

Abstract
Background
Evolutionary theory suggests that males and females may evolve sexually dimorphic phenotypic and biochemical traits concordant with each sex having different optimal strategies of resource investment to maximise reproductive success and fitness. Such sexual dimorphism would result in sex biased gene expression patterns in non-floral organs for autosomal genes associated with the control and development of such phenotypic traits.
Results
We examined morphological, biochemical and herbivory traits to test for sexually dimorphic resource allocation strategies within collections of sexually mature and immature Populus tremula (European aspen) trees. In addition we profiled gene expression in mature leaves of sexually mature wild trees using whole-genome oligonucleotide microarrays and RNA-Sequencing.
Conclusions
We found no evidence of sexual dimorphism or differential resource investment strategies between males and females in either sexually immature or mature trees. Similarly, single-gene differential expression and machine learning approaches revealed no evidence of large-scale sex biased gene expression. However, two significantly differentially expressed genes were identified from the RNA-Seq data, one of which is a robust diagnostic marker of sex in P. tremula.
Electronic supplementary material
The online version of this article (doi:10.1186/s12870-014-0276-5) contains supplementary material, which is available to authorized users.
Keywords: Sexual dimorphism, RNA-Sequencing, transcriptomics, Populus tremula, dioecious
Background
Sexual dimorphism, the differentiation of both primary (i.e. gonads) and secondary (other morphological, behavioural and physiological) sex characteristics is the norm in animal systems [1]. In angiosperms the majority of extant species are co-sexual, being either monoecious or hermaphroditic (i.e. they bear separate male and female flowers or have either flowers containing both sexual organs, respectively). However, ~4% of plant species are dioecious [2,3], with different individuals producing only male or female flowers, and it is thought that dioecy evolved from ancestral hermaphrodites, which inherently lack sex chromosomes [4]. In several animal systems including nematodes, insects and mammals, sex determination is well characterised [5], whereas the molecular mechanisms underlying dioecious sex determination in plants remain largely unresolved [4,6]. The emergence of dioecy appears to have occurred relatively recently in many plant species, with sex determining loci being located in small regions of reduced recombination where there may not yet have been adequate time for heteromorphic sex chromosomes to have evolved [4].
Evolutionary theory suggests that sexual dimorphism arises after release from a co-sexual state as each sex adapts to a new fitness optimum following the removal of constraints previously imparted by the other sex – i.e. that trade-offs necessarily exist between the male and female functions in a monoecious state [4,7,8]. With the exception of sex-determining loci (or chromosomes), males and females share the same genome. Thus sexually dimorphic phenotypes that are not controlled by genes within the sex determining loci/chromosome must result from differential expression regulation of autosomal genes involved in the development and control of those traits [1]. Examples of expected sexual trade-offs include differential optimal strategies of resource allocation to growth and secondary metabolites (such as phenolic compounds) given production of either pollen or seeds; for example, females may allocate more carbon to secondary metabolites at the expense of stem growth in order to protect seeds from predators and pathogens [9,10], resulting in males and females experiencing contrasting selective pressures [8,11].
The genus Populus includes poplars, aspens, and cottonwoods and is a well-established model system [12] with a high quality genome sequence available for P. trichocarpa [13,14]. Populus species and hybrids have numerous industrial and silvicultural uses [15,16] and are often keystone species [17,18]. In Populus, dioecy is the common condition with the only exception being the monoecious, hermaphroditic P. lasiocarpa (see citations in [19]). There are also rare cases of gender reversion, perfect (bisexual) flower formation and even mature seed catkin formation on male trees [19-21 and citations in 22]. Populus species do not have heteromorphic sex specific chromosomes [22], and the molecular mechanism of sex determination remains undetermined, although sex is genetically determined [23]. In P. trichocarpa there is substantial evidence that the sex-determining locus is located in the peritelomeric region of chromosome 19 [22,23]. For all Populus genetic maps where sex has been included as a marker during map construction, there is always a single sex-linked locus that is located on chromosome 19. However, its location on that chromosome varies in different sections of the genus. There are also contrasting reports as to which sex is heterogametic [22,24-26]. In the aspens it is now well established that the sex determination locus is located in the pericentromeric region of chromosome 19 [24-28]. Pakull et al. [28] recently identified that Potri.019G047300, a gene that the same group had previously identified as a candidate in the sex determination locus [24], is either completely or partially deleted specifically in females, a finding that we independently discovered and detail below.
There is a current lack of knowledge of whether global or specific patterns of sex biased gene expression exist in non-reproductive tissues of dioecious plant species [4]. To date, this has been investigated in a single study of Silene latifolia [29], which considered only 22 ESTs. Here we addressed this question using P. tremula, which produces high amounts of phenolic-based secondary metabolites that have been implicated in defence against herbivores and pathogens [30,31] making it a suitable model system to test for sexually dimorphic differences in resource allocation to growth and defence. We explored global gene expression patterns in combination with a set of diagnostic phenotypes in non-reproductive tissues (leaves) of sexually mature P. tremula. The same phenotypes were additionally assayed in sexually immature trees. Gene expression was profiled using both whole genome oligonucleotide microarrays and RNA-Sequencing (RNA-Seq). The expression data were used for both individual gene differential expression tests as well as a machine learning approach to test for genomic regions containing combinations of genes exhibiting sex-related expression differences.
Results
Phenotypic analysis reveals no evidence of sexual dimorphism in P. tremula
We found no evidence of sexual dimorphism in tree height or diameter (Additional file 1) in either the Umeå Aspen collection (UmAsp; [32]) or the Swedish Aspen (SwAsp; [33]) samples or for height increment, a measure of vigour, in the juvenile SwAsp samples (Figure 1a, Additional file 1). Similarly, we found no statistical evidence of sexual dimorphism for leaf area (Figure 1b), leaf nutritional quality (nitrogen and carbon content and their ratio, Figure 1c) or specific secondary metabolites (total phenolics and condensed tannins, Figure 2a-b) in either the UmAsp or SwAsp samples (Additional file 1). All SwAsp phenotypic data except carbon and nitrogen concentration were generated by Robinson et al. [34], who showed that these, and other, traits had a substantial degree of heritability (clonal repeatability), a result that could only be obtained from high quality phenotypic data, negating the possibility that the observed lack of significant sexual dimorphism resulted from low data quality.
Figure 1.
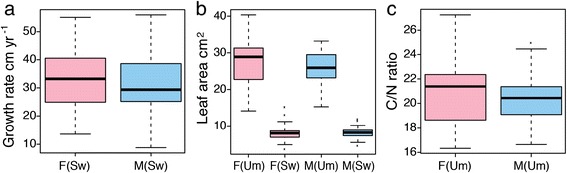
Growth and resource allocation in male and female Populus temula trees. Boxes representing females (F) are coloured pink and males (M) coloured blue, in the Umeå Aspen (Um) and Swedish Aspen (Sw) collections. (a) Growth rate calculated as height increment over five years in SwAsp. Analysis of Variance (ANOVA) results showed no significant sex differences (F1,45 = 0.448, P =0.507) (b) Individual leaf area in UmAsp and SwAsp. ANOVA results showed no significant sex differences for samples from either the Um (F1,38 = 0.958, P =0.334) or Sw (F1,44 = 0.012, P =0.914) collections. (c) Foliar carbon/nitrogen ratio in Um. ANOVA results showed no significant sex differences (F1,38 = 0.631, P =0.432).
Figure 2.
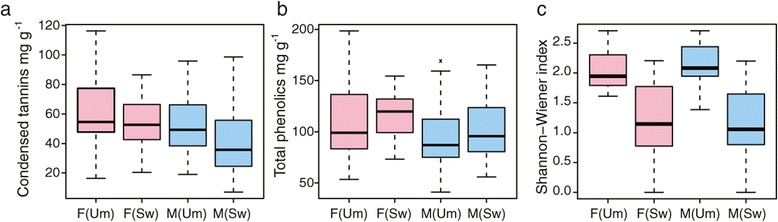
Secondary metabolite and herbivory phenotypes in male and female Populus tremula in the Umeå Aspen (Um) and Swedish Aspen (Sw) collections. Boxes representing females (F) are coloured pink and males (M) coloured blue. (a) Foliar condensed tannins. Analysis of Variance (ANOVA) results showed no significant sex differences for samples from either the Um (F1,38 = 1.667, P =0.203) or Sw (F1,45 = 2.764, P =0.103) collections (b) Foliar total phenolic concentrations. ANOVA results showed no significant sex differences for samples from either the Um (F1,38 = 01941, P =0.172) or Sw (F1,45 = 2.561, P =0.117) collections. (c) Shannon-Wiener index of arthropod herbivore diversity. ANOVA results showed no significant sex differences for samples from either the Um (F1,38 = 0.659 P =0.422) or Sw (F1,45 = 0.074, P =0.787) collections.
