

Abstract
Hyperoxaluria can cause not only nephrolithiasis and nephrocalcinosis, but also renal parenchymal disease histologically characterized by deposition of calcium oxalate crystals throughout the renal parenchyma, profound tubular damage and interstitial inflammation and fibrosis. Hyperoxaluric nephropathy presents clinically as acute or chronic renal failure that may progress to end-stage renal disease (ESRD). This sequence of events, well recognized in the past in primary and enteric hyperoxalurias, has also been documented in a few cases of dietary hyperoxaluria. Estimates of oxalate intake in patients with chronic dietary hyperoxaluria who developed chronic kidney disease or ESRD were comparable to the reported average oxalate content of the diets of certain populations worldwide, thus raising the question whether dietary hyperoxaluria is a primary cause of ESRD in these regions. Studies addressing this question have the potential of improving population health and should be undertaken, alongside ongoing studies which are yielding fresh insights into the mechanisms of intestinal absorption and renal excretion of oxalate, and into the mechanisms of development of oxalate-induced renal parenchymal disease. Novel preventive and therapeutic strategies for treating all types of hyperoxaluria are expected to develop from these studies.
Keywords: Dietary hyperoxaluria, Chronic oxalate nephropathy, Acute oxalate nephropathy, Acute tubular necrosis, Interstitial nephritis, Nephrocalcinosis, Calcium oxalate nephrolithiasis, Oxalate transporters, Inflammasomes
Core tip: Chronic nephropathy secondary to dietary hyperoxaluria has been reported in a limited number of patients. Dietary oxalate intake in these patients was lower than the average intake in certain parts of the world. This raises the question whether dietary hyperoxaluria has been a neglected cause of chronic kidney disease. This question along with recent findings elucidating the pathogenesis of oxalate nephropathy calls for further research in epidemiology, prevention and treatment of hyperoxaluria.
INTRODUCTION
Oxaluria has been extensively studied in the context of nephrolithiasis[1-15]. While hyperoxaluria from various causes represents a definitive risk for calcium oxalate nephrolithiasis[1,2], lacking is convincing epidemiological evidence that oxaluria is a risk factor for idiopathic renal stone formation[9,10]. In addition to nephrolithiasis, hyperoxaluria can also cause nephrocalcinosis involving the renal cortex, the renal medulla, or both[16-21], acute kidney injury (AKI) and chronic kidney disease (CKD). Oxaluria has two sources: oxalate formed endogenously from metabolism of its precursors and oxalate absorbed from the gastrointestinal tract. Increased rate of formation or increased rate of absorption of oxalate can lead to hyperoxaluria. The principal aim of this review is to address various aspects of hyperoxaluric AKI and CKD with emphasis on nephropathy secondary to high dietary intake of oxalate. This topic was selected because of its potential epidemiologic importance. In addition, interest to the topic is enhanced by important recent developments in the pathogenesis of hyperoxaluric CKD and the relative paucity of published information on renal parenchymal disease from dietary hyperoxaluria.
This review will analyze in sequence the biochemistry of oxalate and oxalate stones, the pathways of hepatic synthesis of oxalate, the gastrointestinal absorption and renal excretion of oxalate, the various types of hyperoxaluria with emphasis on the dietary variety, and the histologic types of oxalate nephropathy and their pathogenesis. The final section focuses on future research avenues that may illuminate the topic of dietary hyperoxaluria. The potential benefit from this research could be a reduction of the incidence of end-stage renal disease (ESRD)[22].
CHEMISTRY AND PROPERTIES OF OXALIC ACID AND OXALATE STONES
Oxalic acid is a two-carbon dicarboxylic acid (HOOC-COOH). For a long time it was thought that oxalate stones were comprised of mono- and di-hydrates of calcium oxalate, with some contribution from trihydrates. However, recent studies have led to a picture in which some non-oxalate preformed particle such as a crystal of uric acid, phosphate salts, drugs or drug metabolite act as the heterogeneous nucleus for formation of the oxalate calculus[23].
Oxalic acid is a moderately strong acid with pKa values of 1.23 and 4.19. In its full ionic form it is called oxalate. Whereas oxalic acid is relatively soluble in water (8700 mg/dL; pH 7, 20 °C), calcium oxalate is three to four orders of magnitude less soluble (0.67 mg/dL; pH 7.0, 20 °C) and crystallizes readily. By way of comparison, calcium urate is about 400-fold more soluble than calcium oxalate[24]. Oxalate also forms crystals with other polyvalent ions, including magnesium, ferrous iron and zinc. The water solubility (expressed as mg/dL) of these complexes at 18 °C to 20 °C is as follows: magnesium oxalate 70.0, ferrous oxalate 22.0 and zinc oxalate 0.79, respectively. The solubility of calcium oxalate increases slightly with increasing pH; however, hydrogen ion changes in the physiological range have only a small effect on calcium oxalate solubility.
Oxalic acid is a toxic substance. It is not known whether oxalic acid and oxalate are themselves toxic before they react with calcium to form calcium oxalate. Under normal circumstances the concentration of oxalate in the blood and urine depends on the content of oxalic acid in foods and on metabolic conversion of endogenous oxalate precursors largely by oxidative reactions. Furthermore, dietary factors and substances other than oxalic acid per se can influence the tendency for oxalate crystals to form; these factors include: the amino acids 4-hydroxyproline, serine and glycine, calcium, and possibly ascorbic acid and fructose.
EXOGENOUS SOURCES OF OXALIC ACID
In Nature oxalic acid occurs in the free form but more commonly as the salt of sodium, potassium, calcium, magnesium or iron. The oxalate content of dietary items consumed by several populations has been analyzed[6,25-31]. Widely consumed foods that are rich in preformed oxalic acid include vegetables, nuts, cocoa, tea, and fruits high in vitamin C. Red meats, fish, poultry, eggs and dairy products contain relatively small amounts of oxalic acid. Items in Western diets that significantly increase urinary oxalate excretion include spinach, rhubarb, beets, nuts, chocolate, tea, wheat bran, and strawberries[6]. The bioavailability of ingested oxalate is influenced by other ingested items[32]. Oxalate content of various diets, its relation to nephrolithiasis, and guidelines for oxalate intake have been reported[13-15,33-37]. One set of guidelines for prevention of nephrolithiasis proposed a maximal daily oxalate intake of 200 mg daily[33]. We found no epidemiological reports relating dietary oxalate intake to oxalate nephropathy and no guidelines for prevention of this nephropathy.
Table 1 shows estimates of dietary oxalate intake in six countries[13,35,36,38-42]. Oxalate intake varies greatly between countries and regions of the same country. For example, daily oxalate intake in Western diets ranges between 44 and 930 mg[13]. The seasonal variation of oxalate intake in a rural population in India is extreme (Table 1). Very high consumption of oxalate in the context of dietary intake can be comparable to some reported lethal doses of the compound. Although the average lethal dose (LD50) of oxalate was estimated at 375 mg/kg[43], or 26.3 g for a 70 kg person, much lower doses of oxalate can be lethal. An intravenous dose of 1.2 g of sodium oxalate, which is equivalent to 0.8 g of oxalate, was lethal in one reported case[44]. Of note also is that most studies cited in Table 1[13,35,36,39,40,42] as well as other large epidemiological studies[45] analyzed dietary oxalate intake to evaluate the risk of nephrolithiasis and no study addressed the risk of CKD from dietary hyperoxaluria.
Table 1.
Daily dietary oxalate intake in various countries and regions
| Country-region | Subjects | Subject number | Oxalate intake (mg/24-h) | Ref. |
| Brazil, Sao Paolo | +Stones | 70 (M:42, F:28) | 98 ± 1373 | [13] |
| Healthy controls | 41 (M:14, F:27) | 108 ± 1333 | ||
| England | Hospital diet | Not reported | 118 | [38] |
| Germany | +Stones, ↑oxaluria | 93 (M:73, F:20) | 130 ± 1813 | [39] |
| +Stones, →oxaluria | 93 (M:73, F:20) | 101 ± 1453 | ||
| India, Rajasthan | Rural “common” diet | Not reported | 78 | [40] |
| Rural rainy season | Not reported | 2045 | ||
| Urban, upper income | Not reported | 606 | ||
| Urban, lower income | Not reported | 169 | ||
| Hospital diet | Not reported | 139 | ||
| India, Pune | Boys, upper income | 100 | 193 (116-309)4 | [41] |
| Boys, lower income | 100 | 169 (102-354)4 | ||
| Girls, upper income | 100 | 168 (115-209)4 | ||
| Girls, lower income | 100 | 133 (87-209)4 | ||
| Italy | Normal subjects1 | 12 (M:8, F:4) | 335 | [42] |
| Normal subjects2 | 12 (M:8, F:4) | 18 | ||
| United States, South | F, 50-79 yr, +Stones | 1.179 | 330 ± 1613 | [35] |
| F, 50-79 yr. –Stones | 1.179 | 345 ± 1663 | ||
| United States | M, +Stones | 1.627 | 214 ± 1173 | [36] |
| M, -Stones | 44.358 | 214 ± 1213 | ||
| F, older, +Stones | 1.414 | 184 ± 1093 | ||
| F, older, -Stones | 91.358 | 185 ± 1123 | ||
| F, younger, +Stones | 1.564 | 179 ± 1213 | ||
| F, younger, -Stones | 100.260 | 183 ± 1213 |
Diet containing fruits and vegetables;
Diet without fruits and vegetables;
Mean ± SD;
Mean (25th-75th percentile). +Stones: History of urinary stones; -Stones: Absence of history of urinary stones; M: Male; F: Female.
SOURCES OF OXALIC ACID IN THE BODY
The body burden of oxalic acid has two sources, endogenous production in the liver and absorption from the gastrointestinal tract. The pathways of hepatic production and gastrointestinal absorption of oxalic acid are discussed below.
Hepatic production of oxalic acid
Oxalate is synthesized in the liver but is not metabolized further in humans. Oxalic acid produced by catabolism of ingested oxalate precursors by means of normal metabolic pathways contributes significantly to the body’s burden of oxalate. Earlier reports estimated that only 10% of the urinary oxalate was derived from dietary oxalate, while the remaining 90% was derived equally from metabolism of other oxalate precursors, including ascorbic acid[46].
Figure 1 shows the metabolism of oxalate, with emphasis on the pathways of primary hyperoxaluria and of metabolism of ethylene glycol, which is a major cause of acute oxalate intoxication. The major precursors of oxalate under normal circumstances appear to be the amino acids hydroxyproline, glycine and serine (Figure 1). Glycine and serine are present in all food proteins. Oxalate is also the end-product of the metabolism of ingested ethylene glycol, the main component of antifreeze, which is encountered usually in the setting of attempted suicide. In order to facilitate understanding of the these endogenous pathways, it may be helpful to consult Figure 1 which relates the major two-and three-carbon compounds that are relevant to this discussion. The key player in this story is glyoxylate: it is the nexus of pathways that lead to and away from oxalate.
Figure 1.
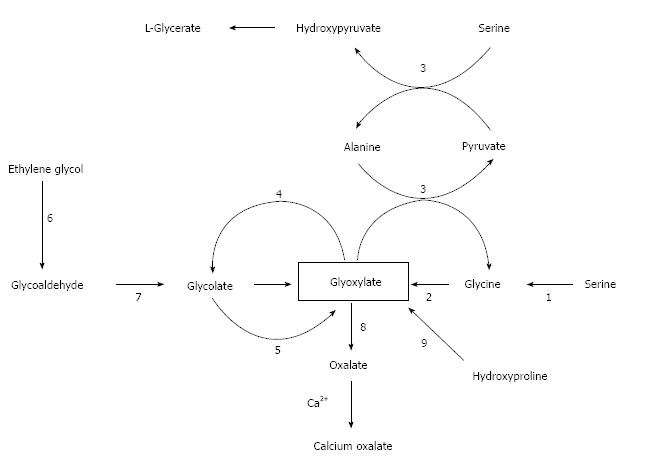
Biosynthesis of calcium oxalate. Glyoxylate is the main precursor of oxalate which combines spontaneously with calcium ions to form calcium oxalate. Names of enzymes: 1, serine hydroxymethyltransferase; 2, D-amino acid oxidase; 3, alanine:glyoxylate aminotransferase (AGT); 4, glyoxylate reductase-hydroxypyruvate reductase (GRHPR); 5, glycolate oxidase; 6, alcohol dehydrogenase; 7, aldehyde dehydrogenase; 8, lactate dehydrogenase; and 9, five enzyme-catalyzed reactions. PH1 results from mutations in AGT which is a hepatic peroxisomal enzyme. PH2 results from mutations in GRHPR which is a cytosolic enzyme found in several tissues, but primarily the liver. PH3 results from defects in the hepatic mitochondrial enzyme 4-hydroxy-2-oxoglutarate (HOG) aldolase which converts HOG and glyoxylate to pyruvate (reaction not shown), the last step in hydroxyproline catabolism. The reason why a deficiency of HOG aldolase activity increases oxalate production is obscure.
Hydroxyproline is one of the most abundant amino acids in collagen. It is present in collagen-containing meat products, including gelatin, and is one of the most abundant proteins in the human body. In fact, collagen accounts for about 30% of total animal proteins and contains about 13% hydroxyproline[47]. Glyoxylate is the two-carbon end-product of hydroxyproline catabolism (pyruvate is the other product). The conversion of glyoxylate to oxalate is catalyzed by lactate dehydrogenase. Each day the human body turns over 2-3 g of collagen. In the process 240-420 mg of hydroxyproline are released with the concomitant production of 140-240 mg of glyoxylate[48].
Knight et al[48] demonstrated using healthy volunteers that daily ingestion of 30 g of collagen for three days increased glycolate and oxalate excretion by 43% and 5.3-fold, respectively. Glycolate is produced when glyoxylate is acted on by glyoxylate reductase which in the literature is also identified as hydroxypyruvate reductase and D-glycerate dehydrogenase. However, only 5% of the ingested hydroxyproline was recovered as glyoxylate plus oxalate, thereby indicating that most of the glyoxylate resulting from hydroxyproline catabolism was probably diverted to glycine synthesis in the reaction catalyzed by alanine:glyoxylate aminotransferase (AGT). The means of directing glyoxylate away from oxalate synthesis is the glyoxylate reductase reaction that converts glyoxylate into glycolate. Since oxalate is not oxidized to carbon dioxide and water or otherwise metabolized by humans, its only route of disposal is urinary excretion. Quantitatively, transamination of glycine and oxidation of glycine by D-amino acid oxidase are much less important than catabolism of hydroxyproline as sources of oxalate.
Since the metabolism of serine and glycine are so intimately linked in humans and because they are interconvertible, it is reasonable to expect that if one of these amino acids is metabolized to glyoxylate, the other too should be a precursor of glyoxylate, and that both should be sources of oxalate. Such is the case. The enzyme that catalyzes the serine-glycine interconversion is folate-dependent serine hydroxymethyl transferase. Another enzyme, namely D- amino acid oxidase, also converts glycine to glyoxylate.
Although the underlying metabolic link between ascorbic acid and oxalic acid is obscure, there is evidence that a high oral or intravenous intake of ascorbic acid can result in a moderate increase in urinary oxalic acid[8,39,49,50]. With regard to parenteral feeding, Robitaille et al[51] found that, on average, 80 mg of a 105 g infused dose of ascorbic acid was recovered as urinary oxalic acid in elderly adults with normal kidney function. Furthermore, intravenous ascorbic acid administration increased urinary oxalic acid excretion in a dose-dependent manner. These authors cautioned against high-dose infusions of ascorbic acid for individuals already at high risk of oxalate stones.
Epidemiologic studies that have addressed the relation between fructose intake and increased risk for oxalate stones have yielded conflicting results: however, a large epidemiological study found a significant association between high consumption of fructose and risk of kidney stones[52]. On the other hand, studies of urinary oxalate excretion in humans administered high amounts of fructose orally[53] or intravenously[54] have produced equivalent results. A 2010 investigation of the relationship between fructose consumption and urinary oxalate in healthy subjects found that urinary excretion of oxalate and glyoxylate, which is a marker of oxalate synthesis, did not change when the fructose content of the diet was raised as high as 21% of calories[55]. A possible effect of fructose on the absorption of dietary oxalate or calcium excretion was not assessed in that study. Furthermore, lacking is evidence that humans metabolize fructose to oxalate. However, fructose could affect the serum oxalate level indirectly by affecting events in the gastrointestinal tract. For example, hyperabsorption of oxalate caused by a low intake of calcium for complexation with oxalate in the GI tract can exacerbate hyperoxaluria[39].
Gastrointestinal absorption of oxalate
The contribution of oxalate absorbed from the gastrointestinal tract to the total body burden of oxalate depends on the oxalate content of the diet. Recently, in a study of normal volunteers consuming diets with varying oxalate content, Holmes and associates[56] showed that oxalate excretion in urine depends significantly on the dietary oxalate intake. Dietary oxalate intake accounted for 24.4% of the urinary oxalate excretion when the diet contained 10 mg of oxalate per 2500 kcal. Urinary oxalate excretion and the percent of urinary oxalate derived from dietary oxalate increased progressively with progressive rises in dietary oxalate content, reached a value of 41.5% of the urine oxalate when the diet contained 250 mg of oxalate per 2500 kcal, and increased further to 52.6% of the urine oxalate when the diet contained both 250 mg of oxalate per 2500 kcal and a low calcium intake. In the same study, although urinary excretion of oxalate increased substantially with increasing oxalate intake, estimated fractional absorption of oxalate from the gastrointestinal tract decreased from 55.4% at the lowest oxalate intake to 5.8% at the highest intake and then increased to 9.7% at the highest oxalate intake combined with low calcium intake[56]. These findings are important in the context of dietary hyperoxaluria.
