

Abstract
The human gyrovirus derived protein Apoptin (HGV-Apoptin) a homologue of the chicken anemia virus Apoptin (CAV-Apoptin), a protein with high cancer cells selective toxicity, triggers apoptosis selectively in cancer cells. In this paper, we show that HGV-Apoptin acts independently from the death receptor pathway as it induces apoptosis in similar rates in Jurkat cells deficient in either FADD (fas-associated death domain) function or caspase-8 (key players of the extrinsic pathway) and their parental clones. HGV-Apoptin induces apoptosis via the activation of the mitochondrial intrinsic pathway. It induces both mitochondrial inner and outer membrane permebilization, characterized by the loss of the mitochondrial potential and the release into cytoplasm of the pro-apoptotic molecules including apoptosis inducing factor and cytochrome c. HGV-Apoptin acts via the apoptosome, as lack of expression of apoptotic protease-activating factor 1 in murine embryonic fibroblast strongly protected the cells from HGV-Apoptin–induced apoptosis. Moreover, QVD-oph a broad-spectrum caspase inhibitor delayed HGV-Apoptin–induced death. On the other hand, overexpression of the anti-apoptotic BCL-XL confers resistance to HGV-Apoptin–induced cell death. In contrast, cells that lack the expression of the pro-apoptotic BAX and BAK are protected from HGV-Apoptin induced apoptosis. Furthermore, HGV-Apoptin acts independently from p53 signal but triggers the cytoplasmic translocation of Nur77. Taking together these data indicate that HGV-Apoptin acts through the mitochondrial pathway, in a caspase-dependent manner but independently from the death receptor pathway.
Abbreviations: 7AAD, 7-amino-actinomycin D; AIF, Apoptosis inducing factor; BCL-XL, B-cell lymphoma extra-large; CAV-Apoptin, Chicken anemia virus apoptin; cyt c, cytochrome c; DISC, Death-inducing signal complex; FADD, Fas-associated death domain; HGV-Apoptin, Human gyrovirus apoptin; MEF, Mouse embryonic fibroblast; MOMP, Mitochondrial outer membrane permeabilization; TMRM, Tetramethylrhodamine methyl ester perchlorate0
Introduction
Apoptosis is the process whereby individual cells of multicellular organisms undergo systematic self-destruction in response to a wide variety of stimuli [1]. Apoptosis is a genetically encoded program that is involved in normal development and homeostasis and in diverse patho-physiological processes [2]. Apoptosis functions to eliminate cells during development when they become redundant or as an emergency response after radiation damage, viral infection, or aberrant growth induced by the activation of oncogenes [1]. The morphology of apoptosis is orchestrated by the proteolytic activity of the caspase proteases [3], [4], [5] through which the cleavage of many proteins largely orchestrate apoptotic process [6]. In vertebrate cells, apoptosis typically occurs through one of two the major signaling pathways termed the extrinsic/cell death receptor pathway and the intrinsic/mitochondrial-initiator pathway [7]. In the extrinsic pathway, the ligation of the death receptors leads to the recruitment of the adaptor molecule FADD (fas-associated death domain) that bind, trimerize, and activate an initiator caspase (caspase-8), that in turn directly cleaves and activates the apoptosis executioner caspases (caspase-3 and -7) [2], [7], [8]. In the intrinsic pathway, the mitochondria responds to apoptotic stimuli through mitochondrial outer membrane permeabilization (MOMP). MOMP leads to the release of pro-apoptotic proteins from the mitochondrial intermembrane space. Following its release, cytochrome c (cyt c) binds apoptotic protease-activating factor 1 (APAF1), inducing its conformational change and oligomerization and leading to the formation of a caspase activation platform termed apoptosome. The apoptosome recruits, dimerizes, and activates an initiator caspase, caspase-9, which, in turn, cleaves and activates caspase-3 and -7 [2], [7], [8]. Thus the caspase cascade activation result from the remarkable MOMP and its subsequent intermembrane space mitochondrial proteins release. MOMP is highly regulated by the B cell lymphoma 2 (BCL-2) family members [2] which have been classified into 3 classes [9], [10]. One class inhibits apoptosis (BCL-2, BCL-XL, MCL-1, etc), the second class promotes apoptosis (BAX, BAK), and a third class termed the BH3-only proteins (BAD, BIK, BID, BIM, BOK, etc.) binds and regulates the anti-apoptotic BCL-2 proteins to promote apoptosis [4]. While the pro-apoptotic family members BAX and BAK are crucial for the induction of MOMP and the release of the pro-apoptotic molecules, the anti-apoptotic family members BCL-2 and BCL-XL inhibit BAX and BAK [4], [11]. Following MOMP, the mitochondrial transmembrane potential is dissipated through caspase-dependent and caspase-independent means [2], [12], [13]. The intrinsic death pathway is induced by many different stress signals including DNA-damaging agents, viral and cellular oncogenes, and transcriptional blockade [12], [14]. The stimuli are transmitted from the nucleus to the mitochondria by two main molecules: the tumor suppressor gene p53 and the orphan steroid receptor Nur77 [15].
Apoptosis plays an important role in the treatment of cancer as it is induced by many treatments [16]. While the most used strategies aim at targeting the apoptotic defects [16], some of the emerging strategies aim at the development of cancer selective therapies by molecules that target and kill preferentially cancer cells. One of the potential tools for cancer selective therapy is CAV-Apoptin as it induces apoptosis selectively in cancer cells [17], [18]. CAV-Apoptin is a viral protein of 14 kDa derived from the chicken anemia virus [19], [20]. The selective toxicity of CAV-Apoptin is associated at least in part to its tumor specific nuclear localization and its tumor specific phosphorylation at Theorine-108, which are essential for its nuclear accumulation and its induction of apoptosis [21], [22]. Recently, the human homolog of the CAV named the human gyrovirus (HGV) has been identified [23]. Its genome presents an overall organization similar to that of CAV [23], [24], it consists of a single negative-strand circular DNA of 2315 nucleotides. HGV has a similar organization of the promoter region and the encoded proteins as the CAV as revealed by both virus sequence alignment. It encodes a 125 amino-acid homologue of the CAV-Apoptin VP3 protein that despite a low overall identity has conserved important sites including nuclear localization and export signals and phosphorylation sites [23], [25]. HGV-Apoptin has the same subcellular distribution as the CAV-Apoptin, it localizes in the nuclei of cancer cells where it shows a granular distribution that later clusters to form aggregates while it remains in the cytoplasm of normal human cells [25]. Like CAV-Apoptin, HGV-Apoptin induces apoptosis selectively in cancer cells but not in normal cells [25] and is therefore a potential biologics anti-tumor candidate.
In this paper, we focus on the molecular mechanisms of HGV-Apoptin selective toxicity. Using cells with defective FADD or caspase-8 (key players in death receptor signaling), APAF1 deficient cells, BAK/BAX-deficient cells, and other molecular tools, we demonstrate that HGV-Apoptin induces apoptosis independently of the death receptor pathway. Hence, it triggers the activation of the mitochondrial death pathway via MOMP and the release of cyt c, and apoptosis-inducing factor (AIF) from mitochondria, in a caspase dependent manner. HGV-Apoptin induced apoptosis is modulated by the BCL-2 family members, is independent from p53 signal and causes the cytoplasmic translocation of Nur77.
Material and Methods
Chemotherapeutics Inhibitors
Staurosporine (2.5 μM) was purchased from Roche Diagnostics, Mannheim, Germany. The broad-range caspase inhibitor QVD-OPh was from Enzyme Systems (Dublin, CA, USA) and was added to the cells at a concentration of 25 μM immediately after transfection.
Antibodies
The following primary antibodies were used: mouse anti-active caspase-8 (Cell Signaling Technology, Beverly, USA), rabbit anti FAS, and anti FADD antibodies (Santa Cruz Biotechnology), murine activating anti-CD95/FAS (50 ng/ml) from upstate signaling, rabbit anti-AIF IgG (Sigma-Aldrich), mouse anti-p53 antibody (Millipore), Rabbit anti-Flag (Thermo/Fisher Scientific), rabbit anti-nur77 IgG (Santa Cruz), mouse anti–β-actin (Abcam). The following secondary antibodies were used: Infrared dye 800cw goat, anti-rabbit antibody, Infrared dye 680cw goat, anti-mouse antibody (Licor), Rhodamine redX anti-rabbit antibody (Life Technologies), cy5 anti-murine antibody (Abcam).
Cell Culture and Reagents
Jurkat (T-cell leukaemia) cells, Jurkat clones stably transfected with FADD-DN (a dominant-negative FADD mutant lacking the N-terminal death-effector domain), caspase-8 deficient Jurkat cells, Jurkat cells overexpressing BCL-XL and MCF7 (breast adenocarcinoma), MCF7 expressing caspase-3, MCF7 overexpressing BCL-XL cells, and MCF7 expressing GFP–cyt c were grown in RPMI-1640 medium supplemented with 10% fetal calf serum (Hyclone), 100 μg/ml penicillin and 0.1 μg/ml streptomycin (Gibco BRL). HCT116 (colon carcinoma), MEF (mouse embryonic fibroblasts) immortalized by retroviral transduction with a temperature-sensitive simian virus 40 large T antigen as described in [26], MEF-APAF1–/–, and MEF-BAX-BAK–/– were grown in DMEM medium supplemented with 10% fetal calf serum (Hyclone), 100 μg/ml penicillin and 0.1 μg/ml streptomycin (Gibco BRL). Human primary fibroblasts were grown in FibroGRO media for culture of human fibroblast (Millipore). Cells were grown at 37 °C with 5% CO2 in a humidified incubator.
