

Abstract
Apoptosis is an intrinsic immune defense mechanism in the host response to microbial infection. Not surprisingly, many pathogens have evolved various strategies to manipulate this important pathway to benefit their own survival and dissemination in the host during infection. To our knowledge, no attempts have been made to explore the host cell survival signals modulated by the bacterium Enterococcus faecalis. Here, we show for the first time that during early stages of infection, internalized enterococci can prevent host cell (RAW264.7 cells, primary macrophages, and mouse embryonic fibroblasts [MEFs]) apoptosis induced by a wide spectrum of proapoptotic stimuli. Activation of caspase 3 and cleavage of the caspase 3 substrate poly(ADP-ribose) polymerase were inhibited in E. faecalis-infected cells, indicating that E. faecalis protects macrophages from apoptosis by inhibiting caspase 3 activation. This antiapoptotic activity in E. faecalis-infected cells was dependent on the activation of the phosphatidylinositol 3-kinase (PI3K)/Akt signaling pathway, which resulted in the increased expression of the antiapoptotic factor Bcl-2 and decreased expression of the proapoptotic factor Bax. Further analysis revealed that active E. faecalis physiology was important for inhibition of host cell apoptosis, and this feature seemed to be a strain-independent trait among E. faecalis isolates. Employing a mouse peritonitis model, we also determined that cells collected from the peritoneal lavage fluid of E. faecalis-infected mice showed reduced levels of apoptosis compared to cells from uninfected mice. These results show early modulation of apoptosis during infection and have important implications for enterococcal pathogenesis.
INTRODUCTION
Enterococci are ubiquitous in the gastrointestinal tracts of most complex metazoans, where they occur as part of the commensal flora at approximately 104 to 107 organisms per gram of feces. Despite being a commensal, Enterococcus faecalis is endowed with traits that make it an opportunistic pathogen, especially in immunocompromised hosts (1, 2). Enterococci have emerged as a leading cause of antibiotic-resistant infections and now rank among the most common nosocomial pathogens infecting the bloodstream, surgical sites, and urinary tract (3, 4). While substantial progress has been made in recent years in understanding the genetic makeup of these organisms, much remains to be learned regarding how they interact with host cells during enterococcal infection.
It has been previously reported that E. faecalis can translocate through the intact epithelial cell monolayer in vitro (5) and disseminate from the intestinal lumen into the bloodstream, liver, and spleen in an antibiotic-treated murine model of superinfection (6). Enterococci that gain access to the sterile abdominal cavity, either from the endogenous flora or as a result of surgical intervention, encounter tissue and peritoneal macrophages involved in the first line of host defense. For these commensals to cause systemic infection, they must, at least in part, escape the bactericidal activity of macrophages to enter draining lymph nodes or the bloodstream. Certain strains of E. faecalis are known to survive within macrophages for extended periods (7–9). The resulting failure of macrophages to kill intracellular E. faecalis likely promotes the systemic spread of infection (6). Therefore, elucidation of E. faecalis survival mechanisms in macrophages, and the effect on host signaling pathways, may provide new ways to prevent enterococcal infection.
Apoptosis is a highly conserved pathway designed to maintain tissue homeostasis by elimination of aged and damaged cells and also serves as an important defense mechanism to control bacteria, viruses, and parasites during infection (10, 11). There are two means by which apoptosis is triggered: the extrinsic and intrinsic pathways. Stimulation of the transmembrane death receptors with their cognate ligands can activate the extrinsic pathway (12, 13). Upon stimulation, these receptors transmit external apoptotic signals and result in the activation of caspase 3 within the cell. In contrast, the intrinsic pathway is initiated by signaling factors released from the mitochondria (14). Various intrinsic stimuli activate Bcl-2 homology 3 (BH3)-only proteins, which overcome the inhibitory effects of antiapoptotic Bcl-2 proteins (15, 16). The activated BH3-only proteins promote the oligomerization of proapoptotic proteins, such as Bax and Bak, in the mitochondrial outer membrane, which results in the release of cytochrome c into the cytoplasm. Cytochrome c induces the formation of the apoptosome, a multimeric protein complex that serves as a scaffold for caspase activation and proteolytically activates procaspase 9. The activated caspase 9 then cleaves and activates other caspases to induce apoptosis (17).
Numerous studies have shown that many pathogens can block or delay host cell death to promote their intracellular survival (18, 19). Chlamydia infection activates phosphatidylinositol 3-kinase (PI3K), which leads to Akt activation and subsequent phosphorylation of the proapoptotic protein Bad (20). Helicobacter pylori inhibits gut epithelial cell apoptosis to dampen its self-renewal and to enhance gastric colonization (21). Cells infected by Toxoplasma gondii show reduced activation of apoptotic cascades, and this effect is mainly dependent on the transcription factor NF-κB, which regulates the host prosurvival machinery (22). The precise molecular mechanisms of apoptosis inhibition vary depending on the infectious agent (11).
In this study, we investigated the interaction of enterococci with macrophages and found that E. faecalis-infected host cells were profoundly resistant to apoptosis induced by either exogenous or intrinsic apoptotic stimuli. The antiapoptotic activity correlated with the Enterococcus-induced blockade of caspase 3 activation. The molecular mechanism for E. faecalis-infected cells inhibiting apoptosis in vitro was PI3K-dependent activation of the antiapoptotic factor Akt. Activation of Akt resulted in increased expression of the antiapoptotic factor Bcl-2 and decreased expression of the proapoptotic factor Bax. In agreement with these data, results from a mouse peritonitis model confirmed that peritoneal cells from E. faecalis-infected mice had reduced apoptosis scores compared to those from uninfected mice. To our knowledge, this is the first report of E. faecalis modulating apoptosis in infected macrophages.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
The E. faecalis strains used in this study included E99, a clinical isolate from a urinary tract infection (UTI), multilocus sequence type 4 (MLST4) (23); OG1RF, a clinical oral isolate free of any plasmids and a strain used commonly in the laboratory (24); MMH594, a clinical isolate that caused multiple infections in a hospital ward outbreak and the prototype for the pathogenicity island (1); and V583, the first clinical isolate with vancomycin resistance identified in the United States (25). All the E. faecalis strains were grown for 16 h in Todd-Hewitt broth (THB) containing 1% glucose supplemented with the appropriate antibiotics (kanamycin [25 μg/ml] for E99, gentamicin [500 μg/ml] for MMH594 and V583, and rifampin [25 μg/ml] and fusidic acid [10 μg/ml] for OG1RF), and bacteria were counted by serial dilution and plating. Before use, the bacteria were pelleted by centrifugation and washed with phosphate-buffered saline (PBS).
Cell culture and infection.
RAW264.7 macrophages and mouse embryonic fibroblasts (MEFs) were cultivated initially in Dulbecco's modified Eagle medium (DMEM) plus 10% fetal bovine serum (FBS) to confluence in T-25 flasks. Bone marrow-derived macrophages (BMDMs) were isolated according to the method of a previous study (26). The cells were seeded in 6-well culture dishes and then infected with E. faecalis for 1 h. For longer periods of incubation, the cells were washed thrice with PBS to remove unbound bacteria and further incubated with medium containing vancomycin (16 μg/ml) and gentamicin (150 μg/ml) to kill all extracellular bacteria. For some experiments, host cells were infected with E. faecalis for various periods and then treated with apoptosis inducers in medium containing gentamicin and vancomycin. To inhibit bacterial internalization by RAW264.7 cells, the cells were pretreated with cytochalasin D (2.5 μg/ml) for 1 h and then infected with E99 at a multiplicity of infection (MOI) of 10 for 1 h, followed by removal of cytochalasin D and extracellular bacteria. The uninfected cells were treated in a manner identical to the experimental groups in all respects.
