

Abstract
Motivation: Accurately predicting protein secondary structure and relative solvent accessibility is important for the study of protein evolution, structure and function and as a component of protein 3D structure prediction pipelines. Most predictors use a combination of machine learning and profiles, and thus must be retrained and assessed periodically as the number of available protein sequences and structures continues to grow.
Results: We present newly trained modular versions of the SSpro and ACCpro predictors of secondary structure and relative solvent accessibility together with their multi-class variants SSpro8 and ACCpro20. We introduce a sharp distinction between the use of sequence similarity alone, typically in the form of sequence profiles at the input level, and the additional use of sequence-based structural similarity, which uses similarity to sequences in the Protein Data Bank to infer annotations at the output level, and study their relative contributions to modern predictors. Using sequence similarity alone, SSpro’s accuracy is between 79 and 80% (79% for ACCpro) and no other predictor seems to exceed 82%. However, when sequence-based structural similarity is added, the accuracy of SSpro rises to 92.9% (90% for ACCpro). Thus, by combining both approaches, these problems appear now to be essentially solved, as an accuracy of 100% cannot be expected for several well-known reasons. These results point also to several open technical challenges, including (i) achieving on the order of ≥80% accuracy, without using any similarity with known proteins and (ii) achieving on the order of ≥85% accuracy, using sequence similarity alone.
Availability and implementation: SSpro, SSpro8, ACCpro and ACCpro20 programs, data and web servers are available through the SCRATCH suite of protein structure predictors at http://scratch.proteomics.ics.uci.edu.
Contact: pfbaldi@uci.edu
1 INTRODUCTION
The prediction of protein structural features, such as secondary structure and relative solvent accessibility, are useful for the study of protein evolution, structure and function and as modular components of protein 3D structure prediction pipelines. Most of the best predictors use a combination of machine learning and evolutionary information, in the form of multiple alignment profiles, and thus must be retrained and assessed periodically as the available protein data continues to grow. Here we present the results obtained by modifying and retraining SSpro and ACCpro (Cheng et al., 2005; Pollastri et al., 2001, 2002), two widely used predictors of secondary structure and relative solvent accessibility, respectively, and broadly assessing prediction performance in these fields.
It has been known for two decades that evolutionary information in the form of profiles calculated on similar sequences helps predictors. In the case of secondary structure prediction, for instance, performance accuracy improves by roughly 2 percentage points when profiles are used in the input, as opposed to raw sequences alone. It is also known that using profiles in the output, by predicting the secondary structure of each sequence in an alignment and taking the ‘majority’ of each column, leads to a smaller improvement (<1%) over using the raw sequences alone. Furthermore, using profiles in both the input and the output usually leads to no significant improvement over using profiles in the input alone.
Machine learning-based predictors developed during the past two decades have been almost exclusively focused and evaluated on their ability to predict the secondary structure or relative solvent accessibility from the sequence or the profile alone. The past decade of work has not produced major prediction improvements, resulting in a set of competing predictors with similar methods, training datasets, protocols and, most importantly, similar prediction performances—for instance, roughly around 80% for secondary structure prediction. This is despite the number of experimentally solved structure deposited in the Protein Data Bank (PDB) (Berman et al., 2000), which has significantly increased over the same period (Fig. 1) and continues to increase faster each year (Fig. 2).
Fig. 1.

Number of protein chains in the PDB from 1990 to 2013
Fig. 2.

Number of protein chains deposited every month in the PDB from 1990 to 2013
However, besides using sequence similarity to create input profiles, there is potentially a second way of using sequence similarity to leverage the growth of the PDB and improve predictions. Namely, if a portion of a query sequence is similar to a sequence in the PDB, it may be possible to use the annotation of the PDB sequence to annotate the query sequence, in lieu or in combination with the prediction produced by the machine learning methods. This is what we call sequence-based structural similarity. Although the correlation between sequence similarity and structure similarity is not perfect (Kosloff and Kolodny, 2008), it is well known that two domains with similar sequences will in general have similar structures (Kaczanowski and Zielenkiewicz, 2010).
