

Abstract
The precise, temporal order of gene expression during development is critical to ensure proper lineage commitment, cell fate determination, and ultimately, organogenesis. Epigenetic regulation of chromatin structure is fundamental to the activation or repression of genes during embryonic development. In recent years there has been an explosion of research relating to various modes of epigenetic regulation such as DNA methylation, posttranslational histone tail modifications, non-coding RNA control of chromatin structure, and nucleosome remodeling. Technological advances in genome wide epigenetic profiling and pluripotent stem cell (PSC) differentiation have been primary drivers for elucidating the epigenetic control of cellular identity during development and nuclear reprogramming. Not only do epigenetic mechanisms regulate transcriptional states in a cell type-specific manner but they also establish higher-order genomic topology and nuclear architecture. Here, we review the epigenetic control of pluripotency and changes associated with PSC differentiation. We focus on DNA methylation and demethylation, and common histone tail modifications. Finally, we briefly discuss epigenetic heterogeneity among PSC lines and the influence of epigenetic patterns on genome topology.
Keywords: epigenetic, pluripotent, differentiation, methylation, topology
Introduction
The growing use of human pluripotent stem cells (hPSCs) in biomedical research, coupled with advances in techniques to probe the epigenome and genome, have shown that a complex interplay of DNA elements and dynamic epigenetic processes underlie the coordinately regulated patterns of gene expression during human development. Here we provide a global overview of the current knowledge of epigenetic regulation in pluripotent cells and during mammalian development. We focus on two of the major epigenetic controls, DNA methylation and histone modifications, assessing both studies performed in the mouse and with human cells.
Human embryonic stem cells (hESCs) are self-renewing and pluripotent, with the capacity to differentiate into any somatic cell type. These qualities are valuable, as they can provide an inexhaustible source of cells for drug screening and potentially for a wide range of cell replacement therapies. Recent work has shown that hESC differentiation can recapitulate endogenous developmental processes such as neocortical development 1-3, and can even generate structures resembling immature organs4-6. Therefore, hESCs provide the possibility of unprecedented insight into human development. With the discovery that somatic cells could be reprogrammed into induced pluripotent stem cells (iPSCs), which harbor a developmental potential indistinguishable from ESCs7, 8, it became possible to generate disease-specific hiPSC lines by reprogramming cells from individuals suffering from the disease of interest. Further, the refinement of methods for reprogramming and culturing human pluripotent stem cells (i.e. hESCs and hiPSCs; henceforth, hPSCs), has allowed labs across the world to integrate these cells into their research programs.
As this widespread adoption of hPSC technologies has placed hPSCs in the spotlight, early successes or failures with these cells will have profound effects on hESC-based disease modeling and regenerative medicine. Irrespective of the specific disease or developmental process of interest in any given study, it is of critical importance to ensure that hPSC-derived cell types are well defined and biologically relevant. In this regard, determining epigenetic and transcriptional profiles of the population of cells under study may be the best readout of biological relevance and cellular identity.
Epigenetics is classically defined as a “stably heritable phenotype resulting from changes in a chromosome without alterations in the DNA sequence”9. Adrian Bird has revised this definition to “the structural adaptation of chromosomal regions so as to register, signal or perpetuate altered activity states”10. The different modes of epigenetic regulation include DNA modifications such as 5-methylcytosine (5mC) and 5-hydroxymethylcytosine (5hmC), posttranslational histone tail modifications, energy dependent nucleosomal remodeling, and long non-coding RNA regulation of local chromatin structure and chromosomal organization. It has been known for more than three decades that epigenetic modification of DNA by methylation of deoxycytidine residues has a powerful impact on gene expression11-13. More recently, with the growing understanding of histone-mediated chromatin remodeling (the “histone code”14) the complex interplay of different epigenetic modes is beginning to be revealed. Epigenetic modes that involve DNA or histone modifications include the enzymes that catalyze the particular modification (“writers”), proteins that recognize and bind the modification (“readers”), and the enzymes that remove the modification (“erasers”). For example, histone 3 lysine 27 trimethylation (H3K27me3) is catalyzed by the histone methyltransferase (HMTase) enhancer of zeste homolog 2 (EZH2), read by chromobox homolog 7 (CBX7) and erased by the lysine-specific demethylase UTX. Readers possess characteristic recognition motifs such as the methyl-CpG binding domain for 5mC and the bromodomain for lysine acetylation. A number of different domains recognize methylated lysines or arginines in a residue/modification-specific manner. They include chromodomains and tudor, WD40 repeat, and PHD finger domains15. See Figure 1 for a list of common readers, writers and erasers pertaining to the epigenetic marks reviewed here.
Figure 1.

Proteins involved in the regulation of common epigenetic modifications. “Writer” refers to the enzyme(s) that catalyze and establish a particular modification. “Readers” are proteins or multiprotein complexes that recognize and bind the modification, and “Erasers” are a group of enzymes that catalyze removal of the mark. The coordinated action of these three groups not only ensures the faithful maintenance of epigenetic heritability but also determines the dynamic nature of epigenetic regulation during development. The list of histone modification writers, readers and erasers is exceedingly long. Therefore, we have listed only a few well-studied examples for each modification. New nomenclature guidelines have been proposed for many of these proteins9; however, we have used the old nomenclature because it is more widely used in the literature. Proteins denoted in blue text exhibit heart defects when mutated. See also10. DPF3B – Tetralogy of Fallot, MLL2- Kabuki Syndrome, CHD7 - CHARGE Syndrome, WHSC1 - Wolf-Hirschhorn Syndrome.
In this review, we provide an overview of DNA methylation and histone modifications and their crosstalk on how they affect specific regulatory elements, namely promoters and enhancers. We will then review available knowledge pertaining to epigenetic dynamics during the developmental processes of differentiation and X-chromosome inactivation, including the role that epigenetics plays on nuclear organization and genome topology. Where appropriate we provide examples pertaining to cardiovascular research and direct the reader to more in depth reviews of the topics we discuss.
DNA methylation
DNA methylation is a heritable, yet reversible, epigenetic modification that plays a central role in transcriptional repression, suppression of retroelement transposition, genomic imprinting, X-chromosome inactivation, and higher-order chromatin organization. DNA methyltransferases (DNMTs) catalyze the transfer of a methyl group from cofactor S-adenosylmethionine to carbon 5 of the cytosine ring to generate 5-methylcytosine (5mC)16. In mammals, the de novo DNMTs (DNMT3A, DNMT3B, and cofactor DNMT3L) are responsible for establishing post-fertilization genomic methylation patterns, while DNMT1, together with its cofactor UHRF1 (ubiquitin-like with PHD and ring finger domains 1) (Figure 1), localize to the replication fork in order to maintain methylation patterns on the newly synthesized DNA stand during replication. Although the minimal sequence requirement for DNA methylation is that the target cytosine typically resides within a CpG dinucleotide, sequence preferences have been identified for the de novo DNMTs that appear to be conserved from mice to humans17-19.
In mouse development, after fertilization and before pronuclear fusion, zygotic genome activation and the first cell division, the paternal genome is demethylated by enzymatic modification of 5mC (discussed later), whereas the maternal genome is passively demethylated during subsequent rounds of replication via nuclear exclusion of an oocyte specific Dnmt1 isoform (reviewed in 20). Thus, the totipotent zygote is essentially devoid of DNA methylation except at imprinted regions. The genome is gradually re-methylated during subsequent cleavage divisions that generate the morula and early blastocyst. The genomes of pluripotent stem cells (PSCs) derived from the inner cell mass/epiblast are highly methylated20. Erasure of gametic methylation patterns and genomic re-methylation in the developing zygote are necessary for specification of initial lineage commitment. DNA methylation, however, is not required for pluripotency as the genomes of Dnmt3a, Dnmt3b, and Dnmt1 triple-knockout mouse ESCs are hypomethylated, yet the cells retain self-renewal and pluripotency, but exhibit a host of differentiation defects21. This suggests that establishment of zygotic methylation patterns is crucial for germ layer specification and lineage commitment. Underscoring the importance of DNA methylation for cellular differentiation and embryonic development, Dnmt1 and Dnmt3b knockout mice exhibit embryonic lethality, while Dnmt3a knockouts display stunted growth and neonatal mortality22, 23.
The DNA methylation machinery works in conjunction with other modes of epigenetic regulation to regulate gene expression via local chromatin structure and higher-order genomic topology. Methyl-DNA binding proteins (MBD1, 2, 3; MECP2; and ZBTB33 (Kaiso)) bind 5mC and recruit histone deacetylases, repressive histone methyltransferases and other chromatin remodeling proteins such as ATP-dependent chromatin remodeling complexes to actively repress transcription24.
The genomic location of DNA methylation affects its role on transcription and higher-order chromatin structure. The highly and constitutively methylated centromeric and pericentromeric DNA satellite repeats play a large role in heterochromatin organization at these regions. Indeed, the heterochromatic centromeres from many chromosomes aggregate to form structures called chromocenters that play roles in overall nuclear architecture. Many studies have demonstrated that promoter CpG methylation is inversely correlated with gene expression. Genes regulated by methylation usually contain a low density of promoter CpG sites. Most low CpG density promoters are methylated in ESCs and subsequently demethylated and expressed in a lineage or cell type-specific manner during differentiation25-27. Areas termed “CpG islands”, which are regions of high CpG density found within or near proximal promoters or transcription start sites, are typically devoid of DNA methylation28.
Recent studies suggest that promoter hypomethylation coupled with methylation within gene bodies strongly correlates with expression29, 30. Differentially methylated regions (DMRs), generally found at regulatory elements such as enhancers and promoters, display lineage- or cell type-specific methylation patterns. DMRs can be used to predict whether a cell belongs to a certain lineage or tissue based on the genes they regulate27. As a general rule the extent of genomic methylation exhibits an inverse relationship to developmental potential such that pluripotent genomes are highly methylated whereas differentiated cells from a variety of lineages display reduced levels of global DNA methylation29, 31. These data indicate that there is a progressive loss of methylation as differentiation proceeds27, 29. Inhibition of DNA methylation has been shown to result in strong induction of cardiomyocyte differentiation32. In addition, a subset of genes involved in cardiac structure undergo cardiomyocyte-specific demethylation during development33. There are, however, specific examples of methylation gains at DMRs during differentiation. For example, promoter methylation and silencing of the pluripotency regulators, POU5F1 and NANOG, occurs during lineage specification, and the HOX gene cluster becomes progressively methylated as development proceeds.
Non-CpG methylation
Non-CpG methylation (CpH, where H = A, T or C), is enriched in PSCs and lost upon differentiation in all cell-types/tissues except the brain, where it accumulates in neurons during postnatal and adolescent development, coinciding with periods of synaptogenesis and synaptic pruning34, 35. It is far less abundant than CpG methylation in somatic cells, constituting only 0.02% of total 5mC in somatic cells, but in hESCs, 25% of 5mC are in CpH29, 35. The most prevalent non-CpG methylation is found at CpA dinucleotides with mCpT and mCpC constituting a very small fraction of CpH methylation29, 34, 36, 37. Non-CpG methylation appears to be catalyzed by DNMT3A34, 36, 37 and DNMT3B29. The functions of non-CpG methylation remain elusive, but high-resolution global-profiling techniques suggest it may have a myriad of functions. For example, non-CpG and CpG methylation have been implicated in the regulation of RNA splicing in ESCs and neurons, respectively29, 38, and enrichment of CpH methylation on the template strand within gene bodies in ESCs correlates with high expression35. However, in the brain CpH methylation is associated with transcriptional repression18, 34. Interestingly, CpH methylation, but not CpG methylation, is depleted at pluripotent-specific enhancers in ESCs35, whereas CpG methylation is lost at cell type-specific enhancers during differentiation27. Thus, it appears PSCs use the DNA methylation machinery differently than somatic cells for gene regulation.
Oxidation of 5mC and DNA demethylation
Recently it was discovered that the Ten Eleven Translocation (TET) family of 2OG-Fe(II) dioxygenases catalyze the hydroxylation of 5mC to generate 5-hydroxymethylcytosine (5hmC)39. Genomic 5hmC was first characterized in cerebellar Purkinje neurons where it is nearly 40% as abundant as 5mC40. The genomes of PSCs also possess high levels of 5hmC39, 41, 42. At low resolution, 5hmC and 5mC exhibit mutually exclusive localization patterns: centromeric and pericentromeric regions tend to have higher concentrations of 5mC while 5hmC is more enriched on chromosome arms42. High-resolution global mapping demonstrates an anti-correlation between 5mC and 5hmC levels at regulatory elements. Moreover, 5hmC signatures are enriched at sites of DNaseI hypersensitivity, which are indicative of genomic regions bound by regulatory proteins, whereas 5mC is generally depleted at sites of DNA-protein interaction43. 5hmC is particularly enriched at bivalent domains (discussed later), near transcription start sites, at CpG-rich proximal promoters, and at active and poised enhancers41, 43, 44. Interestingly, both 5mC and 5hmC are enriched within the gene bodies of actively transcribed genes30, 41-43, 45.