E. faecalis cell-free culture supernatants.
E. faecalis E99 was inoculated into DMEM plus 10% fetal bovine serum in 6-well plates without RAW264.7 cells at the same inoculum used for infection experiments. After 1 h, the supernatants were collected by centrifugation at 6,000 × g for 10 min and filtered twice using 0.22-μm syringe filters before being used to replace the culture media of 6-well dishes that had confluent RAW264.7 cells. After 12 h, the cells were stimulated with 1 μM staurosporine (STS) for 5 h to induce apoptosis.
Western blotting.
Following infection with E. faecalis strains as described above, the cells were washed with cold PBS and placed on ice. The cells in each well were harvested by scraping into lysis buffer consisting of 50 mM Tris (pH 7.4), 1.0% NP-40, 150 mM sodium chloride, 1 mM EDTA, 1 mM sodium fluoride, 1 mM sodium orthovanadate, and 1 mM phenylmethylsulfonyl fluoride (PMSF) for 15 min. The lysate was centrifuged at 14,000 × g for 10 min, and the protein concentration of the supernatant was determined using the Bio-Rad protein assay. Total protein (25 μg) was subjected to polyacrylamide gel electrophoresis on either 10 or 12% gels by using a Bio-Rad Mini Protean II apparatus and transferred electrophoretically onto a polyvinylidene difluoride (PVDF) membrane, which was then blocked with 5% skim milk. The PVDF membrane was incubated for 1 h with primary antibodies (dilution, 1:1,000) in 3% skim milk. The blots were further incubated with appropriate secondary antibodies conjugated to horseradish peroxidase (dilution, 1:3,000). Following three washes with Tris-buffered saline containing 0.1% Tween 20 (TBST), the substrate was detected using a chemiluminescence detection kit (Thermo Scientific, Rockford, IL). Densitometric calculations were conducted on the images of blots with Image J software.
Bacterial viability within RAW264.7 cells.
Survival of E. faecalis within macrophages was assessed as described previously (27). Briefly, RAW264.7 macrophages were seeded onto six-well plates at approximately 106 cells/well and incubated at 37°C under 5% CO2 for 24 h prior to infection. Triplicate wells of RAW264.7 cells were infected with E99 at an MOI of 10 for 1 h at 37°C under 5% CO2. The cells were then washed thrice with PBS and further incubated with DMEM plus 10% FBS containing vancomycin (16 μg/ml) and gentamicin (150 μg/ml) to kill all extracellular bacteria. At 3, 6, 12, 24, 48, and 72 h, the macrophages were washed twice with PBS and harvested in 1 ml of PBS. The viability and cell count were assessed by trypan blue staining using a TC10 Automated Cell Counter (Bio-Rad, Hercules, CA). The macrophages were then lysed by adding 1/10 volume of a saponin cell lysis solution (saponin [40 mg/ml], polypropylene glycol [P-2000; 8 ml/liter], and sodium polyanetholsulfonate [9.6 mg/ml]) to release intracellular bacteria. The bacteria were quantified by serial dilution and plating. The number of viable bacteria at each time point was expressed as CFU per 105 macrophages. Experiments were performed in triplicate three independent times, and the means and standard errors were determined for each time point.
Apoptosis assays.
For analysis of apoptosis, RAW264.7 cells seeded on glass coverslips in 24-well plates 24 h prior to infection were infected with E99 at an MOI of 10 for 1 h at 37°C under 5% CO2. Subsequently the monolayers were washed three times with PBS and then incubated in fresh culture medium containing 10% FBS, vancomycin (16 μg/ml), and gentamicin (150 μg/ml) for 12 h to kill all extracellular bacteria. The cells were then stimulated with 0.5 mg/ml actinomycin D for 8 h, cycloheximide (CHX) (10 μg/ml) plus tumor necrosis factor alpha (TNF-α) (25 ng/ml) for 5 h, or 1 μM staurosporine for 3 h. Levels of apoptosis in infected RAW264.7 cells were measured by the terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling (TUNEL) assay with the DeadEnd Fluorometric TUNEL System (Promega, Madison, WI) according to the manufacturer's instructions. Cells from five random fields for each sample were counted using a 40× objective lens. The percentage of apoptotic cells was calculated as follows: number of apoptotic cells/number of total cells counted × 100.
Mouse peritonitis model.
In vivo studies were conducted under an animal protocol approved by the University of Oklahoma Health Sciences Center Institutional Animal Care and Use Committee and adapted from previous reports (27, 28). Bacteria were cultivated overnight at 37°C in THB containing 1% glucose supplemented with appropriate antibiotics. The bacteria were collected by centrifugation at 6,000 × g for 10 min, washed twice in PBS (pH 7.4), and resuspended in 5% hog gastric mucin (pH 7.0; Pfaltz and Bauer, Waterbury, CT) at a concentration of 1.0 × 109 CFU/ml. Six- to 8-week-old female C57BL/6 mice (Harlan, Indianapolis, IN) were administered 200 μl of the suspension (2.0 × 108 CFU) via intraperitoneal injection. Control mice were injected with 200 μl of 5% hog gastric mucin without bacteria. The control animals were treated in a manner identical to the experimental groups in all respects. Groups of 3 mice were euthanized 6 h or 12 h postinfection by isoflurane overdose, followed by cervical dislocation. Three milliliters of PBS was injected into the peritoneal cavity of each mouse, and the peritoneal lavage fluid was collected from each group and mixed together. The cells in the lavage fluid were subjected to Western blot analysis by using anti-cleaved caspase 3 antibody according to the method described above.
Statistical analyses.
The statistical significance of results was determined by using Student's t test. Differences between experimental groups were considered significant when P values were ≤0.05.
RESULTS
Resistance to killing is accompanied by antiapoptotic signals in host macrophages early during E. faecalis infection.
To characterize the survival of E. faecalis in macrophages in vitro, we infected RAW264.7 cells with E. faecalis strain E99 at an MOI of 10. Examination of the number of viable intracellular bacteria during 72 h postinfection revealed that the number remained constant until about 12 h postinfection, followed by a significant decrease by 24 h, and declined gradually thereafter (Fig. 1A). Viable E. faecalis bacteria were recovered up to 72 h postinfection, in contrast to Escherichia coli DH5α (control), which could be eliminated from macrophages to undetectable levels by 24 h (data not shown). Taken together with our previous results (27) and those of others (7), which showed that infected macrophages essentially remain intact at least until 24 h after infection, these observations suggested that intracellular E. faecalis may be able to modulate host cell signaling and apoptosis early during infection to promote survival.
FIG 1.
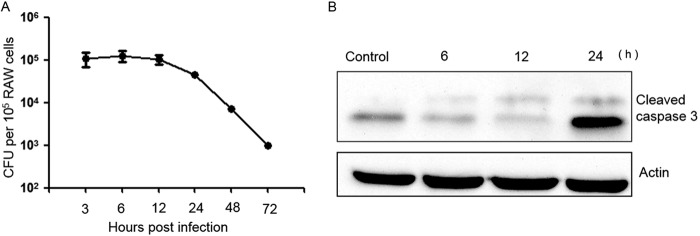
Intracellular survival of E. faecalis strain E99 and effect on caspase 3 processing. (A) RAW264.7 cells were infected as described in Materials and Methods, and the number of viable E. faecalis bacteria at each time point is expressed as CFU per 105 macrophages. Experiments were performed three times, and the means and standard errors are reported for each time point. (B) Western blot of lysates at different time points from uninfected RAW264.7 cells (control) or infected with E. faecalis E99, processed using antibody against cleaved caspase 3.
