

Summary
The proper folding of proteins is continuously challenged by intrinsic and extrinsic stresses, and the accumulation of toxic misfolded proteins is associated with many human diseases. Eukaryotic cells have evolved a complex network of protein quality control pathways to protect the proteome, and these pathways are specialized for each subcellular compartment. While many details have been elucidated for how the cytosol and endoplasmic reticulum counteract proteotoxic stress, relatively little is known about the pathways protecting the nucleus from protein misfolding. Here, we offer a conceptual framework for how proteostasis is maintained in this organelle. We define the particular requirements that must be considered for the nucleus to manage proteotoxic stress, summarize the known and implicated pathways of nuclear protein quality control, and identify the unresolved questions in the field. Proper maintenance of nuclear proteostasis has important implications in preserving genomic integrity, as well as for aging and disease.
Introduction
Proteins are the essential ‘workhorses’ in the cell that must fold into unique three-dimensional structures to properly function for all aspects of cell growth and vitality [1]. A multitude of proteotoxic stresses, including genetic mutations, biosynthetic errors, and physiological and environmental insults, constantly challenge the proper folding and function of the proteome. Many of these proteotoxic stresses are compounded by age, and aberrantly folded proteins are associated with a variety of diseases, including type II diabetes, cancer, and many neurodegenerative diseases [2]. To counteract this, cells have evolved elaborate pathways to protect against protein misfolding and aggregation to maintain protein homeostasis (proteostasis). These pathways are collectively called the proteostasis network, and include machineries that maintain functional protein conformations folding, assembly, and disaggregation mechanisms; clearance pathways that recognize and dispose of terminally misfolded proteins; as well as secondary defense mechanisms that minimize protein aggregate toxicity (Figure 1; [2]). The relative amounts of these protein quality control (PQC) machineries are controlled by adaptive stress responses, which transcriptionally tune the cell’s folding capacity under fluctuating proteotoxic stress conditions [3–5].
Figure 1. The proteostasis network maintains a functional proteome.

Molecular chaperones and the ubiquitin-proteasome system (UPS) cooperate in pathways of protein folding, refolding, disaggregation, and degradation. At the cellular level, the accumulation of protein aggregates is also managed by autophagic degradation, mitotic clearance, and physical sequestration pathways.
Eukaryotic cells are physically and functionally compartmentalized by membrane-bound organelles, and PQC pathways have become specialized for specific compartments, including the cytoplasm, endoplasmic reticulum (ER), and mitochondria [6, 7]. Many illuminating studies have begun to precisely define how proteostasis in these compartments is maintained. Surprisingly, relatively less is known about proteostasis in the nucleus, although this organelle has a critical role in cellular homeostasis by protecting genomic expression and integrity. The importance of understanding nuclear protein folding and quality control mechanisms is underscored not only by their implied responsibility in maintaining the functionality of proteins that control gene expression fidelity, but also by the fact that a multitude of neurodegenerative diseases-- including polyglutamine-expanded diseases such as Huntington’s Disease, the spinocerebellar ataxias, and amyotrophic lateral sclerosis- are pathologically associated with nuclear protein misfolding and aggregation [8–12].
In this review, we examine how the nucleus maintains proteostasis. While certain aspects for how the nuclear proteome is protected from proteotoxic stress are not elucidated, we offer a conceptual framework to define this problem. General concepts of PQC are summarized to provide context to how the unique characteristics of the nucleus influences how the proteostasis network is established in this organelle. We examine known, as well as implicated, pathways important for nuclear proteostasis, and also consider the functional implications of a dysregulated nuclear proteostasis network in aging and disease.
General concepts of protein quality control and homeostasis
The functional folding of proteins is accomplished by molecular chaperones, a diverse class of proteins belonging to a number of different protein families that include the Hsp60, Hsp70, Hsp90, Hsp100 and sHSP families [1]. Chaperones have multiple roles to preserve proteostasis, and different members promote the folding of nascent polypeptides, refolding of damaged proteins, disassembly of protein aggregates, as well as the assembly and disassembly of functional protein complexes. In general, chaperones interact with exposed hydrophobic protein patches, and many utilize ATP hydrolysis to drive successive rounds of substrate binding and release to promote folding. Others, such as sHSPs, act as ATP-independent ‘holdases’ that bind to misfolded proteins to maintain their solubility. Chaperone activity is further fine-tuned by co-chaperones, which control rates of chaperone ATPase activity, provide substrate specificity, as well as impart bridging mechanisms to couple separate chaperone systems together or with degradation machineries. This is illustrated by the large family of Hsp40/DNAJ proteins that modulate Hsp70 function [13]. In addition, different chaperone systems often functionally cooperate with each other to form specialized chaperone machines. This is exemplified by the Hsp70:90 as well as the metazoan Hsp70:Hsp110 and yeast Hsp70:Hsp104 systems, that are important for the maturation of specific client proteins and for protein disaggregation, respectively [6, 14]. Chaperones are highly abundant, essential for viability, and are found in virtually all compartments of the cell [1, 5, 6, 15, 16].
When proteins become terminally misfolded, they can be recognized and destroyed by proteolysis. The destruction of most misfolded proteins occurs through the ubiquitin-proteasome system (UPS), which degrades up to ~90% of all proteins [17]. Most misfolded substrates are recognized and polyubiquitylated by an enzymatic cascade involving E1–E3 enzymes [18]. The latter of these, the ubiquitin E3 ligases, are prolific in number, recognize different degradation motifs (degrons), and localize to various subcellular compartments to confer substrate specificity to the UPS [19]. Misfolding recognition by E3 ligases is often coupled to chaperone systems, as with the E3 ligase CHIP, which interacts with Hsp70 and Hsp90 to ubiquitylate chaperone-bound substrates [20]. Once polyubiquitylated, substrates are degraded by the proteasome, a ~2.5 MDa proteolytic machine composed of the activating 19S regulatory particle and proteolytic 20S core particle [21]. Specificity for substrate degradation can be further fine-tuned at the level of the proteasome by association of the 20S core particle with distinct activators, as well as by modulating the proteasome’s subcellular localization [19, 22]. Protein degradation by the UPS is not limited to misfolded proteins; UPS-dependent destruction of many proteins serves as important regulatory mechanisms to control processes such as transcription, DNA repair, and the cell cycle [23, 24].
Although chaperones and the UPS can prevent the accumulation of misfolded proteins under optimal conditions, protein aggregation occurs when these pathways become overwhelmed. Often, aggregates cannot be recognized or efficiently cleared by the UPS [25], and an emerging concept in proteostasis is the existence of mechanisms that specifically manage protein aggregation [26, 27]. These strategies include spatial partitioning and packaging aggregates within the cell to minimize cytotoxicity [28, 29], asymmetric segregation of aggregates into only one cell during cell division [26] [30], as well as clearance by macroautophagy [31].
In addition to PQC machineries that maintain proteomic integrity, cells employ a number of stress responses to sense and promote cellular adaptation to proteotoxic stress by increasing relative folding and degradation capacities. Several of these pathways primarily respond to protein misfolding in a compartment-specific manner. These include the heat shock response, which senses cytoplasmic protein misfolding, as well as the unfolded protein responses of the ER and mitochondria that independently respond to misfolding within these respective organelles. Activation of individual response pathways can be distinctly triggered by environmental stresses—such as elevated temperatures and oxidants for the cytoplasm and reducing agents for the ER— given the unique composition of the organelle’s proteome [6]; however, integration amongst stress responses also exist [32]. The initiation of stress responses leads to a wide range of cellular events, including transient attenuation of translation and nuclear transport of mRNAs and proteins, as well as transcriptional induction of multiple genes encoding proteostasis network components [3, 4].
Underlying principles for preserving nuclear proteostasis
The organization and contents of the nucleus present unique challenges for preserving proteostasis in this organelle. The identity of the nucleus is preserved by the nuclear envelope, a membrane sheet composed of a double lipid bilayer that extends from, and is continuous with, the ER [33]. The nuclear envelope is embedded with nuclear pore complexes, which form aqueous channels that facilitate the exchange of components between the nucleus and the cytoplasm. By separating the genome and mRNA transcription from protein synthesis, the nuclear envelope and pores serve as a critical nexus in the regulation and diversification of gene expression [33].
The nucleoplasm is also further compartmentalized. In metazoan cells, the nuclear lamina, a protein matrix of intermediate filaments, lies underneath the nuclear envelope. The lamina is physically linked to the nuclear envelope through direct interactions with inner nuclear membrane proteins and acts as both a structural and epigenetic scaffold, defining the organelle’s shape as well as binding to chromatin to regulate gene expression [34]. Within the nucleoplasm, different nuclear bodies also exist that are associated with numerous specialized functions involving gene expression [35]. The most prominent of these is the nucleolus, which forms around ribosomal DNA and is the site for the coordinated assembly of ribosome subunits [35]. This complex process of assembling four rRNAs and ~80 ribosomal proteins into two functional ribosomal subunits requires ~200 auxiliary proteins as well as small nucleolar RNAs [36], suggesting that this subcompartment may particularly rely on nuclear PQC pathways to alleviate misassembly errors.
The yeast nucleus lacks a nuclear lamina and contains only the nucleolus as a distinct subcompartment, although less defined functional compartmentalization still occurs [37]. One unique aspect of the yeast nucleus is the spindle pole body, which is the equivalent to mammalian centrosomes and acts as the microtubule-organizing center [38]. Unlike metazoans, yeast mitosis occurs without nuclear envelope breakdown (i.e., closed mitosis), and the mitotic spindle forms within the nucleus for chromosome segregation. The spindle pole body is integrally embedded into the nuclear envelope, where components that nucleate assembly of both cytoplasmic microtubules and the mitotic spindle are poised in the cytosol and nucleoplasm [38].
