

Abstract
Background
The success of herbivorous insects has been shaped largely by their association with microbes. Seed parasitism is an insect feeding strategy involving intimate contact and manipulation of a plant host. Little is known about the microbial associates of seed-parasitic insects. We characterized the bacterial symbionts of Megastigmus (Hymenoptera: Torymidae), a lineage of seed-parasitic chalcid wasps, with the goal of identifying microbes that might play an important role in aiding development within seeds, including supplementing insect nutrition or manipulating host trees. We screened multiple populations of seven species for common facultative inherited symbionts. We also performed culture independent surveys of larvae, pupae, and adults of M. spermotrophus using 454 pyrosequencing. This major pest of Douglas-fir is the best-studied Megastigmus, and was previously shown to manipulate its tree host into redirecting resources towards unfertilized ovules. Douglas-fir ovules and the parasitoid Eurytoma sp. were also surveyed using pyrosequencing to help elucidate possible transmission mechanisms of the microbial associates of M. spermotrophus.
Results
Three wasp species harboured Rickettsia; two of these also harboured Wolbachia. Males and females were infected at similar frequencies, suggesting that these bacteria do not distort sex ratios. The M. spermotrophus microbiome is dominated by five bacterial OTUs, including lineages commonly found in other insect microbiomes and in environmental samples. The bacterial community associated with M. spermotrophus remained constant throughout wasp development and was dominated by a single OTU – a strain of Ralstonia, in the Betaproteobacteria, comprising over 55% of all bacterial OTUs from Megastigmus samples. This strain was also present in unparasitized ovules.
Conclusions
This is the first report of Ralstonia being an abundant and potentially important member of an insect microbiome, although other closely-related Betaproteobacteria, such as Burkholderia, are important insect symbionts. We speculate that Ralstonia might play a role in nutrient recycling, perhaps by redirecting nitrogen. The developing wasp larva feeds on megagametophyte tissue, which contains the seed storage reserves and is especially rich in nitrogen. Future studies using Ralstonia-specific markers will determine its distribution in other Megastigmus species, its mode of transmission, and its role in wasp nutrition.
Electronic supplementary material
The online version of this article (doi:10.1186/s12866-014-0224-4) contains supplementary material, which is available to authorized users.
Keywords: Burkholderia, Endophytophagy, Galls, Microbiome, Ralstonia, Rickettsia, Seed parasitism, Symbiosis, Wolbachia
Background
One of the major reasons that insects are the most diverse and abundant animals on Earth is due to their coevolution with plants and the myriad strategies they have evolved to successfully feed on them [1]. Only recently have we come to appreciate that microbial endosymbionts of phytophagous insects have played a important role in this success [2,3], for example by providing essential metabolites and vitamins [4-8], breaking down cell wall components, such as lignocellulose [9], recycling nitrogenous waste [10] and detoxifying plant secondary metabolites [11,12]. Maternally transmitted intracellular symbionts are extremely common in herbivorous insects [3]. Obligate nutritional symbionts are usually found within specialized host-derived organs called bacteriomes and they often exhibit co-speciation with their host lineages, indicative of an ancient association stabilized by strict vertical transmission from mother to offspring [13,14]. In addition, many insects harbour facultative heritable endosymbionts that are not necessary for the development and reproduction of the host [14]. These symbionts have evolved diverse strategies to persist in their hosts, including manipulating reproduction, for example by inducing parthenogenesis [15]. Other facultative symbionts increase host fitness under certain conditions, and it is in this regard that they are potentially important in mediating plant-insect interactions [3,16,17]. For example, facultative inherited symbionts of pea aphids have been implicated in facilitating the colonization of novel host plants [18,19].
Gut microbes also play critical roles in plant-insect interactions. Some herbivorous insects are associated with essential communities of microbes found within gut chambers (e.g. termite, cockroach) [20,21] or crypts (e.g. true bugs) [22]. Several posthatch transmission mechanisms have evolved to ensure transmission of gut associates from generation to generation, such as egg-smearing [23], coprophagy [24] and capsule-mediated transmission [25]. In addition, some true bugs acquire their gut microbes de novo every generation from the environment [26-28]. Gut bacteria can affect a herbivore’s host range. For example, when the symbiont capsule from a stinkbug pest of soybean, Megacopta punctatissima, is exchanged with a non-pest species, M. cribraria, there is an increase in fitness of this species on soybean and a decrease in fitness of the pest species on soybean [29]. This implies that the obligate symbiont dictates the pest status of the host. Since some of the major lineages of gut symbionts have only recently been discovered and characterized, we are still in early days in our understanding of how associated microbial communities are able to shape plant-insect interactions [16].
There are many examples of nutritional symbiosis among phytophagous hymenopterans. Xylophagous woodwasps and horntails rely on a symbiotic fungus for cellulose-digestion and/or nutrition during larval stages [30,31] and woodwasps have also been found to be associated with cellulose degrading bacteria [32]. Leaf-cutter ants have also formed a symbiotic relationship with fungi, in which the ants cultivate and consume a mutualistic fungus on a substrate of foraged leaf fragments [33]. The honeybee, Apis mellifera, is known to be associated with a distinct microbiota [34-39], that is thought to be important for both bee health and nutrition [35,38], including pollen coat digestion. Arboreal herbivorous ants that subsist mainly on a nutrient-poor diet of sugary plant exudates and hemipteran honeydew secretions harbour gut symbionts, which aid in nutrition. These symbiotic gut microbes include bacteria that are related to nitrogen-fixing root-nodule bacteria [40-42]. Carpenter ants in the genus Camponotus have an obligate endosymbiont, the gammaproteobacterium Blochmannia, which is found in host-derived bacteriomes [43]. Sequencing of the Blochmannia genome suggests that this symbiont provides its host with essential amino acids [44,45]. There is also evidence that Blochmannia plays a role in nitrogen recycling by encoding urease [46].
Many insects have independently evolved the ability to feed from within plant issues, for example, as seed-feeders, gallers, or leaf-miners. This feeding style permits the larval stage access to internal plant tissues with relatively high nutrient content and low defence response, and often involves complex physiological and morphological modifications of host plant tissue, including differentiation of additional tissues (gall formation), in situ up-regulation and synthesis of proteins and sugars, translocation of nutrients to the insect feeding site and the formation of green islands (photosynthetically active areas surrounding leaf-mining insects during leaf senescence) [47-50]. However, the mechanisms controlling these complex modifications are not well understood; it remains an open question whether symbiotic microbes might have a role in these systems. An interesting study recently implicated bacterial symbionts in insect endophytophagy. Feeding by leaf-mining Phyllonorycter blancardella caterpillars prevents leaf senescence, resulting in characteristic islands of green tissue. These green islands are associated with increased levels of plant hormones [47,48,51], including cytokinins similar to those used by bacteria to manipulate plant physiology [52-54]. When leaf-miners were treated with antibiotics, the green-island phenotype failed to appear, suggesting that bacterial symbionts of P. blancardella might be involved in manipulation of the plant [51,55].
Seed chalcid wasps of the genus Megastigmus (Hymenoptera: Torymidae) provide an interesting system to explore the role of microbes in nutrition and host manipulation of endophytophagous insects. The genus Megastigmus contains 134 described species, of which more than 72 are tree and shrub seed feeders; the remaining species are thought to be mainly parasitoids of gall insects [56,57]. Seed infesting species of Megastigmus undergo their development within the seeds of plants, obtaining nourishment from the developing embryo and storage reserves within the megagametophyte [58]. The best-studied species, M. spermotrophus, is a major pest of Douglas-fir (Pseudotsuga menziesii). This insect has the ability to manipulate the seed development of Douglas-fir for its own reproductive success [59,60]. First, M. spermotrophus can re-direct unfertilized ovules that normally abort to continue developing. Ovules do not redirect resources back to the mother plant, but instead feed the insect [59]. Second, the developing larva acts like a ‘surrogate’ embryo, causing the continued accumulation of storage reserves in the megagametophyte, which provides nourishment for the larva [60]. The re-direction of unfertilized ovule development by the presence of the parasite can be partially explained by changes in seed hormone levels, especially cytokinins [61]. It is suspected that all Megastigmus species infesting Pinaceae hosts can manipulate seed development [62].
Do Megastigmus wasps contain bacterial associates, and if so could they play an important role in the endophytophagous lifestyle of the host? In this paper, we used two approaches to characterize the microbial symbionts of Megastigmus, with the long-term goal of understanding their role in host nutrition and manipulation. Using symbiont-specific primers we screened a large sample of sexual Megastigmus species and two parasitoids of M. spermotrophus for common insect facultative heritable endosymbionts [63]. We also used 16S rRNA bacterial amplicon pyrosequencing to perform an unbiased and in-depth survey of the microbes associated with different developmental stages of M. spermotrophus (the best-studied Megastigmus species and an important pest of Douglas-fir), Douglas-fir ovules and the parasitoid Eurytoma sp. There have not been any studies on the microbial associates of Megastigmus except for a recent study that showed that thelytokous parthenogenesis in Megastigmus is caused by the reproductive parasite Wolbachia [64].
Results
Common heritable endosymbiont infections in Megastigmus
Three species tested positive in our inherited symbiont screens, with infection frequencies ranging from 33–100% (Table 1). Megastigmus milleri harbours a strain of Rickettsia from the bellii clade (Figure 1) [GenBank:KJ353735]. Megastigmus amicorum and M. bipunctatus harbour a strain of Rickettsia that is allied with R. felis, i.e. in the ‘transitional’ group [65]. Rickettsia citrate synthase sequences from these two hosts were identical [GenBank:KJ353732 - KJ353734]. These two hosts also harboured supergroup A Wolbachia infections (Figure 2) [GenBank:KJ353723 - KJ353731]. M. amicorum collected from different host plants and locations (Juniperus oxycedrus from Corsica and J. phoenicea from mainland France) were 2% divergent in mitochondrial COI [GenBank:KJ535736 - KJ535737] and infected with different Wolbachia strains. There was no significant difference in the frequency of infection in males and females, nor did we find an association between Wolbachia and Rickettsia in coinfected species (Fisher’s exact tests, data not shown). Arsenophonus, Cardinium, and Spiroplasma were not detected in Megastigmus samples screened using PCR with symbiont-specific primers.
Table 1.
