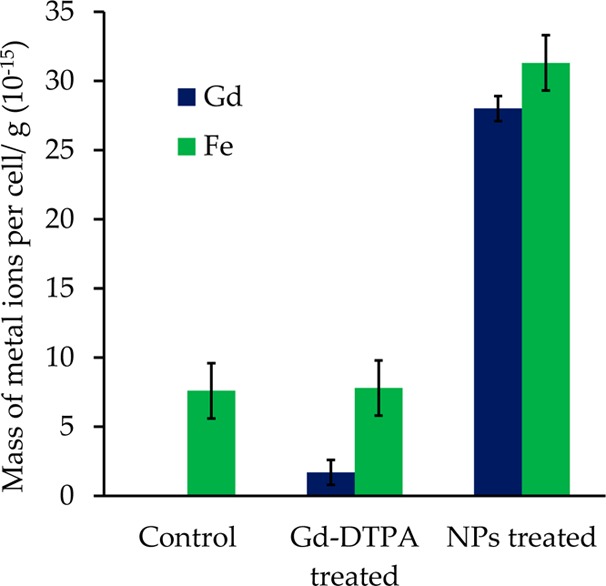
Gadolinium and iron concentrations of NP-treated (0.25 mM) vs Gd-DTPA-treated HeLa (0.25 mM) cells (5 h incubation) along with untreated HeLa cells as the control.
The cytotoxicity of the NPs in HeLa cells was evaluated using the MTT assay with various concentrations of the NPs and two different incubation times to study both the dose response and time effect. As shown in Figure 8, more than 90% of the cells were still viable after 48 h of incubation with 1.00 mM PVP-C-KGdFeCN NPs, indicating the nontoxic nature of such NPs. We attribute the low cytotoxicity of the NPs to the proper surface coating and the high structural integrity of the coordination polymer itself. To further confirm the stability of such NPs against the leaching of toxic CN– and Gd3+ ions, we carried out a series of leaching experiments under physiologically relevant conditions. Specifically, the solution concentrations of the CN– and Gd3+ ions leached out of the NPs were determined using a static method in which the NPs that were sealed in a membrane dialysis bag were allowed to equilibrate with different media that are physiologically relevant. The resulting solutions were then analyzed for free CN– and Gd3+ ions by a fluorometric method based on the Konig reaction39 and ICP-MS analysis, respectively. Figure 9 shows the concentrations of the CN– ions released from the NPs under different conditions. The highest cyanide concentration was found in an extremely acidic, albeit physiologically less relevant, solution of pH 1 to be approximately 3.5 ppm, and the cyanide concentration released from the NPs was found to be ∼0.8 ppm in a saline solution and ∼0.5 ppm in distilled water and other media containing a biologically relevant divalent metal ion at pH 7.4. In comparison, the maximum allowable level of cyanide in drinking water is 0.2 ppm as set by the environmental protection agency (EPA).40 It should be noted that in cigarette smokers, the cyanide concentration in blood can be as high as 35–65 ppm right after a cigarette is smoked.41 However, under all of the tested conditions, the concentrations of the Gd3+ ions released from the NPs were all below 1 ppm (or <6 μM), except in the acidic solution of pH 1 where the concentration of the Gd3+ ions was found to be approximately 3 ppm (i.e., ∼20 μM, Figure 10). These results suggest that the PVP-C-KGdFeCN NPs are stable against dissociation to release both Gd3+ and CN– ions under thermodynamic equilibrium conditions and kinetically inert to metal substitution reactions. This remarkable stability of the coordination polymer stems from the fact that as a ligand, the hexacyanoferrate(II) anion, [Fe(CN)6]4–, possesses the highest-possible ligand field stabilization energy (LFSE) for any complex containing the low-spin Fe(II) center and that the formation of an extended 3D coordination network structure results in high lattice energy for the coordination polymer. Consequently, both the CN– group and the Gd3+ ion are completely locked in their corresponding lattice positions and cannot be released from the structure by self-dissociation or ion exchange, confirming that the NPs are extremely stable and resistant to the displacement of gadolinium in the presence of the biologically relevant divalent metal ions. In sharp contrast, some small-molecule-based CAs are susceptible to in vivo transmetalation reactions with endogenous Zn2+ and Ca2+ to release Gd3+ ions. The latter was linked to the development of nephrogenic systemic fibrosis (NSF) in some renally impaired patients,42 which prompted the U.S. Food and Drug Administration (FDA) in 2007 to issue a public health advisory regarding the use of gadolinium-containing CAs. Currently, the manufacturers of such CAs are required by the FDA to include new boxed warnings and new warning sections in the labels to describe the possible link between the use of such CAs and the development of NSF.43
Figure 8.