The identity of the nuclear proteome is largely established by facilitated transport through the nuclear pore complexes. Unlike other membrane protein transport channels, the nuclear pores are much larger complexes comprised of multiple copies of over thirty different proteins [39, 40]. Pore selectivity is established by phenylalanine-glycine repeat domains of nuceloporin proteins lining the channel interior. Protein transport through the nuclear pores is less stringent than for other protein-conducting membrane channels, but nevertheless tightly regulated depending on the cargo’s molecular size [41, 42]. Whereas globular proteins smaller than ~30 kDa can passively diffuse through the pore, larger proteins must enter or exit the nucleus through energy-driven facilitated transport. Nuclear protein transport is largely driven by a family of RanGTP-dependent nuclear transport receptors, called the importins and exportins. The importins mediate nuclear entry by binding nuclear localization signals within their cargo in the cytosol to traverse the nuclear pore complex. Cargo release occurs in the nucleoplasm upon importin binding to the highly abundant RanGTP. Nuclear export of cargo via the exportins occurs in the opposite manner, where RanGTP binding promotes, and dissociation attenuates, exportin-cargo interaction. Nuclear export of mRNA-protein complexes occurs through an independent mechanism facilitated by the Mex67-Mtr2/TAP-p15 complex, but this pathway still depends on RanGTP-dependent mechanisms for the proper localization of these factors [43].
The fidelities of the nuclear envelope, nuclear pores, and nuclear transport mechanisms are crucial for protecting this organelle from proteotoxic stress (Figure 2). By establishing the nucleus as a posttranslational compartment, the nuclear envelope and pores shield the nuclear proteome from protein misfolding in the cytoplasm, particularly of newly synthesized polypeptides that are especially aggregation-prone [1, 44]. The specific PQC machineries that protect against misfolding in the nucleus may be either nuclear residents or PQC components of the ER or cytoplasm imported in during times of proteotoxic stress. In all cases, proper transport will be critical in establishing the nuclear proteostasis network, particularly for components that are shared between the nucleocytoplasmic compartments. Given the compartmentalization of the nucleus, the localization of PQC machineries, misfolding sensors, and aggregates within the nucleoplasm must also be considered. Transport pathways will be especially important in preserving nuclear proteostasis in yeast and post-mitotic metazoan cells, where nuclear envelope breakdown does not occur. On the other hand, the semi-permeable nature of the nuclear pores would also constantly challenge the integrity of the nuclear proteome and the pathways that protect it, and defects in nuclear pore complexes or transport pathways may pose as significant risk factors in maintaining nuclear proteostasis during aging and disease [45].
Figure 2. The integrities of the nuclear envelope, nuclear pore complexes, and transport pathways are critical for preserving proteostasis in the nucleus.

These three factors are speculated to protect the nuclear proteome by (A) restricting access of aggregation-prone nascent polypeptide synthesis and folding processes; (B) transporting protein quality control (PQC) machineries into the nucleoplasm to establish the nuclear proteostasis network; and (C) possibly sensing nuclear protein misfolding to signal for increased import of PQC components (1), as well as clearing the nucleus of misfolded or aggregated protein (2). Molecular pathways that sense and respond to nuclear protein misfolding are currently unknown. ONM, outer nuclear membrane; INM, inner nuclear membrane; HSP, heat shock protein/chaperone; E3, E3 ubiquitin ligase.
Molecular chaperones in the nucleus
Many chaperones and co-chaperones found in the cytoplasm also localize to the nucleus under various conditions. Here, we confine our definition of chaperones to the protein families involved in general folding and misfolding, as discussed above [1]. We will not discuss specialized chaperones that assemble specific complexes, such as those that package nucleosomes or ribosomal particles, but many of these are important for nuclear homeostasis and are examined in detail by others [36, 46, 47].
In metazoan cells, shuttling between the nucleus and cytoplasm appears to be a prevalent feature of chaperones involved in general protein folding representing many of the major structural families, including Hsp70, Hsp90, sHSPs, and the co-chaperones Hsp40 and HOP/mSti1 [48–57]. In general, these chaperones are enriched in the cytoplasm but can transiently translocate into the nucleus under various conditions, including different cell cycle phases and upon exposure to acute proteotoxic stresses such as heat shock (Figures 3A–B). In the latter case, chaperones further accumulate in the nucleoli. The molecular function of chaperones in the nucleus/nucleolus during stress remains undetermined, but nucleolar accumulation of chaperones may prevent the aggregation of unassembled ribosomal proteins at this site (see below for further discussion).
Figure 3. Cytoplasmic chaperones shuttle into the nucleus under various conditions.

(A) Several cytoplasmic chaperones contain nuclear-targeting peptide signals and can be imported into the nucleus by RanGTP-mediated mechanisms in ambient growth conditions. (B) Cytoplasmic chaperones shuttle into the nucleus upon acute environmental stress. For Hsp70, this transport occurs independently of RanGTP and is mediated by the Hikeshi protein. (C) Chaperones are found associated with nuclear aggregates composed of disease-associated proteins, i.e. chronic nuclear misfolding stress. It is thus far unclear how these chaperones are transported into the nucleus under these conditions.
Given the molecular size of chaperones and chaperone complexes, their import would require active transport. Some chaperones possess nuclear localization signals, which may be utilized for import during the cell cycle [56]. Hsp70 and Hsp90 in particular also promote nuclear import of several proteins [58, 59], and could thus ‘piggyback’ into the nucleus this way. RanGTP transport pathways are transiently attenuated during many acute proteotoxic stresses [60, 61], thus presenting a paradox for maintaining nuclear proteostasis. Inhibition of import would limit nuclear entrance of cytosolic misfolded proteins, but would also block the transport of chaperones that prevent nuclear misfolding damage.
Recently, an alternate pathway has been identified that imports Hsp70 into the nucleus during acute heat stress. Import occurs through Hsp70 binding to Hikeshi, a conserved, soluble protein, which appears to transport through the nuclear pore independently of RanGTP [62]. Hikeshi preferentially interacts with and transports ATP-bound Hsp70. This proposed model implies a large increase in the ATP-bound, and by extension substrate-free, pool of Hsp70 under acute proteotoxic stress, but how this might occur is unclear and likely involves additional or upstream factors. For example, Hsp70 nuclear translocation is attenuated in heat-stressed cells treated with phosphatase inhibitors [63]. Whether Hikeshi transports chaperones other than Hsp70 remains to be established.
Beyond acute environmental stresses, multiple chaperones, including Hsp70, Hsp110, and Hsp40, co-localize with nuclear aggregates formed by different disease-associated proteins such as mutant Huntingtin, ataxin-1, and TDP-43, which are linked to Huntington’s disesae, spinocerebellar ataxia 1, and amyotrophic lateral sclerosis, respectively (Figure 3C; [11, 64]). The physical association of chaperones with aggregates appears to be transient, suggesting that chaperones may be actively recognizing aggregates for disaggregation and refolding [65]. The presence of Hsp70 and Hsp110, which together possess disaggregase activity [14], supports this concept. This in turn raises the possibility that mechanisms sensing nuclear misfolding to modulate chaperone import under chronic misfolding stress may exist, but these pathways remain to be determined.
Nuclear chaperone enrichment also occurs in yeast, where chaperones from multiple families, including Hsp70, Hsp90, Hsp100, and sHSPs, concentrate in the nucleus under various metabolic and proteotoxic stress conditions (Figure 3A; [66–70]). Several of these contain nuclear localization signals, but, as in metazoans, acute stress-induced translocation occurs independently of RanGTP-mediated transport. A difference in yeast cells, however, is that many chaperones-- representing most major families-- are already enriched in the nucleus under non-stress conditions [15, 16, 71]. The purpose of having high nuclear chaperone concentrations during normal growth is unclear, but may relate to closed mitosis. For example, nuclear Hsp110/Sse1 appears necessary for proper mitotic spindle assembly [72]. Whether Hikeshi orthologs modulate nuclear localization or enrichment of chaperones in yeast remains to be established.
UPS in the nucleus
Nuclear UPS in yeast
The specificity for misfolded substrate recognition and degradation by proteasomes is largely imparted by ubiquitin E3 ligases. This particular arm of nuclear proteostasis is most established by experimental evidence in yeast, where several E3 ligases have been identified to participate in nuclear PQC (Figure 4A).
Figure 4. The nuclear UPS in budding yeast.

(A) Misfolded substrates within the nucleus are polyubiqitinated for degradation by nuclear resident E3 ligases San1 and Slx5-Slx8, and the integral membrane protein Doa10. San1 also polyubiquitnates cytoplasmic misfolded substrates targeted to the nucleus. The E3 ligase Ubr1 and its S. pombe homolog Ubr11 (not shown) also mediate degradation of proteins that reside in or sample the nucleus. The different colored misfolded proteins represent substrates with different misfolded moieties. (B) Proteasomes are imported into the nucleus by RanGTP-dependent transport pathways. Several individual 20S subunits possess nuclear localization-like signals and are transported by the importins; the assembled 20S core particle can also be imported by the proteasomal activator Blm10. Cut8/Sts1 retains proteasomes in the nucleoplasm. Note that the role for nuclear proteasomes in nuclear proteostasis has not been formally established. See text for a discussion on nuclear UPS in higher eukaryotes.
The primary nuclear ubiquitin ligase is San1, which ubiquitylates misfolded polypeptide substrates for degradation in both budding and fission yeasts [73–75]. San1 localizes to the nucleus under stress and nonstress conditions [74, 75], and ubiquitylates a large variety of misfolded substrates, including destabilized endogenous mutant proteins, short synthetic polypeptides, exogenous model misfolded proteins, as well as misfolded ER substrates lacking their ER-targeting sequences [74, 76–81]. The subcellular localizations of these polypeptides are diverse, where they are found in the nucleus but, perhaps more surprisingly, also in the cytoplasm as soluble or ER membrane-associated states (more on this below). San1’s ability to ubiquitylate diverse substrates occurs through its disordered N- and C-termini that flexibly interact with disordered polypeptides. These domains recognize exposed, hydrophobic polypeptide stretches in a postulated mechanism analogous to how chaperones, particularly sHSPs, interact with misfolded proteins [76–78].