To date there are three identified TET isoforms that play different roles during development. TET3 primarily functions early in embryogenesis, where it rapidly converts 5mC to 5hmC in the male pronucleus. The other two isoforms, TET1 and TET2, share some targets but also have mutually exclusive functions during development. Depletion of Tet1 or Tet2 in ESCs results in globally reduced levels of 5hmC but has no discernible effect on pluripotency. Tet1 or Tet2 single knockouts display different differentiation defects – Tet1 knockout (KO) differentiation is skewed toward trophectoderm (in agreement with in vitro differentiation data using Tet1 knockdown mESCs), and Tet2 KO animals possess dysregulated hematopoietic stem cells46-49. The majority of double Tet1/Tet2 KO animals die at birth; however, a small percentage of double knockout animals are phenotypically normal and fertile but possess imprinting abnormalities50, thereby implicating TET proteins and 5hmC in the proper maintenance of genomic imprints. These results suggest that TET1 and TET2 have non-overlapping albeit mild functions during development: TET1 helps maintain pluripotency by suppression of trophectoderm differentiation and TET2 maintains normal homeostasis in the hematopoietic lineage.
Originally it was thought that 5hmC was merely an intermediate in the demethylation pathway, but its putative role in gene regulation has grown, along with increased controversy about the details of its function. Recent studies indicate that it might be a stable mark complete with its own “readers”51 some of which appear to overlap with MBDs (methyl-DNA binding proteins). Since the discovery of 5hmC, a significant question is whether MBDs also recognize 5hmC. The literature is contradictory on whether proteins that recognize 5mC also function as 5hmC readers. For example, several studies have shown that many of the proteins that directly bind methylated DNA (e.g. MBD1, MBD2b, MBD3, MBD4, MeCP2 and UHRF1) either do not recognize and cannot bind 5hmC, or exhibit very low and variable affinity for 5hmC51-54. However, another report indicates that MBD3 does bind 5hmC and influences the expression of genes marked with 5hmC55. Furthermore, there are conflicting reports as to whether MeCP2 is able to recognize and bind 5hmC. Much emphasis has been placed on MeCP2's putative role in reading 5hmC because of the high levels of both MeCP2 and 5hmC in the brain, and MeCP2's role in the neurodevelopmental autistic disorder, Rett Syndrome. Again, there are conflicting reports as to whether MeCP2 is able to recognize and bind 5hmC. Mellen et al identify 5hmC as a high affinity substrate of MeCP2, and observe MeCP2 and 5hmC co-localization at genes expressed in the brain56. Szulwach et al, however, demonstrate an inverse relationship between MeCP2 and 5hmC levels in the brain44. Moreover, other reports suggest that 5hmC is a poor physical substrate for MeCP2 in vitro54. While recombinant MeCP2 may possess low affinity for 5hmC in vitro, its affinity in vivo may be enhanced by either accessory proteins or by posttranslational modifications. It will be important to rectify these differences, and to further identify molecules that read 5hmC and its oxidized derivatives.
DNA demethylation has both fascinated and frustrated scientists for decades. The most dramatic example of DNA demethylation occurs early in development when the paternal genome is rapidly demethylated before pronuclear fusion and the first cell division occurs57, 58. Other examples of demethylation are more localized such as the de-repression of genes during germ layer formation, lineage specification and terminal differentiation, and the activity-dependent epigenetic remodeling events related to synaptic plasticity and memory formation27, 38, 59-61.
The process of active, or enzymatic, DNA demethylation has been the subject of speculation for years62. Recent studies indicate that active demethylation involves a DNA repair event such as base excision repair (BER) (reviewed in 63). However, none of the glycosylases that initiate BER (TDG, MBD4 or SMUG1) possess high affinity for 5mC as a substrate. Therefore, in order for demethylation to proceed through a BER-based mechanism, 5mC must be modified into a form that is efficiently recognized by the BER glycosylases. Spontaneous deamination of 5mC to thymidine yields a T:G mismatch and deamination of 5hmC results in a 5-hydroxyuracil (5hmU):G mismatch, both of which are recognized by either of two mammalian BER glycosylases; thymine-DNA glycosylase (TDG) or methyl-DNA binding protein 4 (MDB4). TDG and MBD4 have both been implicated in DNA demethylation64 and recent studies have defined a clear role for TDG in BER-mediated DNA demethylation65, 66. Deamination of 5hmC by activation-induced deaminase (AID) results in demethylation of reporter constructs containing 5hmC in mammalian cells38 and AID has been shown to be important for the demethylation events that occur during somatic cell reprogramming to pluripotency67. The precise role of AID in active DNA demethylation remains unclear because recently it has been shown that recombinant AID displays very little activity toward 5mC and no activity toward 5hmC as substrates68, although unidentified cofactors may enhance AID substrate affinity in vivo.
Currently, experimental evidence suggests a model of active DNA demethylation that can be summarized as follows (Figure 2): TET-mediated hydroxylation of 5mC generates 5hmC which is recognized by a complex containing TDG and AID (and possibly GADD4569). AID then deaminates 5hmC to 5hmU thereby generating a suitable substrate for TDG, which initiates the BER cascade ultimately ending with restoration of a CpG dinucleotide. Two other DNA glycosylases (Mpg and Neil3) have been recently identified as 5hmC readers, but their role in DNA demethylation remains to be determined51.
Figure 2.

DNA methylation dynamics. Cytosine is methylated at carbon 5 of the pyrimidine ring by DNA methyltransferases (DNMT) to generate 5-methylcytosine (5mC), which can be hydroxylated to 5-hydroxymethylcytosine (5hmC) by TET dioxygenases. 5hmC can be deaminated by the cytidine deaminase, AID, to generate 5-hydroxymethyluracil (5hmU), or further oxidized by TETs to 5-formylcytosine (5fC) or 5-carboxylcytosine (5caC). 5hmU, 5fC and 5caC are all substrates for thymine DNA glycosylase (TDG); the primary glycosylase that initiates base excision repair-mediated DNA demethylation.
It has also been shown that TET enzymes possess the ability to further oxidize 5hmC in a step-wise manner to generate 5-formylcytosine (5fC) and 5-carboxylcytosine (5caC)70. 5caC is a high affinity substrate for purified TDG but not MBD4,70, 71 and TDG has been shown to rapidly excise both 5fC and 5caC72. Depletion or knockdown of Tdg in ESCs results in drastic reduction or ablation of glycosylase activity toward 5caC, and increased levels of 5fC and 5caC at promoters also marked with 5hmC71, 73, 74. Interestingly, 5fC also appears to be preferentially enriched at a specific class of regulatory elements, called “poised enhancers” (discussed later), concomitant with increased levels of 5hmC and decreased 5mC levels. Furthermore, increased levels of 5fC are observed at poised enhancers in Tdg null ESCs suggesting Tdg-mediated demethylation occurs at these regulatory elements74.
To return to the most striking example of rapid demethylation, the genome-wide demethylation of DNA in the male pronucleus of a zygote57, 58, evidence indicates that this occurs by TET3-mediated conversion of 5mC to 5hmC rather than a BER-mediated mechanism, mainly for two reasons: 1) 5mC is symmetric at CpG sites, where the cytosine on each strand is methylated; therefore, BER of a symmetrically methylated CpG site would result in a double strand break. Genome-wide demethylation within a short temporal window would generate hundreds of thousands of double strand breaks and likely trigger apoptosis. 2) BER is a laborious and time-consuming enzymatic cascade catalyzed by multiple proteins. In the original studies documenting rapid demethylation it appeared that the paternal genome was actively demethylated because the antibody-based methods used to visualize 5mC did not recognize 5hmC. It has more recently been demonstrated that 5mC in the paternal genome is rapidly converted to 5hmC by TET3, which is then passively lost during subsequent rounds of replication75-77.
The Histone Code
Posttranslational modification of histone tails results in a combinatorial readout that affects gene expression through the regulation of local chromatin structure14. A comprehensive review of all histone tail modifications, which include acetylation, methylation, citrullination (also known as deimination), phosphorylation, ubiquitylation, sumoylation, and biotinylation, is beyond the scope of this article. The precise role of histone modifications such as phosphorylation, sumoylation and biotinylation, and their effects on chromatin structure and transcriptional state remain unclear and warrant further investigation78. Interestingly, citrullination, the enzymatic conversion of arginine to citrulline, has recently been implicated in the establishment and maintenance of pluripotency79. Here, we will focus on the five most commonly studied modifications of histone 3 (H3), specifically H3 lysine 4 methylation (H3K4me), H3 lysine 9 methylation (H3K9me), H3 lysine 27 acetylation (H3K27ac), H3 lysine 27 methylation (H3K27me) and H3 lysine 36 methylation (H3K36me3), as they have been shown to correlate with and accurately predict chromatin/transcriptional states of regulatory elements in many different cell types.
High-resolution, genome-wide studies have elucidated common themes of gene regulation by modified histones. Genomic regulatory regions that respond to developmental and environmental stimuli (i.e. enhancers and promoters) are marked by histone modifications that confer transcriptionally permissive (euchromatin) or repressive (heterochromatin) chromatin states, which are mediated by the Trithorax group (TrxG) proteins and Polycomb group (PcG) proteins, respectively. Common euchromatin modifications are H3K4me3, H3K9ac, H3K27ac and H3K36me3. They are found primarily at active enhancers (H3K9ac, H3K27ac), promoters (H3K4me3) and within the bodies of actively transcribed genes (H3K36me3)80. H3K4me3 displays a punctate localization pattern within 1-2 kb of active promoters that contain CpG islands28. It promotes transcription by recruiting nucleosome remodeling complexes and histone acetylases.
The two most studied modifications found at repressed chromatin are H3K9me3 and H3K27me3; generally localized to constitutive and facultative heterochromatin, respectively. The histone methyltransferase that catalyzes H327me3 is EZH2 (enhancer of zeste homolog 2), which is a member of the multi-subunit Polycomb group complex 2 (PRC2) that also contains EED (embryonic ectoderm development), SUZ12 (suppressor of zeste 12) and RBBP4/7 (retinoblastoma binding proteins 4 and 7). PRC2 is targeted to genomic regions in response to developmental cues where it catalyzes H3K27me3. This modification recruits another multi-protein complex, PRC1, which contains members of the chromobox (CBX) family that recognize H3K27me3. Also included in PRC1 is the ring finger protein, RING1B, an ubiquitin ligase that catalyzes monoubiquitination of histone H2A, a modification that impedes RNA polymerase II (RNAPII) elongation, resulting in transcriptional repression. Originally identified in Drosophila, the SUV39 (suppressor of variegation 3-9) family of histone methyltransferases, which do not associate with PRC complexes, catalyze methylation of H3 to H3K9me3. This modification is heavily enriched at centromeric and pericentromeric DNA where, together with the DNA methylation machinery, it mediates the constitutive higher-order heterochromatin structure necessary for mitotic spindle assembly.
PRC complexes play many roles regarding determination of cell identity81. Abolishment of either PRC1 or PRC2 activity in ESCs results in differentiation, albeit skewed toward ectoderm and endoderm, respectively. However, double knockouts (Eed-/-, Ring1B-/-) completely lose the ability to initiate differentiation82, 83. These results indicate the two complexes have both redundant and non-overlapping functions during development and the maintenance of PSC identity. Furthermore, in an excellent example of epigenetic crosstalk, PRC2 has been physically coupled to long non-coding RNA regulation of chromatin structure during cardiomyocyte differentiation84.
In general the genome of pluripotent stem cells is enriched for “open” or transcriptionally permissive/poised chromatin and contains less heterochromatin relative to somatic cells (see below). PSCs contain more acetylated chromatin and smaller regions of H3K9me3 and H3K27me3 relative to differentiated cells. Knockout of the histone acetyltransferase MYST1 (also known as MOF), a member of the core pluripotency network, results in enhanced heterochromatin formation and loss of differentiation potential in ESCs85.
Bivalent domains
Traditionally it was thought that genes required for downstream developmental process were actively repressed until needed. The discovery of bivalent domains in ESCs changed the way we view gene regulation in pluripotency and differentiation28. Bivalent domains were first identified as broad H3K27me regions that also contain smaller, narrowly defined regions of H3K4me localized at transcriptional start sites 28. Bivalent domains can be further categorized based on histone modifications and PcG (Polycomb group proteins) occupancy. Domains that contain PRC2 only are weakly conserved and generally correspond to membrane proteins or genes of unknown function86. However, domains containing both PRC1 and PRC2 complexes are highly conserved evolutionarily and show strong enrichment for many developmentally regulated transcription factors and other factors involved in morphogenesis and in cell signaling. Polycomb targets are commonly associated with CpG-rich DNA, and approximately half of PRC2 binding sites correspond to CpG islands86. Interestingly, PRC2 is present at nearly all bivalent regions, whereas PRC1 occupies less than half (39%) of bivalent promoters86. The majority of bivalent domains in ESCs directly overlap transcription start sites of genes that encode transcription factors and approximately half of the bivalent domains contain binding sites for at least one of the pluripotency transcription factors28.