There are good reasons to suspect that sequence-based structural similarity may be effective. A simple analysis of the PDB entries released between 1996 and 2013 (Figs 3 and 4) reveals that as of today, only 6% of the amino acids deposited in the PDB each month belong to protein regions with no similarity to sequences previously deposited in the PDB (Fig. 3). Furthermore, this percentage is decreasing year after year. Finally, when we examine the large, redundancy reduced and representative set of all proteins UNIREF50 (Suzek et al., 2007), we find that 62% of its sequences have at least one domain with a similarity of ≥30% to a sequence in the PDB.
Fig. 3.

Mean number of previously deposited chains found with sequence similarity of at least 30% with respect to newly deposited sequences in the PDB, computed on a monthly basis from 1996 to 2013
Fig. 4.

Percentage of amino acids in newly deposited sequences covered by previously deposited chains with sequence similarity of at least 30% in the PDB, computed on a monthly basis from 1996 to 2013
Thus, in short, the main purposes of this article, besides the upgrade of existing predictors, is to study in detail the effectiveness of sequence-based structural similarity for secondary structure and relative solvent accessibility prediction alone, and how it can be combined with predictions derived by machine learning methods with profiles to improve the overall state-of-the-art.
2 METHODS
2.1 Training datasets
Three datasets are curated to train and evaluate the four predictors (SSpro, ACCpro, SSpro8 and ACCpro20). The first dataset (pdb_full) is derived from the PDB structures released before August 20, 2013. Protein structures solved by X-ray crystallography with a resolution of at least 2.5 angstroms, with no chain breaks, with less than five unknown amino acids and of length at least 30 are first extracted from the database. Redundancy of the selected protein chains is first reduced at the 25% sequence identity level using CD-HIT (Li and Godzik, 2006) and Blastclust (Altschul et al., 1997), and then further reduced using an HSSP cutoff distance of 5 using UniqueProt (Mika and Rost, 2003) resulting in 5772 protein chains. The second dataset (pdb_ante) is derived from the PDB entries released before May 1, 2012 following the same protocol than pdb_full and contains 5310 protein chains. The third dataset (pdb_post) contains the 11 213 protein chains passing the filtering criteria described above deposited in the PDB after May 1, 2012 and before August 20, 2013. Redundancy in this last dataset is not reduced because its purpose is precisely to allow a realistic assessment of post-performance given pre-training data.
The secondary structure and solvent accessibility values assigned by DSSP (Kabsch and Sander, 1983) to each amino acid in these datasets are used to determine the target classes for the four predictors: secondary structure in 3 and 8 classes, and relative solvent accessibility for 20 different thresholds from 0 to 95% in 5% increments.
2.2 Three-stage prediction
The four predictors share the same three-stage prediction workflow depicted in Figure 5. The first step is similar to other predictors and consists in using three iterated steps of PSI-BLAST with the UNIREF50 database (Suzek et al., 2007) to derive multiple sequence alignment and profile probabilities. The second step is similar to previous versions of SSpro or ACCpro, and uses an ensemble of 100 Bidirectional Recursive Neural Networks (BRNNs) trained on the data to generate a first set of probability predictions for each secondary structure or solvent accessibility class. Any other machine learning approach can be substituted in this step. The third stage replaces the previous predictions with predictions derived using sequence-based structural similarity in regions where similar sequences can be found in the PDB. Similar sequences extracted using BLAST are filtered such that at least 10 amino acids are aligned to the target protein without any gap (30 amino acids for relative solvent accessibility), the BLAST expectation value is <10−9 and the aligned regions must have at least 45% identical amino acids and 55% positive substitutions (70 and 75%, respectively, for relative solvent accessibility predictions). The most frequent DSSP-assigned class in the set of proteins selected for a given position in the target protein is selected as the final prediction for that position. The BRNN predictions from the second stage are used for all positions that do not have similar regions in the PDB, as well as those with similar sequences in the PDB but no dominant DSSP class.