Herbivorous insects display no sexual preference
Arthropods are common folivores on P. tremula and numerous aspen-associated morphospecies have been recorded [34]. We found no statistically significant sex-related differences for arthropod abundance, species richness, feeding guild abundances, or the Shannon-Wiener diversity index in either the UmAsp or SwAsp samples (Figure 2c, Additional file 1). We also found no statistically significant sex-related differences in the arthropod community of UmAsp and SwAsp analysed by non-parametric Multivarite Analysis of Variance (MANOVA; UmAsp: F1,38 = 0.325, P =0.808; SwAsp: F1,45 = 0.825, P =0.5, Additional file 1).
Transcript profiling reveals no global patterns of sex-biased expression
We profiled gene expression in mature leaves of male and female P. tremula from the UmAsp collection using whole genome oligonucleotide microarrays (Figure 3) and RNA-Sequencing (RNA-Seq; (Figure 4). The samples used for RNA-Seq profiling were collected in two years and a Principle Component Analysis (PCA) analysis revealed clear differences between samples from the two years (Figure 4a). A total of 1,138 genes were identified as significantly differentially expressed between years (Figure 5).
Figure 3.
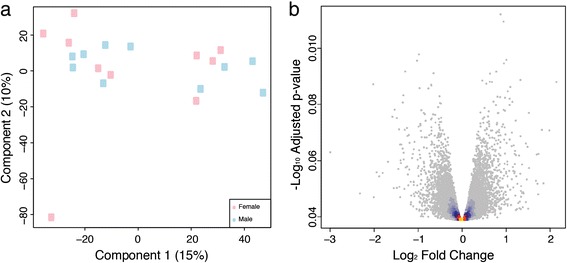
Overview of microarray gene expression patterns in male and female Populus tremula trees from the Umeå Aspen collection. (a) Principal Component Analysis plot of the microarray data with samples classified by sex (male in blue, female in pink). The percentage variance explained by each component is shown in parenthesis for each axis. The female sample shown at the bottom left of the plot was classified as an outlier and excluded from statistical analyses. (b) Volcano plot of the negative log10 p-value (y-axis) plotted against log2-fold change (x-axis) showing the results of differential expression analysis comparing male to female trees assayed using whole-genome Agilent oligonucleotide microarrays. Technical noise was accounted for in the statistical model by including factors for slide and sub-array within slide and the effect of sex was tested after removal of variance due to those technical effects. Non-significant genes are coloured to indicate density, which is shaded from yellow (high) to blue (low). No genes were significant (note that 0.01 on the y-axis corresponds to a p-value of 0.977).
Figure 4.
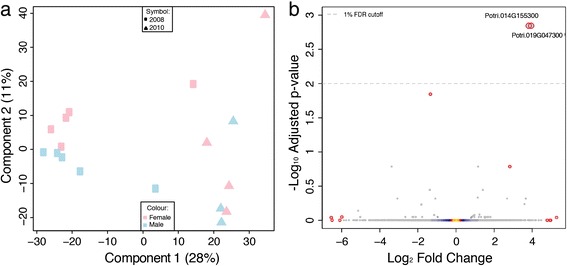
Overview of RNA-Sequencing gene expression patterns in male and female Populus tremula trees from the Umeå Aspen collection. (a) Principal Component Analysis plot of the RNA-Sequencing (RNA-Seq) expression data with samples classified by sex (male in blue, female in pink) and by year of sampling (2008 as squares and 2010 as triangles). The percentage variance explained by each component is shown in parenthesis for each axis. (b) Volcano plot of the negative log10 p-value (y-axis) (i.e. the log odds ratio) plotted against log2-fold change (x-axis) showing the results of differential expression analysis assayed using RNA-Seq comparing male to female trees. The statistical model included factors for year of sampling and sex and the effect of sex was tested after removal of the year effect. Significant genes are shown in blue where expression was higher in males. For the two significant genes at a 1% False Discover Rate (FDR) cut-off, the obtained p-value was <1e-10 and was therefore set to 0. As a result the log odds value is infinite and was therefore replaced with the next largest log odds +1. Non-significant genes are coloured to indicate density, which is shaded from yellow (high) to blue (low). The dashed horizontal line represents a 1% FDR. The four genes with the smallest p-values (regardless of significance) and the four genes with the highest and lowest non-significant fold change values are circled in red. These genes are represented in Additional file 2. The gene identifiers for the two statistically significant genes are shown (identifiers refer to V3 of the P. trichocarpa genome).
Figure 5.
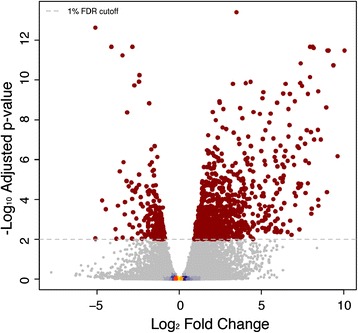
Differential expression of year of sampling effect. Volcano plot of the negative log10 p-value (y-axis) - i.e. log odds ratio - plotted against log2-fold change (x-axis) showing the results of differential expression analysis assayed using RNA-Sequencing comparing samples collected from trees in 2008 and 2010. Gene expression was assayed using samples collected from trees in 2008 and 2010 by RNA-Sequencing. Significant genes are shown as larger brown points. Non-significant genes are coloured to indicate density, which is shaded from yellow (high) to blue (low). The dashed horizontal line represents a 1% False Discovery Rate.
Despite many genes having relatively high mean fold-changes between sexes in the RNA-Seq data (Figure 4b), the within-sex variation for those genes was high resulting in non-significant statistical test results. To further explore this, we examined the variance among samples for the four genes with the lowest and highest fold change values and for the four genes with the smallest p values regardless of fold change (of which only two were statistically significant) in the RNA-Seq data. Variance for genes with high between-sex fold-change values was high (Additional file 2) and only two genes (see below) were statistically significantly differentially expressed between males and females.
We applied a machine learning approach, support vector machines (SVMs), to sliding windows of contiguous genes in the P trichocarpa genome to identify any regions where the combination of expression patterns for all genes within the window were predictive of sex. No statistically significant gene combinations that were predictive of sex were identified.
Potri.019G047300 is not present in females and is located in the sex determination locus
In contrast to the clear influence resulting from year of sampling, differential expression analysis identified only two statistically significant sexually dimorphic differences in the RNA-Seq dataset (Figure 4b; Potri.014G155300, FDR adjusted p-value 0.00: Potri.019G047300, FDR adjusted p-value 0.00) and none in the microarray dataset (Figure 3b). These two genes were not represented in the v1.1 genome annotation that was used for the array design, therefore excluding the possibility to cross-validate the result in the microarray dataset. However, Pakull et al. [28] provide an excellent and completely independent confirmation of this finding for Potri.019G047300.
Potri.014G155300 has no functional annotation but contains Pfam (Protein family) domains associated with cellulose synthase activity. This gene has highest sequence similarity to the Arabidopsis thaliana homolog AT2G32540, which is annotated as “Cellulose synthase-like B4”. More interestingly, the second gene (Potri.019G047300) is one of seven candidate genes identified within the sex determination locus of P. tremuloides by Kersten et al. [24] and was recently shown by the same authors to have a partial or complete deletion in female aspens [28] resulting in expression only being observed in males. The gene has no current functional description in poplar but contains WD40 domains and shows highest sequence similarity based homology to the A. thaliana gene AT5G16750 (TORMOZEMBRYO DEFECTIVE, TOZ). In A. thaliana this gene is required for regulated division planes and embryo development [35] and is thought to be involved in 18S rRNA biogenesis and RNA methylation. We examined the expression of the seven candidates highlighted by Kersten et al. [24] within our data, revealing that this was the only gene displaying any evidence of differential expression between sexes (Figure 6). The gene was expressed more highly in male than female trees. Examination of Affymetrix gene expression microarray data represented at the poplar eFP resource (http://bar.utoronto.ca/efppop/cgi-bin/efpWeb.cgi; [36]) shows that this gene has high expression in male catkins and low expression in female catkins for the three array probes representing this gene (PtpAffx.113801.1.S1_s_at, PtpAffx.212175.1.S1_at; probe-to-gene links were obtained from PopArray [37], http://aspendb.uga.edu/). However, as these data represent expression in P. balsamifera and as this gene is not deleted in female P. trichocarpa trees (as suggested by the presence of the complete gene structure in the assembled genome sequence) these results require caution for extrapolation to the aspens.
Figure 6.
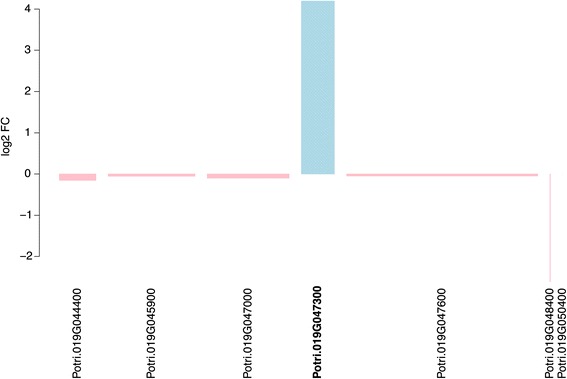
Bar graph showing differential expression of seven candidate genes from the sex determination locus of Populus tremuloides [ 24 ]. The width of each bar indicates the mean expression level of each gene relative to the gene with the highest expression (Potri.019G047600), which had an expression value of 240, such that narrow bars represent low expression and wide bars high expression. The y-axis indicates the log2 fold-change between male and female trees. Expression values represent variance stabilising transformation normalised read counts. Genes displaying higher expression in females are shown in pink and those with higher expression in males in blue. The gene model identifier for the only one of these genes that was identified as significantly differentially expressed in the RNA-Seq data is marked in bold text.