To examine host cell apoptosis during E. faecalis infection, we studied two major hallmarks of apoptosis, namely, DNA fragmentation and caspase 3 cleavage in infected cells (29). TUNEL staining revealed DNA fragmentation in the nuclei, and based on this staining pattern, we quantitated the apoptotic cells in culture, with or without E. faecalis infection, at defined time points. At 6 and 12 h postinfection, a level of <1% apoptotic cells was detected in either the infected or uninfected cell populations from most culture samples, with fewer apoptotic cells in infected samples than in the uninfected cell population, and the difference between the two groups was not statistically significant (P > 0.05). By 24 h, there were increased apoptotic events in infected cultures compared with uninfected cultures (data not shown). Immunoblotting with an anti-cleaved caspase 3 antibody showed RAW264.7 cells infected with E99 to have lower levels of cleaved caspase 3 at 6 or 12 h postinfection, and the activation of caspase 3 was detected at 24 h, which is consistent with the TUNEL-staining assay (Fig. 1B). In contrast, RAW264.7 cells infected with E. coli DH5α showed an increase in cleaved caspase 3 at 6 or 12 h postinfection (see Fig. S1A in the supplemental material). These results showed that enterococcus-infected RAW264.7 macrophages are not apoptotic at the early stage of infection and implied that a survival signal might be triggered during the early stage of E. faecalis infection.
E. faecalis-infected cells are resistant to stimulus-induced apoptosis.
To evaluate how E. faecalis-infected cells respond to apoptosis inducers, RAW264.7 cells with or without bacterial infection were treated with the kinase inhibitor STS, a potent apoptosis inducer. We found that STS could efficiently induce uninfected RAW264.7 cells to become apoptotic, as revealed by TUNEL staining (∼30%). However, RAW264.7 cells infected with E. faecalis significantly resisted STS-induced apoptosis (∼5%) (Fig. 2A and B). We also found similar antiapoptotic activity in RAW264.7 cells infected with E. faecalis strains V583 and OR1GF (data not shown), suggesting that antiapoptotic activity may be a strain-independent trait in E. faecalis. Besides analysis of apoptosis induced by STS, we also evaluated the effect of E. faecalis infection on apoptosis initiated by TNF-α and actinomycin D. Enterococcus infection also significantly inhibited host cell apoptosis induced by these two stimuli, although the induction potencies of the different agents varied (Fig. 2A and B). Because the reagents use different pathways to induce apoptosis, these observations suggested that E. faecalis infection may regulate a common step or block multiple steps leading to host cell apoptosis.
FIG 2.

Effects of E. faecalis infection on host cell apoptosis induced by proapoptotic stimuli. RAW264.7 cells, alone or infected with E. faecalis E99 for 1 h, were treated with 0.5 mg/ml actinomycin D for 8 h, with 10 μg/ml CHX and 25 ng/ml TNF-α for 5 h, or with 1 μM STS for 3 h. (A) Representative fluorescence microscope images of TUNEL staining. (B) Average percentages of TUNEL-positive stained cells. Cells from five random fields were counted under a 40× objective lens, and the percentage of apoptotic cells was calculated. The graph shows the results of three independent experiments and standard deviations (SD). *, P < 0.05.
Activation of caspase 3 is inhibited by intracellular E. faecalis.
Since caspase 3 is a critical executioner of apoptosis and cleavage of caspase 3 often happens in the late stages of cellular apoptosis, we hypothesized that E. faecalis might inhibit host cell apoptosis through inhibition of caspase 3 activation. As shown in Fig. 3A, when antibody to cleaved caspase 3 was used to detect endogenous levels of activated caspase 3, the results revealed that the processing of caspase 3 induced by STS or TNF-α in E. faecalis-infected RAW264.7 cells was significantly inhibited. In contrast, E. coli-infected RAW264.7 cells did not significantly inhibit STS-induced caspase 3 activation (see Fig. S1B in the supplemental material). We next evaluated the caspase enzymatic activity by measuring the cleavage of the caspase 3 substrate poly(ADP-ribose) polymerase (PARP). PARP p112 was reduced to the fragment p86 in RAW264.7 cells treated with STS or TNF-α, and this was blocked in E. faecalis-infected cells. These observations suggested that antiapoptotic factors of E. faecalis may function by preventing the activation of caspase 3. To test whether these observed effects were cell line dependent, we evaluated the effects of E. faecalis infection on apoptosis progression in BMDMs and MEFs, again by measuring cleavage of caspase 3. Similar to RAW264.7 cells, inhibition of caspase 3 cleavage was noted during E. faecalis infection of BMDMs and MEFs (Fig. 3B and C). Finally, a murine peritonitis model was used to evaluate the status of caspase 3 during enterococcal infection. In the peritoneal cells of E99-infected mice, a lower level of cleaved caspase 3 was observed than in mice infected with vehicle alone (Fig. 3D).
FIG 3.

Inhibition of caspase 3 activity in E. faecalis-infected cells. (A to C) RAW264.7 cells (A), BMDMs (B), and MEFs (C) were infected as described in Materials and Methods. After treatment or not with apoptotic stimuli, the samples were lysed for Western blot analysis using antibodies against cleaved caspase 3 or PARP. (D) C57BL/6 mice were injected intraperitoneally (i.p.) with 2.0 × 108 CFU of E. faecalis E99 suspended in 5% hog gastric mucin or with 5% hog gastric mucin alone, and the peritoneal lavage fluid was collected at 6 or 12 h postinfection. After centrifugation, the pelleted cells were lysed and subjected to Western blot analysis.
E. faecalis-induced antiapoptotic activity is dose and time dependent.
To understand the mechanism of E. faecalis-induced antiapoptotic activity, we evaluated the kinetic relationship between the infectious dose and antiapoptotic activity. As shown in Fig. 4A, E. faecalis-induced antiapoptotic activity correlated with an increasing bacterial MOI in a dose-dependent manner. The inhibition of apoptosis, as a function of time, appeared as early as 2 h after infection and persisted for ∼24 h (Fig. 4B), when apoptotic activity became evident. Since E. faecalis survival patterns in RAW264.7 cells showed that virtually all bacteria remained viable at least up to 12 h postinfection, these observations suggested that active intracellular resistance to killing is required for antiapoptotic activity in RAW264.7 cells.
FIG 4.

Effects of dose and duration of infection on host cell antiapoptotic activity. (A) RAW264.7 cells were infected as described in Materials and Methods with E. faecalis E99 at an MOI of 0.5, 1, 10, or 20. Apoptosis was induced at 12 h postinfection with 1 μM staurosporine for 5 h. (B) RAW264.7 cells were infected with E99 at an MOI of 10, and apoptosis was induced at 2, 12, and 24 h postinfection with 1 μM staurosporine for 5 h. Western blots of cell lysates at the indicated time points were processed for detection of cleaved caspase 3 levels. Cleaved caspase 3 levels were normalized by actin densitometry, and fold activations relative to untreated samples were determined. The graphs show the relative ratios of cleaved caspase 3, which are averages of three independent experiments and SD.
Inhibition of host cell apoptosis is strain independent but requires active E. faecalis physiology.