Fig. 5.
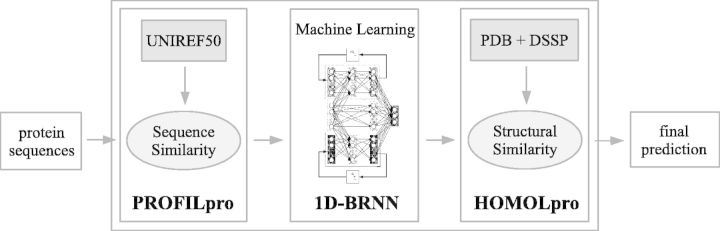
General workflow for the SSpro, SSpro8, ACCpro and ACCpro20 predictors. Sequence and structural similarity analyses are performed by stand-alone modules (PROFILpro and HOMOLpro, respectively). BRNN models are trained to predict the features from the profiles and combined in an ensemble
2.3 Evaluation procedure
The four predictors are evaluated following two distinct protocols.
The first protocol (P1) aims to estimate the accuracy of the final predictors made available online. A double 10-fold cross validation is performed on pdb_full where the data is first randomly divided into 10 folds. For each cross validation fold, the remaining 90% of the data is further subdivided into 10 sub-folds, and 10 BRNN models are trained, respectively, on each set of nine sub-folds. The 10 models are tested together on the remaining 10% of the data. The resulting ensemble of 100 BRNN models is used to produce the final predictor. Predictions using sequence-based structural similarity are added to the BRNN predictions after each step of the cross validation. To provide a complete but fair evaluation of the sequence-based structural similarity predictions, protein chains identical to any protein sequence in pdb_full are removed from the PDB for this evaluation. However, sequences that are similar but not identical to sequences in pdb_full are not removed, regardless of their similarity level, precisely to evaluate the prediction accuracy as a function of sequence similarity.
The second protocol (P2) aims to estimate how the method performs over time by training the BRNN models following protocol P1 but using the smaller set of PDB entries released before May 1, 2012 (dataset pdb_ante), and testing the resulting predictor on all the protein entries released after May 1, 2012 (dataset pdb_post). Sequence-based structural similarity predictions in this case use all the PDB entries released before May 1, 2012, thus enabling the assessment of the performance of the combined predictor over a 16 month period without retraining.
2.4 Comparison with other predictors
Fair comparisons with existing predictors are difficult for several reasons. First, the various predictors are trained on different datasets, at different times, using different versions of the PDB. Often, the datasets are not available and, even when they are, the details of the cross validation datasets are not preventing a perfect comparison requiring training and testing on the same folds. Second, retraining third-party predictors using our own data is also not feasible because the trainable version of these predictors is not publicly available. Furthermore, retraining third-party software is subject to criticism owing to the variability resulting from the fine-tuning of hyperparameters and related matters. Third, even the most popular predictors still actively updated, like DISTILL (Mirabello and Pollastri, 2013) or PSIPRED (Buchan et al., 2013), do not come with a sequence-based structural similarity module.
Nevertheless, we provide some measure of comparison by reporting the accuracy numbers for DISTILL and PSIPRED, using the most recent version of these programs kindly made available to us by the authors. We compute these accuracy numbers on the same datasets used to evaluate SSpro and ACCpro, giving the other predictors the benefit of the doubt, as some of the proteins in our test sets are likely to be in the training sets of the other predictors. Because both DISTILL and PSIPRED use profile inputs and are currently provided without corresponding sequence profile generators, we used our own generator PROFILpro to generate all the necessary sequence profiles. Note that PROFILpro is available for download, together with all our other predictors.
3 RESULTS
Results obtained following the two protocols described in Section 2.3 are summarized in Table 1. The accuracy of the predictors on the pdb_full and pdb_ante datasets is obtained following the P1 protocol, whereas the accuracy of the predictors on the pdb_post dataset is obtained following the P2 protocol.