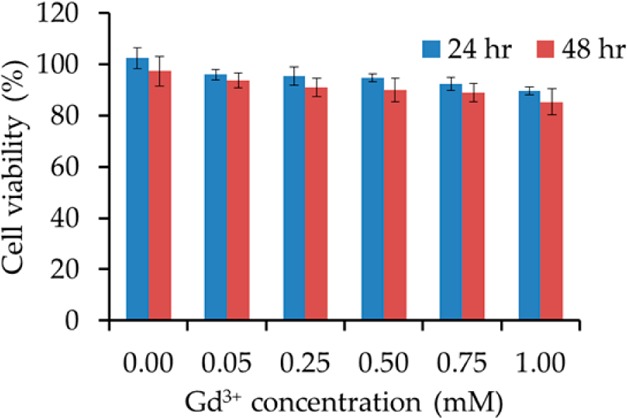
Viability of HeLa cells after incubation with PVP-C-KGdFeCN NPs for 24 and 48 h.
Figure 9.
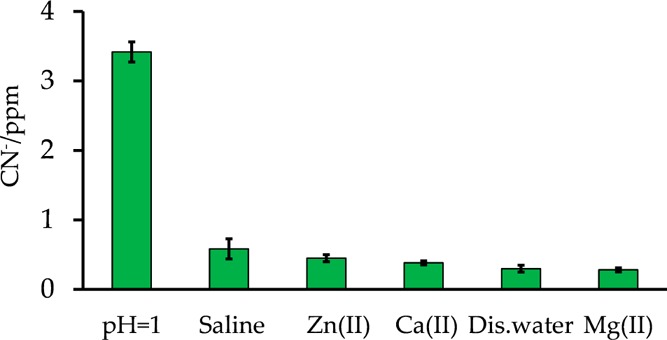
Cyanide-releasing test for different conditions.
Figure 10.
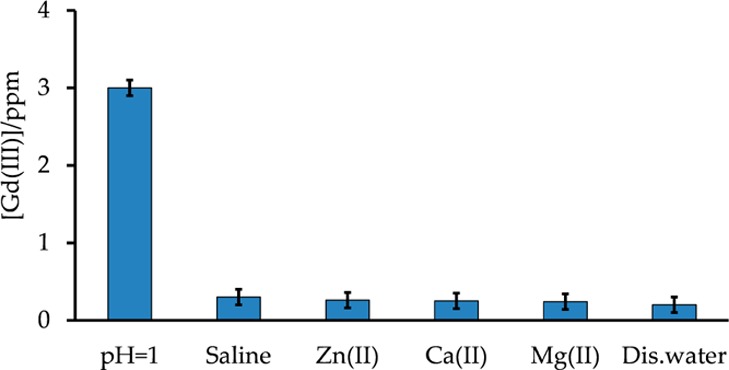
Gadolinium-releasing test for different conditions.
Cellular MR Imaging Studies Using Nanoparticles
To determine whether the internalized PVP-C-KGdFeCN NPs could enhance the T1-weighted MRI contrast of cells, we incubated PC3 cells with various concentrations of PVP-C-KGdFeCN NPs and examined the T1-weighted image for each sample at 37 °C using a spin-echo saturation recovery sequence on a Bruker 9.4-T MRI scanner. As shown in Figure 11, there is a considerable change in image brightness in the pellets of the PC3 cells incubated with the NPs. In particular, the cells treated with the NPs at a concentration equivalent to 0.25 mM Gd3+ ions prior to imaging exhibited a strong MRI signal brightening effect. These results demonstrate that PVP-C-KGdFeCN NPs have the potential to be used as an effective T1-weighted CA for cellular imaging at a high magnetic field.
Figure 11.

T1-weighted MRI phantoms of PC3 cells incubated in PBS buffer (left), 0.13 mM NPs (central), and 0.25 mM NPs (right) for 6 h. The images were collected using a Bruker 9.4-T scanner.