Megastigmus spp. and parasitoids screened for common heritable symbionts using PCR
| Species | Host plant | Year | Location | N | Sample type | Wolbachia positive | Rickettsia positive |
|---|---|---|---|---|---|---|---|
| Family: Pinaceae | |||||||
| M. schimitscheki | Cedrus atlantica | 2010 | Petit Luberon, FR | 15 | Female | ||
| M. schimitscheki | Cedrus atlantica | 2009 | Mont Ventoux, FR | 14 | Female | ||
| M. schimitscheki | Cedrus atlantica | 2010 | Saou, FR | 14 | Female | ||
| M. schimitscheki | Cedrus atlantica | 2010 | Gap, FR | 15 | Female | ||
| M. schimitscheki | Cedrus atlantica | 2008 | Barjac, FR | 15 | Female | ||
| M. schimitscheki | Cedrus libani | 2005 | Turkey | 9 | Female | ||
| M. rafni | Abies alba | 2009 | Lespinassière, FR | 15 | Female | ||
| M. rafni | Abies alba | 2009 | Pardailhan, FR | 15 | Female | ||
| M. rafni | Abies alba | 2010 | Ventouret, FR | 15 | Female | ||
| M. rafni | Abies alba | 2004 | Doubs, FR | 9 | Female | ||
| M. rafni | Abies nordmanniana | 2000 | Rold Skov, DK | 9 | Female | ||
| M. rafni | Abies grandis | 2012 | Vancouver Island, CAN | 16 | Female | ||
| M. rafni | Abies grandis | 2012 | Vancouver Island, CAN | 10 | Male | ||
| M. milleri | Abies grandis | 2012 | Vancouver Island, CAN | 16 | Female | 75% (12) | |
| M. milleri | Abies grandis | 2012 | Vancouver Island, CAN | 10 | Male | 90% (9) | |
| M. spermotrophus | Pseudotsuga menziesii | 2011 | British Columbia, CAN | 26 | Female | ||
| M. spermotrophus | Pseudotsuga menziesii | 2011 | British Columbia, CAN | 10 | Larvae | ||
| Family: Cupressaceae | |||||||
| M. watchli | Cupressus sempervirens | 2011 | Sallèles du Bosc, FR | 15 | Female | ||
| M. watchli | Cupressus sempervirens | 2011 | Monfavet, FR | 15 | Female | ||
| M. watchli | Cupressus sempervirens | 2011 | Ruscas, FR | 16 | Female | ||
| M. watchli | Cupressus sempervirens | 1997 | Aghois Ioannis, GR | 10 | Female | ||
| M. bipuncatatus | Juniperus sabina | 2011 | Briançon, FR | 10 | Female | 90% (9) | 100% (10) |
| M. bipuncatatus | Juniperus sabina | 2011 | Pallon, FR | 13 | Female | 38% (5) | 54% (7) |
| M. bipuncatatus | Juniperus sabina | 2011 | Pallon, FR | 10 | Male | 50% (5) | 60% (6) |
| M. amicorum | Juniperus phoenicea | 2011 | Petit Luberon, FR | 8 | Female | 100% (8) | 100% (8) |
| M. amicorum | Juniperus phoenicea | 2011 | Luberon, FR | 15 | Female | 100% (15) | 93% (14) |
| M. amicorum | Juniperus phoenicea | 2011 | Luberon, FR | 10 | Male | 80% (8) | 70% (7) |
| M. amicorum | Juniperus oxycedrus | 2009 | Corsica, FR | 10 | Female | 70% (7) | 80% (8) |
| M. amicorum | Juniperus oxycedrus | 2011 | Corsica, FR | 10 | Female | 80% (8) | 100% (10) |
| M. amicorum | Juniperus oxycedrus | 2011 | Corsica, FR | 9 | Male | 33% (3) | 56% (5) |
| Parasitoids of M. spermotrophus | |||||||
| Eurytoma sp. | - | 2011 | British Columbia, CAN | 7 | - | ||
| Mesopolobus sp. | - | 2011 | British Columbia, CAN | 16 | - | ||
These species did not host Arsenophonus, Cardinium, or Spiroplasma. Spiroplasma was identified from Eurytoma sp. using 16S rRNA pyrosequencing.
Figure 1.
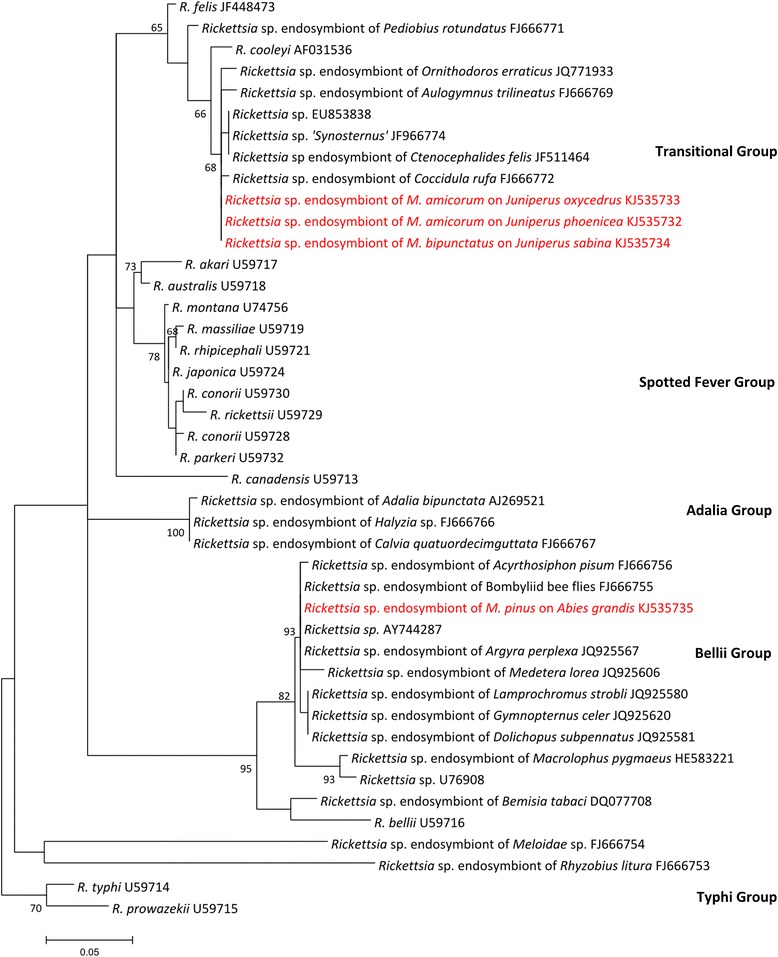
Maximum likelihood phylogeny for Rickettsia citrate synthase sequence constructed using the Tamura 3-parameter plus gamma distributed rates among sites model of nucleotide substitution. The sequences generated by this study are highlighted in red. Numbers next to the nodes indicate percentage of bootstrap support from 500 bootstrap replicates. Nodes without numbers received less than 65% bootstrap support.
Figure 2.
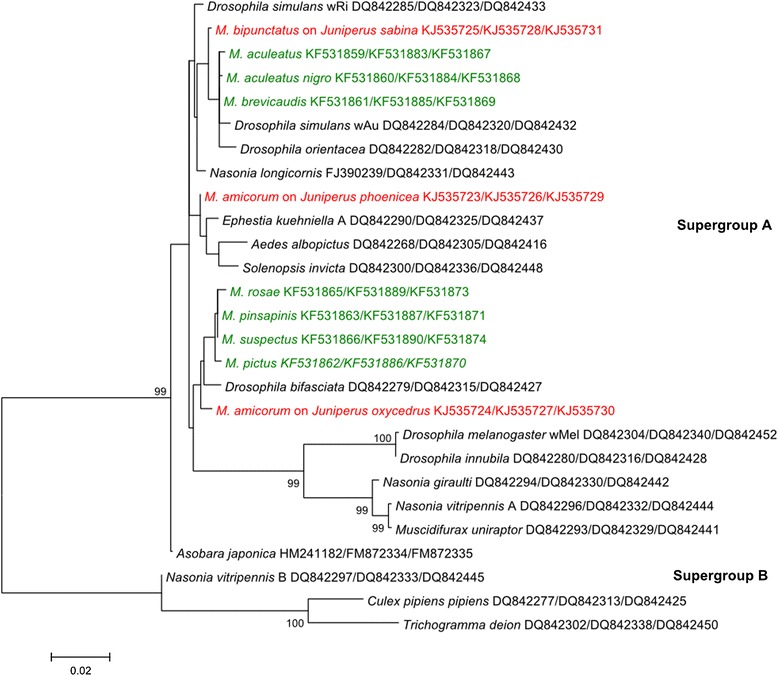
Concatenated maximum likelihood phylogeny for Wolbachia coxA, ftsZ and gatB sequence constructed using the Tamura 3-parameter plus gamma distributed rates among sites model of nucleotide substitution. Sequences generated by this study are red and sequences previously obtained from parthenogenetic Megastigmus are green [64]. Numbers next to the nodes indicate percentage of bootstrap support from 500 bootstrap replicates. Nodes without numbers received less than 65% bootstrap support.
Microbial associates of M. spermotrophus
16S rRNA bacterial amplicon pyrosequencing of M. spermotrophus (adult females, larvae and pupae), adult Eurytoma sp. and P. menziesii ovules generated 81,207 raw reads with an average length of 422 bp (see Additional file 1) [BioProject: PRJNA239784]. Quality and chimera filtering removed approximately 27% of the reads. The assignment of operational taxonomic units (OTUs) resulted in 352 unique bacterial clusters after the removal of singletons. A total of 160 OTUs were assigned to the genus level. The average sequencing depth was 3,616 sequences per sample (minimum and maximum of 1,962 and 6,130 sequences per sample). Rarefaction analysis showed that for most of the M. spermotrophus samples the number of observed OTUs no longer exponentially increased after an approximate sampling depth of 3,000 sequences (see Additional file 2) and the average number of observed species was 60 ± 13 and the average Chao1 species diversity estimate was 71 ± 25.
Fifteen major OTUs form the core bacterial microbiome of M. spermotrophus, i.e. having a total relative abundance of 0.5% or greater (Table 2). These OTUs are from five bacterial classes: Betaproteobacteria, Gammaproteobacteria, Actinobacteria, Firmicutes and Alphaproteobacteria. Over 60% of the sequences from the M. spermotrophus samples were assigned to the genus Ralstonia spp. (61.57%). Other major OTUs were assigned to the genera Acinetobacter and Corynebacterium representing 17.20% and 4.44% of total relative abundance, respectively. Further investigation using BLAST searches against the Ribosomal Database Project (http://rdp.cme.msu.edu/) and GenBank’s 16S ribosomal RNA sequence database revealed that all but one of the major OTUs not assigned to the genus level were actually Acinetobacter, Corynebacterium, or Ralstonia. The unknown Firmicutes is most closely related to Turicibacter, a strictly anaerobic gram-positive bacteria in the family Erysipelotrichaceae [66]; this OTU represents 0.74% of the total relative abundance of the 16S rRNA sequences in the M. spermotrophus samples.
Table 2.
