


Abstract
The structural organization of chromosomes is essential for their correct function and dynamics during the cell cycle. The assembly of DNA into chromatin provides the substrate for topoisomerases and condensins, which introduce the different levels of superhelical torsion required for DNA metabolism. In particular, Top2 and condensin are directly involved in both the resolution of precatenanes that form during replication and the formation of the intramolecular loop that detects tension at the centromeric chromatin during chromosome biorientation. Here we show that histone depletion activates the spindle assembly checkpoint (SAC) and impairs sister chromatid decatenation, leading to chromosome mis-segregation and lethality in the absence of the SAC. We demonstrate that histone depletion impairs chromosome biorientation and activates the Aurora-dependent pathway, which detects tension problems at the kinetochore. Interestingly, SAC activation is suppressed by the absence of Top2 and Smc2, an essential component of condensin. Indeed, smc2-8 suppresses catenanes accumulation, mitotic arrest and growth defects induced by histone depletion at semi-permissive temperature. Remarkably, SAC activation by histone depletion is associated with condensin-mediated alterations of the centromeric chromatin. Therefore, our results reveal the importance of a precise interplay between histone supply and condensin/Top2 for pericentric chromatin structure, precatenanes resolution and centromere biorientation.
INTRODUCTION
The assembly of DNA into nucleosomes provides the first level of chromosome organization and the substrate for a plethora of enzymatic and structural factors that build chromosomes. Nucleosomes, topoisomerase II (Top2 in yeast) and condensin are the main determinants of the level of DNA supercoiling and chromosome compaction. Not surprisingly, they play essential roles in DNA metabolic processes, which involve the continuous accumulation and release of torsional stress.
Chromosome packaging starts with the assembly of newly replicated DNA into chromatin during S phase. The nucleosome—the repetitive unit of chromatin—is formed by ∼146 base pairs of DNA wrapped 1.65 times around an octamer of histones. This octamer is formed by a core (H3/H4)2 tetramer to which an H2A/H2B dimer binds on each side (1). In a highly regulated process, histone chaperones and chromatin assembly factors interact physically and genetically with components of the replisome to ensure a rapid and correct supply of histones at the replication fork (2). Nucleosome assembly introduces an accumulation of DNA supercoiling that is further modulated by the activities of DNA topoisomerases and condensins, which play fundamental roles in the structural and functional organization of chromosomes. In particular, Top2 can untangle catenated molecules and resolve both positive and negative supercoils. Condensins are multisubunit complexes formed by two ‘structural maintenance of chromosomes’ (SMCs, Smc2 and Smc4 in yeast) ATPases and three non-SMC subunits, which form a V-shaped structure able to trap DNA and introduce positive superhelical tension. Top2 and condensin are essential for proper chromosome condensation and segregation and play important roles in processes such as transcription, recombination and DNA repair (3,4). Top2 and condensin are also essential for resolving the precatenanes that result from the advance of the replication forks (5–8). Finally, they provide the tensile properties of the centromeric chromatin, which are essential for the activation of the spindle assembly checkpoint (SAC) in response to problems in the attachment of the microtubules to the kinetochores (9–14).
Chromatin is directly involved in the response to DNA damage and chromosome segregation. Specifically, subtle changes in chromatin structure caused by a deficit or an excess in the pool of available histones have deleterious consequences on genome integrity (15–18). In the case of histone depletion, yeast mutants display replication fork instability and accumulation of recombinogenic DNA damage, phenotypes that are reminiscent to those displayed by chromatin assembly mutants lacking either the histone chaperone Asf1 or the chromatin assembly factors Cac1 and Rtt106 (17–20). In addition, cells expressing different mutants of histone H4 or lacking Cac1 and the histone chaperone Hir1 display defects in the centromeric chromatin and the kinetochores that are associated with increased rates of chromosome mis-segregation and the activation of the SAC (21–23). Not surprisingly, nucleosome depletion by histone loss causes a G2/M arrest (18,24). Here we show that, unexpectedly, the DNA damage checkpoint does not contribute to this arrest; instead, histone depletion activates the SAC. We show that histone depletion affects chromosome biorientation and activates the Aurora-dependent branch of the SAC, which detects problems of tension at the kinetochore (25,26). We also show that histone depletion impairs sister chromatid decatenation, leading to chromosome mis-segregation and cell lethality in SAC-defective cells. Importantly, histone depletion-mediated SAC activation is associated with condensin-mediated alterations of the centromeric chromatin, and accordingly, it is suppressed by the absence of Smc2 and Top2; likewise, catenanes accumulation is suppressed by the absence of Smc2. Altogether, our results suggest that histone deposition and condensin/Top2 cooperate to assemble a structural and functional chromatin structure at the centromeric region.
MATERIALS AND METHODS
Yeast strains and plasmids
Yeast strains used in this study are listed in Supplementary Table S1. Tagged strains and deletion mutants were constructed by a polymerase chain reaction (PCR)-based strategy (27). chr-G::HHF1 strains were constructed by replacing in a hhf2Δ strain the HHF1 promoter with a PCR fragment containing the GAL1 promoter and the URA3 marker from pARSGLB-IN (28). G::HHF2 and M::HHF2 strains were constructed by replacing plasmid p413TARtetH4 with plasmid pUK421 (G::HHF2) or p416MetH4 (M::HHF2). p413TARtetH4 (18), pUK421 (24) and p416MetH4 are HIS3-, TRP1- and URA3-based centromeric plasmids expressing histone H4 from tet, GAL1 and MET25 promoter, respectively. p416MetH4 was constructed in two steps: first, a BamHI-XhoI PCR fragment containing the ORF of HHF2 was inserted at the XbaI (made blunt)-XhoI site of p426Met25 (29); then, the generated PvuII fragment containing the MET25p::HHF2::CYC1t construct was inserted at PvuII of pRS416. pWJ1344 (R. Rothstein, Columbia University) and p314R52YFP are LEU2- and TRP1-based centromeric plasmids expressing RAD52-YFP. p314R52YFP was constructed by inserting a SacI-XhoI fragment from pWJ1213 containing the construct RAD52-YFP at the SacI-XhoI site of pRS314. pRS416 (30) and YCp50 (31) are URA3-based centromeric plasmids; YCpPDED1T2 is a URA3-based centromeric plasmid expressing Top2 from the DED1 promoter (32).
Growth conditions
Yeast cells were grown at 30°C—unless otherwise stated—in supplemented minimal medium (SMM), except for the analyses of Mad2-GFP in Figure 2B and Brn1-Pk9 in Figures 4D, 5A and 5B, which required nocodazole (NCD) treatment and were performed in YPD-rich medium. For G1 synchronization, cells were grown to mid-log-phase and α-factor was added twice at 60-min intervals at either 2 (BAR1 strains) or 0.5 μg/ml (bar1Δ strains), except for t::HHF2/G::HHF2/M::HHF2 strains, which were treated at 90 (t::HHF2 and G::HHF2) or 120 (M::HHF2) min intervals at either 5 (BAR1) or 1 μg/ml (bar1Δ). Cells were then washed three times and released into fresh medium with 50 μg/ml pronase. To induce histone depletion, t::HHF2 cells growing in the presence of 5 μg/ml doxycycline were shifted to 0.25 μg/ml during G1 synchronization and release; G::HHF2 cells growing in the presence of 2% galactose were shifted to 2% glucose during the second incubation with α-factor and after release, except for Figure 6D where they were shifted to 0.05% galactose; chr-G::HHF1 cells growing in the presence of 2% galactose were shifted to 2% glucose after α-factor release; M::HHF2 cells growing in the absence of methionine were shifted to medium with 100 μM methionine during G1 synchronization and release. To induce Top2 degradation during G1 synchronization in top2dg strains, mid-log phase cells growing at 26°C in SMM with 2% raffinose were shifted to 2% raffinose with α-factor for 2 h at 26°C, then shifted to 1.5% raffinose/2% galactose with 50 ng/ml doxycycline and α-factor for 1 h at 26°C, and finally shifted to 1.5% raffinose/2% galactose with 50 ng/ml doxycycline and α-factor for 1 h at 37°C. Cell growth analyses were performed by plating 10-fold serial dilutions from the same number of mid-log phase cells in SMM plates containing 0.25 μg/ml doxycycline.
Figure 2.
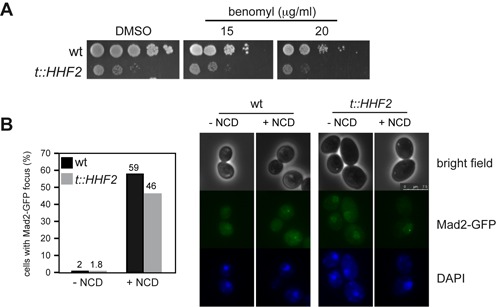
Histone depletion does not affect the SAC response to microtubule depolymerization. (A) Benomyl sensitivity of wild-type and t::HHF2 cells as determined by 10-fold serial dilutions. (B) SAC activation by histone loss is not associated with the formation of a Mad2-GFP focus. The percentage of wild-type and t::HHF2 cells with a Mad2-GFP focus was determined after incubation with or without nocodazole (NCD) at 15 μg/ml for 2 h under conditions of histone depletion (0.25 μg/ml). A total number of 200 cells from two independent experiments were analyzed for each strain and condition. Representative images are shown on the right.