Table 1.
SSpro, ACCpro, SSpro8 and ACCpro20 prediction accuracy evaluated following the protocols described in Section 2.3
| Predictor | pdb_full (%) | pdb_ante (%) | pdb_post (%) |
|---|---|---|---|
| SSpro | 92.91 | 92.92 | 91.74 |
| SSpro8 | 87.92 | 87.92 | 85.88 |
| ACCpro | 90.02 | 90.13 | 88.22 |
| ACCpro20 | 89.92 | 90.02 | 87.98 |
| SSpro (2005) | 83.21 | 84.04 | 85.68 |
| SSpro8 (2005) | 63.38 | 63.59 | 63.50 |
| ACCpro (2005) | 80.71 | 81.17 | 81.50 |
| ACCpro20 (2005) | 76.31 | 76.39 | 77.73 |
| DISTILL Porter 4.0 | 82.56 | 82.74 | 81.62 |
| DISTILL PaleAle 4.0 | 80.43 | 80.51 | 81.28 |
| PSIPRED 3.3 | 80.59 | 80.74 | 79.60% |
Note: Accuracy reported for the pdb_full and pdb_ante datasets is obtained following protocol P1, and accuracy reported for the pdb_post dataset is obtained following protocol P2. Accuracy reported for ACCpro20 is the accuracy at the 25% accessibility threshold. Accuracy of the newly trained versions of SSpro and ACCpro are reported in bold font. Previous published accuracy of the predictors is also reported (Cheng et al., 2005). Accuracy of DISTILL (Mirabello and Pollastri, 2013) and PSIPRED (Buchan et al., 2013) are reported for the same datasets using the downloadable packages made available by the authors (see Section 2.4).
Using both sequence similarity and sequence-based structural similarity, SSpro’s accuracy is 92.91% on pdb_full and 91.74% when trained on pdb_ante (PDB entries released before May 1, 2012) and evaluated on the 11 213 recent PDB entries in pdb_post (released after May 1, 2012). This is considerably better than any other existing predictor, including the previous release of SSpro (Table 1). This result, combined with the initial analysis performed on the PDB and UNIREF50 databases (see Section 1), highlights the interest of systematically combining sequence-based structural similarity with profile-based machine learning methods. Without using sequence-based structural similarity, the accuracy of the best secondary structure predictors (including SSpro) is estimated to be between 79 and 82% (Mirabello and Pollastri, 2013), thus 10% lower than the accuracy of SSpro after combining the raw predictions with the ones based on known protein structures. It is useful to contrast the combination of both approaches with the use of sequence-based structural similarity alone. For instance, using sequence-based structural similarity alone, predictions can be made for 75% of the amino acids in the dataset pdb_post, with an overall accuracy of 96%.
SSpro’s accuracy as a function of sequence similarity and identity is reported in Figure 6, showing that as soon as similar sequences can be found in the PDB, even with low sequence identity or similarity with the target protein, the prediction accuracy significantly increases. SSpro is also the only secondary structure predictor made publicly available for download, which includes both profile generation and sequence-based structural similarity modules.
Fig. 6.

SSpro prediction accuracy on the pdb_full and pdb_post datasets calculated as a function of the percentage of sequence identity or similarity (BLAST positive substitutions) with proteins found in the PDB. Cases where no similar sequence is found in the PDB for a given residue position, and thus predicted without using sequence-based structural similarity, are included in the 0% sequence identity or similarity case
Similarly, at the 25% accessibility threshold, ACCpro accuracy is 90.02% on pdb_full and 88.22% on pdb_post, above any other predictor, and about 8% better than the accuracy of the previous ACCpro release. A similar detailed evaluation of the predictor’s accuracy, as a function of sequence identity and similarity, is provided in Figure 7.
Fig. 7.