Conclusions
We have developed a simple one-step method for preparing extremely stable and biocompatible NPs of the gadolinium ferrocyanide coordination polymer. Such NPs can readily penetrate the cell membrane and exhibit no cytotoxicity. Furthermore, we have demonstrated that such NPs exhibit extremely high T1-weighted relaxivity, suggesting the potential of this coordination-polymer structural platform in the development of new-generation T1-weighted cellular MR probes for biological receptors or markers within the cell to study molecular events as well as for in vivo MR imaging in biomedical research and clinical applications.
Acknowledgments
We thank NIH-NCI for financial support (1R21CA143408-01A1). The TEM data were obtained using the Cryo TEM Facility at the Liquid Crystal Institute, KSU, supported by the Ohio Research Scholars Program.
Supporting Information Available
X-ray crystallographic data in CIF format. Results of the thermogravimetric analysis (TGA) on the bulk sample. Energy-dispersive X-ray spectrum of NPs. Fluorescence spectra of carboxyfluorescein dye and dye-labeled NPs. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
The manuscript was written through the contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Lauffer R. B. Paramagnetic metal complexes as water proton relaxation agents for NMR imaging: theory and design. Chem. Rev. 1987, 87, 901–927. [Google Scholar]
- Caravan P.; Ellison J. J.; McMurry T. J.; Lauffer R.B. Gadolinium(III) Chelates as MRI Contrast Agents: Structure, Dynamics, and Applications. Chem. Rev. 1999, 99, 2293–2352. [DOI] [PubMed] [Google Scholar]
- Aime S.; Botta M.; Terreno E. Gd(III)-based contrast agents for MRI. Adv. Inorg. Chem. 2005, 57, 173–237. [Google Scholar]
- Aime S.; Botta M.; Fasano M.; Terreno E. Lanthanide (III) chelates for NMR biomedical applications. Chem. Soc. Rev. 1998, 27, 19–29. [Google Scholar]
- Aime S.; Gianolio E.; Terreno E.; Giovenzana G. B.; Pagliarin R.; Sisti M.; Palmisano G.; Botta M.; Lowe M. P.; Parker D. Ternary Gd(III)L-HSA Adducts: Evidence for the Replacement of Inner-Sphere Water Molecules by Coordinating Groups of the Protein. Implications for the Design of Contrast Agents for MRI. J. Biol. Inorg. Chem. 2000, 5, 488–497. [DOI] [PubMed] [Google Scholar]
- Kang H.W.; Josephson L.; Petrovsky A.; Weissleder R.; Bogdanov A. Jr. Magnetic resonance imaging of inducible E-selectin expression in human endothelial cell culture. Bioconjugate Chem. 2002, 13, 122–127. [DOI] [PubMed] [Google Scholar]
- Caravan P. Protein-Targeted Gadolinium-Based Magnetic Resonance Imaging (MRI) Contrast Agents: Design and Mechanism of Action. Acc. Chem. Res. 2009, 42, 851–862. [DOI] [PubMed] [Google Scholar]
- Louie A. MRI biosensors: A short primer. J. Mag. Reson. Imaging 2013, 38, 530–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merbach A. E.; Helm L.; Tóth E.. The Chemistry of Contrast Agents in Medical Magnetic Resonance Imaging, 2nd ed.; John Wiley and Sons: Chichester, 2013. [Google Scholar]
- Kowalewski J.; Nordenskioeld L.; Benetis N.; Westlund P. O. Theory of nuclear spin relaxation in paramagnetic systems in solution. Prog. Nucl. Magn. Reson. Spectrosc. 1985, 17, 141–185. [Google Scholar]
- Caravan P. Strategies for increasing the sensitivity of gadolinium based MRI contrast agents. Chem. Soc. Rev. 2006, 35, 512–523. [DOI] [PubMed] [Google Scholar]
- Blamire A. M. The technology of MRI--the next 10 years?. Br. J. Radiol. 2008, 81, 601–617. [DOI] [PubMed] [Google Scholar]