We used genomic re-sequencing data (collected for another study, but available on request) from two of the assayed trees, one male and one female, to further explore this locus. Genomic DNA sequencing reads (2x100 bp paired-end reads generated from a 300 bp insert library and sequenced using standard procedures on the Illumina HiSeq 2000 platform) were aligned to the reference P. trichocarpa genome sequence and only uniquely mapping reads were considered. This revealed that there is a deletion of this region in the female individual (Figure 7), which is in agreement with the results recently reported by Pakull et al. [28] and that explains the lack of any RNA-Seq reads being produced from female individuals in this region. As such females appear to be homozygous for absence of this locus. Corresponding plots based on RNA-Seq reads from all individuals assayed are available in Additional file 3. A single female individual (226.1) showed expression of the TOZ gene. We have been unable to confirm the sex of this tree as it has not flowered again since sex was originally determined. Repeating the above analyses with or without this individual did not affect the results obtained (see the R analysis HTML report on the PopGenIE FTP site [38]).
Figure 7.
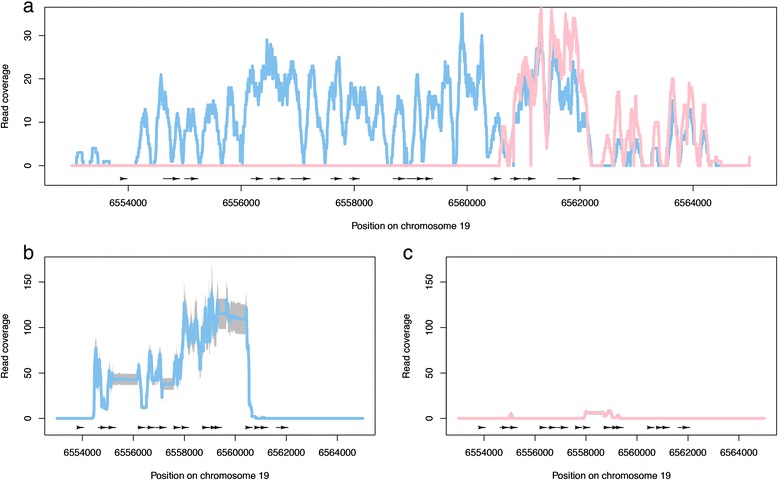
Genomic DNA and RNA-Sequencing read coverage (y axis) for the region of chromosome 19 (x axis) including Potri.019G047300. (a) Read coverage of uniquely mapping genomic DNA reads from a male (blue, 229.1) and female (pink, 349.2) individual. Black arrows represent exons with arrow direction indicating strand. (b,c) Read coverage of uniquely mapping RNA-Sequencing reads for male (b, n = 8) and female (c, n = 9) individuals. The coloured line represents the average per base pair read coverage across all individuals with grey indicating ± two standard deviations.
No evidence of biased sex ratio in P. tremula
We observed no sex bias in the P. tremula collections studied. The sex ratio of the SwAsp samples was 1:1 (female:male, where 52 trees of a total 116 in the collection are of known sex, Additional file 4). In the UmAsp samples the sex ratio was 1:1.1 (where 42 trees of 350 are of known sex, Additional file 4).
Discussion
In dioecious species, evolutionary theory suggests that males and females may have contrasting optimal strategies of resource investment to maximise reproductive success. As a result, natural selection would result in the emergence of sexual dimorphism in phenotypic, biochemical and ecological traits associated with contrasting resource allocation and utilisation as each sex evolves towards fitness optima. If phenotypic sexual dimorphism does arise, there will be concomitant dimorphism in gene expression patterns in the corresponding tissue(s) associated with those phenotypic traits. Such dimorphic gene expression patterns will be independent of any differential gene expression associated with sex determination and the control of reproductive tissue development. As such, although those genes may in some cases be located within the sex determination region or chromosome, it is likely that many such regulated genes will be autosomal.
In the current study our primary interest was to test the hypothesis that male and female P. tremula individuals invest resources differentially, resulting in sexual dimorphism. To this end a number of morphological and biochemical traits (Additional file 1) were selected to be diagnostic of such dimorphism in leaves sampled from a set of wild-growing, sexually mature P. tremula individuals (the UmAsp collection) and a set of common-garden, sexually immature and clonally replicated individuals (the SwAsp collection, see materials and methods). We focused on leaves as these are the primary point of interaction between aspens and the majority of their associated herbivores as well as representing the site of energy assimilation and therefore carbohydrate production for utilisation in primary (growth-associated) and secondary metabolism.
P. tremula shows no phenotypic evidence of sexual dimorphism
Height and diameter are often used as proxies for fitness based on the assumption that faster growing and larger individuals are better equipped to out-compete their neighbours, allowing greater resource acquisition that can be invested in sexual reproduction [39]. We found no statistical evidence supporting phenotypic differences between males and females for any of the phenotypic traits that we assayed in either the sexually mature UmAsp or sexually immature SwAsp samples. These results contrast with observations in Pauley [40] who reported a strong male biased sex ratio within a collection of superior-growth individuals of five North American Populus species. This was interpreted as potential evidence that males may display more vigorous growth. In P. euphratica growth traits showed variable differences between sexes among sample plots with no consistent statistically significant difference between sexes for assayed growth traits [41]. In the cross-species meta-analysis presented in Cornelissen & Stillin [10], males in general exhibited larger leaves, lower concentrations of secondary metabolites and higher growth rates. However, and in agreement with our results, there was no sexual dimorphism for height or nutrient concentrations. In P. deltoides, Farmer [42] observed that males were taller than females but did not have greater stem diameter. Citations within Farmer detail observations that the height of P. tremula x P. tremuloides seedling cohorts was correlated to the proportion of males, but also that no differences in vigour between sexes had been identified in P. tremuloides. Our results are also in agreement with those reported for P. tremuloides by Mitton & Grant [43] and Stevens & Esser [44]. Based on the current limited number of publications examining sexual dimorphism we would conclude that it is not yet possible to ascertain whether any generalisations can be formed regarding the presence or absence of sexual dimorphism for growth or defence related traits in Populus.
Several studies have additionally reported higher herbivore loads associated with increased growth in males [45-48], however we found no such reports in Populus. Although the meta-analysis presented in Cornelissen & Stiling [10] found that, in general, males suffered higher arthropod abundances, showed evidence of reduced levels of secondary metabolites and increased growth rates, it is not possible to extrapolate such generalised findings as being relevant to a specific species. Our own data identified no statistical evidence of sexual dimorphism in arthropod abundance, diversity or folivore herbivory damage in P. tremula in concordance with a lack of dimorphism in assayed growth and defence related phenotypes.
The majority of current evidence for sexual dimorphism in Populus has been identified in response to stressful environmental conditions, for example under drought, salinity [49-51], UV-B radiation [52], chilling stress [53], or differential nutrient availability [54-57] where females were found to be more sensitive. However, these studies typically used small sample sizes, in some cases being restricted to only a single individual of either sex. They also profile response to short term, acute stress exposure in most cases. This is in contrast to the approach taken here where we sample a collection of wild-growing trees. In these conditions individuals would have been exposed to various short to long-term stress events. We were interested to know whether evidence of dimorphism is present under such conditions in addition to knowing if there is evidence of sexual dimorphism for resource allocation to growth in sexually immature trees. In Salix it has been reported that evidence for sexual dimorphism varies through the growing season [11]. Such reports can lead to the general impression that sexual dimorphism is common or expected. However, bias against the publication of negative results potentially means that many such examples of a lack of dimorphism have remained unreported. The variable presence of evidence for sexual dimorphism also cautions against over-extrapolation of such results until multiple conditions and seasonal sampling points have been considered for each species and each geographic area of interest.
At both the national (SwAsp) and local (UmAsp) scales we believe that our sampling represents an unbiased representation of wild-growing mature trees, with sampling taking place with no knowledge of, or consideration for, sex or the presence of flowering. It is, of course, possible that studies testing more specific hypotheses, for example along an elevational cline (as reported for Salix [11]), may uncover evidence for shifting sex ratios or for sexual dimorphism. Indeed we see weak evidence for this within the SwAsp collection (Additional file 4) suggesting that further studies are needed in P. tremula before general conclusions can be drawn. We would caution against extrapolation of these findings beyond P. tremula growing in natural conditions within the geographic range covered by our sampling. To allow more general conclusions to be drawn for other Populus species, members of the Salicaeae and, more widely, other dioecious herbaceous species, will require equivalently detailed investigation and publication.