Promoters
Promoters surround transcriptional start sites and serve as the platform on which the basal transcription machinery and RNAPII assemble during transcription initiation. They are generally categorized based on CpG content/DNA methylation status and histone modifications as active, repressed, or poised (Figure 3). Active genes possess promoters enriched for H3K4me3, H3K9ac, H3K27ac, and 5hmC, and are typically devoid of 5mC. Conversely, the promoters of repressed genes are marked by H3K27me3 and/or H3K9me3, and 5mC. Poised genes, which are either activated or repressed during development, have bivalent promoters, marked by the presence of both H3K4me3 and H3K27me3. Poised genes are generally associated with more complex expression patterns and include key developmental transcription factors, morphogens and cell surface molecules. In addition, several bivalent promoters appear to regulate transcript for lineage-specific microRNAs26.
Figure 3.

Epigenetic control and remodeling of regulatory elements during development. (A) Poised enhancers/promoters are typically found in pluripotent stem cells. They possess epigenetic modifications indicative of both transcriptionally active (green marks) and repressed (red mark) chromatin. For instance, the Trithorax Group (TrxG) proteins and Polycomb Group (PcG) proteins, which catalyze H3K4me3 and H3K27me3 respectively, localize to poised promoters. Poised regulatory elements are also generally devoid of 5mC and enriched for 5hmC. Poised enhancers can be distinguished from poised promoters by the presence of the enhancer-specific mark, H3K4me1, and bound Mediator complex. Chromatin loops organized by CTCF binding localizes distal poised (or active) enhancers and promoters within close physical proximity to facilitate transcription. (B) The transition from poised to active regulatory elements involves loss of PcG localization, and demethylation and concomitant acetylation of H3K27. Gains of H3K36me3 within gene bodies are also observed upon gene activation. (C) Heterochromatin formation is associated with gene repression. This is mediated in part by erasure of activating histone modifications (H3K4me1, H3K27ac, 5hmC), establishment of repressive marks such as H3K27me3 and 5mC, and nucleosomal compaction. MBD, methyl-DNA binding domain protein; DNMT, DNA methyltransferase; TET, ten eleven translocation dioxygenase; HAT, histone acetyltransferase; HDAC, histone deacetylase; PcG, polycomb group complex; TrxG, trithorax group complex; TF, transcription factor; MED, Mediator complex; POL II, RNA polymerase II.
Highly expressed genes, such as metabolic and housekeeping genes, usually contain hypomethylated CpG islands within their promoters. It is estimated that 95% of transcriptional start sites marked by H3K4me3 contain CpG islands28. Promoters with low CpG content are more sensitive to regulation by DNA methylation and are typically found at genes expressed in a highly tissue-specific manner87. In ESCs only a small percentage of low CpG promoters contain significant levels of H3K4me3 and essentially none are marked by H3K27me326.
Ren and colleagues have proposed that the epigenetic mechanisms regulating a given promoter may be influenced by the promoter's sequence88. In support of this, they find that genes preferentially expressed in ESCs and early cell types during differentiation (NPCs and mesendoderm) are CpG-rich and contain CpG islands whereas a much smaller percentage of genes containing CpG islands are expressed in tissue-specific manner. Furthermore, they find that developmentally regulated genes are regulated by promoters with high CpG content and are typically marked by H3K27me3, in contrast to tissue-specific promoters that possess low CpG density and are preferentially enriched for DNA methylation88.
Enhancers
Approximately 400,000 putative enhancers have been defined in the human genome based on analysis of histone modifications and proximity to transcription start sites89. Enhancers are typically 200-300 bp in length and exhibit nucleosomal depletion compared to flanking chromatin90. They are enriched for DNaseI hypersensitivity sites and the histone acetyltransferase, EP300 (E1A binding protein p300), suggesting an overall open chromatin structure90. Indeed, most enhancers are bound by multiple transcription factors. The Mediator complex, a transcriptional co-activator, is also commonly found at enhancers. The Mediator complex is a large multi-protein complex that is necessary for RNAPII recruitment to the promoters of enhancer-regulated target genes91. In ESCs, co-occupancy by Mediator proteins, Oct4, Sox2 and Nanog is predictive of an enhancer92, 93.
Enhancers can be distinguished from promoters by their unique histone modification signatures and, similar to promoters, these histone modifications can be used to classify them as active, poised or repressed (Figure 3). For example, while both promoters and enhancers typically possess H3K27ac, enhancers are characteristically enriched for H3K4me1, in contrast to the H3K4me3 seen at promoters80, 94, 95. Wysocka and colleagues have categorized bivalent enhancers based on histone modification signatures; class I elements represent active enhancers and are enriched for H3K4me1 and H3K27ac90, 95 and the Mediator complex. Class II elements exhibit high levels of H3K27me3 in addition to H3K4me1 and are considered to be poised enhancers. Repressed enhancers are devoid of both H3K4me1 and H3K27ac, and instead are marked by H3K27me3. Class I elements are expressed at a higher level than class II elements, which agrees with the RNAPII occupancy observed at class I elements but not at class II enhancers95. Active enhancers are also enriched for 5hmC and depleted of 5mC; whereas inactive and developmentally poised enhancers are marked by 5mC and 5hmC, respectively41, 42, 80.
Enhancers are highly variable between lineages and cell types, and confer cell type-specific or lineage-restricted gene expression88, 94, 96, 97. Interestingly, only a small percentage of either class I or II enhancers overlap with CpG islands, and it is estimated that 94% of lineage-restricted enhancers are CpG poor and depleted for 5mC88, 95. Furthermore, most lineage-restricted enhancers have low CpG density and are marked by H3K27ac in ESCs and cells from the early germ layers. These same enhancers are enriched for H3K27me3 and DNA methylation in terminally differentiated cells, which negatively correlates with expression of their target genes88.
“Super enhancers” are a recently identified class of enhancers that play a major role in determining cellular identity97, 98. They are characterized as large genomic regions (up to 50 kb) possessing enhancer-like features: enrichment for H3K4me1, H3K27ac, Mediator complex and transcription factors. However, unlike typical enhancers, they contain unusually high levels of Mediator complex proteins and transcription factors, and display enrichment for cohesin and Nipbl (Nipped-B homolog), a protein involved in enhancer-promoter communication97. The ∼200 super enhancers are often observed as clusters of smaller enhancers enriched for transcription factor motifs involved in cell identity97, 98. Therefore, genes regulated by super enhancers are highly cell type-specific and are usually expressed at higher levels than genes regulated by typical enhancers. For example, nearly all genes associated with ESC identity (e.g. POU5F1 (OCT4), SOX2, NANOG, KLF4) are regulated by super enhancers98.
Epigenetic dynamics during differentiation
The epigenome of PSCs is dramatically different from that of somatic cells. When comparing histone modification and DNA methylation patterns between differentiated cells and ESCs, Hawkins et al estimate that approximately one third of the genome differs in chromatin structure99. Many of these differences can be attributed to differential transcription factor binding and chromatin state remodeling during differentiation96, 99. For example, differentiated cells have larger regions marked by H3K9me3 and H3K27me3 compared to ESCs99. Most of these H3K9me3 and H3K27me3 domains are significantly larger (2-3 fold) in fibroblasts and T lymphocytes compared to ESCs, while H3K36me3 domains are similar between ESCs and differentiated cells99, 100. These large expanded H3K27me3 domains are remodeled to an ESC-like state during reprogramming100. This suggests that expansion of H3K27me3 domains accompanies cellular differentiation.
Bivalent domains in ESCs often transition to H3K27me3 domains that expand in lineage-committed cells such that H3K27me3 affects ∼40% of the genome in differentiated cells compared to only ∼8% of the ESC genome100. Differentiation results in a progressive, global H3K27me3 enrichment that occurs in a cell type-specific manner. These results identified Polycomb-mediated repression as an important mechanism for cell fate determination and lend support to the longstanding idea that differentiation results in an increasingly restrictive chromatin landscape88, 99, 100. The majority of bivalent domains observed in undifferentiated ESCs resolve to a monovalent status in cells committed to a given lineage26, 96. Bivalent domains found in promoters encoding transcription factors typically resolve to either H3K4me3 only or H3K27me3 only in a lineage-specific manner96. A clear example of this can be found during cardiomyocyte differentiation from ESCs. Many key transcription factors such as GATA6, GATA4, NKX2.5, HAND2, TBX5, and signaling molecules (BMP2, WNT5A) involved in cardiac development, gradually lose H3K27me3 and gain H3K4me3 concomitant with transcription in a stage-specific manner as differentiation proceeds101, 102. Conversely, later stages of cardiac differentiation require EZH2-mediated epigenetic repression of progenitor-specific transcription factors for cardiomyocyte maturation102, 103. Loss of H3K4me1/me3 is usually accompanied by the gain of DNA methylation96. Transcription factor motifs are particularly enriched for gains of DNA methylation during germ layer specification. For example, the number of transcription factor motifs that gain DNA methylation are ∼2-fold greater in the definitive endoderm than in either the early ectoderm or mesoderm96. Loss of transcription factor occupancy during differentiation may trigger DNA methylation; for example, putative regulatory elements downstream of the pro-neural gene DBX1 (developing brain homeobox 1) gain DNA methylation in the endoderm and mesoderm, but not in the ectoderm where this gene is expressed96.
In contrast, many poised enhancers become active (assessed by the loss of H3K27me3, gain of H3K27ac and acquisition of RNAPII) during the transition from pluripotency to germ layer specification95. Boyer and colleagues have found that the majority of enhancers are poised at any given stage during cardiomyocyte differentiation. However, a small group of enhancers become active in a cell type-specific manner. For example, the enhancers that become active in cardiac progenitors regulate genes essential for heart morphogenesis and development whereas later during differentiation there is enrichment for genes encoding cardiac structural and functional proteins104. Loss of DNA methylation at regulatory elements is also common during germ layer formation and lineage commitment, and is especially prevalent in ectoderm formation relative to the other germ layers27, 96. Similar to the idea of poised regulatory elements there are examples of lineage-specific epigenetic priming during development. Specifically, many regions that are associated with fetal and adult brain development exhibit high levels of DNA methylation in ESCs. These regions are demethylated and resolve to an H3K4me1 state early during ectoderm differentiation yet remain methylated in the other lineages96. Many of the genes that acquire H3K4me1 do not show immediate expression changes, and therefore can be said to be primed for transcription at later stages. There are similar examples of epigenetic priming in cardiogenesis and lymphogenesis suggesting that epigenetic priming is a common feature of development104, 105. Other examples of priming include distal CpG-poor regions that exhibit high DNA methylation levels in ESCs are demethylated and transition to H3K27me3 only in a lineage-specific manner. These regions are generally required at later stages of development in that particular lineage96, further emphasizing the dynamic nature of histone-mediated epigenetic regulation compared to DNA methylation. Genes that temporally regulate cellular identity during differentiation can also be subject to cyclical methylation events. For instance, a subset of genes necessary for neuronal progenitor fates are demethylated in neural precursors, expressed in progenitors and then re-methylated and repressed in later cell types such as neurons.
Epigenetic effects on nuclear organization and genome topology
At the most basic level, chromatin can be viewed as “beads on a string” where DNA (string) is coiled around nucleosomes (beads) with ∼165 bp periodicity to form the 11 nm fiber. Subsequent compaction generates the 30 nm and 300 nm fibers, ∼700 nm chromosomal domains and ∼1400 nm mitotic chromosomes106. During interphase each chromosome occupies its own defined space within the nucleus, referred to as a “chromosome territory”. This allows non-random juxtapositions or physical interactions between genomic regions both in cis (intrachromosomal) and trans (interchromosomal). These interactions are characterized using chromosome conformation capture (3C) techniques and their derivatives such as 4C, which permits interrogation of multiple genomic contacts at a single locus, and Hi-C or Chromatin Interaction Analysis by Paired-End Tag Sequencing (ChIA-PET), which have the power to identify many associations, thereby providing information on genome topology107. Chromosomal regions of high gene density and/or high transcriptional activity tend to loop out of their respective chromosome territories and indiscriminately associate in trans with each other. Unlike associations of active regions, subnuclear movement of inactive regions is more locally constrained and hence these interactions tend to be intrachromosomal within a chromosome territory core108.
Somatic nuclei possess a nuclear organization different from pluripotent cells and these differences are largely reflected by chromatin state109. Pluripotent cells contain less heterochromatin in general and less chromatin associated with the nuclear lamina and nucleoli than differentiated cells. Lamina-associated domains (LADs) are a class of stable chromatin domains found at the nuclear periphery that typically possesses low levels of gene expression110. There are approximately 1,300 LADs in mammalian cells ranging in size from 0.1-10 MB. Coincident with their transcriptional status, LADs are enriched for the heterochromatic marks, H3K9me2/3 and H3K27me3, and their borders usually contain CpG islands and/or binding sites for the chromatin insulator, CTCF (CCCTC-binding factor)110. The interaction pattern and number of LADs appears to be cell-type specific; however, some LADs are common across cell types111. During differentiation, specific LADs containing genes destined for activation in the differentiated progeny translocate from the nuclear periphery to the interior. Conversely, genes to be repressed in the next cell type condense and are often relocated to the periphery where they interact with the nuclear lamina111. Interestingly, genome reorganization occurs sequentially during differentiation. For example, specific genomic regions are relocated from the nuclear lamina during differentiation of ESCs to neural precursor cells111. These regions remain spatially constant during the differentiation of NPCs to astrocytes, but a new, distinct group of LADs relocates away from the periphery during this subsequent differentiation step. Repressed genes that relocate away from the nuclear lamina become “unlocked” and are primed/poised for activation later during differentiation, whereas active genes that shuttle to the nuclear lamina become “locked” and stably repressed.
