

Abstract
RNA–protein interactions differ from DNA–protein interactions because of the central role of RNA secondary structure. Some RNA-binding domains (RBDs) recognize their target sites mainly by their shape and geometry and others are sequence-specific but are sensitive to secondary structure context. A number of small- and large-scale experimental approaches have been developed to measure RNAs associated in vitro and in vivo with RNA-binding proteins (RBPs). Generalizing outside of the experimental conditions tested by these assays requires computational motif finding. Often RBP motif finding is done by adapting DNA motif finding methods; but modeling secondary structure context leads to better recovery of RBP-binding preferences. Genome-wide assessment of mRNA secondary structure has recently become possible, but these data must be combined with computational predictions of secondary structure before they add value in predicting in vivo binding. There are two main approaches to incorporating structural information into motif models: supplementing primary sequence motif models with preferred secondary structure contexts (e.g., MEMERIS and RNAcontext) and directly modeling secondary structure recognized by the RBP using stochastic context-free grammars (e.g., CMfinder and RNApromo). The former better reconstruct known binding preferences for sequence-specific RBPs but are not suitable for modeling RBPs that recognize shape and geometry of RNAs. Future work in RBP motif finding should incorporate interactions between multiple RBDs and multiple RBPs in binding to RNA. WIREs RNA 2014, 5:111–130. doi: 10.1002/wrna.1201
INTRODUCTION
Eukaryotic genomes encode hundreds of RNA-binding proteins (RBPs) with diverse functions in co- and post-transcriptional regulation of RNA metabolism. Recent studies have revealed that RBPs typically have hundreds of targets and multiple RBPs coordinately regulate populations of functionally related mRNAs.1–4 Identification of RBP target sites is an important step toward understanding the mechanisms by which they conduct post-transcriptional regulation.
In this article, we review computational and experimental methodologies for identifying the binding sites of RBPs. We pay special attention to the role of RNA secondary structure and its impact on binding-site selection. This attention naturally leads to discussions of how mRNA secondary structure is experimentally assessed and computationally predicted. Surprisingly, existing large-scale experimental methods for assaying secondary structure are no better than computational methods at predicting RBP binding. Having established that the secondary structure context of putative binding sites can be determined, we also review the major computational methods for generating motif models for RBPs, which incorporate both primary and secondary structure preferences. This review closes with some discussion of open questions in this field and open computational problems in RBP motif finding.
HOW RNA-BINDING PROTEINS BIND RNA
Primary sequence specificity is often critical for binding-site recognition by both RNA- and DNA-binding proteins; however, RNA–protein interactions differ from DNA–protein interactions because double-stranded RNA (dsRNA) typically adopts the A-form helical structure whose major groove is deeper and narrower than that of the B-form helix of dsDNA. As such, base-specific interactions by amino acid side chains are rare in dsRNA,5,6 and sequence-specific RBPs are likely to require at least some of their binding sites to be single-stranded.7 There is substantial evidence that this is the almost exclusive form of interaction between sequence-specific RBPs and their targets. Indeed, the two most common RBDs in eukaryotes, the RNA recognition motif (RRM) and the hnRNP K-homology (KH) domains bind single-stranded RNA.8,9 Early surveys10–12 of RBP–RNA complexes deposited in the Protein Data Bank (PDB) have reported that base-specific interactions between RBPs and RNA only occur in or near regions of single-stranded RNA (ssRNA). Subsequent surveys of RRM–RNA complexes13 and solved co-complex structures of Pum-homology domains (PUM-HD)14 and even a dsRNA binding protein (ADAR2,15 see below) have added further support to this well-established tendency. As examples of RBP interactions with ssRNA, Figure 1(a) and (b) shows structures of an RRM and a PUM-HD in complex with their ssRNA targets.
Figure 1.