Environment affected gene expression more than sex
We profiled gene expression in leaves of sexually mature P. tremula individuals from the UmAsp collection to test the hypothesis that sexually dimorphic phenotypic traits would also be revealed by concomitant differential gene expression between males and females in non-reproductive tissues for genes associated with those phenotypes. In agreement with the above morphological and biochemical phenotypic results, we found no reliable evidence of large-scale sexually dimorphic (sex-biased) differential expression (Figures 3 and 4). In contrast, clear evidence of an effect of sampling collection was found (Figures 4a and 5). As samples from the two years were collected on different dates and from different heights within the canopy we cannot determine whether environmental/climatic variation between years or height in the canopy accounted for this difference. Significantly differentially expressed genes between the sample collections were over-represented for Gene Ontology (GO) biological process categories primarily involved in cellulose biosynthesis and glucan and lipid metabolism, most likely reflecting the slightly different sampling dates, with year-to-year variance in climatic conditions affecting the rate of leaf development and maturity. This exemplifies that in P. tremula leaves, changes in environmental conditions influence expression to a greater extent than the sex of an individual and that our expression data was of sufficient quality to identify biological effects influencing gene expression patterns.
The primary aim of this study was to identify patterns of sexually dimorphic gene expression associated with the morphological and biochemical traits profiled. As such, we would have expected relatively large numbers of genes to be involved should dimorphism have been present. For example, if females invest more resources into chemical defences produced via secondary metabolism, there would be corresponding sexually dimorphic differences in the expression of genes involved in secondary metabolism. Here we present gene expression results generated using P. tremula RNA-Seq read alignments to the P. trichocarpa reference genome. On the basis of a number of considered factors we do not believe that this biased our results: firstly, the vast majority - over 90% - of RNA-Seq reads aligned to the P. trichocarpa genome, suggesting that the two species have an almost entirely overlapping gene space and that sequence divergence within coding regions is not high enough to impact read alignment; secondly, we have also used a draft assembly of the P. tremula genome (available at the PopGenIE FTP resource [38]; ftp://popgenie.org/popgenie/UPSC_genomes/UPSC_Draft_Assemblies/Current/Genome/) to confirm that the vast majority of annotated CDS regions in P. trichocarpa can be aligned to the draft assembly and that analysis of the RNA-Seq data aligned to this draft genome does not produce different results; lastly, alignment of P. tremuloides and P. tremula x P. tremuloides genetic maps to the P. trichocarpa chromosomes suggests that there have been no major genome rearrangements between aspens and P. trichocarpa [24,27], although micro-synteny has not been examined to date. As such, although there may be a small number of genes unique to, or highly variable between, each species, differences between the two species are not sufficient to affect the results of global-scale expression pattern analyses. We would caution that studies aiming specifically to identify the gene(s) underlying sex determination, where genetic mapping suggests a single locus is involved and for which a single or small number of genes are likely involved, could substantially benefit from use of species-specific genome sequences.
Potri.019G047300 is absent in females and is located in the sex determination locus
The proposed peritelomeric sex determination locus on chromosome 19 of P. trichocarpa represents a region of reduced recombination [23]. Kersten et al. [24] recently provided evidence of a similar region of reduced recombination in the pericentromeric sex-linked locus of chromosome 19 in P. tremuloides. One of the two genes that we identified as being highly, and exclusively, significantly differentially expressed between sexes in the RNA-Seq data (Potri.019G047300) is located in that identified sex determination locus of P. tremuloides. It is one of seven candidate genes identified by Kersten et al. [24] on the basis of Gene Ontology and other annotation evidence as having the potential to be involved in sex determination, primarily due to annotated involvement in floral organ development. This was the only one of those seven genes with evidence of differential expression between sexes in our data (Figure 6). Pakull et al. [28] recently refined this finding, reporting a complete or partial deletion of this gene in female P. tremuloides and P. tremula individuals. Here we present independent confirmation of this finding, supported by both genomic DNA and RNA-Seq results (Figure 7). It is unclear what the biological influence of differential expression of the gene in leaves might be. Our results clearly show that this single gene did not result in any larger-scale downstream patterns of sex-biased expression and examination of expression evidence at the PopGenIE [58] org and poplar eFP resources showed that expression of this gene varies between tissues and through the growth cycle, suggesting that expression is not merely constitutively fixed in males. This is certainly a finding that deserves future attention.
Due to reduced recombination rates in sex-determination loci, all genes within a locus will, on average, be co-inherited [22]. Such a case could be identifiable as a region of the genome where a contiguous set of genes would have consistently sex-biased expression, resulting from either presence/absence differences for genes present only in the W-linked (or Y-linked) haplotype, or expression level differences for genes present in both haplotypes, but with fixed cis-acting differences between the Z and W (or X and Y) haplotypes. As the degree of expression bias may be small on a gene-by-gene basis, single gene analysis methods may lack the sensitivity to detect such differences but methods considering combinations of genes may succeed. For example, such a situation could have been possible for all seven of the candidate genes in the P. tremuloides sex determination locus discussed above. We therefore applied a machine learning approach to identify any sets of collinear genes (within sliding windows) that were predictive of sex. However, no statistically significant combinations of weakly predictive genes or synergistically predictive genes were identified.
Conclusions
We present an assessment of sex ratio and the lack of sexual dimorphism based on two independent samplings of Swedish P. tremula. Our sample of 87 was more comprehensive than almost all previous such assessments in Populus and, as such, we feel that the results obtained are an accurate representation for P. tremula. We identified no evidence that sex has served as a significant selective pressure affecting gross-scale morphological, biochemical or herbivorous insect interaction traits expected to be diagnostic of differential resource investment and allocation strategies. Correspondingly, there was no evidence for sex-biased patterns of gene expression associated with those, or any other, traits.
Although no evidence of large-scale patterns of sexually dimorphic gene expression patterns were identified, a previously identified candidate gene for sex determination in P. tremuloides [24] showed exclusive expression in males due to the homozygous absence of the locus in female individuals, an observation warranting future attention.
Methods
Phenotypic, morphological and biochemical traits
We examined the incidence of flowering in a collection of (sexually mature) wild, mature aspen (Populus tremula L.) trees in Sweden, the Umeå Aspen collection (UmAsp; [32]). In addition we used sexually immature clonal copies of trees of known sex from the Swedish Aspen (SwAsp) collection growing in a common garden experiment near Sävar, Umeå in Sweden, that were propagated and planted as described previously [33].
Umeå aspen collection (UmAsp)
Twenty-two trees bore male flowers and 20 trees bore female flowers in the spring of 2007. Tree sex was determined by visual examination of catkins and was confirmed by returning to each tree to record whether female trees retained catkins post pollination when male catkins had died. A description of trees and their geographic coordinates, together with sampling dates, is provided in Additional file 5. Tree height was measured in 2007 (when the collection was established) using a vertex dendrometer and trunk circumference was measured at breast height (1.3 m). Sampling took place on 22-25 June 2008. Six branches, each bearing approximately 60 leaves, were cut 4-5 m above ground level, in a transect from east to west across the canopy, or the nearest feasible positions, for morphological and herbivore community analyses. Sampled branches were sealed into plastic bags and kept at 4°C prior to morphological and arthropod analyses. A second sample of ten undamaged leaves from the west of the canopy was frozen in liquid nitrogen and stored at -80°C prior to RNA extraction. Following RNA extraction, samples were freeze-dried and used in assays of total phenolics and condensed tannins as described in [34] and leaf carbon and nitrogen against aspartame, wheat and atropine standards (Flash EA 1112 NC Soil Analyser, Thermo Fisher Scientific, Milan). Ten undamaged leaves were taken from each branch sample and scanned for image analysis conducted with LAMINA [59] to obtain leaf area. The same ten leaves were dried and weighed to calculate specific leaf area. From each sample bag, forty leaves were removed at random and examined for arthropod herbivore specimens and leaf modifications caused by known arthropods on aspen [34], from which arthropod species richness was calculated as the total number of morphospecies and arthropod abundance as the total number of individuals. Herbivores were also classified and summed by feeding guilds based on utilisation of the plant tissue: leaf-chewers, leaf-miners, gall-makers and leaf-rollers. Arthropod herbivore diversity was calculated for each genotype with the Shannon-Wiener index [60] using the diversity function from the package vegan [61] in R [62].
Swedish aspen collection (SwAsp)
In the Sävar common garden, clonal copies of 23 genotypes within the SwAsp collection originated from female and 24 from male trees. Sex was determined based on available flowering observations of the original trees from which the common garden trees were cloned. To determine sex, catkins were removed and examined using a binocular microscope. The cloned trees in this common garden experiment have not yet reached sexual maturity. Trees were measured annually in autumn for height with a measuring pole and for diameter at 30 cm from ground level using digital calipers. Growth rate over a five year period was calculated as (Height (2011) – Height (2006))/5, when the trees were between two and seven years old. Ten undamaged, mature leaves were harvested for measurement of leaf area and specific leaf area (leaf area/dry mass), and a further ten leaves were harvested, dried and assayed for condensed tannins and total phenolics as described in [34]. Leaf nitrogen and carbon content were analysed on an available subset of six male and six female genotypes harvested on 29 June 2010. Counts of all arthropod herbivores on each tree in the SwAsp common garden were conducted on 27 – 29 June 2008 as described in [34]. Morphospecies of folivorous arthropods were summed from the replicates of each genotype. Arthropod species richness was calculated as the sum of arthropod morphospecies on each SwAsp genotype. Arthropod herbivore diversity (Shannon-Wiener index), abundance, species richness and feeding guild abundances were calculated on each genotype using the same methods as the UmAsp samples. Details of clone geographic origins, sex, replication in the common garden and phenotypic data collected are provided in Additional file 5.