Plasmids and Transient Transfections
The expression vectors of HGV-Apoptin GFP-HGV-APT and FLAG- HGV-APT were provided by Dr M. Tavassoli [25]. The empty vector pEGFPC1 was used as negative control. Cells were transfected using XtremeGENE HP DNA Transfection Reagent according to the manufacturer's instructions (Roche), Jurkat cells were transfected by electroporation using a BIO-RAD electroporator at a density of 107 cells per electroporation with 60 μg of DNA. The expression of GFP and GFP-HGV-Apoptin was confirmed by fluorescence microscopy. The broad-range caspase inhibitor QVD-oph was added to the cells at a concentration of 25 μM immediately after transfection.
Measurement of Apoptosis by Flow Cytometry
Apoptosis was quantified at the indicated time points after transfection using PO-PRO and 7-amino-actinomycin D (7AAD) staining and according to the manufacture's instructions (Invitrogen). Briefly, at the indicated time points, cells were harvested and washed twice with PBS, then stained in PBS with PO-PRO and 7AAD for 30 min on ice. Cells were analysed using a Gallios flow-cytometer. The population, of GFP positive cells that corresponds to the cells transfected with either the control vector pEGFPC1 or with GFP-HGV-APT were gated and analyzed among the whole population of cells, by staining cells with PO-PRO™-1 and 7-AAD apoptotic cells show violet/blue fluorescence, dead cells show violet/blue and red fluorescence, and live cells show little or no fluorescence.
Measurement of Mitochondrial Membrane Potential
Mitochondrial permeability transition was determined by staining the cells with tetramethylrhodamine methyl ester (TMRM) (Life Technologies). The cells were harvested and washed once with PBS then stained with TMRM 15 min at 37 °C and mitochondrial membrane potential was quantified by flow cytometric determination of the FL2 fluorescence of the cells. Data were collected and analyzed using a flow analysis Beckman-Coulter software (Kaluza).
Immunocytochemistry and Confocal Imaging
Cells were grown overnight on coverslips and then transfected with pEGFPC1, FLAG, GFP-HGV-APT, and FLAG-HGV-APT. After the indicated time points, cells were washed with PBS and then fixed in 4% paraformaldehyde for 20 min then permeabilized with 0.1% Triton X-100 and blocked in 0.1% BSA. To detect AIF release, cells were incubated with anti-AIF rabbit IgG (Sigma-Aldrich); diluted 1:500. Followed by three wash steps with PBS, the anti-AIF-antibody complexes were stained with the corresponding Rhodamine redX secondary antibody (diluted 1:200) then washed 3 times with PBS. To visualize nuclei, cells were stained with DAPI (10 μg/ml). The same was done for Nur77 using anti-Nur77 rabbit IgG (Santa-Cruz). The fluorescent images were then observed and analyzed with a Zeiss LSM 510 inverted laser-scanning confocal fluorescence microscope using 63 × (NA 5 1.4) oil planochromat objective (Carl Zeiss, Thornwood, NY).
Cell Extracts and Immunoblotting
The activation of caspase8 and the level of protein expression of Fas and FADD, were detected by immunoblotting. Briefly, 2×105 cells were transfected with pEGFPC1 vector alone or with GFP-HGV-APT. The expression of GFP and GFP-HGV-APT was confirmed by fluorescence microscopy. At the indicated time periods green cells were sorted using BD FACSAria III cell sorter (Becton Dickinson, Palo Alto CA, USA), whole cell lysates were prepared from green fluorescent cells using RIPA Buffer and according to the manufacture's instructions (Thermo-Scientific). Proteins (30 μg) were separated by denaturing SDS-PAGE and then transferred onto a PVDF membrane. The membranes were blocked in 5% non-fat dry milk in TBS 0.1% Tween and then incubated overnight with the following primary antibodies: anti-active caspase8 (Cell Signaling Technology, Beverly, USA), anti FAS, and anti FADD antibodies (Santa Cruz Biotechnology, Inc.) at 4 °C. Then, the blots were incubated with the corresponding InfraRed dye secondary antibodies (Licor). The visualization of the membrane was carried using Licor membrane scanner.
Detection and Quantification of Apoptotic Cells Using Laser Scanning Cytometry
Cells grown on cover slips and transfected with pEGFPC1 control vector or GFP- HGV-APT were stained at the indicated time points post-transfection with DAPI and then analysed by Laser scanning cytometry, using the one primary (DAPI fluorescence) and one sub (GFP fluorescence) protocol. For the detection of the chromatin condensation, three parameters were used, the nuclear area, the green maxpixel, and the blue maxpixel values. In this case, the nuclear area was set on the blue channel, cells with high blue maxpixel values (condensed chromatin) [27] are considered apoptotic.
Results
HGV-Apoptin Induce Apoptosis Independently from the Death Receptor Pathway
Both CD95 ligand and its receptor are rapidly expressed following T cell activation. In both cases, the soluble CD95L can act either in an autocrine manner and trigger apoptosis at a single cell level or can also mediate at high concentrations apoptosis of short distanced neighboring cells in a paracrine manner. CD95L can also act at the membrane bound form and also trigger apoptosis in an autocrine/paracrine manner.
To examine the role of the CD95 system in HGV-Apoptin induced apoptosis, we have tested the role of components of the death-inducing signal complex (DISC) which is critical for initiation of death-receptor-mediated apoptosis [28]. DISC is a complex of proteins formed upon the recruitment of the initiator caspase, pro-caspase-8, through its pro-domain together with FADD, a bipartite molecule with a death effector domain, to the death receptor [28], [29], [30]. This leads to the activation of the initiator caspase-8 that will directly mediate the down-stream caspase cascade and the execution of apoptosis through the death receptor pathway [30], [31].
First, we have tested the role of caspase-8 using Jurkat T cells that lacked the expression of caspase-8 (Jurkat-caspase-8−/−). Transiently expressed HGV-Apoptin in these cells accumulates in the nucleus similar to that of their wild type counterparts, and induces apoptotic morphological changes including cell contraction and nuclear condensation (Figure 1A). Thus, suggesting that the lack of caspase-8 does not affect the ability of HGV-Apoptin to induce apoptosis in Jurkat cells.
Figure 1.