Figure 4.
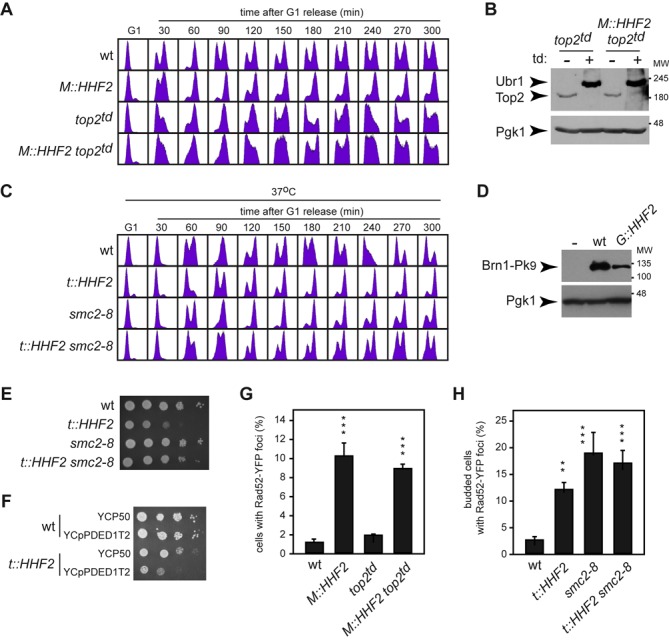
Top2 and condensin are required for SAC activation independently of DNA damage in cells partially depleted of histones. (A) Top2 is required for SAC activation by histone depletion. Cell cycle progression was analyzed by flow cytometry of wild-type, M::HHF2, top2td and M::HHF2 top2td cells synchronized in G1 under restrictive conditions to deplete Top2 and then released into fresh medium under conditions of histone depletion. (B) Western blot analysis of Top2 and Ubr1 in top2td and M::HHF2 top2td G1 synchronized cells under restrictive conditions for Top2 and H4. Both proteins were labeled with Myc epitopes. Pgk1 was used as loading control. MW, molecular weight marker. (C) Condensin is required for SAC activation by histone depletion. Cell cycle progression was analyzed by flow cytometry of wild-type, t::HHF2, smc2-8 and t::HHF2 smc2-8 cells synchronized in G1 at permissive temperature (26°C) and released into fresh medium at restrictive temperature (37°C) under conditions of histone depletion. (D) Western blot analysis of Brn1 in wild-type and G::HHF2 cells synchronized in G1 and released into fresh medium under conditions of histone depletion in the presence of 15 μg/ml NCD for 90 min. Pgk1 was used as loading control. (E) A reduction in condensin activity partially suppresses t::HHF2 growth defects at 26°C. (F) Cells partially depleted of histone H4 are sensitive to Top2 overexpression, as determined by cell growth for wild-type and t::HHF2 cells transformed with plasmids YCp50 (empty) or YCpPDED1T2 (Top2). (G) Rad52 foci accumulation by histone depletion is independent of Top2. Rad52-YFP foci accumulation was determined in wild-type, M::HHF2, top2td and M::HHF2 top2td cells transformed with p314R52YFP (RAD52-YFP), synchronized in G1 under restrictive conditions to deplete Top2 and released into fresh medium for 60 min under conditions of histone depletion. (H) Rad52 foci accumulation in asynchronous cultures of wild-type, t::HHF2, smc2-8 and t::HHF2 smc2-8 cells transformed with pWJ1344 (RAD52-YFP) and grown at permissive temperature (26°C) under conditions of histone depletion (0.25 μg/ml). (G, H) A total number of 100 cells were analyzed for each strain and experiment. The average and SEM of three independent experiments performed with three different transformants from two independent transformation of each strain are shown. Asterisks indicate statistically significant differences (M::HHF2 and M::HHF2 top2td relative to wild type and top2td, and t::HHF2, smc2-8 and t::HHF2 smc2-8 relative to wild type) according to a one-way ANOVA test, where two and three asterisks indicate P < 0.01 and <0.001, respectively.
Figure 5.
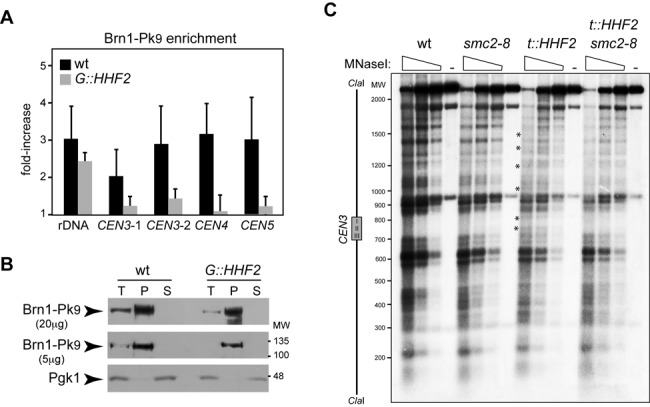
Interplay between histone supply and condensin. (A) Condensin enrichment at different positions of the pericentric chromatin and the ribosomal DNA by ChIP analysis of Brn1-Pk9 in wild-type and G::HHF2 cells grown as indicated in Figure 4D. The analyzed regions lie at the 5′ untranslated region of RDN37 (rDNA) and at 3.35 (CEN3-1), 1.74 (CEN3-2), 0.5 (CEN4) and 2.6 kb (CEN5) from the corresponding centromere. The average and SEM of three independent experiments are shown. The differences are not statistically significant. (B) Western analysis of Brn1 after chromatin fractionation of wild-type and G::HHF2 cells grown as indicated in Figure 4D. Twenty (upper panel) or five micrograms (middle panel) of total (T), pellet (P) and soluble (S) proteins were analyzed. Pgk1 was used as loading control (lower panel). (C) Defective centromeric chromatin structure by histone depletion depends on condensin. MNaseI digestion and indirect-end labeling of CEN3 in wild-type, smc2-8, t::HHF2 and t::HHF2 smc2-8 cells synchronized in G1 and released into fresh medium at restrictive temperature (37°C) until G2/M (90 min for wild-type and smc2-8 and 120 min for t::HHF2 and t::HHF2 smc2-8) under conditions of histone depletion. A scheme with the position of the CDE I-II-III sequence of CEN3 is shown on the left. Asterisks show sites of hypersensitivity to MNaseI. The experiment was repeated twice with similar results.
Figure 6.
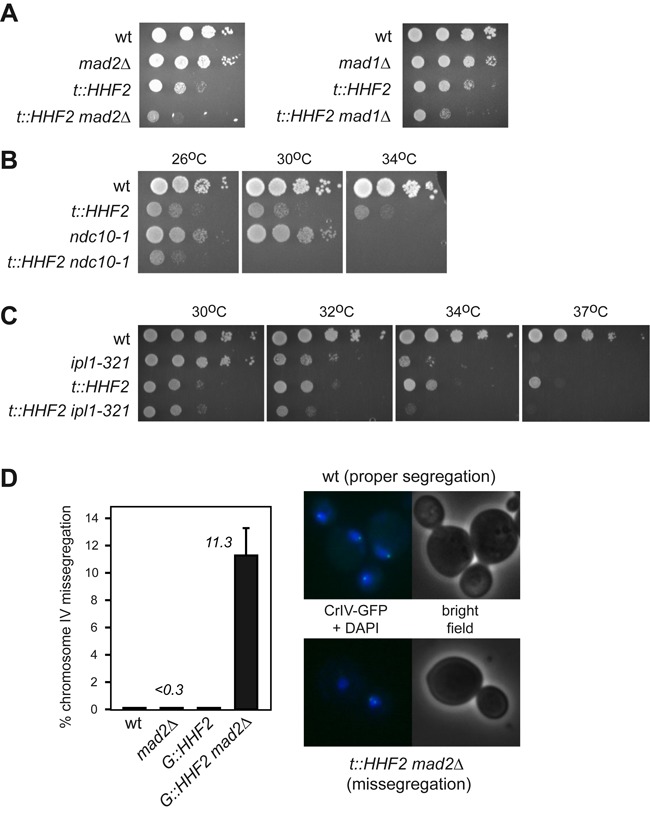
The SAC prevents cell lethality by chromosome mis-segregation. (A–C) SAC is required for cell viability under conditions of histone depletion. The heterodimer Mad2/Mad1 (A), a functional kinetochore (B) and Aurora kinase (C) are required for t::HHF2 growth, as determined by synthetic growth defects of t::HHF2 and mad2Δ, mad1Δ, ndc10-1 or ipl1-321. (D) SAC prevents chromosome mis-segregation in cells partially depleted of histones. Chromosome segregation was followed in cells harboring an array of tetO sites labeled with tetR-GFP at 1.4 kb from the centromere. Asynchronous wild-type and G::HHF2 cultures were shifted from 2% galactose to 0.05% galactose medium for 4 h, and the fate of chromosome IV was followed by fluorescence microscopy in anaphase cells (as determined by DAPI staining). The right panel shows images of proper (one dot in the mother and one dot in the bud) and incorrect (both dots in either the mother or the bud) chromosome segregation. A total of 90–100 cells were analyzed for each strain and experiment. The average and SEM of three independent experiments are shown.