The functions involved in the disposition of dietary oxalate are exclusively absorption from the intestines and renal excretion[57]. In the intestines, oxalate is absorbed passively by means of a paracellular pathway. Whereas unbound oxalate is absorbable, oxalate salts of divalent cations such as calcium and magnesium are insoluble in water and therefore not absorbable. Oxalate transporters in the enteric[58,59] and renal epithelial cells have been identified and are discussed in some detail in the following subsection.
The magnitude of oxalate absorption is affected by various dietary substances and the gastrointestinal milieu. Dietary oxalate content is an important determinant of oxalate absorption that is particularly relevant to this review. The fact that urinary oxalate is derived from two sources, absorption of dietary oxalate and endogenously produced oxalate, complicates the study of oxalate absorption in the gastrointestinal tract. A reliable method for estimating oxalate absorption is by labelling oxalate with a stable carbon isotope (13C), ingesting a known quantity of labelled oxalate, and measuring the fractional (or percent) excretion of the labelled oxalate in the urine[60,61]. The method assumes that absorbed oxalate is excreted exclusively in the urine. In one study conducted in normal subjects, oxalate absorption was evaluated by this method when total dietary intake of oxalate was low (63 mg daily) and high (600 mg daily). Mean daily urine oxalate was 25 mg at the low oxalate intake and 43 mg at the high intake, while the percent absorption of ingested oxalate increased from 7.9% at the lower intake to 14.7% at the higher oxalate intake[62].
The dietary content of certain divalent cations has clinically important effects on oxalate absorption. High dietary contents of calcium[63-65] and magnesium[66] inhibit oxalate absorption. The mechanism of this inhibition is formation of insoluble and poorly absorbable oxalate salts of these two divalent cations when they are in abundance in the enteric lumen. Fatty acids have an opposite effect from divalent cations on oxalate absorption. High intake of the 20-carbon polyunsaturated fatty acid arachidonic acid was shown to be associated with increased urinary excretion of oxalate[67]. Fatty acids bind bivalent cations, thereby decreasing the latter’s availability for binding oxalate in the intestinal lumen. This effect of fatty acids on oxalate absorption also has clinical implications (see below).
Several anaerobic bacteria, including Oxalobacter formigenes, Eubacterium lentum, Enterococcus faecalis and Lactobacillus acidofilus, metabolize oxalate in the gut[68]. Administration of probiotics containing one or more of these bacteria to healthy subjects and, particularly, subjects with high baseline levels of oxalate absorption, decreases oxalate absorption[68,69]. Conditions that are known to affect oxalate absorption include the pH of the intestinal fluids and intestinal transit time[62]. Whether these conditions have clinical significance or not is unclear.
RENAL EXCRETION OF OXALATE
Oxalate is eliminated almost exclusively by the kidneys. In two studies involving subjects with normal renal function, more than 90% of injected radiolabelled oxalate was recovered in the urine[70,71]. Circulating oxalate is almost 100% ultrafilterable and it is filtered in the glomeruli[72] and excreted in the proximal tubules[73,74]. The basolateral membrane of proximal tubular cells contains a transporter, SLC26A1 that exchanges oxalate for bicarbonate or sulfate[75]. Exchangers of the SLC26 family, including SLC26A6, SLC26A7, SLC26A8, and SLC26A9, have been identified on the plasma membrane of cells that transport oxalate[76,77]. The SLC26A6 transporter has also been localized to the brush border of proximal tubular cells[78]. Holmes and Assimos hypothesized that increases in plasma concentration of oxalate activate the basolateral SLC26A1 transporter which facilitates entry of oxalate into proximal tubular cells, which is then followed by oxalate efflux into the tubular lumen[79]. Tubular secretion of oxalate may have clinical significance. One study found enhanced tubular secretion of oxalate in hyperoxaluric patients compared to controls with normal oxalate excretion[4].
Oxalate transfer in the enteric epithelial cells gut is similar to that in the renal tubular cells. Oxalate transfer through the enteric tight junction is driven by a lumen-to blood concentration gradient and by water absorption. Soluble oxalate is secreted back into the enteric lumen through SLC26A1 and SLC26A6. SLC26A1 is located in the basolateral membrane and transfers oxalate from the paracellular space to the intracellular compartment. SLC26A6 is located in the apical membrane and returns oxalate to the enteric lumen. The transfer of oxalate through the anion transporters back into the enteric lumen modulates the absorption of this toxic compound[59].
In renal failure, oxalate excretion decreases roughly in proportion to the decrease in renal function and serum oxalate concentration increases[80]. As a compensatory mechanism, elimination of oxalate through the gastrointestinal tract is increased in renal failure[81,82]. A study by Hatch and colleagues provided evidence that the increased intestinal excretion of oxalate in renal failure is mediated, at least in part, by angiotensin II[83]. Renal failure, therefore, is one condition in which oxalate is not eliminated in its entirety by the kidneys. Diuresis and body size are two factors that affect urinary oxalate excretion. In normal subjects, oxalate elimination in the urine increases in parallel to urinary flow rate[84,85]. The clinical significance of this finding is obscure because urinary oxalate concentration decreases in parallel as urinary flow increases[85]. Large body size is associated with a high urinary oxalate excretion rate[86,87]. This finding is clinically relevant because obesity is a risk factor for nephrolithiasis[88]. Finally, urinary oxalate excretion shows seasonal variations[89] that can have clinical importance.
CLINICAL TYPES OF HYPEROXALURIA
Hyperoxaluria can result from excessive endogenous production of oxalate, excessive absorption of dietary oxalate, excessive dietary or parenteral intake of oxalate, or a combination of these processes. Four main categories of hyperoxaluria are recognized: primary hyperoxaluria, absorptive or intestinal hyperoxaluria, idiopathic mild hyperoxaluria and dietary hyperoxaluria.
Primary hyperoxaluria
Primary hyperoxaluria (PH) consists of a family of autosomal recessive inherited disorders characterized by endogenous overproduction of oxalate[90-94]. Mutations in three enzymes involved in oxalate synthesis lead to three distinct PH subtypes, PH1[95], PH2[96,97] and PH3[98-100].
PH1 accounts for about 80% of all PH cases. PH1 results from mutations in the hepatic peroxisomal enzyme AGT[93-95]. The gene encoding AGT (AGTX) is located on chromosome 2q37.3[91]. AGT is pyridoxal-5-phosphate dependent[93,94] and catalyzes the transamination of glyoxylate to glycine[94,95]. PH1 mutations result in in accumulation of glyoxylate and excessive production of oxalate and glycolate[94]. Figure 1 illustrates these relationships.
As of 2013, 178 different AGT mutations had been discovered[94]. Phenotypes vary from nephrocalcinosis, failure to thrive and advanced renal failure in early childhood to recurrent or even occasional nephrolithiasis in adulthood[96,97]. As renal failure progresses, high plasma levels of oxalate result in supersaturation and precipitation of calcium oxalate crystals in various organs (oxalosis). Blood vessel walls, bones, joints, retinae, skin, bone marrow, cardiac tissue and the nervous system are sites affected in oxalosis[90-95]. Life-threatening clinical manifestations accompany the deposition of oxalate crystals in vital organs[97].
The diagnosis of PH1 is assisted by finding elevated levels of oxalate and glycolate in the urine. It should be noted, however, that approximately one quarter of subjects with PH1 do not have elevated glycolate levels in the urine[95]. Renal failure consistently decreases urinary oxalate excretion which can cause diagnostic problems[90]. In the past, liver biopsy for assessment of AGT activity was required for the diagnosis of PH1. Nowadays, however, the diagnosis relies on molecular genetic testing including DNA sequencing, deletion/duplication analysis and targeted mutation analysis[95].
The management of PH1 follows some of the same principles of management of urinary stones in general. Fluid intake to ensure large urinary volumes is recommended for patients without advanced renal failure. Calcium supplements and other measures to reduce gastrointestinal absorption of oxalate have limited effectiveness in treating PH. Potassium citrate or, in cases of advanced renal failure, sodium citrate may reduce the tendency to form stones[95]. Pyridoxine administration reduces oxalate formation in 10% to 30% of the patients with PH1[91]. Effectiveness of pyridoxine has been linked to the AGT genotypes Gly170R and Phe152lle, which are associated with some residual activity of the enzyme[91,94]. Combined liver-kidney transplantation is the method of choice for PH1 patients with advanced renal failure[95].
PH2 is found in about 10% of the PH cases. PH2 results from mutations of the cytosolic enzyme glyoxylate reductase/hydroxypyruvate reductase (GRHPR)[94,96]. The gene for GRHPR is located on in chromosome 9p13.2[94]. GRHPR is present in tissues throughout the body and catalyzes the conversion of glyoxylate to glycolate and hydroxypyruvate to D-glycerate[94]. Reduced or absent GRHPR activity leads to increased availability of lactate and hydroxypyruvate for conversion to oxalate and L-glycerate. Urinary excretion of high levels of L-glycerate is a characteristic of PH2[96]. Nephrolithiasis, nephrocalcinosis, end-stage renal failure and oxalosis in advanced renal failure are the clinical hallmarks of PH2[92,96]. The severity of these manifestations is less than in PH1: nephrocalcinosis is rare and end-stage renal failure develops later in life[92]. The diagnosis can be made by assay of GRHPR in blood mononuclear cells[97]. The treatment of PH2 is similar to that of PH1, with two exceptions: pyridoxine is not effective in PH2; and renal transplantation has been used for treatment of end-stage renal failure, while combined liver-kidney transplantation has not been used in PH2[96].
PH3 accounts for 2.5% of PH cases. PH3 results from mutation of the hepatic mitochondrial enzyme 4-hydroxy-2-oxoglutarate aldolase (HOGA1)[98,99]. HOGA1 catalyzes the last step in the conversion of hydroxyproline to oxalate. The chromosomal location of the gene responsible for PH3 is in 10q242[94]. The mechanism by which non-functioning mutations of HOGA1 lead to hyperoxaluria is an enigma. Intuitively, decreased HOGA1 activity should lead to decreased production of oxalate through the hydroxyproline pathway. A hypothesis for the pathogenesis of hyperoxaluria in PH3 was recently proposed by Belostotsky and associates[100]. These investigators identified a cytosolic 4-hydroxy-2-oxoglutarate aldolase distinct from mitochondrial HOGA1 in human hepatocytes. They speculated that individuals with PH3 accumulate 4-hydroxy-2-oxoglutarate in mitochondria and that following transfer of this compound into the cytosol it is converted to glyoxylate by the cytosolic aldolase[100]. Oxaluria is less marked in PH3 than in PH1 or PH2 and the clinical manifestations are less severe. Urolithiasis is the main clinical manifestation in PH3. Furthermore, nephrocalcinosis and renal failure are uncommon, and oxalosis has not been described in PH3[94].
Surveys of primary hyperoxaluria in various countries[101-105] have identified prolonged delays in the diagnosis of PH. Delays in the diagnosis have been observed also in enteric hyperoxaluria and could be present also in dietary hyperoxaluria (see Figure 2 below).
Figure 2.
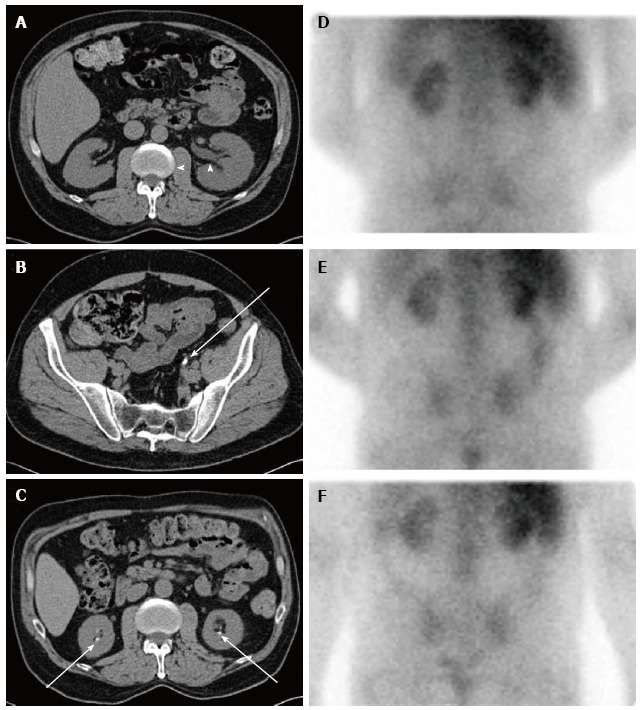
Sequential imaging studies of a not yet reported patient with chronic kidney disease from dietary hyperoxaluria. Axial computed tomography (CT) images obtained two years before the hyperoxaluria diagnosis show (A) mild left hydronephrosis (arrowheads) caused by (B) a left distal ureteral calculus (arrow). Axial CT image obtained around the time of the hyperoxaluria diagnosis shows (C) bilateral nephrolithiasis (arrows). Nuclear medicine gallium-67 citrate scan images were also obtained around the time of diagnosis, including (D) 4-, (E) 24-, and (F) 48 h after administration. These show abnormal, persistent bilateral renal activity at all time points, indicative of interstitial nephritis. Gallium scanning has classically been used to distinguish acute interstitial nephritis from acute tubular necrosis and other causes of acute renal failure[216-218]. In this patient chronic interstitial nephritis associated with hyperoxaluria led to this positive scan. The patient’s diet for several years was based on nuts with estimated oxalate consumption ≥ 800 mg daily. During high oxalate intake, urine oxalate excretion was > 200 mg/24-h in several measurements obtained at serum creatinine levels > 3.5 mg/dL. After resumption of a diet low in oxalate and improvement of renal function to serum creatinine levels < 3.0 mg/dL, urine oxalate excretion decreased to normal levels.
Enteric hyperoxaluria
Table 2 lists some of the conditions and surgical interventions in the gastrointestinal tract, including medical diseases of the gastrointestinal tract, medical or surgical conditions outside the gastrointestinal tract, and medications, that are associated with hyperoxaluria secondary to excessive intestinal absorption of oxalate[106-132]. A common characteristic of the conditions listed in Table 2 is the presence of steatorrhea with excessive amounts of fatty acids in the enteric lumen which bind divalent cations, especially calcium, thereby increasing the availability of the unbound oxalate for absorption[109]. In certain morbid conditions, such as cystic fibrosis, solid organ transplants or octreotide administration, frequent use of antibiotics causing alterations in the intestinal flora and lack of colonization by oxalate-consuming bacteria increases the availability of oxalate for absorption[109,122-129]. In recipients of organ transplants, use of anti-rejection drugs (e.g., mycofenolate) that cause diarrhea and steatorrhea can contribute to hyperoxaluria[127].
Table 2.
Surgical procedures and medical conditions associated with enteric hyperoxaluria
| Surgical conditions | Medical gastrointestinal conditions | Other medical/surgical conditions | Drugs |
| Jejunoileal bypass[106,108,110] | Crohn’s disease[109,119] | Morbid obesity[112] | Orlistat[130,131] |
| Roux-en-y gastric bypass[111,113] | Diabetic gastroenteropathy[115,116] | Cystic fibrosis[122,123] | Octreotide[132] |
| Small bowel resection[108,109] | Sprue[117] | Organ transplants[124-129] | |
| Partial gastrectomy[108] | Primary biliary cirrhosis[109] | ||
| Pancreatectomy[109] | Chronic pancreatitis[118] | ||
| External biliary drainage[114] | Intestinal lymphangiectasia[120] | ||
| Clostridium difficile colitis[121] |
Studies conducted more than 30 years ago documented that the colon was the primary site of oxalate absorption and suggested that an intact colon is necessary for the development of enteric hyperoxaluria[133,134]. However, enteric hyperoxaluria has also been noted in patients with partial colon resection[119]. In patients with enteric hyperoxaluria, diarrhea causes volume depletion and metabolic acidosis leading to low urinary pH and hypocitraturia. In conjunction with hyperoxaluria, these conditions facilitate precipitation of calcium oxalate in renal tissues and promote the development of renal stones, nephrocalcinosis and oxalate nephropathy[109]. In patients with primary hyperoxaluria, the renal failure that follows the development of nephrolithiasis, hydronephrosis, nephrocalcinosis and particularly parenchymal oxalate nephropathy is chronic. Enteric hyperoxaluria can cause new-onset acute renal failure (acute oxalate nephropathy)[121,124-126,129,130,132,135-140], acute renal failure superimposed on pre-existing chronic kidney disease[116,118], or chronic oxalate nephropathy[110,113,119,120,128].