Major bacterial OTUs associated with M. spermotrophus (greater than 0.5% average relative abundance) based on 16S rRNA amplicons from pyrosequencing
| Phylum | Class | Order | Family | Genus | Percent total relative abundance |
|---|---|---|---|---|---|
| Proteobacteria | Betaproteobacteria | Burkholderiales | Oxalobacteraceae | Ralstonia | 55.86 |
| Proteobacteria | Gammaproteobacteria | Pseudomonadales | Moraxellaceae | Acinetobacter | 16.28 |
| Actinobacteria | Actinobacteria | Actinomycetales | Corynebacteriaceae | Corynebacterium | 3.41 |
| Proteobacteria | Betaproteobacteria | Burkholderiales | Oxalobacteraceae | Ralstonia | 3.12 |
| Proteobacteria | Betaproteobacteria | Burkholderiales | Oxalobacteraceae | Ralstonia | 2.59 |
| Proteobacteria | 1.29 | ||||
| Actinobacteria | Actinobacteria | Actinomycetales | Corynebacteriaceae | Corynebacterium | 1.03 |
| Actinobacteria | Actinobacteria | Actinomycetales | 0.95 | ||
| Proteobacteria | Gammaproteobacteria | Pseudomonadales | Moraxellaceae | Acinetobacter | 0.92 |
| Firmicutes | Clostridia | Clostridiales | Clostridiaceae | Anaerococcus | 0.79 |
| Firmicutes | 0.74 | ||||
| Proteobacteria | 0.73 | ||||
| Proteobacteria | Betaproteobacteria | 0.72 | |||
| Firmicutes | Clostridia | Clostridiales | Clostridiaceae | Anaerococcus | 0.52 |
| Proteobacteria | Alphaproteobacteria | Rhizobiales | Bradyrhizobiaceae | 0.50 |
The relative abundance of the major OTUs from the different developmental stages of M. spermotrophus was mostly conserved (Figure 3), and there was no difference in the core microbiomes of the different developmental stages, based on principle coordinate analysis of weighted or unweighted UniFrac phylogenetic distances (see Additional file 3). The total relative abundance of OTUs from the class Betaproteobacteria (all in the genus Ralstonia) ranged from 46.4 - 72.3%. One female sample contained only a very small proportion of OTUs assigned to the class Gammaproteobacteria (0.36% relative abundance) while the total relative abundance of Gammaproteobacteria ranged from 12.7 - 33.1% in the remaining samples. The total relative abundance of all OTUs within the class Actinobacteria (all in the genus Corynebacterium) ranged from 1.9 - 7.1%.
Figure 3.
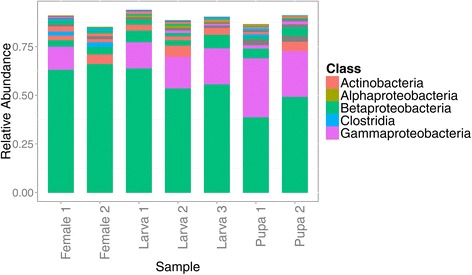
Relative abundance of major bacterial OTUs associated with larvae, pupae and adult M . spermotrophus (total relative abundance greater than or equal to 0.5%) based on 16 rRNA sequence from pyrosequence. Unknown classes are coloured grey.
A maximum likelihood phylogeny for Ralstonia was created using 16S rRNA sequence from the most abundant Ralstonia OTU in the pyrosequencing data set (Figure 4). Strong bootstrap support (0.99) clusters the Ralstonia isolated from M. spermotrophus with the human pathogen R. pickettii (sequence divergence = 3.3%).
Figure 4.
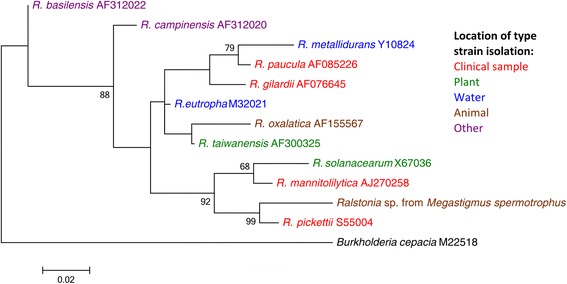
Maximum likelihood phylogeny for Ralstonia 16S rRNA sequence constructed using the Tamura-Nei with invariant sites and gamma distributed rate among sites model of nucleotide substitution. Numbers next to the nodes indicate percentage of bootstrap support from 500 bootstrap replicates. Nodes without numbers received less than 65% bootstrap support.
Ovule samples were dominated by chloroplast rRNA (99.0%); the remaining OTUs included Ralstonia (0.8%) and Acinetobacter (0.2%). The Eurytoma parasitoid samples were dominated by one OTU, which is allied with inherited Spiroplasma in the Ixodetis group (see Additional file 4) [GenBank:KJ535740], (99.6%). The remaining OTUs were Ralstonia.
Discussion
Common heritable endosymbiont infections in Megastigmus
We found three sexual Megastigmus species infected with Rickettsia, and two of these same species infected with Wolbachia. None of the species was infected with Arsenophonus, Spiroplasma, or Cardinium. From this patchy distribution (i.e. high prevalence in some host populations and low prevalence or absence in others), we can likely conclude that none of these inherited symbionts are essential in host nutrition and/or manipulation.
It is not surprising that Wolbachia was detected, as it is the most common intracellular bacterial symbiont of insects [67]. Wolbachia are transmitted maternally, in the egg cytoplasm, and many strains have evolved strategies to increase the frequency of infected female hosts in the population. Reproductive manipulating strains of Wolbachia have been show to either cause cytoplasmic incompatibility or distort sex ratios by killing males or inducing parthenogenetic reproduction (i.e. clonal production of females) or feminization [68]. Parthenogenesis-inducing Wolbachia are common in Hymenoptera and have been characterized in several parasitoid [69] and cynipid gall wasps [70,71]. A recent study implicated Wolbachia in parthenogenetic reproduction in Megastigmus, with 10/10 asexual species infected [64]. Treating M. pinsapinis with the antibiotic tetracycline restored the production of males, strongly suggesting that Wolbachia is the causative agent of thelytoky in asexual Megastigmus. No sexual Megastigmus species were infected with Wolbachia in the Boivin et al. study [64]; however, we found infections in M. amicorum and M. bipunctatus. The Wolbachia strains that we identified from sexual Megastigmus are closely allied with those in asexual Megastigmus. It would be interesting to determine if parthenogenesis-induction in Megastigmus is due to the host or the particular Wolbachia strain.
Rickettsia infections were discovered in three species. Bacteria in the genus Rickettsia are well known for being insect-vectored vertebrate pathogens, such as the causal agents of Rocky Mountain spotted fever (R. rickettsiae) and typhus (R. typhi). However, recent surveys have uncovered many Rickettsia that are vertically transmitted symbionts of diverse arthropods, most of which do not feed on vertebrates [72]. Some Rickettsia symbionts have been shown to distort host sex ratios via male-killing [73] or parthenogenesis-induction [74]. The presence of Rickettsia and Wolbachia in males likely rules out sex ratio distortion in our study. Alternatively, facultative symbionts may benefit their hosts under some circumstances. For example, some Wolbachia and Rickettsia increase host fitness by providing protection against natural enemies [75,76].
Phylogenetic analysis shows that closely related Rickettsia and Wolbachia infect distantly related Megastigmus (Figures 1 and 2). This provides strong evidence of horizontal transmission over evolutionary timescales, and is a common pattern in facultative inherited symbionts of insects [14]. In most cases, it is not known how inherited symbionts colonize novel hosts; shared hosts and shared natural enemies have both been implicated [77-80]. Interestingly, for some inherited symbionts, horizontal transmission over ecological timescales may be quite common [19,81]. It would be useful to sequence more rapidly evolving Rickettsia genes, to determine if there was very recent transmission between M. amicorum and M. bipunctatus. Since both these species develop in junipers, we could speculate that horizontal transmission occurs via shared host plants; the Boivin et al. study of Wolbachia in asexual Megastigmus also found evidence for such host-plant-mediated transmission [64]. Plant-mediated transmission may be an important and underappreciated way for symbionts to colonize hosts. Indeed, a recent study showed that an inherited Rickettsia in the sweet potato whitefly can be transmitted via phloem [61]. Two strains of Arsenophonus that infect planthoppers are transmitted both transovarially and via plants, and both have been implicated in plant disease [82,83]. However, as far as we are aware, interspecific transmission via plants has not yet been demonstrated in any inherited symbionts.
Microbial associates of M. spermotrophus
Our estimate of M. spermotrophus microbial species richness (60 ± 13 OTUs) fell within the range of other studies of insect microbiomes. Pollenivorous and predacious Hymenoptera (bees and wasps) harbour distinct bacterial communities with the lowest level of species richness (11.0 ± 5.4 OTUs/sample), while termites harbour the highest species diversity (89.5 ± 61.2 OTUs/sample), based on a recent meta-analysis [84]. A recent study estimated the diversity of bacteria associated with parasitoid wasps from the genus Nasonia ranged from 14 to 38 bacterial OTUs [85]. Pyrosequencing has been show to detect a greater number of OTUs compared to traditional methods, such as 16S rRNA clone sequencing [86]. This might explain why the estimated bacterial diversity associated with M. spermotrophus is comparably high because the Nasonia study and many previous insect microbiome surveys were done using 16S rRNA clone sequencing.
Despite a relatively high overall richness, only fifteen major OTUs are present with a total relative abundance of 0.5% or greater. The core bacterial community of M. spermotrophus can thus be considered to have a somewhat low diversity, characterized by bacterial OTUs that are commonly found associated with insect guts. The major OTUs associated with M. spermotrophus can be grouped into five distinct phylotypes: Betaproteobacteria (mostly Ralstonia), Gammaproteobacteria (mostly Acinetobacter), Actinobacteria (Corynebacterium), Firmicutes (mostly Anaerococcus) and Alphaproteobacteria (family Bradyrhizobiales). Most of these OTUs are related to bacteria that have been previously reported in insect guts, with Acinetobacter and Corynebacterium especially common (e.g. [85,87]). All of the major OTUs identified below the order level are bacteria that commonly occur in the environment, such as in soil [88] and in the rhizospere [89]. Similar results are commonly found with microbial associates of insects. For example, the microbial symbionts of Tetraponera ants are closely related to nitrogen-fixing root nodule bacteria [40]. The giant mesquite bug, Thasus neocalifornicus acquires an important mutualistic gut symbiont de novo every generation from the soil [27]. The presence of the same major OTUs in M. spermotrophus in ovule and even Eurytoma samples provides clues to the distribution and transmission of the Megastigmus microbiome; it suggests that it is derived from the environment, which, for the developing wasp, is the ovule. Acinetobacter and Corynebacterium have been previously cultured from within surface-sterilized seeds and ovules [90-92].
The M. spermotrophus microbiome appears to be highly conserved across development, as demonstrated by the UniFrac analysis, with all of the samples tightly grouped. This contrasts with a recent survey of microbial associates of three Nasonia species that found that bacterial species richness increased with development [85]. Like most higher Hymenoptera, the larvae of M. spermotrophus have a blind digestive system with the midgut and hind gut only uniting during the last larval instar. Prior to pupation all of the built-up wastes are voided in a fecal pellet, termed the meconium [93]. During metamorphosis the larval midgut epithelium is discarded and replaced by a new pupal epithelium [94]. If these bacteria are associated with the gut, how M. spermotrophus maintains its major associates throughout development is not known. Some insects, like true bugs, termites and cockroaches, have crypts or paunches associated with the gut that are thought to enhance persistence of the microbiota [6]. This physiological feature is not well characterized in the Hymenoptera, with the exception of some ants [95].