In addition to LADs, mammalian genomes contain approximately 2,000 large domains called Topologically Associating Domains (TADs), which are megabase-sized local interaction domains that inhibit spreading of heterochromatin in cis112. They are generally bounded by invariant CTCF binding sites, tRNAs or small interspersed elements (SINEs), and contain coordinately regulated enhancers and promoters that cluster together. The CTCF binding sites that demarcate the boundaries of a large number of TADs are highly conserved between human and mouse, and between cell types. Thus, they are unlikely to play a role in the establishment of cell type-specific genomic topology during differentiation. Subregions within each TAD, however, seem to be dynamic112. These dynamic regions may confer cell type-specific genome organization via transcription factor complexes bound at the regulatory elements of active genes (Figure 4).
Figure 4.
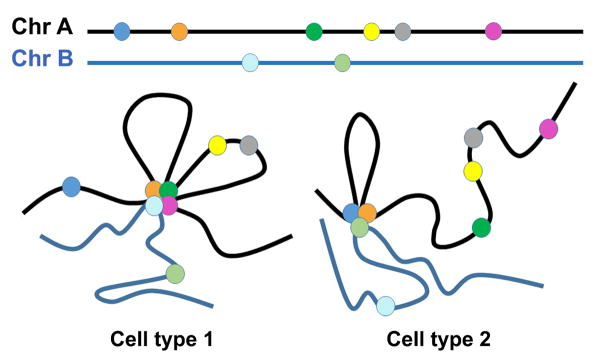
Schematic of cell type-specific genome topology. Genetic loci (depicted as colored circles) cluster together making both intra- (Chr A: Chr A) and inter- (Chr A: Chr B) chromosomal interactions in a cell type-specific manner. This establishes coregulated transcription of multigene networks that confer cellular identity. Differing topological interaction patterns are expected to be associated with different cell types such as ESCs (Cell type 1) and progenitor cells (Cell type 2).
The genome of PSCs is divided into a large complex pattern of chromatin loops bounded by CTCF binding sites113. These loops can be classified according to epigenetic modifications found within the loop and at the loop boundary113. Loops are generally enriched for either active or repressive histone modifications inside the loop in a mutually exclusive manner, although some loops do not appear to be enriched for any particular histone modification. Interestingly, ∼20% of loops are enriched for enhancer marks (H3K4me1/2) within the loop, promoter marks (H3K4me3) at the loop boundary, and marks for active transcription (H3K36me3) and repressed chromatin (H3K27me3) outside the loops on opposite sides. CTCF and cohesin function together to form chromatin loops that segregate heterochromatin from euchromatin113, 114. An example of chromatin dynamics that occur during differentiation can be found at the HOXA locus. In ESCs the HOXA locus consists of bivalent chromatin114. Upon differentiation, chromatin reorganization results in demarcation of a euchromatic and a heterochromatic domain at a highly conserved CTCF binding site bound by both CTCF and cohesin114.
It has become increasing clear that interactions between transcription factors and Mediator complex bound to regulatory elements influence genome topology through their interactions with CTCF and/or cohesin. For example, Oct4 has been shown to interact with CTCF during the establishment of X-chromosome inactivation and the maintenance of bivalent chromatin114, 115. Conversely, loss of Oct4 expression during differentiation permits the CTCF/cohesin-mediated heterochromatin formation at the HOXA locus114 mentioned earlier. Interactions between Mediator and cohesin at enhancers have been shown to regulate gene expression in a cell type-specific manner by regulating the formation of chromatin loops that bring distal enhancers into close proximity with promoters of active genes93. Interestingly, both active and poised enhancers participate in looping interactions in differentiated cells, suggesting that chromatin looping may proceed and/or influence epigenetic priming116 during differentiation. Promoter clusters play a central role in chromosome topology but their contribution appears to not be tissue-specific as promoters have an equally probable chance of interaction in ESCs as well in somatic cells. Surprisingly, high resolution analyses have determined that promoter states are largely invariant and highly stable between diverse cell types80, 90. Furthermore, the majority of promoter clusters exhibit activity in multiple cell types80. Enhancers, on the other hand, contribute to genome topology in a more tissue-specific manner93, 117, 118. For example, in ESCs the SOX2 promoter interacts with a cluster of enhancers that are specifically active in ESCs, whereas a different set of enhancers are associated with the SOX2 promoter in neural progenitor cells118, 119. In general, cell-specific enhancers are highly methylated in cells of other lineages and a direct correlation exists between DNA demethylation and enhancer activation118. Inactive enhancers are highly methylated and become demethylated in a tissue-specific manner during development. Interestingly, in addition to the cell type-specific transcription programs regulated by enhancers, differential enhancer usage may also regulate common sets of promoters. Indeed, cell type-specific enhancers have been shown to interact with promoter clusters common to multiple cell types, suggesting that different cell types use specific enhancers to regulate broadly expressed housekeeping genes118, 119.
During reprogramming to iPSCs, cells acquire a spatial genomic organization specific to pluripotent cells. In ESCs, genes involved in the maintenance of pluripotency (i.e. Dppa5, Zfp42, Zfp281, Lefty, Lin28) make interchromosomal contacts with the Nanog locus, suggesting that pluripotency genes aggregate together117. Reprogramming-induced reactivation of the Nanog locus results in the establishment of pluripotency-associated gene contacts that are not observed at the Nanog locus in somatic cells117. Furthermore, in ESCs it has been shown that the majority of genes associated with reprogramming are spatially connected to the SOX2 locus within one large gene cluster, implying they may be coordinately regulated within an active structure termed a “transcription factory”119.
Imprinting and X-chromosome inactivation
Two specialized modes of epigenetic regulation are imprinting and X-chromosome inactivation (XCI). Both rely on combinatorial epigenetic silencing, that is, a complex interplay of repressive histone modifications, CpG methylation and non-coding RNA modulation of chromatin structure. Genomic imprinting is parent-of-origin allele-specific expression believed to have arisen in order to allocate extra-embryonic nutrient resources to the developing fetus120. The number of imprinted genes is a matter of debate. Estimates in mice range from ∼150 to >1000 imprinted genes121, but recent work indicates there are probably not many more than ∼20018, 122. The majority of imprinted genes contain DMRs that possess parent-of-origin-specific DNA methylation patterns (CpG and non-CpG) at enhancers and/or promoters that confer allele-specific expression. Many imprinted genes are expressed in the brain in a temporal manner to regulate neurodevelopment. Loss of imprinting (LOI), either through gain or loss of DNA methylation (or genetic deletion of regulatory elements/DMRs), results in biallelic gene silencing or expression that has been implicated in cancer and a number of neurodevelopmental disorders including Beckwith-Wiederman Syndrome, Prader-Willi Syndrome and Angelman Syndrome123. Genomic imprints are known to be unstable in mESCs. Recent studies examining a large number of hPSC lines come to the same conclusion. Tissue samples as well as androgenetic and parthenogenetic hPSC lines have been used to establish the “normal” DNA methylation status of a number of imprinted genes. Many hPSC (ESC and iPSC) lines exhibit both and gains and losses of methylation at imprinted loci leading to loss of imprinting and either gene silencing or biallelic expression. These same loci appeared to be epigenetically stable in primary cell lines and tissues. Moreover, certain imprinted genes (PEG3, MEG3, H19) exhibit LOI via hypermethylation and gene silencing in several hPSC lines. More importantly, aberrations in genomic imprints and XCI in undifferentiated hPSCs are faithfully maintained during differentiation and cannot be reset with further reprogramming27, 124.
X-chromosome inactivation (XCI) is an excellent example of many epigenetic modes acting synergistically to control gene expression via chromatin structure and chromosome topology. XCI is a means of dosage compensation between XY males and XX females. Shortly before embryo implantation, one of the X-chromosomes is randomly chosen for transcriptional repression. The earliest event in XCI is expression of XIST, a long non-coding RNA expressed from the X-inactivation center (Xic) on the X-chromosome marked for inactivation. XIST then spreads along the X chromosome beginning with regions either interacting with or in close proximity to the XIST locus followed by spreading to more distant regions. Specific domains of XIST recruit PRC2 to establish transcriptional repression. Together, PRC1 and DNA methylation maintain heritable XCI in somatic cells. In the naïve state of undifferentiated mESCs both X chromosomes are active (Xa/Xa). Human PSCs, on the other hand, generally exhibit XCI (Xa/Xi) upon derivation. Recently, it has been shown that the inactive X in hPSCs often becomes reactivated over time in culture27, 124. Once reactivated, XCI is not re-established by differentiation124. Erosion of XCI during time in culture can have profound effects on the utility of female hPSCs for cell therapy, disease modeling or drug screening125. Careful analysis using SNP genotyping combined with allele-specific RT-qPCR, demonstrate that erosion or loss of XCI is common among late passage female hPSCs27. Loss of XCI in undifferentiated hPSCs is associated with XIST repression, loss of H3K27me3 and de-repression of genes silenced on the Xi. Once lost, the active state is stable and XCI cannot be re-established by differentiation124.
Epigenetic heterogeneity among PSCs
Reprogramming of a somatic cell fate to a pluripotent state is largely an epigenetic process126. Indeed, removal of certain epigenetic “roadblocks” either by chemical inhibition of DNA methylation or knockdown of MBDs during reprogramming enhances iPSC generation127, 128. Additionally, inclusion of histone deacetylase inhibitors during exogenous transcription factor-based reprogramming and somatic cell nuclear transfer also increases the proficiency of PSC derivation129, 130. The three different types of pluripotent stem cells, ESCs, iPSCs and SCNT ESCs (derived by somatic cell nuclear transfer), display global methylation patterns that are very similar to each other and very different from somatic cells. The first in-depth study to examine the epigenomic differences among the three classes of mouse PSCs found that SCNT ESC global DNA methylation patterns were more similar to ESCs than iPSC patterns were to ESCs131. This suggests the ooplasm is more robust at reprogramming somatic genomes than transcription factor-based reprogramming. Now that human SCNT ESC lines are available132-134, it will be interesting to compare the three human PSC epigenomes and contrast them with data generated from mouse-derived PSC types to determine whether conclusions drawn from this study are applicable to humans.
It is generally accepted that transcription factor-based reprogramming does not completely remodel the epigenome, at least initially. Multiple studies have shown that a certain degree of epigenetic memory is retained in early passage iPSCs, and this can lead to biased differentiation propensities depending on the donor cell source131, 135-137. This somatic memory is attributable to incomplete silencing of donor (source) cell-specific expression patterns. DNA methylation patterns account for the majority of somatic memory131, 138, as histone modification patterns between iPSCs and ESCs exhibit minimal differences139, 140. During reprogramming, large regions of the genome fail to reset non-CpG methylation patterns, and many DMRs are specific to and common across iPSC lines compared to ESCs17. Recently however, it has been demonstrated that these epigenetic memories account for only a small percentage of the variability among iPSCs and ESCs141. In addition, it has been shown that expansion of iPSCs and time in culture diminishes the epigenetic differences between ESCs and iPSCs135, 142. This suggests that iPSC global epigenetic patterns stabilize over time, although certain hotspots of variation remain, particularly at imprinted regions27, 142, 143. A closer examination of global methylation patterns and gene expression profiles reveals that the variability among ESCs and iPSCs is largely due to variations among individual cell lines rather than differences between classes of PSCs27, 141. Genes with the highest variability in expression are genes induced upon differentiation, and genes exhibiting the most epigenetic variability in PSCs are imprinted genes such as H19, PEG3 and MEG327, 141. Furthermore, when DNA methylation patterns and gene expression profiles are considered together, the majority of iPSC lines cannot be distinguished from the majority of ESC lines27,140, 141. Together these studies suggest that the epigenetic heterogeneity observed in iPSC lines is not substantially different from the heterogeneity seen in ESC lines. Thus, when compared to ESC epigenetic patterns established during preimplantation development, it appears that iPSCs display a small amount of epigenetic variation, which can be attributed to reprogramming; however, hiPSCs and hESCs are essentially identical in their epigenetic profiles and their differentiation abilities.
Concluding remarks
We have discussed several of the best-understood epigenetic mechanisms and linked them to current knowledge of pluripotent stem cells and cardiac lineage development wherever possible. We did not touch upon nucleosome remodeling, histone variants, histone replacement or non-coding RNA regulation of chromatin. This is a very fast moving field, so our intention in this review is to provide a general background for appreciating future studies.