Idiopathic (mild) hyperoxaluria
Idiopathic hyperoxaluria is a condition characterized by hyperoxaluria that is much less severe than primary on enteric hyperoxaluria and recurrent calcium oxalate stone formation[5,141,142]. This entity is encountered in subjects without any of the known types of enteric or primary hyperoxaluria. Increased synthesis, increased gastrointestinal absorption, or increased renal tubular secretion of oxalate are the only known mechanisms of hyperoxaluria. All three mechanisms have been implicated in idiopathic hyperoxaluria. Increased absorption of oxalate by patients with idiopathic hyperoxaluria, especially when the dietary content of calcium is low, has been reported[143-145]. In other studies in patients with the same entity, reduction of hyperoxaluria by large doses of pyridoxine was noted, suggesting that these subjects had excessive production of oxalate[146,147]. In another set of studies subjects with idiopathic hyperoxaluria developed higher levels of oxaluria than control subjects after ascorbate loads[148] or following meat ingestion[12,149]. This set of studies also pointed towards increased endogenous production of oxalate as the source of idiopathic hyperoxaluria. Finally, another study found enhanced tubular secretion in idiopathic hyperoxaluria[4]. Therefore, it is unclear whether idiopathic hyperoxaluria represents one or more types of hyperoxaluria. Further research is needed to clarify the mechanism(s) of hyperoxaluria in this particular condition.
Dietary hyperoxaluria
This section addresses dietary hyperoxaluria and hyperoxaluria secondary to medications or overdoses. The clinical and histological manifestations of these three categories of hyperoxaluria are similar. The reports of nephropathy from dietary hyperoxaluria, especially its chronic variety, are few and contain, in many instances, incomplete information. Clinical and histological findings associated with the last two categories complete the picture of nephropathy in dietary hyperoxaluria.
Dietary hyperoxaluria should be differentiated from the other three categories of hyperoxaluria, since its treatment, which consists of reducing the dietary oxalate, is relatively simple. Elimination of the diagnostic option of primary hyperoxaluria may require genetic testing, but this is usually not required. A careful history should eliminate the possibility of enteric hyperoxaluria. Routine laboratory findings, such as normal serum albumin and electrolyte levels, may assist in eliminating this diagnosis. Differentiating between dietary and idiopathic hyperoxaluria can be difficult. Features establishing the diagnosis of dietary hyperoxaluria include: absence of primary or enteric hyperoxaluria; ingestion of large amounts of oxalate, usually found after the patient’s oxalate-induced end organ damage has become manifest; documented hyperoxaluria associated with a high oxalate diet; and reduction of the oxaluria to within normal levels after normalization of the dietary oxalate. The evaluation of oxaluria is complicated in patients with impaired renal function, which, as noted earlier, decreases urinary oxalate excretion.
Dietary hyperoxaluria can cause renal disease and systemic oxalosis. Earlier studies focused mainly on the association between dietary hyperoxaluria and nephrolithiasis. A study by Neuhaus et al[11] established this association. More recently, several case reports of renal parenchymal disease manifested as either AKI or CKD[150-158] and oxalosis with primary neurological manifestations from dietary hyperoxaluria[159-163] have been published. Identified causes of dietary hyperoxaluria include ingestion of large amounts of the following: peanuts[150]; rhubarb[151]; Chaga mushroom powder[152]; Irumban puli (Averrhoa bilimbi), which is a fruit in the same family as star fruit[153]; juice made of celery, carrots, parsley, beets with greens, and spinach[154]; and, ingestion of star fruit (Averrhoa carambola), which has a very high content of oxalate[155-163]. Star fruit-induced oxalate nephropathy has also been investigated in experimental animals[164,165].
Table 3 shows estimates of oxalate intake and urinary excretion, type of clinical renal syndrome induced by oxalate (AKI vs CKD), peak serum creatinine concentration, whether dialysis was performed or not, and outcomes of patients with dietary hyperoxaluria-induced deterioration of renal function. The estimates of oxalate intake are approximations because estimates of the oxalate content of the same dietary item often vary widely[27,166-168]. We recorded in Table 3 either the oxalate intake reported in a study, or, if this intake was not reported directly, an estimate calculated from the amount of the dietary item consumed and the average oxalate content of this item.
Table 3.
Reports of parenchymal renal disease induced by dietary hyperoxaluria
| Ref. | Daily oxalate intake (mg), duration | Urine oxalate (mg/24 per hour) | Peak SCr (mg/dL) | Clinical diagnosis, course, outcome, final SCr (mg/dL) |
| 150 | 310, many mo | 16.61 | 1.8 | CKD with SCr 1.7-1.8 |
| 151 | 1880, 4 wk | 34.22 | AKI on diabetic CKD. Progression to ESRD | |
| 152 | 2240-2800, 6 mo | - | 8.08 | CKD. Progression to ESRD |
| 153a | 9000, 4 d | 603 | 6.4 | AKI, HDx10 days. SCr 0.9 in 6 wk |
| 153b | 4500, 5 d | - | 9.3 | AKI, HDx6 times. SCr 1.3 in 5 wk |
| 153c | 3600, NS | - | 6 | AKI, No HD. SCr 1.0 in 4 wk |
| 153d | 1800, NS | - | 5.5 | AKI, No HD. SCr 0.8 in 2 wk |
| 153e | 5400-6300, NS | - | 12.3 | AKI, HD. SCr 2.1 in 4 wk |
| 153f | 6300-7200, NS. | - | 6.7 | AKI, no HD. SCr 1.1 in 6 wk |
| 153g | 4500-5400, NS | - | 9.8 | AKI, HD. SCr 1.2 in 6 wk |
| 153h | 6300, NS | - | 6.6 | AKI, HD. SCr 1.1 in 4 wk |
| 153i | 2700-3600, NS | - | 5.2 | AKI, HD. SCr 0.8 in 2 wk |
| 153j | 7200 NS | - | 10.4 | AKI, HD. SCr 1.5 in 6 wk |
| 154 | 1260, 6 wk | - | 7.9 | CKD on CKD from HTN. SCr 1.9 in 4 mo |
| 155a | 13120, once | 74 | 12 | AKI, HDx2 times. SCr 1.3 in 1 yr |
| 155b | 9240, once | 74 | 11.7 | AKI, no HD. SCr 1.3 in 4 mo |
| 156 | 450-660, > 3 yr | - | 6.9 | CKD on other CKD, no HD. SCr 3.4 in 3 mo |
| 157a | 3725, once | - | - | AKI, no HD. Final SCr 1.1 |
| 157b | 4360, once | - | 6.3 | AKI, no HD. Final SCr 1.1 NS |
| 157c | 7545, once | - | 6.1 | AKI, no HD. Final SCr 1.2 |
| 157d | 1300, once | - | 5.7 | AKI, no HD. Final SCr 1.0 |
| 157e | 2170, once | - | 4.5 | AKI, no HD. Final SCr 1.1 |
| 158 | 6830, once | - | 16.4 | AKI, no HD. SCr 0.9 mg/dL in 1 mo |
During recovery. SCr approximately 1.7-1.8 mg/dL;
Post-ingestion. SCr approximately 3.6 mg/dL;
During AKI. SCr approximately 6.4 mg/dL;
Post recovery. a,b,c,d,e,f,g,h,i,j,k in Ref. 153 and a,b,c,d,e in Ref. 157 represent the numerical sequence of the patients in these references (1st, 2nd, etc). SCr 1.3 mg/dL. SCr: Serum creatinine; AKI: Acute kidney injury; CKD: Chronic kidney disease; ESRD: End-stage renal disease; HD: Hemodialysis; NS: Not specified duration of intake.
Data regarding urinary oxalate excretion were missing from the majority of the published cases presented in Table 3. Even when urine oxalate excretion was reported, the findings were complicated by the presence of advanced renal failure, which, as noted above, decreases urinary oxalate excretion, or by the fact that oxalate excretion was measured in the recovery period after oxalate intake had been reduced. An elevation of urinary oxalate excretion rate was reported only in one patient, who also had advanced renal failure[153]. Urinalysis findings varied: Proteinuria was absent in a few patients, modest in most patients, and as high as 3.7 gm/24 h in one patient who also had diabetes mellitus[151]. Hematuria and sterile pyuria were reported in several patients. Crystaluria was absent in several patients.
Oxalate nephropathy in subjects who briefly consumed food items containing very large amounts of oxalate tended to present as AKI, which was severe enough to require hemodialysis in some cases, but appeared to be reversible in all of them (Table 3). A few patients with chronic intake of oxalate at levels substantially lower than those causing AKI did develop CKD; their kidney function improved but did not normalize after reducing their dietary intake of oxalate[150,154,156].
The paucity of reported cases of chronic nephropathy secondary to dietary hyperoxaluria and of measurement of urinary oxalate in those cases led us to investigate other clinical states of temporary hyperoxaluria caused by excessive intake or formation of oxalate. These states include intake of ascorbic acid, drugs containing oxalate and intoxication with ethylene glycol.
As in dietary hyperoxaluria, excessive intake of ascorbate was initially linked to an increased risk of nephrolithiasis[169,170]. Recently, renal parenchymal disease from oxalate nephropathy causing AKI or CKD has been reported in patients with excessive oral[171-178] or parenteral[179-183] intake of ascorbate. An elevated urinary oxalate excretion rate at the time of ingestion of large amounts of ascorbate and decrease in oxaluria to within or close to its normal range was reported in several cases[171,172,175,179]. Severe AKI was present in most cases[171,173,174,179-183]. Several of these patients required hemodialysis for various periods of time and recovery of renal function was complete[171,174,179-183] or partial[173]. CKD was noted in four patients[175-178]. These patients were ingesting ascorbate chronically but usually in quantities substantially lower than the amounts of ascorbate that cause AKI. Two of these patients developed ESRD[176,178] and one of them died[178].
Many cases of severe AKI after accidental or suicidal ingestion of oxalate[184,185] or ethylene glycol[186-198] have been reported. AKI had a protracted course in many of these patients and in most instances dialysis was required. Patients with severe ethylene glycol poisoning had significant mortality, especially in decades past[186]. Renal function did not return in several patients with AKI, although some did recover completely. Hyperoxaluria and calcium oxalate nephrolithiasis[199] or oxalate nephropathy with AKI or CKD were reported with the use of two medications used as vasodilators, namely pyridoxilate[200,201] and Praxilene[202-204]. Pyridoxilate is a combination of glyoxylate with pyridoxine. Pyridoxine was intended to redirect glycine formation away from glyoxylate. Nevertheless, at least a portion of the administered glyoxylate was still metabolized to oxalate. Praxilene’s common name is naftidrofyryl oxalate. When this salt dissociates in the body oxalate is released. Finally, hyperoxaluria and oxalate nephropathy has been seen with the use of the anesthetic agent, methoxyfluorane[205]. The clinical and histologic features of drug-induced hyperoxaluria have been studied more extensively than those of dietary hyperoxaluria.
Urinary oxalate excretion rates differ between oxaluric states and can provide clues for the differential diagnosis between these states[95]. Table 4 summarizes reported daily rates of urinary excretion of oxalate in various clinical states. The table includes only representative studies for all types of hyperoxaluria, except dietary hyperoxaluria. For this last category of hyperoxaluria, we included in Table 4 all the reports providing measurements of oxalate excretion in patients with oxalate nephropathy that we could find. The degree of renal function has a major impact on urinary oxalate excretion. Primary hyperoxaluria, particularly PH1, is associated with very high rates of urine oxalate excretion[90,95,98,99]. However, even in primary hyperoxaluria, the renal oxalate excretion rate was within the normal range in patients with advanced renal failure[90,99]. Oxalate excretion rates in enteric hyperoxaluria depend on dietary oxalate content; the rate is generally less than in the primary variety, but can be within the range seen in primary hyperoxalurias[1,109,118,206].
Table 4.
Daily urinary oxalate excretion in various hyperoxaluric states
| Oxaluric state | Urinary oxalate, mg/24-h |
| Normal range | < 45, < 301 |
| PH1 | > 90[95], > 63[94], 25-492[90], 26-530[99] |
| PH2 | > 42[95], 44-520[99] |
| PH3 | 80-194[98], 35-120[99] |
| Enteric | > 90[95], 30-110[1]1, 63 ± 13[2], 130[109], 52-92[118], 77 ± 44[123], 48-90[206] |
| Oral ascorbic acid | 98[171], 37[172], 84[175] |
| Parenteral ascorbic acid | 76[179], 100[180], 176[181], 88[182] |
| Ethylene glycol | 29[190], 10[195] |
| Methoxyfluorane | 96-480[205] |
| Idiopathic | < 63[95], 56 ± 15[39], 38-50[206], 48[207] |
| Dietary | < 54[95], 16.6[150], 34.2[151], 60[153] |
Oxalate excretion is presented as a single number representing the mean or median of the study (not specified in several studies), range (interquartile range in reference 120), or mean ± SD. For patients with two or more sequential measurements of urinary oxalate excretion rate, the Table reports the highest oxalate excretion.
Pediatric values. PH1: Primary hyperoxaluria, type 1; PH2: Primary hyperoxaluria type 2; PH3: Primary hyperoxaluria type 3.
Reported excretion rates of oxalate are comparable in idiopathic[35,39,95,206] and dietary[95,150,151,153] hyperoxaluria and substantially lower than in the primary varieties of hyperoxaluria. However, the degree of renal failure differs greatly between the reports of idiopathic and those of dietary hyperoxaluria. Determination of oxaluria in subjects with the dietary variety was usually performed in patients with AKI or advanced CKD whereas idiopathic hyperoxaluria was studied in the context of nephrolithiasis. The urinary oxalate excretion rate of patients with dietary hyperoxaluria may be in the range of subjects with the idiopathic variety (see the legend of Figure 2). Daily urinary oxalate excretion rates exceeding 90 mg (1 mmol) were considered primary or enteric hyperoxaluria[95]. We suggest that dietary hyperoxaluria can also cause oxalate excretion rates similar to those observed in primary hyperoxaluria.
RENAL PATHOLOGY AND PATHOPHYSIOLOGY IN HYPEROXALURIA
The chronic histologic lesions in the kidneys are indistinguishable between all categories of hyperoxaluria. Histologic lesions are also indistinguishable between AKI cases of enteric hyperoxaluria[115,121,125,126] and AKI cases of hyperoxaluria that have dietary, toxic or pharmacologic causes. Hyperoxaluric renal parenchymal disease is classified as a crystalline nephropathy[207], because it is widely acknowledged that oxalate injury to renal tissues begins with the deposition of abundant calcium oxalate crystals[208] in the lumen of renal tubules, the renal interstitium, and the walls of the renal vessels in all categories of hyperoxaluria[90,209-211].
Although finding calcium oxalate crystals in kidney biopsy specimens is necessary for the diagnosis of oxalate nephropathy, it is not a specific finding. Oxalate crystals are found in the kidneys in all conditions that elevate the plasma oxalate level. Principal among these conditions are all types of acute and chronic renal failure[212].
Extensive tubular damage with epithelial necrosis and tubular dilatation is the second cardinal characteristic of both acute and chronic oxalate nephropathy, while the involvement of glomeruli is inconsistent. The histologic features of renal tubules in hyperoxaluric AKI have the characteristics of acute tubular necrosis[115,121,151,153,158,159,164,171,174,195,213]. Changes in the renal interstitium are the other histologic characteristic of oxalate nephropathy. Profound interstitial fibrosis is present in chronic cases of oxalate nephropathy[90]. Tubulointerstitial nephritis with interstitial collection of mononuclear cells is a prominent characteristic of both chronic[90] and acute[175] cases of oxalate nephropathy. In some instances, interstitial nephritis takes the form of granuloma[150,214]. Oxalate-induced AKI may[157,164] or may not[153,155] exhibit interstitial nephritis in addition to acute tubular necrosis. Features of acute tubular injury, namely tubular simplification, flattening of tubular epithelial cells and dilatation of the tubular lumen are the earliest histological changes observed in kidneys of animals with experimental dietary acute oxalate nephropathy[165]. In addition to the kidneys, calcium oxalate crystals can be found in bone, skin, vessels and joints in patients with oxalosis[215]. Radiological and histologic features of nephropathy in a patient with dietary hyperoxaluria are shown in Figures 2 and 3 respectively.
Figure 3.
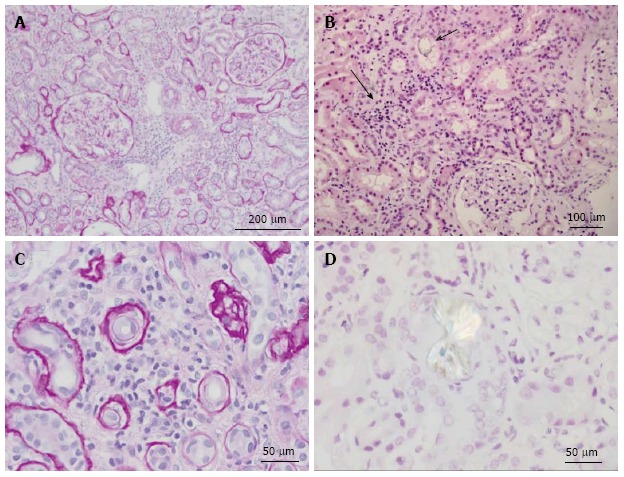
Renal histology in the patient depicted in Figure 2. A: Low power view of kidney showing two complete glomeruli and expansion of the interstitium by lymphocytes and edema. Periodic acid-Schiff (PAS) stain highlights the basement membranes of the tubules and Bowman’s capsule. PAS stain; B: Low power view of renal parenchyma showing tubulointerstitial nephritis (solid arrow) and oxalate crystal within tubule (open arrow). H and E stain; C: High power view showing interstitium expanded by lymphocytic infiltrates and tubular atrophy. PAS stain; D: High power view of calcium oxalate crystal under polarized light. H and E stain.