A single OTU assigned to the genus Ralstonia comprised over 55% of all sequences from the M. spermotrophus samples. The high abundance and persistence of Ralstonia throughout host development is a strong indicator that this bacterium is an important associate of M. spermotrophus. Ralstonia was also found to be associated with Douglas-fir ovules and the parasitoid Eurytoma. The genus Ralstonia contains species from ecological diverse niches, such as the plant pathogen R. solanacearum, the opportunistic human pathogen R. pickettii and the environmental isolate R. eurytropha [96]. A maximum likelihood phylogeny placed M. spermotrophus associated Ralstonia in a cluster with the human pathogen R. pickettii (Figure 4). To our knowledge, this is the first report of Ralstonia being a very abundant and potentially important component of an insect microbiome, although Ralstonia spp. have been previously reported from microbial surveys of insects, including the cotton bollworm (not published; accession # EU124821), Bartonella-positive fleas [97], an omnivorous carabid beetle [98] and the Potato Psyllid (as well as the faucet water used to water the potato plants) [99]. Recently, Husnik et al. also report the horizontal transfer of one Ralstonia gene into the genome of the mealybug Planococcus citri [100]. Also, R. oxalatica was isolated from the alimentary canal of an Indian earthworm [101].
A recent meta-analysis of 16S clone-library studies of insect associated microbes found that Betaproteobacteria contributed over 50% to all sequences from Hymenoptera [84]. The most common bacterial phylotype identified from solitary bee species, was a Betaproteobacteria from the genus Burkholderia [35], which is closely related to Ralstonia. Burkholderia spp. have also been identified as important mutualists of some phytophagous true bugs (suborder Heteroptera), where they reside in gut crypts [26-28,102,103].
The developing M. spermotrophus larva feeds on megagametophyte tissue, which contains all of the seed storage reserves, primarily in the form of starch, triacylglycerols, and nitrogen rich proteins [104,105]. Therefore, Ralstonia and other microbial associates of M. spermotrophus would not likely play a role in supplementing this already rich diet with missing essential nutrients but instead may play a role in nutrient recycling. Parasitism by M. spermotrophus results in the formation of a nutrient sink, in which the larva and associated microbes are nourished by storage reserves of the megagametophyte. The reserves are intended to provide nourishment for the developing seedling or to be re-absorbed by the mother plant in the event of megagametophyte abortion. In loblolly pine, more than half of the nitrogen in megagametophytes comes from the amino acid arginine [106]. Insects use the enzyme arginase to hydrolyze arginine into ornithine and urea [107]. Excretion of urea would result in the substantial loss of nitrogen, especially since larvae must undergo extended periods of diapause. Very few insects are known to produce urease, the enzyme required to convert urea into ammonium for subsequent amino acid biosynthesis [108]. We speculate that Ralstonia or other microbial associates of M. spermotrophus might play an important role in nitrogen recycling by producing urease or other key enzymes missing from the host genome. Many insect symbionts have been suggested to promote increased availability of nitrogen in a variety of ways [5]. For example, Blochmannia and Blattabacterium, the obligate nutritional symbionts of carpenter ants and cockroaches, respectively, use ureases to recycle nitrogen from urea [109,110]. Nitrogen recycling by symbionts has also been shown to be important during diapause in the shield bug, Parastrachia japonensi [111].
It is also tempting to speculate that Ralstonia could potentially play a role in plant manipulation. Another Ralstonia species, R. taiwanensi, has been shown to be capable of nodulating and fixing nitrogen in Mimosa spp. [112], which implies an ability to manipulate plant physiology. Alternatively, Ralstonia may not be a key associate of Megastigmus species in general, but rather a microbe that is found in the seed environment that encodes enzymes required for the catabolism of seed storage molecules or other essential pathways required for the seed feeding lifestyle of M. spermotrophus.
Now that Ralstonia has been identified as a likely symbiont of M. spermotrophus, further targeted surveys using Ralstonia-specific PCR primers would be helpful in determining its prevalence in other populations of M. spermotrophus, in other Megastigmus species, and in associated plants. The development of strain-specific markers for fluorescence in situ hybridization would also be useful for localizing Ralstonia on or within M. spermotrophus and the ovule, and following its transmission throughout its life cycle. It would also be interesting to examine Ralstonia’s role in nitrogen recycling, for example by identifying and following the expression of ureases and other key enzymes during M. spermotrophus development.
Conclusions
In this study two different approaches were used to survey Megastigmus for microbial symbionts. The directed PCR screens identified the presence of two common heritable symbionts, Wolbachia and Rickettsia; these are not likely distorting sex ratios in the sexual Megastigmus species surveyed in this study. Pyrosequencing was used to characterize the core microbiome of the Douglas-fir seed chalcid, M. spermotrophus, which is dominated by Ralstonia, a microbe that has not been previosly characterized as an important microbial associated of an insect. Interestingly, Ralstonia was also present in ovule and Eurytoma samples, indicating its prevalence within the niche of the ovule and potential horizontal transmission route from host to parasitoid.
This initial characterization of microbial associates of Megastigmus did not provide any insight into the potential involvement in host manipulation, although the maintenance of a consistent microbiome from larvae to adult suggests that microbes may be vital to the development and reproduction of M. spermotrophus. Many new questions are inspired by these findings, such as, how is the microbiome of M. spermotrophus maintained and transmitted? How widespread is the association with Ralstonia? What is the effect of heritable symbionts in sexual Megastigmus?
Methods
Insect samples
Several species of Megastigmus and their parasitoids were screened for common heritable symbionts using PCR. Adult insects were reared from seeds that were collected from forest stands in France, Greece, Denmark and Turkey from 1997 to 2011; detailed information on sample species is listed in Table 1. Also, larvae of M. spermotrophus were dissected from infested seed collected in 2011 from seed orchards located throughout British Columbia. Adult M. spermotrophus were reared from this same seed. Any Eurytoma sp. parasitoids that emerged were also collected. Wild adult female M. spermotrophus were collected from trees located on the University of Victoria campus in Victoria, BC. Whole insect samples were stored in 95% ethanol at −20°C until DNA extraction.
For 16S rRNA bacterial amplicon pyrosequencing, M. spermotrophus and their parasitoids were obtained in 2011 from heavily infested seed from the Mt. Newton Seed Orchard, located in Saanichton, BC. The seeds were placed at room temperature to hasten the development of larvae and adult emergence. Larvae as well as approximately one-week-old pupae were extracted from surface-sterilized seeds. Adult female M. spermotrophus and adult Eurytoma sp. were collected upon emergence about two and three weeks later, respectively. Samples of uninfested ovules were also collected from surface-sterilized seeds.
DNA extraction
Whole insects were rinsed several times with sterile water and allowed to air dry. The samples were then placed individually into 2 mL Micro tubes (Sarstedt) with 100 μL of PrepMan Ultra Reagent (Applied Biosystems, USA) and approximately twenty 1.0 mm diameter zirconia or silica beads (BioSpec Products). Samples were homogenized using the Mini-Beadbeater-16 (BioSpec Products) on maximum (3450 oscillations/min) for two 20–30 second cycles separated by 30 seconds of centrifugation at 13,000 × g. The samples were then incubated at 100°C for ten minutes, then cooled to room temperature for one minute, then centrifuged for three minutes at 13,000 × g and transferred into new Eppendorf tubes. DNA samples used for pyrosequencing were purified by precipitation in cold isopropanol and then washed with 70% ethanol and re-suspended in TE buffer (pH = 7.5). A NanoDrop 2000 Spectrophotometer (Thermo Scientific) was used to determine the DNA concentration and quality. The quality of the DNA extract was also checked by successful PCR amplification of the mitochondrial cytochrome oxidase subunit I (COI) gene using standard primers for invertebrates (see Additional file 5). All DNA extracts were stored at −20°C.
Directed PCR
Directed PCRs were conducted using either Invitrogen or ABM PCR Taq and reagents. Symbiont-specific primer-pairs were used to screen the samples for the presence of common heritable symbionts (see Additional file 5) with the following infected insects used as positive controls: Drosophila neotestacea (Wolbachia and Spiroplasma positive), Macrosteles quadrilineatus (Arsenophonus and Cardinium positive), and Ctenocephalides felis (Rickettsia positive). Sterile water was used as a negative control. Positive PCR products were separated on 1% agarose gel, stained with eithidium bromide and visualized under UV light.
Five microlitres of DNA from each individual extraction within a sample subset (individuals of the same species, sample type, location and year) were pooled (total of 32 pooled samples) and then screened using each primer set. If a positive PCR product was amplified from a pooled sample then each individual sample was screened for presence or absence of the corresponding symbiont using the same primer set. Positive PCR products were validated by sequencing representative amplicons in both directions. Purification and sequencing of PCR products were completed at Macrogen USA (Maryland). Forward and reverse sequences were aligned using MUSCLE and manually edited using the software Geneious (v6.1.3) (Biomatters) to create high-quality consensus sequences. A portion of mitochondrial COI was sequenced from one representative female of every symbiont-positive population, using Megastigmus-specific primers (see Additional file 5) and compared with other Megastigmus sequences deposited in GenBank. Percent divergence between COI sequences from M. amicorum populations was calculated using MEGA 5.1 [113].
Phylogenetic analysis of Rickettsia and Wolbachia infecting Megastigmus
A number of additional symbiont genes were amplified via PCR and sequenced: citrate synthase gene (gltA) for Rickettsia, and coxA, and gatB for Wolbachia (see Additional file 5). Phylogenies were re-constructed using sequences generated in this study and a sample of sequences obtained from GenBank. For Wolbachia, a sample of sequences obtained from an independent study of Wolbachia in parthenogenetic Megastigmus was also included [64]. Sequences were aligned using ClustalW, visually inspected and trimmed when necessary. A maximum-likelihood tree was generated using the Tamura 3-parameter model plus gamma distributed rates among sites (best substitution model identified by MEGA), with MEGA 5.1 [113], bootstrapped 500 times.
Bacterial tag-encoded FLX amplicon pyrosequencing
Three replicates of five sample types were submitted for bacterial tag-encoded FLX 454-pyrosequencing (bTEFAP): M. spermotrophus larvae, pupae and adult females, Eurytoma sp. adults and P. menziesii ovules. Although the 27 F/519R primer set is not ideal for characterizing bacterial 16S rRNA sequence from plant tissue due to chloroplast DNA contamination [114,115], we included ovule samples in order to see if any trace endophytic bacteria could be found after post-sequencing removal of plastid sequences. Inhibitor removal and bTEFAP were completed by MR. DNA Laboratories (Shallowater, TX). Inhibitor removal involved the use of the PowerClean DNA Clean-up kit (MO BIO Laboratories, Inc., Carlsbad, CA) according to the manufacturer’s protocol. The methods used for bTEFAP are previously described in Palavesam et al. (2012) and Shange et al. (2012) [116,117] and were originally described by Dowd et al. (2008) [118]. Briefly, a single-step PCR was done using the following temperature profile: 94°C for 3 minutes, followed by 28 cycles of 94°C for 30 seconds, 53°C for 40 seconds and 72°C for 1 minute, with a final elongation step at 72°C for 5 minutes using HotStarTaq Plus Master Mix Kit (Qiagen, Valencia, CA). The 16S universal bacterial primers 27Fmod (5’-AGRGTTTGATCMTGGCTCAG-3’) and 519Rmodbio (5’-GTNTTACNGCGGCKGCTG-3’) were used to amplify a 500 bp region of the 16S rRNA gene spanning the V1-V3 regions. The PCR products from each of the different samples were mixed in equal concentrations and then purified using Agencourt Ampure beads (Agencourt Bioscience Corporation, MA, USA). Following the manufacturer’s guidelines, sequencing was conducted using the Roche 454 FLX titanium platform (Roche, Indianapolis, IN).