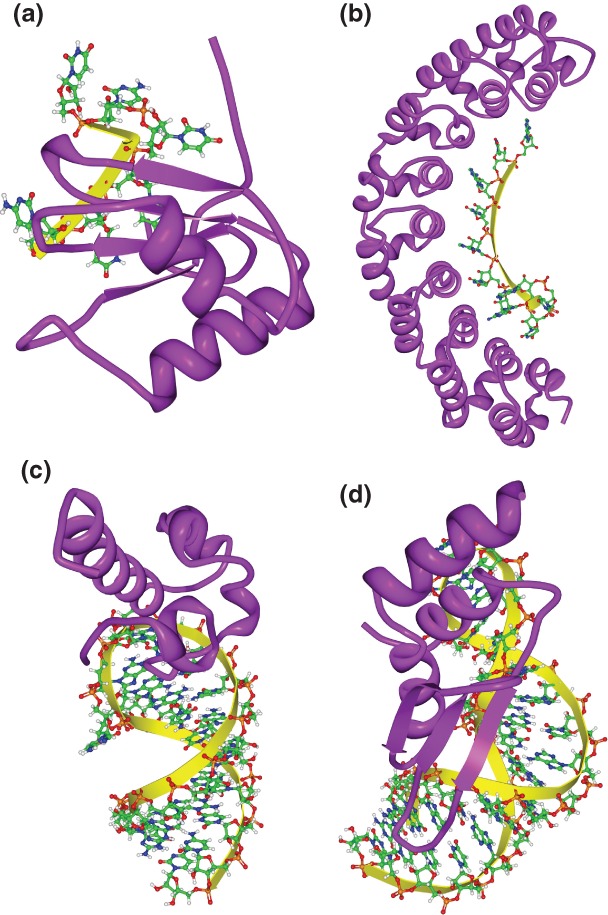
Three-dimensional structures of RNA-binding domain (RBD)–RNA complexes. (a) Solution structure of polypyrimidine tract binding (PTB) protein RBD1 in complex with CUCUCU RNA [Protein Data Bank (PDB): 2AD9]. PTB RBD1 binds a YCU site (Y indicating pyrimidine) through β4, β1, and β2, respectively. (b) Co-crystal structure of the PUM-homology domain (PUM-HD) in human Pum1 complexed with a 10-nucleotide single-stranded RNA, 5′-AUUGUACAUA where the last eight nucleotides (UGUACAUA) are individually recognized by three conserved amino acids in Puf repeats 8 to 1, respectively14 (PDB: 1M8Y). (c) Solution structure of the Vts1p sterile-α motif (specific affinity matrix, SAM) domain in complex with a 5′-CUGGC-3′ pentaloop as part of a 19nt hairpin (PDB: 2ESE). The specific interaction between the Vts1p SAM domain and the target RNA is stabilized by both the direct interaction to the third guanosine base in the RNA pentaloop and the contacts to the unique backbone structure.16–18 (d) Solution structure of dsRBD of yeast Rnt1p in complex with the 5′ terminal AGNN tetraloop of snR47 precursor RNA (PDB: 1T4l). Neither A nor G are recognized by specific hydrogen bonds; instead, the N-terminal helix of the Rnt1p dsRBD interacts with the backbone and the two nonconserved tetraloop bases, by snugly fitting into the minor groove side of the RNA tetraloop and extending into the minor groove at the top of the stem.19
Not all interactions with ssRNA occur in the same context: ssRNA can occur outside of any RNA loops (called ‘external’), in a hairpin loop, in an internal/bulge loop, or in more complex loop structures (called ‘multiloops’); and RBPs can vary in their preference for these different ‘structural contexts’ of ssRNA. For example, yeast Vts1p and its Drosophila homolog, Smaug, have strong preferences for binding CNGG within a hairpin loop16–18 (Figure 1(c)).
On the other hand, some RBDs recognize their target sites mainly by their shape and geometry and not by their sequence content. For example, proteins with double-stranded RNA-binding domains (dsRBDs) bind stems of dsRNA with at least 10 base pairs (bps), mainly through the interactions of the 2′-hydroxyl groups of the ribose sugars and direct (or water-mediated) interactions with the non-bridging oxygen residues of the phosphodiester backbone, rather than specific interactions with the bases. Examples of dsRBD-containing proteins with structures that have been solved in co-complex with RNA include Xenopus Xlrbpa,20 Drosophila Staufen,21 and yeast Rnt1p (Figure 1(d)). As a counterexample, ADAR2 binds its targets through dsRBD–RNA interactions that include sequence-specific contacts.15 However, two of the four sequence-specific interactions are to unpaired bases in bulge loops, and the other two are near disruptions in the dsRNA helical structure that expand the minor groove.15
EXPERIMENTAL METHODS TO DETECT RNA–PROTEIN INTERACTIONS
Identification of the RNAs bound by each RBP is the key for understanding the interactions governing post-transcriptional regulation. A number of low- and high-throughput experimental methods have been developed to assess the in vitro sequence-binding preferences of RBPs, as well as to identify the in vivo binding sites for RBPs in particular cellular contexts.