The initial event in the development of oxalate nephropathy is the formation of calcium oxalate crystals in the lumen of proximal tubules[219]. Details of the mechanism of crystal formation, which have been reviewed extensively in the literature on stone formation, are outside the scope of this report. Randall’s plaque (apatite collections in the interstitium of the papillae) was noticed in abundance in several hyperoxaluric states and may play a role in stone formation[220].
Adhesion of calcium oxalate crystals to the surface of tubular epithelial cells follows formation of the crystals. The mechanisms of adhesion have been extensively studied recently[221-227]. Coating with urine proteins, facilitated by low urinary pH, was shown to reduce the attachment of calcium oxalate crystals to renal inner medullary epithelial cells[221]. Calcium oxalate binding proteins that promote oxalate nephropathy have also been identified. Calcium oxalate monohydrate binding protein, one of these promoters, was shown to be upregulated by oxalate-induced oxidative stress[223]. A dual role was suggested for osteopontin, which inhibits calcium oxalate crystal formation and tubular retention[222], but also increases adhesion of these crystals to carboxylate ions that would promote oxalate-induced renal disease[225]. Prostaglandin E2 inhibits binding of calcium oxalate crystals to renal epithelial cells[224,226]. In a recent report, 26 oxalate-binding proteins were identified the kidney[227]. Further studies are needed to clarify the role of each of these proteins in oxalate-induced renal disease.
Evidence of the direct toxicity of supraphysiologic concentrations of oxalate to renal tubular cells was found in studies using cultured cells[228]. Both inhibition of cell proliferation and apoptosis have been identified as mechanisms of this nephrotoxicity. Studies in epithelial, endothelial and interstitial renal cell cultures found that exposure to sodium oxalate leads to reduced cell survival through inhibition of cell proliferation[229]. Evidence of oxalate-induced toxicity to renal cells was provided by finding increased levels of protein and mRNA of kidney injury molecule-1 in both human cell cultures and experimental animals[230]. In experimental animals hyperoxaluria increased production of TNF-α, FAS and FAS ligand, and apoptosis[231].
Research involving the mechanisms of innate immunity has shed considerable light on the molecular mediators and histologic features of oxalate nephropathy[232-243]. A role for toll-like receptors, NOD-like receptors and inflammasomes in AKI secondary to ischemia and sepsis has been documented[232]. A growing body of evidence has given inflammasomes a central place in our understanding of complex diseases (e.g., metabolic syndromes, carcinogenesis) and physiological processes (e.g., regulation of intestinal microbiome) and has identified them as important players of the intracellular surveillance system. Recent emphasis was also placed on the role of inflammasomes in various renal disease categories, including crystalline nephropathies[233].
Inflammasomes are part of the innate immune system. As their name suggests, inflammasomes represent large multimolecular cytosolic complexes that assemble into a platform for the activation of pro-inflammatory caspase 1[234-236]. Inflammasomes are important mediators of apoptosis, interstitial inflammation and fibrosis in various types of renal disease[237,238]. Of great importance in the context of oxalate nephropathy is the nucleotide-binding domain, leucine-rich repeat inflammasome (NALP3 or NLRP3). When activated, NALP3 proteins oligomerize and form a protein complex with caspase-1. This process activates caspase 1 which cleaves the inactive precursors of IL-1β and IL-18 to generate active cytokines that promote inflammation. The NALP3 inflammasome has been implicated in the molecular mechanism of nephropathy caused by urate crystals[239]. More recent studies detail the functional significance of the inflammasome and the IL-1β/IL-18 axis as an important factor in interstitial inflammation and fibrosis, as well as progression of renal failure, in oxalate nephropathy[240-242] and other kidney diseases[243]. In experimental models, genetic deletions of antagonists of the NALP3 inflammasome pathway have decreased the severity of oxalate nephropathy[240-242].
MANAGEMENT OF NEPHROPATHY IN ACQUIRED HYPEROXALURIAS
The general principles of management of oxalate-related nephropathies are the same in all categories of acquired hyperoxaluric nephropathy and include a diet low in oxalate and relatively high in calcium, fluid intake exceeding 1.5 L per m2 body surface area per day, treatment with probiotics containing oxalate degrading bacteria, and medications to increase urinary solubility of crystals (e.g., potassium citrate)[244]. Studies on the effect of probiotics on oxaluria have produced conflicting results. Intake of probiotics led to significant reduction of oxaluria in some studies[245,246], but had no effect on oxaluria in several other studies[247-249].
Specific measures targeted to the mechanism of hypercalciuria can be effective in patients with enteric hypercalciuria[244,250]. It is possible that probiotics may be useful in certain categories of patients with enteric hyperoxaluria, in particular, those who have altered enteric flora because of protracted courses of antibiotics, but this will require further study. A study by Toblli et al[251] reported that the angiotensin-converting enzyme inhibitor enalapril had a protective effect on the formation of tubulointerstitial lesions in rats fed ethylene glycol. Studies in humans with hyperoxaluria are needed to determine the effectiveness of this drug. Further studies are also needed to objectively assess the effectiveness of traditional herbal medications used for prevention or treatment of renal stones[252,253].
FUTURE RESEARCH
Our main reason for undertaking this review was to underscore the need for epidemiologic, biochemical and histologic studies of the effects of dietary hyperoxaluria on the development of CKD and end-stage renal disease (ESRD) across the globe. Occasional intake of nutritional foods high in oxalate has been advocated[254]. While doing so may have merit, neither the highest “safe” dose of oxalate nor whether this dose differs between individuals has been determined. However, the main concern is not with brief ingestion of a relatively high dose of oxalate, but instead with the effects of chronic ingestion of high doses of oxalate on renal function, which is common in several parts of the world (Table 1). Interestingly, several patients with documented CKD due to chronic dietary hyperoxaluria had ingested amounts of oxalate comparable to or even lower than the average values reported in certain parts of the world (Tables 1 and 3). Difficulties and delays with the recognition of hyperoxaluria as the cause of CKD and ESRD have been documented, even for the primary hyperoxalurias[101,103,105,255], where early appearance of symptoms and renal failure, oxalosis and a family history of recurrent nephrolithiasis, renal failure and oxalosis should lead one to the diagnosis. That retention of oxalate in patients with CKD from any etiology may result in renal deposition of calcium oxalate, secondary deterioration of the renal function and systemic toxicities has been recognized[256]. However, in a recent comprehensive review excessive dietary oxalate intake was not listed among the primary risk factors for CKD[257]. Appropriate studies in populations with high dietary oxalate intake have the potential to reduce the rates of CKD and ESRD by simple dietetic interventions (e.g., fluid intake, leaching of oxalate by soaking). Such studies should be encouraged.
Related to the need of studying the effects of oxalate intake on the development of CKD in various areas of the globe is the need to continue performing studies on genetic influences on oxalate absorption and excretion. Clinical and epidemiologic studies suggested that genetic influences can affect oxalate absorption and excretion[254,258-261]. Ongoing studies of genetic differences in intestinal and renal oxalate transporters[262-266] and of factors related to calcium metabolism[267] have the potential of leading to novel preventive and therapeutic modalities.
Future research should also include enzymologic and protein-structure studies aimed at identifying potential drugs that would either promote reductive metabolism of glyoxylate, the immediate precursor of oxalate, or inhibit oxidative enzyme-catalyzed reactions that increase oxalate production, for example the LDH reaction. Inhibiting LDH activity would reduce oxalate production and increase the levels of calcium glyoxylate and calcium glycolate which are 3 to 4 orders of magnitude more soluble in water than calcium oxalate. This approach is analogous to the treatment of gout where allopurinol inhibits xanthine oxidase activity, thereby reducing uric acid production and increasing the levels of much more water soluble xanthine oxidase substrates (e.g., hypoxanthine). The inflammasome NLP3 is an emerging potential target for new drug development NLP3[268].
CONCLUSION
Hyperoxaluria, regardless of its mechanism, can cause not only nephrolithiasis and nephrocalcinosis, but also AKI, CKD and ESRD. Research to verify or reject the hypothesis that chronic dietary hyperoxaluria is underrecognized as a cause of CKD and ESRD, particularly in global areas with high dietary oxalate consumption, has the potential of improving health, well-being and economy in these areas. This research should be combined with research on the genetics of oxalate transport, oxalate-induced mechanisms of disease and development of medications affecting these processes.
Footnotes
P- Reviewer: Lehtonen SH, Sakhaee K S- Editor: Wen LL L- Editor: A E- Editor: Lu YJ
References
- 1.Ogilvie D, McCollum JP, Packer S, Manning J, Oyesiku J, Muller DP, Harries JT. Urinary outputs of oxalate, calcium, and magnesium in children with intestinal disorders. Potential cause of renal calculi. Arch Dis Child. 1976;51:790–795. doi: 10.1136/adc.51.10.790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pak CY, Britton F, Peterson R, Ward D, Northcutt C, Breslau NA, McGuire J, Sakhaee K, Bush S, Nicar M, et al. Ambulatory evaluation of nephrolithiasis. Classification, clinical presentation and diagnostic criteria. Am J Med. 1980;69:19–30. doi: 10.1016/0002-9343(80)90495-7. [DOI] [PubMed] [Google Scholar]
- 3.Larking P, Lovell-Smith CJ, Hocken AG. Urine oxalate levels in a New Zealand reference population and renal stone formers. N Z Med J. 1983;96:606–607. [PubMed] [Google Scholar]
- 4.Lindsjö M, Fellström B, Danielson BG, Kasidas GP, Rose GA, Ljunghall S. Hyperoxaluria or hypercalciuria in nephrolithiasis: the importance of renal tubular functions. Eur J Clin Invest. 1990;20:546–554. doi: 10.1111/j.1365-2362.1990.tb01900.x. [DOI] [PubMed] [Google Scholar]
- 5.Laminski NA, Meyers AM, Kruger M, Sonnekus MI, Margolius LP. Hyperoxaluria in patients with recurrent calcium oxalate calculi: dietary and other risk factors. Br J Urol. 1991;68:454–458. doi: 10.1111/j.1464-410x.1991.tb15383.x. [DOI] [PubMed] [Google Scholar]
- 6.Massey LK, Roman-Smith H, Sutton RA. Effect of dietary oxalate and calcium on urinary oxalate and risk of formation of calcium oxalate kidney stones. J Am Diet Assoc. 1993;93:901–906. doi: 10.1016/0002-8223(93)91530-4. [DOI] [PubMed] [Google Scholar]
- 7.Verkoelen CF, Romijn JC. Oxalate transport and calcium oxalate renal stone disease. Urol Res. 1996;24:183–191. doi: 10.1007/BF00295891. [DOI] [PubMed] [Google Scholar]
- 8.Trinchieri A, Ostini F, Nespoli R, Rovera F, Zanetti G, Pisani E. Hyperoxaluria in patients with idiopathic calcium nephrolithiasis. J Nephrol. 1998;11 Suppl 1:70–72. [PubMed] [Google Scholar]
- 9.Curhan GC. Epidemiologic evidence for the role of oxalate in idiopathic nephrolithiasis. J Endourol. 1999;13:629–631. doi: 10.1089/end.1999.13.629. [DOI] [PubMed] [Google Scholar]
- 10.Osther PJ. Hyperoxaluria in idiopathic calcium nephrolithiasis--what are the limits? Scand J Urol Nephrol. 1999;33:368–371. [PubMed] [Google Scholar]
- 11.Neuhaus TJ, Belzer T, Blau N, Hoppe B, Sidhu H, Leumann E. Urinary oxalate excretion in urolithiasis and nephrocalcinosis. Arch Dis Child. 2000;82:322–326. doi: 10.1136/adc.82.4.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nguyen QV, Kälin A, Drouve U, Casez JP, Jaeger P. Sensitivity to meat protein intake and hyperoxaluria in idiopathic calcium stone formers. Kidney Int. 2001;59:2273–2281. doi: 10.1046/j.1523-1755.2001.00744.x. [DOI] [PubMed] [Google Scholar]
- 13.de O G Mendonça C, Martini LA, Baxmann AC, Nishiura JL, Cuppari L, Sigulem DM, Heilberg IP. Effects of an oxalate load on urinary oxalate excretion in calcium stone formers. J Ren Nutr. 2003;13:39–46. doi: 10.1053/jren.2003.50002. [DOI] [PubMed] [Google Scholar]
- 14.Robertson WG. Renal stones in the tropics. Semin Nephrol. 2003;23:77–87. doi: 10.1053/snep.2003.50007. [DOI] [PubMed] [Google Scholar]
- 15.Agarwal MM, Singh SK, Mavuduru R, Mandal AK. Preventive fluid and dietary therapy for urolithiasis: An appraisal of strength, controversies and lacunae of current literature. Indian J Urol. 2011;27:310–319. doi: 10.4103/0970-1591.85423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Malek RS, Kelalis PP. Nephrocalcinosis in infancy and childhood. J Urol. 1975;114:441–443. doi: 10.1016/s0022-5347(17)67052-6. [DOI] [PubMed] [Google Scholar]
- 17.Pyrah LN, Anderson CK, Hodgkinson A, Zarembski PM. A case of oxalate nephrocalcinosis and primary hyperoxaluria. Br J Urol. 1959;31:235–248. doi: 10.1111/j.1464-410x.1959.tb09413.x. [DOI] [PubMed] [Google Scholar]
- 18.Monserrat JL, Rapado A, Castrillo JM, Diaz Curiel M, Traba ML. [Nephrocalcinosis as a clinical syndrome. Study of 77 cases (author’s transl)] Med Clin (Barc) 1979;73:305–311. [PubMed] [Google Scholar]
- 19.Wilson DA, Wenzl JE, Altshuler GP. Ultrasound demonstration of diffuse cortical nephrocalcinosis in a case of primary hyperoxaluria. AJR Am J Roentgenol. 1979;132:659–661. doi: 10.2214/ajr.132.4.659. [DOI] [PubMed] [Google Scholar]
- 20.Aziz S, Callen PW, Vincenti F, Hirose R. Rapidly developing nephrocalcinosis in a patient with end-stage liver disease who received a domino liver transplant from a patient with known congenital oxalosis. J Ultrasound Med. 2005;24:1449–1452. doi: 10.7863/jum.2005.24.10.1449. [DOI] [PubMed] [Google Scholar]
- 21.Mantan M, Bagga A, Virdi VS, Menon S, Hari P. Etiology of nephrocalcinosis in northern Indian children. Pediatr Nephrol. 2007;22:829–833. doi: 10.1007/s00467-006-0425-7. [DOI] [PubMed] [Google Scholar]
- 22.Mandal AK. What is the purpose of launching the World Journal of Nephrology? World J Nephrol. 2012;1:1–3. doi: 10.5527/wjn.v1.i1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grases F, March JG, Conte A, Costa-Bauzá A. New aspects on the composition, structure and origin of calcium oxalate monohydrate calculi. Eur Urol. 1993;24:381–386. doi: 10.1159/000474333. [DOI] [PubMed] [Google Scholar]
- 24.Hodgkinson A, Zarembski PM. Oxalic acid metabolism in man: a review. Calcif Tissue Res. 1968;2:115–132. doi: 10.1007/BF02279201. [DOI] [PubMed] [Google Scholar]
- 25.Isong EU, Idiong UI. Comparative studies on the nutritional and toxic composition of three varieties of Lesianthera africana. Plant Foods Hum Nutr. 1997;51:79–84. doi: 10.1023/a:1007922308985. [DOI] [PubMed] [Google Scholar]
- 26.Noonan SC, Savage GP. Oxalate content of foods and its effect on humans. Asia Pac J Clin Nutr. 1999;8:64–74. [PubMed] [Google Scholar]
- 27.Holmes RP, Kennedy M. Estimation of oxalate content of foods and daily oxalate intake. Kidney Int. 2000;57:1662–1667. doi: 10.1046/j.1523-1755.2000.00010.x. [DOI] [PubMed] [Google Scholar]
- 28.Kumar A. Influence of radish consumption on urinary calcium oxalate excretion. Nepal Med Coll J. 2004;6:41–44. [PubMed] [Google Scholar]
- 29.Siener R, Hönow R, Voss S, Seidler A, Hesse A. Oxalate content of cereals and cereal products. J Agric Food Chem. 2006;54:3008–3011. doi: 10.1021/jf052776v. [DOI] [PubMed] [Google Scholar]
- 30.Kynast-Gales SA, Massey LK. Food oxalate: an international database. J Am Diet Assoc. 2007;107:1099. doi: 10.1016/j.jada.2007.05.027. [DOI] [PubMed] [Google Scholar]
- 31. Available from: http://www.spokane.wsu.edu/research&service/HREC/FoodOxalate.asp.
