

Abstract
Strain superinfection occurs when a second pathogen strain infects a host already infected with a primary strain. The selective pressures that drive strain divergence, which underlies superinfection, and allow penetration of a new strain into a host population are critical knowledge gaps relevant to shifts in infectious disease epidemiology. In regions of endemicity with a high prevalence of infection, broad population immunity develops against Anaplasma marginale, a highly antigenically variant rickettsial pathogen, and creates strong selective pressure for emergence of and superinfection with strains that differ in their Msp2 variant repertoires. The strains may emerge either by msp2 locus duplication and allelic divergence on an existing genomic background or by introduction of a strain with a different msp2 allelic repertoire on a distinct genomic background. To answer this question, we developed a multilocus typing assay based on high-throughput sequencing of non-msp2 target loci to distinguish among strains with different genomic backgrounds. The technical error level was statistically defined based on the percentage of perfect sequence matches of clones of each target locus and validated using experimental single strains and strain pairs. Testing of A. marginale-positive samples from tropical regions where A. marginale infection is endemic identified individual infections that contained unique alleles for all five targeted loci. The data revealed a highly significant difference in the number of strains per animal in the tropical regions compared to infections in temperate regions and strongly supported the hypothesis that transmission of genomically distinct A. marginale strains predominates in high-prevalence areas of endemicity.
INTRODUCTION
Microbial strain structure is dynamic over space and time. For pathogens, shifts in structure characterized by emergence of genotypically unique strains and loss of others results in changing patterns of disease, whether within an individual or a population. However, the scales of space and time differ markedly among pathogens, depending on multiple factors, including pathogen-specific mechanisms of genetic change and effective pathogen population size. For example, RNA viruses such as lentiviruses are capable of rapidly generating mutations throughout the genome due to the large number of progeny emanating from a single virus within an infected cell and the minimal proofreading during replication (1–3). In contrast, bacteria and eukaryotic parasites have both a limited number of progeny per replicative cycle and stricter control of genomic fidelity between generations (4, 5). These differences in the time required to generate genetic variants aside, the successful emergence of a new genotypically unique strain is dependent on the strength of the selective pressure acting on the pathogen population (6).
Immunity is a strong selective pressure for newly emergent genetic variants. Within an individual host, the immune response selects for new variants: for viral pathogens, this includes true genomic variants, whereas higher-order microbial pathogens most typically use differential expression within a more stable genome (3–5, 7, 8). However, at the host population level, immunity is also a strong selective pressure and has been postulated to be a driver for diversification in strain structure across the spectrum of microbial pathogens (9, 10). In this model, the development of immunity within a temporally and spatially defined host population against an existing strain provides strong selection for an antigenically distinct strain that infects the host population regardless of the preexisting immunity (11).
We have been investigating how infection prevalence and associated population immunity drive strain structure using Anaplasma marginale, an alphaproteobacterium that infects wild and domestic ruminants (12). A. marginale establishes persistent infection within an animal by antigenic variation in the immunodominant surface protein, major surface protein 2 (MSP2), which is generated via gene conversion using a genomic repertoire of distinct msp2 alleles (13–16). Genomic comparison of multiple strains revealed that although A. marginale has a “closed core” genome with essentially the same gene content among all strains, the repertoire of msp2 alleles is distinct (17, 18). Importantly, strains with a distinct msp2 allelic repertoire have been shown experimentally to be capable of strain superinfection, in which a second strain establishes infection in an animal already infected with a primary strain (15, 19, 20). Superinfection requires that the second strain contain at least one unique msp2 allele and that this be expressed at superinfection (19, 21). Consistent with these experimental observations, we have more recently identified a significantly higher number and diversity in expressed msp2 alleles in A. marginale infections from tropical regions with high prevalence and high population immunity compared to temperate regions with low prevalence and low population immunity (22).
The increase in msp2 allelic diversity in the high-prevalence tropical regions may arise from two different scenarios (Fig. 1). In the first, the theoretical strain A establishes broad population immunity and thus selects for a distinct strain B that has a unique msp2 allelic repertoire. Alternatively, strain A may expand its allelic repertoire on the same genomic background to generate a closely related strain, A′. The latter possibility is supported by msp2 allelic gene duplication within a single strain and the presence of different numbers of msp2 loci and alleles among strains (16–19). Either strain A′ or B would generate the observed increase in the number and diversity of expressed msp2 alleles within the high-prevalence tropical regions (22). In the present study, we address this question by developing and validating a multilocus strain typing approach and testing whether strain superinfection in tropical regions is linked to strains with the same or different genomic backgrounds.
FIG 1.