ACCpro prediction accuracy on the pdb_full and pdb_post datasets calculated as a function of the percentage of sequence identity or similarity (BLAST positive substitutions) with proteins found in the PDB. Cases where no similar sequence is found in the PDB for a given residue position, and thus predicted without using sequence-based structural similarity, are included in the 0% sequence identity or similarity case
Results for SSpro and ACCpro multi-class variants SSpro8 and ACCpro20 also show significant improvements. SSpro8 is the eight-class version of SSpro, predicting the protein secondary structure using the eight different classes assigned by DSSP. The sequence-based structural similarity analysis is available for the first time for this kind of predictor and results in an accuracy of 87.92% on pdb_full and 85.88% on pdb_post, a 22% improvement over the last published accuracy of the SSpro8 predictor. ACCpro20 is the 20-class version of ACCpro predicting if an amino acid is buried or exposed for 20 different relative solvent accessibility thresholds. ACCpro20 prediction accuracy is 89.92% in the hard case (25% accessibility threshold) and is now comparable with the accuracy of ACCpro. Note that both predictors have no other existing method to be directly compared with.
Finally, redundancy in the SSpro and ACCpro training sets was reduced using first a 25% sequence identity threshold. This is a common practice in the field, established over the years through many trial and errors and aimed at balancing multiple constraints: in particular redundancy reduction versus training set size. However, there is nothing fundamental about such a threshold, and one might be concerned by the existence of proteins in the training and test sets that are remotely homologous, with a level of sequence identity <25%, which could lead to overestimating the accuracy of the predictors. To address this point, first note that we used a more stringent redundancy reduction procedure than a simple 25% identity cutoff by using an additional HSSP distance cutoff (see Section 2). Second, we conducted one additional set of tests. For each test protein in pdb_full, we used PSI-BLAST to extract any segment of length at least 10 with sequence identity of at least 18% with any other protein in the training set. In total, 58% of amino acids in the test set were found to occur in such segments, leaving 42% of amino acids in the test set with no similarity to the training set (at the 18% identity level). The accuracy of SSpro on both groups of amino acids is almost identical: 92.98% on the first group (58% of all amino acids) and 92.81% on the second, highly non-redundant, group (42% of all amino acids), with a combined accuracy of 92.91% as reported above. Interestingly, using a much higher and more permissive identity threshold of 50% over at least 10 amino acids, the predictor’s accuracy is 92.61% on the redundant group (7% of all amino acids) and 92.93% on the least redundant group (93% of all amino acids). Similar results are observed for ACCpro. Thus, these results show that the small residual redundancy between the training and test sets does not lead to overestimating the performance of the predictors. Within reasonable ranges, this accuracy is roughly constant regardless of the residual level of similarity, providing additional confirmation of the validity of the training protocol used here.
4 DISCUSSION AND CONCLUSION
SSpro and ACCpro achieve a performance accuracy above 92 and 90%, respectively, when presented with samples of proteins either in the PDB (pdb_full) or in the process of being added to the PDB (pdb_post). Barring surprises from proteins that yet remain to be discovered, one may conjecture that the problems of protein secondary structure and relative solvent accessibility prediction are close to being solved.
It is well known that there are fundamental reasons why an accuracy of 100% should not be expected, including (i) the presence of disordered regions; (ii) the ambiguities inherent in the definitions of secondary structure or relative solvent accessibility, as reflected by the imperfect correlation between several programs for determining these features from PDB files; (iii) the errors and uncertainties contained in the PDB; and (iv) the role of the solvent and other molecules, from ions to chaperone proteins, in determining structure, and which are not taken into consideration by most present methods.
As the PDB continues to grow, so will the coverage provided by sequence-based structural similarity methods. Systematically combining profiles, machine learning methods and sequence-based structural similarity seems to be the best strategy, and this is one of the reasons we are providing separate modules for each one of these three tasks. Because protein structures are more conserved than protein sequences, in the future further small improvements may be possible by using methods capable of detecting remote structural similarity, not readily visible in the sequences alone. However, the best existing such methods use predicted secondary structure and relative solvent accessibility to detect remote homology. To avoid any circularity, in the present study we have striven to separate the detection of structural similarity from the prediction of secondary structure.