The integral ER membrane E3 ligase, Doa10, also ubiquitylates several soluble and membrane-bound proteins localized to the nucleoplasm or inner nuclear membrane [82–85]. Doa10 has a well-characterized role in ER-associated degradation of proteins with misfolded cytoplasmic domains, but is able to access the inner nuclear membrane to mediate nuclear PQC [82]. Experiments with a destabilized component of the kinetochore complex indicate that Doa10 also recognizes misfolded substrates that have exposed hydrophobicity, although the particular motifs appear different from San1’s [85]. Doa10 also recognizes N-terminally acetylated proteins as part of the N-end rule pathway, but how this contributes to nuclear proteostasis is unexplored [86]. Of the substrates tested, those recognized by San1 and Doa10 do not widely overlap (reviewed in [80]), and Doa10 may preferentially recognize misfolding of integral membrane proteins [87].
Slx5-Slx8, a nuclear E3 ligase complex that recognizes sumoylated proteins, has also been implicated in nuclear PQC [88]. Slx5-Slx8 mediates degradation of a temperature-sensitive mutant of the transcriptional regulator Mot1, and degradation of this protein also requires its sumoylation. These results suggest that this posttranslational modification pathway may additionally contribute to nuclear proteostasis, but the extent to which sumoylation plays a role is currently unknown (further discussed in [11]).
Ubr1, an E3 ligase that mediates degradation in the N-end rule pathway, also ubiqutiylates several San1 substrates for degradation in S. cerevisiae [79–81], as does its homolog Ubr11 in S. pombe [89]. However, Ubr1 function is proposed to be confined to cytoplasmic PQC [79], and it is unclear whether or how the cytoplasmic and nuclear PQC pathways coordinate substrate degradation.
Cooperation between nuclear E3 ligases and chaperones in yeast
Given its ability to directly bind misfolded proteins, San1 has been proposed to generically scan the nucleoplasmic volume for misfolding. However, this would imply a direct competition with chaperones for misfolded substrate, and the abundance of molecular chaperones far exceeds that of San1 [90, 91]. Perhaps this chaperone excess provides the misfolded substrate a greater chance to refold before being terminally degraded by the UPS. Regardless, additional triage criteria likely exist to determine whether a misfolded substrate should be refolded or destroyed, and this may occur through a more intimate interplay between chaperones and the nuclear UPS.
Cooperation between the two systems is supported by experiments with cytosolic misfolded substrates, which are targeted to the nucleus by Hsp70 for San1-mediated degradation. The targeting ability of Hsp70/Ssa1 depends on its ATPase activity, requiring the nucleotide exchange factor Hsp110/Sse1 and the Hsp40 co-chaperones Ydj1 and/or Sis1 (reviewed in [80]). Sis1 may have additional roles targeting misfolded cytoplasmic substrates to the nucleus for degradation [92]. Doa10-driven degradation of mutant Ndc10 also requires Hsp70/Ssa1 [85], which suggests a role for chaperone-dependent degradation of nuclear substrates by this E3 ligase. In addition, recent work in S. pombe indicates the involvement of bag102, an Hsp70 nucleotide exchange factor tethered to the ER, in the degradation of several misfolded kinetochore proteins [89].
It is unclear to what extent physical coupling occurs between the two systems. For San1, Hsp70/Ssa1facilitates this E3 ligase’s ability to bind at least one substrate [80]. Other studies indicate that chaperones do not readily interact with San1 [78]. Future experiments will be necessary to elucidate how E3 ligases and chaperones might coordinate their activities in nuclear PQC.
Nuclear proteasomes in yeast
Once polyubiquitinated, misfolded substrates are generally delivered to the proteasome through interactions of the E3 ligase or adaptor proteins with this proteolytic machine, where the substrates are then deubiquitylated by associated ubiquitin hydrolases and degraded [21]. How and to what extent these processes occur in the nucleus remains to be determined, but proteasomes are enriched in yeast nuclei [93, 94]. This localization appears to rely on RanGTP-dependent transport (Figure 4B). Several individual proteasomal subunits possess nuclear localization signals; in addition, Blm10, an alternate activator of the proteolytic 20S core particle, can also direct nuclear localization of the 20S core particle itself [95].
The nuclear retention of proteasomes depends on Cut8/Sts1 in fission and budding yeasts ([96–98]; Figure 4B). This nuclear protein appears to directly tether proteasomes to inner nuclear membranes [99], although the molecular details of this tethering process is unclear. The regulated degradation of the nuclear proteins Cdc13/cyclin and Cut3/securin requires Cut8 for cell cycle progression, and this protein is also essential for viability during heat stress, where its expression levels are elevated [97]. These results suggest the hypothesis that Cut8-- and by extension, nuclear proteasomes— is involved in maintaining nuclear proteostasis, but more work is necessary to establish how this occurs.
Nuclear UPS in higher eukaryotes
The identity of nuclear UPS pathways in higher eukaryotes is less understood, and many of the nuclear UPS pathways described above are unique to yeast. For example, San1 is not conserved in metazoans, and to date, no functional orthologs have been identified. Homologs of Doa10 and Slx5-Slx8 exist but their involvement in nuclear PQC has not been established. In addition, Cut8/Sts1 is not found in most metazoans, although a homolog appears to exist in Drosophila [96].
Nevertheless, multiple studies support the presence of an active UPS within the nucleus [100, 101]. A nuclear-targeted reporter for proteasomal clearance, GFP fused to the CL1 degron, is degraded rapidly [102], and nuclear ubiquitylation for degradation appears to be a global feature in transcriptional regulation [103]. More notably for PQC, 20S proteasomal core particles have been implicated in the degradation of oxidatively damaged histones [104]. Other studies report that newly synthesized ribosomal proteins at stoichiometric excess of other ribosomal components are still imported into the nucleus and associate with the nucleolus, but are then degraded [105, 106]. How these misfolded histone and ribosomal proteins are recognized for proteasomal degradation remains to be determined, but certain E3 ligases have been independently identified to degrade certain nuclear misfolded disease proteins. These include Uhrf-2 and PML IV, which increase the turnover of mutant polyglutamine-expanded proteins localized in the nucleus [107, 108]. It is unclear how these E3 ligases recognize mutant polyglutamine proteins, and thus the specificity or generality of misfolding recognition by these E3 ligases is unknown. As in yeast, proteasome particles are also actively imported into and enriched in metazoan nuclei [101, 109].
Spatial control of misfolded substrate engagement and degradation
A central unresolved question regarding nuclear PQC is not only on how recognition and degradation of protein misfolding occur, but where each of these steps happen in context to cellular location. As presented above, the nuclear localization of E3 ligases and the proteasome implies that recognition, targeting, and clearance of nuclear misfolded substrates can all take place within this compartment, and multiple lines of evidence suggest this to be the case. Perhaps surprisingly, beyond nuclear misfolded proteins, multiple cytoplasmic misfolded proteins also appear to be actively targeted to the nucleus for degradation. Studies indicate that this targeting is dependent on Hsp70/Ssa1 [80, 81, 87], as well as the DNAJ co-chaperone Sis1 and the RanGTP gradient [92]. A similar situation for cytoplasmic misfolded polypeptides has been described in mammalian cells, where a model misfolded protein, mutant firefly luciferase, is proposed to be imported into the nucleus by the Sis1 ortholog DNAJB1 for degradation [92]. For luciferase, the UPS components responsible for degradation remain unidentified.
The nuclear enrichment of proteasomes has been given as a major reason for why cytoplasmic substrates are degraded in the nucleus [81, 92], but many questions remain on how and why this phenomenon occurs. The nuclear import factors that transport these large chaperone:substrate complexes through the nuclear pores remains unknown. It is also unclear whether nuclear substrate targeting is coupled to the relative concentration of proteasomes in the nucleus. An accumulation of misfolded substrates in the nucleus could occur otherwise during conditions when proteasomes are depleted from the nucleus, lading to toxic aggregation.
Contradicting evidence that nuclear misfolded proteins are degraded in the cytoplasm also exists. Ubp3, a proteasome-associated deubiquitylating enzyme involved in the clearance of mutant nuclear kinetochore proteins in S. pombe [89], is largely cytosolic [110]. Nuclear export-based degradation pathways also have been described in mammalian cells. The regulated degradation of certain factors such as p53, albeit not misfolded, occurs by first being exported to the cytoplasm for destruction [111]. Nuclear export defects of an aggregation-prone mutant Huntingtin fragment have also been proposed in the toxic nuclear accumulation of this disease protein [112]. Thus, a clear picture on the general ‘rules’ of where misfolded substrates are engaged and ultimately destroyed has not yet emerged, making this rich ground for future research.
Spatial management of toxic protein aggregation in the nucleus
If protein degradation pathways become dysregulated, misfolded proteins can accumulate as toxic aggregates. Within the cytoplasm, cells are known to employ several tactics of aggregate containment and clearance to prevent this from occurring [26–29]. These include sequestration of misfolded or aggregated proteins in certain locales to minimize toxicity, as well as clearance mechanisms during mitosis or via macroautophagy. Little is known about analogous pathways that might manage nuclear protein aggregation, but certain nuclear subcompartments have been implicated in this process in mammalian cells. Beyond its purported role in ubiquitylation, PML protein relocalizes around nuclear aggregates of mutant polyglutamine-expanded proteins to form cage-like structures [113, 114]. These PML bodies are postulated to have a protective effect by sequestering aggregates from the rest of the nucleoplasm. More generally, PML bodies contain ubiquitin, proteasomal 20S core particles, multiple chaperones, as well as misfolded or metastable proteins [108]. Similar nuclear bodies enriched with PQC components and aggregation-prone proteins, with or without PML protein, have also been observed during Herpesvirus replication, where they are proposed to act as nuclear PQC centers [108, 115].