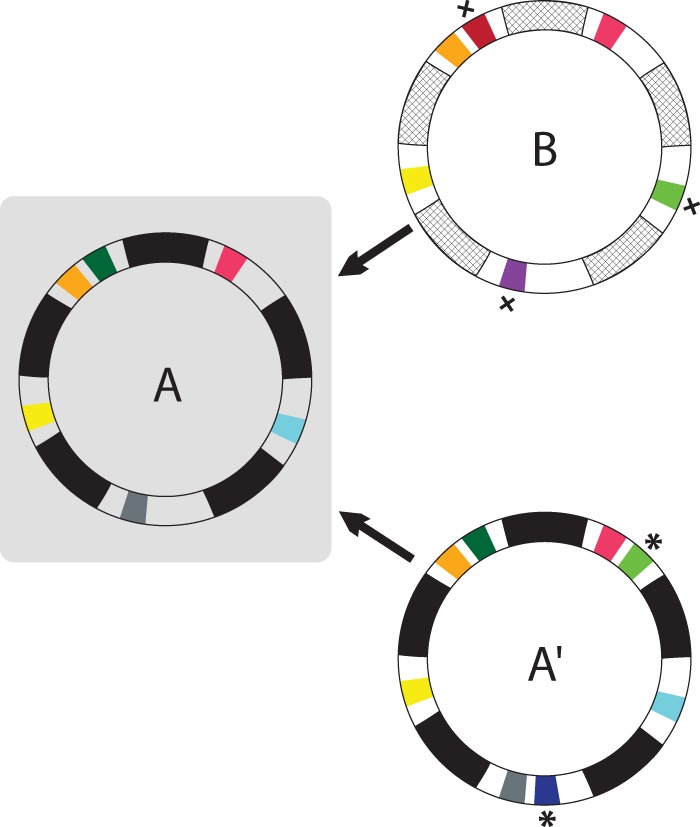
The increased msp2 allelic diversity in high-infection-prevalence populations may arise from superinfection by strains on the same or distinct genomic backgrounds. The gray box represents either an individual or population infected with a predominant strain, A; arrows represent the introduction of either a closely related strain, A′, or a strain B on a distinct genomic background. The multiple chromosomal msp2 alleles are represented by colored segments; each color represents an allele encoding a structurally and antigenically unique Msp2 variant. Strain A′ shares the same genomic background (indicated by the black segments) as strain A but differs in msp2 alleles generated by locus duplication and mutation (*). In contrast, strain B differs in genomic background, indicated by the hatched segments, as well as containing unique msp2 alleles (x).
MATERIALS AND METHODS
Identification of loci with strain-specific alleles.
Target loci that had previously been shown to be broadly conserved among strains (17, 18, 23) were initially screened for strain-specific alleles using the complete genome sequences of the St. Maries (GenBank accession no. CP000030) and Florida (CP001079) strains. Candidates were then screened against the whole-genome shotgun data contigs from the Oklahoma (AFMX00000000.1), Washington-Okanogan (AFMW00000000.1), and South Idaho (AFMY00000000.1) strains with the corresponding target loci using blastn, blastx, and tblastn analyses. Target sequences across the five strains were compiled using the VECTOR NTI software suite (Invitrogen), and single nucleotide polymorphisms (SNPs) and insertions/deletions (indels) were identified. Forward and reverse primers were designed for the identical conserved regions flanking the most informative SNPs and indels: omp5, 5′-CTGGAAAGCTGCACAGGATG-3′ and 5′-CGACGCTTCCGCAAACATAC-3′; omp9, 5′-GGCAATTCCAATCATGTGCG-3′ and 5′-CAAGCTGTGAAGTCACTACACG-3′; omp12, 5′-CTAGCGCTATGTTGCATGCATC-3′ and 5′-ACGCAAATTCAGATCACAGGG-3′; omp13, 5′-CAAGCAGATCCACAGCATCAATTC-3′ and 5′-GTGACGCCCTCATTGACC-3′; and omp14, 5′-GCAGAAGGAGTTGTCCAAGC-3′ and 5′-CCACTTATTTCCACAATCTCCATGC-3′ (see Fig. S1 in the supplemental material). The targeted regions are all <350 bp, allowing double coverage in the sequencing platform.
Sequencing.
DNA was extracted from either stabilates of the test strains (St. Maries, Washington-Okanogan, and Florida [24, 25]) or blood obtained directly from naturally infected A. marginale bacteremic animals using the Qiagen DNeasy blood and tissue kit following the manufacturer's instructions. Forward and reverse primers containing overhang adapters were added for each target locus and amplified under the following conditions: 95°C for 2 min; 30 cycles of 95°C for 20 s, 54°C for 30 s, and 68°C for 30 s; final extension at 68°C for 3 min; and holding at 4°C. Each amplicon pool was purified using Agencourt AMPure XP (Beckman Coulter), prepared using the Truseq protocol (Illumina), and sequenced using a MiSeq sequencer (Illumina). Runs were set to the “generate FASTQ only” work flow in Illumina Experiment Manager, and 500-cycle MiSeq v.2 reagent cartridges were used to sequence libraries with paired-end 251 cycles per read. Sequencing run monitoring was achieved through BaseSpace beta, and the demultiplexed data were analyzed using Sequencher 5.0 (Gene Codes). Consensus sequences were generated from paired ends using the “assemble by handle” method using the 100% sequence match criterion. Each consensus sequence was trimmed internal to the PCR primer sites, assigned to one of the target loci by BLAST, and then aligned to generate population reports. Within each sample, proportions of the different target alleles were calculated using Excel (Microsoft).
Technical error level.