Statistical analysis
Statistical analyses were conducted and figures generated in R conducted [R Core Development Team reference]. Statistical significance for all tests was determined at α ≤0.05. Dependent variables (tree phenotypes) were tested for normality and homogeneity of variance using Anderson-Darling and equal variance (Bartlett) tests to meet the assumptions of analysis of variance (ANOVA). Where transformation using Box-Cox powers or log-transformation did not result in improvement of the distribution of a dependent variable, a two-tailed Mann–Whitney U-test was applied. In SwAsp, the latitude of origin for each genotype was initially applied as a covariate, to account for phenotypic variation associated with latitude, however no significant effect of sex was identified for any response variable (P >0.1), therefore final analyses were conducted without a covariate. ANOVA or Mann–Whitney U-tests tested the effect of sex (independent variable) on each phenotypic trait (response variable). To test for potential environmental influences partitioned by sex (independent variable) in UmAsp trees, the response variables latitude, longitude, and elevation were used in separate one-way ANOVAs but sex had no significant effect on the responses (P >0.5 in all cases), therefore environmental factors were not considered necessary in analyses of phenotypic traits. In each of UmAsp and SwAsp, arthropod community composition was compared between male and female trees using non-parametric multivariate analysis of variance (npMANOVA; [63]). A Bray-Curtis dissimilarity matrix constructed from counts of arthropod herbivores on aspen genotypes (response variable) and tested for effects of tree sex (independent variable) using npMANOVA in the adonis function implemented in the R package vegan [61]. The p-value for significance was determined from 999 permutations of the data matrix.
Gene expression analysis
Sample collection for microarray and RNA-Seq analysis
Sample collection from the UmAsp trees is described above and sample details are given in Additional file 5. Briefly, ten mature leaves produced from pre-formed, overwintered buds were collected per tree, from ten male and ten female trees on June 29 2009 and used to perform whole genome oligonucleotide microarray hybridisations. For RNA-Seq analysis we used a combination of a set of samples that had been collected in 2008 (five male and five female individuals collected 22-25 June) and additional samples collected in 2010 (three male and four female individuals collected 11 August). All samples consist of pools of ten leaves collected from ten buds (one leaf per bud avoiding the first and last emergent leaf) collected by removing a length of branch from either the base of the tree canopy (2009 and 2010 samples) or from a branch at a height of 4-5 m (2008 samples).
RNA extraction
Total RNA was extracted from 0.5 g tissue using a modified version of the CTAB method [64] as described in [65]. Briefly, the ten sampled leaves were ground under liquid nitrogen using a pestle and mortar and 0.5 g of ground material was then used for RNA extraction. Precipitated RNA was further purified using an RNeasy Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s protocol. RNA concentration and purity were measured using a NanoDrop 2000 spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA) and integrity was analysed on an Agilent 2100 Bioanalyzer (Agilent Technologies, Waldbronn, Germany). For each set of samples (i.e. all samples used for microarray or RNA-Seq analysis) all RNA extractions were performed together on the same day with the order of male and female samples randomised.
Microarray hybridisation and analysis
We used the Agilent v1.0 4x44k Populus gene expression oligonucleotide microarray (Agilent Technologies, Waldbronn, Germany), as detailed in the Gene Expression Omnibus platform ID GPL16040. We used the cDNA synthesis, amplification, microarray hybridisation and washing protocols supplied by Agilent (Agilent Technologies, Waldbronn, Germany) with no modifications. All hybridisations were performed using only one sample and using Cy3. Ten male and ten female individuals were profiled and the respective samples were randomised on arrays with two male and female samples run on each slide and with the position of males and females randomised between the four array sections per array slide. Arrays were scanned at 5 μm resolution, using a Scanarray 4000 microarray analysis system scanner (Perkin-Elmer, Boston, MA, USA). Spot data were extracted using GenePix (v5, Axon Instruments Inc, Union City, CA, USA). Microarray normalisation and analyses were performed using the Bioconductor [66] limma package [67] in R [62]. Microarray annotations were obtained from the PopArray resource [37] and were based on V2 of the genome annotation. The microarrays were first background corrected using the normexp method implemented in the backgroundCorrect function. Then, a between microarray quantile normalisation was performed using the normalizeBetweenArrays function. A Principle Component Analysis (PCA) plot was used for quality control and this identified one sub array assaying a female individual as a clear outlier and this sample was therefore eliminated and not used for the statistical analyses. These were conducted by fitting a linear model taking into account batch effects for slide and position of sub-array within slide to the data in order to identify genes with a high probability of differential expression between sexes. FDR-adjusted P values were used to assess the significance of differential expression.
RNA sequencing and analysis
Total RNA preparations were sent to the Science for Life Laboratory (SciLifeLab, Stockholm, Sweden) for sequencing. Paired-end (2 × 100 bp) RNA-Seq data were generated using standard Illumina protocols and kits (TruSeq SBS KIT-HS v3, FC-401-3001; TruSeq PE Cluster Kit v3, PE-401-3001) and all sequencing was performed using the Illumina HiSeq 2000 platform. We generated data from 8 male individuals (five sampled in 2008 and three in 2010) and 9 female individuals (five sampled in 2008 and four in 2010). For sequencing, samples were recoded (from 1-17) with males and females randomised to avoid bias due to sample handling order. Samples were multiplexed by the addition of a unique barcode sequence and all samples were profiled on two lanes of the same flowcell with male and female samples and samples from 2008 and 2010 randomised between the two lanes. Briefly, the sequencing protocol involved DNase 1 digestion of total RNA, mRNA isolation by use of oligo(dT) beads, mRNA fragmentation, first and second strand cDNA synthesis, end-repair, A-tailing, bar-coded adapter ligation and PCR amplification. Sequencing libraries were quality checked using an Agilent 2100 Bioanalyzer (Agilent Technologies, Waldbronn, Germany) before sequencing. The quality of the raw sequence data was assessed using FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Data were then filtered to remove adapters and trimmed for quality using Trimmomatic (v0.32; [68]; settings TruSeq3-PE-2.fa:2:30:10 LEADING:3 SLIDINGWINDOW:5:20 MINLEN:50). Residual ribosomal RNA (rRNA) contamination was assessed and filtered using SortMeRNA (v1.9; [69]; settings -n 6 -a 8 -v) using the rRNA sequences provided with SortMeRNA (rfam-5 s-database-id98.fasta, rfam-5.8 s-database-id98.fasta, silva-bac-16 s-database-id85.fasta, silva-euk-18 s-database-id95.fasta, silva-bac-23 s-database-id98.fasta and silva-euk-28 s-database-id98.fasta). After both filtering steps, FastQC was run again to ensure that no technical artefacts were introduced. Filtered reads were aligned to v3.0 of the P. trichocarpa genome (retrieved from the Phytozome [70] resource) using STAR (v2.3.1e [71]; non default settings: --OutQSconversion -31 --outReadsUnmapped Fastx --alignIntronMax 11000). The annotations obtained from the P. trichocarpa v3.0 GFF file were modified to generate ‘synthetic’ gene models; i.e. for each gene a non-redundant set of all exons from all transcripts was defined, with overlapping exons merged where necessary. This gene-model GFF file and the OSA read alignments were used as input to the HTSeq (http://www-huber.embl.de/users/anders/HTSeq/doc/overview.html) htseq-count python utility to calculate exon-based read count values. The htseq-count utility takes only uniquely mapping reads into account. Statistical analysis of single-gene differential expression between sexes was performed in R (v3.1.0 [62]) using the Bioconductor (v2.14 [66]) DESeq and DESeq2 packages (v1.16.0 [72] and v1.4.5 [73]). For the DESeq/DESeq2 analyses, a two-factor linear model was fitted with the factors Sex and Year where Year was included as a blocking factor and the effect of Sex was tested after removal of the Year effect. FDR adjusted p-values were used to assess significance. The normalised read counts obtained from DESeq2 were used for all subsequent expression analyses, e.g. PCA, which were performed in R, with the exception of the differential gene expression analyses, which were performed using DESeq as it has been shown to be the most conservative of the currently available methods with the lowest false discovery rate [74]. An overview of the data, including raw and post-QC read counts and alignment rates is given in Additional file 6.
We analysed the RNA-Seq dataset using read alignments to both v2.0 and v3.0 of the P. trichocarpa genome assembly and annotation, yielding similar results in both cases. Similarly we analysed the microarray dataset using probe annotations based on v1.0 and v2.0 of the genome and assembly with similar gene-level results in both cases. We have also analysed the microarray data at the probe level, again yielding similar results.