The nucleolus is also implicated in nuclear aggregation. Nucleolar accumulation of misfolded and aggregated proteins occurs with heat stress or proteasomal inhibition [116, 117], and many chaperones and UPS components localize to the nucleolus [48, 49, 105]. The presence of misfolded proteins and PQC components at the nucleolus may mean that nascent ribosomal proteins or the auxiliary nucleolar proteins themselves are simply aggregation-prone, whereby accumulation of misfolded and nuclear PQC proteins is simply coincident (further discussed below). However, Hsp70 appears to be actively recruited to nucleoli by stress-induced noncoding nucleolar RNAs [118]. More studies are necessary to determine whether PML bodies or nucleoli have abilities to actively sequester or disaggregate nuclear protein aggregates.
Aside from sequestration, cells employ various pathways to clear cytoplasmic aggregation [26], and we speculate that analogous methods for clearing cells of nuclear aggregates may also exist (Figure 5). One strategy for clearing aggregation is during mitosis, where cytoplasmic protein aggregates can be segregated asymmetrically into one cell, thus keeping the other cell pristine [26, 30]. This uneven inheritance is proposed to contribute to the aging of the cell acquiring the damage [26, 119], and has been best studied in budding yeast, which naturally divide asymmetrically to produce mother and daughter cells.
Figure 5. Hypothetical mechanisms for clearance of nuclear aggregates.
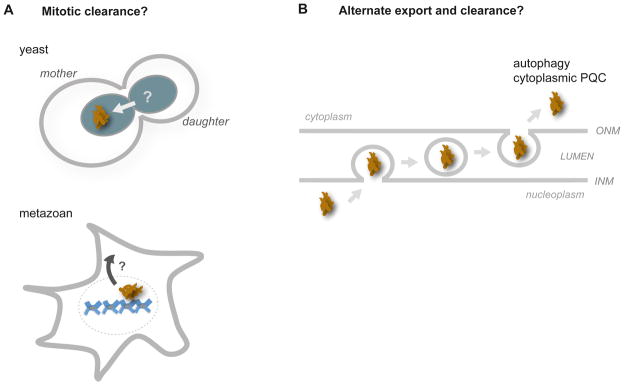
(A) Nuclear aggregates may be cleared during mitosis through asymmetric inheritance, as in yeast (top panel). In metazoans, the dissolution of the nuclear envelope may expose nuclear aggregates to cytoplasmic PQC machineries (bottom panel). (B) Large nuclear aggregates have been proposed to actively transport through the nuclear envelope to the cytoplasm for disaggregation or degradation, analogous to nuclear egress pathways of Herpesvirus and mRNA-protein granules; adapted from a model proposed by Schlieker and colleagues [125]. These models require experimental validation.
Several nuclear elements are known to segregate asymmetrically during mitosis in S. cerevisiae, raising the possibility that nuclear aggregates are also cleared this way (Figure 5A). Extrachromosomal rDNA circles are retained in the mother cell and this strongly correlates with aging [120]. On the other hand, older nuclear pore complexes appear to predominantly segregate to the younger daughter cell, as does the older spindle pole body [121–123]. Segregation of nuclear aggregates into one cell could occur through physical association with any of these nuclear elements, especially with spindle pole bodies, given that cytosolic aggregates accumulate around these structures [25, 28]. Asymmetric clearance could also occur through the mechanisms that segregate these elements themselves. In higher eukaryotes, nuclear envelope breakdown during mitosis could provide an additional opportunity for sequestering, disaggregase, or autophagy machineries in the cytoplasm to directly access nuclear aggregates.
Cytoplasmic aggregates can also be directly degraded through macroautophagy. However, studies in mammalian cells and mice indicate that nuclear aggregates of mutant Huntingtin or ataxin-1 are cleared ineffectively by this pathway [124]. Given this, an alternative strategy for nuclear aggregate degradation has been hypothesized by Schlieker and colleagues, where nuclear aggregates first exit the nucleus independently of nuclear pores for autophagic clearance (Figure 5B; [125]). This provocative model proposes transport of aggregates through analogous pathways that mediate nuclear egress of Herpesvirus capsids or of mRNA-protein granules [126, 127], and would require the vesicular budding of nuclear aggregates through the double membrane of the nuclear envelope.
It should be stressed that the above pathways for asymmetric segregation and aggregate export are purely speculative, and exploring the existence of such pathways deserves future attention.
Nuclear proteostasis: The intersection of multiple quality control pathways and stress responses?
Given the unique organization and composition of the nucleus, crosstalk may exist between nuclear PQC pathways and those that protect the integrity of DNA and RNA. Many of the speculated nuclear PQC components discussed above in yeast are also involved in, or genetically interact with, pathways controlling DNA replication and repair, cell cycle, and transcription [84, 88, 97, 128, 129]. It would seem reasonable that decisions for replication and transcription of the genome could be tightly linked to nuclear PQC and the quality of the nuclear proteome. The possible coordination between mechanisms protecting protein and nucleic acid fidelity may be particularly important during ribosome subunit assembly, a complex multi-step process prone to error [130]. As stated previously, ribosomal proteins in excess are degraded [105, 106], and in yeast, the ribosomal RNA of misassembled pre-ribosomal particles is cleared at the nucleolus by the exosome, an RNA degradation machine [130, 131]. The coordination between RNA degradation and the stability of ribosomal proteins has yet to be established, but the presence of chaperones and UPS machinery at the nucleolus raises the possibility of cooperation between RNA and protein quality control pathways within the nucleus.
Crosstalk of mechanisms protecting nuclear protein folding and other nuclear processes may extend to the transcriptional stress responses that protect this organelle. The stress response(s) that sense and transcriptionally respond to nuclear protein misfolding is presently unknown but likely exist. For example, S. cerevisiae cells lacking San1 show increased transcription of several chaperone genes [78], indicating that compensatory transcriptional pathways exist to increase proteostasis capacity. While a novel transcriptional pathway is possible, it is likely that transcription factors that modulate cytoplasmic and ER stress responses are involved, given the many components that are shared among these different compartments. The less-established auxiliary components important for the localization of essential PQC machinery to the nucleus—Hikeshi and Cut8—show increased expression with heat stress and are necessary for viability at elevated temperatures [62, 97], suggesting a role for the cytoplasmic heat shock response. Given the participation of the membrane-bound E3 ligase Doa10, the unfolded protein response of the ER may sense and respond to nuclear protein misfolding along the nuclear envelope. The common susceptibility of both proteins and nucleic acids to oxidative damage suggests that the oxidative stress response may also be activated. The elucidation of how discrete nuclear quality control and transcriptional pathways might coordinate to sense and adapt to nuclear proteotoxic stress will be important in understanding the pathways protecting nuclear proteostasis.
Consequences of dysregulated nuclear proteostasis network in aging and disease
We predict that the functional consequences of a dysregulated nuclear proteostasis network are far-reaching and may impact genomic expression, integrity, and epigenetics, as well as contribute to the pathologies of age-dependent protein conformational diseases. A direct link between genomic and proteomic stabilities is beginning to emerge, suggesting that the proper maintenance of one affects the stability of the other. Yeast cells that experience widespread protein misfolding induce DNA mutagenesis [132], and chronic inhibition of Hsp90 induces aberrant chromosomal number (aneuploidy) [133]. On the other hand, aneuploid yeast cells reportedly have impaired proteostasis capacities, which may occur from stoichiometric protein complex imbalances caused by uneven gene dosage [134, 135]. The ability of chronic proteotoxic stress to stimulate genetic error has been proposed to drive the evolutionary adaptation of cancer cells. Whether the nuclear proteostasis network might promote or attenuate this adaptation is presently unclear.
The fidelity of the nuclear proteostasis network likely impacts aging as well. Several histone and nuclear pore complex components are extremely long-lived proteins that do not turn over in post-mitotic cells, instead persisting for the lifespan of the cell [45, 136]. Why these proteins are not degraded is unclear but may indicate nuclear UPS selectivity or deficiencies, especially given that mechanisms appear to exist for histone degradation [104]. The long-lived nature of histones and nuclear pore complex components underscores the importance of nuclear-localized chaperones and their transport pathways to protect them from age-related damage. Inevitably, the selective porosity of old nuclear pores becomes leaky in aged cells, and cytoplasmic proteins can accumulate within the nucleus [45]. We postulate that this likewise leads to a decline in the fidelity of transport pathways that establish a robust proteostasis network in the nucleus, thus exposing this organelle to the consequences of protein misfolding seen in age-related proteinopathies.
The accumulation of misfolded and aggregated disease proteins is increasingly recognized as a common pathological mechanism in many neurodegenerative disorders [137]. The toxicity of aggregation is largely attributed to gain-of-function effects caused by the aberrant association of various chaperones, UPS components, and other metastable proteins with these aggregates. Nuclear protein misfolding and aggregation of disease-associated proteins are prominent features in several of these diseases [11, 12]; specific examples include TDP-43, huntingtin, and ataxin-1, which are associated with amyotrophic lateral sclerosis, Huntington’s Disease, and spinocerebellar ataxia 1, respectively [8–10]. The nuclear localization of aggregates is suggested to be important in disease pathology, as shown with huntingtin and ataxin-1 [9, 138]. While aspects of PQC have been studied in context to these disease-associated proteins within the nucleus as described above, we predict that global mechanisms that sense the need for, transport, and establish PQC machineries into this organelle likely play important roles in the molecular pathology of these diseases as well.
In addition, the robustness of nuclear PQC pathways may differ among distinct cell types. Certain neurons show decreased sensitivity for the heat-induced nuclear translocation of chaperones [139], and the long-lived nature of neurons suggests that these cells may be particularly susceptible to protein folding and degradation dysfunction, dependent on age. Gaining an understanding of nuclear proteostasis mechanisms in neurons is important, given the multitude of protein conformational diseases that primarily affect the function of these cells.