To investigate whether inhibition of apoptosis by E. faecalis E99 was unique to this particular E. faecalis isolate, we evaluated strains V583, MMH594, and OG1RF, with differing phenotypic characteristics (24). Our results showed that there were no significant differences among the strains (Fig. 5A and B), indicating that the antiapoptotic activity induced by E. faecalis is strain independent. To further investigate whether intact bacterial physiology was critical for the ability to inhibit host cell apoptosis, we exposed RAW264.7 cells to either live bacteria or bacteria that were killed by 4% paraformaldehyde. E. faecalis-induced antiapoptotic activity was significantly reduced for the killed bacteria, but not completely abolished (Fig. 5C). We tested the importance of bacterial internalization for inhibition of host cell apoptosis using cytochalasin D and found a significant reduction in antiapoptotic activity when bacterial uptake was prevented. Furthermore, supernatants from bacterial conditioned medium failed to inhibit host cell apoptosis, suggesting that no soluble factor secreted by E. faecalis was responsible for inhibiting host cell apoptosis (Fig. 5D).
FIG 5.

Multiple E. faecalis strains can induce host cell antiapoptotic response. (A and B) RAW264.7 cells were infected with E. faecalis strains OG1RF, V583, MMH594, and E99 as described in Materials and Methods before treatment (Treat) with staurosporine (A) or TNF-α (B) and analysis of cleaved caspase 3 by Western blotting. (C) The levels of cleaved caspase 3 in RAW264.7 cells were detected after different treatments, i.e., conditioned medium from E99 (Medium), killed E99 (KE99), and cytochalasin D plus E99 (CytD&E99), and compared to cells treated with live E99 (E99) used as a control. The images are representative of three independent experiments. (D) Cleaved caspase 3 levels were normalized by actin densitometry, and fold activations relative to untreated samples were determined. Each data point in the graph represents the mean and SD of three independent experiments. *, P < 0.05; **, P < 0.01.
E. faecalis inhibits apoptosis via the PI3K/Akt pathway.
Several studies suggest that the PI3K/Akt signaling pathway is important for host cell survival and protects apoptotic cell death induced by various stresses (19). Hence, we evaluated the effect of E. faecalis infection on the PI3K/Akt signaling pathway in RAW264.7 cells by using antibody specific to phosphorylated Akt (phospho-Akt) (Ser473). As shown in Fig. 6A, phosphorylated Akt was significantly elevated in E. faecalis-infected cells at 6 and 12 h postinfection. We further determined that the activation of Akt was dependent on PI3K activity, since the PI3K inhibitor LY294002 blocked E. faecalis-induced Akt phosphorylation (data not shown).
FIG 6.

Blockade of PI3K activation reverses E. faecalis-induced antiapoptotic activity. (A) RAW264.7 cells were infected with E. faecalis E99 as described in Materials and Methods and analyzed for pAkt levels at 6, 12, and 24 h by Western blotting. (B) RAW264.7 cells were pretreated with LY294002 (20 μM) for 30 min and then infected with enterococcus (MOI = 10). After 1 h of incubation, the cells were washed and replenished with fresh medium containing vancomycin plus gentamicin and LY294002 (20 μM) for 12 h and then treated with STS to induce apoptosis. TUNEL staining was used to measure cell apoptosis. (C) Cell lysates after different treatments were harvested for Western blotting. Cleaved caspase 3 levels were normalized by actin densitometry, and fold activations relative to untreated samples were determined. Each data point in the graph represents the mean and SD of three independent experiments. *, P < 0.05; ns, not significant.
We next investigated whether the upregulation of Akt phosphorylation during E. faecalis infection was associated with inhibition of host cell apoptosis. As shown in Fig. 6B, treatment of RAW264.7 cells with the PI3K-specific inhibitor LY294002 significantly reversed E. faecalis-induced apoptosis inhibition as measured by TUNEL assays. When cleavage of caspase 3 was used as a marker to monitor host cell apoptosis, the inhibition of apoptosis induced by STS was reversed in the presence of the PI3K inhibitor LY294002 in E. faecalis-infected RAW264.7 cells (Fig. 6C). LY294002 had no effect on macrophage phagocytosis of E. faecalis and did not directly affect bacterial growth or viability in culture medium (data not shown). Therefore, these results indicate that infection with E. faecalis activates the PI3K/Akt pathway to delay apoptosis caused by the pathogen. These observations were not limited to the RAW264.7 cells, since LY294002 similarly reversed E. faecalis-induced antiapoptotic activity in MEFs (data not shown).
Since the contact of bacteria with cells before internalization may induce the PI3K/Akt pathway, we investigated whether enterococci could activate PI3K/Akt signaling after internalization. When the cells were coincubated with the PI3K inhibitor for 1 h during infection, the inhibitor completely inhibited Akt phosphorylation during internalization of E. faecalis by RAW264.7 cells (data not shown). After bacterial internalization and removal of the inhibitor, PI3K/Akt activation was evident at ∼12 h postinfection and subsided by 24 h (Fig. 7A). These results show that internalized E. faecalis induces Akt phosphorylation in RAW264.7 cells. To further confirm the correlation between antiapoptotic activity and PI3K/Akt activation induced by internalized E. faecalis, we employed TUNEL staining and cleavage of caspase 3 to quantify apoptosis. As shown in Fig. 7B and C, inhibition of PI3K activation during bacterium-cell contact still led to apoptosis inhibition in RAW264.7 cells containing internalized E. faecalis. This inhibition, however, was lost when killed E. faecalis was used. It has been previously shown that cytokines secreted during infection can delay host cell apoptosis in an autocrine fashion (30). To address this issue, RAW264.7 cells were cultured in supernatants collected from E. faecalis-infected and uninfected macrophages in culture. No difference in RAW264.7 cell apoptosis was observed between the two groups (Fig. 7D), implying that the cytokines induced by E. faecalis are not likely involved in the antiapoptotic activity seen in these assays.
FIG 7.

Internalized live E. faecalis bacteria can activate the PI3K/Akt pathway and inhibit apoptosis induced by STS. RAW264.7 cells were pretreated with LY294002 (20 μM) for 30 min and then infected with live or killed E99 (KE99) at an MOI of 10. After 1 h of incubation, the cells were washed and replenished with fresh medium containing vancomycin and gentamicin without LY294002. (A) The macrophages were further incubated for 6, 12, and 24 h and then processed for Western blotting. (B and C) The macrophages were further incubated for 12 h, and then STS was used to induce host cell apoptosis, which was measured by Western blotting with anti-cleaved caspase 3 antibody (B) and TUNEL staining (C). (D) The supernatants from live-E99-infected (+) or uninfected (−) RAW264.7 cells treated as for panel A for 12 h were collected and, after centrifugation, separately added to freshly prepared RAW264.7 cells for 12 h before induction of apoptosis by STS. *, P < 0.05; ns, not significant. The error bars indicate SD.
E. faecalis infection regulates the abundance of Bax and Bcl-2, which are dependent on the PI3K-Akt pathway.
Since Akt directly affects the apoptotic machinery by regulating the expression of the proapoptotic protein Bax and the antiapoptotic protein Bcl-2 (31), we wished to examine the expression of these factors in RAW264.7 cells during E. faecalis infection. The level of Bax decreased following infection, while Bcl-2 increased prior to 24 h postinfection (Fig. 8A, B, and C). Blocking PI3K activity using LY294002 increased Bax and decreased Bcl-2 in RAW264.7 cells containing internalized E. faecalis (Fig. 8D, E, and F). Thus, these results suggest that Bax and Bcl-2 are downstream targets of the PI3K/Akt pathway and that they may promote antiapoptotic responses in macrophages following phagocytosis of viable E. faecalis.
FIG 8.