Human pluripotent stem cells are an ideal model for studying the dynamic epigenetic changes that accompany the development of cellular diversity. High-resolution global analyses of epigenetic dynamics during hPSC differentiation have greatly advanced our understanding of the mechanisms that regulate aspects of human embryonic development. In the last few years, gene expression signatures have become a routine means to define specific cell types that in the past were identified by histology or expression of a few antigens. Epigenetic technologies hold the promise to characterize cells by their epigenetic landscape, which can give tremendous insight into the functional aspects of cell type specificity. Global epigenetic profiling of heterogeneous cell populations such as those found in tissues have thus far been useful to establish the general themes of epigenetic transcriptional regulation. Such analyses, however, are ultimately limited and epigenetic events important for rare cell types will undoubtedly be missed. Further refinements in cellular differentiation and characterization have permitted the study of nearly homogenous populations of cells. As genome-wide, single cell profiling methods and technologies become more sensitive and inexpensive, there will be far more fine-grained information related to human development and stem cell differentiation.
New discoveries (e.g. 5hmC and its oxidized derivatives) have begun to shed light on some of developmental biology's long-standing questions, such as the epigenomic remodeling events that occur in the earliest stages of development. While epigenetic research has advanced greatly in the last two decades, much remains to be discovered. For example, evidence suggests the existence of epigenetic readers able to simultaneously recognize multiple histone marks144, but we have a limited grasp of the combinatorial nature of histone modifications on the same histone and between histone tails.
The potentially the most powerful epigenomic processes involve changes in genome topology: precise rearrangements of inter- and intra-chromosomal associations that occur in response to developmental or environmental stimuli. Genomic association studies have shown that growth factor induction results in hierarchical gene expression networks mediated by the spatial relocalization of genes145. It will be exciting to examine the role of epigenetic changes in chromosomal interactions that result in gene network activation or repression during differentiation.
Understanding epigenetic regulation of differentiation and cellular identify is critically important for use of hPSCs and their derivatives in clinical applications. Evolving in vitro differentiation protocols increasingly incorporate knowledge about the normal processes of embryogenesis. As protocols are fine-tuned to mimic morphogen gradients and subtle signaling events in the developing embryo, researchers are learning to develop hPSC-derived cell types that are more physiologically similar to their in vivo counterparts. Knowledge of cellular epigenetic states will allow researchers to better develop cell types or combinations of cell types that faithfully represent human cells and tissues. Ultimately this knowledge will lead to tremendous improvements in predictive in vitro assays for drug toxicity and efficacy, which will greatly enhance the effectiveness and safety of clinical trials. As regenerative medicine, in the form of cell replacement therapies, begins to be a viable medical treatment, epigenetic profiling will allow exquisite quality control of cells used for transplantation.
Acknowledgments
Due to the broad nature of this review and to length restrictions, we apologize to those investigators whose work was not cited. We thank the manuscript reviewers for insightful suggestions.
Sources of funding: We thank members of the Loring lab for helpful comments and suggestions. This work was supported by NIH grant 5R33MH087925 and California Institute for Regenerative Medicine grants RM1-01717, CL1-00502, RT1-01108, and TR1-01250 to JFL.
Non-standard Abbreviations and Acronyms
- PSC
Pluripotent stem cells
- ESC
Embryonic stem cells
- iPSC
Induced pluripotent stem cells
- DNMT
DNA methyltransferase
- 5mC
5-methylcytosine
- 5hmC
5-hydroxymethylcytosine
- PcG
Polycomb group
- PRC
Polycomb group complex
- LAD
Lamina associated domains
- TAD
Topologically associating domains
Footnotes
Disclosures: None.
References
- 1.Gaspard N, Bouschet T, Hourez R, Dimidschstein J, Naeije G, van den Ameele J, Espuny-Camacho I, Herpoel A, Passante L, Schiffmann SN, Gaillard A, Vanderhaeghen P. An intrinsic mechanism of corticogenesis from embryonic stem cells. Nature. 2008;455:351–357. doi: 10.1038/nature07287. [DOI] [PubMed] [Google Scholar]
- 2.Shi Y, Kirwan P, Smith J, Robinson HP, Livesey FJ. Human cerebral cortex development from pluripotent stem cells to functional excitatory synapses. Nature neuroscience. 2012;15:477–486. S471. doi: 10.1038/nn.3041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Espuny-Camacho I, Michelsen Kimmo A, Gall D, Linaro D, Hasche A, Bonnefont J, Bali C, Orduz D, Bilheu A, Herpoel A, Lambert N, Gaspard N, Péron S, Schiffmann Serge N, Giugliano M, Gaillard A, Vanderhaeghen P. Pyramidal neurons derived from human pluripotent stem cells integrate efficiently into mouse brain circuits in vivo. Neuron. 2013;77:440–456. doi: 10.1016/j.neuron.2012.12.011. [DOI] [PubMed] [Google Scholar]
- 4.Eiraku M, Takata N, Ishibashi H, Kawada M, Sakakura E, Okuda S, Sekiguchi K, Adachi T, Sasai Y. Self-organizing optic-cup morphogenesis in three-dimensional culture. Nature. 2011;472:51–56. doi: 10.1038/nature09941. [DOI] [PubMed] [Google Scholar]
- 5.Lancaster MA, Renner M, Martin CA, Wenzel D, Bicknell LS, Hurles ME, Homfray T, Penninger JM, Jackson AP, Knoblich JA. Cerebral organoids model human brain development and microcephaly. Nature. 2013;501:373–379. doi: 10.1038/nature12517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Takebe T, Sekine K, Enomura M, Koike H, Kimura M, Ogaeri T, Zhang RR, Ueno Y, Zheng YW, Koike N, Aoyama S, Adachi Y, Taniguchi H. Vascularized and functional human liver from an iPSC-derived organ bud transplant. Nature. 2013;499:481–484. doi: 10.1038/nature12271. [DOI] [PubMed] [Google Scholar]
- 7.Boland MJ, Hazen JL, Nazor KL, Rodriguez AR, Gifford W, Martin G, Kupriyanov S, Baldwin KK. Adult mice generated from induced pluripotent stem cells. Nature. 2009;461:91–94. doi: 10.1038/nature08310. [DOI] [PubMed] [Google Scholar]
- 8.Zhao XY, Li W, Lv Z, Liu L, Tong M, Hai T, Hao J, Guo CL, Ma QW, Wang L, Zeng F, Zhou Q. Ips cells produce viable mice through tetraploid complementation. Nature. 2009;461:86–90. doi: 10.1038/nature08267. [DOI] [PubMed] [Google Scholar]
- 9.Berger SL, Kouzarides T, Shiekhattar R, Shilatifard A. An operational definition of epigenetics. Genes & development. 2009;23:781–783. doi: 10.1101/gad.1787609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bird A. Perceptions of epigenetics. Nature. 2007;447:396–398. doi: 10.1038/nature05913. [DOI] [PubMed] [Google Scholar]
- 11.Christman JK, Price P, Pedrinan L, Acs G. Correlation between hypomethylation of DNA and expression of globin genes in friend erythroleukemia cells. European journal of biochemistry / FEBS. 1977;81:53–61. doi: 10.1111/j.1432-1033.1977.tb11926.x. [DOI] [PubMed] [Google Scholar]
- 12.McGhee JD, Ginder GD. Specific DNA methylation sites in the vicinity of the chicken beta-globin genes. Nature. 1979;280:419–420. doi: 10.1038/280419a0. [DOI] [PubMed] [Google Scholar]
- 13.Jones PA, Taylor SM. Cellular differentiation, cytidine analogs and DNA methylation. Cell. 1980;20:85–93. doi: 10.1016/0092-8674(80)90237-8. [DOI] [PubMed] [Google Scholar]
- 14.Jenuwein T, Allis CD. Translating the histone code. Science (New York, NY) 2001;293:1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 15.Taverna SD, Li H, Ruthenburg AJ, Allis CD, Patel DJ. How chromatin-binding modules interpret histone modifications: Lessons from professional pocket pickers. Nature structural & molecular biology. 2007;14:1025–1040. doi: 10.1038/nsmb1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Christman JK. 5-azacytidine and 5-aza-2′-deoxycytidine as inhibitors of DNA methylation: Mechanistic studies and their implications for cancer therapy. Oncogene. 2002;21:5483–5495. doi: 10.1038/sj.onc.1205699. [DOI] [PubMed] [Google Scholar]
- 17.Lister R, Pelizzola M, Kida YS, Hawkins RD, Nery JR, Hon G, Antosiewicz-Bourget J, O'Malley R, Castanon R, Klugman S, Downes M, Yu R, Stewart R, Ren B, Thomson JA, Evans RM, Ecker JR. Hotspots of aberrant epigenomic reprogramming in human induced pluripotent stem cells. Nature. 2011;471:68–73. doi: 10.1038/nature09798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xie W, Barr Cathy L, Kim A, Yue F, Lee Ah Y, Eubanks J, Dempster Emma L, Ren B. Base-resolution analyses of sequence and parent-of-origin dependent DNA methylation in the mouse genome. Cell. 2012;148:816–831. doi: 10.1016/j.cell.2011.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wienholz BL, Kareta MS, Moarefi AH, Gordon CA, Ginno PA, Chedin F. Dnmt3l modulates significant and distinct flanking sequence preference for DNA methylation by dnmt3a and dnmt3b in vivo. PLoS genetics. 2010;6:e1001106. doi: 10.1371/journal.pgen.1001106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li E. Chromatin modification and epigenetic reprogramming in mammalian development. Nature reviews. Genetics. 2002;3:662–673. doi: 10.1038/nrg887. [DOI] [PubMed] [Google Scholar]
- 21.Tsumura A, Hayakawa T, Kumaki Y, Takebayashi S, Sakaue M, Matsuoka C, Shimotohno K, Ishikawa F, Li E, Ueda HR, Nakayama J, Okano M. Maintenance of self-renewal ability of mouse embryonic stem cells in the absence of DNA methyltransferases dnmt1, dnmt3a and dnmt3b. Genes to cells : devoted to molecular & cellular mechanisms. 2006;11:805–814. doi: 10.1111/j.1365-2443.2006.00984.x. [DOI] [PubMed] [Google Scholar]
- 22.Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases dnmt3a and dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99:247–257. doi: 10.1016/s0092-8674(00)81656-6. [DOI] [PubMed] [Google Scholar]
- 23.Li E, Bestor TH, Jaenisch R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell. 1992;69:915–926. doi: 10.1016/0092-8674(92)90611-f. [DOI] [PubMed] [Google Scholar]
- 24.Klose RJ, Bird AP. Genomic DNA methylation: The mark and its mediators. Trends in Biochemical Sciences. 2006;31:89–97. doi: 10.1016/j.tibs.2005.12.008. [DOI] [PubMed] [Google Scholar]
- 25.Fouse SD, Shen Y, Pellegrini M, Cole S, Meissner A, Van Neste L, Jaenisch R, Fan G. Promoter cpg methylation contributes to es cell gene regulation in parallel with oct4/nanog, pcg complex, and histone h3 k4/k27 trimethylation. Cell stem cell. 2008;2:160–169. doi: 10.1016/j.stem.2007.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mikkelsen TS, Ku M, Jaffe DB, Issac B, Lieberman E, Giannoukos G, Alvarez P, Brockman W, Kim TK, Koche RP, Lee W, Mendenhall E, O'Donovan A, Presser A, Russ C, Xie X, Meissner A, Wernig M, Jaenisch R, Nusbaum C, Lander ES, Bernstein BE. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature. 2007;448:553–560. doi: 10.1038/nature06008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nazor KL, Altun G, Lynch C, Tran H, Harness JV, Slavin I, Garitaonandia I, Muller FJ, Wang YC, Boscolo FS, Fakunle E, Dumevska B, Lee S, Park HS, Olee T, D'Lima DD, Semechkin R, Parast MM, Galat V, Laslett AL, Schmidt U, Keirstead HS, Loring JF, Laurent LC. Recurrent variations in DNA methylation in human pluripotent stem cells and their differentiated derivatives. Cell stem cell. 2012;10:620–634. doi: 10.1016/j.stem.2012.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, Fry B, Meissner A, Wernig M, Plath K, Jaenisch R, Wagschal A, Feil R, Schreiber SL, Lander ES. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006;125:315–326. doi: 10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]