Perspectives
Many exciting questions remain to be addressed on the molecular pathways that protect the nucleus from protein misfolding. A comprehensive identification of the components of the nuclear proteostasis network is necessary, especially in higher eukaryotic systems. Elucidation of the molecular signals, transport pathways, and transcription responses that sense the need for and establish robust nuclear proteostasis mechanisms is vital, especially in context to chronic misfolding stresses. Finally, an understanding of how nuclear PQC may crosstalk with other nuclear pathways such as those mediating ribosome assembly, DNA replication, mRNA transcription and quality control is required. Grasping how all of these pathways coordinate in postmitotic cells and on the organismal level will be essential in understanding how this remarkable organelle functions in health and disease.
Acknowledgments
Y.S. is an HHMI postdoctoral fellow of the Damon Runyon Cancer Foundation (DRG 2086-11). R.I.M. is supported by the Ellison Medical Foundation, the Daniel F. and Ada L. Rice Foundation, the Chicago Biomedical Consortium, and NIH grants GM038109, GM081192, AG026647 and NS047331.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hartl FU, Bracher A, Hayer-Hartl M. Molecular chaperones in protein folding and proteostasis. Nature. 2011;475:324–332. doi: 10.1038/nature10317. [DOI] [PubMed] [Google Scholar]
- 2.Powers ET, Morimoto RI, Dillin A, Kelly JW, Balch WE. Biological and chemical approaches to diseases of proteostasis deficiency. Annual review of biochemistry. 2009;78:959–991. doi: 10.1146/annurev.biochem.052308.114844. [DOI] [PubMed] [Google Scholar]
- 3.Morimoto RI. Proteotoxic stress and inducible chaperone networks in neurodegenerative disease and aging. Genes & development. 2008;22:1427–1438. doi: 10.1101/gad.1657108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nature reviews Molecular cell biology. 2007;8:519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- 5.Baker BM, Haynes CM. Mitochondrial protein quality control during biogenesis and aging. Trends in biochemical sciences. 2011;36:254–261. doi: 10.1016/j.tibs.2011.01.004. [DOI] [PubMed] [Google Scholar]
- 6.Buchberger A, Bukau B, Sommer T. Protein quality control in the cytosol and the endoplasmic reticulum: brothers in arms. Molecular cell. 2010;40:238–252. doi: 10.1016/j.molcel.2010.10.001. [DOI] [PubMed] [Google Scholar]
- 7.Haynes CM, Ron D. The mitochondrial UPR - protecting organelle protein homeostasis. Journal of cell science. 2010;123:3849–3855. doi: 10.1242/jcs.075119. [DOI] [PubMed] [Google Scholar]
- 8.Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science (New York, NY) 2006;314:130–133. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- 9.Klement IA, Skinner PJ, Kaytor MD, Yi H, Hersch SM, Clark HB, Zoghbi HY, Orr HT. Ataxin-1 nuclear localization and aggregation: role in polyglutamine-induced disease in SCA1 transgenic mice. Cell. 1998;95:41–53. doi: 10.1016/s0092-8674(00)81781-x. [DOI] [PubMed] [Google Scholar]
- 10.Davies SW, Turmaine M, Cozens BA, DiFiglia M, Sharp AH, Ross CA, Scherzinger E, Wanker EE, Mangiarini L, Bates GP. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell. 1997;90:537–548. doi: 10.1016/s0092-8674(00)80513-9. [DOI] [PubMed] [Google Scholar]
- 11.Gallagher PS, Oeser ML, Abraham AC, Kaganovich D, Gardner RG. Cellular maintenance of nuclear protein homeostasis. Cellular and molecular life sciences : CMLS. 2013 doi: 10.1007/s00018-013-1530-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Woulfe JM. Abnormalities of the nucleus and nuclear inclusions in neurodegenerative disease: a work in progress. Neuropathology and applied neurobiology. 2007;33:2–42. doi: 10.1111/j.1365-2990.2006.00819.x. [DOI] [PubMed] [Google Scholar]
- 13.Kampinga HH, Craig EA. The HSP70 chaperone machinery: J proteins as drivers of functional specificity. Nature reviews Molecular cell biology. 2010;11:579–592. doi: 10.1038/nrm2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rampelt H, Kirstein-Miles J, Nillegoda NB, Chi K, Scholz SR, Morimoto RI, Bukau B. Metazoan Hsp70 machines use Hsp110 to power protein disaggregation. The EMBO journal. 2012;31:4221–4235. doi: 10.1038/emboj.2012.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Matsuyama A, Arai R, Yashiroda Y, Shirai A, Kamata A, Sekido S, Kobayashi Y, Hashimoto A, Hamamoto M, Hiraoka Y, et al. ORFeome cloning and global analysis of protein localization in the fission yeast Schizosaccharomyces pombe. Nature biotechnology. 2006;24:841–847. doi: 10.1038/nbt1222. [DOI] [PubMed] [Google Scholar]
- 16.Huh WK, Falvo JV, Gerke LC, Carroll AS, Howson RW, Weissman JS, O’Shea EK. Global analysis of protein localization in budding yeast. Nature. 2003;425:686–691. doi: 10.1038/nature02026. [DOI] [PubMed] [Google Scholar]
- 17.Lee DH, Goldberg AL. Proteasome inhibitors: valuable new tools for cell biologists. Trends in cell biology. 1998;8:397–403. doi: 10.1016/s0962-8924(98)01346-4. [DOI] [PubMed] [Google Scholar]
- 18.Komander D, Rape M. The ubiquitin code. Annual review of biochemistry. 2012;81:203–229. doi: 10.1146/annurev-biochem-060310-170328. [DOI] [PubMed] [Google Scholar]
- 19.Pines J, Lindon C. Proteolysis: anytime, any place, anywhere? Nature cell biology. 2005;7:731–735. doi: 10.1038/ncb0805-731. [DOI] [PubMed] [Google Scholar]
- 20.Arndt V, Rogon C, Hohfeld J. To be, or not to be--molecular chaperones in protein degradation. Cellular and molecular life sciences : CMLS. 2007;64:2525–2541. doi: 10.1007/s00018-007-7188-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Elsasser S, Finley D. Delivery of ubiquitinated substrates to protein-unfolding machines. Nature cell biology. 2005;7:742–749. doi: 10.1038/ncb0805-742. [DOI] [PubMed] [Google Scholar]
- 22.Hanna J, Finley D. A proteasome for all occasions. FEBS letters. 2007;581:2854–2861. doi: 10.1016/j.febslet.2007.03.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Varshavsky A. The ubiquitin system, an immense realm. Annual review of biochemistry. 2012;81:167–176. doi: 10.1146/annurev-biochem-051910-094049. [DOI] [PubMed] [Google Scholar]
- 24.Weissman AM, Shabek N, Ciechanover A. The predator becomes the prey: regulating the ubiquitin system by ubiquitylation and degradation. Nature reviews Molecular cell biology. 2011;12:605–620. doi: 10.1038/nrm3173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kopito RR. Aggresomes, inclusion bodies and protein aggregation. Trends in cell biology. 2000;10:524–530. doi: 10.1016/s0962-8924(00)01852-3. [DOI] [PubMed] [Google Scholar]
- 26.Tyedmers J, Mogk A, Bukau B. Cellular strategies for controlling protein aggregation. Nature reviews Molecular cell biology. 2010;11:777–788. doi: 10.1038/nrm2993. [DOI] [PubMed] [Google Scholar]
- 27.Sontag EM, Vonk WI, Frydman J. Sorting out the trash: the spatial nature of eukaryotic protein quality control. Current opinion in cell biology. 2014;26C:139–146. doi: 10.1016/j.ceb.2013.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kaganovich D, Kopito R, Frydman J. Misfolded proteins partition between two distinct quality control compartments. Nature. 2008;454:1088–1095. doi: 10.1038/nature07195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Escusa-Toret S, Vonk WI, Frydman J. Spatial sequestration of misfolded proteins by a dynamic chaperone pathway enhances cellular fitness during stress. Nature cell biology. 2013;15:1231–1243. doi: 10.1038/ncb2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Spokoini R, Moldavski O, Nahmias Y, England JL, Schuldiner M, Kaganovich D. Confinement to organelle-associated inclusion structures mediates asymmetric inheritance of aggregated protein in budding yeast. Cell reports. 2012;2:738–747. doi: 10.1016/j.celrep.2012.08.024. [DOI] [PubMed] [Google Scholar]
- 31.Williams A, Jahreiss L, Sarkar S, Saiki S, Menzies FM, Ravikumar B, Rubinsztein DC. Aggregate-prone proteins are cleared from the cytosol by autophagy: therapeutic implications. Current topics in developmental biology. 2006;76:89–101. doi: 10.1016/S0070-2153(06)76003-3. [DOI] [PubMed] [Google Scholar]
- 32.Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M, Sadri N, Yun C, Popko B, Paules R, et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Molecular cell. 2003;11:619–633. doi: 10.1016/s1097-2765(03)00105-9. [DOI] [PubMed] [Google Scholar]
- 33.Hetzer MW, Walther TC, Mattaj IW. Pushing the envelope: structure, function, and dynamics of the nuclear periphery. Annual review of cell and developmental biology. 2005;21:347–380. doi: 10.1146/annurev.cellbio.21.090704.151152. [DOI] [PubMed] [Google Scholar]
- 34.Burke B, Stewart CL. The nuclear lamins: flexibility in function. Nature reviews Molecular cell biology. 2013;14:13–24. doi: 10.1038/nrm3488. [DOI] [PubMed] [Google Scholar]
- 35.Spector DL. Nuclear domains. Journal of cell science. 2001;114:2891–2893. doi: 10.1242/jcs.114.16.2891. [DOI] [PubMed] [Google Scholar]