Role of PI3K in regulating antiapoptotic and proapoptotic proteins in E. faecalis-infected cells. (A, B, and C) Lysates from infected RAW264.7 cells were prepared for Western blot analysis to detect the relative levels of Bax and Bcl-2 proteins using anti-Bax and anti-Bcl-2 antibodies, respectively. (D, E, and F) Untreated RAW264.7 cells or cells pretreated for 30 min with the PI3K inhibitor LY294002 (20 μM) were infected with E. faecalis for 1 h, and then the cells were washed and replenished with fresh medium containing vancomycin plus gentamicin and LY294002 (20 μM) for 12 h. The cells were harvested for Western blot analysis using Bax- and Bcl-2-specific antibodies. The relative densities of Bax and Bcl-2 protein bands on Western blots were compared to that of actin in each group, and fold activations relative to untreated samples were determined. Each data point in the graph represents the mean and SD of three independent experiments. *, P < 0.05 compared with control; ns, not significant compared with control.
DISCUSSION
In this study, we demonstrate for the first time, that phagocytosis of E. faecalis by macrophages induces a dose- and time-dependent antiapoptotic response characterized by reduced DNA fragmentation and caspase 3 activation. Enterococci are able to survive for prolonged periods in cultured macrophages, unlike other Gram-positive cocci, such as Lactococcus lactis, but the exact mechanism by which this occurs remains unclear. Previous data from our laboratory (27) and that of others (7) have shown that most strains of E. faecalis are rapidly phagocytized by macrophages and appear within phagolysosomes within the first 24 h postinfection. Subsequently, based on electron microscopic examination of infected cells at later time points, intracellular enterococci are found in the cytoplasm at 24 to 48 h and eventually lead to lysis and fragmentation of host macrophages by 48 to 72 h postinfection. Consistent with these previous observations, our data presented here further show that during early infection of macrophages by E. faecalis, the bacteria effectively resist killing, as their numbers remain constant for at least up to 12 h, and during this time, the viable intracellular E. faecalis bacteria can interfere with host cell apoptotic signaling pathways. These results also imply that at the early stage of infection, E. faecalis has characteristics similar to those of some intracellular bacteria that can communicate with host cells to benefit bacterial survival and dissemination.
Apoptosis is a highly regulated host process and a prominent feature of macrophage biology in the context of infectious disease and inflammation. Different pathogens potentially use various mechanisms to induce or inhibit apoptosis in macrophages and thereby influence the disease pathology (32). For instance, macrophage death caused by Shigella and Salmonella is linked to release of proinflammatory cytokines, and the ensuing inflammation leads to the spread of bacteria within host tissues (33). Likewise, Yersinia spp. induce apoptosis by suppressing NF-κB activation and TNF-α synthesis, which are essential to pathogen clearance (34). Highly adapted intracellular bacterial pathogens, such as Mycobacterium tuberculosis, Legionella pneumophila, and Brucella spp., use differing mechanisms to block apoptosis in order to use the host cell as a vehicle for in vivo dissemination (18).
Gram-positive pathogens known to modulate apoptosis in host cells include Streptococcus, Staphylococcus, Bacillus, Listeria, and Clostridia species, although there appears to be limited conservation of the mechanisms involved (35). Among those more closely related to Enterococcus, group A streptococcus (GAS) induces rapid, dose-dependent apoptosis in primary and cultured macrophages and neutrophils, and it has been shown that the pore-forming cytolysin streptolysin O is necessary and sufficient for the apoptosis-inducing phenotype (36). Induction of host cell death by group B streptococcus (GBS) includes apoptotic mechanisms in several cell types, including macrophages, and was initially attributed to β-hemolysin production, leading to caspase-dependent apoptosis (37, 38). Similarly, dual mechanisms are observed during apoptosis of Streptococcus pneumoniae-infected cell types mediated by the pore-forming exotoxin pneumolysin. In tissue macrophages and respiratory epithelial cells, pneumolysin contributes to a caspase-dependent pathway of apoptosis (39, 40). Interestingly, in macrophages, Staphylococcus aureus induces strong caspase 3 activation concomitant with activation of the antiapoptotic machinery (41). The ability of S. aureus to persist within host macrophages before escaping the intracellular niche has been proposed as a mechanism for systemic dissemination, similar to the case for Listeria monocytogenes (42, 43). In contrast to the conversion to small-colony variants (SCVs), which allows survival for months or years (44), the limited survival within macrophages is considered a short-term solution.
Enterococci are occupants of the gastrointestinal tract as part of the commensal flora, and it is conceivable that they may use macrophages as vehicles for systemic dissemination. While our observations here show that E. faecalis resists apoptosis of macrophages early during infection, we (27) and others (7) have shown in previous studies that viable E. faecalis bacteria can be recovered at significantly later time points, beyond when apoptosis is evident (24 h). Electron micrographs of E. faecalis-infected macrophages further suggested that some bacteria at these later time points appear capable of cytoplasmic existence and continued survival, eventually leading to macrophage disintegration and lysis (7). Thus, it appears that E. faecalis may employ two distinct strategies during macrophage infection, similar to those described for S. aureus (41). One is to delay cell death during early infection to allow intracellular adaptation and dissemination, while the other is to induce later cell death to promote bacterial egress from infected macrophages. While the molecular mechanisms involved in these plausible scenarios require further investigation, a question raised by the viable intracellular E. faecalis bacteria regards their genetic makeup. Do these bacteria coordinate the expression of genes specifically required for intracellular survival, likely genes that are on the E. faecalis core genome, or do they represent genetic variants of the parent strain that are better adapted to survive inside the host cell? Although the transcriptional changes occurring in the E. faecalis genome in response to the intramacrophage environment has not been reported, SCVs of E. faecalis isolated from clinical specimens have been described (45). The SCV phenotype has been characterized as cells of different sizes with aberrant shapes forming smaller colonies than the clonally related normal strain on culture media, growing with an extended lag phase with delayed entry into stationary phase, failing to grow on simple media without the addition of blood, and showing changes in metabolic pathways. Along these lines, internalization of Salmonella enterica serovar Typhimurium by macrophages has been reported to induce the formation of nonreplicating persisters (46). Multiple toxin-antitoxin (TA) modules contributed to the formation of intracellular persisters, and the macrophage environment was found to induce phenotypic heterogeneity, leading to either bacterial replication or the formation of nonreplicating persisters that could provide a reservoir for relapsing infection. The exact nature of E. faecalis isolates that survive the antimicrobial activities of macrophages remains to be investigated.
It is also important to consider apoptosis in the context of nonphagocytic cells, such as epithelial cells, because our studies show that enterococcus infection can induce antiapoptotic activity in both professional phagocytes and nonphagocytic cells. Apoptosis is a normal occurrence for epithelial cells in order to maintain a critical balance of cells in the crypt and villus during self-renewal of the intestinal lining, but it may also function as a mechanism to eliminate infected cells so as to prevent pathogen proliferation and access to deeper tissues (47, 48). The same strategy is evident in superficial epithelial cells lining the urinary bladder. Uropathogens, such as Proteus mirabilis and certain strains of E. coli, effectively invade the urothelium, form intracellular reservoirs that evade antibiotics and the immune response, and allow recurring infections (49). The host response to such infections is sloughing of cells into the urine (50). E. faecalis accounts for a significant proportion of chronic bladder infections worldwide and is also a leading cause of catheter-associated UTIs (51). Recent studies have shown that E. faecalis bacteria are harbored within urothelial cells shed from the bladders of patients with chronic UTIs (52). The ability to persist in epithelial cells lining the intestine or the bladder and to resist apoptosis and shedding of these cells may provide time for E. faecalis to adapt to the intracellular environment before emerging from this niche to disseminate and cause systemic infection (48). Experiments to determine whether E. faecalis-infected epithelial cells respond to proapoptotic stimuli in the same manner as macrophages are under way in our laboratory.