The intrinsic variation associated with the sequencing procedure was determined using clones of targeted alleles, which eliminated any “true” genotypic variation within a strain or in a strain mixture. The five targeted outer membrane protein (omp) alleles were amplified from St. Maries genomic DNA and cloned using the Zero Blunt TOPO PCR cloning kit into pCR-Blunt II-TOPO plasmid vector (Invitrogen). Plasmid DNA was extracted from individual clones and Sanger sequenced using an ABI 3130x sequencer to ensure fidelity of the clone. PCR, library preparation, and MiSeq sequencing were then carried out as described above. The technical error level was calculated by determining the percentage of perfect sequence matches (100% identity with the reference clone) for each of three independent sequencing runs.
Detection of single strains and strain pairs.
Stabilates prepared from animals previously experimentally infected (18, 21) with single strains (St. Maries, Florida, or Washington-Okanogan) were either analyzed individually or mixed to create three defined strain pairs with equal representation of each strain based on bacteremia levels. DNA was extracted from the single strains or mixed strain pairs and then processed and sequenced as described above. Independent replicates of an uncloned strain (St. Maries; n = 3) were used to determine the variation observed within a single strain. The criterion for identifying an allele was set at a proportion of perfect sequence matches that was greater than the technical error level for the respective cloned omp + 3 standard deviations.
Strain composition in naturally occurring superinfections.
Blood was collected from adult cattle (>18 months old) pastured in high-prevalence tropical regions in the Mexican states of Veracruz and Chiapas (26, 27). Samples were collected in Medellin, Veracruz (19°03′N, 96°09′W; annual precipitation, 1,418 mm), La Concordia, Chiapas (16°07′N, 92°41′W; annual precipitation, 1,450 mm), and Tonala, Chiapas (16°06′N, 93°45′W; annual precipitation, 1,953 mm). Samples from temperate areas were obtained from adult cattle (>18 months) pastured in temperate areas in the United States: Tonasket, WA (48°42′N, 119°26′W; annual precipitation, 383 mm), and Burns, OR (43°35′N, 119°03′W; annual precipitation, 278 mm). Cattle had not been vaccinated to prevent A. marginale infection or treated to clear infection. All samples were confirmed positive using either direct detection of A. marginale bacteremia or by serological screening using an msp5 competitive enzyme-linked immunosorbent assay (CELISA) (VMRD, Inc.). DNA was extracted, processed, and sequenced as described above.
RESULTS
Identification of loci with strain-specific alleles.
Loci that would define the stable genomic background of a strain independent of the variable msp2 allelic repertoire were identified by screening the complete genome sequences of the St. Maries (GenBank accession no. CP000030) and Florida (CP001079) strains. Five omp loci were identified that encoded alleles specific to each strain (18, 23). The single nucleotide substitutions between the two strains ranged from 1 to 46, with the number of nucleotide differences due to indels ranging from 3 to 21 (Table 1). Each of these omp genes had previously been shown to be invariant within a strain during long-term persistent infection and among different animals infected with the same strain (18). The candidate loci were then compared among three additional strains (South Idaho, Washington-Okanogan, and Oklahoma) using sequences that had been generated by shotgun sequencing and previously deposited in GenBank (accession no. AFMY00000000.1, AFMX00000000.1, and AFMW00000000.1). While allelic identity was observed among individual loci between specific strain pairs, the inclusion of five target loci ensured that there were differences at multiple loci between any given strain pair (Table 1). Importantly, three of these strains (St. Maries, South Idaho, and Washington-Okanogan) are from within the same region in the northwest United States and are transmitted both naturally and experimentally by the same vector, Dermacentor andersoni (25, 28–30), suggesting that the ability of the five targets to distinguish among strains is conserved even among closely related strains. Primers were designed in the conserved regions among these five strains to encompass the largest number of strain-specific SNPs and indels (Table 2; see Fig. S1 in the supplemental material).
TABLE 1.
Strain-specific alleles for multilocus typing
| Locus | Strain | No. of nucleotide differences (substitutions/indels) for strain: |
||||
|---|---|---|---|---|---|---|
| St. Maries | Florida | South Idaho | Washington-Okanogan | Oklahoma | ||
| omp5 | St. Maries | 46/21 | 1/0 | 0/0 | 27/21 | |
| Florida | 46/21 | 45/21 | 46/21 | 19/0 | ||
| South Idaho | 1/0 | 45/21 | 1/0 | 26/21 | ||
| Washington-Okanogan | 0/0 | 46/21 | 1/0 | 27/21 | ||
| Oklahoma | 27/21 | 19/0 | 26/21 | 27/21 | ||
| omp9 | St. Maries | 1/0 | 44/21 | 44/21 | 4/0 | |
| Florida | 1/0 | 3/0 | 44/12 | 3/0 | ||
| South Idaho | 44/21 | 3/0 | 0/0 | 40/21 | ||
| Washington-Okanogan | 44/21 | 44/12 | 0/0 | 40/21 | ||
| Oklahoma | 4/0 | 3/0 | 40/21 | 40/21 | ||
| omp12 | St. Maries | 1/0 | 9/0 | 1/0 | 2/0 | |
| Florida | 1/0 | 10/0 | 0/0 | 1/0 | ||
| South Idaho | 9/0 | 10/0 | 10/0 | 11/0 | ||
| Washington-Okanogan | 1/0 | 0/0 | 10/0 | 1/0 | ||
| Oklahoma | 2/0 | 1/0 | 11/0 | 1/0 | ||
| omp13 | St. Maries | 4/0 | 13/18 | 4/0 | 3/0 | |
| Florida | 4/0 | 17/13 | 0/0 | 7/0 | ||
| South Idaho | 13/18 | 17/13 | 17/13 | 14/13 | ||
| Washington-Okanogan | 4/0 | 0/0 | 17/13 | 7/0 | ||
| Oklahoma | 3/0 | 7/0 | 14/13 | 7/0 | ||
| omp14 | St. Maries | 17/0 | 23/03 | 15/0 | 17/0 | |
| Florida | 17/0 | 36/3 | 2/0 | 0/0 | ||
| South Idaho | 23/3 | 36/3 | 38/3 | 36/3 | ||
| Washington-Okanogan | 15/0 | 2/0 | 38/3 | 2/0 | ||
| Oklahoma | 17/0 | 0/0 | 36/3 | 7/0 | ||
TABLE 2.