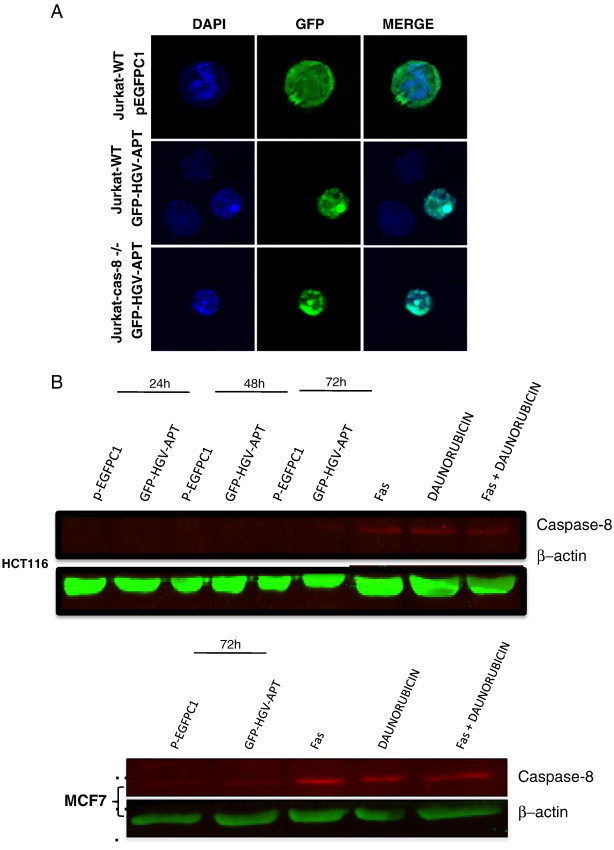
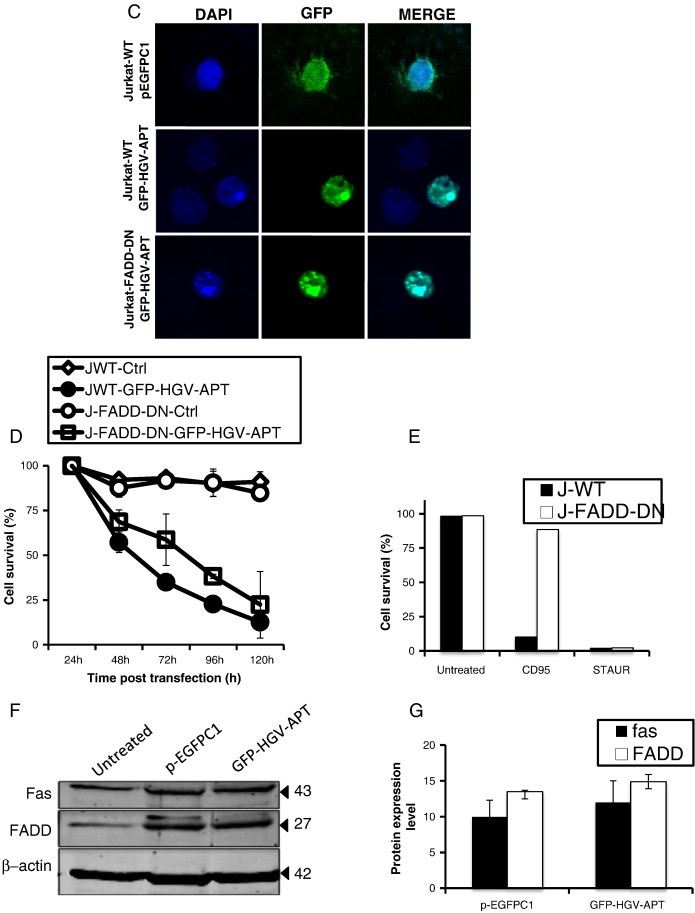
HGV-Apoptin mediated cell death is independent of the death receptor pathway. A) Cellular localization and morphological changes induced by HGV-Apoptin in Jurakt WT or Jurkat caspase-8-/- cells. Cells were transfected by electroporation with GFP-HGV-APT and the corresponding pEGFPC1 control plasmid, cells were fixed 48 h post transfection and stained with DAPI for the detection of nuclear morphology. B) HGV-Apoptin does not activate caspase-8. Western blot analysis of cell lysates from HCT116 (colon carcinoma) and MCF7 cells transfected either with pEGFPC1 or with GFP-HGV-APT for caspase-8 activation. At the indicated time points post-transfection, GFP positive cells were sorted by flow cytometry; cells treated with anti-FAS activating antibodies, or, Daunorubicin or both represent a positive control. C) Detection of apoptosis induced by HGV-Apoptin in Jurkat WT cells or Jurkat FADD-DN. Cells were transfected by electroporation with GFP-HGV-APT or the corresponding pEGFPC1 control plasmid. 48 h later, transfected Jurkat cells were fixed and stained with DAPI for the detection of nuclear morphology. D) Quantification of cell survival in Jurkat WT and Jurkat FADD-DN as percentage of PO-PRO/7AAD double negative cells (cell survival) in the GFP-positive population from 24 to 120 h post-transfection. E) Control experiment to assess cell death mediated by CD95/FAS, both Jurkat WT and FADD-DN cells were treated with anti-FAS (IgM) antibody or with Staurosporine (STAUR), and cell death was measured by flow cytometry after 12 h. F) Western blot analysis of FAS and FADD level of expression in Jurkat cells 48 h post transfection with GFP-HGV-APT or the corresponding pEGFPC1 control vector. In panels “B” and “F” β − actin was used as a protein loading control. G) Quantification of the level of expression of FAS and FADD 48 h post-transfection (Western blot shown in “G”).
To further confirm the above findings, caspase-8 activation was monitored by Western blot of the cell lysates from GFP-positive MCF7 and HCT116 at the indicated time points post-transfection with GFP-HGV-Apoptin and its corresponding pEGFPC1 control vector. Up to 72 hours post-transfection, caspase-8 activation was not detected in both cell lines in response to HGV-Apoptin transient expression unlike the cells that were treated with anti-FAS antibody or daunorubucin or both activators of the death receptor pathway and caspase-8 (Figure 1B). Taking together this data show that caspase-8 is neither required nor activated during HGV-Apoptin induced apoptosis.
Next, we tested the role of the FADD adaptor molecule in HGV-Apoptin triggered apoptosis in Jurkat T cells that are overexpressing a truncated, dominant-negative form of FADD (FADD-DN). The expression of a dominant negative form of FADD prevents the formation of a functional DISC and will block the signaling via the extrinsic pathway. Jurkat-FADD-DN cells show apoptotic morphology similar to the Jurkat WT cells (Jurkat expressing the intact DADD) after transient expression of HGV-Apoptin (Figure 1C). Moreover the monitoring of apoptosis induced by HGV-Apoptin in both Jurkat-WT and Jurkat-FADD-DN cells from 24 to 120 hours post-transfection by flow cytometry shows that both WT and FADD-DN cells have similar sensitivity to HGV-Apoptin (Figure 1D). Thus, the state of FADD has no effect in HGV-Apoptin induced apoptosis.
In a control experiment, Jurkat WT and FADD-DN were treated for 12 hours with the anti-FAS human activating antibody, which induce cell death via the recruitment of FADD and the activation of caspase-8. After 12 hours of treatment, while almost all the Jurkat WT cells were dead, the Jurkat-FADD-DN were completely resistant to treatment with FAS (Figure 1E).
The above observations were further confirmed by monitoring the expression of FAS and FADD in GFP-HGV-Apoptin transiently expressing Jurkat cells by Western blot. The protein expression profiles of these apoptotic regulators were monitored in cell lysates of the GFP-positive cells at 48 h post-transfection. Expression of FAS and FADD were detected in all the samples with no significant increase in response to HGV-Apoptin expression. (Figure 1, F and G) Taking together this data indicates that HGV-Apoptin similar to its homolog CAV-Apoptin induce apoptosis independently from the death receptor pathway.
HGV-Apoptin Triggers the Mitochondrial Pathway of Cell Death and Induces Mitochondrial Alterations
To gain more insight into the mechanism of apoptotic signaling triggered by HGV-Apoptin, we investigated the role of the mitochondrial intrinsic pathway in HGV-Apoptin induced apoptosis. The intrinsic signaling pathways that initiate apoptosis involve diverse non-receptor mediated stimuli that produce intracellular signals. These signals act directly on targets within the cell and are mitochondrial-initiated events [32] that lead to mitochondrial alterations and loss of the mitochondrial membrane potential [33]. Thus in order to assess changes in the mitochondrial potential induced by HGV-Apoptin we have used the TMRM dye that is rapidly sequestrated by healthy mitochondria while the depolarized mitochondria does not accumulate the dye [34]. Indeed, cells transfected with HGV-apoptin show severe decrease in TMRM fluorescence (Figure 2A) compared to control treated cells. Moreover monitoring of the mitochondrial potential by flow cytometry in HCT116 shows a huge decrease in the mitochondrial potential among HGV-Apoptin transient expressing cells (Figure 2B). Similar results were obtained in Jurkat WT cells along with the mutant Jurkat FADD-DN cells after transfection with HGV-Apoptin showing that FADD have no effect on the loss of the mitochondrial potential caused by HGV-Apoptin (Figure 2C). This data indicates that HGV-Apoptin triggered apoptosis is associated with the loss of the mitochondrial potential and further confirms that this occurs independently from the death receptor pathway.
Figure 2.
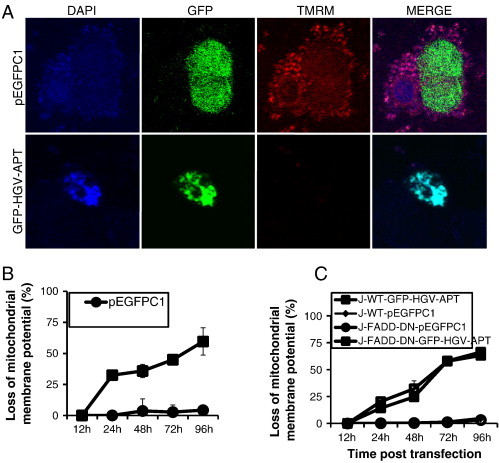
Mitochondrial membrane potential is lost in response to transient expression of HGV-Apoptin. A) Mitochondrial staining of HCT116 cells transfected with GFP-HGV-APT or the corresponding pEGFPC1 control plasmid. Cells were stained with TMRM dye for 15 min, then fixed and counterstained with DAPI. B) Monitoring of mitochondrial membrane potential (Ψm) in HCT116, either transfected with the HGV-Apoptin tagged to GFP: GFP-HGV-APT or the control plasmid: pEGFPC1, for the indicated time periods. Ψm was determined using TMRM dye, which shows a decrease in the red fluorescence upon loss of the mitochondrial potential. C) Monitoring of the mitochondrial potential in Jurkat WT and Jurkat-FADD-DN from 12 to 96 h post-transfection with GFP-HGV-APT and pEGFPC1.