Qiime pipeline
The 454 generated Standard Format Flowgram (SFF) file was converted into a SFF text file using Mothur (v1.23.0) [119]. The open source software package Quantitative Insights Into Microbial Ecology (QIIME v1.6.0) was used to process the sequence data [120]. The raw sequencing data was filtered using the following parameters: minimum sequence length of 100 bp, maximum sequence length of 2,000 bp and maximum homopolymer region of eight. Also, any sequences with an average quality score below 25 or any ambiguous bases were discarded. This filtering step reduced the number of total sequences from 81,207 to 60,543. The 454 data were then denoised to reduce the number of erroneous OTUs [121]. Chimera detection was done independently of QIIME by implementing UCHIME through the USEARCH (v6.0.307) program [122]. The sequences were compared against the Gold database (http://www.drive5.com/usearch/manual/otupipe.html, downloaded February 13, 2013). Chimeric sequences (1,190 or 1.97%) were gleaned from the data set.
OTUs were picked with the UCLUST method with the optimal option indicated. Similar sequences were clustered at the default level of 0.97 [123]. Taxonomy was assigned to representative sequences using the RDP Classifier 2.2 method at the 0.9 confidence level [124]. Taxonomies were based on the Greengenes database (ftp://greengenes.microbio.me/greengenes_release/gg_12_10/, downloaded February 1, 2013) [125,126].
Originally, the PyNast method was used to align the representative sequences to a pre-aligned database; however, this method resulted in poor overall alignment. Alternatively, representative sequences were aligned to a Stockholm format reference of pre-aligned sequences and secondary structures using Infernal [127]. The aligned sequences were filtered to remove common gap positions, with the gap filter threshold set to 0.8 and the entropy threshold set to 0.10. An approximately-maximum-likelihood phylogenetic tree was created using FastTree 2.1.3 [128]. An OTU table in Biom format was created and then split at the highest taxonomic ranking to remove unclassified OTUs (likely remnant chimeric sequences). Singletons were removed from the Biom table. Alpha diversity results were generated using a rarefaction depth of 5,000. In order to identify possible outliers (i.e., samples that contain unusual or unexpected OTUs), the microbiome data were visualized using a correspondence analysis biplot [129]. One pupal sample (P1) and one female sample (F4) were found to be associated with distinct OTUs that did not cluster with the remaining samples. Sample P1 had a relatively elevated species richness compared to the other samples, likely originating from environmental contamination (data not shown). Sample F4 contained bacteria typical of human contamination. Subsequently these two samples were removed from further analysis.
Data exploration, visualization and analyses were performed in R (v3.0.1) [130] on RStudio (v0.97.336) (www.rstudio.com, downloaded August 5, 2013), mainly using the Phyloseq R-package (v1.5.19) [131]. Data were rarefied to an equal sampling depth of 1,962 prior to community analysis. Initial correspondence analysis and biplots were generated using the Ade4 R-package (v1.5-2) [132]. Principle component analysis was completed using unweighted and weighted UniFrac distances [133,134].
In order to obtain longer 16S rRNA fragments for phylogenetic analysis from the Spiroplasma strain infecting Eurytoma, general 16S rRNA amplicons were generated using the primers 63 F (5’-CAGGCCTAACACATGCAAGTC-3’) [135] and 907R (5’-CCGTCAATTCCTTTRAGTTT-3’) [136]. Amplicons were then cloned using the Strataclone kit with Solopack Competent cells (Stratagene). Transformation was validated with PCR using M13F (5’- CACGACGTTGTAAAACGAC-3’) and M13R (5’-GGATAACAATTTCACACAGG-3’). Eight clones were sent for sequencing and one representative Spiroplasma 16S rRNA sequence was used for further analysis. Attempts to clone longer Ralstonia 16S rRNA fragments were not successful.
Ralstonia sequence from the most abundant OTU in the pyrosequencing data was used to generate a 16S rRNA phylogeny, along with representative Ralstonia species and outgroup sequences, obtained from GenBank. Maximum likelihood analysis was performed as above, except using the Tamura-Nei model with invariant sites and gamma rate distribution among sites.
Acknowledgements
We would like to thank Dave Koletelo at the Surrey Seed Centre, Don Piggott at Yellow Point Propagation Ltd, and orchard managers Tim Crowder (Mt. Newton Seed Orchard) and the late Tim Lee (Vernon Seed Orchard) for providing infested seed. We would like to thank Marie-Anne Auger-Rozenberg and Thomas Boivin for very helpful discussions and for sharing unpublished data. We would like to thank Marie-Anne Auger-Rozenberg, Thomas Boivin, Alain Roques, Alain Chalon and colleagues from INRA for collecting and rearing infested seed in Europe. We would also like to thank Belaid Moa for assistance with the HPC on Westgrid. Jean-Noël Candau provided guidance on rearing Megastigmus. This work was funded by the Strategic Project Partnership Grants Program of NSERC and the Agence Nationale de Recherche - Programme Blanc International. SJP acknowledges support from the Canadian Institute for Advanced Research, Integrated Microbial Biodiversity Program.
Additional files
Summary of 454 16S rRNA sequence data. Summary of sequence data from tag encoded FLX 454-pyrosequencing of 16S rRNA from M. spermotrophus, Eurytoma sp. and P. menziesii ovule samples.
Observed species and Chao1 species diversity estimator rarefaction curves. Observed species richness and Chao1 species diversity estimator rarefaction curves for bacteria associated with different life stages of M. spermotrophus, based on 16S rRNA pyrosequencing.
Analysis of phylogenetic distances. Analysis of phylogenetic distances (UniFrac) for all OTUs associated with different developmental stages of M. spermotrophus based on 16S rRNA amplicon pyrosequence.
Maximum likelihood phylogeny for Spiroplasma 16S rRNA. Maximum likelihood phylogeny for Spiroplasma 16S rRNA sequence constructed using the general time reversible model of nucleotide substitution with gamma distributed rates among sites. The sequence generated in this study is highlighted in red. Numbers next to the nodes indicate percentage of bootstrap support from 500 bootstrap replicates. Nodes without numbers received less than 65% bootstrap support.
List of PCR primers and reactions conditions. List of PCR primers and reactions conditions used to generate COI sequence from Megastigmus spp. and screen for common heritable symbiont infections.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
AP designed experiments, collected and analyzed data, and wrote the paper; PvA conceived the project, designed experiments and commented on the manuscript; SP conceived the project, designed experiments and wrote the paper. All authors read and approved the final manuscript.
Contributor Information
Amber R Paulson, Email: apaulson@shaw.ca.
Patrick von Aderkas, Email: pvonader@uvic.ca.
Steve J Perlman, Email: stevep@uvic.ca.
References
- 1.Schoonhoven LM, van Loon JJA, Dicke M. Insect-Plant Biology 2nd Edition. New York, USA: Oxford University Press; 2005. [Google Scholar]
- 2.Janson EM, Stireman JO, Singer MS, Abbot P. Phytophagous insect-microbe mutualisms and adaptive evolutionary diversification. Evolution. 2008;62:997–1012. doi: 10.1111/j.1558-5646.2008.00348.x. [DOI] [PubMed] [Google Scholar]
- 3.Feldhaar H. Bacterial symbionts as mediators of ecologically important traits of insect hosts. Ecol Entomol. 2011;36:533–543. doi: 10.1111/j.1365-2311.2011.01318.x. [DOI] [Google Scholar]
- 4.Dillon RJ, Dillon VM. The gut bacteria of insects: nonpathogenic interactions. Annu Rev Entomol. 2004;49:71–92. doi: 10.1146/annurev.ento.49.061802.123416. [DOI] [PubMed] [Google Scholar]
- 5.Douglas AE. The microbial dimension in insect nutritional ecology. Funct Ecol. 2009;23:38–47. doi: 10.1111/j.1365-2435.2008.01442.x. [DOI] [Google Scholar]
- 6.Engel P, Moran NA. The gut microbiota of insects - diversity in structure and function. FEMS Microbiol Rev. 2013;37:699–735. doi: 10.1111/1574-6976.12025. [DOI] [PubMed] [Google Scholar]
- 7.Nakabachi A, Ishikawa H. Provision of riboflavin to the host aphid, Acyrthosiphon pisum, by endosymbiotic bacteria, Buchnera. J Insect Physiol. 1999;45:1–6. doi: 10.1016/S0022-1910(98)00104-8. [DOI] [PubMed] [Google Scholar]
- 8.McCutcheon JP, Moran NA. Parallel genomic evolution and metabolic interdependence in an ancient symbiosis. Proc Natl Acad Sci. 2007;104:19392–19397. doi: 10.1073/pnas.0708855104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Warnecke F, Luginbühl P, Ivanova N, Ghassemian M, Richardson TH, Stege JT, Cayouette M, McHardy AC, Djordjevic G, Aboushadi N, Sorek R, Tringe SG, Podar M, Martin HG, Kunin V, Dalevi D, Madejska J, Kirton E, Platt D, Szeto E, Salamov A, Barry K, Mikhailova N, Kyrpides NC, Matson EG, Ottesen EA, Zhang X, Hernández M, Murillo C, Acosta LG, et al. Metagenomic and functional analysis of hindgut microbiota of a wood-feeding higher termite. Nature. 2007;450:560–565. doi: 10.1038/nature06269. [DOI] [PubMed] [Google Scholar]
- 10.Whitehead LF, Wilkinson TL, Douglas AE. Nitrogen recycling in the pea aphid (Acyrtosiphon pisum) symbiosis. Proc R Soc London B Biol Sci. 1992;250:115–117. doi: 10.1098/rspb.1992.0138. [DOI] [Google Scholar]