Targeted loci with strain-specific allelesa
| Gene (locus tag no.) | Target length (bp) | Coordinates |
|
|---|---|---|---|
| Genome | Gene | ||
| omp5 (AM1166) | 310 | 1042525–1042834 | 543–852 |
| omp9 (AM1222) | 318 | 1091404–1091721 | 405–722 |
| omp12 (AM1257) | 347 | 1117250–1117596 | 398–744 |
| omp13 (AM1258) | 306 | 1118171–1118476 | 254–559 |
| omp14 (AM075) | 327 | 63573–63899 | 518–844 |
The gene and locus tag designations, genome coordinates, and gene coordinates are based on the St. Maries strain.
Identification of single-strain infections.
The identity of each strain was accurately detected by high-throughput sequencing of the target alleles with the predicted allele represented by perfect matches. The numbers of paired-read consensus sequences obtained for each allele ranged from 2,229 to 8,080: omp5, 2,769 sequences in the Florida strain to 5,931 in the St. Maries strain; omp9, from 2,229 in the Florida strain to 3,083 in the Washington-Okanogan strain; omp12, from 2,908 in the St. Maries strain to 4,101 in the Florida strain; omp13, from 3,342 in the Florida strain to 4,072 in the St. Maries strain; and omp14, from 4,371 in the Washington-Okanogan strain to 8,080 in the Florida strain. Perfect matches (those without a single nucleotide mismatch) were >90% for all alleles in all strains, with a range from 90.4% for omp12 to 94.9% for omp9, both in the Washington-Okanogan strain. The percentage of perfect matches was independent of the number of sequences generated: for the Florida strain: there were 94.2% perfect match sequences out of 2,100 total for omp9 and 92.7% perfect match sequences out of 8,080 total for omp14. These percentages of perfect match sequences were higher than previously reported for a comparison set of four microbial genomes using MiSeq, reflecting the inclusion of only consensus sequences with no disagreement in overlap (31). To determine the technical error level associated with sequencing of the five loci, clones of each target allele were obtained for the St. Maries strain and sequenced using the same procedure in three biological replicates. The percentages of perfect match sequences for the cloned alleles were as follows: omp5, 93.8% ± 0.9%; omp9, 93.9% ± 1.3%; omp12, 92.4% ± 0.4%; omp13, 93.8% ± 0.5%; and omp14, 93.8% ± 1.1%. Thus, the higher percentage of perfect match sequences for the five alleles compared to previously published genomes (31) is reflected in the higher percentage using cloned omps, consistent with the designed sequence lengths being appropriate for strain differentiation by a multilocus approach. The percentages of perfect match sequences for each allele were not significantly different between cloned omp and the same omp in the uncloned St. Maries strain: omp5, P = 0.2; omp9, P = 0.2; omp12, P = 0.4; omp13, P = 0.1; and omp14, P = 0.2 (two-sample t test assuming unequal variances). The criterion for identifying an allele was set at a proportion of perfect match sequences that exceeded the technical error level by a minimum of 3 standard deviations.
Identification of multiple strain infections.
To determine if the multilocus approach would distinguish among strains, strain pairs were mixed to mimic strain superinfection and then sequenced. In each of the three strain pairs St. Maries and Florida, St. Maries and Washington-Okanogan, and Florida and Washington-Okanogan, each strain could be definitively identified (Table 3). When the omp alleles differed between the two paired strains, the two alleles each were detected with a percentage of perfect matches that exceeded the technical error level in all cases. When the omp alleles were common between the two paired strains (omp5 in the St. Maries and Washington-Okanogan strains and omp12 and omp13 in the Florida and Washington-Okanogan strains), the common allele was detected with a percentage of perfect match sequences that exceeded 90% (Table 3).
TABLE 3.
Detection of strain-specific alleles in experimental strain pairs
| Locus | Strains | Allele identifieda |
|||
|---|---|---|---|---|---|
| St. Maries | Florida | Washington-Okanogan | Common allele | ||
| omp5 | St. Maries and Florida | + | + | No | |
| St. Maries and Washington-Okanogan | + | + | Yes | ||
| Florida and Washington-Okanogan | + | + | No | ||
| omp9 | St. Maries and Florida | + | + | No | |
| St. Maries and Washington-Okanogan | + | + | No | ||
| Florida and Washington-Okanogan | + | + | No | ||
| omp12 | St. Maries and Florida | + | + | No | |
| St. Maries and Washington-Okanogan | + | + | No | ||
| Florida and Washington-Okanogan | + | + | Yes | ||
| omp13 | St. Maries and Florida | + | + | No | |
| St. Maries and Washington-Okanogan | + | + | No | ||
| Florida and Washington-Okanogan | + | + | Yes | ||
| omp14 | St. Maries and Florida | + | + | No | |
| St. Maries and Washington-Okanogan | + | + | No | ||
| Florida and Washington-Okanogan | + | + | No | ||
Identification based on a percentage of sequences perfectly matching the specific allele greater than 3 standard deviations above the technical error level based on identically sequenced cloned omp loci.