HGV-Apoptin Induces the Release of the Mitochondrial Components Cytochrome c and AIF and Triggers the Apoptosome Pathway
Most pro-apoptotic stimuli that involve mitochondrial outer membrane permeabilization leading to intra-cytosolic release of mitochondria intermembrane space proteins such as cytochrome c (cyt c) will trigger caspase activation and/or AIF mediated caspase independent death pathway [35]. Activation of both pathways often coexists. Thus to further examine the effect of HGV-Apoptin on mitochondria, we have first checked cyt c release. For that purpose we have used MCF7 cells stably transfected with GFP–cyt c fusion protein (MCF7-cytc-GFP). As shown in Figure 3A, the transient expression of FLAG-HGV-APT in MCF7-cytc-GFP leads to the release of cyt c into the cytoplasm but not the transient expression of the FLAG empty vector (please note the selective loss of the green granular pattern in cells transfected with FLAG-HGV-APT).
Figure 3.
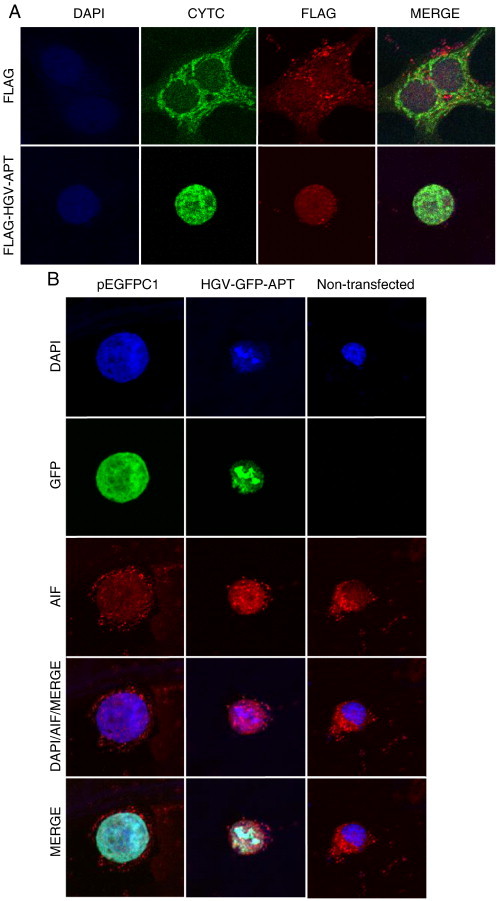
HGV-Apoptin triggers the release of pro-apoptotic mitochondrial proteins. A) HGV-Apoptin induces cyt c release from transformed cells. MCF7-cyt c-GFP cell line was transiently transfected with FLAG-HGV-APT and the corresponding FLAG control vector, at 48 h post-transfection cells were fixed, stained with a primary rabbit anti-FLAG and secondary Red-X anti-rabbit antibody and counterstained with DAPI. B) The cellular localization of AIF in HCT116 cells as determined by confocal microscopy. HCT116 cells were transiently transfected with GFP-HGV-APT and the corresponding pEGFPC1 control plasmid, after 48 h cells were fixed, stained with a primary rabbit anti-AIF and secondary Red-X anti-rabbit antibody and counterstained with DAPI for nuclear visualization (the non-transfected cells along with p-EGFPC1 transfected cells were used as negative controls).
Next, we checked for the release of AIF. Similar to cyt c release, the release of AIF from mitochondria was also detected by confocal microscopy in HCT116 in response to GFP-HGV-Apoptin transient expression (Figure 3B). This data show that HGV-Apoptin triggers the release of the mitochondrial components cyt c and AIF.
While AIF can cause apoptosis through high molecular weight DNA fragmentation and chromation condensation independently from caspase activity [35], [36], cyt c, along with dATP binds APAF1 and form the death signaling platform: apoptosome. This event is a key step for caspase-9 activation and is crucial for the initiation of apoptosis via the intrinsic pathway [37]. Therefore, in order to give more insight into the role of the apoptosome in HGV-Apoptin induced cell death we have checked the role of APAF1 using transformed mouse embryonic fibroblast genetically modified to lack the expression of APAF1 in transformed mouse embryonic fibroblast (MEF-APAF1−/−). Expression of GFP-HGV-Apoptin in MEF-APAF1−/−is non-toxic as compared to the WT cells (Figure 4, A and B). These results show that HGV-Apoptin induce apoptosis via the apoptosome and further confirm the implication of the mitochondrial intrinsic pathway in HGV-Apoptin induced apoptosis.
Figure 4.
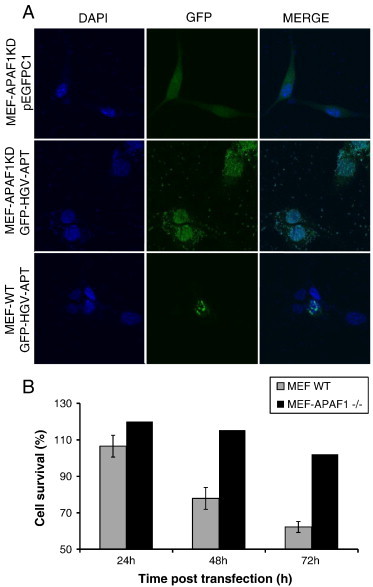
Requirement of APAF1 for HGV-Apoptin induced apoptosis. A) Immortalized APAF1 knockout mouse fibroblasts were transfected with GFP-HGV-Apoptin or the corresponding pEGFPC1 control plasmid and the relative confocal images of MEF WT and MEF-APAF1−/−48 h post-transfection, cells were fixed and stained with DAPI for the detection of nuclear morphology. B) Quantification of cell survival as percentage of PO-PRO/7AAD double negative cells in the GFP-positive population from 24 to 72 h post-transfection of MEF-WT and MEF-APAF1-/- cells.
HGV-Apoptin Mediated Cell Death is Caspase Dependent
To further elucidate the HGV-Apoptin triggered death pathway, we have checked the role of caspases, the key mediator of the apoptotic program [5]. Upon apoptosis triggering, the upstream caspases (caspase-8, caspase-9) cleave and activate downstream caspases (caspase-3, caspase-7) leading to the execution of apoptosis. For this purpose, Jurkat T cells were transfected with GFP-HGV-APT in the presence or the absence of 25 μM of the broad-range caspase inhibitor (QVD-oph). At 24 hours post-electroporation cells were examined microscopically for protein expression and the effects of QVD-oph. In the presence of QVD-oph, cells that express HGV-Apoptin were partially protected from cell death. In contrast, QVD-oph non-treated cells were highly sensitivity to HGV-Apoptin. To investigate the effect of caspase inhibition in detail, apoptosis was monitored in both Jurkat non-treated cells and Jurkat cells treated with caspase inhibitor QVD-oph from 24 to 120 h post-transfection. As shown in Figure 5A, the inhibition of caspase activity by QVD-oph delayed HGV-Apoptin induced apoptosis in Jurkat cells.
Figure 5.
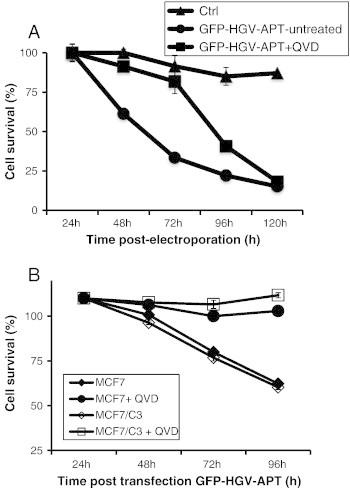
HGV-Apoptin mediated cell death is caspase-dependent. A) Quantification of cell survival as percentage of PO-PRO/7AAD double negative cells in the GFP-positive population from 24 to 120 h post-transfection. Jurkat cells were transfected either with GFP-HGV-APT or with pEGFPC1 control plasmid and then either left untreated or incubated with 25 μM of QVD-oph and then stained with PO-PRO/7AAD. B) Quantification of cell survival as percentage of PO-PRO/7AAD double negative cells in the GFP-positive population from 24 to 96 h post-transfection. Wild-type caspase-3 deficient MCF7 cells and MCF7 cells expressing caspase-3 (MCF7/C3) were transiently transfected either with GFP-HGV-APT or with pEGFPC1 control plasmid and then either left untreated or incubated with 25 μM of QVD-oph. Cells were then stained with PO-PRO/7AAD and analyzed by flow cytometry.
To further investigate whether the inhibition of caspase could block HGV-Apoptin induced apoptosis, cell death was monitored among caspase-3-deficient MCF7 cells and MCF7 cells stably overexpressing caspase-3 (MCF7/C3) either left untreated or treated with 25 μM of the caspase inhibitor QVD. As shown in Figure 5B, the ectopic expression of caspase-3 renders MCF7 more sensitivity to HGV-Apoptin. Moreover, treatment with the caspase inhibitor QVD-oph strongly inhibited HGV-Apoptin induced apoptosis in both MCF7 and MCF7/C3 cells, these results further confirm that caspase activity is required for HGV-Apoptin induced apoptosis.