- 11.Adams AS, Aylward FO, Adams SM, Erbilgin N, Aukema BH, Currie CR, Suen G, Raffa KF. Mountain pine beetles colonizing historical and naïve host trees are associated with a bacterial community highly enriched in genes contributing to terpene metabolism. Appl Environ Microbiol. 2013;79:3468–3475. doi: 10.1128/AEM.00068-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Genta FA, Dillon RJ, Terra WR, Ferreira C. Potential role for gut microbiota in cell wall digestion and glucoside detoxification in Tenebrio molitor larvae. J Insect Physiol. 2006;52:593–601. doi: 10.1016/j.jinsphys.2006.02.007. [DOI] [PubMed] [Google Scholar]
- 13.Baumann P. Biology bacteriocyte-associated endosymbionts of plant sap-sucking insects. Annu Rev Microbiol. 2005;59:155–189. doi: 10.1146/annurev.micro.59.030804.121041. [DOI] [PubMed] [Google Scholar]
- 14.Moran NA, McCutcheon JP, Nakabachi A. Genomics and evolution of heritable bacterial symbionts. Annu Rev Genet. 2008;42:165–190. doi: 10.1146/annurev.genet.41.110306.130119. [DOI] [PubMed] [Google Scholar]
- 15.Stouthamer R, Breeuwer JAJ, Hurst GDD. Wolbachia pipientis: microbial manipulator of arthropod reproduction. Annu Rev Microbiol. 1999;53:71–102. doi: 10.1146/annurev.micro.53.1.71. [DOI] [PubMed] [Google Scholar]
- 16.Frago E, Dicke M, Godfray HCJ. Insect symbionts as hidden players in insect-plant interactions. Trends Ecol Evol. 2012;27:705–711. doi: 10.1016/j.tree.2012.08.013. [DOI] [PubMed] [Google Scholar]
- 17.Oliver KM, Degnan PH, Burke GR, Moran NA. Facultative symbionts in aphids and the horizontal transfer of ecologically important traits. Annu Rev Entomol. 2010;55:247–266. doi: 10.1146/annurev-ento-112408-085305. [DOI] [PubMed] [Google Scholar]
- 18.Tsuchida T, Koga R, Fukatsu T: Host plant specialization governed by facultative symbiont.Science 2004, 303:1989. [DOI] [PubMed]
- 19.Henry LM, Peccoud J, Simon J-C, Hadfield JD, Maiden MJC, Ferrari J, Godfray HCJ. Horizontally transmitted symbionts and host colonization of ecological niches. Curr Biol. 2013;23:1–5. doi: 10.1016/j.cub.2013.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Breznak JA. Intestinal microbiota of termites and other xylophagous insects. Annu Rev Microbiol. 1982;36:323–343. doi: 10.1146/annurev.mi.36.100182.001543. [DOI] [PubMed] [Google Scholar]
- 21.Bracke JW, Cruden DL, Markovetz AJ. Intestinal microbial flora of the of the American cockroach, Periplaneta americana L. Appl Environ Microbiol. 1979;38:945–955. doi: 10.1128/aem.38.5.945-955.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Glasgow H. The gastric caeca and the caecal bacteria of the Heteroptera. Biol Bull. 1914;26:101–170. doi: 10.2307/1536004. [DOI] [Google Scholar]
- 23.Jones KG, Dowd PF, Blackwell M. Polyphyletic origins of yeast-like endocytobionts from anobiid and cerambycid beetles. Mycol Res. 1999;103:542–546. doi: 10.1017/S0953756298007308. [DOI] [Google Scholar]
- 24.Nalepa CA, Bignell DE, Bandi C. Detritivory, coprophagy, and the evolution of digestive mutualisms in Dictyoptera. Insectes Soc. 2001;48:194–201. doi: 10.1007/PL00001767. [DOI] [Google Scholar]
- 25.Hosokawa T, Kikuchi Y, Meng XY, Fukatsu T. The making of symbiont capsule in the plataspid stinkbug Megacopta punctatissima. FEMS Microbiol Ecol. 2005;54:471–477. doi: 10.1016/j.femsec.2005.06.002. [DOI] [PubMed] [Google Scholar]
- 26.Shibata TF, Maeda T, Nikoh N, Yamaguchi K, Oshima K, Hattori M, Nishiyama T, Hasebe M, Fukatsu T, Kikuchi Y, Shigenobu S. Bacterial symbiont of the Bean bug Riptortus pedestris. Genome Announc. 2013;1:1–2. doi: 10.1128/genomeA.00441-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Olivier-Espejel AS, Sabree ZL, Noge K, Becerra JX. Gut microbiota in nymph and adults of the giant mesquite bug (Thasus neocalifornicus) (Heteroptera: Coreidae) is dominated by Burkholderia acquired de novo every generation. Environ Entomol. 2011;40:1102–1110. doi: 10.1603/EN10309. [DOI] [PubMed] [Google Scholar]
- 28.Kikuchi Y, Hosokawa T, Fukatsu T. Insect-microbe mutualism without vertical transmission: a stinkbug acquires a beneficial gut symbiont from the environment every generation. Appl Environ Microbiol. 2007;73:4308–4316. doi: 10.1128/AEM.00067-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hosokawa T, Kikuchi Y, Shimada M, Fukatsu T. Obligate symbiont involved in pest status of host insect. Proc R Soc B Biol Sci. 2007;274:1979–1984. doi: 10.1098/rspb.2007.0620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kukor JJ, Martin MM. Acquisition of digestive enzymes by siricid woodwasps from their fungal symbiont. Science. 1983;220:1161–1163. doi: 10.1126/science.220.4602.1161. [DOI] [PubMed] [Google Scholar]
- 31.Šrůtka P, Pažoutová S, Kolařík M. Daldinia decipiens and Entonaema cinnabarina as fungal symbionts of Xiphydria wood wasps. Mycol Res. 2007;111(Pt 2):224–231. doi: 10.1016/j.mycres.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 32.Adams AS, Jordan MS, Adams SM, Suen G, Goodwin LA, Davenport KW, Currie CR, Raffa KF. Cellulose-degrading bacteria associated with the invasive woodwasp Sirex noctilio. ISME J. 2011;5:1323–1331. doi: 10.1038/ismej.2011.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Weber NA. Fungus-growing ants. Science. 1966;153:587–604. doi: 10.1126/science.153.3736.587. [DOI] [PubMed] [Google Scholar]
- 34.Martinson VG, Moy J, Moran NA. Establishment of characteristic gut bacteria during development of the honeybee worker. Appl Environ Microbiol. 2012;78:2830–2840. doi: 10.1128/AEM.07810-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Martinson VG, Danforth BN, Minckley RL, Rueppell O, Tingek S, Moran NA. A simple and distinctive microbiota associated with honey bees and bumble bees. Mol Ecol. 2011;20:619–628. doi: 10.1111/j.1365-294X.2010.04959.x. [DOI] [PubMed] [Google Scholar]
- 36.Jeyaprakash A, Hoy MA, Allsopp MH. Bacterial diversity in worker adults of Apis mellifera capensis and Apis mellifera scutellata (Insecta: Hymenoptera) assessed using 16S rRNA sequences. J Invertebr Pathol. 2003;84:96–103. doi: 10.1016/j.jip.2003.08.007. [DOI] [PubMed] [Google Scholar]
- 37.Mohr KI, Tebbe CC. Diversity and phylotype consistency of bacteria in the guts of three bee species (Apoidea) at an oilseed rape field. Environ Microbiol. 2006;8:258–272. doi: 10.1111/j.1462-2920.2005.00893.x. [DOI] [PubMed] [Google Scholar]
- 38.Olofsson TC, Vásquez A. Detection and identification of a novel lactic acid bacterial flora within the honey stomach of the honeybee Apis mellifera. Curr Microbiol. 2008;57:356–363. doi: 10.1007/s00284-008-9202-0. [DOI] [PubMed] [Google Scholar]
- 39.Moran NA, Hansen AK, Powell JE, Sabree ZL: Distinctive gut microbiota of honey bees assessed using deep sampling from individual worker bees.PLoS One 2012, 7:e36393. [DOI] [PMC free article] [PubMed]
- 40.Van Borm S, Buschinger A, Boomsma JJ, Billen J. Tetraponera ants have gut symbionts related to nitrogen-fixing root-nodule bacteria. Proc R Soc London B Biol Sci. 2002;269:2023–2027. doi: 10.1098/rspb.2002.2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Anderson KE, Russell JA, Moreau CS, Kautz S, Sullam KE, Hu Y, Basinger U, Mott BM, Buck N, Wheeler DE. Highly similar microbial communities are shared among related and trophically similar ant species. Mol Ecol. 2012;21:2282–2296. doi: 10.1111/j.1365-294X.2011.05464.x. [DOI] [PubMed] [Google Scholar]
- 42.Russell JA, Moreau CS, Goldman-huertas B, Fujiwara M, Lohman DJ, Pierce NE. Bacterial gut symbionts are tightly linked with the evolution of herbivory in ants. Proc Natl Acad Sci. 2009;106:21236–21241. doi: 10.1073/pnas.0907926106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Degnan P, Lazarus A, Brock C, Wernegreen J. Host-symbiont stability and fast evolutionary rates in an ant-bacterium association: Cospeciation of Camponotus species and their endosymbionts, Candidatus Blochmannia. Syst Biol. 2004;53:95–110. doi: 10.1080/10635150490264842. [DOI] [PubMed] [Google Scholar]
- 44.Gil R, Silva FJ, Zientz E, Delmotte F, González-Candelas F, Latorre A, Rausell C, Kamerbeek J, Gadau J, Hölldobler B, van Ham RCHJ, Gross R, Moya A. The genome sequence of blochmannia floridanus: comparative analysis of reduced genomes. Proc Natl Acad Sci. 2003;100:9388–9393. doi: 10.1073/pnas.1533499100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Degnan PH, Lazarus AB, Wernegreen JJ. Genome sequence of Blochmannia pennsylvanicus indicates parallel evolutionary trends among bacterial mutualists of insects. Genome Res. 2005;15:1023–1033. doi: 10.1101/gr.3771305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Feldhaar H, Straka J, Krischke M, Berthold K, Stoll S, Mueller MJ, Gross R: Nutritional upgrading for omnivorous carpenter ants by the endosymbiontBlochmannia.BMC Biol 2007, 5:48. [DOI] [PMC free article] [PubMed]
- 47.Giron D, Kaiser W, Imbault N, Casas J. Cytokinin-mediated leaf manipulation by a leafminer caterpillar. Biol Lett. 2007;3:340–343. doi: 10.1098/rsbl.2007.0051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Giron D, Frago E, Glevarec G, Pieterse CMJ, Dicke M. Cytokinins as key regulators in plant-microbe-insect interactions: connecting plant growth and defence. Funct Ecol. 2013;27:599–609. doi: 10.1111/1365-2435.12042. [DOI] [Google Scholar]
- 49.Stone GN, Schönrogge K. The adaptive significance of insect gall morphology. Trends Ecol Evol. 2003;18:512–522. doi: 10.1016/S0169-5347(03)00247-7. [DOI] [Google Scholar]
- 50.Schwachtje J, Baldwin IT. Why does herbivore attack reconfigure primary metabolism? Plant Physiol. 2008;146:845–851. doi: 10.1104/pp.107.112490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kaiser W, Huguet E, Casas J, Commin C, Giron D. Plant green-island phenotype induced by leaf-miners is mediated by bacterial symbionts. Proc R Soc B Biol Sci. 2010;277:2311–2319. doi: 10.1098/rspb.2010.0214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jameson P. Cytokinins and auxins in plant-pathogen interactions – An overview. Plant Growth Regul. 2000;32:369–380. doi: 10.1023/A:1010733617543. [DOI] [Google Scholar]