Strain composition in naturally occurring superinfections.
Infections from two tropical regions in Mexico were examined—Veracruz and Chiapas. The infection prevalence within these high-humidity, high-temperature regions has been reported as the highest in Mexico, consistent with strong selective pressure for strain superinfection (26, 27). Multilocus typing revealed multiple omp alleles in at least one locus for 21/23 tropical region infections, 16/16 for all infections obtained in Veracruz, and 5/7 for those from Chiapas (see Table S1 in the supplemental material). The numbers of unique alleles per infection, both for each locus and collectively across all loci, were significantly greater for tropical region infections than for temperate region infections (Table 4). This reflected both an increased percentage of loci with more than a single allele and an increase in the number of alleles at each locus (Table 4). Correspondingly, the maximal mean number of strains per animal in the tropical region was 3.3 ± 1.2, with up to 6 strains in a single infected animal. This was significantly higher (P = 0.0004, Student's unpaired t test) in the tropical regions compared to infections in two temperate regions, with a maximal mean of 1.3 ± 0.5 with a maximum of two strains in an individual infected animal.
TABLE 4.
Detection of strain-specific alleles in natural infection
| Locusa | Region | Mean no. of alleles detected/infection (maximum) | No. of infections/total with: |
|
|---|---|---|---|---|
| Multiple alleles at locus | >2 alleles at locus | |||
| omp5 | Tropical | 2.4 ± 1.0 (4) | 19/23 | 10/23 |
| Temperate | 1.3 ± 0.5 (2) | 3/8 | 0/8 | |
| omp9 | Tropical | 2.8 ± 1.2 (5) | 19/23 | 13/23 |
| Temperate | 1.0 ± 0.0 (1) | 0/4 | 0/4 | |
| omp12 | Tropical | 2.7 ± 1.1 (5) | 18/23 | 13/23 |
| Temperate | 1.1 ± 0.4 (2) | 1/8 | 0/8 | |
| omp13 | Tropical | 2.2 ± 1.0 (4) | 18/23 | 8/23 |
| Temperate | 1.1 ± 0.4 (2) | 1/8 | 0/8 | |
| omp14 | Tropical | 2.7 ± 1.4 (6) | 17/23 | 12/23 |
| Temperate | 1.1 ± 0.4 (2) | 1/8 | 0/8 | |
The greater number of alleles per tropical region infection compared to the temperate region infection was statistically significant for all loci collectively at P < 0.0001 and for each locus as follows: omp5, P = 0.006; omp9, P = 0.007; omp12, P = 0.0004; omp13, P = 0.006; and omp14, P = 0.004.
DISCUSSION
We accept that individual animals within regions of endemicity are infected with multiple strains of different genetic backgrounds. The following four lines of evidence support this conclusion. First, the multilocus typing assay detected only one allele per target omp locus when applied to experimental single-strain infections. Second, the assay detected the two predicted alleles in experimental paired-strain infections, and when there was a common allele between two strains, only the common allele was detected. Unique alleles were detected with percentages of perfect sequence matches that exceeded the mean + 3 standard deviations of the technical error level as determined for cloned omps and as represented by the omps in experimental single-strain infections. Third, individual infections from the high-prevalence tropical regions were identified that contained multiple unique alleles for all five target omp loci. As each omp locus in itself represents an independent detection event, the probability that the multiple unique alleles for all five target loci reflects sequencing error is extraordinarily low (P < 2 × 10−13). Finally, the number of alleles detected was significantly greater in the tropical region field infections than that in the temperate region infections for the cumulative set of loci and for each locus (Table 4). Sequencing errors would be expected to be equal between unknown samples from temperate and tropical infections that were handled identically using the same procedure, instrument, personnel, and analysis. The higher number of genetically distinct strains within individual animal infections within the tropical region compared to the temperate region (P = 0.0004) is consistent with prior independent studies using single-locus msp1α typing and msp2 expression site diversity (22). Typing using msp1α has provided significant epidemiologic information but is limited in its ability to define genetic background due to being a single locus and variations in the number of very closely related nucleotide repeats (17, 18, 32–37), which gives rise to errors in repeat numbers in replicate assays.
The previously detected increase in msp2 variants and their diversity in infections from tropical regions (22) may arise from a new strain, B, with a unique repertoire of msp2 alleles superinfecting an animal already carrying a primary strain, A, or, alternatively, via msp2 locus duplication and divergence in strain A to create A′, which is also capable of superinfection (Fig. 1) (11). Our findings strongly support the hypothesis that A. marginale superinfection in high-prevalence areas of endemicity is predominantly due to transmission of genomically distinct strains, supporting the proposed strain B scenario. How these “B” strains enter into the population is unknown: movement of infected domestic and/or wild animal reservoirs and introduction of different vector populations are likely routes. Once introduced, a strain with a unique msp2 repertoire has a competitive advantage in a population of animals with high prevalence of infection and consequent population immunity to the existing predominant strain, A (11, 21, 35). Although studies indicate that expression of a unique msp2 allele is required for strain superinfection (19, 21), that other OMPs encoded by unique alleles, such as those used in the multilocus typing assay, may contribute to competitive advantage remains an unexplored possibility.