Flow cytometry
DNA content analysis was performed by Flow cytometry as reported previously (18). Each cell cycle progression analysis by Flow cytometry was repeated two to three times with similar results.
Immunofluorescence analysis
Immunolocalization of the spindles was performed by immunofluorescence using antibodies anti-tubulin (Abcam) and anti-rat fluorescein isothiocyanate (Jackson ImmunoResearch Laboratories, Inc.) as previously described (33), except that formaldehyde fixation was overnight. Each cell cycle progression analysis by immunofluorescence against tubulin was repeated twice with similar results. Rad52-YFP, Mad2-GFP and tetR-GFP signals were detected in cells fixed with formaldehyde as described (34). Microscope preparations were visualized with a Leica CTR6000 fluorescence microscope.
Western blot and chromatin fractionation
Yeast protein extracts—except for chromatin fractionation—were prepared using the trichloroacetic acid protocol as described (18). For chromatin fractionation in Figure 5, total, soluble and pellet (chromatin) proteins were isolated as previously detailed for young yeast cells (35). Yeast proteins were run on an 8% (HA-Pds1, Brn1-Pk9 and Pgk1), 6% (Myc-Top2 and Myc-Ubr1) or 15% (H4 and Pgk1) sodium dodecyl sulfate-polyacrilamyde gel. HA-Pds1, Myc-Top2, Myc-Ubr1, Pgk1, Brn1-Pk9 and H4 were detected with the mouse monoclonal HA.11 (Covance), the mouse monoclonal 9E10 (Covance), the mouse polyclonal 22C5D8 (Invitrogen), the mouse monoclonal SV5-Pk1 (Serotec) and the rabbit polyclonal ab10158 (Abcam) antibodies, respectively. Peroxidase conjugate (Figures 1D, 4B, 4D and 5B) and fluorophore conjugate (Supplementary Figures S1A and S4A) secondary antibodies were used.
Figure 1.
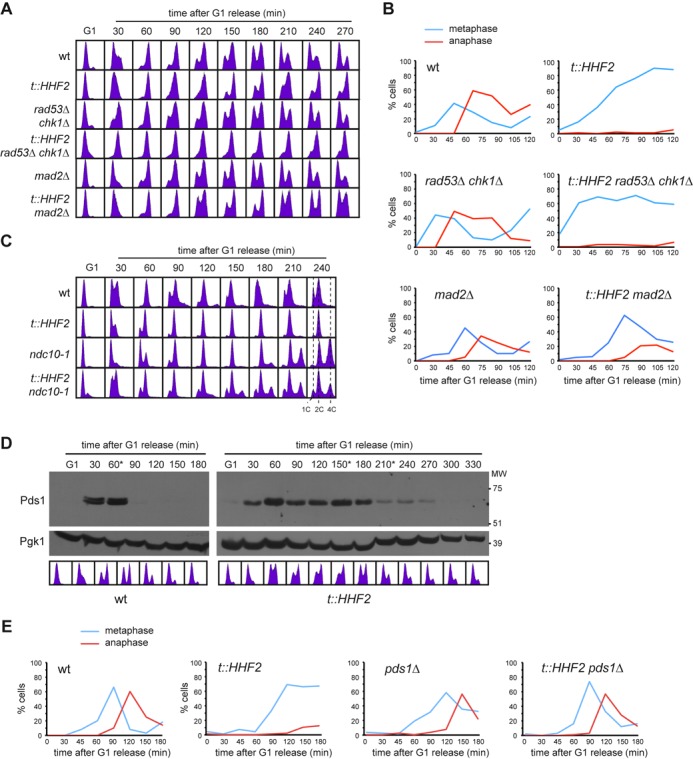
Histone depletion activates the SAC. (A, B) Metaphase arrest by histone loss is Mad2-dependent and Rad53 and Chk1-independent. Cell cycle progression analysis by flow cytometry (A) and nuclear and spindle morphologies by DAPI and immunofluorescence against tubulin (B) of wild-type, t::HHF2, sml1Δ rad53Δ chk1Δ, sml1Δ t::HHF2 rad53Δ chk1Δ, mad2Δ and t::HHF2 mad2Δ cells synchronized in G1 and released into fresh medium under conditions of histone depletion. sml1Δ did not affect cell cycle progression of wild type and t::HHF2 (data not shown). (C) A functional kinetochore is required for SAC activation in t::HHF2. DNA content was analyzed by flow cytometry of wild-type, ndc10-1, t::HHF2 and t::HHF2 ndc10-1 cells synchronized in G1 at permissive temperature (26°C) and released into fresh medium at restrictive temperature (37°C) under conditions of histone depletion. (D) Securine/Pds1 remains undegraded during SAC activation in t::HHF2 cells. Pds1 accumulation was determined by western analysis of wild-type and t::HHF2 cells synchronized in G1 and released into fresh medium for one cell cycle under conditions of histone depletion. Asterisks show the time at which α-factor was added to prevent cells from re-entering a new mitosis. MW, molecular weight marker. Cell cycle progression by DNA content analysis is shown below. (E) Securine/Pds1 is required for SAC activation in t::HHF2 cells. Cell cycle progression was analyzed by immunofluorescence against tubulin (spindle morphology) for wild-type, pds1Δ, t::HHF2 and t::HHF2 pds1Δ cells synchronized in G1 and released into fresh medium at 23°C under conditions of histone depletion.
Chromatin immunoprecipitation analysis
Chromatin immunoprecipitation (ChIP) assays were performed as described (36) with the anti-Pk antibody SV5-Pk1 (Serotec) for Brn1-Pk9. Oligonucleotide sequences for the real-time PCR amplifications performed on purified DNA before (input; I) or after (immunoprecipitated; IP) immunoprecipitation are shown in Supplementary Table S2. Protein enrichment at each specific region was calculated as the ratio between the IP and the I in the tagged strain relative to the same ratio in the untagged strain. The average and SEM of three independent experiments are shown.
Chromosome decatenation analysis
Total DNA was extracted by standard protocols (37). DNA samples were run in 0.8% TAE (40 mM Tris, 40 mM acetic acid, 1 mM EDTA) agarose gels for 19 h at 2 V/cm. Gels were blotted onto HybondTM-XL membranes and hybridized with a 32P-labeled URA3 fragment. All signals were quantified in a Fuji FLA5100 with the ImageGauge analysis program.
Chromatin analysis by MNase digestion
Nucleosome positioning was determined by micrococcal nuclease (MNaseI) digestion and indirect end-labeling as previously reported (18). MNaseI-treated DNA was digested with ClaI, resolved in a 1.5% agarose gel, blotted onto a HybondTM-XL membrane and probed with a 246-bp 32P-labeled PCR fragment located at 101 bp from the ClaI site.
RESULTS
Metaphase arrest by histone depletion is independent of the S phase checkpoints
Histone H4 depletion, which can be induced from regulatable promoters such as GAL1 or tet in a single-cell cycle (Supplementary Figure S1A; (17,18)), causes an arrest in G2/M as determined by cell morphology—large budded cells with a single nucleus—and DNA content (18,24). Monitoring the nuclear and spindle morphologies in asynchronous cultures by DAPI (4' ,6'-diamidino-2-phenylindole) and immunofluorescence with antibodies against tubulin showed that histone loss in cells expressing H4 from the doxycycline-regulatable tet promoter (t::HHF2 mutants) led to an accumulation of cells in metaphase (Supplementary Figure S1B).
A reduction in the amount of available histones causes the collapse and breakage of advancing replication forks, which are then rescued by recombination. Consequently, t::HHF2 cells are hyper-recombinant and accumulate markers of DNA damage checkpoint activation such as histone H2A phosphorylation and Ddc2 foci (17,18). Therefore, we first determined whether the S phase checkpoints were responsible for the delay in the anaphase onset. These checkpoints rely on the activation of the effector kinases Rad53 and Chk1 by the sensor Mec1 (38). Analyses of cell cycle progression in cells synchronized in G1 and released into fresh medium under conditions of histone depletion showed that t::HHF2 cells remained arrested in metaphase in the absence of Rad53 and Chk1 (Figure 1A and B). Accordingly, t::HHF2 viability was not affected by rad53Δ chk1Δ or mec1Δ (Supplementary Figure S2A). Likewise, Mec1-dependent H2A phosphorylation was not required for cell cycle arrest in t::HHF2 (Supplementary Figure S2B). We conclude that the mitotic arrest induced by nucleosome depletion is not mediated by the S phase checkpoints.
Nucleosome depletion activates the SAC
The SAC inhibits progression into anaphase if it detects unattached kinetochores or erroneous kinetochore–microtubule attachments that compromise proper chromosome biorientation and segregation (39). We addressed whether Mad2, an essential component of the SAC, is required for the mitotic arrest induced by histone depletion. Cell cycle analyses by DNA content and nuclear and spindle morphologies showed that the absence of Mad2 relieved the mitotic arrest (Figure 1A and B and Supplementary Figure S2C). A similar Mad2-dependent metaphase arrest was observed in chr-G::HHF1 cells, which express histone H4 under the control of the GAL1 promoter from the HHF1 locus (Supplementary Figure S2D).