An important finding of this study is that E. faecalis in RAW264.7 cells activates the PI3K/Akt pathway, although the mechanism by which this occurs is not fully understood. As shown by previous studies, cytokines induced during infection can stimulate PI3K/Akt activation (30). Enterococcal infection can induce the robust production of proinflammatory cytokines and chemokines from macrophages (9, 53). However, culture medium from E. faecalis-infected macrophages did not inhibit host cell apoptosis induced by proapoptotic stimuli. While we cannot completely rule out a role for cytokines in E. faecalis-induced antiapoptotic activity, it does not appear to be a significant mechanism for our observations. A more plausible explanation could be that E. faecalis produces one or more novel antiapoptotic factors in the host cell to manipulate the PI3K/Akt pathway directly. This strategy is widely used by many intracellular pathogens to manipulate host cell kinase signaling, and a number of viral and bacterial antiapoptotic factors have been identified (54, 55). Interestingly, a novel mode of modulation of apoptosis and release of intracellular bacteria from infected epithelial cells has recently been described for S. aureus, which is primarily an extracellular pathogen (56). This strategy involves the ESAT-6-like secretion system (Ess), which is similar to the Esx-1 secretion system described for M. tuberculosis and is apparently conserved in the genomes of many Gram-positive bacteria, including Bacillus subtilis, Bacillus anthracis, and L. monocytogenes (57, 58). The proteins EsxA and EsxB, substrates of the S. aureus Ess, belong to the WXG100 motif superfamily (58) and were shown to be important during intracellular S. aureus infection (56). EsxA interfered with S. aureus-induced apoptosis in human epithelial cells in vitro and required unimpeded secretion of the protein. Overexpression of EsxA in transfected cells increased protection from apoptotic cell death. Further, it was shown that EsxA acted in concert with EsxB to mediate release of S. aureus from host cells. While orthologous Esx proteins in M. tuberculosis have been implicated in several aspects of pathogenesis, including apoptosis and host cell lysis (59), the functions of other orthologs, such as those in L. monocytogenes, remain unclear (60). Enterococcal genomes also appear to encode EsxA-like proteins, but their contribution to enterococcal physiology and pathogenesis remains to be studied (61, 62). Although their precise role(s) during infection remains to be determined, the studies described above raise the prospect that the Esx proteins in Gram-positive bacteria may represent conserved factors that control host cell apoptosis and cell lysis. In any case, the results obtained with the Esx proteins of S. aureus demonstrate that bacterial proteins are active in host cells and can regulate apoptosis in infected cells. Consistent with this notion, we found that nonviable E. faecalis bacteria after internalization could not protect cells from apoptosis, suggesting that a newly synthesized enterococcal factor(s) might be responsible for the inhibition of apoptosis. While our data show that the activation of the PI3K/Akt pathway modulates expression of the key apoptosis effectors Bax and Bcl-2, the fact that enterococcal infection inhibited apoptosis induced by a wide range of stimuli makes it likely that other apoptosis effectors are affected, as well.
In conclusion, we have shown that E. faecalis can effectively manipulate host cell signaling in infected macrophages to delay apoptosis. In this regard, this commensal organism appears to mimic classic intracellular pathogens that hijack cell death pathways to promote survival in the intracellular niche. Enterococcal antiapoptotic activity may have evolved to suppress the host apoptotic response to initial intracellular invasion as a means to modulate the host immune response and may be regarded as a “gain-in-time strategy” for adaptation of the bacterium to its new environment. Understandably, this strategy has important consequences for enterococcal pathogenesis, and a better understanding of the factors and mechanisms involved may provide new avenues to prevent and treat enterococcal infections.
Supplementary Material
ACKNOWLEDGMENTS
This project was funded, in part, by a President's Associates Presidential Professorship awarded by the University of Oklahoma to N.S.
We gratefully acknowledge the assistance of Mark Huycke and Xingmin Wang for critical reading of the manuscript.
Footnotes
Published ahead of print 29 September 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.02426-14.
REFERENCES
- 1.Shankar N, Baghdayan AS, Gilmore MS. 2002. Modulation of virulence within a pathogenicity island in vancomycin-resistant Enterococcus faecalis. Nature 417:746–750. 10.1038/nature00802. [DOI] [PubMed] [Google Scholar]
- 2.Linden P. 2003. Can enterococcal infections initiate sepsis syndrome? Curr. Infect. Dis. Rep. 5:372–378. 10.1007/s11908-003-0016-8. [DOI] [PubMed] [Google Scholar]
- 3.Richards MJ, Edwards JR, Culver DH, Gaynes RP. 2000. Nosocomial infections in combined medical-surgical intensive care units in the United States. Infect. Control Hosp. Epidemiol. 21:510–515. 10.1086/501795. [DOI] [PubMed] [Google Scholar]
- 4.Shepard BD, Gilmore MS. 2002. Antibiotic-resistant enterococci: the mechanisms and dynamics of drug introduction and resistance. Microbes Infect. 4:215–224. 10.1016/S1286-4579(01)01530-1. [DOI] [PubMed] [Google Scholar]
- 5.Zeng J, Teng F, Weinstock GM, Murray BE. 2004. Translocation of Enterococcus faecalis strains across a monolayer of polarized human enterocyte-like T84 cells. J. Clin. Microbiol. 42:1149–1154. 10.1128/JCM.42.3.1149-1154.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wells CL, Jechorek RP, Maddaus MA, Simmons RL. 1988. Effects of clindamycin and metronidazole on the intestinal colonization and translocation of enterococci in mice. Antimicrob. Agents Chemother. 32:1769–1775. 10.1128/AAC.32.12.1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gentry-Weeks CR, Karkhoff-Schweizer R, Pikis A, Estay M, Keith JM. 1999. Survival of Enterococcus faecalis in mouse peritoneal macrophages. Infect. Immun. 67:2160–2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Giard JC, Riboulet E, Verneuil N, Sanguinetti M, Auffray Y, Hartke A. 2006. Characterization of Ers, a PrfA-like regulator of Enterococcus faecalis. FEMS Immunol. Med. Microbiol. 46:410–418. 10.1111/j.1574-695X.2005.00049.x. [DOI] [PubMed] [Google Scholar]
- 9.Leendertse M, Willems RJ, Giebelen IA, Roelofs JJ, van Rooijen N, Bonten MJ, van der Poll T. 2009. Peritoneal macrophages are important for the early containment of Enterococcus faecium peritonitis in mice. Innate Immun. 15:3–12. 10.1177/1753425908100238. [DOI] [PubMed] [Google Scholar]
- 10.Williams GT. 1994. Programmed cell death: a fundamental protective response to pathogens. Trends Microbiol. 2:463–464. 10.1016/0966-842X(94)90648-3. [DOI] [PubMed] [Google Scholar]