Although the present study confirms the presence of strains on distinct genomic backgrounds, the alternative epidemiologic mechanism, emergence of a strain, A′, due to msp2 gene duplication and divergence, cannot be ruled out. The numbers of msp2 loci differ among strains, and there is genomic evidence of gene duplication and divergence (16–18). The significant increase in the number of msp2 variants in tropical versus temperate region, ∼3.5× (22), may arise from both an increase in the number of alleles per strain and the consequent increase in the number of segmental recombinants (15) and thus cannot be directly compared to the increase (∼2.2×) in the number of omp alleles between tropical and temperate infections in the present study. The full range of mechanisms by which new strains emerge, either in the msp2-centric scenario of strain A′ or the broader genomic changes in the strain B scenario, remain as significant knowledge gaps in our understanding of strain evolution.
Multiple genotypes have been detected within a related vaccine strain, A. marginale subsp. centrale (38). In contrast, the present data using multiple loci and comparing the percentages of perfect match sequences in cloned alleles with those in alleles in the uncloned A. marginale strains suggests that wild type strains, specifically the St. Maries, Florida, and Washington-Okanogan strains, are not composed of multiple genotypes. This is consistent with genomic assembly data and supports that A. marginale subsp. centrale is an outlier, postulated to reflect its 100-year passage history as a vaccine strain (38–40).
The dynamic relationship among population immunity, intrinsic microbial replication fitness, and antigenic variation is central to pathogen strain structure. These relationships are especially notable for arthropod-vector borne pathogens, including Anaplasma, African trypanosomes, relapsing fever Borrelia, and Plasmodium (4, 5, 41). Defining the underlying genetic change and the forces that shape the genome and gene expression will enable understanding of both how new strains emerge and their consequences for epidemiology and disease control.
Supplementary Material
ACKNOWLEDGMENTS
This work is dedicated to the memory of Francisco Alpirez Mendoza, who died 9 August 2014. F. Alpirez Mendoza was dedicated to tropical infectious diseases research and to improving the lives and livelihoods in his community.
This research was supported by National Institutes of Health grant R37 AI44005, U.S. Department of Agriculture grants ARS 5348-32000-033-00D and CSREES 35604-15440, and Programa de Salud Animal, INIFAP-CIRGOC. Eduardo Vallejo Esquerra was supported by a scholarship through the National Council of Science and Technology (CONACyT) and the Council of Science and Technology for the State of Querétaro (CONCYTEQ).
We appreciate the excellent technical support of Beverly Hunter and Lisa Orfe.
Footnotes
Published ahead of print 6 October 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.02537-14.
REFERENCES
- 1.Holland J, Spindler K, Horodyski F, Grabau E, Nichol S, VandePol S. 1982. Rapid evolution of RNA genomes. Science 215:1577–1585. 10.1126/science.7041255. [DOI] [PubMed] [Google Scholar]
- 2.Kato N. 2000. Genome of hepatitis C virus (HCV): gene organization, sequence diversity, and variation. Microb. Comp. Genomics 5:129–151. 10.1089/omi.1.2000.5.129. [DOI] [PubMed] [Google Scholar]
- 3.Lauring AS, Andino R. 2010. Quasispecies theory and the behavior of RNA viruses. PLoS Pathog. 6:e1001005. 10.1371/journal.ppat.1001005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Deitsch KW, Lukehart SA, Stringer JR. 2009. Common strategies for antigenic variation by bacterial, fungal and protozoan pathogens. Nat. Rev. Microbiol. 7:493–503. 10.1038/nrmicro2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Palmer GH, Bankhead T, Lukehart SA. 2009. Nothing is permanent but change: antigenic variation in persistent bacterial pathogens. Cell. Microbiol. 11:1697–1705. 10.1111/j.1462-5822.2009.01366.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Adams B, Sasaki A. 2009. Antigenic distance and cross-immunity, invasibility and coexistence of pathogen strains in an epidemiological model with discrete antigenic space. Theor. Popul. Biol. 76:157–167. 10.1016/j.tpb.2009.06.001. [DOI] [PubMed] [Google Scholar]
- 7.Simmonds P. 2004. Genetic diversity and evolution of hepatitis C virus—15 years on. J. Gen. Virol. 85:3173–3180. 10.1099/vir.0.80401-0. [DOI] [PubMed] [Google Scholar]
- 8.Smith DM, Richman DD, Little SJ. 2005. HIV superinfection. J. Infect. Dis. 192:438–444. 10.1086/431682. [DOI] [PubMed] [Google Scholar]
- 9.Hethcote HW. 2000. The mathematics of infectious diseases. SIAM Rev. 42:599–653. 10.1137/S0036144500371907. [DOI] [Google Scholar]