- 53.Sakakibara H. Cytokinins: activity, biosynthesis, and translocation. Annu Rev Plant Biol. 2006;57:431–449. doi: 10.1146/annurev.arplant.57.032905.105231. [DOI] [PubMed] [Google Scholar]
- 54.Frugier F, Kosuta S, Murray JD, Crespi M, Szczyglowski K. Cytokinin: secret agent of symbiosis. Trends Plant Sci. 2008;13:115–120. doi: 10.1016/j.tplants.2008.01.003. [DOI] [PubMed] [Google Scholar]
- 55.Body M, Kaiser W, Dubreuil G, Casas J, Giron D. Leaf-miners co-opt microorganisms to enhance their nutritional environment. J Chem Ecol. 2013;39:969–977. doi: 10.1007/s10886-013-0307-y. [DOI] [PubMed] [Google Scholar]
- 56.Auger-Rozenberg M-A, Roques A. Seed wasp invasions promoted by unregulated seed trade affect vegetal and animal biodiversity. Integr Zool. 2012;7:228–246. doi: 10.1111/j.1749-4877.2012.00308.x. [DOI] [PubMed] [Google Scholar]
- 57.Grissell EE. An annotated catalog of world Megastigminae (Hymenoptera: Chalcidoidea: Torymidae) Contrib Am Entomol Inst. 1999;31:1–92. [Google Scholar]
- 58.Roques A, Skrzypczyńska M. Seed-infesting chalcids of the genus Megastigmus Dalman, 1820 (Hymenoptera: Torymidae) native and introduced to the West Palearctic region: Taxonomy, host specificity and distribution. J Nat Hist. 2003;37:127–238. doi: 10.1080/713834669. [DOI] [Google Scholar]
- 59.von Aderkas P, Rouault G, Wagner R, Rohr R, Roques A. Seed parasitism redirects ovule development in Douglas fir. Proc R Soc B Biol Sci. 2005;272:1491–1496. doi: 10.1098/rspb.2005.3061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.von Aderkas P, Rouault G, Wagner R, Chiwocha S, Roques A. Multinucleate storage cells in Douglas-fir (Pseudotsuga menziesii (Mirbel) Franco) and the effect of seed parasitism by the chalcid Megastigmus spermotrophus Wachtl. Heredity. 2005;94:616–622. doi: 10.1038/sj.hdy.6800670. [DOI] [PubMed] [Google Scholar]
- 61.Chiwocha S, Rouault G, Abrams S, von Aderkas P. Parasitism of seed of Douglas fir (Pseudotsuga menziesii) by the seed chalcid, Megastigmus spermotrophus, and its influence on seed hormone physiology. Sex Plant Reprod. 2007;20:19–25. doi: 10.1007/s00497-006-0039-z. [DOI] [Google Scholar]
- 62.Rouault G, Turgeon J, Candau J-N, Roques A, von Aderkas P. Oviposition strategies of conifer seed chalcids in relation to host phenology. Naturwissenschaften. 2004;91:472–480. doi: 10.1007/s00114-004-0554-4. [DOI] [PubMed] [Google Scholar]
- 63.Duron O, Bouchon D, Boutin S, Bellamy L, Zhou L, Engelstädter J, Hurst GD: The diversity of reproductive parasites among arthropods:Wolbachiado not walk alone.BMC Biol 2008, 6:27. [DOI] [PMC free article] [PubMed]
- 64.Boivin T, Henri H, Vaver F, Gidoin C, Candau J-N, Magnoux E, Roques A, Auger-Rozenbert M-A. Epidemiology of thelytoky induced by the endosymbiotic Wolbachia across seed-specialized wasp species: host plant specialization matters. Mol Ecol. 2014;23:2362–2375. doi: 10.1111/mec.12737. [DOI] [PubMed] [Google Scholar]
- 65.Weinert LA, Werren JH, Aebi A, Stone GN, Jiggins FM: Evolution and diversity ofRickettsiabacteria.BMC Biol 2009, 7:6. [DOI] [PMC free article] [PubMed]
- 66.Ludwig W, Schleifer K-H, Whitman WB. Bergey’s Manual of Systematic Bacteriology Vol 3. New York: Springer; 2008. Revised road map to the phylum Firmicutes. [Google Scholar]
- 67.Zug R, Hammerstein P: Still a host of hosts forWolbachia: analysis of recent data suggests that 40% of terrestrial arthropod species are infected.PLoS One 2012, 7:e38544. [DOI] [PMC free article] [PubMed]
- 68.Werren JH, Baldo L, Clark ME. Wolbachia: master manipulators of invertebrate biology. Nat Rev Microbiol. 2008;6:741–751. doi: 10.1038/nrmicro1969. [DOI] [PubMed] [Google Scholar]
- 69.Stouthamer R. Wolbachia-induced parthenogenesis. In: O’Neill SL, Werren JH, editors. Influential Passengers. New York, USA: Oxford University Press; 1997. pp. 102–124. [Google Scholar]
- 70.Rokas A, Atkinson RJ, Nieves-Aldrey J-L, West SA, Stone GN. The incidence and diversity of Wolbachia in gallwasps (Hymenoptera; Cynipidae) on oak. Mol Ecol. 2002;11:1815–1829. doi: 10.1046/j.1365-294X.2002.01556.x. [DOI] [PubMed] [Google Scholar]
- 71.Plantard O, Rasplus J, Clainche Le I, Solignac M. Wolbachia-induced thelytoky in the rose gallwasp Diplolepis spinosissimae (Giraud) (Hymenoptera: Cynipidae), and its consequences on the genetic structure of its host. Proc R Soc London B Biol Sci. 1998;265:1075–1080. doi: 10.1098/rspb.1998.0401. [DOI] [Google Scholar]
- 72.Perlman SJ, Hunter MS, Zchori-Fein E. The emerging diversity of Rickettsia. Proc R Soc B Biol Sci. 2006;273:2097–2106. doi: 10.1098/rspb.2006.3541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Werren JH, Hurst GD, Zhang W, Breeuwer JA, Stouthamer R, Majerus ME. Rickettsial relative associated with male killing in the ladybird beetle (Adalia bipunctata) J Bacteriol. 1994;176:388–394. doi: 10.1128/jb.176.2.388-394.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hagimori T, Abe Y, Date S, Miura K. The first finding of a Rickettsia bacterium associated with parthenogenesis induction among insects. Curr Microbiol. 2006;52:97–101. doi: 10.1007/s00284-005-0092-0. [DOI] [PubMed] [Google Scholar]
- 75.Teixeira L, Ferreira A, Ashburner M: The bacterial symbiontWolbachiainduces resistance to RNA viral infections inDrosophila melanogaster.PLoS Biol 2008, 6:e2. [DOI] [PMC free article] [PubMed]
- 76.Łukasik P, van Asch M, Guo H, Ferrari J, Godfray HCJ. Unrelated facultative endosymbionts protect aphids against a fungal pathogen. Ecol Lett. 2013;16:214–218. doi: 10.1111/ele.12031. [DOI] [PubMed] [Google Scholar]
- 77.Gehrer L, Vorburger C. Parasitoids as vectors of facultative bacterial endosymbionts in aphids. Biol Lett. 2012;8:613–615. doi: 10.1098/rsbl.2012.0144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Duron O, Wilkes TE, Hurst GDD. Interspecific transmission of a male-killing bacterium on an ecological timescale. Ecol Lett. 2010;13:1139–1148. doi: 10.1111/j.1461-0248.2010.01502.x. [DOI] [PubMed] [Google Scholar]
- 79.Bressan A, Sémétey O, Arneodo J, Lherminier J, Boudon-Padieu E. Vector transmission of a plant-pathogenic bacterium in the Arsenophonus clade sharing ecological traits with facultative insect endosymbionts. Phytopathology. 2009;99:1289–1296. doi: 10.1094/PHYTO-99-11-1289. [DOI] [PubMed] [Google Scholar]
- 80.Jaenike J, Polak M, Fiskin A, Helou M, Minhas M. Interspecific transmission of endosymbiotic Spiroplasma by mites. Biol Lett. 2007;3:23–25. doi: 10.1098/rsbl.2006.0577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Moran NA, Dunbar HE. Sexual acquisition of beneficial symbionts in aphids. Proc Natl Acad Sci U S A. 2006;103:12803–12806. doi: 10.1073/pnas.0605772103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gatineau F, Jacob N, Vautrin S, Larrue J, Lherminier J, Richard-Molard M, Boudon-Padieu E. Association with the syndrome “Basses Richesses” of sugar beet of a Phytoplasma and a bacterium-like organism transmitted by a Pentastiridius sp. Phytopathology. 2002;92:384–392. doi: 10.1094/PHYTO.2002.92.4.384. [DOI] [PubMed] [Google Scholar]
- 83.Danet J, Foissac X, Zreik L, Salar P, Verdin E, Nourrisseau J, Garnier M. “Candidatus Phlomobacter fragariae ” is the prevalent agent of marginal chlorosis of strawberry in French production fields and is transmitted by the planthopper Cixius wagneri (China) Phytopathology. 2002;93:644–649. doi: 10.1094/PHYTO.2003.93.6.644. [DOI] [PubMed] [Google Scholar]
- 84.Colman DR, Toolson EC, Takacs-Vesbach CD. Do diet and taxonomy influence insect gut bacterial communities? Mol Ecol. 2012;20:5124–5137. doi: 10.1111/j.1365-294X.2012.05752.x. [DOI] [PubMed] [Google Scholar]
- 85.Brucker RM, Bordenstein SR. The roles of host evolutionary relationships (genus: Nasonia) and development in structuring microbial communities. Evolution. 2012;66:349–362. doi: 10.1111/j.1558-5646.2011.01454.x. [DOI] [PubMed] [Google Scholar]
- 86.Kautz S, Rubin BER, Russell JA, Moreau CS. Surveying the microbiome of ants: comparing 454 pyrosequencing with traditional methods to uncover bacterial diversity. Appl Environ Microbiol. 2013;79:525–534. doi: 10.1128/AEM.03107-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ishak HD, Plowes R, Sen R, Kellner K, Meyer E, Estrada DA, Dowd SE, Mueller UG. Bacterial diversity in Solenopsis invicta and Solenopsis geminata ant colonies characterized by 16S amplicon 454 pyrosequencing. Microb Ecol. 2011;61:821–831. doi: 10.1007/s00248-010-9793-4. [DOI] [PubMed] [Google Scholar]
- 88.Janssen PH. Identifying the dominant soil bacterial taxa in libraries of 16S rRNA and 16S rRNA genes. Appl Environ Microbiol. 2006;72:1719–1728. doi: 10.1128/AEM.72.3.1719-1728.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Da Rocha UN, van Overbeek L, van Elsas JD. Exploration of hitherto-uncultured bacteria from the rhizosphere. FEMS Microbiol Ecol. 2009;69:313–328. doi: 10.1111/j.1574-6941.2009.00702.x. [DOI] [PubMed] [Google Scholar]
- 90.Mundt JO, Hinkle NF. Bacteria within ovules and seeds. Appl Environ Microbiol. 1976;32:694–698. doi: 10.1128/aem.32.5.694-698.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Mukhopadhyay K, Garrison NK, Hinton DM, Bacon CW, Khush GS, Peck HD, Datta N. Identification and characterization of bacterial endophytes of rice. Mycopathologia. 1996;134:151–159. doi: 10.1007/BF00436723. [DOI] [PubMed] [Google Scholar]
- 92.Hallmann J, Quadt-Hallmann A, Mahaffee WF, Kloepper JW. Bacterial endophytes in agricultural crops. Can J Microbiol. 1997;43:895–914. doi: 10.1139/m97-131. [DOI] [Google Scholar]
- 93.Sharkey MJ. Phylogeny and classification of Hymenoptera. Zootaxa. 2007;1668:521–548. [Google Scholar]