SAC activation occurs through targeting of the heterodimer Mad1/Mad2 to unattached kinetochores to catalyze the inactivation of Cdc20, a cofactor of the anaphase-promoting complex (APC). APC/Cdc20 promotes sister chromatid segregation and mitotic exit by ubiquitination and subsequent proteasome-dependent degradation of the mitotic cyclins and securin/Pds1, an inhibitor of separase/Esp1 that cleaves the cohesin complex holding sister chromatids together (39). However, non-canonical activation of the SAC has been reported; for instance, in response to methyl-methane sulfonate (MMS), the DNA damage checkpoint-mediated arrest is reinforced by SAC independently of the kinetochore (40). Also, a kinetochore- and Pds1-independent SAC pathway monitors the strand passage reaction of Top2 (41,42). To assess the relevance of the kinetochore in the mitotic arrest induced by histone loss, we used ndc10-1, a thermosensitive mutant of an essential component of the kinetochore (43). At restrictive temperature, ndc10-1 cells do not form a functional kinetochore, and the spindle cannot pull the sister chromatids apart. Consequently, DNA replication in the next cell cycle duplicates the DNA content (Figure 1C). Ndc10 inactivation in t::HHF2 cells also led to an increase in ploidy, indicating that they progressed into the next cell cycle and that therefore the checkpoint was not activated. Thus, SAC activation by histone depletion requires a functional kinetochore.
Next, we addressed Pds1 degradation in t::HHF2 cells. For this, G1-synchronyzed cells were released into fresh medium and treated with α-factor during G2/M to arrest them in the following G1 and prevent a second mitosis. As shown in Figure 1D, Pds1 was not degraded as long as cells remained in mitosis, suggesting that SAC activation proceeded through inhibition of Pds1 degradation. To further demonstrate this, we followed cell cycle progression by spindle morphology in the absence of Pds1; this experiment was performed at 23°C because pds1Δ cells lack sufficient Esp1 in the nucleus at higher temperatures (44). As shown in Figure 1E, the absence of Pds1 prevented t::HHF2 cells to arrest in metaphase. Altogether, these results indicate that histone depletion activates the SAC.
SAC activation by histone depletion requires Aurora/Ipl1 kinase and is associated with defective centromere biorientation
Unattached kinetochores are the primary signal to activate the SAC and can be followed by the formation of a Mad2-GFP focus (45). If the interaction between microtubule and kinetochore were affected by histone depletion, we would expect t::HHF2 cells to be sensitive to microtubule-depolymerizing drugs such as benomyl and NCD. In contrast, t::HHF2 cells were resistant to benomyl (Figure 2A; note that t::HHF2 displays a similar growth defect as compared with the wild type both with and without benomyl) and accumulated wild-type levels of Mad2-GFP foci in the presence of NCD (Figure 2B). Thus, histone depletion seems not to affect microtubule attachment to kinetochores. Intriguingly, Mad2-GFP foci were not detected in t::HHF2 in the absence of drug even though histone depletion activated the SAC in a kinetochore-dependent manner, suggesting that the binding of Mad2-GFP is transient.
Transient binding of Mad2 to vertebrate kinetochores occurs in response to lack of tension (46). Thus, we studied whether SAC activation in t::HHF2 is due to a problem of tension at the kinetochore. Proper tension as a consequence of the attachment of the sister kinetochores to opposite spindle pole bodies (SPBs) (amphitelic attachments) leads to a separation of the centromeric chromatin during metaphase (centromere biorientation) that is essential for chromosome segregation. Therefore, defective attachments that do not generate enough tension (e.g. syntelic attachments, where both sister kinetochores attach microtubules from the same SPB) need to be corrected. This is carried out by Aurora/Ipl1, which destabilizes kinetochore–microtubule interactions and generates unattached kinetochores that activate the SAC (25,26). We first assessed the relevance of Ipl1 in SAC activation by histone loss by using a thermosensitive ipl1-321 mutant. Analysis of cell cycle progression showed that the lack of Ipl1 activity at restrictive temperature prevents t::HHF2 cells to arrest in mitosis (Figure 3A).
Figure 3.
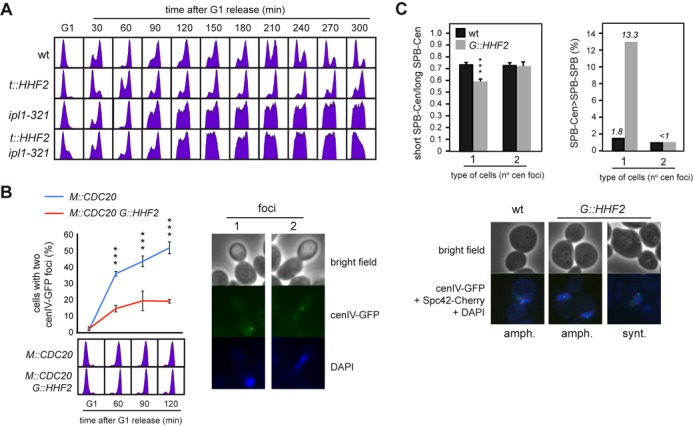
SAC activation by histone depletion requires Ipl1/Aurora kinase and is associated with defective centromere biorientation. (A) Ipl1/Aurora kinase is required for SAC activation by histone depletion. DNA content analysis by flow cytometry of wild-type, ipl1-321, t::HHF2 and t::HHF2 ipl1-321 cells synchronized in G1 at permissive temperature (26°C) and released into fresh medium at semi-permissive temperature (34°C) under conditions of histone depletion. (B) Histone depletion impairs chromosome biorientation. Chromosome biorientation was determined by fluorescence microscopy based on the accumulation of metaphase M::CDC20 and M::CDC20 G::HHF2 cells with two cenIV-GFP foci. For this, cultures were synchronized in G1 and released under conditions of histone depletion into fresh medium containing 8 mM methionine, which switches off the MET3 promoter and arrests cells in metaphase through Cdc20 depletion. The DNA content profile for each sample is shown at the bottom. A total number of 100 cells were analyzed for each time point. The average and SEM of three independent experiments are shown. Three asterisks indicate a statistically significant difference according to a two-way Anova test (P-value <0.001). (C) Histone depletion alters the symmetry of the centromere relative to the SPBs in cells with a single cenIV-GFP dot but not in cells with two cenIV-GFP dots. This symmetry was determined as the ratio between the short and long distances from the centromere (detected as a cenIV-GFP dot) to each SPB (detected as two Spc42-Cherry dots) (left), and the frequency of cells in which the longest SPB-centromere distance is higher than the SPB–SPB distance (right). Three asterisks indicate a statistically significant difference according to a one-way Anova test (P-value <0.001). The analysis was performed in M::CDC20 and M::CDC20 G::HHF2 cells synchronized in G1 and released into fresh medium for 120 min following the growth conditions indicated in (B). A total number of 250 cells for each strain from two independent experiments were analyzed. Representative images for (B) and (C) are shown.
Centromere biorientation can be followed in strains with a green-fluorescence protein (GFP)-labeled centromere by the appearance in metaphase of two proximal foci (Figure 3B, right) (47). Thus, we used tetR-GFP expressing cells that harbor an array of tetO sites at 1.4 kb from the centromere of chromosome IV (CenIV-GFP) (34). Since the presence of doxycycline prevents tetR-GFP from binding to tetO, the tet promoter expressing histone H4 in t::HHF2 was replaced with the GAL1 promoter in G::HHF2, to induce histone depletion by addition of glucose (Supplementary Figure S1A; (24)). Additionally, and given that histone depletion activates the SAC, we induced a metaphase arrest by Cdc20 depletion to follow the kinetics of centromere biorientation under similar cell cycle conditions for the mutant and the wild type. Thus, synchronous cultures were first arrested in G1, where all cells displayed a single GFP dot, and then released in the presence of methionine to switch off the MET3 promoter that drives Cdc20 expression (M::CDC20). As shown in Figure 3B, cells displayed two closed GFP dots as they accumulated in metaphase. Notably, only ∼20% of the metaphase arrested G::HHF2 cells showed two dots as compared with the ∼50% detected in the wild type, indicating that histone depletion affects chromosome biorientation.
To further evaluate whether histone depletion-induced defective chromosome biorientation was associated with syntelic attachments, the distance between CenIV and each of the SPBs was measured in M::CDC20 and M::CDC20 G::HHF2 metaphase cells expressing both CenIV-GFP and the SPB protein Spc42 fused to Cherry. In accordance with previous results (48), we observed that histone depletion increased the length of the spindle, in particular in cells with a single CenIV-GFP dot (Supplementary Figure S3A). As expected if histone depletion led to the formation of syntelic attachments, the ratio between the shorter and the longer CenIV-SPB distance was lower in the mutant than in the wild type for cells with a single CenIV-GFP dot, while it was similar for cells with two CenIV-GFP dots (Figure 3C, left, and Supplementary Figure S3B). In addition, the spindle length was shorter than the distance from the centromere to one of the SPBs in 13.3% of histone-depleted cells with a single CenIV-GFP dot (Figure 3C, right). Altogether these results suggest that histone depletion gives rise to syntelic attachments.