- 36.Kressler D, Hurt E, Bassler J. Driving ribosome assembly. Biochimica et biophysica acta. 2010;1803:673–683. doi: 10.1016/j.bbamcr.2009.10.009. [DOI] [PubMed] [Google Scholar]
- 37.Taddei A, Schober H, Gasser SM. The budding yeast nucleus. Cold Spring Harbor perspectives in biology. 2010;2:a000612. doi: 10.1101/cshperspect.a000612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jaspersen SL, Winey M. The budding yeast spindle pole body: structure, duplication, and function. Annual review of cell and developmental biology. 2004;20:1–28. doi: 10.1146/annurev.cellbio.20.022003.114106. [DOI] [PubMed] [Google Scholar]
- 39.Hoelz A, Debler EW, Blobel G. The structure of the nuclear pore complex. Annual review of biochemistry. 2011;80:613–643. doi: 10.1146/annurev-biochem-060109-151030. [DOI] [PubMed] [Google Scholar]
- 40.Bilokapic S, Schwartz TU. 3D ultrastructure of the nuclear pore complex. Current opinion in cell biology. 2012;24:86–91. doi: 10.1016/j.ceb.2011.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fried H, Kutay U. Nucleocytoplasmic transport: taking an inventory. Cellular and molecular life sciences : CMLS. 2003;60:1659–1688. doi: 10.1007/s00018-003-3070-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Guttler T, Gorlich D. Ran-dependent nuclear export mediators: a structural perspective. The EMBO journal. 2011;30:3457–3474. doi: 10.1038/emboj.2011.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rodriguez-Navarro S, Hurt E. Linking gene regulation to mRNA production and export. Current opinion in cell biology. 2011;23:302–309. doi: 10.1016/j.ceb.2010.12.002. [DOI] [PubMed] [Google Scholar]
- 44.Pechmann S, Willmund F, Frydman J. The ribosome as a hub for protein quality control. Molecular cell. 2013;49:411–421. doi: 10.1016/j.molcel.2013.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.D’Angelo MA, Raices M, Panowski SH, Hetzer MW. Age-dependent deterioration of nuclear pore complexes causes a loss of nuclear integrity in postmitotic cells. Cell. 2009;136:284–295. doi: 10.1016/j.cell.2008.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ellis RJ. Molecular chaperones: assisting assembly in addition to folding. Trends in biochemical sciences. 2006;31:395–401. doi: 10.1016/j.tibs.2006.05.001. [DOI] [PubMed] [Google Scholar]
- 47.Philpott A, Krude T, Laskey RA. Nuclear chaperones. Seminars in cell & developmental biology. 2000;11:7–14. doi: 10.1006/scdb.1999.0346. [DOI] [PubMed] [Google Scholar]
- 48.Velazquez JM, Lindquist S. hsp70: nuclear concentration during environmental stress and cytoplasmic storage during recovery. Cell. 1984;36:655–662. doi: 10.1016/0092-8674(84)90345-3. [DOI] [PubMed] [Google Scholar]
- 49.Welch WJ, Feramisco JR. Nuclear and nucleolar localization of the 72,000-dalton heat shock protein in heat-shocked mammalian cells. The Journal of biological chemistry. 1984;259:4501–4513. [PubMed] [Google Scholar]
- 50.Welch WJ, Mizzen LA. Characterization of the thermotolerant cell. II. Effects on the intracellular distribution of heat-shock protein 70, intermediate filaments, and small nuclear ribonucleoprotein complexes. The Journal of cell biology. 1988;106:1117–1130. doi: 10.1083/jcb.106.4.1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Akner G, Mossberg K, Sundqvist KG, Gustafsson JA, Wikstrom AC. Evidence for reversible, non-microtubule and non-microfilament-dependent nuclear translocation of hsp90 after heat shock in human fibroblasts. European journal of cell biology. 1992;58:356–364. [PubMed] [Google Scholar]
- 52.Langer T, Rosmus S, Fasold H. Intracellular localization of the 90 kDA heat shock protein (HSP90alpha) determined by expression of a EGFP-HSP90alpha-fusion protein in unstressed and heat stressed 3T3 cells. Cell biology international. 2003;27:47–52. doi: 10.1016/s1065-6995(02)00256-1. [DOI] [PubMed] [Google Scholar]
- 53.Willsie JK, Clegg JS. Small heat shock protein p26 associates with nuclear lamins and HSP70 in nuclei and nuclear matrix fractions from stressed cells. Journal of cellular biochemistry. 2002;84:601–614. [PubMed] [Google Scholar]
- 54.Hattori H, Kaneda T, Lokeshwar B, Laszlo A, Ohtsuka K. A stress-inducible 40 kDa protein (hsp40): purification by modified two-dimensional gel electrophoresis and co-localization with hsc70(p73) in heat-shocked HeLa cells. Journal of cell science. 1993;104(Pt 3):629–638. doi: 10.1242/jcs.104.3.629. [DOI] [PubMed] [Google Scholar]
- 55.Daniel S, Bradley G, Longshaw VM, Soti C, Csermely P, Blatch GL. Nuclear translocation of the phosphoprotein Hop (Hsp70/Hsp90 organizing protein) occurs under heat shock, and its proposed nuclear localization signal is involved in Hsp90 binding. Biochimica et biophysica acta. 2008;1783:1003–1014. doi: 10.1016/j.bbamcr.2008.01.014. [DOI] [PubMed] [Google Scholar]
- 56.Longshaw VM, Chapple JP, Balda MS, Cheetham ME, Blatch GL. Nuclear translocation of the Hsp70/Hsp90 organizing protein mSTI1 is regulated by cell cycle kinases. Journal of cell science. 2004;117:701–710. doi: 10.1242/jcs.00905. [DOI] [PubMed] [Google Scholar]
- 57.Milarski KL, Morimoto RI. Expression of human HSP70 during the synthetic phase of the cell cycle. Proceedings of the National Academy of Sciences of the United States of America. 1986;83:9517–9521. doi: 10.1073/pnas.83.24.9517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Melchior F, Gerace L. Mechanisms of nuclear protein import. Current opinion in cell biology. 1995;7:310–318. doi: 10.1016/0955-0674(95)80084-0. [DOI] [PubMed] [Google Scholar]
- 59.Dezwaan DC, Freeman BC. HSP90: the Rosetta stone for cellular protein dynamics? Cell cycle (Georgetown, Tex) 2008;7:1006–1012. doi: 10.4161/cc.7.8.5723. [DOI] [PubMed] [Google Scholar]
- 60.Furuta M, Kose S, Koike M, Shimi T, Hiraoka Y, Yoneda Y, Haraguchi T, Imamoto N. Heat-shock induced nuclear retention and recycling inhibition of importin alpha. Genes to cells : devoted to molecular & cellular mechanisms. 2004;9:429–441. doi: 10.1111/j.1356-9597.2004.00734.x. [DOI] [PubMed] [Google Scholar]
- 61.Miyamoto Y, Saiwaki T, Yamashita J, Yasuda Y, Kotera I, Shibata S, Shigeta M, Hiraoka Y, Haraguchi T, Yoneda Y. Cellular stresses induce the nuclear accumulation of importin alpha and cause a conventional nuclear import block. The Journal of cell biology. 2004;165:617–623. doi: 10.1083/jcb.200312008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kose S, Furuta M, Imamoto N. Hikeshi, a nuclear import carrier for Hsp70s, protects cells from heat shock-induced nuclear damage. Cell. 2012;149:578–589. doi: 10.1016/j.cell.2012.02.058. [DOI] [PubMed] [Google Scholar]
- 63.Chu A, Matusiewicz N, Stochaj U. Heat-induced nuclear accumulation of hsc70s is regulated by phosphorylation and inhibited in confluent cells. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2001;15:1478–1480. doi: 10.1096/fj.00-0680fje. [DOI] [PubMed] [Google Scholar]
- 64.Udan-Johns M, Bengoechea R, Bell S, Shao J, Diamond MI, True HL, Weihl CC, Baloh RH. Prion-like nuclear aggregation of TDP-43 during heat shock is regulated by HSP40/70 chaperones. Human molecular genetics. 2014;23:157–170. doi: 10.1093/hmg/ddt408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kim S, Nollen EA, Kitagawa K, Bindokas VP, Morimoto RI. Polyglutamine protein aggregates are dynamic. Nature cell biology. 2002;4:826–831. doi: 10.1038/ncb863. [DOI] [PubMed] [Google Scholar]
- 66.Tapia H, Morano KA. Hsp90 nuclear accumulation in quiescence is linked to chaperone function and spore development in yeast. Molecular biology of the cell. 2010;21:63–72. doi: 10.1091/mbc.E09-05-0376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chughtai ZS, Rassadi R, Matusiewicz N, Stochaj U. Starvation promotes nuclear accumulation of the hsp70 Ssa4p in yeast cells. The Journal of biological chemistry. 2001;276:20261–20266. doi: 10.1074/jbc.M100364200. [DOI] [PubMed] [Google Scholar]
- 68.Quan X, Rassadi R, Rabie B, Matusiewicz N, Stochaj U. Regulated nuclear accumulation of the yeast hsp70 Ssa4p in ethanol-stressed cells is mediated by the N-terminal domain, requires the nuclear carrier Nmd5p and protein kinase C. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2004;18:899–901. doi: 10.1096/fj.03-0947fje. [DOI] [PubMed] [Google Scholar]
- 69.Tkach JM, Glover JR. Nucleocytoplasmic trafficking of the molecular chaperone Hsp104 in unstressed and heat-shocked cells. Traffic (Copenhagen, Denmark) 2008;9:39–56. doi: 10.1111/j.1600-0854.2007.00666.x. [DOI] [PubMed] [Google Scholar]
- 70.Rossi JM, Lindquist S. The intracellular location of yeast heat-shock protein 26 varies with metabolism. The Journal of cell biology. 1989;108:425–439. doi: 10.1083/jcb.108.2.425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tkach JM, Yimit A, Lee AY, Riffle M, Costanzo M, Jaschob D, Hendry JA, Ou J, Moffat J, Boone C, et al. Dissecting DNA damage response pathways by analysing protein localization and abundance changes during DNA replication stress. Nature cell biology. 2012;14:966–976. doi: 10.1038/ncb2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Makhnevych T, Wong P, Pogoutse O, Vizeacoumar FJ, Greenblatt JF, Emili A, Houry WA. Hsp110 is required for spindle length control. The Journal of cell biology. 2012;198:623–636. doi: 10.1083/jcb.201111105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Dasgupta A, Ramsey KL, Smith JS, Auble DT. Sir Antagonist 1 (San1) is a ubiquitin ligase. The Journal of biological chemistry. 2004;279:26830–26838. doi: 10.1074/jbc.M400894200. [DOI] [PubMed] [Google Scholar]