- 11.Ashida H, Mimuro H, Ogawa M, Kobayashi T, Sanada T, Kim M, Sasakawa C. 2011. Cell death and infection: a double-edged sword for host and pathogen survival. J. Cell Biol. 195:931–942. 10.1083/jcb.201108081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ashkenazi A, Dixit VM. 1998. Death receptors: signaling and modulation. Science 281:1305–1308. 10.1126/science.281.5381.1305. [DOI] [PubMed] [Google Scholar]
- 13.Zimmermann KC, Bonzon C, Green DR. 2001. The machinery of programmed cell death. Pharmacol. Ther. 92:57–70. 10.1016/S0163-7258(01)00159-0. [DOI] [PubMed] [Google Scholar]
- 14.Green DR, Reed JC. 1998. Mitochondria and apoptosis. Science 281:1309–1312. 10.1126/science.281.5381.1309. [DOI] [PubMed] [Google Scholar]
- 15.Adams JM, Cory S. 1998. The Bcl-2 protein family: arbiters of cell survival. Science 281:1322–1326. 10.1126/science.281.5381.1322. [DOI] [PubMed] [Google Scholar]
- 16.Gross A, McDonnell JM, Korsmeyer SJ. 1999. BCL-2 family members and the mitochondria in apoptosis. Genes Dev. 13:1899–1911. 10.1101/gad.13.15.1899. [DOI] [PubMed] [Google Scholar]
- 17.Arnoult D, Carneiro L, Tattoli I, Girardin SE. 2009. The role of mitochondria in cellular defense against microbial infection. Semin. Immunol. 21:223–232. 10.1016/j.smim.2009.05.009. [DOI] [PubMed] [Google Scholar]
- 18.Faherty CS, Maurelli AT. 2008. Staying alive: bacterial inhibition of apoptosis during infection. Trends Microbiol. 16:173–180. 10.1016/j.tim.2008.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Franke TF, Hornik CP, Segev L, Shostak GA, Sugimoto C. 2003. PI3K/Akt and apoptosis: size matters. Oncogene 22:8983–8998. 10.1038/sj.onc.1207115. [DOI] [PubMed] [Google Scholar]
- 20.Verbeke P, Welter-Stahl L, Ying S, Hansen J, Hacker G, Darville T, Ojcius DM. 2006. Recruitment of BAD by the Chlamydia trachomatis vacuole correlates with host-cell survival. PLoS Pathog. 2:e45. 10.1371/journal.ppat.0020045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mimuro H, Suzuki T, Nagai S, Rieder G, Suzuki M, Nagai T, Fujita Y, Nagamatsu K, Ishijima N, Koyasu S, Haas R, Sasakawa C. 2007. Helicobacter pylori dampens gut epithelial self-renewal by inhibiting apoptosis, a bacterial strategy to enhance colonization of the stomach. Cell Host Microbe 2:250–263. 10.1016/j.chom.2007.09.005. [DOI] [PubMed] [Google Scholar]
- 22.Payne TM, Molestina RE, Sinai AP. 2003. Inhibition of caspase activation and a requirement for NF-kappaB function in the Toxoplasma gondii-mediated blockade of host apoptosis. J. Cell Sci. 116:4345–4358. 10.1242/jcs.00756. [DOI] [PubMed] [Google Scholar]
- 23.Tendolkar PM, Baghdayan AS, Shankar N. 2006. Putative surface proteins encoded within a novel transferable locus confer a high-biofilm phenotype to Enterococcus faecalis. J. Bacteriol. 188:2063–2072. 10.1128/JB.188.6.2063-2072.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McBride SM, Fischetti VA, Leblanc DJ, Moellering RC, Jr, Gilmore MS. 2007. Genetic diversity among Enterococcus faecalis. PLoS One 2:e582. 10.1371/journal.pone.0000582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Paulsen IT, Banerjei L, Myers GS, Nelson KE, Seshadri R, Read TD, Fouts DE, Eisen JA, Gill SR, Heidelberg JF, Tettelin H, Dodson RJ, Umayam L, Brinkac L, Beanan M, Daugherty S, DeBoy RT, Durkin S, Kolonay J, Madupu R, Nelson W, Vamathevan J, Tran B, Upton J, Hansen T, Shetty J, Khouri H, Utterback T, Radune D, Ketchum KA, Dougherty BA, Fraser CM. 2003. Role of mobile DNA in the evolution of vancomycin-resistant Enterococcus faecalis. Science 299:2071–2074. 10.1126/science.1080613. [DOI] [PubMed] [Google Scholar]
- 26.Celada A, Gray PW, Rinderknecht E, Schreiber RD. 1984. Evidence for a gamma-interferon receptor that regulates macrophage tumoricidal activity. J. Exp. Med. 160:55–74. 10.1084/jem.160.1.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Coburn PS, Baghdayan AS, Dolan GT, Shankar N. 2008. An AraC-type transcriptional regulator encoded on the Enterococcus faecalis pathogenicity island contributes to pathogenesis and intracellular macrophage survival. Infect. Immun. 76:5668–5676. 10.1128/IAI.00930-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kajfasz JK, Mendoza JE, Gaca AO, Miller JH, Koselny KA, Giambiagi-Demarval M, Wellington M, Abranches J, Lemos JA. 2012. The Spx regulator modulates stress responses and virulence in Enterococcus faecalis. Infect. Immun. 80:2265–2275. 10.1128/IAI.00026-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Greene W, Xiao Y, Huang Y, McClarty G, Zhong G. 2004. Chlamydia-infected cells continue to undergo mitosis and resist induction of apoptosis. Infect. Immun. 72:451–460. 10.1128/IAI.72.1.451-460.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sarkar A, Hellberg L, Bhattacharyya A, Behnen M, Wang K, Lord JM, Moller S, Kohler M, Solbach W, Laskay T. 2012. Infection with Anaplasma phagocytophilum activates the phosphatidylinositol 3-kinase/Akt and NF-kappaB survival pathways in neutrophil granulocytes. Infect. Immun. 80:1615–1623. 10.1128/IAI.05219-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Downward J. 2004. PI 3-kinase, Akt and cell survival. Semin. Cell Dev. Biol. 15:177–182. 10.1016/j.semcdb.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 32.Navarre WW, Zychlinsky A. 2000. Pathogen-induced apoptosis of macrophages: a common end for different pathogenic strategies. Cell. Microbiol. 2:265–273. 10.1046/j.1462-5822.2000.00056.x. [DOI] [PubMed] [Google Scholar]
- 33.Falkow S. 1997. Perspectives series: host/pathogen interactions. Invasion and intracellular sorting of bacteria: searching for bacterial genes expressed during host/pathogen interactions. J. Clin. Invest. 100:239–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Aepfelbacher M, Zumbihl R, Ruckdeschel K, Jacobi CA, Barz C, Heesemann J. 1999. The tranquilizing injection of Yersinia proteins: a pathogen's strategy to resist host defense. Biol. Chem. 380:795–802. [DOI] [PubMed] [Google Scholar]
- 35.Ulett GC, Adderson EE. 2006. Regulation of apoptosis by Gram-positive bacteria: mechanistic diversity and consequences for immunity. Curr. Immunol. Rev. 2:119–141. 10.2174/157339506776843033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Timmer AM, Timmer JC, Pence MA, Hsu LC, Ghochani M, Frey TG, Karin M, Salvesen GS, Nizet V. 2009. Streptolysin O promotes group A Streptococcus immune evasion by accelerated macrophage apoptosis. J. Biol. Chem. 284:862–871. 10.1074/jbc.M804632200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fettucciari K, Rosati E, Scaringi L, Cornacchione P, Migliorati G, Sabatini R, Fetriconi I, Rossi R, Marconi P. 2000. Group B Streptococcus induces apoptosis in macrophages. J. Immunol. 165:3923–3933. 10.4049/jimmunol.165.7.3923. [DOI] [PubMed] [Google Scholar]