- 29.Laurent L, Wong E, Li G, Huynh T, Tsirigos A, Ong CT, Low HM, Kin Sung KW, Rigoutsos I, Loring J, Wei CL. Dynamic changes in the human methylome during differentiation. Genome research. 2010;20:320–331. doi: 10.1101/gr.101907.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ball MP, Li JB, Gao Y, Lee JH, LeProust EM, Park IH, Xie B, Daley GQ, Church GM. Targeted and genome-scale strategies reveal gene-body methylation signatures in human cells. Nature biotechnology. 2009;27:361–368. doi: 10.1038/nbt.1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bibikova M, Chudin E, Wu B, Zhou L, Garcia EW, Liu Y, Shin S, Plaia TW, Auerbach JM, Arking DE, Gonzalez R, Crook J, Davidson B, Schulz TC, Robins A, Khanna A, Sartipy P, Hyllner J, Vanguri P, Savant-Bhonsale S, Smith AK, Chakravarti A, Maitra A, Rao M, Barker DL, Loring JF, Fan JB. Human embryonic stem cells have a unique epigenetic signature. Genome research. 2006;16:1075–1083. doi: 10.1101/gr.5319906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Abbey D, Seshagiri PB. Aza-induced cardiomyocyte differentiation of p19 ec-cells by epigenetic co-regulation and erk signaling. Gene. 2013;526:364–373. doi: 10.1016/j.gene.2013.05.044. [DOI] [PubMed] [Google Scholar]
- 33.Gu Y, Liu GH, Plongthongkum N, Benner C, Yi F, Qu J, Suzuki K, Yang J, Zhang W, Li M, Montserrat N, Crespo I, Del Sol A, Esteban CR, Zhang K, Izpisua Belmonte JC. Global DNA methylation and transcriptional analyses of human esc-derived cardiomyocytes. Protein & cell. 2014;5:59–68. doi: 10.1007/s13238-013-0016-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lister R, Mukamel EA, Nery JR, Urich M, Puddifoot CA, Johnson ND, Lucero J, Huang Y, Dwork AJ, Schultz MD, Yu M, Tonti-Filippini J, Heyn H, Hu S, Wu JC, Rao A, Esteller M, He C, Haghighi FG, Sejnowski TJ, Behrens MM, Ecker JR. Global epigenomic reconfiguration during mammalian brain development. Science (New York, NY) 2013;341:1237905. doi: 10.1126/science.1237905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, Tonti-Filippini J, Nery JR, Lee L, Ye Z, Ngo QM, Edsall L, Antosiewicz-Bourget J, Stewart R, Ruotti V, Millar AH, Thomson JA, Ren B, Ecker JR. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462:315–322. doi: 10.1038/nature08514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gowher H, Jeltsch A. Enzymatic properties of recombinant dnmt3a DNA methyltransferase from mouse: The enzyme modifies DNA in a non-processive manner and also methylates non-cpg [correction of non-cpa] sites. Journal of molecular biology. 2001;309:1201–1208. doi: 10.1006/jmbi.2001.4710. [DOI] [PubMed] [Google Scholar]
- 37.Ramsahoye BH, Biniszkiewicz D, Lyko F, Clark V, Bird AP, Jaenisch R. Non-cpg methylation is prevalent in embryonic stem cells and may be mediated by DNA methyltransferase 3a. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:5237–5242. doi: 10.1073/pnas.97.10.5237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guo Junjie U, Su Y, Zhong C, Ming Gl, Song H. Hydroxylation of 5-methylcytosine by tet1 promotes active DNA demethylation in the adult brain. Cell. 2011;145:423–434. doi: 10.1016/j.cell.2011.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, Agarwal S, Iyer LM, Liu DR, Aravind L, Rao A. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by mll partner tet1. Science (New York, NY) 2009;324:930–935. doi: 10.1126/science.1170116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kriaucionis S, Heintz N. The nuclear DNA base 5-hydroxymethylcytosine is present in purkinje neurons and the brain. Science (New York, NY) 2009;324:929–930. doi: 10.1126/science.1169786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pastor WA, Pape UJ, Huang Y, Henderson HR, Lister R, Ko M, McLoughlin EM, Brudno Y, Mahapatra S, Kapranov P, Tahiliani M, Daley GQ, Liu XS, Ecker JR, Milos PM, Agarwal S, Rao A. Genome-wide mapping of 5-hydroxymethylcytosine in embryonic stem cells. Nature. 2011;473:394–397. doi: 10.1038/nature10102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Szulwach KE, Li X, Li Y, Song CX, Han JW, Kim S, Namburi S, Hermetz K, Kim JJ, Rudd MK, Yoon YS, Ren B, He C, Jin P. Integrating 5-hydroxymethylcytosine into the epigenomic landscape of human embryonic stem cells. PLoS genetics. 2011;7:e1002154. doi: 10.1371/journal.pgen.1002154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wu H, D'Alessio AC, Ito S, Wang Z, Cui K, Zhao K, Sun YE, Zhang Y. Genome-wide analysis of 5-hydroxymethylcytosine distribution reveals its dual function in transcriptional regulation in mouse embryonic stem cells. Genes & development. 2011;25:679–684. doi: 10.1101/gad.2036011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Szulwach KE, Li X, Li Y, Song CX, Wu H, Dai Q, Irier H, Upadhyay AK, Gearing M, Levey AI, Vasanthakumar A, Godley LA, Chang Q, Cheng X, He C, Jin P. 5-hmc-mediated epigenetic dynamics during postnatal neurodevelopment and aging. Nature neuroscience. 2011;14:1607–1616. doi: 10.1038/nn.2959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wu H, Coskun V, Tao J, Xie W, Ge W, Yoshikawa K, Li E, Zhang Y, Sun YE. Dnmt3a-dependent nonpromoter DNA methylation facilitates transcription of neurogenic genes. Science (New York, NY) 2010;329:444–448. doi: 10.1126/science.1190485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dawlaty MM, Ganz K, Powell BE, Hu YC, Markoulaki S, Cheng AW, Gao Q, Kim J, Choi SW, Page DC, Jaenisch R. Tet1 is dispensable for maintaining pluripotency and its loss is compatible with embryonic and postnatal development. Cell stem cell. 2011;9:166–175. doi: 10.1016/j.stem.2011.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Koh KP, Yabuuchi A, Rao S, Huang Y, Cunniff K, Nardone J, Laiho A, Tahiliani M, Sommer CA, Mostoslavsky G, Lahesmaa R, Orkin SH, Rodig SJ, Daley GQ, Rao A. Tet1 and tet2 regulate 5-hydroxymethylcytosine production and cell lineage specification in mouse embryonic stem cells. Cell stem cell. 2011;8:200–213. doi: 10.1016/j.stem.2011.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ko M, Bandukwala HS, An J, Lamperti ED, Thompson EC, Hastie R, Tsangaratou A, Rajewsky K, Koralov SB, Rao A. Ten-eleven-translocation 2 (tet2) negatively regulates homeostasis and differentiation of hematopoietic stem cells in mice. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:14566–14571. doi: 10.1073/pnas.1112317108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Quivoron C, Couronne L, Della Valle V, Lopez CK, Plo I, Wagner-Ballon O, Do Cruzeiro M, Delhommeau F, Arnulf B, Stern MH, Godley L, Opolon P, Tilly H, Solary E, Duffourd Y, Dessen P, Merle-Beral H, Nguyen-Khac F, Fontenay M, Vainchenker W, Bastard C, Mercher T, Bernard OA. Tet2 inactivation results in pleiotropic hematopoietic abnormalities in mouse and is a recurrent event during human lymphomagenesis. Cancer cell. 2011;20:25–38. doi: 10.1016/j.ccr.2011.06.003. [DOI] [PubMed] [Google Scholar]
- 50.Dawlaty MM, Breiling A, Le T, Raddatz G, Barrasa MI, Cheng AW, Gao Q, Powell BE, Li Z, Xu M, Faull KF, Lyko F, Jaenisch R. Combined deficiency of tet1 and tet2 causes epigenetic abnormalities but is compatible with postnatal development. Developmental cell. 2013;24:310–323. doi: 10.1016/j.devcel.2012.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Spruijt Cornelia G, Gnerlich F, Smits Arne H, Pfaffeneder T, Jansen Pascal WTC, Bauer C, Münzel M, Wagner M, Müller M, Khan F, Eberl HC, Mensinga A, Brinkman Arie B, Lephikov K, Müller U, Walter J, Boelens R, van Ingen H, Leonhardt H, Carell T, Vermeulen M. Dynamic readers for 5-(hydroxy)methylcytosine and its oxidized derivatives. Cell. 2013;152:1146–1159. doi: 10.1016/j.cell.2013.02.004. [DOI] [PubMed] [Google Scholar]
- 52.Hashimoto H, Liu Y, Upadhyay AK, Chang Y, Howerton SB, Vertino PM, Zhang X, Cheng X. Recognition and potential mechanisms for replication and erasure of cytosine hydroxymethylation. Nucleic acids research. 2012;40:4841–4849. doi: 10.1093/nar/gks155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jin SG, Kadam S, Pfeifer GP. Examination of the specificity of DNA methylation profiling techniques towards 5-methylcytosine and 5-hydroxymethylcytosine. Nucleic acids research. 2010;38:e125. doi: 10.1093/nar/gkq223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Valinluck V, Tsai HH, Rogstad DK, Burdzy A, Bird A, Sowers LC. Oxidative damage to methyl-cpg sequences inhibits the binding of the methyl-cpg binding domain (mbd) of methyl-cpg binding protein 2 (mecp2) Nucleic acids research. 2004;32:4100–4108. doi: 10.1093/nar/gkh739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yildirim O, Li R, Hung JH, Chen Poshen B, Dong X, Ee LS, Weng Z, Rando Oliver J, Fazzio Thomas G. Mbd3/nurd complex regulates expression of 5-hydroxymethylcytosine marked genes in embryonic stem cells. Cell. 2011;147:1498–1510. doi: 10.1016/j.cell.2011.11.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mellen M, Ayata P, Dewell S, Kriaucionis S, Heintz N. Mecp2 binds to 5hmc enriched within active genes and accessible chromatin in the nervous system. Cell. 2012;151:1417–1430. doi: 10.1016/j.cell.2012.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mayer W, Niveleau A, Walter J, Fundele R, Haaf T. Demethylation of the zygotic paternal genome. Nature. 2000;403:501–502. doi: 10.1038/35000656. [DOI] [PubMed] [Google Scholar]
- 58.Oswald J, Engemann S, Lane N, Mayer W, Olek A, Fundele R, Dean W, Reik W, Walter J. Active demethylation of the paternal genome in the mouse zygote. Current biology : CB. 2000;10:475–478. doi: 10.1016/s0960-9822(00)00448-6. [DOI] [PubMed] [Google Scholar]
- 59.Kaas Garrett A, Zhong C, Eason Dawn E, Ross Daniel L, Vachhani Raj V, Ming Gl, King Jennifer R, Song H, Sweatt JD. Tet1 controls cns 5-methylcytosine hydroxylation, active DNA demethylation, gene transcription, and memory formation. Neuron. 2013;79:1086–1093. doi: 10.1016/j.neuron.2013.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ma DK, Jang MH, Guo JU, Kitabatake Y, Chang ML, Pow-Anpongkul N, Flavell RA, Lu B, Ming GL, Song H. Neuronal activity-induced gadd45b promotes epigenetic DNA demethylation and adult neurogenesis. Science (New York, NY) 2009;323:1074–1077. doi: 10.1126/science.1166859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rudenko A, Dawlaty MM, Seo J, Cheng AW, Meng J, Le T, Faull KF, Jaenisch R, Tsai LH. Tet1 is critical for neuronal activity-regulated gene expression and memory extinction. Neuron. 2013;79:1109–1122. doi: 10.1016/j.neuron.2013.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gehring M, Reik W, Henikoff S. DNA demethylation by DNA repair. Trends in genetics : TIG. 2009;25:82–90. doi: 10.1016/j.tig.2008.12.001. [DOI] [PubMed] [Google Scholar]
- 63.Bhutani N, Burns DM, Blau HM. DNA demethylation dynamics. Cell. 2011;146:866–872. doi: 10.1016/j.cell.2011.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sjolund AB, Senejani AG, Sweasy JB. Mbd4 and tdg: Multifaceted DNA glycosylases with ever expanding biological roles. Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis. 2013:743–744. 12–25. doi: 10.1016/j.mrfmmm.2012.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cortellino S, Xu J, Sannai M, Moore R, Caretti E, Cigliano A, Le Coz M, Devarajan K, Wessels A, Soprano D, Abramowitz LK, Bartolomei MS, Rambow F, Bassi MR, Bruno T, Fanciulli M, Renner C, Klein-Szanto AJ, Matsumoto Y, Kobi D, Davidson I, Alberti C, Larue L, Bellacosa A. Thymine DNA glycosylase is essential for active DNA demethylation by linked deamination-base excision repair. Cell. 2011;146:67–79. doi: 10.1016/j.cell.2011.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Metivier R, Gallais R, Tiffoche C, Le Peron C, Jurkowska RZ, Carmouche RP, Ibberson D, Barath P, Demay F, Reid G, Benes V, Jeltsch A, Gannon F, Salbert G. Cyclical DNA methylation of a transcriptionally active promoter. Nature. 2008;452:45–50. doi: 10.1038/nature06544. [DOI] [PubMed] [Google Scholar]