- 74.Gardner RG, Nelson ZW, Gottschling DE. Degradation-mediated protein quality control in the nucleus. Cell. 2005;120:803–815. doi: 10.1016/j.cell.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 75.Matsuo Y, Kishimoto H, Tanae K, Kitamura K, Katayama S, Kawamukai M. Nuclear protein quality is regulated by the ubiquitin-proteasome system through the activity of Ubc4 and San1 in fission yeast. The Journal of biological chemistry. 2011;286:13775–13790. doi: 10.1074/jbc.M110.169953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Fredrickson EK, Gallagher PS, Clowes Candadai SV, Gardner RG. Substrate recognition in nuclear protein quality control degradation is governed by exposed hydrophobicity that correlates with aggregation and insolubility. The Journal of biological chemistry. 2013;288:6130–6139. doi: 10.1074/jbc.M112.406710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Fredrickson EK, Rosenbaum JC, Locke MN, Milac TI, Gardner RG. Exposed hydrophobicity is a key determinant of nuclear quality control degradation. Molecular biology of the cell. 2011;22:2384–2395. doi: 10.1091/mbc.E11-03-0256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Rosenbaum JC, Fredrickson EK, Oeser ML, Garrett-Engele CM, Locke MN, Richardson LA, Nelson ZW, Hetrick ED, Milac TI, Gottschling DE, et al. Disorder targets misorder in nuclear quality control degradation: a disordered ubiquitin ligase directly recognizes its misfolded substrates. Molecular cell. 2011;41:93–106. doi: 10.1016/j.molcel.2010.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Heck JW, Cheung SK, Hampton RY. Cytoplasmic protein quality control degradation mediated by parallel actions of the E3 ubiquitin ligases Ubr1 and San1. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:1106–1111. doi: 10.1073/pnas.0910591107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Guerriero CJ, Weiberth KF, Brodsky JL. Hsp70 targets a cytoplasmic quality control substrate to the San1p ubiquitin ligase. The Journal of biological chemistry. 2013;288:18506–18520. doi: 10.1074/jbc.M113.475905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Prasad R, Kawaguchi S, Ng DT. A nucleus-based quality control mechanism for cytosolic proteins. Molecular biology of the cell. 2010;21:2117–2127. doi: 10.1091/mbc.E10-02-0111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Deng M, Hochstrasser M. Spatially regulated ubiquitin ligation by an ER/nuclear membrane ligase. Nature. 2006;443:827–831. doi: 10.1038/nature05170. [DOI] [PubMed] [Google Scholar]
- 83.Ravid T, Kreft SG, Hochstrasser M. Membrane and soluble substrates of the Doa10 ubiquitin ligase are degraded by distinct pathways. The EMBO journal. 2006;25:533–543. doi: 10.1038/sj.emboj.7600946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Swanson R, Locher M, Hochstrasser M. A conserved ubiquitin ligase of the nuclear envelope/endoplasmic reticulum that functions in both ER-associated and Matalpha2 repressor degradation. Genes & development. 2001;15:2660–2674. doi: 10.1101/gad.933301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Furth N, Gertman O, Shiber A, Alfassy OS, Cohen I, Rosenberg MM, Doron NK, Friedler A, Ravid T. Exposure of bipartite hydrophobic signal triggers nuclear quality control of Ndc10 at the endoplasmic reticulum/nuclear envelope. Molecular biology of the cell. 2011;22:4726–4739. doi: 10.1091/mbc.E11-05-0463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hwang CS, Shemorry A, Varshavsky A. N-terminal acetylation of cellular proteins creates specific degradation signals. Science (New York, NY) 2010;327:973–977. doi: 10.1126/science.1183147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Prasad R, Kawaguchi S, Ng DT. Biosynthetic mode can determine the mechanism of protein quality control. Biochemical and biophysical research communications. 2012;425:689–695. doi: 10.1016/j.bbrc.2012.07.080. [DOI] [PubMed] [Google Scholar]
- 88.Wang Z, Prelich G. Quality control of a transcriptional regulator by SUMO-targeted degradation. Molecular and cellular biology. 2009;29:1694–1706. doi: 10.1128/MCB.01470-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kriegenburg F, Jakopec V, Poulsen EG, Nielsen SV, Roguev A, Krogan N, Gordon C, Fleig U, Hartmann-Petersen R. A chaperone-assisted degradation pathway targets kinetochore proteins to ensure genome stability. PLoS genetics. 2014;10:e1004140. doi: 10.1371/journal.pgen.1004140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ghaemmaghami S, Huh WK, Bower K, Howson RW, Belle A, Dephoure N, O’Shea EK, Weissman JS. Global analysis of protein expression in yeast. Nature. 2003;425:737–741. doi: 10.1038/nature02046. [DOI] [PubMed] [Google Scholar]
- 91.Marguerat S, Schmidt A, Codlin S, Chen W, Aebersold R, Bahler J. Quantitative analysis of fission yeast transcriptomes and proteomes in proliferating and quiescent cells. Cell. 2012;151:671–683. doi: 10.1016/j.cell.2012.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Park SH, Kukushkin Y, Gupta R, Chen T, Konagai A, Hipp MS, Hayer-Hartl M, Hartl FU. PolyQ proteins interfere with nuclear degradation of cytosolic proteins by sequestering the Sis1p chaperone. Cell. 2013;154:134–145. doi: 10.1016/j.cell.2013.06.003. [DOI] [PubMed] [Google Scholar]
- 93.Russell SJ, Steger KA, Johnston SA. Subcellular localization, stoichiometry, and protein levels of 26 S proteasome subunits in yeast. The Journal of biological chemistry. 1999;274:21943–21952. doi: 10.1074/jbc.274.31.21943. [DOI] [PubMed] [Google Scholar]
- 94.Laporte D, Salin B, Daignan-Fornier B, Sagot I. Reversible cytoplasmic localization of the proteasome in quiescent yeast cells. The Journal of cell biology. 2008;181:737–745. doi: 10.1083/jcb.200711154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Weberruss MH, Savulescu AF, Jando J, Bissinger T, Harel A, Glickman MH, Enenkel C. Blm10 facilitates nuclear import of proteasome core particles. The EMBO journal. 2013;32:2697–2707. doi: 10.1038/emboj.2013.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Takeda K, Yanagida M. Regulation of nuclear proteasome by Rhp6/Ubc2 through ubiquitination and destruction of the sensor and anchor Cut8. Cell. 2005;122:393–405. doi: 10.1016/j.cell.2005.05.023. [DOI] [PubMed] [Google Scholar]
- 97.Tatebe H, Yanagida M. Cut8, essential for anaphase, controls localization of 26S proteasome, facilitating destruction of cyclin and Cut2. Current biology : CB. 2000;10:1329–1338. doi: 10.1016/s0960-9822(00)00773-9. [DOI] [PubMed] [Google Scholar]
- 98.Chen L, Romero L, Chuang SM, Tournier V, Joshi KK, Lee JA, Kovvali G, Madura K. Sts1 plays a key role in targeting proteasomes to the nucleus. The Journal of biological chemistry. 2011;286:3104–3118. doi: 10.1074/jbc.M110.135863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Takeda K, Tonthat NK, Glover T, Xu W, Koonin EV, Yanagida M, Schumacher MA. Implications for proteasome nuclear localization revealed by the structure of the nuclear proteasome tether protein Cut8. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:16950–16955. doi: 10.1073/pnas.1103617108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Bader N, Jung T, Grune T. The proteasome and its role in nuclear protein maintenance. Experimental gerontology. 2007;42:864–870. doi: 10.1016/j.exger.2007.03.010. [DOI] [PubMed] [Google Scholar]
- 101.von Mikecz A. The nuclear ubiquitin-proteasome system. Journal of cell science. 2006;119:1977–1984. doi: 10.1242/jcs.03008. [DOI] [PubMed] [Google Scholar]
- 102.Bennett EJ, Bence NF, Jayakumar R, Kopito RR. Global impairment of the ubiquitin-proteasome system by nuclear or cytoplasmic protein aggregates precedes inclusion body formation. Molecular cell. 2005;17:351–365. doi: 10.1016/j.molcel.2004.12.021. [DOI] [PubMed] [Google Scholar]
- 103.Catic A, Suh CY, Hill CT, Daheron L, Henkel T, Orford KW, Dombkowski DM, Liu T, Liu XS, Scadden DT. Genome-wide Map of Nuclear Protein Degradation Shows NCoR1 Turnover as a Key to Mitochondrial Gene Regulation. Cell. 2013;155:1380–1395. doi: 10.1016/j.cell.2013.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ullrich O, Reinheckel T, Sitte N, Hass R, Grune T, Davies KJ. Poly-ADP ribose polymerase activates nuclear proteasome to degrade oxidatively damaged histones. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:6223–6228. doi: 10.1073/pnas.96.11.6223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Andersen JS, Lam YW, Leung AK, Ong SE, Lyon CE, Lamond AI, Mann M. Nucleolar proteome dynamics. Nature. 2005;433:77–83. doi: 10.1038/nature03207. [DOI] [PubMed] [Google Scholar]