Top2 and condensin are required for SAC activation in response to histone depletion
The assembly of DNA into chromatin provides a major source of topological constraints that demand the activity of topoisomerases during chromosome dynamics. In fact, Top2 binds preferentially to genes with low nucleosome density in histone-depleted cells (49). Thus, we decided to assess the role of Top2 in SAC activation by histone loss. Since TOP2 is essential (50), we used a ‘degron’ allele of TOP2 that expresses a Top2 protein that can be rapidly degraded in the presence of doxycycline and galactose at 37°C (6). Under these conditions Top2 expression from the tet promoter is repressed, and Top2 degradation is promoted by the E3 ubiquitin ligase Ubr1 expressed from the GAL1 promoter. This background forced us to replace the promoter expressing histone H4 with the MET25 promoter, which represses H4 in response to methionine (Supplementary Figure S1A; strain M::HHF2). As shown for t::HHF2 and G::HHF2, M::HHF2 cells arrested in metaphase when histone expression was repressed with 100 μM methionine (Figure 4A). We verified that Top2 was degraded in G1-arrested cells prior to their release into fresh medium under restrictive conditions (Figure 4B). As previously demonstrated, the lack of Top2 did not prevent progression through mitosis even though top2td cells entered into the following cell cycle with an aberrant DNA content profile due to chromosome mis-segregation by defective sister chromatid decatenation (Figure 4A) (6). Importantly, M::HHF2 top2td progressed through the cell cycle as did top2td, indicating that the absence of Top2 relieved the SAC-mediated arrest induced by histone depletion. As shown in Figure 4B, the expression of Top2 was not affected by histone depletion, indicating that the arrest was not due to increased levels of Top2 in the mutant.
Proper chromosome segregation depends on the accuracy of two processes that require Top2, namely, sister chromatid decatenation and chromosome condensation (4). These two processes also require condensin (5,8,51,52). Thus, we addressed the relevance of Smc2, an essential component of condensin, in SAC activation by histone loss. Wild-type and t::HHF2 cells expressing either SMC2 or a thermosensitive smc2-8 allele were synchronized in G1 and released into fresh medium at restrictive temperature. In accordance with previous results (14), smc2-8 cells arrested in metaphase at 37°C (Figure 4C). Remarkably, t::HHF2 smc2-8 progressed faster through mitosis than the single mutants, indicating that condensin is required for SAC activation by histone depletion. As previously shown for Top2, SAC activation was not due to higher levels of condensin; indeed, histone depletion reduced the total amount of the condensin subunit Brn1 (Figure 4D). Likewise, suppression of the mitotic arrest by the absence of Top2 and condensin was not due to an increase in the levels of histone H4 (Supplementary Figure S4A). Therefore, Top2 and condensin are required for histone depletion-induced SAC activation.
Notably, a reduction of condensin activity in smc2-8 at permissive temperature alleviated the mitotic arrest and partially suppressed the growth defects associated with histone loss (Figure 4E and Supplementary Figure S4B). This suppression was not observed with other conditions that cause DNA damage; in fact, smc2-8 was sensitive to hydroxyurea (HU) and MMS and arrested in G2/M in response to HU (Supplementary Figure S4C and S4D). These results suggest that condensin generates a chromosomal problem in histone-depleted cells that activates the SAC. Growth analyses could not be performed with top2td because it is lethal at restrictive conditions; however, if Top2 causes a chromosomal problem detected by the SAC, t::HHF2 cells should be sensitive to high levels of Top2. Consistently, t::HHF2 cells transformed with a plasmid that increases Top2 by a factor of 10 (32) displayed growth defects (Figure 4F).
Although the SAC and not the S phase checkpoints arrests t::HHF2 cells, we cannot rule out the possibility that the SAC is activated by DNA damage in this mutant. We thereby tested if DNA damage induced by histone depletion depends on Top2 and/or condensin by following the accumulation of recombinogenic DNA damage (Rad52-YFP foci). Consistent with the fact that DNA damage by Top2 depletion requires cytokinesis (6), top2td cells released into S phase without Top2 did not accumulate Rad52-YFP foci (Figure 4G). Importantly, Top2 was not responsible for DNA damage in t::HHF2, as evidenced by the accumulation of Rad52-YFP foci in t::HHF2 top2td. When we analyzed the effect of condensin in Rad52-YFP foci, we observed that smc2-8 cells accumulated DNA damage even at permissive temperature, as previously shown for other condensin mutants (53), and that this accumulation was maintained in t::HHF2 smc2-8 cells (Figure 4H). Thus, we cannot establish the relevance of condensin in the accumulation of DNA damage by histone loss. Nevertheless, the fact that smc2-8 suppresses the growth defects of t::HHF2 despite the cells accumulating DNA damage indicates that DNA damage does not activate the SAC in histone-depleted cells.
Defective centromeric chromatin structure by histone depletion depends on condensin
Histone depletion causes alterations at the centromeric chromatin which make DNA more accessible to the micrococcal nuclease (MNaseI), which cuts preferentially at nucleosome-free DNA (54). These chromatin modifications might alter the occupancy/activity of condensin and Top2 thus impairing the tensile properties of the centromeric chromatin. An alternative but not mutually exclusive possibility is that condensin and Top2 might be required to modify the centromeric chromatin structure in cells with reduced levels of histones. To explore these possibilities, we first analyzed Brn1 binding to previously identified regions (55). The results suggested that histone depletion causes a reduction in the amount of condensin bound at the pericentric chromatin but not at the ribosomal chromatin, even though the differences were not statistically significant (Figure 5A). Likewise, the total amount of Brn1 bound to chromatin was slightly reduced in the mutant as determined by chromatin fractionation (P lane; Figure 5B).
Next, we analyzed the chromatin structure of CEN3 by MNaseI digestion and indirect-end labeling in mitotic cells (Figure 5C). While the lack of condensin activity in smc2-8 cells at restrictive temperature did not affect the chromatin structure of CEN3, histone depletion increased the accessibility of MNaseI to both the centromere DNA elements (CDEs I, II and III)—which are wrapped around an specialized nucleosome—and proximal DNA sequences (Figure 5C; asterisks (54)). Remarkably, these nucleosome alterations were not detected in t::HHF2 smc2-8. Therefore, histone depletion by itself is not sufficient to alter the centromeric chromatin; condensin activity is required for histone depletion-induced chromatin alterations. This result suggests interplay between histone supply and condensin.
The SAC prevents chromosome mis-segregation by condensin-dependent defective decatenation in response to histone depletion
As the biological aim of the checkpoints is to provide time to resolve chromosome-associated problems, the lack of a checkpoint may lead to a loss of viability. Accordingly, the absence of the heterodimer Mad1/Mad2 led to a loss of viability in t::HHF2 cells (Figure 6A). Likewise, t::HHF2 ndc10-1 and t::HHF2 ipl1-321 were lethal at semipermissive temperature (Figure 6B and C). Therefore, the SAC is required for t::HHF2 viability.
Cell growth analyses also showed that the lack of either the sensor Mec1 or the effectors Rad53 and Chk1 does not affect the viability of t::HHF2 mad2Δ cells (Supplementary Figure S5A and B), indicating that the SAC is not masking a putative effect of the S phase checkpoints on histone depletion-dependent mitotic arrest.
As a major genetic consequence of defective SAC activation is chromosome mis-segregation, we followed the fate of CrIV in G::HHF2. To prevent a complete metaphase block by addition of glucose (24), wild-type and G::HHF2 cells were shifted from 2% galactose to 0.05% galactose for 4 h. Proper chromosome segregation led to anaphase cells with a dot in the mother and a dot in the bud, while chromosome mis-segregation led to anaphase cells with the two dots either in the mother or in the bud (Figure 6D, right panel). Whereas chromosome IV mis-segregation in G::HHF2, mad2Δ and wild-type cells was barely detectable (<0.3%), it increased to 11.3% in G::HHF2 mad2Δ cells (Figure 6D), indicating that SAC activation ensures proper chromosome segregation in response to histone depletion.
Next, we asked whether chromosome mis-segregation in t::HHF2 was due to defective sister chromatid decatenation. DNA unwinding during replication generates positive torsional stress ahead the fork, part of which is diffused across the replicated DNA by swiveling of the fork leading to the formation of intertwined sister chromatids (precatenanes). Resolution of precatenanes, which occurs right after DNA synthesis and during mitosis as sister chromatids move apart, is carried out by Top2 and condensin, and it is essential for proper chromosome segregation (5–8). Formation and resolution of precatenanes during cell cycle can be followed by studying the accumulation of the different intermediates of a centromeric plasmid (8). Wild-type cells transiently accumulated different intermediates (Figure 7A; CI, CII and CIII), which represent catenated molecules as inferred by their accumulation in cells lacking Top2 (Supplementary Figure S6). Most plasmids were monomeric in G1-arrested t::HHF2 cells (rM and SC(-)M intermediates), indicating that most precatenanes were properly resolved under conditions of histone depletion (note that only a small fraction of catenated plasmids was detected as compared to the wild type, which might reflect minor resolution defects and/or incomplete synchronization; see fluorescence-activated cell sorting profiles). However, the catenanes accumulated in t::HHF2 relative to the wild type during S and G2/M, suggesting that histone depletion impaired decatenation (Figure 7A). While Top2 rapidly resolves most precatenanes after their formation, a fraction remains until mitosis, during which condensins and spindle tension facilitate Top2 activity; accordingly, checkpoint activation prevents their resolution (5,8). Indeed, the entry into mitosis by lack of Mad2 reduced the fraction of catenanes at late times, but it did not affect the accumulation of early catenanes in t::HHF2 (Figure 7B). Next, we analyzed the role of condensins in the accumulation of these catenated plasmids, taking advantage of the fact that the effect of condensin inactivation is barely detectable in small plasmids (8). As shown in Figure 7C, the accumulation of catenanes in t::HHF2 cells was suppressed by reducing the activity of condensins.