- 38.Liu GY, Doran KS, Lawrence T, Turkson N, Puliti M, Tissi L, Nizet V. 2004. Sword and shield: linked group B streptococcal beta-hemolysin/cytolysin and carotenoid pigment function to subvert host phagocyte defense. Proc. Natl. Acad. Sci. U. S. A. 101:14491–14496. 10.1073/pnas.0406143101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Marriott HM, Bingle CD, Read RC, Braley KE, Kroemer G, Hellewell PG, Craig RW, Whyte MK, Dockrell DH. 2005. Dynamic changes in Mcl-1 expression regulate macrophage viability or commitment to apoptosis during bacterial clearance. J. Clin. Invest. 115:359–368. 10.1172/JCI200521766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Srivastava A, Henneke P, Visintin A, Morse SC, Martin V, Watkins C, Paton JC, Wessels MR, Golenbock DT, Malley R. 2005. The apoptotic response to pneumolysin is Toll-like receptor 4 dependent and protects against pneumococcal disease. Infect. Immun. 73:6479–6487. 10.1128/IAI.73.10.6479-6487.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Koziel J, Maciag-Gudowska A, Mikolajczyk T, Bzowska M, Sturdevant DE, Whitney AR, Shaw LN, DeLeo FR, Potempa J. 2009. Phagocytosis of Staphylococcus aureus by macrophages exerts cytoprotective effects manifested by the upregulation of antiapoptotic factors. PLoS One 4:e5210. 10.1371/journal.pone.0005210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Drevets DA. 1999. Dissemination of Listeria monocytogenes by infected phagocytes. Infect. Immun. 67:3512–3517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kubica M, Guzik K, Koziel J, Zarebski M, Richter W, Gajkowska B, Golda A, Maciag-Gudowska A, Brix K, Shaw L, Foster T, Potempa J. 2008. A potential new pathway for Staphylococcus aureus dissemination: the silent survival of S. aureus phagocytosed by human monocyte-derived macrophages. PLoS One 3:e1409. 10.1371/journal.pone.0001409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Proctor RA, von Eiff C, Kahl BC, Becker K, McNamara P, Herrmann M, Peters G. 2006. Small colony variants: a pathogenic form of bacteria that facilitates persistent and recurrent infections. Nat. Rev. Microbiol. 4:295–305. 10.1038/nrmicro1384. [DOI] [PubMed] [Google Scholar]
- 45.Wellinghausen N, Chatterjee I, Berger A, Niederfuehr A, Proctor RA, Kahl BC. 2009. Characterization of clinical Enterococcus faecalis small-colony variants. J. Clin. Microbiol. 47:2802–2811. 10.1128/JCM.00485-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Helaine S, Cheverton AM, Watson KG, Faure LM, Matthews SA, Holden DW. 2014. Internalization of Salmonella by macrophages induces formation of nonreplicating persisters. Science 343:204–208. 10.1126/science.1244705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hall PA, Coates PJ, Ansari B, Hopwood D. 1994. Regulation of cell number in the mammalian gastrointestinal tract: the importance of apoptosis. J. Cell Sci. 107:3569–3577. [DOI] [PubMed] [Google Scholar]
- 48.Kim JM, Eckmann L, Savidge TC, Lowe DC, Witthoft T, Kagnoff MF. 1998. Apoptosis of human intestinal epithelial cells after bacterial invasion. J. Clin. Invest. 102:1815–1823. 10.1172/JCI2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Anderson GG, Martin SM, Hultgren SJ. 2004. Host subversion by formation of intracellular bacterial communities in the urinary tract. Microbes Infect. 6:1094–1101. 10.1016/j.micinf.2004.05.023. [DOI] [PubMed] [Google Scholar]
- 50.Mysorekar IU, Hultgren SJ. 2006. Mechanisms of uropathogenic Escherichia coli persistence and eradication from the urinary tract. Proc. Natl. Acad. Sci. U. S. A. 103:14170–14175. 10.1073/pnas.0602136103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Low DE, Keller N, Barth A, Jones RN. 2001. Clinical prevalence, antimicrobial susceptibility, and geographic resistance patterns of enterococci: results from the SENTRY Antimicrobial Surveillance Program, 1997–1999. Clin. Infect. Dis. 32(Suppl 2):S133–S145. 10.1086/320185. [DOI] [PubMed] [Google Scholar]
- 52.Horsley H, Malone-Lee J, Holland D, Tuz M, Hibbert A, Kelsey M, Kupelian A, Rohn JL. 2013. Enterococcus faecalis subverts and invades the host urothelium in patients with chronic urinary tract infection. PLoS One 8:e83637. 10.1371/journal.pone.0083637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Daw K, Baghdayan AS, Awasthi S, Shankar N. 2012. Biofilm and planktonic Enterococcus faecalis elicit different responses from host phagocytes in vitro. FEMS Immunol. Med. Microbiol. 65:270–282. 10.1111/j.1574-695X.2012.00944.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pirbhai M, Dong F, Zhong Y, Pan KZ, Zhong G. 2006. The secreted protease factor CPAF is responsible for degrading pro-apoptotic BH3-only proteins in Chlamydia trachomatis-infected cells. J. Biol. Chem. 281:31495–31501. 10.1074/jbc.M602796200. [DOI] [PubMed] [Google Scholar]
- 55.Laguna RK, Creasey EA, Li Z, Valtz N, Isberg RR. 2006. A Legionella pneumophila-translocated substrate that is required for growth within macrophages and protection from host cell death. Proc. Natl. Acad. Sci. U. S. A. 103:18745–18750. 10.1073/pnas.0609012103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Korea CG, Balsamo G, Pezzicoli A, Merakou C, Tavarini S, Bagnoli F, Serruto D, Unnikrishnan M. 2014. The staphylococcal Esx proteins modulate apoptosis and release of intracellular Staphylococcus aureus during infection in epithelial cells. Infect. Immun. 82:4144–4153. 10.1128/IAI.01576-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Burts ML, Williams WA, DeBord K, Missiakas DM. 2005. EsxA and EsxB are secreted by an ESAT-6-like system that is required for the pathogenesis of Staphylococcus aureus infections. Proc. Natl. Acad. Sci. U. S. A. 102:1169–1174. 10.1073/pnas.0405620102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pallen MJ. 2002. The ESAT-6/WXG100 superfamily—and a new Gram-positive secretion system? Trends Microbiol. 10:209–212. 10.1016/S0966-842X(02)02345-4. [DOI] [PubMed] [Google Scholar]
- 59.Derrick SC, Morris SL. 2007. The ESAT6 protein of Mycobacterium tuberculosis induces apoptosis of macrophages by activating caspase expression. Cell. Microbiol. 9:1547–1555. 10.1111/j.1462-5822.2007.00892.x. [DOI] [PubMed] [Google Scholar]
- 60.Way SS, Wilson CB. 2005. The Mycobacterium tuberculosis ESAT-6 homologue in Listeria monocytogenes is dispensable for growth in vitro and in vivo. Infect. Immun. 73:6151–6153. 10.1128/IAI.73.9.6151-6153.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bourgogne A, Garsin DA, Qin X, Singh KV, Sillanpaa J, Yerrapragada S, Ding Y, Dugan-Rocha S, Buhay C, Shen H, Chen G, Williams G, Muzny D, Maadani A, Fox KA, Gioia J, Chen L, Shang Y, Arias CA, Nallapareddy SR, Zhao M, Prakash VP, Chowdhury S, Jiang H, Gibbs RA, Murray BE, Highlander SK, Weinstock GM. 2008. Large scale variation in Enterococcus faecalis illustrated by the genome analysis of strain OG1RF. Genome Biol. 9:R110. 10.1186/gb-2008-9-7-r110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Palmer KL, Carniol K, Manson JM, Heiman D, Shea T, Young S, Zeng Q, Gevers D, Feldgarden M, Birren B, Gilmore MS. 2010. High-quality draft genome sequences of 28 Enterococcus sp. isolates. J. Bacteriol. 192:2469–2470. 10.1128/JB.00153-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.