- 32.Brogren M, Savage GP. Bioavailability of soluble oxalate from spinach eaten with and without milk products. Asia Pac J Clin Nutr. 2003;12:219–224. [PubMed] [Google Scholar]
- 33.Meschi T, Schianchi T, Ridolo E, Adorni G, Allegri F, Guerra A, Novarini A, Borghi L. Body weight, diet and water intake in preventing stone disease. Urol Int. 2004;72 Suppl 1:29–33. doi: 10.1159/000076588. [DOI] [PubMed] [Google Scholar]
- 34.Hess B. ‘Bad dietary habits’ and recurrent calcium oxalate nephrolithiasis. Nephrol Dial Transplant. 1998;13:1033–1038. doi: 10.1093/ndt/13.4.1033. [DOI] [PubMed] [Google Scholar]
- 35.Hall WD, Pettinger M, Oberman A, Watts NB, Johnson KC, Paskett ED, Limacher MC, Hays J. Risk factors for kidney stones in older women in the southern United States. Am J Med Sci. 2001;322:12–18. doi: 10.1097/00000441-200107000-00003. [DOI] [PubMed] [Google Scholar]
- 36.Taylor EN, Curhan GC. Oxalate intake and the risk for nephrolithiasis. J Am Soc Nephrol. 2007;18:2198–2204. doi: 10.1681/ASN.2007020219. [DOI] [PubMed] [Google Scholar]
- 37.Alaya A, Sakly R, Nouri A, Najjar MF. Nutritional aspects of idiopathic nephrolithiasis in Tunisian children. Arch Ital Urol Androl. 2011;83:136–140. [PubMed] [Google Scholar]
- 38.Zarembski PM, Hodgkinson A. The oxalic acid content of English diets. Br J Nutr. 1962;16:627–634. doi: 10.1079/bjn19620061. [DOI] [PubMed] [Google Scholar]
- 39.Siener R, Ebert D, Nicolay C, Hesse A. Dietary risk factors for hyperoxaluria in calcium oxalate stone formers. Kidney Int. 2003;63:1037–1043. doi: 10.1046/j.1523-1755.2003.00807.x. [DOI] [PubMed] [Google Scholar]
- 40.Singh PP, Kothari LK, Sharma DC, Saxena SN. Nutritional value of foods in relation to their oxalic acid content. Am J Clin Nutr. 1972;25:1147–1152. doi: 10.1093/ajcn/25.11.1147. [DOI] [PubMed] [Google Scholar]
- 41.Sanwalka NJ, Khadilkar AV, Mughal MZ, Sayyad MG, Khadilkar VV, Shirole SC, Divate UP, Bhandari DR. A study of calcium intake and sources of calcium in adolescent boys and girls from two socioeconomic strata, in Pune, India. Asia Pac J Clin Nutr. 2010;19:324–329. [PubMed] [Google Scholar]
- 42.Meschi T, Maggiore U, Fiaccadori E, Schianchi T, Bosi S, Adorni G, Ridolo E, Guerra A, Allegri F, Novarini A, et al. The effect of fruits and vegetables on urinary stone risk factors. Kidney Int. 2004;66:2402–2410. doi: 10.1111/j.1523-1755.2004.66029.x. [DOI] [PubMed] [Google Scholar]
- 43.Diggers Australia PTY Limited. Material safety data Sheet oxalic acid. 2003. Available from: http://www.diggersaust.com.au/files/Oxalic Acid.pdf. [Google Scholar]
- 44.Dvorácková I. [Fatal poisoning following intravenous administration of sodium oxalate] Arch Toxikol. 1966;22:63–67. [PubMed] [Google Scholar]
- 45.Taylor EN, Fung TT, Curhan GC. DASH-style diet associates with reduced risk for kidney stones. J Am Soc Nephrol. 2009;20:2253–2259. doi: 10.1681/ASN.2009030276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Williams HE, Wandzilak TR. Oxalate synthesis, transport and the hyperoxaluric syndromes. J Urol. 1989;141:742–749. doi: 10.1016/s0022-5347(17)40999-2. [DOI] [PubMed] [Google Scholar]
- 47.Ichiyama A, Xue HH, Oda T, Uchida C, Sugiyama T, Maeda-Nakai E, Sato K, Nagai E, Watanabe S, Takayama T. Oxalate synthesis in mammals: properties and subcellular distribution of serine: pyruvate/alanine: glyoxylate aminotransferase in the liver. Mol Urol. 2000;4:333–340. [PubMed] [Google Scholar]
- 48.Knight J, Jiang J, Assimos DG, Holmes RP. Hydroxyproline ingestion and urinary oxalate and glycolate excretion. Kidney Int. 2006;70:1929–1934. doi: 10.1038/sj.ki.5001906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Urivetzky M, Kessaris D, Smith AD. Ascorbic acid overdosing: a risk factor for calcium oxalate nephrolithiasis. J Urol. 1992;147:1215–1218. doi: 10.1016/s0022-5347(17)37521-3. [DOI] [PubMed] [Google Scholar]
- 50.Peña de la Vega L, Lieske JC, Milliner D, Gonyea J, Kelly DG. Urinary oxalate excretion increases in home parenteral nutrition patients on a higher intravenous ascorbic acid dose. JPEN J Parenter Enteral Nutr. 2004;28:435–438. doi: 10.1177/0148607104028006435. [DOI] [PubMed] [Google Scholar]
- 51.Robitaille L, Mamer OA, Miller WH, Levine M, Assouline S, Melnychuk D, Rousseau C, Hoffer LJ. Oxalic acid excretion after intravenous ascorbic acid administration. Metabolism. 2009;58:263–269. doi: 10.1016/j.metabol.2008.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Taylor EN, Curhan GC. Fructose consumption and the risk of kidney stones. Kidney Int. 2008;73:207–212. doi: 10.1038/sj.ki.5002588. [DOI] [PubMed] [Google Scholar]
- 53.Nguyen NU, Dumoulin G, Wolf JP, Berthelay S. Urinary calcium and oxalate excretion during oral fructose or glucose load in man. Horm Metab Res. 1989;21:96–99. doi: 10.1055/s-2007-1009160. [DOI] [PubMed] [Google Scholar]
- 54.Nguyen NU, Dumoulin G, Henriet MT, Regnard J. Increase in urinary calcium and oxalate after fructose infusion. Horm Metab Res. 1995;27:155–158. doi: 10.1055/s-2007-979929. [DOI] [PubMed] [Google Scholar]
- 55.Knight J, Assimos DG, Easter L, Holmes RP. Metabolism of fructose to oxalate and glycolate. Horm Metab Res. 2010;42:868–873. doi: 10.1055/s-0030-1265145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Holmes RP, Goodman HO, Assimos DG. Contribution of dietary oxalate to urinary oxalate excretion. Kidney Int. 2001;59:270–276. doi: 10.1046/j.1523-1755.2001.00488.x. [DOI] [PubMed] [Google Scholar]
- 57.Robijn S, Hoppe B, Vervaet BA, D’Haese PC, Verhulst A. Hyperoxaluria: a gut-kidney axis? Kidney Int. 2011;80:1146–1158. doi: 10.1038/ki.2011.287. [DOI] [PubMed] [Google Scholar]
- 58.Lien YH. Juicing is not all juicy. Am J Med. 2013;126:755–756. doi: 10.1016/j.amjmed.2013.04.007. [DOI] [PubMed] [Google Scholar]
- 59.Knauf F, Ko N, Jiang Z, Robertson WG, Van Itallie CM, Anderson JM, Aronson PS. Net intestinal transport of oxalate reflects passive absorption and SLC26A6-mediated secretion. J Am Soc Nephrol. 2011;22:2247–2255. doi: 10.1681/ASN.2011040433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.von Unruh GE, Voss S, Sauerbruch T, Hesse A. Reference range for gastrointestinal oxalate absorption measured with a standardized [13C2]oxalate absorption test. J Urol. 2003;169:687–690. doi: 10.1097/01.ju.0000051637.63068.92. [DOI] [PubMed] [Google Scholar]
- 61.Sikora P, von Unruh GE, Beck B, Feldkötter M, Zajaczkowska M, Hesse A, Hoppe B. [13C2]oxalate absorption in children with idiopathic calcium oxalate urolithiasis or primary hyperoxaluria. Kidney Int. 2008;73:1181–1186. doi: 10.1038/ki.2008.63. [DOI] [PubMed] [Google Scholar]
- 62.Zimmermann DJ, Hesse A, von Unruh GE. Influence of a high-oxalate diet on intestinal oxalate absorption. World J Urol. 2005;23:324–329. doi: 10.1007/s00345-005-0028-0. [DOI] [PubMed] [Google Scholar]
- 63.Liebman M, Chai W. Effect of dietary calcium on urinary oxalate excretion after oxalate loads. Am J Clin Nutr. 1997;65:1453–1459. doi: 10.1093/ajcn/65.5.1453. [DOI] [PubMed] [Google Scholar]
- 64.Hess B, Jost C, Zipperle L, Takkinen R, Jaeger P. High-calcium intake abolishes hyperoxaluria and reduces urinary crystallization during a 20-fold normal oxalate load in humans. Nephrol Dial Transplant. 1998;13:2241–2247. doi: 10.1093/ndt/13.9.2241. [DOI] [PubMed] [Google Scholar]
- 65.von Unruh GE, Voss S, Sauerbruch T, Hesse A. Dependence of oxalate absorption on the daily calcium intake. J Am Soc Nephrol. 2004;15:1567–1573. doi: 10.1097/01.asn.0000127864.26968.7f. [DOI] [PubMed] [Google Scholar]
- 66.Zimmermann DJ, Voss S, von Unruh GE, Hesse A. Importance of magnesium in absorption and excretion of oxalate. Urol Int. 2005;74:262–267. doi: 10.1159/000083560. [DOI] [PubMed] [Google Scholar]
- 67.Naya Y, Ito H, Masai M, Yamaguchi K. Association of dietary fatty acids with urinary oxalate excretion in calcium oxalate stone-formers in their fourth decade. BJU Int. 2002;89:842–846. doi: 10.1046/j.1464-410x.2002.02740.x. [DOI] [PubMed] [Google Scholar]
- 68.Okombo J, Liebman M. Probiotic-induced reduction of gastrointestinal oxalate absorption in healthy subjects. Urol Res. 2010;38:169–178. doi: 10.1007/s00240-010-0262-9. [DOI] [PubMed] [Google Scholar]
- 69.Al-Wahsh I, Wu Y, Liebman M. Acute probiotic ingestion reduces gastrointestinal oxalate absorption in healthy subjects. Urol Res. 2012;40:191–196. doi: 10.1007/s00240-011-0421-7. [DOI] [PubMed] [Google Scholar]
- 70.Hodgkinson A, Wilkinson R. Plasma oxalate concentration and renal excretion of oxalate in man. Clin Sci Mol Med. 1974;46:61–73. doi: 10.1042/cs0460061. [DOI] [PubMed] [Google Scholar]
- 71.Prenen JA, Boer P, Mees EJ, Endeman HJ, Spoor SM, Oei HY. Renal clearance of [14C]oxalate: comparison of constant-infusion with single-injection techniques. Clin Sci (Lond) 1982;63:47–51. doi: 10.1042/cs0630047. [DOI] [PubMed] [Google Scholar]
- 72.Schwille PO, Manoharan M, Rümenapf G, Wölfel G, Berens H. Oxalate measurement in the picomol range by ion chromatography: values in fasting plasma and urine of controls and patients with idiopathic calcium urolithiasis. J Clin Chem Clin Biochem. 1989;27:87–96. doi: 10.1515/cclm.1989.27.2.87. [DOI] [PubMed] [Google Scholar]
- 73.Williams HE, Johnson GA, Smith LH. The renal clearance of oxalate in normal subjects and patients with primary hyperoxaluria. Clin Sci. 1971;41:213–218. doi: 10.1042/cs0410213. [DOI] [PubMed] [Google Scholar]
- 74.Knight TF, Sansom SC, Senekjian HO, Weinman EJ. Oxalate secretion in the rat proximal tubule. Am J Physiol. 1981;240:F295–F298. doi: 10.1152/ajprenal.1981.240.4.F295. [DOI] [PubMed] [Google Scholar]
- 75.Karniski LP, Lötscher M, Fucentese M, Hilfiker H, Biber J, Murer H. Immunolocalization of sat-1 sulfate/oxalate/bicarbonate anion exchanger in the rat kidney. Am J Physiol. 1998;275:F79–F87. doi: 10.1152/ajprenal.1998.275.1.F79. [DOI] [PubMed] [Google Scholar]
- 76.Lohi H, Kujala M, Makela S, Lehtonen E, Kestila M, Saarialho-Kere U, Markovich D, Kere J. Functional characterization of three novel tissue-specific anion exchangers SLC26A7, -A8, and -A9. J Biol Chem. 2002;277:14246–14254. doi: 10.1074/jbc.M111802200. [DOI] [PubMed] [Google Scholar]
- 77.Xie Q, Welch R, Mercado A, Romero MF, Mount DB. Molecular characterization of the murine Slc26a6 anion exchanger: functional comparison with Slc26a1. Am J Physiol Renal Physiol. 2002;283:F826–F838. doi: 10.1152/ajprenal.00079.2002. [DOI] [PubMed] [Google Scholar]
- 78.Knauf F, Yang CL, Thomson RB, Mentone SA, Giebisch G, Aronson PS. Identification of a chloride-formate exchanger expressed on the brush border membrane of renal proximal tubule cells. Proc Natl Acad Sci USA. 2001;98:9425–9430. doi: 10.1073/pnas.141241098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Holmes RP, Assimos DG. The impact of dietary oxalate on kidney stone formation. Urol Res. 2004;32:311–316. doi: 10.1007/s00240-004-0437-3. [DOI] [PubMed] [Google Scholar]
- 80.Camici M, Balestri PL, Lupetti S, Colizzi V, Falcone G. Urinary excretion of oxalate in renal failure. Nephron. 1982;30:269–270. doi: 10.1159/000182486. [DOI] [PubMed] [Google Scholar]
- 81.Costello JF, Smith M, Stolarski C, Sadovnic MJ. Extrarenal clearance of oxalate increases with progression of renal failure in the rat. J Am Soc Nephrol. 1992;3:1098–1104. doi: 10.1681/ASN.V351098. [DOI] [PubMed] [Google Scholar]
- 82.Hatch M, Freel RW, Vaziri ND. Intestinal excretion of oxalate in chronic renal failure. J Am Soc Nephrol. 1994;5:1339–1343. doi: 10.1681/ASN.V561339. [DOI] [PubMed] [Google Scholar]
- 83.Hatch M, Freel RW, Vaziri ND. Regulatory aspects of oxalate secretion in enteric oxalate elimination. J Am Soc Nephrol. 1999;10 Suppl 14:S324–S328. [PubMed] [Google Scholar]
- 84.Oreopoulos DG, Husdan H, Leung M, Reid AD, Rapoport A. Urine oxalic acid: relation to urine flow. Ann Intern Med. 1976;85:617–618. doi: 10.7326/0003-4819-85-5-617. [DOI] [PubMed] [Google Scholar]
- 85.Klän R, Butz M. Effect of diuresis on the oxalate excretion and the calcium-oxalate product in non-stone formers. Urol Int. 1987;42:19–22. doi: 10.1159/000281844. [DOI] [PubMed] [Google Scholar]
- 86.Lemann J, Pleuss JA, Worcester EM, Hornick L, Schrab D, Hoffmann RG. Urinary oxalate excretion increases with body size and decreases with increasing dietary calcium intake among healthy adults. Kidney Int. 1996;49:200–208. doi: 10.1038/ki.1996.27. [DOI] [PubMed] [Google Scholar]
- 87.Taylor EN, Curhan GC. Body size and 24-hour urine composition. Am J Kidney Dis. 2006;48:905–915. doi: 10.1053/j.ajkd.2006.09.004. [DOI] [PubMed] [Google Scholar]
- 88.Taylor EN, Stampfer MJ, Curhan GC. Obesity, weight gain, and the risk of kidney stones. JAMA. 2005;293:455–462. doi: 10.1001/jama.293.4.455. [DOI] [PubMed] [Google Scholar]
- 89.Hallson PC, Kasidas GP, Rose AL. Seasonal variations in urinary excretion of calcium and oxalate in normal subjects in patients with idiopathic hyperclaciuria. Br J Urol. 1977;49:1–10. doi: 10.1111/j.1464-410x.1977.tb04513.x. [DOI] [PubMed] [Google Scholar]
- 90.Hockaday TD, Clayton JE, Frederick EW, Smith LH. Primary hyperoxaluria. Medicine (Baltimore) 1964;43:315–345. doi: 10.1097/00005792-196405000-00010. [DOI] [PubMed] [Google Scholar]
- 91.Bobrowski AE, Langman CB. The primary hyperoxalurias. Semin Nephrol. 2008;28:152–162. doi: 10.1016/j.semnephrol.2008.01.008. [DOI] [PubMed] [Google Scholar]
- 92.Hoppe B, Beck BB, Milliner DS. The primary hyperoxalurias. Kidney Int. 2009;75:1264–1271. doi: 10.1038/ki.2009.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Harambat J, Fargue S, Bacchetta J, Acquaviva C, Cochat P. Primary hyperoxaluria. Int J Nephrol. 2011;2011:864580. doi: 10.4061/2011/864580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Cochat P, Rumsby G. Primary hyperoxaluria. N Engl J Med. 2013;369:649–658. doi: 10.1056/NEJMra1301564. [DOI] [PubMed] [Google Scholar]