Figure 7.
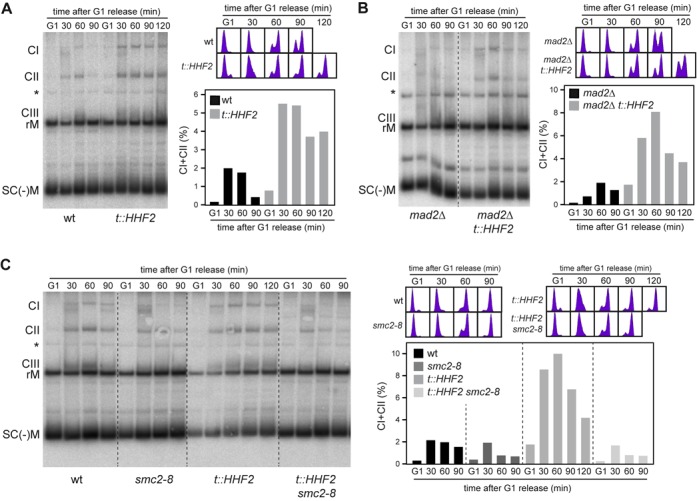
Histone depletion induces condensin-mediated defective decatenation. (A,B) Histone depletion impairs plasmid decatenation regardless of checkpoint activation. The formation and resolution of catenanes were followed by southern analysis of the centromeric plasmid pRS416 in wild-type, t::HHF2 (A), mad2Δ and mad2Δ t::HHF2 (B) transformants synchronized in G1 and released into fresh medium under conditions of histone depletion. CI, CII and CIII show different catenated intermediates as determined in Supplementary Figure S6; rM and SC(-)M show relaxed and negatively supercoiled monomers as determined by running purified pRS314 (data not shown). The asterisk marks an unspecific signal. The percentage of catenanes relative to the total amount of plasmid ([CI+CII/rM+SC(-)M+CI+CII]x100) and the DNA content profiles are shown on the right. Only catenanes CI and CII were quantified because CIII and rM intermediates often overlapped. (C) Condensin impairs plasmid decatenation in t::HHF2. The formation and resolution of catenanes was followed as in (A) in wild-type, smc2-8, t::HHF2 and t::HHF2 smc2-8 transformants synchronized in G1 at permissive temperature (26°C) and released into fresh medium at semi-permissive temperature (30°C) under conditions of histone depletion. The decatenation analyses were repeated twice with similar results.
DISCUSSION
Here we show that histone depletion causes an unscheduled activation of the SAC by Top2 and condensins, as well as an accumulation of catenanes by condensins. Since catenanes accumulation in the absence of Top2 does not arrest yeast cells (6,42), it is unlikely that checkpoint activation results as a consequence of defective decatenation in histone-depleted cells. Instead, SAC activation is associated with problems of chromosome biorientation. Nevertheless, SAC activation provides time to resolve the problems generated by condensins on a nucleosome-depleted chromatin. Accordingly, a reduction of condensin activity partially rescues t::HHF2 growth defects (Figure 4E), while the lack of a functional SAC aggravates them as a consequence of chromosome mis-segregation (Figure 6).
It has been shown by genome-wide ChIP analyses that Top2 and condensin preferentially bind nucleosome-free DNA (49,56). Indeed, histone depletion increases Top2 binding to genes with low nucleosome density (49). Unexpectedly, though, histone depletion does not increase condensin binding to pericentric chromatin (Figure 5A). Instead, altered chromatin structure at centromeres by histone depletion requires condensin (Figure 5C), indicating that a precise interplay between histone deposition and condensin/Top2 is necessary for centromeric chromatin structure, chromosome biorientation and precatenanes resolution.
Histone depletion causes Top2 and condensin-dependent SAC activation
Our results demonstrate that under conditions of histone depletion Top2 and condensin impair chromosome biorientation and activate the Ipl1-dependent branch of the SAC, which detects microtubule attachments that do not generate tension at the kinetochores (25,26). Top2 and condensin are enriched in the pericentric chromatin, where they work together with cohesins to form an intramolecular loop that protrudes ∼12.5 kb toward the spindle, leaving the kinetochore at its tip (Figure 8A) (10–13,57). Importantly, Top2 and condensins are responsible for the axial compaction of this loop, as shown by an increase in spindle length and sister-centromere distance, and the stretching of LacO-labeled centromeric chromatin in metaphase cells defective in Top2 or condensin activity (12,13). This compaction provides the tensile properties of this molecular spring, generating the inward forces that counterbalance the pulling forces of the microtubules. In fact, chromatin has also been proposed to contribute to the biophysical properties of the intramolecular loop, as histone depletion also increased the distance between the centromeres without affecting the proportion of metaphase cells with two separated LacO-labeled centromeres (12,48). We notice that those results are in apparent contradiction with ours; however, they were performed with asynchronous cultures, and therefore the centromeres of the histone-depleted but not of the wild-type metaphase cells were tensioned by SAC activation. Indeed, a metaphase arrest increases the spindle length in wild-type cells (12). In contrast, when the analysis was performed with both mutant and wild-type cells arrested in metaphase by CDC20 repression, histone depletion strongly reduced the number of metaphase cells with two separated LacO-labeled centromeres (Figure 3B), a defect in chromosome biorientation that is consistent with the requirement of Ipl1 for SAC activation (Figure 3A) and the accumulation of syntelic attachments (Figure 3C) in histone-depleted cells.
Figure 8.
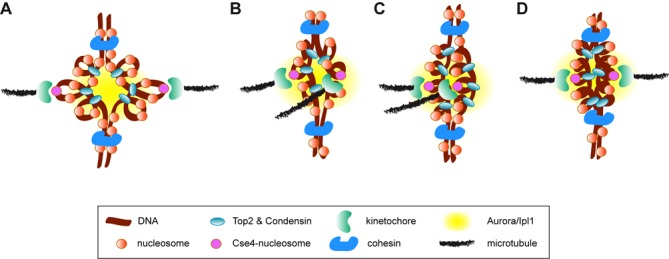
Models for histone depletion-induced SAC activation. At the pericentric chromatin, Top2 and condensin are responsible for the axial compaction of an intramolecular loop that provides the tensile properties of the centromere (A). Condensin/Top2-dependent alterations of the pericentric chromatin might affect the geometry of the sister kinetochores leading to syntelic attachments (B). An accumulation of torsional stress at the pericentric chromatin might also contribute to SAC activation by either altering the geometry of the sister kinetochores (C) or overly tightening the intramolecular loop (D). All situations would leave the kinetochores close to the inner centromere and thereby to Ipl1, which would destabilize the attachments and activate the SAC.
How do condensin and Top2 activate the SAC under conditions of defective histone supply? Some chromatin mutants that activate the SAC build defective kinetochores and undergo chromosome mis-segregation (21–23). In fact, reducing histone H4 levels affects the integrity of the kinetochores (48), which is consistent with a loss of positioning/integrity of the nucleosome that interacts with the kinetochore at CEN3 (Figure 5C). However, this loss of kinetochore integrity is not observed by histone H3 depletion—likely because the centromere nucleosome contains the histone H3 variant Cse4—and yet causes a similar mitotic arrest (48). Furthermore, histone-depleted cells neither are more sensitive nor display a higher activation of the SAC in response to drugs that inhibit microtubules polymerization (Figure 2), suggesting that the interaction between kinetochore and microtubule is not affected.
Defective nucleosome organization by condensin under conditions of histone depletion could also alter the geometry of the pericentric chromatin and promote syntelic attachments (Figure 8B). Additionally, an excess of topological tension at the centromeric region could also contribute to SAC activation. Indeed, the fact that t::HHF2 smc2-8 progressed faster through mitosis than the single mutants (Figure 4C and Supplementary Figure S4B) indicates not only that condensin is required for SAC activation by histone depletion, but also that histone depletion relieves the arrest mediated by loss of condensin activity. A simple explanation for this result is that histone depletion caused an excess of condensin activity. However, our ChIP analyses argue against this possibility, as we do not detect more condensin at the centromeres; indeed, it seems that histone depletion reduces condensin binding to chromatin (Figure 5A and B), even though a more exhaustive analysis is required. Nevertheless, an excess of topological tension could also result from the loss of nucleosome integrity observed at the centromeric chromatin. In fact, the loss of nucleosome integrity by histone depletion increases plasmid superhelical density (18,24). Such excess of topological tension at the centromere might also alter the geometry of the pericentric chromatin in a way that favors syntelic attachments (Figure 8C). An alternative but not mutually exclusive possibility is related to the mechanism of action of Ipl1 during SAC activation. This mechanism is based on the proximity of Ipl1 (located at the inner centromere) to the outer kinetochore, which contains several targets of Ipl1. Ipl1-mediated phosphorylation of these substrates reduces microtubule-binding affinity. If the attachment is correct, the microtubules pull the kinetochore apart, and binding is stabilized; in contrast, if the attachment is erroneous and the tension is low, the kinetochore remains close to Ipl1, and binding is destabilized (39). Given that the distance between Ipl1 and the kinetochore is essential for SAC activation, an excess of torsional stress might overly tighten the intramolecular loop and leave the kinetochores close to the inner centromere, even though the attachments were correct (Figure 8D).