- 67.Bhutani N, Brady JJ, Damian M, Sacco A, Corbel SY, Blau HM. Reprogramming towards pluripotency requires aid-dependent DNA demethylation. Nature. 2010;463:1042–1047. doi: 10.1038/nature08752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nabel CS, Jia H, Ye Y, Shen L, Goldschmidt HL, Stivers JT, Zhang Y, Kohli RM. Aid/apobec deaminases disfavor modified cytosines implicated in DNA demethylation. Nature chemical biology. 2012;8:751–758. doi: 10.1038/nchembio.1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Niehrs C, Schafer A. Active DNA demethylation by gadd45 and DNA repair. Trends in cell biology. 2012;22:220–227. doi: 10.1016/j.tcb.2012.01.002. [DOI] [PubMed] [Google Scholar]
- 70.Ito S, Shen L, Dai Q, Wu SC, Collins LB, Swenberg JA, He C, Zhang Y. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science (New York, NY) 2011;333:1300–1303. doi: 10.1126/science.1210597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.He YF, Li BZ, Li Z, Liu P, Wang Y, Tang Q, Ding J, Jia Y, Chen Z, Li L, Sun Y, Li X, Dai Q, Song CX, Zhang K, He C, Xu GL. Tet-mediated formation of 5-carboxylcytosine and its excision by tdg in mammalian DNA. Science (New York, NY) 2011;333:1303–1307. doi: 10.1126/science.1210944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Maiti A, Drohat AC. Thymine DNA glycosylase can rapidly excise 5-formylcytosine and 5-carboxylcytosine: Potential implications for active demethylation of cpg sites. The Journal of biological chemistry. 2011;286:35334–35338. doi: 10.1074/jbc.C111.284620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Shen L, Wu H, Diep D, Yamaguchi S, D'Alessio AC, Fung HL, Zhang K, Zhang Y. Genome-wide analysis reveals tet- and tdg-dependent 5-methylcytosine oxidation dynamics. Cell. 2013;153:692–706. doi: 10.1016/j.cell.2013.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Song CX, Szulwach KE, Dai Q, Fu Y, Mao SQ, Lin L, Street C, Li Y, Poidevin M, Wu H, Gao J, Liu P, Li L, Xu GL, Jin P, He C. Genome-wide profiling of 5-formylcytosine reveals its roles in epigenetic priming. Cell. 2013;153:678–691. doi: 10.1016/j.cell.2013.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gu TP, Guo F, Yang H, Wu HP, Xu GF, Liu W, Xie ZG, Shi L, He X, Jin SG, Iqbal K, Shi YG, Deng Z, Szabo PE, Pfeifer GP, Li J, Xu GL. The role of tet3 DNA dioxygenase in epigenetic reprogramming by oocytes. Nature. 2011;477:606–610. doi: 10.1038/nature10443. [DOI] [PubMed] [Google Scholar]
- 76.Iqbal K, Jin SG, Pfeifer GP, Szabo PE. Reprogramming of the paternal genome upon fertilization involves genome-wide oxidation of 5-methylcytosine. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:3642–3647. doi: 10.1073/pnas.1014033108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wossidlo M, Nakamura T, Lepikhov K, Marques CJ, Zakhartchenko V, Boiani M, Arand J, Nakano T, Reik W, Walter J. 5-hydroxymethylcytosine in the mammalian zygote is linked with epigenetic reprogramming. Nature communications. 2011;2:241. doi: 10.1038/ncomms1240. [DOI] [PubMed] [Google Scholar]
- 78.Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell research. 2011;21:381–395. doi: 10.1038/cr.2011.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Christophorou MA, Castelo-Branco G, Halley-Stott RP, Oliveira CS, Loos R, Radzisheuskaya A, Mowen KA, Bertone P, Silva JC, Zernicka-Goetz M, Nielsen ML, Gurdon JB, Kouzarides T. Citrullination regulates pluripotency and histone h1 binding to chromatin. Nature. 2014;507:104–108. doi: 10.1038/nature12942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ernst J, Kheradpour P, Mikkelsen TS, Shoresh N, Ward LD, Epstein CB, Zhang X, Wang L, Issner R, Coyne M, Ku M, Durham T, Kellis M, Bernstein BE. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature. 2011;473:43–49. doi: 10.1038/nature09906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Aloia L, Di Stefano B, Di Croce L. Polycomb complexes in stem cells and embryonic development. Development. 2013;140:2525–2534. doi: 10.1242/dev.091553. [DOI] [PubMed] [Google Scholar]
- 82.Leeb M, Pasini D, Novatchkova M, Jaritz M, Helin K, Wutz A. Polycomb complexes act redundantly to repress genomic repeats and genes. Genes & development. 2010;24:265–276. doi: 10.1101/gad.544410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Leeb M, Wutz A. Ring1b is crucial for the regulation of developmental control genes and prc1 proteins but not x inactivation in embryonic cells. The Journal of cell biology. 2007;178:219–229. doi: 10.1083/jcb.200612127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Klattenhoff CA, Scheuermann JC, Surface LE, Bradley RK, Fields PA, Steinhauser ML, Ding H, Butty VL, Torrey L, Haas S, Abo R, Tabebordbar M, Lee RT, Burge CB, Boyer LA. Braveheart, a long noncoding rna required for cardiovascular lineage commitment. Cell. 2013;152:570–583. doi: 10.1016/j.cell.2013.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Li X, Li L, Pandey R, Byun JS, Gardner K, Qin Z, Dou Y. The histone acetyltransferase mof is a key regulator of the embryonic stem cell core transcriptional network. Cell stem cell. 2012;11:163–178. doi: 10.1016/j.stem.2012.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ku M, Koche RP, Rheinbay E, Mendenhall EM, Endoh M, Mikkelsen TS, Presser A, Nusbaum C, Xie X, Chi AS, Adli M, Kasif S, Ptaszek LM, Cowan CA, Lander ES, Koseki H, Bernstein BE. Genomewide analysis of prc1 and prc2 occupancy identifies two classes of bivalent domains. PLoS genetics. 2008;4:e1000242. doi: 10.1371/journal.pgen.1000242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Barrera LO, Li Z, Smith AD, Arden KC, Cavenee WK, Zhang MQ, Green RD, Ren B. Genome-wide mapping and analysis of active promoters in mouse embryonic stem cells and adult organs. Genome research. 2008;18:46–59. doi: 10.1101/gr.6654808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Xie W, Schultz MD, Lister R, Hou Z, Rajagopal N, Ray P, Whitaker JW, Tian S, Hawkins RD, Leung D, Yang H, Wang T, Lee AY, Swanson SA, Zhang J, Zhu Y, Kim A, Nery JR, Urich MA, Kuan S, Yen CA, Klugman S, Yu P, Suknuntha K, Propson NE, Chen H, Edsall LE, Wagner U, Li Y, Ye Z, Kulkarni A, Xuan Z, Chung WY, Chi NC, Antosiewicz-Bourget JE, Slukvin I, Stewart R, Zhang MQ, Wang W, Thomson JA, Ecker JR, Ren B. Epigenomic analysis of multilineage differentiation of human embryonic stem cells. Cell. 2013;153:1134–1148. doi: 10.1016/j.cell.2013.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Consortium EP, Bernstein BE, Birney E, Dunham I, Green ED, Gunter C, Snyder M. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Heintzman ND, Stuart RK, Hon G, Fu Y, Ching CW, Hawkins RD, Barrera LO, Van Calcar S, Qu C, Ching KA, Wang W, Weng Z, Green RD, Crawford GE, Ren B. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nature genetics. 2007;39:311–318. doi: 10.1038/ng1966. [DOI] [PubMed] [Google Scholar]
- 91.Kornberg RD. Mediator and the mechanism of transcriptional activation. Trends Biochem Sci. 2005;30:235–239. doi: 10.1016/j.tibs.2005.03.011. [DOI] [PubMed] [Google Scholar]
- 92.Chen X, Xu H, Yuan P, Fang F, Huss M, Vega VB, Wong E, Orlov YL, Zhang W, Jiang J, Loh YH, Yeo HC, Yeo ZX, Narang V, Govindarajan KR, Leong B, Shahab A, Ruan Y, Bourque G, Sung WK, Clarke ND, Wei CL, Ng HH. Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell. 2008;133:1106–1117. doi: 10.1016/j.cell.2008.04.043. [DOI] [PubMed] [Google Scholar]
- 93.Kagey MH, Newman JJ, Bilodeau S, Zhan Y, Orlando DA, van Berkum NL, Ebmeier CC, Goossens J, Rahl PB, Levine SS, Taatjes DJ, Dekker J, Young RA. Mediator and cohesin connect gene expression and chromatin architecture. Nature. 2010;467:430–435. doi: 10.1038/nature09380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Heintzman ND, Hon GC, Hawkins RD, Kheradpour P, Stark A, Harp LF, Ye Z, Lee LK, Stuart RK, Ching CW, Ching KA, Antosiewicz-Bourget JE, Liu H, Zhang X, Green RD, Lobanenkov VV, Stewart R, Thomson JA, Crawford GE, Kellis M, Ren B. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature. 2009;459:108–112. doi: 10.1038/nature07829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Rada-Iglesias A, Bajpai R, Swigut T, Brugmann SA, Flynn RA, Wysocka J. A unique chromatin signature uncovers early developmental enhancers in humans. Nature. 2011;470:279–283. doi: 10.1038/nature09692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Gifford CA, Ziller MJ, Gu H, Trapnell C, Donaghey J, Tsankov A, Shalek AK, Kelley DR, Shishkin AA, Issner R, Zhang X, Coyne M, Fostel JL, Holmes L, Meldrim J, Guttman M, Epstein C, Park H, Kohlbacher O, Rinn J, Gnirke A, Lander ES, Bernstein BE, Meissner A. Transcriptional and epigenetic dynamics during specification of human embryonic stem cells. Cell. 2013;153:1149–1163. doi: 10.1016/j.cell.2013.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hnisz D, Abraham BJ, Lee TI, Lau A, Saint-Andre V, Sigova AA, Hoke HA, Young RA. Super-enhancers in the control of cell identity and disease. Cell. 2013;155:934–947. doi: 10.1016/j.cell.2013.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, Rahl PB, Lee TI, Young RA. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 2013;153:307–319. doi: 10.1016/j.cell.2013.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hawkins RD, Hon GC, Lee LK, Ngo Q, Lister R, Pelizzola M, Edsall LE, Kuan S, Luu Y, Klugman S, Antosiewicz-Bourget J, Ye Z, Espinoza C, Agarwahl S, Shen L, Ruotti V, Wang W, Stewart R, Thomson JA, Ecker JR, Ren B. Distinct epigenomic landscapes of pluripotent and lineage-committed human cells. Cell stem cell. 2010;6:479–491. doi: 10.1016/j.stem.2010.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zhu J, Adli M, Zou JY, Verstappen G, Coyne M, Zhang X, Durham T, Miri M, Deshpande V, De Jager PL, Bennett DA, Houmard JA, Muoio DM, Onder TT, Camahort R, Cowan CA, Meissner A, Epstein CB, Shoresh N, Bernstein BE. Genome-wide chromatin state transitions associated with developmental and environmental cues. Cell. 2013;152:642–654. doi: 10.1016/j.cell.2012.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Paige SL, Thomas S, Stoick-Cooper CL, Wang H, Maves L, Sandstrom R, Pabon L, Reinecke H, Pratt G, Keller G, Moon RT, Stamatoyannopoulos J, Murry CE. A temporal chromatin signature in human embryonic stem cells identifies regulators of cardiac development. Cell. 2012;151:221–232. doi: 10.1016/j.cell.2012.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Stergachis AB, Neph S, Reynolds A, Humbert R, Miller B, Paige SL, Vernot B, Cheng JB, Thurman RE, Sandstrom R, Haugen E, Heimfeld S, Murry CE, Akey JM, Stamatoyannopoulos JA. Developmental fate and cellular maturity encoded in human regulatory DNA landscapes. Cell. 2013;154:888–903. doi: 10.1016/j.cell.2013.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Delgado-Olguin P, Huang Y, Li X, Christodoulou D, Seidman CE, Seidman JG, Tarakhovsky A, Bruneau BG. Epigenetic repression of cardiac progenitor gene expression by ezh2 is required for postnatal cardiac homeostasis. Nature genetics. 2012;44:343–347. doi: 10.1038/ng.1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wamstad JA, Alexander JM, Truty RM, Shrikumar A, Li F, Eilertson KE, Ding H, Wylie JN, Pico AR, Capra JA, Erwin G, Kattman SJ, Keller GM, Srivastava D, Levine SS, Pollard KS, Holloway AK, Boyer LA, Bruneau BG. Dynamic and coordinated epigenetic regulation of developmental transitions in the cardiac lineage. Cell. 2012;151:206–220. doi: 10.1016/j.cell.2012.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zhang JA, Mortazavi A, Williams BA, Wold BJ, Rothenberg EV. Dynamic transformations of genome-wide epigenetic marking and transcriptional control establish t cell identity. Cell. 2012;149:467–482. doi: 10.1016/j.cell.2012.01.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Felsenfeld G, Groudine M. Controlling the double helix. Nature. 2003;421:448–453. doi: 10.1038/nature01411. [DOI] [PubMed] [Google Scholar]