- 10.Keeling MJ, Rohani P. 2008. Modeling infectious diseases in humans and animals, p 105–154 Princeton University Press, Princeton, NJ. [Google Scholar]
- 11.Palmer GH, Brayton KA. 2013. Antigenic variation and transmission fitness as drivers of bacterial strain structure. Cell. Microbiol. 15:1969–1975. 10.1111/cmi.12182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dumler JS, Barbet AF, Bekker CPJ, Dasch GA, Palmer GH, Ray SC, Rikihisa Y, Rurangirwa FR. 2001. Reorganization of genera in the families Rickettsiaceae and Anaplasmataceae in the order Rickettsiales; unification of some species of Ehrlichia with Anaplasma, Cowdria with Ehrlichia, and Ehrlichia with Neorickettsia; descriptions of six new species combinations; and designation of Ehrlichia equi and “HGE agent” as subjective synonyms of Ehrlichia phagocytophila. Int. J. Syst. Evol. Microbiol. 51:2145–2165. 10.1099/00207713-51-6-2145. [DOI] [PubMed] [Google Scholar]
- 13.Brayton KA, Knowles DP, McGuire TC, Palmer GH. 2001. Efficient use of small genome to generate antigenic diversity in tick-borne ehrlichial pathogens. Proc. Natl. Acad. Sci. U. S. A. 98:4130–4135. 10.1073/pnas.071056298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brayton KA, Meeus PFM, Barbet AF, Palmer GH. 2003. Simultaneous variation of the immunodominant outer membrane proteins MSP2 and MSP3 during Anaplasma marginale persistence in vivo. Infect. Immun. 71:6627–6632. 10.1128/IAI.71.11.6627-6632.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Futse JE, Brayton KA, Knowles DP, Palmer GH. 2005. Structural basis for segmental gene conversion in generation of Anaplasma marginale outer membrane protein variants. Mol. Microbiol. 57:212–221. 10.1111/j.1365-2958.2005.04670.x. [DOI] [PubMed] [Google Scholar]
- 16.Futse JE, Brayton KA, Nydam SD, Palmer GH. 2003. Generation of antigenic variants by gene conversion: evidence for recombination fitness selection at the locus level in Anaplasma marginale. Infect. Immun. 71:6627–6632. 10.1128/IAI.71.11.6627-6632.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brayton KA, Kappmeyer LS, Herndon DR, Dark MJ, Tibbals DL, Palmer GH, McGuire TC, Knowles DP. 2005. Complete genome sequencing of Anaplasma marginale reveals that the surface is skewed to two superfamilies of outer membrane proteins. Proc. Natl. Acad. Sci. U. S. A. 102:844–849. 10.1073/pnas.0406656102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dark MJ, Herndon D, Kappmeyer LS, Gonzales MP, Nordeen E, Palmer GH, Knowles DP, Brayton KA. 2009. Conservation in the face of diversity: multistrain analysis of an intracellular bacterium. BMC Genomics 10:16–28. 10.1186/1471-2164-10-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rodriguez JL, Palmer GH, Knowles DP, Brayton KA. 2005. Distinctly different msp2 pseudogene repertoires in Anaplasma marginale strains that are capable of superinfection. Gene 361:127–132. 10.1016/j.gene.2005.06.038. [DOI] [PubMed] [Google Scholar]
- 20.Galletti MF, Ueti MW, Knowles DP, Brayton KA, Palmer GH. 2009. Independence of Anaplasma marginale strains with high and low transmission efficiencies in the tick vector following simultaneous acquisition by feeding on a superinfected mammalian reservoir host. Infect. Immun. 77:1459–1464. 10.1128/IAI.01518-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Futse JE, Brayton KA, Dark MJ, Knowles DP, Palmer GH. 2008. Superinfection as a driver of genomic diversification in antigenically variant pathogens. Proc. Natl. Acad. Sci. U. S. A. 105:2123–2127. 10.1073/pnas.0710333105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ueti MW, Tan Y, Broschat SL, Castañeda Ortiz EJ, Camacho-Nuez M, Mosqueda JJ, Scoles GA, Grimes M, Brayton KA, Palmer GH. 2012. Expansion of variant diversity associated with high prevalence of pathogen strain superinfection under conditions of natural transmission. Infect. Immun. 80:2354–2360. 10.1128/IAI.00341-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Noh SM, Brayton KA, Knowles DP, Agnes JT, Dark MJ, Brown WC, Baszler TV, Palmer GH. 2006. Differential expression and sequence conservation of the Anaplasma marginale msp2 gene superfamily outer membrane proteins. Infect. Immun. 74:3471–3479. 10.1128/IAI.01843-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McGuire TC, Palmer GH, Goff WL, Johnson MI, Davis WC. 1984. Common and isolate restricted antigens of Anaplasma marginale detected with monoclonal antibodies. Infect. Immun. 45:697–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Eriks IS, Stiller D, Goff WL, Panton M, Parish SM, McElwain TF, Palmer GH. 1994. Molecular and biological characterization of a new isolated Anaplasma marginale strain. J. Vet. Diagn. Invest. 6:435–441. 10.1177/104063879400600406. [DOI] [PubMed] [Google Scholar]