Support vector machine identification of sex-predictive gene combinations
We used both the microarray data and normalised RNA-Seq expression values to test for the presence of contiguous gene combinations (i.e. windows of genes located next to each other within the genome) that were predictive of sex. We applied a sliding window across the genome with a window size of 10 genes (other window sizes were also tested with similar results). In total our expression data included 30,709 and 20,557 genes in the RNA-Seq and microarray datasets, respectively. The criterion for accepting a gene inside a window was that it had at least 5 samples with non-zero expression values. Furthermore, only windows with at least 4 accepted genes were included. The Python module scikit-learn [75] was used to train SVMs with a radial basis function (RBF) kernel parameterised by C and γ. This approach has previously been shown effective on gene expression data [76]. Since the optimal values of these parameters are not known prior to training, a grid search was performed in a parameter space consisting of γ = {10− 4, 5 ⋅ 10− 4, 10− 3, 5 ⋅ 10− 3, 10− 2, 10− 1, 1} and C = {1, 10, 103, 5 ⋅ 103, 104, 5 ⋅ 104, 105}. For each genomic window, a double cross validation (CV) was performed where the outer CV was a leave-one-out and the inner was a 2-fold CV. The inner CV was used to train the SVM (i.e. estimate the parameters), and parameters with the smallest prediction error were used to predict the test data from the outer CV. The error rate was measured as the fraction of incorrect sex predictions. To validate the error rates, a permutation test was performed where 10,000 random genomic windows from all scaffolds were used in the same machine learning approach, but where the sex assignments were shuffled.
Availability of supporting information
Microarray data has been deposited to the Gene Expression Omnibus (GEO) under the accession ID GSE46219. Raw RNA-Seq data has been deposited to the European Nucleotide Archive (ENA) under the accession ID ERP002471.
Raw RNA-Seq fastq, the synthetic exon GFF3 file used for read alignment and HTSeq analysis, read alignment BAM files and other associated outputs from the gene expression analysis can be downloaded from the PopGenIE (Populus Genome Integrative Explorer; [58]) FTP resource [38]). The FTP site includes RData files for both gene expression datasets as well as an HTML transcript of the analyses performed, which we highly encourage readers to examine as all analysis details are included in addition to a number of summary plots exploring the dataset. To facilitate future meta-analyses, all phenotype data used in this study is also available at the FTP site. The data pre-processing source code is available through our public git repository accessible at https://bioinformatics.upsc.se. The RNA-Seq expression data presented here has been integrated in the exImage and exPlot expression visualisation tools at PopGenIE.org [58], where they are called the “Expression diversity (RNASeq)” dataset.
Acknowledgements
We thank Yvan Fracheboud for use of data on flowering in the UmAsp collection, Agneta Olsson for assistance in collecting samples used for the arthropod analysis. This work was supported by funds from the Swedish Research Council (VR), the Swedish Governmental Agency for Innovation Systems (VINNOVA), The Swedish Research Council (FORMAS) and, in parts, through the UPSC Berzelii Centre for Forest Biotechnology. NRS is supported by the Trees and Crops for the Future (TC4F) project.
Abbreviations
- cDNA
Complementary DNA
- CDS
Coding DNA sequence
- DNA
Deoxyribonucleic acid
- ENA
European nucleotide archive
- FTP
File transfer protocol
- GEO
Gene expression omnibus
- GFF
General feature format
- GO
Gene ontology
- PCA
Principal component analysis
- QA
Quality assessment
- rRNA
Ribosomal RNA
- RNA
Seq – RNA-sequencing
- RNA
Ribonucleic acid
- SVM
Support vector machines
- SwAsp
Swedish aspen
- UmAsp
Umeå aspen
- VST
Variance stabilising transformation
Additional files
Statistical analyses of phenotypic and biochemical traits in the UmAsp and SwAsp samples. Phenotypic trait means and standard deviations for female and male individuals from the UmAsp collection (sheet1) and the SwAsp collection (sheet2), with results of one-way ANalyses Of VAriance (ANOVA). Where data could not be transformed to meet the assumptions of variance structure for ANOVA, a Mann–Whitney U test was conducted. For the extended phenotype of the arthropod community, non-parametric MANOVA (npMANOVA) results are shown for each of the UmAsp collection (sheet 1) and SwAsp collection (sheet 2).
PDF image containing dot plot representations of per-sample expression values of the four genes with the smallest p values (regardless of significance) when testing for the effect of sex (top row), the four genes with the highest fold-change between males and females (middle row) and the lowest fold change (bottom row). Bold text gene identifiers in the top row of plots indicate the two statistically significant genes. The genes represented in these figures are those circled in red in Figure 4b. Expression values represent variance stabilising transformation normalised read counts derived using HTSeq and DESeq2. Black lines represent the median expression value per sex. For each gene male and female samples are plotted separately with males represented by blue dots and females by pink dots. The position along the x-axis of the plot has no meaning and merely separates male from female samples. Note that the y-axis is a log scale, for which a pseudo count was added to every value to avoid infinite values from the log transformation.
PDF file containing individual plots of per base pair read coverage for reads aligning uniquely to the Potri.019G047300 locus.
PDF file containing plots further exploring sex ratio of individuals and populations in the Swedish Aspen collection in relation to elevation, latitude and marker based population structure.
Sex, longitude and latitude of clone origin for UmAsp and SwAsp, and elevation for UmAsp, samples used in the current study. The year of sampling for phenotype, microarray and RNA-Seq analysis is indicated. For UmAsp trees the longitude, latitude and elevation values represent the location of the actual tree sampled. For SwAsp samples they represent the origin of the original clone that was used to establish the clonal common garden experiment at the Skogforsk research station, Sävar, near Umeå, (63.896054°N, 20.549321°E). All UmAsp clones flowered in 2007. For the SwAsp samples, the number of clonal replicates present in the common garden is shown.
Overview of RNA-Seq data including quality control metrics and correspondence between sample and ENA submission IDs.
Footnotes
Kathryn M Robinson and Nicolas Delhomme contributed equally to this work.
Competing interests
All authors declare that they have no competing interests.
Authors’ contributions
KMR and NRS collected all leaf samples. KMR performed all morphological, biochemical and herbivore analyses. NRS performed all RNA extractions and microarray hybridisations. ND and NM performed the RNASeq and microarray expression analyses. NM, JÖ and TRH performed the machine learning analyses. BS performed the analysis of read alignments for the TOZ gene. SJ, PI and BA supervised and designed the project, which was originally conceived by SJ. NRS, KMR and ND prepared the manuscript with assistance from all authors. All authors approved the final manuscript.
Contributor Information
Kathryn M Robinson, Email: kathryn.robinson@umu.se.
Nicolas Delhomme, Email: nicolas.delhomme@umu.se.
Niklas Mähler, Email: niklas.mahler@nmbu.no.
Bastian Schiffthaler, Email: basc0003@student.umu.se.
Jenny Önskog, Email: jenny.onskog@umu.se.
Benedicte R Albrectsen, Email: benedicte.albrecsten@umu.se.
Pär K Ingvarsson, Email: par.ingvarsson@emg.umu.se.
Torgeir R Hvidsten, Email: torgeir.r.hvidsten@nmbu.no.
Stefan Jansson, Email: stefan.jansson@umu.se.
Nathaniel R Street, Email: nathaniel.street@umu.se.