- 95.Coulter-Mackie MB, White CT, Hurley RM, Chew BH, Lange D. Primary hyperoxaluria type 1. Gene ReviewsTM [Internet] In: Pagon RA, Ardinger HH, editors. Adam MP, Bird TD, Dolan CR, Fong CT, Stephens K, editors. Seattle (Washington): University of Washington, 1993-; 2013. [Google Scholar]
- 96.Rumsby G. Primary hyperoxaluria type 2. Gene ReviewsTM [Internet] In: Pagon RA, Ardinger HH, Adam MP, Bird TD, Dolan CR, et al., editors. Seattle (Washington): University of Washington, 1993-; 2013. [PubMed] [Google Scholar]
- 97.Knight J, Holmes RP, Milliner DS, Monico CG, Cramer SD. Glyoxylate reductase activity in blood mononuclear cells and the diagnosis of primary hyperoxaluria type 2. Nephrol Dial Transplant. 2006;21:2292–2295. doi: 10.1093/ndt/gfl142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Monico CG, Persson M, Ford GC, Rumsby G, Milliner DS. Potential mechanisms of marked hyperoxaluria not due to primary hyperoxaluria I or II. Kidney Int. 2002;62:392–400. doi: 10.1046/j.1523-1755.2002.00468.x. [DOI] [PubMed] [Google Scholar]
- 99.Monico CG, Rossetti S, Belostotsky R, Cogal AG, Herges RM, Seide BM, Olson JB, Bergstrahl EJ, Williams HJ, Haley WE, et al. Primary hyperoxaluria type III gene HOGA1 (formerly DHDPSL) as a possible risk factor for idiopathic calcium oxalate urolithiasis. Clin J Am Soc Nephrol. 2011;6:2289–2295. doi: 10.2215/CJN.02760311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Belostotsky R, Pitt JJ, Frishberg Y. Primary hyperoxaluria type III--a model for studying perturbations in glyoxylate metabolism. J Mol Med (Berl) 2012;90:1497–1504. doi: 10.1007/s00109-012-0930-z. [DOI] [PubMed] [Google Scholar]
- 101.Hoppe B, Langman CB. A United States survey on diagnosis, treatment, and outcome of primary hyperoxaluria. Pediatr Nephrol. 2003;18:986–991. doi: 10.1007/s00467-003-1234-x. [DOI] [PubMed] [Google Scholar]
- 102.van Woerden CS, Groothoff JW, Wanders RJ, Davin JC, Wijburg FA. Primary hyperoxaluria type 1 in The Netherlands: prevalence and outcome. Nephrol Dial Transplant. 2003;18:273–279. doi: 10.1093/ndt/18.2.273. [DOI] [PubMed] [Google Scholar]
- 103.Hoppe B, Latta K, von Schnakenburg C, Kemper MJ. Primary hyperoxaluria--the German experience. Am J Nephrol. 2005;25:276–281. doi: 10.1159/000086358. [DOI] [PubMed] [Google Scholar]
- 104.Hoppe B. An update on primary hyperoxaluria. Nat Rev Nephrol. 2012;8:467–475. doi: 10.1038/nrneph.2012.113. [DOI] [PubMed] [Google Scholar]
- 105.van der Hoeven SM, van Woerden CS, Groothoff JW. Primary hyperoxaluria type 1, a too often missed diagnosis and potentially treatable cause of end-stage renal disease in adults: results of the Dutch cohort. Nephrol Dial Transplant. 2012;27:3855–3862. doi: 10.1093/ndt/gfs320. [DOI] [PubMed] [Google Scholar]
- 106.Earnest DL. Perspectives on incidence, etiology, and treatment of enteric hyperoxaluria. Am J Clin Nutr. 1977;30:72–75. doi: 10.1093/ajcn/30.1.72. [DOI] [PubMed] [Google Scholar]
- 107.Parks JH, Worcester EM, O’Connor RC, Coe FL. Urine stone risk factors in nephrolithiasis patients with and without bowel disease. Kidney Int. 2003;63:255–265. doi: 10.1046/j.1523-1755.2003.00725.x. [DOI] [PubMed] [Google Scholar]
- 108.Canos HJ, Hogg GA, Jeffery JR. Oxalate nephropathy due to gastrointestinal disorders. Can Med Assoc J. 1981;124:729–733. [PMC free article] [PubMed] [Google Scholar]
- 109.Siener R, Petzold J, Bitterlich N, Alteheld B, Metzner C. Determinants of urolithiasis in patients with intestinal fat malabsorption. Urology. 2013;81:17–24. doi: 10.1016/j.urology.2012.07.107. [DOI] [PubMed] [Google Scholar]
- 110.Hassan I, Juncos LA, Milliner DS, Sarmiento JM, Sarr MG. Chronic renal failure secondary to oxalate nephropathy: a preventable complication after jejunoileal bypass. Mayo Clin Proc. 2001;76:758–760. doi: 10.4065/76.7.758. [DOI] [PubMed] [Google Scholar]
- 111.Whitson JM, Stackhouse GB, Stoller ML. Hyperoxaluria after modern bariatric surgery: case series and literature review. Int Urol Nephrol. 2010;42:369–374. doi: 10.1007/s11255-009-9602-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Froeder L, Arasaki CH, Malheiros CA, Baxmann AC, Heilberg IP. Response to dietary oxalate after bariatric surgery. Clin J Am Soc Nephrol. 2012;7:2033–2040. doi: 10.2215/CJN.02560312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Nelson WK, Houghton SG, Milliner DS, Lieske JC, Sarr MG. Enteric hyperoxaluria, nephrolithiasis, and oxalate nephropathy: potentially serious and unappreciated complications of Roux-en-Y gastric bypass. Surg Obes Relat Dis. 2005;1:481–485. doi: 10.1016/j.soard.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 114.Kaye MC, Streem SB, Hall PM. Enteric hyperoxaluria associated with external biliary drainage. J Urol. 1994;151:396–397. doi: 10.1016/s0022-5347(17)34959-5. [DOI] [PubMed] [Google Scholar]
- 115.Crook ED, Cook WJ, Bergman SM. Rapid renal deterioration secondary to oxalate in a patient with diabetic gastroenteropathy. Am J Kidney Dis. 1995;26:68–71. doi: 10.1016/0272-6386(95)90156-6. [DOI] [PubMed] [Google Scholar]
- 116.Moutzouris DA, Skaneli G, Margellos V, Apostolou T, Petraki C, Nikolopoulou N. Oxalate nephropathy in a diabetic patient after gastric by-pass. Clin Nephrol. 2011;75 Suppl 1:16–19. [PubMed] [Google Scholar]
- 117.McDonald GB, Earnest DL, Admirand WH. Hyperoxaluria correlates with fat malabsorption in patients with sprue. Gut. 1977;18:561–566. doi: 10.1136/gut.18.7.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Cartery C, Faguer S, Karras A, Cointault O, Buscail L, Modesto A, Ribes D, Rostaing L, Chauveau D, Giraud P. Oxalate nephropathy associated with chronic pancreatitis. Clin J Am Soc Nephrol. 2011;6:1895–1902. doi: 10.2215/CJN.00010111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Hueppelshaeuser R, von Unruh GE, Habbig S, Beck BB, Buderus S, Hesse A, Hoppe B. Enteric hyperoxaluria, recurrent urolithiasis, and systemic oxalosis in patients with Crohn’s disease. Pediatr Nephrol. 2012;27:1103–1109. doi: 10.1007/s00467-012-2126-8. [DOI] [PubMed] [Google Scholar]
- 120.Allen A, Clutterbuck E, Maidment G, Thompson E, Watts R, Pusey C. Enteric hyperoxaluria and renal failure associated with lymphangiectasia. Nephrol Dial Transplant. 1997;12:802–806. doi: 10.1093/ndt/12.4.802. [DOI] [PubMed] [Google Scholar]
- 121.Cohen-Bucay A, Garimella P, Ezeokonkwo C, Bijol V, Strom JA, Jaber BL. Acute oxalate nephropathy associated with Clostridium difficile colitis. Am J Kidney Dis. 2014;63:113–118. doi: 10.1053/j.ajkd.2013.09.010. [DOI] [PubMed] [Google Scholar]
- 122.Gibney EM, Goldfarb DS. The association of nephrolithiasis with cystic fibrosis. Am J Kidney Dis. 2003;42:1–11. doi: 10.1016/s0272-6386(03)00403-7. [DOI] [PubMed] [Google Scholar]
- 123.Hoppe B, von Unruh GE, Blank G, Rietschel E, Sidhu H, Laube N, Hesse A. Absorptive hyperoxaluria leads to an increased risk for urolithiasis or nephrocalcinosis in cystic fibrosis. Am J Kidney Dis. 2005;46:440–445. doi: 10.1053/j.ajkd.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 124.Cuvelier C, Goffin E, Cosyns JP, Wauthier M, de Strihou Cv. Enteric hyperoxaluria: a hidden cause of early renal graft failure in two successive transplants: spontaneous late graft recovery. Am J Kidney Dis. 2002;40:E3. doi: 10.1053/ajkd.2002.33934. [DOI] [PubMed] [Google Scholar]
- 125.Lefaucheur C, Hill GS, Amrein C, Haymann JP, Jacquot C, Glotz D, Nochy D. Acute oxalate nephropathy: A new etiology for acute renal failure following nonrenal solid organ transplantation. Am J Transplant. 2006;6:2516–2521. doi: 10.1111/j.1600-6143.2006.01485.x. [DOI] [PubMed] [Google Scholar]
- 126.Rankin AC, Walsh SB, Summers SA, Owen MP, Mansell MA. Acute oxalate nephropathy causing late renal transplant dysfunction due to enteric hyperoxaluria. Am J Transplant. 2008;8:1755–1758. doi: 10.1111/j.1600-6143.2008.02288.x. [DOI] [PubMed] [Google Scholar]
- 127.Parasuraman R, Venkat KK. Crystal-induced kidney disease in 2 kidney transplant recipients. Am J Kidney Dis. 2010;55:192–197. doi: 10.1053/j.ajkd.2009.08.012. [DOI] [PubMed] [Google Scholar]
- 128.Beloncle F, Sayegh J, Duveau A, Besson V, Croue A, Subra JF, Augusto JF. An unexpected cause of progressive renal failure in a 66-year-old male after liver transplantation: secondary hyperoxaluria. Int Urol Nephrol. 2013;45:1209–1213. doi: 10.1007/s11255-012-0140-1. [DOI] [PubMed] [Google Scholar]
- 129.Dheda S, Swaminathan R, Musk M, Sinniah R, Lawrence S, Irish A. Acute irreversible oxalate nephropathy in a lung transplant recipient treated successfully with a renal transplant. Nephrology (Carlton) 2012;17 Suppl 1:12–15. doi: 10.1111/j.1440-1797.2012.01585.x. [DOI] [PubMed] [Google Scholar]
- 130.Singh A, Sarkar SR, Gaber LW, Perazella MA. Acute oxalate nephropathy associated with orlistat, a gastrointestinal lipase inhibitor. Am J Kidney Dis. 2007;49:153–157. doi: 10.1053/j.ajkd.2006.10.004. [DOI] [PubMed] [Google Scholar]
- 131.Dossabhoy NR, McRight S, Sangha B, Khan S, Adgeh C. Orlistat-induced oxalate nephropathy may be dose-independent and present as a late manifestation. J La State Med Soc. 2013;165:283–285. [PubMed] [Google Scholar]
- 132.Gariani K, de Seigneux S, Courbebaisse M, Lévy M, Moll S, Martin PY. Oxalate nephropathy induced by octreotide treatment for acromegaly: a case report. J Med Case Rep. 2012;6:215. doi: 10.1186/1752-1947-6-215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Dobbins JW, Binder HJ. Importance of the colon in enteric hyperoxaluria. N Engl J Med. 1977;296:298–301. doi: 10.1056/NEJM197702102960602. [DOI] [PubMed] [Google Scholar]
- 134.Modigliani R, Labayle D, Aymes C, Denvil R. Evidence for excessive absorption of oxalate by the colon in enteric hyperoxaluria. Scand J Gastroenterol. 1978;13:187–192. doi: 10.3109/00365527809181746. [DOI] [PubMed] [Google Scholar]
- 135.Mandell I, Krauss E, Millan JC. Oxalate-induced acute renal failure in Crohn’s disease. Am J Med. 1980;69:628–632. doi: 10.1016/0002-9343(80)90479-9. [DOI] [PubMed] [Google Scholar]
- 136.Wharton R, D’Agati V, Magun AM, Whitlock R, Kunis CL, Appel GB. Acute deterioration of renal function associated with enteric hyperoxaluria. Clin Nephrol. 1990;34:116–121. [PubMed] [Google Scholar]
- 137.Sentís A, Quintana LF, Massó E, Peréz NS, Botey Puig A, Campistol Plana JM. Acute renal failure due to oxalate crystal deposition and enteric hyperoxaluria. Nefrologia. 2011;31:121–123. doi: 10.3265/Nefrologia.pre2010.Oct.10644. [DOI] [PubMed] [Google Scholar]
- 138.Chaudhari D, Crisostomo C, Ganote C, Youngberg G. Acute oxalate nephropathy associated with orlistat: a case report with a review of the literature. Case Rep Nephrol. 2013;2013:124604. doi: 10.1155/2013/124604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Kwan TK, Chadban SJ, McKenzie PR, Saunders JR. Acute oxalate nephropathy secondary to orlistat-induced enteric hyperoxaluria. Nephrology (Carlton) 2013;18:241–242. doi: 10.1111/j.1440-1797.2012.01649.x. [DOI] [PubMed] [Google Scholar]
- 140.Tintillier M, Pochet JM, Blackburn D, Delgrange E, Donckier JE. Hyperoxaluria: an underestimated cause of rapidly progressive renal failure. Acta Clin Belg. 2001;56:360–363. doi: 10.1179/acb.2001.054. [DOI] [PubMed] [Google Scholar]
- 141.Sutton RA, Walker VR. Enteric and mild hyperoxaluria. Miner Electrolyte Metab. 1994;20:352–360. [PubMed] [Google Scholar]
- 142.Rendina D, De Filippo G, Zampa G, Muscariello R, Mossetti G, Strazzullo P. Characteristic clinical and biochemical profile of recurrent calcium-oxalate nephrolithiasis in patients with metabolic syndrome. Nephrol Dial Transplant. 2011;26:2256–2263. doi: 10.1093/ndt/gfq664. [DOI] [PubMed] [Google Scholar]
- 143.Marangella M, Fruttero B, Bruno M, Linari F. Hyperoxaluria in idiopathic calcium stone disease: further evidence of intestinal hyperabsorption of oxalate. Clin Sci (Lond) 1982;63:381–385. doi: 10.1042/cs0630381. [DOI] [PubMed] [Google Scholar]
- 144.Krishnamurthy MS, Hruska KA, Chandhoke PS. The urinary response to an oral oxalate load in recurrent calcium stone formers. J Urol. 2003;169:2030–2033. doi: 10.1097/01.ju.0000062527.37579.49. [DOI] [PubMed] [Google Scholar]
- 145.Jaeger P, Portmann L, Jacquet AF, Burckhardt P. Influence of the calcium content of the diet on the incidence of mild hyperoxaluria in idiopathic renal stone formers. Am J Nephrol. 1985;5:40–44. doi: 10.1159/000166901. [DOI] [PubMed] [Google Scholar]
- 146.Mitwalli A, Ayiomamitis A, Grass L, Oreopoulos DG. Control of hyperoxaluria with large doses of pyridoxine in patients with kidney stones. Int Urol Nephrol. 1988;20:353–359. doi: 10.1007/BF02549567. [DOI] [PubMed] [Google Scholar]
- 147.Ortiz-Alvarado O, Miyaoka R, Kriedberg C, Moeding A, Stessman M, Monga M. Pyridoxine and dietary counseling for the management of idiopathic hyperoxaluria in stone-forming patients. Urology. 2011;77:1054–1058. doi: 10.1016/j.urology.2010.08.002. [DOI] [PubMed] [Google Scholar]
- 148.Chai W, Liebman M, Kynast-Gales S, Massey L. Oxalate absorption and endogenous oxalate synthesis from ascorbate in calcium oxalate stone formers and non-stone formers. Am J Kidney Dis. 2004;44:1060–1069. doi: 10.1053/j.ajkd.2004.08.028. [DOI] [PubMed] [Google Scholar]
- 149.Nouvenne A, Meschi T, Guerra A, Allegri F, Prati B, Fiaccadori E, Maggiore U, Borghi L. Diet to reduce mild hyperoxaluria in patients with idiopathic calcium oxalate stone formation: a pilot study. Urology. 2009;73:725–730, 730.e1. doi: 10.1016/j.urology.2008.11.006. [DOI] [PubMed] [Google Scholar]
- 150.Sasaki M, Murakami M, Matsuo K, Matsuo Y, Tanaka S, Ono T, Mori N. Oxalate nephropathy with a granulomatous lesion due to excessive intake of peanuts. Clin Exp Nephrol. 2008;12:305–308. doi: 10.1007/s10157-008-0046-5. [DOI] [PubMed] [Google Scholar]