Histone depletion causes condensin-dependent accumulation of catenanes
The absence of Top2 activity does not prevent nucleosome deposition in Xenopus egg extracts, indicating that nucleosomes can be accommodated into the precatenanes generated during DNA replication (58). The authors of this study thus proposed that replication-coupled nucleosome deposition may facilitate chromatid decatenation by forcing the precatenanes nodes to adopt a ‘hooked’ DNA juxtaposition conformation, which is the preferred substrate for the DNA disentangling activity of Top2 (59,60). According to this hypothesis, defective nucleosome organization by condensin under conditions of histone depletion could alter DNA geometry behind the fork and interfere with Top2-mediated chromatid decatenation. Likewise, the excess of positive supercoiling that is associated with histone depletion (18,24) might interfere with Top2 activity by affecting DNA geometry and thereby the resolution of catenanes. Baxter and Aragón have provided strong evidence for a model in which chromosome decatenation results from an exquisite interplay between the enzymatic activities of Top2 and condensin, with the positive supercoiling introduced by condensins and spindle tension as being necessary for catenanes resolution by Top2 (5,51).
In summary, our results reveal the importance of a correct supply of histones for the accuracy of condensin/Top2-mediated DNA processes. Specifically, we show that a precise interplay between histone deposition and condensin is required for pericentric chromatin structure, precatenanes resolution and centromere biorientation. An attractive possibility that we are currently evaluating is that this interplay modulated global chromatin structure and chromosome condensation. Finally, we anticipate that defects in chromosome dynamics and cell cycle progression may be particularly severe during physiological processes such as the replicative aging, which strongly reduce the density of nucleosomes (35,61).
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
We thank L. Aragón, O. Cohen-Fix, J.F.X. Diffley and J. Nitiss for various strains and reagents, and P. Huertas and F. Cortés-Ledesma for critically reading the manuscript.
FUNDING
Spanish Ministry of Science [BFU2009-09036, BFU2012-38171]; Spanish Government [to M.M.-P., M.J.C.-L.]. Funding for open access charge: Spanish Ministry of Science [BFU2012-38171].
Conflict of interest statement. None declared.
REFERENCES
- 1.Khorasanizadeh S. The nucleosome: from genomic organization to genomic regulation. Cell. 2004;116:259–272. doi: 10.1016/s0092-8674(04)00044-3. [DOI] [PubMed] [Google Scholar]
- 2.Burgess R.J., Zhang Z. Histone chaperones in nucleosome assembly and human disease. Nat. Struct. Mol. Biol. 2013;20:14–22. doi: 10.1038/nsmb.2461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hirano T. Condensins: universal organizers of chromosomes with diverse functions. Genes Dev. 2012;26:1659–1678. doi: 10.1101/gad.194746.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vos S.M., Tretter E.M., Schmidt B.H., Berger J.M. All tangled up: how cells direct, manage and exploit topoisomerase function. Nature. 2011;12:827–841. doi: 10.1038/nrm3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baxter J., Sen N., Martinez V.L., De Carandini M.E.M., Schvartzman J.B., Diffley J.F.X., Aragon L. Positive supercoiling of mitotic DNA drives decatenation by topoisomerase II in eukaryotes. Science. 2011;331:1328–1332. doi: 10.1126/science.1201538. [DOI] [PubMed] [Google Scholar]
- 6.Baxter J., Diffley J.F.X. Topoisomerase II inactivation prevents the completion of DNA replication in budding yeast. Mol. Cell. 2008;30:790–802. doi: 10.1016/j.molcel.2008.04.019. [DOI] [PubMed] [Google Scholar]
- 7.Bermejo R., Doksani Y., Capra T., Katou Y.M., Tanaka H., Shirahige K., Foiani M. Top1- and Top2-mediated topological transitions at replication forks ensure fork progression and stability and prevent DNA damage checkpoint activation. Genes Dev. 2007;21:1921–1936. doi: 10.1101/gad.432107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Charbin A., Bouchoux C., Uhlmann F. Condensin aids sister chromatid decatenation by topoisomerase II. Nucleic Acids Res. 2014;42:340–348. doi: 10.1093/nar/gkt882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Winey M., Bloom K. Mitotic spindle form and function. Genetics. 2012;190:1197–1224. doi: 10.1534/genetics.111.128710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang B.D., Eyre D., Basrai M., Lichten M., Strunnikov A. Condensin binding at distinct and specific chromosomal sites in the Saccharomyces cerevisiae genome. Mol. Cell. Biol. 2005;25:7216–7225. doi: 10.1128/MCB.25.16.7216-7225.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Takahashi Y., Yong-Gonzalez V., Kikuchi Y., Strunnikov A. SIZ1/SIZ2 control of chromosome transmission fidelity is mediated by the sumoylation of topoisomerase II. Genetics. 2006;172:783–794. doi: 10.1534/genetics.105.047167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stephens A.D., Haase J., Vicci L., Taylor R.M., Bloom K. Cohesin, condensin, and the intramolecular centromere loop together generate the mitotic chromatin spring. J. Cell Biol. 2011;193:1167–1180. doi: 10.1083/jcb.201103138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Warsi T.H., Navarro M.S., Bachant J. DNA topoisomerase II is a determinant of the tensile properties of yeast centromeric chromatin and the tension checkpoint. Mol. Biol. Cell. 2008;19:4421–4433. doi: 10.1091/mbc.E08-05-0547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yong-Gonzalez V., Wang B.-D., Butylin P., Ouspenski I., Strunnikov A. Condensin function at centromere chromatin facilitates proper kinetochore tension and ensures correct mitotic segregation of sister chromatids. Genes Cells. 2007;12:1075–1090. doi: 10.1111/j.1365-2443.2007.01109.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Singh R.K., Liang D., Gajjalaiahvari U.R., Kabbaj M.-H.M., Paik J., Gunjan A. Excess histone levels mediate cytotoxicity via multiple mechanisms. Cell Cycle. 2010;9:4236–4244. doi: 10.4161/cc.9.20.13636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Meeks-Wagner D., Hartwell L.H. Normal stoichiometry of histone dimer sets is necessary for high fidelity of mitotic chromosome transmission. Cell. 2003;44:43–52. doi: 10.1016/0092-8674(86)90483-6. [DOI] [PubMed] [Google Scholar]
- 17.Clemente-Ruiz M., Prado F. Chromatin assembly controls replication fork stability. EMBO Rep. 2009;10:790–796. doi: 10.1038/embor.2009.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Prado F., Aguilera A. Partial depletion of histone H4 increases homologous recombination-mediated genetic instability. Mol. Cell. Biol. 2005;25:1526–1536. doi: 10.1128/MCB.25.4.1526-1536.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Clemente-Ruiz M., González-Prieto R., Prado F. Histone H3K56 acetylation, CAF1, and Rtt106 coordinate nucleosome assembly and stability of advancing replication forks. PLoS Genet. 2011;7:e1002376. doi: 10.1371/journal.pgen.1002376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Prado F., Clemente-Ruiz M. Nucleosome assembly and genome integrity: the fork is the link. Bioarchitecture. 2012;2:6–10. doi: 10.4161/bioa.19737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sharp J.A., Franco A.A., Osley M.A., Kaufman P.D. Chromatin assembly factor I and Hir proteins contribute to building functional kinetochores in S. cerevisiae. Genes Dev. 2002;16:85–100. doi: 10.1101/gad.925302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Smith M.M., Yang P., Santisteban M.S., Boone P.W., Goldstein A.T., Megee P.C. A novel histone H4 mutant defective in nuclear division and mitotic chromosome transmission. Mol. Cell. Biol. 1996;16:1017–1026. doi: 10.1128/mcb.16.3.1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yu Y., Srinivasan M., Nakanishi S., Leatherwood J., Shilatifard A., Sternglanz R. A conserved patch near the C terminus of histone H4 is required for genome stability in budding yeast. Mol. Cell. Biol. 2011;31:2311–2325. doi: 10.1128/MCB.01432-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim U.J., Han M., Kayne P., Grunstein M. Effects of histone H4 depletion on the cell cycle and transcription of Saccharomyces cerevisiae. EMBO J. 1988;7:2211–2219. doi: 10.1002/j.1460-2075.1988.tb03060.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Biggins S., Murray A.W. The budding yeast protein kinase Ipl1/Aurora allows the absence of tension to activate the spindle checkpoint. Genes Dev. 2001;15:3118–3129. doi: 10.1101/gad.934801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pinsky B.A., Kung C., Shokat K.M., Biggins S. The Ipl1-Aurora protein kinase activates the spindle checkpoint by creating unattached kinetochores. Nat. Cell Biol. 2005;8:78–83. doi: 10.1038/ncb1341. [DOI] [PubMed] [Google Scholar]