References
- 1.Parsch J, Ellegren H. The evolutionary causes and consequences of sex-biased gene expression. Nat Rev Genet. 2013;14:83–87. doi: 10.1038/nrg3376. [DOI] [PubMed] [Google Scholar]
- 2.Ainsworth C. Boys and girls come out to play: the molecular biology of dioecious plants. Ann Bot. 2000;86:211–221. doi: 10.1006/anbo.2000.1201. [DOI] [Google Scholar]
- 3.Heslop-Harrison JSP, Schwarzacher T. Organisation of the plant genome in chromosomes. Plant J. 2011;66:18–33. doi: 10.1111/j.1365-313X.2011.04544.x. [DOI] [PubMed] [Google Scholar]
- 4.Charlesworth D. Plant sex chromosome evolution. J Exp Bot. 2013;64:405–420. doi: 10.1093/jxb/ers322. [DOI] [PubMed] [Google Scholar]
- 5.Williams T, Carroll S. Genetic and molecular insights into the development and evolution of sexual dimorphism. Nat Rev Genet. 2009;10:797–804. doi: 10.1038/nrg2687. [DOI] [PubMed] [Google Scholar]
- 6.Diggle PK, Di Stilio VS, Gschwend AR, Golenberg EM, Moore RC, Russell JRW, Sinclair JP. Multiple developmental processes underlie sex differentiation in angiosperms. Trends Genet. 2011;27:368–376. doi: 10.1016/j.tig.2011.05.003. [DOI] [PubMed] [Google Scholar]
- 7.Obeso J. The costs of reproduction in plants. New Phytol. 2002;155:321–348. doi: 10.1046/j.1469-8137.2002.00477.x. [DOI] [PubMed] [Google Scholar]
- 8.Shine R. Ecological causes for the evolution of sexual dimorphism: a review of the evidence. Q Rev Biol. 1989;64:419–461. doi: 10.1086/416458. [DOI] [PubMed] [Google Scholar]
- 9.Lloyd D, Webb CJ. Secondary sex characters in plants. Bot Rev. 1977;43:177–216. doi: 10.1007/BF02860717. [DOI] [Google Scholar]
- 10.Cornelissen T, Stiling P. Sex-biased herbivory: a meta-analysis of the effects of gender on plant-herbivore interactions. Oikos. 2005;111:488–500. doi: 10.1111/j.1600-0706.2005.14075.x. [DOI] [Google Scholar]
- 11.Dudley LS. Ecological correlates of secondary sexual dimorphism in Salix glauca (Salicaceae) Am J Bot. 2006;93:1775–1783. doi: 10.3732/ajb.93.12.1775. [DOI] [PubMed] [Google Scholar]
- 12.Jansson S, Douglas CJ. Populus: a model system for plant biology. Annu Rev Plant Biol. 2007;58:435–458. doi: 10.1146/annurev.arplant.58.032806.103956. [DOI] [PubMed] [Google Scholar]
- 13.Tuskan GA, Difazio S, Jansson S, Bohlmann J, Grigoriev I, Hellsten U, Putnam N, Ralph S, Rombauts S, Salamov A, Schein J, Sterck L, Aerts A, Bhalerao RR, Bhalerao RP, Blaudez D, Boerjan W, Brun A, Brunner A, Busov V, Campbell M, Carlson J, Chalot M, Chapman J, Chen G-L, Cooper D, Coutinho PM, Couturier J, Covert S, Cronk Q, et al. The genome of black cottonwood, Populus trichocarpa (Torr. & Gray) Science. 2006;313:1596–1604. doi: 10.1126/science.1128691. [DOI] [PubMed] [Google Scholar]
- 14.Wullschleger SD, Weston DJ, DiFazio SP, Tuskan GA. Revisiting the sequencing of the first tree genome: populus trichocarpa. Tree Physiol. 2013;33:357–364. doi: 10.1093/treephys/tps081. [DOI] [PubMed] [Google Scholar]
- 15.Bradshaw HD, Ceulemans R, Davis J, Stettler R. Emerging model systems in plant biology: poplar (Populus) as a model forest tree. J Plant Growth Regul. 2000;19:306–313. doi: 10.1007/s003440000030. [DOI] [Google Scholar]
- 16.Pulford ID, Watson C. Phytoremediation of heavy metal-contaminated land by trees–a review. Environ Int. 2003;29:529–540. doi: 10.1016/S0160-4120(02)00152-6. [DOI] [PubMed] [Google Scholar]
- 17.Whitham TG, DiFazio SP, Schweitzer JA, Shuster SM, Allan GJ, Bailey JK, Woolbright SA. Extending genomics to natural communities and ecosystems. Science (80-) 2008;320:492–495. doi: 10.1126/science.1153918. [DOI] [PubMed] [Google Scholar]
- 18.Latva-Karjanmaa T, Suvanto L, Leinonen K, Rita H: Sexual reproduction of european aspen (Populus tremula L.) at prescribed burned site: the effects of moisture conditions.New For 2006, 31:14.
- 19.Boes TK, Strauss SH. Floral phenology and morphology of black cottonwood, Populus trichocarpa (Salicaceae) Am J Bot. 1994;81:562–567. doi: 10.2307/2445730. [DOI] [Google Scholar]
- 20.Lester DT. Variation in sex expression in Populus tremuloides Michx. Silvae Genet. 1963;12:141–151. [Google Scholar]
- 21.Rowland DL, Garner ER, Jespersen M. A rare occurrence of seed formation on male branches of the dioecious tree, populus deltoides. Am Midl Nat. 2002;147:185–187. doi: 10.1674/0003-0031(2002)147[0185:AROOSF]2.0.CO;2. [DOI] [Google Scholar]
- 22.Tuskan GA, DiFazio S, Faivre-Rampant P, Gaudet M, Harfouche A, Jorge V, Labbé JL, Ranjan P, Sabatti M, Slavov G, Street N, Tschaplinski TJ, Yin T. The obscure events contributing to the evolution of an incipient sex chromosome in Populus: a retrospective working hypothesis. Tree Genet Genomes. 2012;8:559–571. doi: 10.1007/s11295-012-0495-6. [DOI] [Google Scholar]
- 23.Yin T, DiFazio SP, Gunter LE, Zhang X, Sewell MM, Woolbright SA, Allan GJ, Kelleher CT, Douglas CJ, Wang M, Tuskan GA. Genome structure and emerging evidence of an incipient sex chromosome in Populus. Genome Res. 2008;18:422–430. doi: 10.1101/gr.7076308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kersten B, Pakull B, Groppe K, Lueneburg J, Fladung M. The sex-linked region in Populus tremuloides Turesson 141 corresponds to a pericentromeric region of about two million base pairs on P. trichocarpa chromosome 19. Plant Biol (Stuttg) 2014;16:411–418. doi: 10.1111/plb.12048. [DOI] [PubMed] [Google Scholar]
- 25.Paolucci I, Gaudet M, Jorge V, Beritognolo I, Terzoli S, Kuzminsky E, Muleo R, Scarascia Mugnozza G, Sabatti M. Genetic linkage maps of Populus alba L. and comparative mapping analysis of sex determination across Populus species. Tree Genet Genomes. 2010;6:863–875. doi: 10.1007/s11295-010-0297-7. [DOI] [Google Scholar]
- 26.Pakull B, Groppe K, Meyer M, Markussen T, Fladung M. Genetic linkage mapping in aspen (Populus tremula L. and Populus tremuloides Michx.) Tree Genet Genomes. 2009;5:505–515. doi: 10.1007/s11295-009-0204-2. [DOI] [Google Scholar]
- 27.Pakull B, Groppe K, Mecucci F, Gaudet M, Sabatti M, Fladung M. Genetic mapping of linkage group XIX and identification of sex-linked SSR markers in a Populus tremula × Populus tremuloides cross. Can J For Res. 2011;41:245–253. doi: 10.1139/X10-206. [DOI] [Google Scholar]
- 28.Pakull B, Kersten B, Lüneburg J, Fladung M: A simple PCR-based marker to determine sex in aspen.Plant Biol (Stuttg) 2014, doi:10.1111/plb.12217. [DOI] [PubMed]
- 29.Zluvova J, Zak J, Janousek B, Vyskot B: Dioecious Silene latifolia plants show sexual dimorphism in the vegetative stage.BMC Plant Biol 2010, 10:208. [DOI] [PMC free article] [PubMed]
- 30.Osier T, Lindroth R. Effects of genotype, nutrient availability, and defoliation on aspen phytochemistry and insect performance. J Chem Ecol. 2001;27:1289–1313. doi: 10.1023/A:1010352307301. [DOI] [PubMed] [Google Scholar]
- 31.Boeckler A, Gershenzon J, Unsicker S. Phenolic glycosides of the Salicaceae and their role as anti-herbivore defenses. Phytochemistry. 2011;72:1497–1509. doi: 10.1016/j.phytochem.2011.01.038. [DOI] [PubMed] [Google Scholar]
- 32.Fracheboud Y, Luquez V, Bjorken L, Sjodin A, Tuominen H, Jansson S. The control of autumn senescence in European aspen. Plant Physiol. 2009;149:1982–1991. doi: 10.1104/pp.108.133249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Luquez V, Hall D, Albrectsen BR, Karlsson J, Ingvarsson P, Jansson S. Natural phenological variation in aspen (Populus tremula): the SwAsp collection. Tree Genet Genomes. 2007;4:279–292. doi: 10.1007/s11295-007-0108-y. [DOI] [Google Scholar]
- 34.Robinson K, Ingvarsson P, Jansson S, Albrectsen B: Genetic variation in functional traits influences arthropod community xomposition in aspen (Populus tremula L.).PLoS One 2012, 7:e37679. [DOI] [PMC free article] [PubMed]
- 35.Griffith ME, Mayer U, Capron A, Ngo QA, Surendrarao A, McClinton R, Jürgens G, Sundaresan V. The TORMOZ gene encodes a nucleolar protein required for regulated division planes and embryo development in Arabidopsis. Plant Cell. 2007;19:2246–2263. doi: 10.1105/tpc.106.042697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wilkins O, Nahal H, Foong J, Provart NJ, Campbell MM. Expansion and diversification of the Populus R2R3-MYB family of transcription factors. Plant Physiol. 2009;149:981–993. doi: 10.1104/pp.108.132795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tsai CJ, Ranjan P, DiFazio SP, Tuskan GA, Johnson VE, Joshi CP. Poplar genome microarrays. In: Joshi CP, editor. Genet Genomics Breed Poplar. CRC Press Boca Raton: Science Publishers, Inc; 2011. pp. 112–127. [Google Scholar]
- 38.PopGenIE (Populus Genome Integrative Explorer) File Transfer Protocol site. (ftp://popgenie.org/popgenie).
- 39.Price P: The plant vigor hypothesis and herbivore attack.Oikos 1991, 62:244.