HGV-Apoptin Mediated Cell Death is Modulated by the BCL-2 Family Members
Mitochondrial outer membrane permeabilization, which plays a crucial role in apoptosis induction, is controlled by pro- and anti-apoptotic members of the BCL-2 family [35]. Therefore we examined the role of both pro- and anti-apoptotic BCL-2 family in HGV-Apoptin induced apoptosis. First we checked the role of the anti-apoptotic BCL-XL in HGV-Apoptin induced cell death. We compared the sensitivity towards HGV-Apoptin induced apoptosis between the parental MCF7 cell line and its derivates stably over-expressing BCL-XL. The transient expression of HGV-Apoptin leads to the apoptosis of MCF7 cells within 48 h whereas MCF7-BCL-XL cells remained viable and appeared morphologically healthy. These observations were further confirmed by time-lapse studies. The monitoring of apoptosis induced by HGV-Apoptin by flow cytometry in both MCF7 and MCF7-BCL-XL cells from 24 to 96 h shows that over expression of the anti-apoptotic BCL-XL render the cells resistance to HGV-Apoptin as they remain viable compared to the parental clone (Figure 6A).
Figure 6.
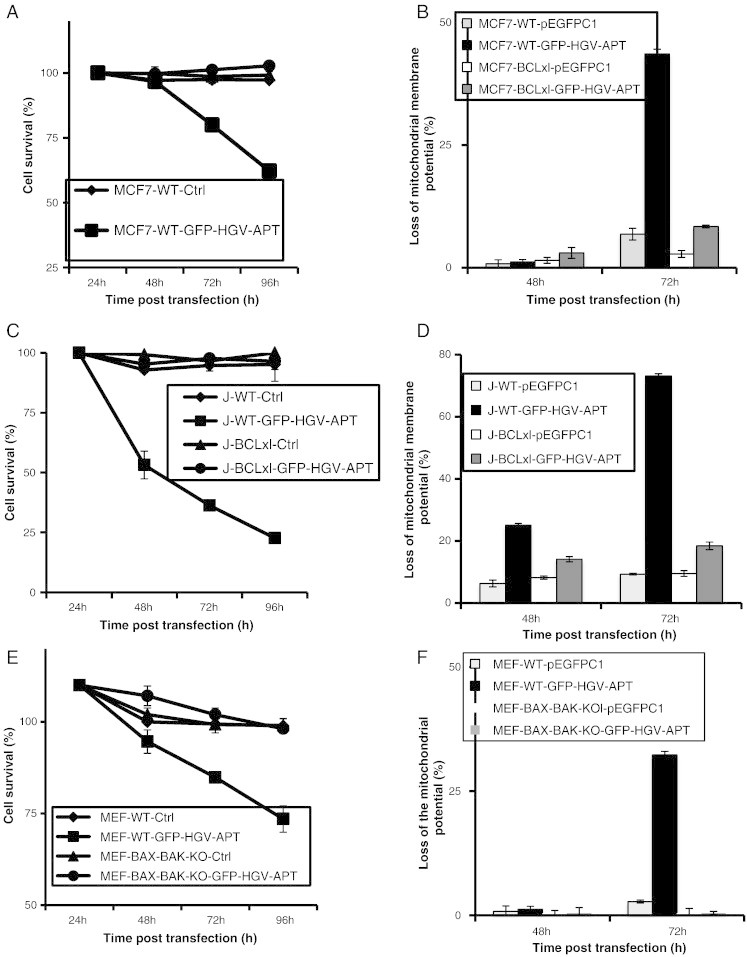
HGV-Apoptin induced apoptosis is regulated by the anti- and pro-apoptotic BCL-2 family members. A) Quantification of cell survival as percentage of PO-PRO/7AAD double negative cells in the GFP-positive population from 24 to 96 h post-transfection. MCF7 cells and MCF7-BCL-XL cells were transfected with GFP-HGV-APT or the corresponding control plasmid pEFPC1 and stained with PO-PRO/7AAD. B) Monitoring of the mitochondrial membrane potential in MCF7 WT and MCF7 BCL-XL cells after transfection with either GFP-HGV-APT or the control plasmid using the TMRM dye. C) Quantification of cell survival in Jurkat wild type and Jurkat-BCL-XL as percentage of PO-PRO/7AAD double negative cells in the GFP-positive population from 24 to 96 h post-transfection. Cells were transfected by electroporation with GFP-HGV-APT or the corresponding control plasmid. D) Monitoring of the mitochondrial membrane potential in Jurkat WT and Jurkat BCL-XL after transfection with either GFP-HGV-APT or the control plasmid using the TMRM dye. E) Quantification of apoptosis in the wild-type murine embryonic fibroblasts (MEF-WT) and MEF BAX and BAK deficient cells (MEF-BAX-BAK-KO). Cells were transfected with GFP-HGV-Apoptin or the corresponding control plasmid pEFPC1. Cell survival was quantified by flow cytometry after PO-PRO/7AAD staining as percentage of PO-PRO/7AAD double negative cells in the GFP-positive population of cells. F) Monitoring of the mitochondrial membrane potential in MEF-WT and MEF-BAX-BAK-KO using the TMRM dye staining. Due to space constrain “BCL-XL” was labeled in some figures as “BCLxl”.
Since the protective effect of BCL-XL against HGV-Apoptin could be cell type specific, we further tested the inhibitory activity of BCL-XL in the Jurkat T cells, and compared the sensitivity towards HGV-Apoptin via apoptosis monitoring by flow cytometry in both the parental clone and Jurkat cells overexpressing BCL-XL. As shown in Figure 6C, over-expression of BCL-XL also protected Jurkat cells from HGV-Apoptin induced apoptosis. This data shows that HGV-Apoptin induced apoptosis is regulated by the anti-apoptotic BCL-XL independent of cell type.
Next, we have tested the role of the pro-apoptotic BAX and BAK in HGV-Apoptin induced apoptosis using MEF cells that lack the expression of BAX and BAK (MEF-BAX-BAK-/-). While MEF WT cells were sensitive to the transient expression of HGV-Apoptin, MEF-BAX-BAK-/- cells remained viable and appeared morphologically healthy (Figure 6E). The monitoring of apoptosis induced by HGV-Apoptin by flow cytometry in both MEF-WT and MEF-BAX-BAK-/- cells from 24 to 72 h post-transfection shows that the lack of expression of the pro-apoptotic family members BAX and BAK protected MEF cells from HGV-Apoptin induced cell death.
To further expand the above observations, we have measured the mitochondrial membrane potential of MCF7-WT, MCF7-BCL-XL (Figure 6B), Jurkat-WT, Jurkat-BCL-XL (Figure 6D) MEF-WT, and MEF-BAX-BAK-/- (Figure 6F) at 48 and 72 hours post transfection with HGV-Apoptin expressing vector. The parental clones showed a decrease in the mitochondrial potential while the BCL-XL-expressing cells have maintained their mitochondrial membrane potential. Also, MEF-BAX-BAK-/- cells failed to respond with the decrease of mitochondrial membrane potential upon the expression of HGV-Apoptin. Taking together, this data show that HGV-Apoptin induced apoptosis is regulated by the BCL-2 family members, both the pro- and the anti-apoptotic ones.
HGV-Apoptin Mediated Cell Death is Independent from p53 Signal and Induces the Cytoplasmic Translocation of Nur77
HGV-Apoptin activates the mitochondrial cell death pathway. p53 and Nur77 are the main molecules capable of transmitting the apoptotic stimuli from nucleus to the cytoplasm. Thus we tested their respective role in HGV-APT induced cell death.
First, we investigated the role of p53, the tumor suppressor protein. p53 functions as a transcriptional regulator of multiple apoptotic genes, also has a direct pro-apoptotic function in the mitochondria by directly interacting with both the anti- and pro-apoptotic BCL-2 family members [15], [34].
As shown in Figure 7A, lack of expression of p53 in HCT116 cells does not prevent HGV-Apoptin from induction of apoptosis as both HCT116 WT and their counterparts lacking p53 both were equally sensitive to HGV-Apoptin expression. Hence, both cell-types show apoptotic morphology in response to HGV-Apoptin expression (48 h time-point). To get further insight in the role of p53 in HGV-Apoptin induced apoptosis, we have performed a time course studies. We have monitored apoptosis in HCT116-WT and their p53-deficient progeny between 24 and 120 h post-transfection with HGV-Apoptin by laser-scanning cytometry. Cells with a condensed chromatin were considered apoptotic and quantified (Figure 7B). These experiments show that HGV-Apoptin does not require p53 for induction of apoptosis.
Figure 7.