- 151.Albersmeyer M, Hilge R, Schröttle A, Weiss M, Sitter T, Vielhauer V. Acute kidney injury after ingestion of rhubarb: secondary oxalate nephropathy in a patient with type 1 diabetes. BMC Nephrol. 2012;13:141. doi: 10.1186/1471-2369-13-141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Kikuchi Y, Seta K, Ogawa Y, Takayama T, Nagata M, Taguchi T, Yahata K. Chaga mushroom-induced oxalate nephropathy. Clin Nephrol. 2014;81:440–444. doi: 10.5414/CN107655. [DOI] [PubMed] [Google Scholar]
- 153.Bakul G, Unni VN, Seethaleksmy NV, Mathew A, Rajesh R, Kurien G, Rajesh J, Jayaraj PM, Kishore DS, Jose PP. Acute oxalate nephropathy due to ‘Averrhoa bilimbi’ fruit juice ingestion. Indian J Nephrol. 2013;23:297–300. doi: 10.4103/0971-4065.114481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Getting JE, Gregoire JR, Phul A, Kasten MJ. Oxalate nephropathy due to ‘juicing’: case report and review. Am J Med. 2013;126:768–772. doi: 10.1016/j.amjmed.2013.03.019. [DOI] [PubMed] [Google Scholar]
- 155.Chen CL, Fang HC, Chou KJ, Wang JS, Chung HM. Acute oxalate nephropathy after ingestion of star fruit. Am J Kidney Dis. 2001;37:418–422. doi: 10.1053/ajkd.2001.21333. [DOI] [PubMed] [Google Scholar]
- 156.Niticharoenpong K, Chalermsanyakorn P, Panvichian R, Kitiyakara C. Acute deterioration of renal function induced by star fruit ingestion in a patient with chronic kidney disease. J Nephrol. 2006;19:682–686. [PubMed] [Google Scholar]
- 157.Neto MM, Silva GEB, Costa RS, Osvaldo M, Neto V, Garcia-Cairasco N, Lopez NB, Haendchen PFC, Silveira C, Mendes AR, et al. Star fruit: simultaneous neurotoxic and nephrotoxic effects in people with previously normal renal function. NDT Plus. 2009;2:485–488. doi: 10.1093/ndtplus/sfp108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Su YJ, Lee CH, Huang SC, Chuang FR. Quiz page April 2011. A woman with oliguria. Acute oxalate nephropathy caused by excess intake of pure carambola juice. Am J Kidney Dis. 2011;57:A23–A25. doi: 10.1053/j.ajkd.2010.11.023. [DOI] [PubMed] [Google Scholar]
- 159.Chang JM, Hwang SJ, Kuo HT, Tsai JC, Guh JY, Chen HC, Tsai JH, Lai YH. Fatal outcome after ingestion of star fruit (Averrhoa carambola) in uremic patients. Am J Kidney Dis. 2000;35:189–193. doi: 10.1016/s0272-6386(00)70325-8. [DOI] [PubMed] [Google Scholar]
- 160.Tse KC, Yip PS, Lam MF, Choy BY, Li FK, Lui SL, Lo WK, Chan TM, Lai KN. Star fruit intoxication in uraemic patients: case series and review of the literature. Intern Med J. 2003;33:314–316. doi: 10.1046/j.1445-5994.2003.00402.x. [DOI] [PubMed] [Google Scholar]
- 161.Neto MM, da Costa JA, Garcia-Cairasco N, Netto JC, Nakagawa B, Dantas M. Intoxication by star fruit (Averrhoa carambola) in 32 uraemic patients: treatment and outcome. Nephrol Dial Transplant. 2003;18:120–125. doi: 10.1093/ndt/18.1.120. [DOI] [PubMed] [Google Scholar]
- 162.Signaté A, Olindo S, Chausson N, Cassinoto C, Edimo Nana M, Saint Vil M, Cabre P, Smadja D. [Star fruit (Averrhoa carambola) toxic encephalopathy] Rev Neurol (Paris) 2009;165:268–272. doi: 10.1016/j.neurol.2008.06.011. [DOI] [PubMed] [Google Scholar]
- 163.Auxiliadora-Martins M, Alkmin Teixeira GC, da Silva GS, Viana JM, Nicolini EA, Martins-Filho OA, Basile-Filho A. Severe encephalopathy after ingestion of star fruit juice in a patient with chronic renal failure admitted to the intensive care unit. Heart Lung. 2010;39:448–452. doi: 10.1016/j.hrtlng.2009.09.003. [DOI] [PubMed] [Google Scholar]
- 164.Fang HC, Chen CL, Wang JS, Chou KJ, Chiou YS, Lee PT, Yeh JH, Yeh MY, Chung HM. Acute oxalate nephropathy induced by star fruit in rats. Am J Kidney Dis. 2001;38:876–880. doi: 10.1053/ajkd.2001.27710. [DOI] [PubMed] [Google Scholar]
- 165.Fang HC, Lee PT, Lu PJ, Chen CL, Chang TY, Hsu CY, Chung HM, Chou KJ. Mechanisms of star fruit-induced acute renal failure. Food Chem Toxicol. 2008;46:1744–1752. doi: 10.1016/j.fct.2008.01.016. [DOI] [PubMed] [Google Scholar]
- 166.Chai W, Liebman M. Oxalate content of legumes, nuts, and grain-based flours. J Food Comp Analysis. 2005;18:723–729. [Google Scholar]
- 167.Massey LK. Food oxalate: factors affecting measurement, biological variation, and bioavailability. J Am Diet Assoc. 2007;107:1191–1194; quiz 1191-1194. doi: 10.1016/j.jada.2007.04.007. [DOI] [PubMed] [Google Scholar]
- 168.Nguyên HVH, Savage GP. Oxalate content of New Zealand grown and imported fruits. J Food Comp Analysis. 2013;31:180–184. [Google Scholar]
- 169.Chalmers AH, Cowley DM, Brown JM. A possible etiological role for ascorbate in calculi formation. Clin Chem. 1986;32:333–336. [PubMed] [Google Scholar]
- 170.Curhan GC, Willett WC, Speizer FE, Stampfer MJ. Intake of vitamins B6 and C and the risk of kidney stones in women. J Am Soc Nephrol. 1999;10:840–845. doi: 10.1681/ASN.V104840. [DOI] [PubMed] [Google Scholar]
- 171.Ramaswamy CR, Williams JD, Griffiths DF. Reversible acute renal failure with calcium oxalate cast nephropathy--possible role of ascorbic acid. Nephrol Dial Transplant. 1993;8:1387–1389. [PubMed] [Google Scholar]
- 172.Auer BL, Auer D, Rodgers AL. Relative hyperoxaluria, crystalluria and haematuria after megadose ingestion of vitamin C. Eur J Clin Invest. 1998;28:695–700. doi: 10.1046/j.1365-2362.1998.00349.x. [DOI] [PubMed] [Google Scholar]
- 173.Mashour S, Turner JF, Merrell R. Acute renal failure, oxalosis, and vitamin C supplementation: a case report and review of the literature. Chest. 2000;118:561–563. doi: 10.1378/chest.118.2.561. [DOI] [PubMed] [Google Scholar]
- 174.Nasr SH, Kashtanova Y, Levchuk V, Markowitz GS. Secondary oxalosis due to excess vitamin C intake. Kidney Int. 2006;70:1672. doi: 10.1038/sj.ki.5001724. [DOI] [PubMed] [Google Scholar]
- 175.Rathi S, Kern W, Lau K. Vitamin C-induced hyperoxaluria causing reversible tubulointerstitial nephritis and chronic renal failure: a case report. J Med Case Rep. 2007;1:155. doi: 10.1186/1752-1947-1-155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.McHugh GJ, Graber ML, Freebairn RC. Fatal vitamin C-associated acute renal failure. Anaesth Intensive Care. 2008;36:585–588. doi: 10.1177/0310057X0803600413. [DOI] [PubMed] [Google Scholar]
- 177.Lamarche J, Nair R, Peguero A, Courville C. Vitamin C-induced oxalate nephropathy. Int J Nephrol. 2011;2011:146927. doi: 10.4061/2011/146927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Gurm H, Sheta MA, Nivera N, Tunkel A. Vitamin C-induced oxalate nephropathy: a case report. J Community Hosp Intern Med Perspect. 2012;2:17718. doi: 10.3402/jchimp.v2i2.17718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Swartz RD, Wesley JR, Somermeyer MG, Lau K. Hyperoxaluria and renal insufficiency due to ascorbic acid administration during total parenteral nutrition. Ann Intern Med. 1984;100:530–531. doi: 10.7326/0003-4819-100-4-530. [DOI] [PubMed] [Google Scholar]
- 180.Lawton JM, Conway LT, Crosson JT, Smith CL, Abraham PA. Acute oxalate nephropathy after massive ascorbic acid administration. Arch Intern Med. 1985;145:950–951. [PubMed] [Google Scholar]
- 181.Wong K, Thomson C, Bailey RR, McDiarmid S, Gardner J. Acute oxalate nephropathy after a massive intravenous dose of vitamin C. Aust N Z J Med. 1994;24:410–411. doi: 10.1111/j.1445-5994.1994.tb01477.x. [DOI] [PubMed] [Google Scholar]
- 182.Alkhunaizi AM, Chan L. Secondary oxalosis: a cause of delayed recovery of renal function in the setting of acute renal failure. J Am Soc Nephrol. 1996;7:2320–2326. doi: 10.1681/ASN.V7112320. [DOI] [PubMed] [Google Scholar]
- 183.Cossey LN, Rahim F, Larsen CP. Oxalate nephropathy and intravenous vitamin C. Am J Kidney Dis. 2013;61:1032–1035. doi: 10.1053/j.ajkd.2013.01.025. [DOI] [PubMed] [Google Scholar]
- 184.Konta T, Yamaoka M, Tanida H, Matsunaga T, Tomoike H. Acute renal failure due to oxalate ingestion. Intern Med. 1998;37:762–765. doi: 10.2169/internalmedicine.37.762. [DOI] [PubMed] [Google Scholar]
- 185.Yamamoto R, Morita S, Aoki H, Nakagawa Y, Yamamoto I, Inokuchi S. Acute renal failure and metabolic acidosis due to oxalic acid intoxication: a case report. Tokai J Exp Clin Med. 2011;36:116–119. [PubMed] [Google Scholar]
- 186.Pons CA, Custer RP. Acute ethylene glycol poisoning; a clinico-pathologic report of eighteen fatal cases. Am J Med Sci. 1946;211:544–552. [PubMed] [Google Scholar]
- 187.Levy RI. Renal failure secondary to ethylene glycol intoxication. JAMA. 1960;173:1210–1213. doi: 10.1001/jama.1960.03020290036007. [DOI] [PubMed] [Google Scholar]
- 188.Friedman EA, Greenberg JB, Merrill JP, Dammin GJ. Consequences of ethylene glycol poisoning. Report of four cases and review of the literature. Am J Med. 1962;32:891–902. doi: 10.1016/0002-9343(62)90035-9. [DOI] [PubMed] [Google Scholar]
- 189.Collins JM, Hennes DM, Holzgang CR, Gourley RT, Porter GA. Recovery after prolonged oliguria due to ethylene glycol intoxication. The prognostic value of serial, percutaneous renal biopsy. Arch Intern Med. 1970;125:1059–1062. [PubMed] [Google Scholar]
- 190.Parry MF, Wallach R. Ethylene glycol poisoning. Am J Med. 1974;57:143–150. doi: 10.1016/0002-9343(74)90780-3. [DOI] [PubMed] [Google Scholar]
- 191.Case records of the Massachusetts General Hospital. Weekly clinicopathological exercises. Case 38-1979. N Engl J Med. 1979;301:650–657. doi: 10.1056/NEJM197909203011208. [DOI] [PubMed] [Google Scholar]
- 192.Frommer JP, Ayus JC. Acute ethylene glycol intoxication. Am J Nephrol. 1982;2:1–5. doi: 10.1159/000166574. [DOI] [PubMed] [Google Scholar]
- 193.Meier M, Nitschke M, Perras B, Steinhoff J. Ethylene glycol intoxication and xylitol infusion--metabolic steps of oxalate-induced acute renal failure. Clin Nephrol. 2005;63:225–228. doi: 10.5414/cnp63225. [DOI] [PubMed] [Google Scholar]
- 194.Stokes MB. Acute oxalate nephropathy due to ethylene glycol ingestion. Kidney Int. 2006;69:203. doi: 10.1038/sj.ki.5000107. [DOI] [PubMed] [Google Scholar]
- 195.Stapenhorst L, Hesse A, Hoppe B. Hyperoxaluria after ethylene glycol poisoning. Pediatr Nephrol. 2008;23:2277–2279. doi: 10.1007/s00467-008-0917-8. [DOI] [PubMed] [Google Scholar]
- 196.Desilva MB, Mueller PS. Renal consequences of long-term, low-dose intentional ingestion of ethylene glycol. Ren Fail. 2009;31:586–588. doi: 10.1080/08860220903003362. [DOI] [PubMed] [Google Scholar]
- 197.Ting SM, Ching I, Nair H, Langman G, Suresh V, Temple RM. Early and late presentations of ethylene glycol poisoning. Am J Kidney Dis. 2009;53:1091–1097. doi: 10.1053/j.ajkd.2008.12.019. [DOI] [PubMed] [Google Scholar]
- 198.Alhamad T, Blandon J, Meza AT, Bilbao JE, Hernandez GT. Acute kidney injury with oxalate deposition in a patient with a high anion gap metabolic acidosis and a normal osmolal gap. J Nephropathol. 2013;2:139–143. doi: 10.12860/JNP.2013.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Daudon M, Reveillaud RJ, Jungers P. Piridoxilate-associated calcium oxalate urinary calculi: a new metabolic drug-induced nephrolithiasis. Lancet. 1985;1:1338. doi: 10.1016/s0140-6736(85)92835-1. [DOI] [PubMed] [Google Scholar]
- 200.Vigeral P, Kenouch S, Chauveau D, Mougenot B, Méry JP. Piridoxilate-associated nephrocalcinosis: a new form of chronic oxalate nephropathy. Nephrol Dial Transplant. 1987;2:275–278. [PubMed] [Google Scholar]
- 201.Mousson C, Justrabo E, Rifle G, Sgro C, Chalopin JM, Gérard C. Piridoxilate-induced oxalate nephropathy can lead to end-stage renal failure. Nephron. 1993;63:104–106. doi: 10.1159/000187151. [DOI] [PubMed] [Google Scholar]
- 202.Cuvelier C, Goffin E, Cosyns JP, Claeys N, Squifflet JP, Pirson Y, de Strihou CV. Acute renal failure due to naftidrofuryl oxalate Praxilène overdose in a kidney transplant recipient. Nephrol Dial Transplant. 1995;10:1756–1758. [PubMed] [Google Scholar]
- 203.Le Meur Y, Moesch C, Rincé M, Aldigier JC, Leroux-Robert C. Potential nephrotoxicity of intravenous infusions of naftidrofuryl oxalate. Nephrol Dial Transplant. 1995;10:1751–1755. [PubMed] [Google Scholar]
- 204.Kim MJ, Lee JS, Kim SW. Acute kidney injury associated with nafronyl oxalate overdose. Clin Exp Nephrol. 2013;17:437–438. doi: 10.1007/s10157-012-0752-x. [DOI] [PubMed] [Google Scholar]
- 205.Frascino JA, Vanamee P, Rosen PP. Renal oxalosis and azotemia after methoxyflurane anesthesia. N Engl J Med. 1970;283:676–679. doi: 10.1056/NEJM197009242831304. [DOI] [PubMed] [Google Scholar]
- 206.Antonelli JA, Langman CB, Odom C, Poindexter J, Huet B, Pearle MS. Defining variation in urinary oxalate in hyperoxaluric stone-formers. J Endourol. 2013:Sep 2; Epub ahead of print. doi: 10.1089/end.2013.0199. [DOI] [PubMed] [Google Scholar]
- 207.Herlitz LC, D’Agati VD, Markowitz GS. Crystalline nephropathies. Arch Pathol Lab Med. 2012;136:713–720. doi: 10.5858/arpa.2011-0565-RA. [DOI] [PubMed] [Google Scholar]
- 208.de Water R, Boevé ER, van Miert PP, Vermaire CP, van Run PR, Cao LC, de Bruijn WC, Schröder FH. Pathological and immunocytochemical changes in chronic calcium oxalate nephrolithiasis in the rat. Scanning Microsc. 1996;10:577–587; discussion 587-590. [PubMed] [Google Scholar]
- 209.SMITH DE. Morphologic lesions due to acute and subacute poisoning with antifreeze (ethylene glycol) AMA Arch Pathol. 1951;51:423–433. [PubMed] [Google Scholar]
- 210.Flanagan P, Libcke JH. Renal biopsy observations following recovery from ethylene glycol nephrosis. Am J Clin Pathol. 1964;41:171–175. doi: 10.1093/ajcp/41.2.171. [DOI] [PubMed] [Google Scholar]
- 211.Bove KE. Ethylene glycol toxicity. Am J Clin Pathol. 1966;45:46–50. doi: 10.1093/ajcp/45.1.46. [DOI] [PubMed] [Google Scholar]
- 212.Mori S, Beppu T. Secondary renal oxalosis. A statistical analysis of its possible causes. Acta Pathol Jpn. 1983;33:661–669. [PubMed] [Google Scholar]