- 40.Pauley SS. Sex and vigor in Populus. Science (80-) 1948;108:302–303. doi: 10.1126/science.108.2803.302-b. [DOI] [PubMed] [Google Scholar]
- 41.Petzold A, Pfeiffer T, Jansen F, Eusemann P, Schnittler M. Sex ratios and clonal growth in dioecious Populus euphratica Oliv., Xinjiang Prov., Western China. Trees. 2012;27:729–744. doi: 10.1007/s00468-012-0828-y. [DOI] [Google Scholar]
- 42.Farmer RE. Sex ratio and sex-related characteristics in eastern cottonwood. Silvae Genet. 1964;13:116–118. [Google Scholar]
- 43.Mitton JB, Grant MC: Observations on the ecology and evolution of quaking aspen, populus tremuloides, in the Colorado Front Range.Am J Bot 1980, 67:202.
- 44.Stevens M, Esser S. Growth–defense tradeoffs differ by gender in dioecious trembling aspen (Populus tremuloides) Biochem Syst Ecol. 2009;37:567–573. doi: 10.1016/j.bse.2009.09.005. [DOI] [Google Scholar]
- 45.Jing S, Coley P: Dioecy and herbivory: the effect of growth rate on plant defense in Acer Negundo.Oikos 1990, 58:369.
- 46.Boecklen WJ, Price PW, Mopper S. Sex and drugs and herbivores: sex-biased herbivory in Arroyo Willow (Salix Lasiolepis) Ecology. 1990;71:581–588. doi: 10.2307/1940311. [DOI] [Google Scholar]
- 47.Hjältén J. Plant sex and hare feeding preferences. Oecologia. 1992;89:253–256. doi: 10.1007/BF00317225. [DOI] [PubMed] [Google Scholar]
- 48.Boecklen W, Hoffman T. Sex-biased herbivory in Ephedra trifurca: the importance of sex-by-environment interactions. Oecologia. 1993;96:49–55. doi: 10.1007/BF00318030. [DOI] [PubMed] [Google Scholar]
- 49.Jiang H, Peng S, Zhang S, Li X, Korpelainen H, Li C. Transcriptional profiling analysis in Populus yunnanensis provides insights into molecular mechanisms of sexual differences in salinity tolerance. J Exp Bot. 2012;63:3709–3726. doi: 10.1093/jxb/ers064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xu X, Yang F, Xiao X, Zhang S, Korpelainen H, Li C. Sex-specific responses of Populus cathayana to drought and elevated temperatures. Plant Cell Environ. 2008;31:850–860. doi: 10.1111/j.1365-3040.2008.01799.x. [DOI] [PubMed] [Google Scholar]
- 51.Chen F, Chen L, Zhao H, Korpelainen H, Li C. Sex-specific responses and tolerances of Populus cathayana to salinity. Physiol Plant. 2010;140:163–173. doi: 10.1111/j.1399-3054.2010.01393.x. [DOI] [PubMed] [Google Scholar]
- 52.Xu X, Zhao H, Zhang X, Hänninen H, Korpelainen H, Li C. Different growth sensitivity to enhanced UV-B radiation between male and female Populus cathayana. Tree Physiol. 2010;30:1489–1498. doi: 10.1093/treephys/tpq094. [DOI] [PubMed] [Google Scholar]
- 53.Zhang S, Jiang H, Peng S, Korpelainen H, Li C. Sex-related differences in morphological, physiological, and ultrastructural responses of Populus cathayana to chilling. J Exp Bot. 2011;62:675–686. doi: 10.1093/jxb/erq306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Randriamanana TR, Nybakken L, Lavola A, Aphalo PJ, Nissinen K, Julkunen-Tiitto R. Sex-related differences in growth and carbon allocation to defence in Populus tremula as explained by current plant defence theories. Tree Physiol. 2014;34:471–487. doi: 10.1093/treephys/tpu034. [DOI] [PubMed] [Google Scholar]
- 55.Zhao H, Li Y, Zhang X, Korpelainen H, Li C. Sex-related and stage-dependent source-to-sink transition in Populus cathayana grown at elevated CO(2) and elevated temperature. Tree Physiol. 2012;32:1325–1338. doi: 10.1093/treephys/tps074. [DOI] [PubMed] [Google Scholar]
- 56.Wang X, Curtis P. Gender-specific responses of Populus tremuloides to atmospheric CO2 enrichment. New Phytol. 2001;150:675–684. doi: 10.1046/j.1469-8137.2001.00138.x. [DOI] [Google Scholar]
- 57.Li L, Zhang Y, Luo J, Korpelainen H, Li C. Sex-specific responses of Populus yunnanensis exposed to elevated CO2 and salinity. Physiol Plant. 2013;147:477–488. doi: 10.1111/j.1399-3054.2012.01676.x. [DOI] [PubMed] [Google Scholar]
- 58.Sjödin A, Street NR, Sandberg G, Gustafsson P, Jansson S. The populus genome integrative explorer (PopGenIE): a new resource for exploring the Populus genome. New Phytol. 2009;182:1013–1025. doi: 10.1111/j.1469-8137.2009.02807.x. [DOI] [PubMed] [Google Scholar]
- 59.Bylesjö M, Segura V, Soolanayakanahally RY, Rae AM, Trygg J, Gustafsson P, Jansson S, Street NR: LAMINA: a tool for rapid quantification of leaf size and shape parameters.BMC Plant Biol 2008, 8:82. [DOI] [PMC free article] [PubMed]
- 60.Whittaker RH. Evolution and measurement of species diversity. Taxon. 1972;21:213–251. doi: 10.2307/1218190. [DOI] [Google Scholar]
- 61.Dixon P. VEGAN, a package of R functions for community ecology. J Veg Sci. 2003;14:927–930. doi: 10.1111/j.1654-1103.2003.tb02228.x. [DOI] [Google Scholar]
- 62.R: A Language and Environment for Statistical Computing. (http://www.r-project.org).
- 63.Anderson MJ. A new method for non-parametric multivariate analysis of variance. Austral Ecol. 2001;26:32–46. [Google Scholar]
- 64.Chang S, Puryear J, Cairney J. A simple and efficient method for isolating RNA from pine trees. Plant Mol Biol Report. 1993;11:113–116. doi: 10.1007/BF02670468. [DOI] [Google Scholar]
- 65.Street NR, Skogström O, Sjödin A, Tucker J, Rodríguez-Acosta M, Nilsson P, Jansson S, Taylor G. The genetics and genomics of the drought response in Populus. Plant J. 2006;48:321–341. doi: 10.1111/j.1365-313X.2006.02864.x. [DOI] [PubMed] [Google Scholar]
- 66.Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, Hornik K, Hothorn T, Huber W, Iacus S, Irizarry R, Leisch F, Li C, Maechler M, Rossini AJ, Sawitzki G, Smith C, Smyth G, Tierney L, Yang JYH, Zhang J: Bioconductor: open software development for computational biology and bioinformatics.Genome Biol 2004, 5:R80. [DOI] [PMC free article] [PubMed]
- 67.Smyth G. limma: Linear Models for Microarray Data. In: Gentleman R, Carey VJ, Huber W, Irizarry RA, Dudoit S, editors. Bioinforma Comput Biol Solut Using R Bioconductor. New York: Springer; 2005. pp. 397–420. [Google Scholar]
- 68.Bolger AM, Lohse M, Usadel B: Trimmomatic: a flexible trimmer for Illumina sequence data.Bioinformatics 2014, 30:btu170. [DOI] [PMC free article] [PubMed]
- 69.Kopylova E, Noé L, Touzet H. SortMeRNA: fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics. 2012;28:3211–3217. doi: 10.1093/bioinformatics/bts611. [DOI] [PubMed] [Google Scholar]
- 70.Goodstein D, Shu S, Howson R, Neupane R, Hayes R, Fazo J, Mitros T, Dirks W, Hellsten U, Putnam N, Rokhsar D. Phytozome: a comparative platform for green plant genomics. Nucleic Acids Res. 2012;40(Database issue):D1178–D1186. doi: 10.1093/nar/gkr944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Anders S, Huber W: Differential expression analysis for sequence count data.Genome Biol 2010, 11:R106. [DOI] [PMC free article] [PubMed]
- 73.Love MI, Huber W, Anders S: Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Cold Spring Harbor Labs Journals; 2014. bioRxiv 2014 doi:10.1101/002832. [DOI] [PMC free article] [PubMed]
- 74.Soneson C, Delorenzi M: A comparison of methods for differential expression analysis of RNA-seq data.BMC Bioinformatics 2013, 14:91. [DOI] [PMC free article] [PubMed]
- 75.Pedregosa F, Varoquaux G, Gramfort A, Michel V, Thirion B, Grisel O, Blondel M, Prettenhofer P, Weiss R, Dubourg V, Vanderplas J, Passos A, Cournapeau D, Brucher M, Perrot M, Duchesnay E. Scikit-learn: machine learning in Python. J Mach Learn Res. 2011;12:2825–2830. [Google Scholar]
- 76.Önskog J, Freyhult E, Landfors M, Rydén P, Hvidsten TR: Classification of microarrays; synergistic effects between normalization, gene selection and machine learning.BMC Bioinformatics 2011, 12:390. [DOI] [PMC free article] [PubMed]