- Aime S.; Crich S. G.; Gianolio E.; Giovenzana G. B.; Tei L.; Terreno E. High sensitivity lanthanide(III) based probes for MR-medical imaging. Coord. Chem. Rev. 2006, 250, 1562–1579. [Google Scholar]
- Weissleder R.; Stark D. D.; Engelstad B. L.; Bacon B. R.; Compton C. C.; White D. L.; Jacobs P.; Lewis J. Superparamagnetic iron oxide: pharmacokinetics and toxicity. Am. J. Roentgenol. 1989, 152, 167–173. [DOI] [PubMed] [Google Scholar]
- Tassa C.; Shaw S. Y.; Weissleder R. Dextran-Coated Iron Oxide Nanoparticles: a Versatile Platform for Targeted Molecular Imaging, Molecular Diagnostics and Therapy. Acc. Chem. Res. 2011, 44, 842–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurent S.; Forge D.; Port M.; Roch A.; Robic C.; Elst L. V.; Muller R. N. Magnetic iron oxide nanoparticles: synthesis, stabilization, vectorization, physicochemical characterizations, and biological applications. Chem. Rev. 2008, 108, 2064–2110. [DOI] [PubMed] [Google Scholar]
- Kim T.; Momin E.; Choi J.; Yuan K.; Zaidi H.; Kim J.; Park M.; Lee N.; McMahon M. T.; Quinones-Hinojosa A.; Bulte J. W. M.; Hyeon T.; Gilad A. A. Mesoporous Silica-Coated Hollow Manganese Oxide Nanoparticles as Positive T1 Contrast Agents for Labeling and MRI Tracking of Adipose-Derived Mesenchymal Stem Cells. J. Am. Chem. Soc. 2011, 133, 2955–2961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Na H. B.; Lee J. H.; An K.; Park Y.; Park M.; Lee I. S.; Nam D.-H.; Kim S. T.; Kim S.-H.; Kim S.-W.; Lim K.-H.; Kim K.-S.; Kim S.-O.; Hyeon T. Development of a T1 Contrast Agent for Magnetic Resonance Imaging Using MnO Nanoparticles. Angew. Chem., Int. Ed. 2007, 46, 5397–5401. [DOI] [PubMed] [Google Scholar]
- Hifumi H.; Yamaoka S.; Tanimoto A.; Citterio D.; Suzuki K. Gadolinium-based hybrid nanoparticles as a positive MR contrast agent. J. Am. Chem. Soc. 2006, 128, 15090–15091. [DOI] [PubMed] [Google Scholar]
- Rieter W. J.; Taylor K. M. L.; An H.; Lin W. Nanoscale metal-organic frameworks as potential multimodal contrast enhancing agents. J. Am. Chem. Soc. 2006, 128, 9024–9025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor K. M. L.; Jin A.; Lin W. Surfactant-assisted synthesis of nanoscale gadolinium metal-organic frameworks for potential multimodal imaging. Angew. Chem., Int. Ed. 2008, 47, 7722–7725. [DOI] [PubMed] [Google Scholar]
- Taylor K. M. L.; Rieter W. J.; Lin W. Manganese-based nanoscale metal-organic frameworks for magnetic resonance imaging. J. Am. Chem. Soc. 2008, 130, 14358–14359. [DOI] [PubMed] [Google Scholar]
- deKrafft K. E.; Xie Z. G.; Cao G. H.; Tran S.; Ma L. Q.; Zhou O. Z.; Lin W. B. Iodinated nanoscale coordination polymers as potential contrast agents for computed tomography. Angew. Chem., Int. Ed. 2009, 48, 9901–9904. [DOI] [PubMed] [Google Scholar]
- Guari Y.; Larionova J.; Corti M.; Lascialfari A.; Marinone M.; Poletti G.; Molvinger K.; Guerin C. Cyano-bridged coordination polymer nanoparticles with high nuclear relaxivity: toward new contrast agents for MRI. Dalton Trans. 2008, 3658–3660. [DOI] [PubMed] [Google Scholar]
- Larionova J.; Guari Y.; Sangregorio C.; Guerina C. Cyano-bridged coordination polymer nanoparticles. New J. Chem. 2009, 33, 1177–1190. [Google Scholar]
- Chelebaeva E.; Larionova J.; Guari Y.; Ferreira R. A. S.; Carlos L. D.; Trifonov A. A.; Kalaivani T.; Lascialfari A.; Guérin Ch.; Molvinger K.; Datas L.; Maynadier M.; Gary-Bobo M.; Garcia M. Nanoscale coordination polymers exhibiting luminescence properties and NMR relaxivity. Nanoscale 2011, 3, 1200–1210. [DOI] [PubMed] [Google Scholar]