- 107.de Wit E, de Laat W. A decade of 3c technologies: Insights into nuclear organization. Genes & development. 2012;26:11–24. doi: 10.1101/gad.179804.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Bickmore Wendy A, van Steensel B. Genome architecture: Domain organization of interphase chromosomes. Cell. 2013;152:1270–1284. doi: 10.1016/j.cell.2013.02.001. [DOI] [PubMed] [Google Scholar]
- 109.Meshorer E, Misteli T. Chromatin in pluripotent embryonic stem cells and differentiation. Nature reviews. Molecular cell biology. 2006;7:540–546. doi: 10.1038/nrm1938. [DOI] [PubMed] [Google Scholar]
- 110.Guelen L, Pagie L, Brasset E, Meuleman W, Faza MB, Talhout W, Eussen BH, de Klein A, Wessels L, de Laat W, van Steensel B. Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions. Nature. 2008;453:948–951. doi: 10.1038/nature06947. [DOI] [PubMed] [Google Scholar]
- 111.Peric-Hupkes D, Meuleman W, Pagie L, Bruggeman SW, Solovei I, Brugman W, Graf S, Flicek P, Kerkhoven RM, van Lohuizen M, Reinders M, Wessels L, van Steensel B. Molecular maps of the reorganization of genome-nuclear lamina interactions during differentiation. Molecular cell. 2010;38:603–613. doi: 10.1016/j.molcel.2010.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS, Ren B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012;485:376–380. doi: 10.1038/nature11082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Handoko L, Xu H, Li G, Ngan CY, Chew E, Schnapp M, Lee CW, Ye C, Ping JL, Mulawadi F, Wong E, Sheng J, Zhang Y, Poh T, Chan CS, Kunarso G, Shahab A, Bourque G, Cacheux-Rataboul V, Sung WK, Ruan Y, Wei CL. Ctcf-mediated functional chromatin interactome in pluripotent cells. Nature genetics. 2011;43:630–638. doi: 10.1038/ng.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kim YJ, Cecchini KR, Kim TH. Conserved, developmentally regulated mechanism couples chromosomal looping and heterochromatin barrier activity at the homeobox gene a locus. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:7391–7396. doi: 10.1073/pnas.1018279108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Donohoe ME, Silva SS, Pinter SF, Xu N, Lee JT. The pluripotency factor oct4 interacts with ctcf and also controls x-chromosome pairing and counting. Nature. 2009;460:128–132. doi: 10.1038/nature08098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Jin F, Li Y, Dixon JR, Selvaraj S, Ye Z, Lee AY, Yen CA, Schmitt AD, Espinoza CA, Ren B. A high-resolution map of the three-dimensional chromatin interactome in human cells. Nature. 2013;503:290–294. doi: 10.1038/nature12644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.de Wit E, Bouwman BA, Zhu Y, Klous P, Splinter E, Verstegen MJ, Krijger PH, Festuccia N, Nora EP, Welling M, Heard E, Geijsen N, Poot RA, Chambers I, de Laat W. The pluripotent genome in three dimensions is shaped around pluripotency factors. Nature. 2013;501:227–231. doi: 10.1038/nature12420. [DOI] [PubMed] [Google Scholar]
- 118.Kieffer-Kwon KR, Tang Z, Mathe E, Qian J, Sung MH, Li G, Resch W, Baek S, Pruett N, Grontved L, Vian L, Nelson S, Zare H, Hakim O, Reyon D, Yamane A, Nakahashi H, Kovalchuk AL, Zou J, Joung JK, Sartorelli V, Wei CL, Ruan X, Hager GL, Ruan Y, Casellas R. Interactome maps of mouse gene regulatory domains reveal basic principles of transcriptional regulation. Cell. 2013;155:1507–1520. doi: 10.1016/j.cell.2013.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Zhang Y, Wong CH, Birnbaum RY, Li G, Favaro R, Ngan CY, Lim J, Tai E, Poh HM, Wong E, Mulawadi FH, Sung WK, Nicolis S, Ahituv N, Ruan Y, Wei CL. Chromatin connectivity maps reveal dynamic promoter-enhancer long-range associations. Nature. 2013;504:306–310. doi: 10.1038/nature12716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Reik W, Walter J. Genomic imprinting: Parental influence on the genome. Nature reviews. Genetics. 2001;2:21–32. doi: 10.1038/35047554. [DOI] [PubMed] [Google Scholar]
- 121.Gregg C, Zhang J, Weissbourd B, Luo S, Schroth GP, Haig D, Dulac C. High-resolution analysis of parent-of-origin allelic expression in the mouse brain. Science (New York, NY) 2010;329:643–648. doi: 10.1126/science.1190830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.DeVeale B, van der Kooy D, Babak T. Critical evaluation of imprinted gene expression by rna–seq: A new perspective. PLoS genetics. 2012;8:e1002600. doi: 10.1371/journal.pgen.1002600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Millan MJ. An epigenetic framework for neurodevelopmental disorders: From pathogenesis to potential therapy. Neuropharmacology. 2013;68:2–82. doi: 10.1016/j.neuropharm.2012.11.015. [DOI] [PubMed] [Google Scholar]
- 124.Mekhoubad S, Bock C, de Boer AS, Kiskinis E, Meissner A, Eggan K. Erosion of dosage compensation impacts human iPSC disease modeling. Cell stem cell. 2012;10:595–609. doi: 10.1016/j.stem.2012.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Shen Y, Matsuno Y, Fouse SD, Rao N, Root S, Xu R, Pellegrini M, Riggs AD, Fan G. X-inactivation in female human embryonic stem cells is in a nonrandom pattern and prone to epigenetic alterations. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:4709–4714. doi: 10.1073/pnas.0712018105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Papp B, Plath K. Epigenetics of reprogramming to induced pluripotency. Cell. 2013;152:1324–1343. doi: 10.1016/j.cell.2013.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Mikkelsen TS, Hanna J, Zhang X, Ku M, Wernig M, Schorderet P, Bernstein BE, Jaenisch R, Lander ES, Meissner A. Dissecting direct reprogramming through integrative genomic analysis. Nature. 2008;454:49–55. doi: 10.1038/nature07056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Rais Y, Zviran A, Geula S, Gafni O, Chomsky E, Viukov S, Mansour AA, Caspi I, Krupalnik V, Zerbib M, Maza I, Mor N, Baran D, Weinberger L, Jaitin DA, Lara-Astiaso D, Blecher-Gonen R, Shipony Z, Mukamel Z, Hagai T, Gilad S, Amann-Zalcenstein D, Tanay A, Amit I, Novershtern N, Hanna JH. Deterministic direct reprogramming of somatic cells to pluripotency. Nature. 2013;502:65–70. doi: 10.1038/nature12587. [DOI] [PubMed] [Google Scholar]
- 129.Huangfu D, Maehr R, Guo W, Eijkelenboom A, Snitow M, Chen AE, Melton DA. Induction of pluripotent stem cells by defined factors is greatly improved by small-molecule compounds. Nature biotechnology. 2008;26:795–797. doi: 10.1038/nbt1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Hai T, Hao J, Wang L, Jouneau A, Zhou Q. Pluripotency maintenance in mouse somatic cell nuclear transfer embryos and its improvement by treatment with the histone deacetylase inhibitor tsa. Cellular reprogramming. 2011;13:47–56. doi: 10.1089/cell.2010.0042. [DOI] [PubMed] [Google Scholar]
- 131.Kim K, Doi A, Wen B, Ng K, Zhao R, Cahan P, Kim J, Aryee MJ, Ji H, Ehrlich LI, Yabuuchi A, Takeuchi A, Cunniff KC, Hongguang H, McKinney-Freeman S, Naveiras O, Yoon TJ, Irizarry RA, Jung N, Seita J, Hanna J, Murakami P, Jaenisch R, Weissleder R, Orkin SH, Weissman IL, Feinberg AP, Daley GQ. Epigenetic memory in induced pluripotent stem cells. Nature. 2010;467:285–290. doi: 10.1038/nature09342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Chung YG, Eum JH, Lee JE, Shim SH, Sepilian V, Hong SW, Lee Y, Treff NR, Choi YH, Kimbrel EA, Dittman RE, Lanza R, Lee DR. Human somatic cell nuclear transfer using adult cells. Cell stem cell. 2014 doi: 10.1016/j.stem.2014.03.015. [DOI] [PubMed] [Google Scholar]
- 133.Tachibana M, Amato P, Sparman M, Gutierrez NM, Tippner-Hedges R, Ma H, Kang E, Fulati A, Lee HS, Sritanaudomchai H, Masterson K, Larson J, Eaton D, Sadler-Fredd K, Battaglia D, Lee D, Wu D, Jensen J, Patton P, Gokhale S, Stouffer RL, Wolf D, Mitalipov S. Human embryonic stem cells derived by somatic cell nuclear transfer. Cell. 2013;153:1228–1238. doi: 10.1016/j.cell.2013.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Yamada M, Johannesson B, Sagi I, Burnett LC, Kort DH, Prosser RW, Paull D, Nestor MW, Freeby M, Greenberg E, Goland RS, Leibel RL, Solomon SL, Benvenisty N, Sauer MV, Egli D. Human oocytes reprogram adult somatic nuclei of a type 1 diabetic to diploid pluripotent stem cells. Nature. 2014 doi: 10.1038/nature13287. [DOI] [PubMed] [Google Scholar]
- 135.Polo JM, Liu S, Figueroa ME, Kulalert W, Eminli S, Tan KY, Apostolou E, Stadtfeld M, Li Y, Shioda T, Natesan S, Wagers AJ, Melnick A, Evans T, Hochedlinger K. Cell type of origin influences the molecular and functional properties of mouse induced pluripotent stem cells. Nature biotechnology. 2010;28:848–855. doi: 10.1038/nbt.1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Kim K, Zhao R, Doi A, Ng K, Unternaehrer J, Cahan P, Huo H, Loh YH, Aryee MJ, Lensch MW, Li H, Collins JJ, Feinberg AP, Daley GQ. Donor cell type can influence the epigenome and differentiation potential of human induced pluripotent stem cells. Nature biotechnology. 2011;29:1117–1119. doi: 10.1038/nbt.2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Bar-Nur O, Russ HA, Efrat S, Benvenisty N. Epigenetic memory and preferential lineage-specific differentiation in induced pluripotent stem cells derived from human pancreatic islet beta cells. Cell stem cell. 2011;9:17–23. doi: 10.1016/j.stem.2011.06.007. [DOI] [PubMed] [Google Scholar]
- 138.Ohi Y, Qin H, Hong C, Blouin L, Polo JM, Guo T, Qi Z, Downey SL, Manos PD, Rossi DJ, Yu J, Hebrok M, Hochedlinger K, Costello JF, Song JS, Ramalho-Santos M. Incomplete DNA methylation underlies a transcriptional memory of somatic cells in human ips cells. Nature cell biology. 2011;13:541–549. doi: 10.1038/ncb2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Chin MH, Pellegrini M, Plath K, Lowry WE. Molecular analyses of human induced pluripotent stem cells and embryonic stem cells. Cell stem cell. 2010;7:263–269. doi: 10.1016/j.stem.2010.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Guenther MG, Frampton GM, Soldner F, Hockemeyer D, Mitalipova M, Jaenisch R, Young RA. Chromatin structure and gene expression programs of human embryonic and induced pluripotent stem cells. Cell stem cell. 2010;7:249–257. doi: 10.1016/j.stem.2010.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Bock C, Kiskinis E, Verstappen G, Gu H, Boulting G, Smith ZD, Ziller M, Croft GF, Amoroso MW, Oakley DH, Gnirke A, Eggan K, Meissner A. Reference maps of human es and ips cell variation enable high-throughput characterization of pluripotent cell lines. Cell. 2011;144:439–452. doi: 10.1016/j.cell.2010.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Nishino K, Toyoda M, Yamazaki-Inoue M, Fukawatase Y, Chikazawa E, Sakaguchi H, Akutsu H, Umezawa A. DNA methylation dynamics in human induced pluripotent stem cells over time. PLoS genetics. 2011;7:e1002085. doi: 10.1371/journal.pgen.1002085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Rugg-Gunn PJ, Ferguson-Smith AC, Pedersen RA. Epigenetic status of human embryonic stem cells. Nature genetics. 2005;37:585–587. doi: 10.1038/ng1556. [DOI] [PubMed] [Google Scholar]
- 144.Ruthenburg AJ, Li H, Milne TA, Dewell S, McGinty RK, Yuen M, Ueberheide B, Dou Y, Muir TW, Patel DJ, Allis CD. Recognition of a mononucleosomal histone modification pattern by bptf via multivalent interactions. Cell. 2011;145:692–706. doi: 10.1016/j.cell.2011.03.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Fanucchi S, Shibayama Y, Burd S, Weinberg MS, Mhlanga MM. Chromosomal contact permits transcription between coregulated genes. Cell. 2013;155:606–620. doi: 10.1016/j.cell.2013.09.051. [DOI] [PubMed] [Google Scholar]