- 94.Hakim RS, Baldwin K, Smagghe G. Regulation of midgut growth, development, and metamorphosis. Annu Rev Entomol. 2010;55:593–608. doi: 10.1146/annurev-ento-112408-085450. [DOI] [PubMed] [Google Scholar]
- 95.Bution ML, Caetano FH. Symbiotic bacteria and the structural specializations in the ileum of Cephalotes ants. Micron. 2010;41:373–381. doi: 10.1016/j.micron.2010.01.004. [DOI] [PubMed] [Google Scholar]
- 96.Brenner DJ, Krieg NR, Staley JT. Bergey’s Manual of Systematic Bacteriology. 2. New York: Springer; 2005. Volume 2, Part C, The Proteobacteria; the Alpha-, Beta-, Delta-, and Epsilonproteobacteria. [Google Scholar]
- 97.Jones RT, McCormick KF, Martin AP. Bacterial communities of Bartonella-positive fleas: diversity and community assembly patterns. Appl Environ Microbiol. 2008;74:1667–1670. doi: 10.1128/AEM.02090-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lundgren JG, Lehman RM: Bacterial gut symbionts contribute to seed digestion in an omnivorous beetle.PLoS One 2010, 5:e10831. [DOI] [PMC free article] [PubMed]
- 99.Hail D, Dowd SE, Bextine B. Identification and location of symbionts associated with potato psyllid (Bactericera cockerelli) Lifestages. Environ Entomol. 2012;41:98–107. doi: 10.1603/EN11198. [DOI] [PubMed] [Google Scholar]
- 100.Husnik F, Nikoh N, Koga R, Ross L, Duncan RP, Fujie M, Tanaka M, Satoh N, Bachtrog D, Wilson ACC, von Dohlen CD, Fukatsu T, McCutcheon JP. Horizontal gene transfer from diverse bacteria to an insect genome enables a tripartite nested mealybug symbiosis. Cell. 2013;153:1567–1578. doi: 10.1016/j.cell.2013.05.040. [DOI] [PubMed] [Google Scholar]
- 101.Vaneechoutte M, Kämpfer P, Thierry DB, Falsen E, Verschraegen G. Wautersia gen. nov., a novel genus accommodating the phylogenetic lineage including Ralstonia eutropha and related species, and proposal of Ralstonia [Pseudomonas] syzygii (Roberts et al. 1990) comb. nov. Int J Syst Evol Microbiol. 2004;54:317–327. doi: 10.1099/ijs.0.02754-0. [DOI] [PubMed] [Google Scholar]
- 102.Kikuchi Y, Hosokawa T, Fukatsu T. An ancient but promiscuous host-symbiont association between Burkholderia gut symbionts and their heteropteran hosts. ISME J. 2011;5:446–460. doi: 10.1038/ismej.2010.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kikuchi Y, Meng X, Fukatsu T. Gut symbiotic bacteria of the genus Burkholderia in the broad-headed bugs Riptortus clavatus and Leptocorisa chinensis (Heteroptera: Alydidae) Appl Environ Microbiol. 2005;71:4035–4043. doi: 10.1128/AEM.71.7.4035-4043.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Stone SL, Gifford DJ. Structural and biochemical changes in Loblolly Pine (Pinus taeda L.) seeds during germination and early seedling growth. II. Storage triacylglycerols and carbohydrates. Int J Plant Sci. 1999;160:663–671. doi: 10.1086/314158. [DOI] [Google Scholar]
- 105.King JE, Gifford DJ. Amino acid utilization in seeds of Loblolly pine during germination and early seedling growth. 1. Arginine and arginase activity. Plant Physiol. 1997;113:1125–1135. doi: 10.1104/pp.113.4.1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Todd CD, Gifford DJ. The role of the megagametophyte in maintaining loblolly pine (Pinus taeda L.) seedling arginase gene expression in vitro. Planta. 2002;215:110–118. doi: 10.1007/s00425-001-0720-2. [DOI] [PubMed] [Google Scholar]
- 107.Pant R. Nitrogen excretion in insects. Proc Anim Sci. 1988;97:379–415. doi: 10.1007/BF03179946. [DOI] [Google Scholar]
- 108.Rosenthal GA, Janzen DH, Dahlman DL, Url S, Carolina N, Hill C. Degradation and detoxification of canavanine by a specialized seed predator. Science. 1977;196:658–660. doi: 10.1126/science.854740. [DOI] [PubMed] [Google Scholar]
- 109.Sabree ZL, Kambhampati S, Moran NA. Nitrogen recycling and nutritional provisioning by Blattabacterium, the cockroach endosymbiont. Proc Natl Acad Sci. 2009;106:19521–19526. doi: 10.1073/pnas.0907504106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Zientz E, Dandekar T, Gross R. Metabolic interdependence of obligate intracellular bacteria and their insect hosts. Microbiol Mol Biol Rev. 2004;68:745–770. doi: 10.1128/MMBR.68.4.745-770.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kashima T, Nakamura T, Tojo S. Uric acid recycling in the shield bug, Parastrachia japonensis (Hemiptera: Parastrachiidae), during diapause. J Insect Physiol. 2006;52:816–825. doi: 10.1016/j.jinsphys.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 112.Chen W-M, James EK, Prescott AR, Kierans M, Sprent JI. Nodulation of Mimosa spp. by the beta-proteobacterium Ralstonia taiwanensis. Mol Plant Microbe Interact. 2003;16:1051–1061. doi: 10.1094/MPMI.2003.16.12.1051. [DOI] [PubMed] [Google Scholar]
- 113.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Wang H-X, Geng Z-L, Zeng Y, Shen Y-M. Enriching plant microbiota for a metagenomic library construction. Environ Microbiol. 2008;10:2684–2691. doi: 10.1111/j.1462-2920.2008.01689.x. [DOI] [PubMed] [Google Scholar]
- 115.Aires T, Marbà N, Serrao EA, Duarte CM, Arnaud-Haond S. Selective elimination of chloroplastidial DNA for metagenomics of bacteria associated with the green algae Caulerpa taxifolia (Bryopsidophyceae) J Phycol. 2012;48:483–490. doi: 10.1111/j.1529-8817.2012.01124.x. [DOI] [PubMed] [Google Scholar]
- 116.Palavesam A, Guerrero FD, Heekin AM, Wang J, Dowd SE, Sun Y, Foil LD, de Pérez León AA: Pyrosequencing-based analysis of the microbiome associated with the Horn Fly,Haematobia irritans.PLoS One 2012, 7:e44390. [DOI] [PMC free article] [PubMed]
- 117.Shange RS, Ankumah RO, Ibekwe AM, Zabawa R, Dowd SE: Distinct soil bacterial communities revealed under a diversely managed agroecosystem.PLoS One 2012, 7:e40338. [DOI] [PMC free article] [PubMed]
- 118.Dowd SE, Callaway TR, Wolcott RD, Sun Y, McKeehan T, Hagevoort RG, Edrington TS: Evaluation of the bacterial diversity in the feces of cattle using 16S rDNA bacterial tag-encoded FLX amplicon pyrosequencing (bTEFAP).BMC Microbiol 2008, 8:125. [DOI] [PMC free article] [PubMed]
- 119.Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stres B, Thallinger GG, Van Horn DJ, Weber CF. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol. 2009;75:7537–7541. doi: 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, Mcdonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7:335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Reeder J, Knight R. Rapidly denoising pyrosequencing amplicon reads by exploiting rank-abundance distributions. Nat Methods. 2010;7:668–669. doi: 10.1038/nmeth0910-668b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics. 2011;27:2194–2200. doi: 10.1093/bioinformatics/btr381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26:2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- 124.Wang Q, Garrity GM, Tiedje JM, Cole JR. Naïve bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol. 2007;73:5261–5267. doi: 10.1128/AEM.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.McDonald D, Price MN, Goodrich J, Nawrocki EP, DeSantis TZ, Probst A, Andersen GL, Knight R, Hugenholtz P. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 2012;6:610–618. doi: 10.1038/ismej.2011.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Werner JJ, Koren O, Hugenholtz P, DeSantis TZ, Walters WA, Caporaso JG, Angenent LT, Knight R, Ley RE. Impact of training sets on classification of high-throughput bacterial 16 s rRNA gene surveys. ISME J. 2012;6:94–103. doi: 10.1038/ismej.2011.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Nawrocki EP, Kolbe DL, Eddy SR. Infernal 1.0: inference of RNA alignments. Bioinformatics. 2009;25:1335–1337. doi: 10.1093/bioinformatics/btp157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Price MN, Dehal PS, Arkin AP: FastTree 2 - Approximately maximum-likelihood trees for large alignments.PLoS One 2010, 5:e9490. [DOI] [PMC free article] [PubMed]
- 129.Sourial N, Wolfson C, Zhu B, Quail J, Fletcher J, Karunananthan S, Bandeen-Roche K, Béland F, Bergman H. Correspondence analysis is a useful tool to uncover the relationships among categorical variables. J Clin Epidemiol. 2010;63:638–646. doi: 10.1016/j.jclinepi.2009.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.R Development Core Team: R . A language and environmnet for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; 2013. [Google Scholar]
- 131.Mcmurdie PJ, Holmes S. Phyloseq: A bioconductor package for handling and analysis of high-throughput phylogenetic sequence data. Pacific Symp Biocomput. 2012;17:235–246. [PMC free article] [PubMed] [Google Scholar]
- 132.Dray S, Dufour A-B. The ade4 package: Implementing the duality diagram for ecologists. J Stat Softw. 2007;22:1–20. [Google Scholar]
- 133.Lozupone C, Knight R. UniFrac: A new phylogenetic method for comparing microbial communities. Appl Environ Microbiol. 2005;71:8228–8235. doi: 10.1128/AEM.71.12.8228-8235.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Hamady M, Lozupone C, Knight R. Fast UniFrac: facilitating high-throughput phylogenetic analyses of microbial communities including analysis of pyrosequencing and PhyloChip data. ISME J. 2010;4:17–27. doi: 10.1038/ismej.2009.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Marchesi JR, Sato T, Weightman AJ, Martin TA, Fry JC, Hiom SJ, Dymock D, Wade WG. Design and evaluation of useful bacterium-specific PCR primers that amplify genes coding for bacterial 16S rRNA. Appl Environ Microbiol. 1998;64:795–799. doi: 10.1128/aem.64.2.795-799.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Schabereiter-Gurtner C, Lubitz W, Rölleke S. Application of broad-range 16S rRNA PCR amplification and DGGE fingerprinting for detection of tick-infecting bacteria. J Microbiol Methods. 2003;52:251–260. doi: 10.1016/S0167-7012(02)00186-0. [DOI] [PubMed] [Google Scholar]