Finally, when proteins fold in vivo or even in vitro, they do not use sequence or structural similarity at all. This suggests that our understanding of protein structural features is far from complete and points to at least two interesting technical challenges for the foreseeable future: (i) predicting structural features with an accuracy of about ≥80%, using no similarity to known proteins at all, i.e. no profiles; (ii) predicting structural features with an accuracy of ≥85%, using sequence similarity alone and no structural similarity. We believe that the key to addressing these challenges is to use machine learning methods that can use information contained in larger input windows, ideally the entire protein lengths. Recent progress in deep learning methods, including the use of new training approaches such as the dropout algorithm (Baldi and Sadowski, 2014) and GPU clusters, may offer some promising directions.
4.1 Software availability
The four predictors are included in the SCRATCH suite of predictors available online at http://scratch.proteomics.ics.uci.edu together with all the data. A stand-alone version of the predictors can be downloaded from the same url and is free for academic use. PROFILpro, the sequence profile generator used by the four predictors, and HOMOLpro, the sequence-based structural similarity secondary structure and relative solvent accessibility predictor, are also made available as stand-alone tools. They can be used in combination with other secondary structure or solvent accessibility predictors.
ACKNOWLEDGEMENTS
We acknowledge the support of the UCI Institute for Genomics and Bioinformatics and a hardware donation by NVIDIA. Additional support of our computational infrastructure has been provided by Yuzo Kanomata.
Funding: This work has been supported by grants NIH LM010235, NIH NLM T15 LM07443 and NSF-IIS-1321053 to P.B.
Conflicts of Interest: none declared.
REFERENCES
- Altschul SF, et al. Gapped blast and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldi P, Sadowski P. The dropout learning algorithm. Artif. Intell. 2014;210:78–122. doi: 10.1016/j.artint.2014.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman HM, et al. The protein data bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchan DW, et al. Scalable web services for the PSIPRED protein analysis workbench. Nucleic Acids Res. 2013;41:W349–W357. doi: 10.1093/nar/gkt381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng J, et al. SCRATCH: a protein structure and structural feature prediction server. Nucleic Acids Res. 2005;33:W72–W76. doi: 10.1093/nar/gki396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W, Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983;22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- Kaczanowski S, Zielenkiewicz P. Why similar protein sequences encode similar three-dimensional structures? Theor. Chem. Acc. 2010;125:643–650. [Google Scholar]
- Kosloff M, Kolodny R. Sequence-similar, structure-dissimilar protein pairs in the PDB. Proteins. 2008;71:891–902. doi: 10.1002/prot.21770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006;22:1658–1659. doi: 10.1093/bioinformatics/btl158. [DOI] [PubMed] [Google Scholar]
- Mika S, Rost B. Uniqueprot: creating representative protein sequence sets. Nucleic Acids Res. 2003;31:3789–3791. doi: 10.1093/nar/gkg620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirabello C, Pollastri G. Porter, PaleAle 4.0: high-accuracy prediction of protein secondary structure and relative solvent accessibility. Bioinformatics. 2013;29:2056–2058. doi: 10.1093/bioinformatics/btt344. [DOI] [PubMed] [Google Scholar]
- Pollastri G, et al. Prediction of coordination number and relative solvent accessibility in proteins. Proteins. 2001;47:142–153. doi: 10.1002/prot.10069. [DOI] [PubMed] [Google Scholar]
- Pollastri G, et al. Improving the prediction of protein secondary structure in three and eight classes using recurrent neural networks and profiles. Proteins. 2002;47:228–235. doi: 10.1002/prot.10082. [DOI] [PubMed] [Google Scholar]
- Suzek BE, et al. Uniref: comprehensive and non-redundant uniprot reference clusters. Bioinformatics. 2007;23:1282–1288. doi: 10.1093/bioinformatics/btm098. [DOI] [PubMed] [Google Scholar]