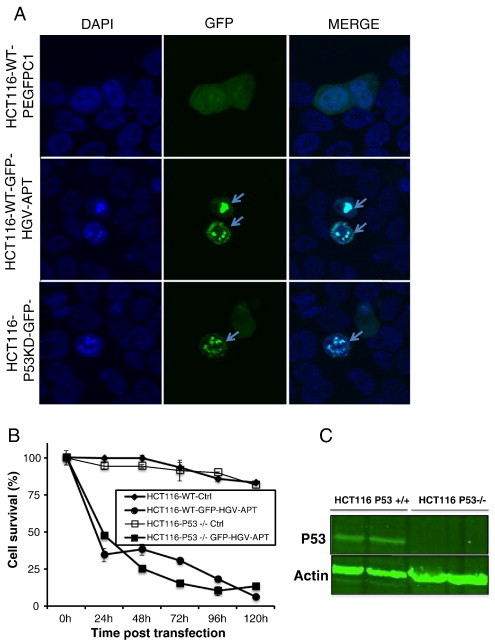
HGV-Apoptin induced apoptosis is independent from p53 signal. A) p53 deficient HCT116 (HCT116-p53−/−) and the wild type (HCT116-WT) colon carcinoma cells were transfected with pEGFPC1 or GFP-HGV-APT. The cells were fixed and stained with DAPI 48 h post-transfection for the detection of the nuclear morphology; arrows indicate apoptotic cells expressing GFP-HGV-APT. B) Quantification of apoptosis in HCT116-WT and HCT116-p53−/− cells using laser-scanning cytometry. Cells were fixed and stained with DAPI then analysed by laser scanning cytometry, cells with a high blue max-pixel values (condensed chromatin) are considered apoptotic. C) Western blot data confirming that HCT116-p53−/− lack p53.
Next, we tested the role of NUR77, an orphan nuclear receptor and member of the steroid/thyroid receptor family, in HGV-Apoptin triggered apoptosis. NUR77 is capable not only of transmitting the apoptotic stimuli from the nucleus to the mitochondria [38], but also can modulate apoptosis via activating the transcription of pro- and anti-apoptotic genes [39]. Expression of HGV-Apoptin leads to the translocation of NUR77 from nucleus to the cytoplasm in both HCT116 (Figure 8A) and MCF7 cells (Figure 8B), indicating that NUR77 is a signaling molecule transmitting apoptotic signal in HGV-Apoptin pathway.
Figure 8.
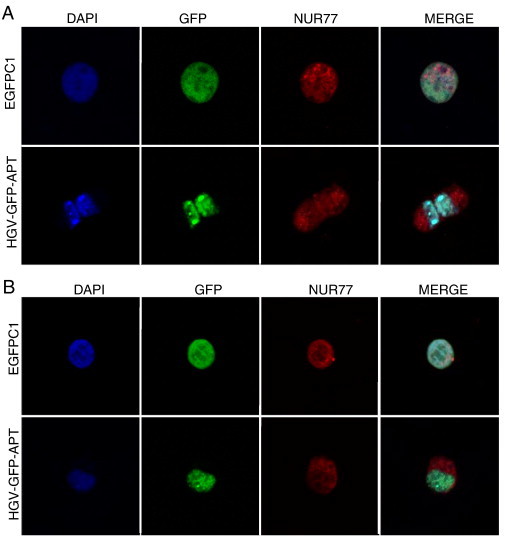
HGV-Apoptin induced cell death involves the nucleo-cytoplasmic translocation of Nur77. Confocal microscopy visualization of Nur77 in A) MCF7 cells, and B) HCT116. Cells were fixed 48 h post-transfection and stained with a primary rabbit anti NUR77 antibody and a secondary anti-rabbit RED-X antibody and then counterstained with DAPI for the visualization of the nuclei.
Discussion
The selective targeting of cancer cells has been a ‘holy grail’ of cancer therapy. Since cancer cells are derived from our own healthy tissues, the task seems very difficult. However, in the past two decades, growing number of proteins have been discovered, that display selective anti-cancer activity [40]. The protoplast of the group is CAV-Apoptin, discovered in the mid-90s by Noteborn and colleagues [41]. CAV-Apoptin causes apoptosis of cultured tumorigenic and transformed human cell lines but is harmless to normal cells [19]. This cancer cell selective toxicity made CAV-Apoptin among the most potential candidate for tumor cells selective therapy. Recently, in 2011, the human homolog of the CAV-Apoptin has been identified. The human gyrovirus-Apoptin (HGV-Apoptin) [23] displays the same differential behavior between normal and tumor cells as its homolog. Recently published studies show that HGV-Apoptin translocates to the nucleus of transformed cells while it remains in the cytoplasm of normal cells, similarly to CAV-Apoptin. Moreover, HGV-Apoptin also induces apoptosis in cancer cells while leaving normal cells intact [25]. This prompted us to deeper the study of cytotoxycity mechanism of HGV-Apoptin. Better understanding of the molecular mechanism of HGV-Apoptin may allow for the development apoptin-protein derivates better suited for the selective killing of cancer cells.
A number of anti-cancer drugs up-regulate CD95L, which in turn may contribute to their anti-cancer activity in an autocrine and/or paracrine manner [42], [43], [44]. Also, activation of T-cells leads to the expression of CD95L that kills T-cells in autocrine or paracrine fashion [45]. Thus, we checked the role of the CD95 signaling pathway in HGV-Apoptin induced apoptosis. FADD and caspase-8 are the most important upstream components of the CD95/Fas signaling pathway, so using cells that overexpress a dominant negative form of FADD and other molecular tools, we show that neither FADD nor caspase-8 are required for the apoptotic activity of HGV-Apoptin. Moreover, since some previous reports indicate that Fas can still signal atypical apoptotic cell death in the absence of caspase-8 [46], we examined by Western blot if HGV-Apoptin upregulates Fas expression. Our results show that in response to HGV-Apoptin expression, no significant increase in Fas level is detectable. Taken together, our data indicate that HGV-Apoptin, like its homolog CAV-Apoptin, kills cancer cells independently from the CD95 system.
The mitochondrial intrinsic death pathway is ultimately activated by a number of anti-cancer drugs and stress stimuli that cause the loss of the mitochondrial membrane potential [47] and the release of a number of pro-apoptotic molecules from the mitochondria. Among others, released are cyt c and AIF [48], [49]. Hence, we have checked the mitochondrial alterations induced by HGV-Apoptin, also the cellular localization of cyt c and AIF. We have observed that HGV-Apoptin induces a decrease in the mitochondrial potential, and release of both cyt c and AIF from the mitochondria.
Although caspase-independent forms of apoptosis have been described [50], among them the nuclear translocation of AIF where it induces apoptosis via DNA fragmentation, caspases play a crucial role in apoptosis and are activated in both the extrinsic and intrinsic pathways. Caspases are activated through the formation of large protein complexes namely ‘death inducing signal complex’ (DISC) within the extrinsic pathway or the apoptosome within the intrinsic pathway [7]. Lack of expression of APAF1 (a major component of the apoptosome), strongly delayed HGV-Apoptin induced apoptosis (up to 96 h post-transfection).
Apoptosome is the molecular platform for caspase-9 activation. Caspase-9 then activates down-stream caspases namely caspase-3 and caspase-7 [51], [52] leading to the execution of apoptosis (Figure 9). So in order to investigate the requirement of caspase activity in HGV-Apoptin induced apoptosis, we have used the broad-spectrum caspase inhibitor QVD-oph that inhibits both up-stream and down-stream caspases. QVD-oph delayed HGV-Apoptin induced cell death, indicating that HGV-Apoptin induced apoptosis occurs in a caspase-dependent manner. The inhibitory effect of QVD on HGV-Apoptin induced cell death was not only observed in Jurkat T cells but also in caspase-3 deficient MCF7 cells and its retransfected derivative MCF7/C3, which were only protected from HGV-Apoptin induced cell death in the presence of the caspase inhibitor. It should be noted that HGV-Apoptin kills MCF7 cells that lack the expression of caspase-3, which indicates that caspase-3 may be dispensable for HGV-Apoptin induced cell death, and substituted by other down-stream caspases such as caspase-6 and caspase-7. Moreover, the ectopic expression of caspase-3 in MCF7 cells renders them slightly more sensitive to HGV-Apoptin. Thus, HGV-Apoptin requires caspase activity to kill its targets.
Figure 9.
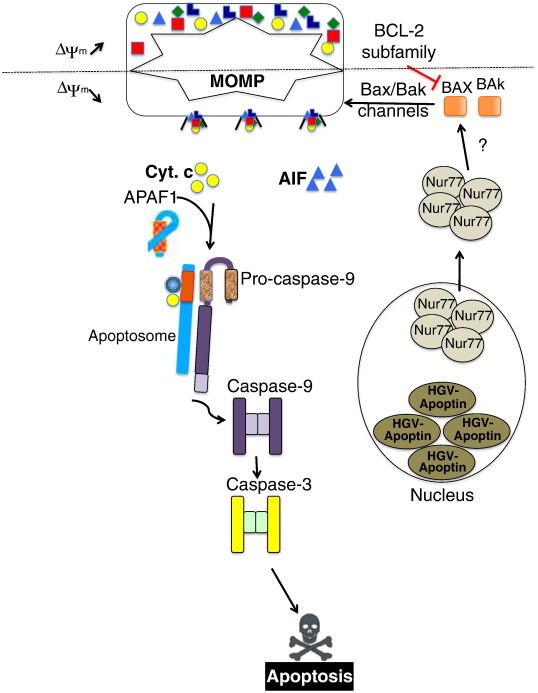
Schematic representation of HGV-Apoptin mechanism of action in cancer cells. HGV-Apoptin activation and nuclear accumulation induces the nuclear export of nur77, which might induce the activation of the BCL-2 family members of proteins, leading to MOMP and the subsequent cytoplasmic release of AIF and cyt c leading to apoptosis through APAF1 activation, the apoptosome formation, and the subsequent caspase activation.