- Perrier M.; Kenouche S.; Long J.; Thangavel K.; Larionova J.; Goze-Bac C.; Lascialfari A.; Mariani M.; Baril N.; Guerin C.; Donnadieu B.; Trifonov A.; Guari Y. Investigation on NMR relaxivity of nano-sized cyano-bridged coordination polymers. Inorg. Chem. 2013, 52, 13402–13414. [DOI] [PubMed] [Google Scholar]
- Shokouhimehr M.; Soehnlen E. S.; Khitrin A.; Basu S.; Huang S. D. Biocompatible Prussian blue nanoparticles: Preparation, stability, cytotoxicity, and potential use as an MRI contrast agent. Inorg. Chem. Commun. 2010, 13, 58–61. [Google Scholar]
- Sholouhimehr M.; Soehnlen E. S.; Hao H. H.; Griswold M.; Flask C.; Fan X. D.; Basilion J. P.; Basu S.; Huang S. P. D. Prussian blue nanoparticles for cellular imaging: Towards a new generation of T1-weighted MRI contrast agents. J. Mater. Chem. 2010, 20, 5251–5259. [Google Scholar]
- Perera V. S.; Hao J.; Gao M.; Gough M.; Zavalij P. Y.; Flask C.; Basilion J. P.; Huang S. D. Nanoparticles of the novel coordination polymer KBi(H2O)2[Fe(CN)6]·H2O as a potential contrast agent for computed tomography. Inorg. Chem. 2011, 50, 7910–7912. [DOI] [PubMed] [Google Scholar]
- Dunbar K. R.; Heintz R. A. Chemistry of transition metal cyanide compounds: modern perspectives. Prog. Inorg. Chem. 1997, 45, 283–391. [Google Scholar]
- Mullica D. F.; Ward J. L.; Sappenfield E. L. Gadolinium Potassium Hexacyanoferrate(II) Trihydrate. Acta Crystallogr. 1996, C52, 2956–2959. [Google Scholar]
- Topas Academic, General Profile and Structure Analysis Software for Powder Diffraction Data; Bruker AXS: Karlsruhe, Germany, 2004.
- Koenig S. H.; Brown R. D. Field-cycling relaxometry of protein solutions and tissue: Implications for MRI. Prog. NMR Spectrosc. 1990, 22, 487–567. [Google Scholar]
- Weissleder R.; Elizondo G.; Wittenberg J.; Rabito C. A.; Bengele H. H.; Josephson L. Ultrasmall superparamagnetic iron oxide: characterization of a new class of contrast agents for MR imaging. Radiology 1990, 175, 489–493. [DOI] [PubMed] [Google Scholar]
- McDonald M. A.; Watkin K. L. Investigations into the physicochemical properties of dextran small particulate gadolinium oxide nanoparticles. Acad. Radiol. 2006, 13, 421–427. [DOI] [PubMed] [Google Scholar]
- Mikawa M.; Kato H.; Okumura M.; Narazaki M.; Kanazawa Y.; Miwa N.; Shinohara H. Paramagnetic water-soluble metallofullerenes having the highest relaxivity for MRI contrast agents. Bioconjugate Chem. 2001, 12, 510–514. [DOI] [PubMed] [Google Scholar]
- Pangburn T. O.; Georgiou K.; Bates F. S.; Kokkoli E. Targeted Polymersome Delivery of siRNA Induces Cell Death of Breast Cancer Cells Dependent upon Orai3 Protein Expression. Langmuir 2012, 28, 12816–12830. [DOI] [PubMed] [Google Scholar]
- Yang Y.; Brownell C.; Sadrieh N.; May J.; Grosso A. D.; Place D.; Leutzinger E.; Duffy E.; He R.; Houn F.; Lyon R.; Faustino P. Quantitative Determination of Cyanide Released from Prussian Blue: An Assessment of Safety. Clin. Toxicol. 2007, 45, 776–781. [DOI] [PubMed] [Google Scholar]
- See http://water.epa.gov/drink/contaminants/index.cfm#List, last accessed on February 24, 2014.
- Vetter J. Plant cyanogenic glycosides. Toxicon 2000, 38, 11–36. [DOI] [PubMed] [Google Scholar]
- Perazella M. A. Current status of gadolinium toxicity in patients with kidney disease. Clin. J. Am. Soc. Nephrol. 2009, 4, 461–469. [DOI] [PubMed] [Google Scholar]
- http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/2007/ucm108919.htm, last accessed on February.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.