- 27.Longtine M.S., McKenzie A., Demarini D.J., Shah N.G., Wach A., Brachat A., Philippsen P., Pringle J.R. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast. 1998;14:953–961. doi: 10.1002/(SICI)1097-0061(199807)14:10<953::AID-YEA293>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 28.Prado F., Aguilera A. Impairment of replication fork progression mediates RNA polII transcription-associated recombination. EMBO J. 2005;24:1267–1276. doi: 10.1038/sj.emboj.7600602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mumberg D., Müller R., Funk M. Regulatable promoters of Saccharomyces cerevisiae: comparison of transcriptional activity and their use for heterologous expression. Nucleic Acids Res. 1994;22:5767–5768. doi: 10.1093/nar/22.25.5767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sikorski R.S., Hieter P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Johnston M., Davis R.W. Sequences that regulate the divergent GAL1-GAL10 promoter in Saccharomyces cerevisiae. Mol. Cell. Biol. 1984;4:1440–1448. doi: 10.1128/mcb.4.8.1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nitiss J.L., Liu Y.X., Harbury P., Jannatipour M., Wasserman R., Wang J.C. Amsacrine and etoposide hypersensitivity of yeast cells overexpressing DNA topoisomerase II. Cancer Res. 1992;52:4467–4472. [PubMed] [Google Scholar]
- 33.Valerio-Santiago M., Monje-Casas F. Tem1 localization to the spindle pole bodies is essential for mitotic exit and impairs spindle checkpoint function. J. Cell Biol. 2011;192:599–614. doi: 10.1083/jcb.201007044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Monje-Casas F., Prabhu V.R., Lee B.H., Boselli M., Amon A. Kinetochore orientation during meiosis is controlled by Aurora B and the monopolin complex. Cell. 2007;128:477–490. doi: 10.1016/j.cell.2006.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Feser J., Truong D., Das C., Carson J.J., Kieft J., Harkness T., Tyler J.K. Elevated histone expression promotes life span extension. Mol. Cell. 2010;39:724–735. doi: 10.1016/j.molcel.2010.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hecht A., Grunstein M. Mapping DNA interaction sites of chromosomal proteins using immunoprecipitation and polymerase chain reaction. Methods Enzymol. 1999;304:399–414. doi: 10.1016/s0076-6879(99)04024-0. [DOI] [PubMed] [Google Scholar]
- 37.Amberg D.C., Burke D.J., Strathern N.J. Methods in Yeast Genetics. Cold Spring Harbor, New York: Cold Spring harbor Laboratory Press; 2005. [Google Scholar]
- 38.Branzei D., Foiani M. The checkpoint response to replication stress. DNA Repair. 2009;8:1038–1046. doi: 10.1016/j.dnarep.2009.04.014. [DOI] [PubMed] [Google Scholar]
- 39.Foley E.A., Kapoor T.M. Microtubule attachment and spindle assembly checkpoint signalling at the kinetochore. Nature. 2013;14:25–37. doi: 10.1038/nrm3494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim E.M., Burke D.J. DNA damage activates the SAC in an ATM/ATR-dependent manner, independently of the kinetochore. PLoS Genet. 2008;4:e1000015. doi: 10.1371/journal.pgen.1000015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Andrews C.A., Vas A.C., Meier B., Giménez-Abián J.F., Díaz-Martínez L.A., Green J., Erickson S.L., Vanderwaal K.E., Hsu W.-S., Clarke D.J. A mitotic topoisomerase II checkpoint in budding yeast is required for genome stability but acts independently of Pds1/securin. Genes Dev. 2006;20:1162–1174. doi: 10.1101/gad.1367206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Furniss K.L., Tsai H.-J., Byl J.A.W., Lane A.B., Vas A.C., Hsu W.-S., Osheroff N., Clarke D.J. Direct monitoring of the strand passage reaction of DNA topoisomerase II triggers checkpoint activation. PLoS Genet. 2013;9:e1003832. doi: 10.1371/journal.pgen.1003832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Phuay-YeeGoh, Kilmartin J.V. NDC10: a gene involved in chromosome segregation in Saccharomyces cerevisiae. J. Cell Biol. 1993;121:503–512. doi: 10.1083/jcb.121.3.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jensen S., Segal M., Clarke D.J., Reed S.I. A novel role of the budding yeast separin Esp1 in anaphase spindle elongation: evidence that proper spindle association of Esp1 is regulated by Pds1. J. Cell Biol. 2001;152:27–40. doi: 10.1083/jcb.152.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Iouk T., Kerscher O., Scott R.J., Basrai M.A., Wozniak R.W. The yeast nuclear pore complex functionally interacts with components of the spindle assembly checkpoint. J. Cell Biol. 2002;159:807–819. doi: 10.1083/jcb.200205068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Waters J.C., Chen R.H., Murray A.W., Salmon E.D. Localization of Mad2 to kinetochores depends on microtubule attachment, not tension. J. Cell Biol. 1998;141:1181–1191. doi: 10.1083/jcb.141.5.1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tanaka T., Fuchs J., Loidl J., Nasmyth K. Cohesin ensures bipolar attachment of microtubules to sister centromeres and resists their precocious separation. Nat. Cell Biol. 2000;2:492–499. doi: 10.1038/35019529. [DOI] [PubMed] [Google Scholar]
- 48.Bouck D.C., Bloom K. Pericentric chromatin is an elastic component of the mitotic spindle. Curr. Biol. 2007;17:741–748. doi: 10.1016/j.cub.2007.03.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sperling A.S., Jeong K.S., Kitada T., Grunstein M. Topoisomerase II binds nucleosome-free DNA and acts redundantly with topoisomerase I to enhance recruitment of RNA Pol II in budding yeast. Proc. Natl Acad. Sci. U.S.A. 2011;108:12693–12698. doi: 10.1073/pnas.1106834108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Holm C., Goto T., Wang J.C., Botstein D. DNA topoisomerase II is required at the time of mitosis in yeast. Cell. 1985;41:553–563. doi: 10.1016/s0092-8674(85)80028-3. [DOI] [PubMed] [Google Scholar]
- 51.Baxter J., Aragón L. A model for chromosome condensation based on the interplay between condensin and topoisomerase II. Trends Genet. 2012;28:110–117. doi: 10.1016/j.tig.2011.11.004. [DOI] [PubMed] [Google Scholar]
- 52.Vas A.C.J., Andrews C.A., Kirkland Matesky K., Clarke D.J. In vivo analysis of chromosome condensation in Saccharomyces cerevisiae. Mol. Biol. Cell. 2007;18:557–568. doi: 10.1091/mbc.E06-05-0454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Huang D. Chromosome integrity in Saccharomyces cerevisiae: the interplay of DNA replication initiation factors, elongation factors, and origins. Genes Dev. 2003;17:1741–1754. doi: 10.1101/gad.1089203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Saunders M.J., Yeh E., Grunstein M., Bloom K. Nucleosome depletion alters the chromatin structure of Saccharomyces cerevisiae centromeres. Mol. Cell. Biol. 1990;10:5721–5727. doi: 10.1128/mcb.10.11.5721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cuylen S., Metz J., Haering C.H. Condensin structures chromosomal DNA through topological links. Nat. Struct. Mol. Biol. 2011;18:894–901. doi: 10.1038/nsmb.2087. [DOI] [PubMed] [Google Scholar]
- 56.Piazza I., Rutkowska A., Ori A., Walczak M., Metz J., Pelechano V., Beck M., Haering C.H. Association of condensin with chromosomes depends on DNA binding by its HEAT-repeat subunits. Nat. Struct. Mol. Biol. 2014;21:560–568. doi: 10.1038/nsmb.2831. [DOI] [PubMed] [Google Scholar]
- 57.Yeh E., Haase J., Paliulis L.V., Joglekar A., Bond L., Bouck D., Salmon E.D., Bloom K.S. Pericentric chromatin is organized into an intramolecular loop in mitosis. Curr. Biol. 2008;18:81–90. doi: 10.1016/j.cub.2007.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Germe T., Hyrien O. Topoisomerase II–DNA complexes trapped by ICRF-193 perturb chromatin structure. EMBO Rep. 2005;6:729–735. doi: 10.1038/sj.embor.7400465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Buck G.R., Lynn Zechiedrich E. DNA disentangling by type-2 topoisomerases. J. Mol. Biol. 2004;340:933–939. doi: 10.1016/j.jmb.2004.05.034. [DOI] [PubMed] [Google Scholar]
- 60.Liu Z., Zechiedrich L., Chan H.S. Action at hooked or twisted–hooked DNA juxtapositions rationalizes unlinking preference of type-2 topoisomerases. J. Mol. Biol. 2010;400:963–982. doi: 10.1016/j.jmb.2010.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.O'Sullivan R.J., Kubicek S., Schreiber S.L., Karlseder J. Reduced histone biosynthesis and chromatin changes arising from a damage signal at telomeres. Nat. Struct. Mol. Biol. 2010;17:1218–1225. doi: 10.1038/nsmb.1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.