The mitochondrial pathway is modulated by the pro- and anti-apoptotic BCL-2 family members [53]. In order to check the role of the BCL-2 members in HGV-Apoptin induced cell death, we have used MCF7 cells that overexpress the anti-apoptotic BCL-XL. Over-expression of BCL-XL inhibits the pro-apoptotic molecules BAX and BAK that in turn, inhibit MOMP and the release of pro-apoptotic molecules from the mitochondria [54]. Over-expression of BCL-XL protected MCF7 cells from HGV-Apoptin induced apoptosis. Moreover, murine embryonic fibroblasts that lack the expression of the pro-apoptotic BAX and BAK are protected from HGV-Apoptin induced apoptosis compared to their parental clones. Thus, both pro- and anti-apoptotic BCL-2 family members regulate HGV-Apoptin induced cell death.
In conclusion, in this paper, we have given an insight into the molecular mechanism of HGV-Apoptin induced cell death (Figure 9). We show that HGV-Apoptin acts via the intrinsic mitochondrial pathway in a caspase-dependent manner, and that its activity is modulated by BCL-2 family members. Elucidating the molecular mechanism of HGV-Apoptin's signaling pathway is of great importance, and it may lead into the development of novel anticancer strategy allowing for selective targeting of cancer cells. Comparative study of HGV-Apoptin and CAV-Apoptin signaling pathways, may lead to the discovery of new molecular targets uniquely available only in cancer cells. HGV-Apoptin does not require p53 tumor suppressor signaling whose inactivation is a frequent event in tumorigenesis [55], and which leads to the resistance to available cancer therapies [56].
Acknowledgments
MJL kindly acknowledges the core/startup support from Linkoping University, from Integrative Regenerative Medicine Center (IGEN), from Cancerfonden (2013/391), and from VR-NanoVision (K2012-99X -22325-01-5). ACP thankfully acknowledges the support from BK/265/RAU1/2014/t.10.
References
- 1.Teodoro JG, Branton PE. Regulation of apoptosis by viral gene products. J Virol. 1997;71:1739–1746. doi: 10.1128/jvi.71.3.1739-1746.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tait SW, Green DR. Mitochondria and cell death: outer membrane permeabilization and beyond. Nat Rev Mol Cell Biol. 2010;11:621–632. doi: 10.1038/nrm2952. [DOI] [PubMed] [Google Scholar]
- 3.Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972;26:239–257. doi: 10.1038/bjc.1972.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Youle RJ, Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol. 2008;9:47–59. doi: 10.1038/nrm2308. [DOI] [PubMed] [Google Scholar]
- 5.Iglesias-Guimarais V, Gil-Guinon E, Sanchez-Osuna M, Casanelles E, Garcia-Belinchon M, Comella JX, Yuste VJ. Chromatin collapse during caspase-dependent apoptotic cell death requires DFF40/CAD-mediated 3′-OH single-strand DNA breaks. J Biol Chem. 2013;288:9200-15. doi: 10.1074/jbc.M112.411371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Taylor RC, Cullen SP, Martin SJ. Apoptosis: controlled demolition at the cellular level. Nat Rev Mol Cell Biol. 2008;9:231–241. doi: 10.1038/nrm2312. [DOI] [PubMed] [Google Scholar]
- 7.Los M, Wesselborg S, Schulze-Osthoff K. The role of caspases in development, immunity, and apoptotic signal transduction: lessons from knockout mice. Immunity. 1999;10:629–639. doi: 10.1016/s1074-7613(00)80062-x. [DOI] [PubMed] [Google Scholar]
- 8.Greil R, Anether G, Johrer K, Tinhofer I. Tracking death dealing by Fas and TRAIL in lymphatic neoplastic disorders: pathways, targets, and therapeutic tools. J Leukoc Biol. 2003;74:311–330. doi: 10.1189/jlb.0802416. [DOI] [PubMed] [Google Scholar]
- 9.Kuwana T, Newmeyer DD. Bcl-2-family proteins and the role of mitochondria in apoptosis. Curr Opin Cell Biol. 2003;15:691–699. doi: 10.1016/j.ceb.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 10.Newmeyer DD, Ferguson-Miller S. Mitochondria: releasing power for life and unleashing the machineries of death. Cell. 2003;112:481–490. doi: 10.1016/s0092-8674(03)00116-8. [DOI] [PubMed] [Google Scholar]
- 11.Chipuk JE, Moldoveanu T, Llambi F, Parsons MJ, Green DR. The BCL-2 family reunion. Mol Cell. 2010;37:299–310. doi: 10.1016/j.molcel.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marchenko ND, Zaika A, Moll UM. Death signal-induced localization of p53 protein to mitochondria. A potential role in apoptotic signaling. J Biol Chem. 2000;275:16202–16212. doi: 10.1074/jbc.275.21.16202. [DOI] [PubMed] [Google Scholar]
- 13.Waterhouse NJ, Goldstein JC, von Ahsen O, Schuler M, Newmeyer DD, Green DR. Cytochrome c maintains mitochondrial transmembrane potential and ATP generation after outer mitochondrial membrane permeabilization during the apoptotic process. J Cell Biol. 2001;153:319–328. doi: 10.1083/jcb.153.2.319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sansome C, Zaika A, Marchenko ND, Moll UM. Hypoxia death stimulus induces translocation of p53 protein to mitochondria. Detection by immunofluorescence on whole cells. FEBS Lett. 2001;488:110–115. doi: 10.1016/s0014-5793(00)02368-1. [DOI] [PubMed] [Google Scholar]
- 15.Moll UM, Marchenko N, Zhang XK. p53 and Nur77/TR3–transcription factors that directly target mitochondria for cell death induction. Oncogene. 2006;25:4725–4743. doi: 10.1038/sj.onc.1209601. [DOI] [PubMed] [Google Scholar]
- 16.Wong RS. Apoptosis in cancer: from pathogenesis to treatment. J Exp Clin Cancer Res. 2011;30:87. doi: 10.1186/1756-9966-30-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Oro C, Jans DA. The tumour specific pro-apoptotic factor apoptin (Vp3) from chicken anaemia virus. Curr Drug Targets. 2004;5:179–190. doi: 10.2174/1389450043490631. [DOI] [PubMed] [Google Scholar]
- 18.Poon IKH, Oro C, Dias MM, Zhang JP, Jans DA. A tumor cell-specific nuclear targeting signal within chicken anemia virus VP3/apoptin. J Virol. 2005;79:1339–1341. doi: 10.1128/JVI.79.2.1339-1341.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Danen-Van Oorschot AA, Fischer DF, Grimbergen JM, Klein B, Zhuang S, Falkenburg JH, Backendorf C, Quax PH, der Eb AJ Van, Noteborn MH. Apoptin induces apoptosis in human transformed and malignant cells but not in normal cells. Proc Natl Acad Sci U S A. 1997;94:5843–5847. doi: 10.1073/pnas.94.11.5843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jangamreddy JR, Panigrahi S, Lotfi K, Yadav M, Maddika S, Tripathi AK, Sanyal S, Los MJ. Mapping of Apoptin-interaction with BCR-ABL1, and development of apoptin-based targeted therapy. Oncotarget. 2014 doi: 10.18632/oncotarget.2278. [in press, 000966–R1] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guelen L, Paterson H, Gaken J, Meyers M, Farzaneh F, Tavassoli M. TAT-apoptin is efficiently delivered and induces apoptosis in cancer cells. Oncogene. 2004;23:1153–1165. doi: 10.1038/sj.onc.1207224. [DOI] [PubMed] [Google Scholar]
- 22.Rohn JL, Zhang YH, Leliveld SR, Danen-van Oorschot AA, Henriquez NV, Abrahams JP, Noteborn MH. Relevance of apoptin's integrity for its functional behavior. J Virol. 2005;79:1337–1338. doi: 10.1128/JVI.79.2.1337-1338.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sauvage V, Cheval J, Foulongne V, Gouilh MA, Pariente K, Manuguerra JC, Richardson J, Dereure O, Lecuit M, Burguiere A. Identification of the first human gyrovirus, a virus related to chicken anemia virus. J Virol. 2011;85:7948–7950. doi: 10.1128/JVI.00639-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rollano Peñaloza OM, Lewandowska M, Stetefeld J, Ossysek K, Madej M, Bereta J, Sobczak M, Shojaei S, Ghavami S, Łos MJ. Apoptins: selective anticancer agents. Trends Mol Med. 2014;20 doi: 10.1016/j.molmed.2014.07.003. [In press] [DOI] [PubMed] [Google Scholar]