- 213.Chintanaboina J, Nadasdy T, Manahan F, Muppidi V. Acute oxalate nephropathy diagnosed by renal biopsy. Intern Med. 2012;51:2249. doi: 10.2169/internalmedicine.51.8183. [DOI] [PubMed] [Google Scholar]
- 214.Hoorens A, Van Der Niepen P, Keuppens F, Vanden Houte K, Klöppel G. Pseudotuberculous pyelonephritis associated with nephrolithiasis. Am J Surg Pathol. 1992;16:522–525. doi: 10.1097/00000478-199205000-00012. [DOI] [PubMed] [Google Scholar]
- 215.Maldonado I, Prasad V, Reginato AJ. Oxalate crystal deposition disease. Curr Rheumatol Rep. 2002;4:257–264. doi: 10.1007/s11926-002-0074-1. [DOI] [PubMed] [Google Scholar]
- 216.Bell EG, McAfee JG, Makhuli ZN. Medical imaging of renal diseases-suggested indication for different modalities. Semin Nucl Med. 1981;11:105–127. doi: 10.1016/s0001-2998(81)80041-4. [DOI] [PubMed] [Google Scholar]
- 217.Helms E, Servilla KS, Hartshorne MF, Harris A, Nichols MJ, Tzamaloukas AH. Tubulointerstitial nephritis and uveitis syndrome: use of gallium scintigraphy in its diagnosis and treatment. Int Urol Nephrol. 2005;37:119–122. doi: 10.1007/s11255-004-2356-1. [DOI] [PubMed] [Google Scholar]
- 218.Joaquim AI, Mendes GE, Ribeiro PF, Baptista MA, Burdmann EA. Ga-67 scintigraphy in the differential diagnosis between acute interstitial nephritis and acute tubular necrosis: an experimental study. Nephrol Dial Transplant. 2010;25:3277–3282. doi: 10.1093/ndt/gfq152. [DOI] [PubMed] [Google Scholar]
- 219.Khan SR, Finlayson B, Hackett RL. Histologic study of the early events in oxalate induced intranephronic calculosis. Invest Urol. 1979;17:199–202. [PubMed] [Google Scholar]
- 220.Evan AP, Lingeman JE, Worcester EM, Bledsoe SB, Sommer AJ, Williams JC, Krambeck AE, Philips CL, Coe FL. Renal histopathology and crystal deposits in patients with small bowel resection and calcium oxalate stone disease. Kidney Int. 2010;78:310–317. doi: 10.1038/ki.2010.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Wiessner JH, Hung LY, Mandel NS. Crystal attachment to injured renal collecting duct cells: influence of urine proteins and pH. Kidney Int. 2003;63:1313–1320. doi: 10.1046/j.1523-1755.2003.00866.x. [DOI] [PubMed] [Google Scholar]
- 222.Wesson JA, Johnson RJ, Mazzali M, Beshensky AM, Stietz S, Giachelli C, Liaw L, Alpers CE, Couser WG, Kleinman JG, et al. Osteopontin is a critical inhibitor of calcium oxalate crystal formation and retention in renal tubules. J Am Soc Nephrol. 2003;14:139–147. doi: 10.1097/01.asn.0000040593.93815.9d. [DOI] [PubMed] [Google Scholar]
- 223.Asokan D, Kalaiselvi P, Muhammed Farooq S, Varalakshmi P. Calcium oxalate monohydrate binding protein: a diagnostic biomarker for calcium oxalate kidney stone formers. Urol Res. 2004;32:357–361. doi: 10.1007/s00240-004-0430-x. [DOI] [PubMed] [Google Scholar]
- 224.Farell G, Huang E, Kim SY, Horstkorte R, Lieske JC. Modulation of proliferating renal epithelial cell affinity for calcium oxalate monohydrate crystals. J Am Soc Nephrol. 2004;15:3052–3062. doi: 10.1097/01.ASN.0000144205.49134.64. [DOI] [PubMed] [Google Scholar]
- 225.Sheng X, Jung T, Wesson JA, Ward MD. Adhesion at calcium oxalate crystal surfaces and the effect of urinary constituents. Proc Natl Acad Sci USA. 2005;102:267–272. doi: 10.1073/pnas.0406835101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Miyazawa K, Takahashi Y, Morita N, Moriyama MT, Kosaka T, Nishio M, Yoshimoto T, Suzuki K. Cyclooxygenase 2 and prostaglandin E2 regulate the attachment of calcium oxalate crystals to renal epithelial cells. Int J Urol. 2012;19:936–943. doi: 10.1111/j.1442-2042.2012.03060.x. [DOI] [PubMed] [Google Scholar]
- 227.Roop-ngam P, Chaiyarit S, Pongsakul N, Thongboonkerd V. Isolation and characterizations of oxalate-binding proteins in the kidney. Biochem Biophys Res Commun. 2012;424:629–634. doi: 10.1016/j.bbrc.2012.07.015. [DOI] [PubMed] [Google Scholar]
- 228.Schepers MS, van Ballegooijen ES, Bangma CH, Verkoelen CF. Oxalate is toxic to renal tubular cells only at supraphysiologic concentrations. Kidney Int. 2005;68:1660–1669. doi: 10.1111/j.1523-1755.2005.00576.x. [DOI] [PubMed] [Google Scholar]
- 229.Knoll T, Steidler A, Trojan L, Sagi S, Schaaf A, Yard B, Michel MS, Alken P. The influence of oxalate on renal epithelial and interstitial cells. Urol Res. 2004;32:304–309. doi: 10.1007/s00240-004-0429-3. [DOI] [PubMed] [Google Scholar]
- 230.Khandrika L, Koul S, Meacham RB, Koul HK. Kidney injury molecule-1 is up-regulated in renal epithelial cells in response to oxalate in vitro and in renal tissues in response to hyperoxaluria in vivo. PLoS One. 2012;7:e44174. doi: 10.1371/journal.pone.0044174. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 231.Horuz R, Göktaş C, Çetinel CA, Akça O, Aydın H, Ekici ID, Albayrak S, Sarıca K. Role of TNF-associated cytokines in renal tubular cell apoptosis induced by hyperoxaluria. Urolithiasis. 2013;41:197–203. doi: 10.1007/s00240-013-0559-6. [DOI] [PubMed] [Google Scholar]
- 232.Gonçalves GM, Castoldi A, Braga TT, Câmara NO. New roles for innate immune response in acute and chronic kidney injuries. Scand J Immunol. 2011;73:428–435. doi: 10.1111/j.1365-3083.2011.02523.x. [DOI] [PubMed] [Google Scholar]
- 233.Mulay SR, Evan A, Anders HJ. Molecular mechanisms of crystal-related kidney inflammation and injury. Implications for cholesterol embolism, crystalline nephropathies and kidney stone disease. Nephrol Dial Transplant. 2014;29:507–514. doi: 10.1093/ndt/gft248. [DOI] [PubMed] [Google Scholar]
- 234.Gross O, Thomas CJ, Guarda G, Tschopp J. The inflammasome: an integrated view. Immunol Rev. 2011;243:136–151. doi: 10.1111/j.1600-065X.2011.01046.x. [DOI] [PubMed] [Google Scholar]
- 235.Dowling JK, O’Neill LA. Biochemical regulation of the inflammasome. Crit Rev Biochem Mol Biol. 2012;47:424–443. doi: 10.3109/10409238.2012.694844. [DOI] [PubMed] [Google Scholar]
- 236.Chai J, Shi Y. Apoptosome and inflammasome: conserved machineries for caspase activation. Natl Sci Rev. 2014;1:101–118. [Google Scholar]
- 237.Anders HJ, Muruve DA. The inflammasomes in kidney disease. J Am Soc Nephrol. 2011;22:1007–1018. doi: 10.1681/ASN.2010080798. [DOI] [PubMed] [Google Scholar]
- 238.Wang W, Wang X, Chun J, Vilaysane A, Clark S, French G, Bracey NA, Trpkov K, Bonni S, Duff HJ, et al. Inflammasome-independent NLRP3 augments TGF-β signaling in kidney epithelium. J Immunol. 2013;190:1239–1249. doi: 10.4049/jimmunol.1201959. [DOI] [PubMed] [Google Scholar]
- 239.Martinon F, Pétrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440:237–241. doi: 10.1038/nature04516. [DOI] [PubMed] [Google Scholar]
- 240.Knauf F, Asplin JR, Granja I, Schmidt IM, Moeckel GW, David RJ, Flavell RA, Aronson PS. NALP3-mediated inflammation is a principal cause of progressive renal failure in oxalate nephropathy. Kidney Int. 2013;84:895–901. doi: 10.1038/ki.2013.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Kurts C. A crystal-clear mechanism of chronic kidney disease. Kidney Int. 2013;84:859–861. doi: 10.1038/ki.2013.251. [DOI] [PubMed] [Google Scholar]
- 242.Mulay SR, Kulkarni OP, Rupanagudi KV, Migliorini A, Darisipudi MN, Vilaysane A, Muruve D, Shi Y, Munro F, Liapis H, et al. Calcium oxalate crystals induce renal inflammation by NLRP3-mediated IL-1β secretion. J Clin Invest. 2013;123:236–246. doi: 10.1172/JCI63679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Lorenz G, Darisipudi MN, Anders HJ. Canonical and non-canonical effects of the NLRP3 inflammasome in kidney inflammation and fibrosis. Nephrol Dial Transplant. 2014;29:41–48. doi: 10.1093/ndt/gft332. [DOI] [PubMed] [Google Scholar]
- 244.Hoppe B, Leumann E, von Unruh G, Laube N, Hesse A. Diagnostic and therapeutic approaches in patients with secondary hyperoxaluria. Front Biosci. 2003;8:e437–e443. doi: 10.2741/1135. [DOI] [PubMed] [Google Scholar]
- 245.Ferraz RR, Marques NC, Froeder L, Menon VB, Siliano PR, Baxmann AC, Heilberg IP. Effects of Lactobacillus casei and Bifidobacterium breve on urinary oxalate excretion in nephrolithiasis patients. Urol Res. 2009;37:95–100. doi: 10.1007/s00240-009-0177-5. [DOI] [PubMed] [Google Scholar]
- 246.Jiang J, Knight J, Easter LH, Neiberg R, Holmes RP, Assimos DG. Impact of dietary calcium and oxalate, and Oxalobacter formigenes colonization on urinary oxalate excretion. J Urol. 2011;186:135–139. doi: 10.1016/j.juro.2011.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Goldfarb DS, Modersitzki F, Asplin JR. A randomized, controlled trial of lactic acid bacteria for idiopathic hyperoxaluria. Clin J Am Soc Nephrol. 2007;2:745–749. doi: 10.2215/CJN.00600207. [DOI] [PubMed] [Google Scholar]
- 248.Lieske JC, Tremaine WJ, De Simone C, O’Connor HM, Li X, Bergstralh EJ, Goldfarb DS. Diet, but not oral probiotics, effectively reduces urinary oxalate excretion and calcium oxalate supersaturation. Kidney Int. 2010;78:1178–1185. doi: 10.1038/ki.2010.310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Siener R, Bade DJ, Hesse A, Hoppe B. Dietary hyperoxaluria is not reduced by treatment with lactic acid bacteria. J Transl Med. 2013;11:306. doi: 10.1186/1479-5876-11-306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Emmett M, Guirl MJ, Santa Ana CA, Porter JL, Neimark S, Hofmann AF, Fordtran JS. Conjugated bile acid replacement therapy reduces urinary oxalate excretion in short bowel syndrome. Am J Kidney Dis. 2003;41:230–237. doi: 10.1053/ajkd.2003.50012. [DOI] [PubMed] [Google Scholar]
- 251.Toblli JE, Ferder L, Stella I, Angerosa M, Inserra F. Protective role of enalapril for chronic tubulointerstitial lesions of hyperoxaluria. J Urol. 2001;166:275–280. [PubMed] [Google Scholar]
- 252.Tsai CH, Chen YC, Chen LD, Pan TC, Ho CY, Lai MT, Tsai FJ, Chen WC. A traditional Chinese herbal antilithic formula, Wulingsan, effectively prevents the renal deposition of calcium oxalate crystal in ethylene glycol-fed rats. Urol Res. 2008;36:17–24. doi: 10.1007/s00240-007-0122-4. [DOI] [PubMed] [Google Scholar]
- 253.Vanachayangkul P, Byer K, Khan S, Butterweck V. An aqueous extract of Ammi visnaga fruits and its constituents khellin and visnagin prevent cell damage caused by oxalate in renal epithelial cells. Phytomedicine. 2010;17:653–658. doi: 10.1016/j.phymed.2009.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Theka T, Rodgers A, Ravenscroft N, Lewandowski S. Intestinal permeability in subjects from two different race groups with diverse stone-risk profiles. Urolithiasis. 2013;41:111–117. doi: 10.1007/s00240-013-0543-1. [DOI] [PubMed] [Google Scholar]
- 255.Chung TT, Summers S, Sheaff M, Cunningham J. Always look beyond the stones: hyperoxaluria overlooked. Clin Nephrol. 2004;62:58–61. doi: 10.5414/cnp62058. [DOI] [PubMed] [Google Scholar]
- 256.Mydlík M, Derzsiová K. Oxalic Acid as a uremic toxin. J Ren Nutr. 2008;18:33–39. doi: 10.1053/j.jrn.2007.10.008. [DOI] [PubMed] [Google Scholar]
- 257.Tangri N, Kitsios GD, Inker LA, Griffith J, Naimark DM, Walker S, Rigatto C, Uhlig K, Kent DM, Levey AS. Risk prediction models for patients with chronic kidney disease: a systematic review. Ann Intern Med. 2013;158:596–603. doi: 10.7326/0003-4819-158-8-201304160-00004. [DOI] [PubMed] [Google Scholar]
- 258.Holmes RP, Assimos DG, Goodman HO. Genetic and dietary influences on urinary oxalate excretion. Urol Res. 1998;26:195–200. doi: 10.1007/s002400050046. [DOI] [PubMed] [Google Scholar]
- 259.Lewandowski S, Rodgers A, Schloss I. The influence of a high-oxalate/low-calcium diet on calcium oxalate renal stone risk factors in non-stone-forming black and white South African subjects. BJU Int. 2001;87:307–311. doi: 10.1046/j.1464-410x.2001.00064.x. [DOI] [PubMed] [Google Scholar]
- 260.Rodgers A, Allie-Hamdulay S, Pinnock D, Baretta G, Trinchieri A. Risk factors for renal calcium stone formation in South African and European young adults. Arch Ital Urol Androl. 2009;81:171–174. [PubMed] [Google Scholar]
- 261.Taylor EN, Curhan GC. Differences in 24-hour urine composition between black and white women. J Am Soc Nephrol. 2007;18:654–659. doi: 10.1681/ASN.2006080854. [DOI] [PubMed] [Google Scholar]
- 262.Clark JS, Vandorpe DH, Chernova MN, Heneghan JF, Stewart AK, Alper SL. Species differences in Cl- affinity and in electrogenicity of SLC26A6-mediated oxalate/Cl- exchange correlate with the distinct human and mouse susceptibilities to nephrolithiasis. J Physiol. 2008;586:1291–1306. doi: 10.1113/jphysiol.2007.143222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Monico CG, Weinstein A, Jiang Z, Rohlinger AL, Cogal AG, Bjornson BB, Olson JB, Bergstralh EJ, Milliner DS, Aronson PS. Phenotypic and functional analysis of human SLC26A6 variants in patients with familial hyperoxaluria and calcium oxalate nephrolithiasis. Am J Kidney Dis. 2008;52:1096–1103. doi: 10.1053/j.ajkd.2008.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Rumsby G. Oxalate transport as contributor to primary hyperoxaluria: the jury is still out. Am J Kidney Dis. 2008;52:1031–1034. doi: 10.1053/j.ajkd.2008.10.004. [DOI] [PubMed] [Google Scholar]
- 265.Hatch M, Freel RW. The roles and mechanisms of intestinal oxalate transport in oxalate homeostasis. Semin Nephrol. 2008;28:143–151. doi: 10.1016/j.semnephrol.2008.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Dawson PA, Sim P, Mudge DW, Cowley D. Human SLC26A1 gene variants: a pilot study. ScientificWorldJournal. 2013;2013:541710. doi: 10.1155/2013/541710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Bid HK, Kumar A, Kapoor R, Mittal RD. Association of vitamin D receptor-gene (FokI) polymorphism with calcium oxalate nephrolithiasis. J Endourol. 2005;19:111–115. doi: 10.1089/end.2005.19.111. [DOI] [PubMed] [Google Scholar]
- 268.Turner CM, Arulkumaran N, Singer M, Unwin RJ, Tam FW. Is the inflammasome a potential therapeutic target in renal disease? BMC Nephrol. 2014;15:21. doi: 10.1186/1471-2369-15-21. [DOI] [PMC free article] [PubMed] [Google Scholar]