- 26.Cossio-Bayúgar BR, Rodriguez SD, Garcia-Ortiz MA, Garcia-Tapia D, Aboytes-Torres R. 1997. Bovine anaplasmosis prevalence in northern Veracruz state, Mexico. Prev. Vet. Med. 32:165–170. 10.1016/S0167-5877(97)00016-0. [DOI] [PubMed] [Google Scholar]
- 27.Garcia-Tapia D, Lopez-Rojas M, Cossio-Bayugar R, Garcia-Vazquez Z, Garcia-Ortiz MA, Dominguez-Jalil P, Aboytes Torres R. 1996. Seroprevalencia de anaplasmosis en explotaciones bovinas de la zona norte de Veracruz. Tec. Pecu. Mex. 34:38–45. [Google Scholar]
- 28.Eriks IS, Stiller D, Palmer GH. 1993. Impact of persistent Anaplasma marginale rickettsemia on tick infection and transmission. J. Clin. Microbiol. 31:2091–2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Scoles GA, Ueti MW, Palmer GH. 2005. Variation among geographically separated populations of Dermacentor andersoni (Acari: Ixodidae) in midgut susceptibility to Anaplasma marginale (Rickettsiales:Anaplasmataceae). J. Med. Entomol. 42:153–162. 10.1603/0022-2585(2005)042[0153:VAGSPO]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 30.Scoles GA, Broce AB, Lysyk TJ, Palmer GH. 2005. Relative efficiency of biological transmission of Anaplasma marginale (Rickettsiales: Anaplasmataceae) by Dermacentor andersoni Stiles (Acari: Ixodidae) compared to mechanical transmission by the stable fly, Stomoxys calcitrans (L.) (Diptera: Muscidae). J. Med. Entomol. 42:668–675. 10.1603/0022-2585(2005)042[0668:REOBTO]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 31.Quail MA, Smith M, Coupland P, Otto TD, Harris SR, Connor TR, Bertoni A, Swerdlow HP, Yong G. 2012. A tale of three next generation sequencing platforms: comparison of Ion Torrent, Pacific Biosciences and Illumina MiSeq sequencers. BMC Genomics 13:341. 10.1186/1471-2164-13-341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Palmer GH, Rurangirwa FR, McElwain TF. 2001. Strain composition of the ehrlichia Anaplasma marginale within persistently infected cattle, a mammalian reservoir for tick-transmission. J. Clin. Microbiol. 39:631–635. 10.1128/JCM.39.2.631-635.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.de la Fuente J, Van Den Bussche RA, Kocan KM. 2001. Molecular phylogeny and biogeography of North American isolates of Anaplasma marginale (Rickettsiaceae: Ehrlichieae). Vet. Parasitol. 97:65–76. 10.1016/S0304-4017(01)00378-8. [DOI] [PubMed] [Google Scholar]
- 34.de la Fuente J, Van Den Bussche RA, Garcia-Garcia JC, Rodríguez SD, García MA, Guglielmone AA, Mangold AJ, Friche Passos LM, Barbosa Ribeiro MF, Blouin EF, Kocan KM. 2002. Phylogeography of New World isolates of Anaplasma marginale based on major surface protein sequences. Vet. Microbiol. 88:275–285. 10.1016/S0378-1135(02)00122-0. [DOI] [PubMed] [Google Scholar]
- 35.Palmer GH, Knowles DP, Rodriguez JL, Gnad DP, Hollis LC, Marston T, Brayton KA. 2004. Stochastic transmission of multiple genotypically distinct Anaplasma marginale strains in a herd with high prevalence of Anaplasma infection. J. Clin. Microbiol. 42:5381–5384. 10.1128/JCM.42.11.5381-5384.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.De la Fuente J, Passos LM, Van Den Bussche RA, Ribeiro MF, Facury-Filho EJ, Kocan KM. 2004. Genetic diversity and molecular phylogeny of Anaplasma marginale isolates from Minas Gerais, Brazil. Vet. Parasitol. 121:307–316. 10.1016/j.vetpar.2004.02.021. [DOI] [PubMed] [Google Scholar]
- 37.Ruybal P, Moretta R, Perez A, Petrigh R, Zimmer P, Alcarez E, Echaide I, Torioni de Echaide S, Kocan KM, de la Fuente J, Farber M. 2009. Genetic diversity of Anaplasma marginale in Argentina. Vet. Parasitol. 162:176–180. 10.1016/j.vetpar.2009.02.006. [DOI] [PubMed] [Google Scholar]
- 38.Herndon DR, Palmer GH, Shkap V, Knowles DP, Jr, Brayton KA. 2010. Complete genome sequence of Anaplasma marginale subsp. centrale. J. Bacteriol. 192:379–380. 10.1128/JB.01330-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Herndon D, Ueti MW, Reif KE, Noh SM, Brayton KA, Agnes JA, Palmer GH. 2013. Identification of multilocus heterogeneity in Anaplasma marginale subsp. centrale and its restriction following tick-borne transmission. Infect. Immun. 81:1852–1858. 10.1128/IAI.00199-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Palmer GH. 2009. Sir Arnold Theiler and the discovery of anaplasmosis: a centennial perspective. Onderstepoort J. Vet. Res. 76:75–79. 10.4102/ojvr.v76i1.68. [DOI] [PubMed] [Google Scholar]
- 41.Hall JPJ, Wang H, Barry JD. 2013. Mosaic VSGs and the scope of Trypanosoma brucei antigenic variation. PLoS Pathog. 9:e1003502. 10.1371/journal.ppat.1003502. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.