- 106.Lam YW, Lamond AI, Mann M, Andersen JS. Analysis of nucleolar protein dynamics reveals the nuclear degradation of ribosomal proteins. Current biology : CB. 2007;17:749–760. doi: 10.1016/j.cub.2007.03.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Iwata A, Nagashima Y, Matsumoto L, Suzuki T, Yamanaka T, Date H, Deoka K, Nukina N, Tsuji S. Intranuclear degradation of polyglutamine aggregates by the ubiquitin-proteasome system. The Journal of biological chemistry. 2009;284:9796–9803. doi: 10.1074/jbc.M809739200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Janer A, Martin E, Muriel MP, Latouche M, Fujigasaki H, Ruberg M, Brice A, Trottier Y, Sittler A. PML clastosomes prevent nuclear accumulation of mutant ataxin-7 and other polyglutamine proteins. The Journal of cell biology. 2006;174:65–76. doi: 10.1083/jcb.200511045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Savulescu AF, Shorer H, Kleifeld O, Cohen I, Gruber R, Glickman MH, Harel A. Nuclear import of an intact preassembled proteasome particle. Molecular biology of the cell. 2011;22:880–891. doi: 10.1091/mbc.E10-07-0595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kouranti I, McLean JR, Feoktistova A, Liang P, Johnson AE, Roberts-Galbraith RH, Gould KL. A global census of fission yeast deubiquitinating enzyme localization and interaction networks reveals distinct compartmentalization profiles and overlapping functions in endocytosis and polarity. PLoS biology. 2010:8. doi: 10.1371/journal.pbio.1000471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Boyd SD, Tsai KY, Jacks T. An intact HDM2 RING-finger domain is required for nuclear exclusion of p53. Nature cell biology. 2000;2:563–568. doi: 10.1038/35023500. [DOI] [PubMed] [Google Scholar]
- 112.Cornett J, Cao F, Wang CE, Ross CA, Bates GP, Li SH, Li XJ. Polyglutamine expansion of huntingtin impairs its nuclear export. Nature genetics. 2005;37:198–204. doi: 10.1038/ng1503. [DOI] [PubMed] [Google Scholar]
- 113.Skinner PJ, Koshy BT, Cummings CJ, Klement IA, Helin K, Servadio A, Zoghbi HY, Orr HT. Ataxin-1 with an expanded glutamine tract alters nuclear matrix-associated structures. Nature. 1997;389:971–974. doi: 10.1038/40153. [DOI] [PubMed] [Google Scholar]
- 114.Reichelt M, Wang L, Sommer M, Perrino J, Nour AM, Sen N, Baiker A, Zerboni L, Arvin AM. Entrapment of viral capsids in nuclear PML cages is an intrinsic antiviral host defense against varicella-zoster virus. PLoS pathogens. 2011;7:e1001266. doi: 10.1371/journal.ppat.1001266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Livingston CM, Ifrim MF, Cowan AE, Weller SK. Virus-Induced Chaperone-Enriched (VICE) domains function as nuclear protein quality control centers during HSV-1 infection. PLoS pathogens. 2009;5:e1000619. doi: 10.1371/journal.ppat.1000619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Latonen L, Moore HM, Bai B, Jaamaa S, Laiho M. Proteasome inhibitors induce nucleolar aggregation of proteasome target proteins and polyadenylated RNA by altering ubiquitin availability. Oncogene. 2011;30:790–805. doi: 10.1038/onc.2010.469. [DOI] [PubMed] [Google Scholar]
- 117.Nollen EA, Salomons FA, Brunsting JF, van der Want JJ, Sibon OC, Kampinga HH. Dynamic changes in the localization of thermally unfolded nuclear proteins associated with chaperone-dependent protection. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:12038–12043. doi: 10.1073/pnas.201112398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Audas TE, Jacob MD, Lee S. Immobilization of proteins in the nucleolus by ribosomal intergenic spacer noncoding RNA. Molecular cell. 2012;45:147–157. doi: 10.1016/j.molcel.2011.12.012. [DOI] [PubMed] [Google Scholar]
- 119.Nystrom T. Spatial protein quality control and the evolution of lineage-specific ageing. Philosophical transactions of the Royal Society of London Series B, Biological sciences. 2011;366:71–75. doi: 10.1098/rstb.2010.0282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Sinclair DA, Guarente L. Extrachromosomal rDNA circles--a cause of aging in yeast. Cell. 1997;91:1033–1042. doi: 10.1016/s0092-8674(00)80493-6. [DOI] [PubMed] [Google Scholar]
- 121.Khmelinskii A, Keller PJ, Lorenz H, Schiebel E, Knop M. Segregation of yeast nuclear pores. Nature. 2010;466:E1. doi: 10.1038/nature09255. [DOI] [PubMed] [Google Scholar]
- 122.Pereira G, Tanaka TU, Nasmyth K, Schiebel E. Modes of spindle pole body inheritance and segregation of the Bfa1p-Bub2p checkpoint protein complex. The EMBO journal. 2001;20:6359–6370. doi: 10.1093/emboj/20.22.6359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Hotz M, Leisner C, Chen D, Manatschal C, Wegleiter T, Ouellet J, Lindstrom D, Gottschling DE, Vogel J, Barral Y. Spindle pole bodies exploit the mitotic exit network in metaphase to drive their age-dependent segregation. Cell. 2012;148:958–972. doi: 10.1016/j.cell.2012.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Iwata A, Christianson JC, Bucci M, Ellerby LM, Nukina N, Forno LS, Kopito RR. Increased susceptibility of cytoplasmic over nuclear polyglutamine aggregates to autophagic degradation. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:13135–13140. doi: 10.1073/pnas.0505801102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Rose A, Schlieker C. Alternative nuclear transport for cellular protein quality control. Trends in cell biology. 2012;22:509–514. doi: 10.1016/j.tcb.2012.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Johnson DC, Baines JD. Herpesviruses remodel host membranes for virus egress. Nature reviews Microbiology. 2011;9:382–394. doi: 10.1038/nrmicro2559. [DOI] [PubMed] [Google Scholar]
- 127.Speese SD, Ashley J, Jokhi V, Nunnari J, Barria R, Li Y, Ataman B, Koon A, Chang YT, Li Q, et al. Nuclear envelope budding enables large ribonucleoprotein particle export during synaptic Wnt signaling. Cell. 2012;149:832–846. doi: 10.1016/j.cell.2012.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Costanzo M, Baryshnikova A, Bellay J, Kim Y, Spear ED, Sevier CS, Ding H, Koh JL, Toufighi K, Mostafavi S, et al. The genetic landscape of a cell. Science (New York, NY ) 2010;327:425–431. doi: 10.1126/science.1180823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Ryan CJ, Roguev A, Patrick K, Xu J, Jahari H, Tong Z, Beltrao P, Shales M, Qu H, Collins SR, et al. Hierarchical modularity and the evolution of genetic interactomes across species. Molecular cell. 2012;46:691–704. doi: 10.1016/j.molcel.2012.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Lafontaine DL. A ‘garbage can’ for ribosomes: how eukaryotes degrade their ribosomes. Trends in biochemical sciences. 2010;35:267–277. doi: 10.1016/j.tibs.2009.12.006. [DOI] [PubMed] [Google Scholar]
- 131.Dez C, Houseley J, Tollervey D. Surveillance of nuclear-restricted pre-ribosomes within a subnucleolar region of Saccharomyces cerevisiae. The EMBO journal. 2006;25:1534–1546. doi: 10.1038/sj.emboj.7601035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Shor E, Fox CA, Broach JR. The yeast environmental stress response regulates mutagenesis induced by proteotoxic stress. PLoS genetics. 2013;9:e1003680. doi: 10.1371/journal.pgen.1003680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Chen G, Bradford WD, Seidel CW, Li R. Hsp90 stress potentiates rapid cellular adaptation through induction of aneuploidy. Nature. 2012;482:246–250. doi: 10.1038/nature10795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Oromendia AB, Dodgson SE, Amon A. Aneuploidy causes proteotoxic stress in yeast. Genes & development. 2012;26:2696–2708. doi: 10.1101/gad.207407.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Pavelka N, Rancati G, Zhu J, Bradford WD, Saraf A, Florens L, Sanderson BW, Hattem GL, Li R. Aneuploidy confers quantitative proteome changes and phenotypic variation in budding yeast. Nature. 2010;468:321–325. doi: 10.1038/nature09529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Toyama BH, Savas JN, Park SK, Harris MS, Ingolia NT, Yates JR, 3rd, Hetzer MW. Identification of long-lived proteins reveals exceptional stability of essential cellular structures. Cell. 2013;154:971–982. doi: 10.1016/j.cell.2013.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Soto C. Unfolding the role of protein misfolding in neurodegenerative diseases. Nature reviews Neuroscience. 2003;4:49–60. doi: 10.1038/nrn1007. [DOI] [PubMed] [Google Scholar]
- 138.Schilling G, Savonenko AV, Klevytska A, Morton JL, Tucker SM, Poirier M, Gale A, Chan N, Gonzales V, Slunt HH, et al. Nuclear-targeting of mutant huntingtin fragments produces Huntington’s disease-like phenotypes in transgenic mice. Human molecular genetics. 2004;13:1599–1610. doi: 10.1093/hmg/ddh175. [DOI] [PubMed] [Google Scholar]
- 139.Manzerra P, Brown IR. The neuronal stress response: nuclear translocation of heat shock proteins as an indicator of hyperthermic stress. Experimental cell research. 1996;229:35–47. doi: 10.1006/excr.1996.0341. [DOI] [PubMed] [Google Scholar]