- 25.Bullenkamp J, Cole D, Malik F, Alkhatabi H, Kulasekararaj A, Odell EW, Farzaneh F, Gaken J, Tavassoli M. Human Gyrovirus Apoptin shows a similar subcellular distribution pattern and apoptosis induction as the chicken anaemia virus derived VP3/Apoptin. Cell Death Differ. 2012;3 doi: 10.1038/cddis.2012.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Almazan G, McKay R. An oligodendrocyte precursor cell line from rat optic nerve. Brain Res. 1992;579:234–245. doi: 10.1016/0006-8993(92)90056-f. [DOI] [PubMed] [Google Scholar]
- 27.Mascetti G, Carrara S, Vergani L. Relationship between chromatin compactness and dye uptake for in situ chromatin stained with DAPI. Cytometry. 2001;44:113–119. doi: 10.1002/1097-0320(20010601)44:2<113::aid-cyto1089>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 28.Hughes MA, Harper N, Butterworth M, Cain K, Cohen GM, MacFarlane M. Reconstitution of the Death-inducing Signaling Comp ex Reveals a Substrate Switch that Determines CD95-Mediated Death or Survival. Mol Cell. 2009;35:265–279. doi: 10.1016/j.molcel.2009.06.012. [DOI] [PubMed] [Google Scholar]
- 29.Xerri L, Devilard E, Bouabdallah R, Stoppa AM, Hassoun J, Birg F. FADD expression and caspase activation in B-cell lymphomas resistant to Fas-mediated apoptosis. Br J Haematol. 1999;106:652–661. doi: 10.1046/j.1365-2141.1999.01586.x. [DOI] [PubMed] [Google Scholar]
- 30.Aggarwal S, Gupta S. Increased activity of caspase 3 and caspase 8 in anti-Fas-induced apoptosis in lymphocytes from ageing humans. Clin Exp Immunol. 1999;117:285–290. doi: 10.1046/j.1365-2249.1999.00957.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim JW, Choi EJ, Joe CO. Activation of death-inducing signaling complex (DISC) by pro-apoptotic C-terminal fragment of RIP. Oncogene. 2000;19:4491–4499. doi: 10.1038/sj.onc.1203796. [DOI] [PubMed] [Google Scholar]
- 32.Elmore S. Apoptosis: A review of programmed cell death. Toxicol Pathol. 2007;35:495–516. doi: 10.1080/01926230701320337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lugli E, Troiano L, Ferraresi R, Roat E, Prada N, Nasi M, Pinti M, Cooper EL, Cossarizza A. Characterization of cells with different mitochondrial membrane potential during apoptosis. Cytometry A. 2005;68:28–35. doi: 10.1002/cyto.a.20188. [DOI] [PubMed] [Google Scholar]
- 34.Vaseva AV, Marchenko ND, Ji K, Tsirka SE, Holzmann S, Moll UM. p53 opens the mitochondrial permeability transition pore to trigger necrosis. Cell. 2012;149:1536–1548. doi: 10.1016/j.cell.2012.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Arnoult D, Parone P, Martinou JC, Antonsson B, Estaquier J, Ameisen JC. Mitochondrial release of apoptosis-inducing factor occurs downstream of cytochrome c release in response to several proapoptotic stimuli. J Cell Biol. 2002;159:923–929. doi: 10.1083/jcb.200207071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Martinou JC, Green DR. Breaking the mitochondrial barrier. Nat Rev Mol Cell Biol. 2001;2:63–67. doi: 10.1038/35048069. [DOI] [PubMed] [Google Scholar]
- 37.Riedl SJ, Salvesen GS. The apoptosome: signalling platform of cell death. Nat Rev Mol Cell Biol. 2007;8:405–413. doi: 10.1038/nrm2153. [DOI] [PubMed] [Google Scholar]
- 38.Erster S, Moll UM. Stress-induced p53 runs a direct mitochondrial death program: its role in physiologic and pathophysiologic stress responses in vivo. Cell Cycle. 2004;3:1492–1495. doi: 10.4161/cc.3.12.1318. [DOI] [PubMed] [Google Scholar]
- 39.Rajpal A, Cho YA, Yelent B, Koza-Taylor PH, Li D, Chen E, Whang M, Kang C, Turi TG, Winoto A. Transcriptional activation of known and novel apoptotic pathways by Nur77 orphan steroid receptor. EMBO J. 2003;22:6526–6536. doi: 10.1093/emboj/cdg620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Noteborn MH. Proteins selectively killing tumor cells. Eur J Pharmacol. 2009;625:165–173. doi: 10.1016/j.ejphar.2009.06.068. [DOI] [PubMed] [Google Scholar]
- 41.Zhuang SM, Landegent JE, Verschueren CA, Falkenburg JH, van Ormondt H, van der Eb AJ, Noteborn MH. Apoptin, a protein encoded by chicken anemia virus, induces cell death in various human hematologic malignant cells in vitro. Leukemia. 1995;9(Suppl. 1):S118–S120. [PubMed] [Google Scholar]
- 42.Dhein J, Walczak H, Baumler C, Debatin KM, Krammer PH. Autocrine T-cell suicide mediated by APO-1/(Fas/CD95) Nature. 1995;373:438–441. doi: 10.1038/373438a0. [DOI] [PubMed] [Google Scholar]
- 43.Dhein J, Walczak H, Westendorp MO, Baumler C, Stricker K, Frank R, Debatin KM, Krammer PH. Molecular mechanisms of APO-1/Fas(CD95)-mediated apoptosis in tolerance and AIDS. Behring Inst Mitt. 1995:13–20. [PubMed] [Google Scholar]
- 44.Fulda S, Los M, Friesen C, Debatin KM. Chemosensitivity of solid tumor cells in vitro is related to activation of the CD95 system. Int J Cancer. 1998;76:105–114. doi: 10.1002/(sici)1097-0215(19980330)76:1<105::aid-ijc17>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 45.Brunner T, Mogil RJ, Laface D, Yoo NJ, Mahboubi A, Echeverri F, Martin SJ, Force WR, Lynch DH, Ware CF. Cell-autonomous Fas (Cd95) Fas-ligand interaction mediates activation-induced apoptosis in T-cell hybridomas. Nature. 1995;373:441–444. doi: 10.1038/373441a0. [DOI] [PubMed] [Google Scholar]
- 46.Holler N, Zaru R, Micheau O, Thome M, Attinger A, Valitutti S, Bodmer JL, Schneider P, Seed B, Tschopp J. Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat Immunol. 2000;1:489–495. doi: 10.1038/82732. [DOI] [PubMed] [Google Scholar]
- 47.Martinou JC, Desagher S, Antonsson B. Cytochrome c release from mitochondria: all or nothing. Nat Cell Biol. 2000;2:E41–E43. doi: 10.1038/35004069. [DOI] [PubMed] [Google Scholar]
- 48.Creagh EM, Martin SJ. Cell stress-associated caspase activation: intrinsically complex? Sci STKE. 2003 doi: 10.1126/stke.2003.175.pe11. [DOI] [PubMed] [Google Scholar]
- 49.Renz A, Berdel WE, Kreuter M, Belka C, Schulze-Osthoff K, Los M. Rapid extracellular release of cytochrome c is specific for apoptosis and marks cell death in vivo. Blood. 2001;98:1542–1548. doi: 10.1182/blood.v98.5.1542. [DOI] [PubMed] [Google Scholar]
- 50.Schulze-Osthoff K, Ferrari D, Los M, Wesselborg S, Peter ME. Apoptosis signaling by death receptors. Eur J Biochem. 1998;254:439–459. doi: 10.1046/j.1432-1327.1998.2540439.x. [DOI] [PubMed] [Google Scholar]
- 51.Bratton SB, Walker G, Srinivasula SM, Sun XM, Butterworth M, Alnemri ES, Cohen GM. Recruitment, activation and retention of caspases-9 and -3 by Apaf-1 apoptosome and associated XIAP complexes. EMBO J. 2001;20:998–1009. doi: 10.1093/emboj/20.5.998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bratton SB, Salvesen GS. Regulation of the Apaf-1-caspase-9 apoptosome. J Cell Sci. 2010;123:3209–3214. doi: 10.1242/jcs.073643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Marsden VS, Strasser A. Control of apoptosis in the immune system: Bcl-2, BH3-only proteins and more. Annu Rev Immunol. 2003;21:71–105. doi: 10.1146/annurev.immunol.21.120601.141029. [DOI] [PubMed] [Google Scholar]
- 54.Lindsten T, Ross AJ, King A, Zong WX, Rathmell JC, Shiels HA, Ulrich E, Waymire KG, Mahar P, Frauwirth K. The combined functions of proapoptotic Bcl-2 family members Bak and Bax are essential for normal development of multiple tissues. Mol Cell. 2000;6:1389–1399. doi: 10.1016/s1097-2765(00)00136-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rivlin N, Brosh R, Oren M, Rotter V. Mutations in the p53 tumor suppressor gene: important milestones at the various steps of tumorigenesis. Genes Cancer. 2011;2:466–474. doi: 10.1177/1947601911408889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wattel E, Preudhomme C, Hecquet B, Vanrumbeke M, Quesnel B, Dervite I, Morel P, Fenaux P. p53 mutations are associated with resistance to chemotherapy and short survival in hematologic malignancies. Blood. 1994;84:3148–3157. [PubMed] [Google Scholar]