SELEX (systematic evolution of ligands by exponential enrichment) is a low-throughput method for in vitro detection of RBP sequence-binding preferences.22 High-affinity binding sequences are selected from a randomized RNA oligonucleotide pool through several sequential rounds of binding to purified protein, each followed by polymerase chain reaction (PCR) amplification. The products are then cloned and sequenced, identifying a set of short sequences preferred by the protein. These short sequences are then analyzed in order to define primary sequence and structural preferences of the RBP. One disadvantage of the SELEX assay is that, because of the multiple rounds of purification and amplification, it reveals only the highest affinity RNA target sites, and does not completely characterize the range and relative affinity of RNA-sequence preferences of an RBP. The recent advent of relatively inexpensive, high-throughput sequencing has facilitated the development of a more quantitative and comprehensive version of this procedure, sometimes called HT-SELEX.23,24 In this procedure, only a single, or a small number of, binding reaction is performed but millions of RNA oligos are sequenced, supporting a more quantitative estimate of the RBP sequence-binding preference.
RNAcompete is a related in vitro method that replaces the large, complex random initial RNA oligo pool used by HT-SELEX with a smaller, designed pool that is synthesized with the help of a custom microarray. The oligo pool contains approximately 244,000 short 30–38nt RNAs whose design is based on modified de Bruijn sequences,9,25,26 ensuring that 7nt RNA sequences appear either in ssRNA or weakly paired RNA in at least 128 oligos. This allows an unbiased measurement of the relative sequence-binding preferences of RBPs. An advantage of RNAcompete is that it is much less expensive than HT-SELEX because its small pool size allows the relative abundances of each oligo to be measured using a custom-designed Agilent microarray. To date, RNA primary sequence preferences for more than 200 RBPs have been reported and these are summarized in the CisBP-RNA website26,27 (Table 1 summarizes the web resources for the RBP binding sites). However, because the RNAcompete pool is depleted for RNAs with stable secondary structure, RBPs with strict structural requirements on their binding sites are less successful in this assay. Nonetheless, RNAcompete is still able to recover the primary sequence binding preferences of some RBPs that have preferences for particular secondary structural contexts for these sequences, such as Vts1p16–18 and Lin28.34
Table 1.
Web Resources for RBP Binding Sites
| Database | Collection | Properties (Features) | Availability | References |
|---|---|---|---|---|
| ARESITE | AU-rich elements (ARE) in vertebrate mRNA UTR sequences | Input gene sequence is searched for enrichment of eight predefined consensus ARE. For each detected motif, conservation patterns and predicted accessibility values are displayed. | http://rna.tbi.univie.ac.at/AREsite/ | 28 |
| CisBP-RNA | RBP motifs identified by RNAcompete and RBPDB | Users can browse or bulk download motifs for all eukaryotic RBPs including direct measured motifs for more than 200 RBPs from RNAcompete or RBPDB, as well as thousands more motifs inferred by homology. Also, scans input RNA sequences for hits to directly motifs. | http://cisbp-rna.ccbr.utoronto.ca/ | 27 |
| CLIPZ | Binding sites from CLIP experiments, including Quaking, Pumilio, Argonautes 1–4, TNRC6 A-C, IGF2BP 1–3 | Users can browse the clusters of genome- or transcript-based reads. Clusters from different experiments can be compared. The transcripts associated with a gene name could be searched for binding sites. There is also a motif enrichment tool that identifies overrepresented k-mers in a set of sequences. | http://www.clipz.unibas.ch/ | 29 |
| doRiNA | RBP and miRNA binding sites identified by CLIP experiments | CLIP-derived peaks for RBPs and miRNAs from humans, mouse, flies, and worms are available. Users can also search overlapping sites between multiple RBPs or between RBPs and miRNAs. | http://dorina.mdc-berlin.de/ | 30 |
| RBPDB | Experiments and observations about RBP binding sites in metazoan genomes | All experiments with binding data related to metazoan RBPs can be retrieved by entering the associated gene name. Input sequences can be scanned for matches with RBP binding sites. Includes motif models for more than 70 RBPs. | http://rbpdb.ccbr.utoronto.ca/ | 31 |
| Rfam | Non-coding RNA genes, structured cis-regulatory elements and self-splicing elements | Each entry includes multiple sequence alignment, a secondary structure, and related references. Please see associated reference for a complete description of available features. | http://rfam.sanger.ac.uk/ | 32 |
| UTRSite | Regulatory elements in 5′ and 3′ UTRs | Each entry summarizes the current knowledge on a regulatory element: location (e.g., 3′UTR), Rfam cross-reference, binding proteins and interactor(s) of binding protein(s) and related references. Tools for searching and scanning are available. | http://utrsite.ba.itb.cnr.it/ | 33 |
There are two major approaches for large-scale assays of RBP binding sites in vivo: Ribonucleoprotein immunoprecipitation (RIP)-based methods, which do not permanently cross-link the RBP to the RNA, and cross-linking and immunoprecipitation (CLIP)-based methods, which do. In RIP-based assays, RNAs associated with the RBP of interest are isolated from cell lysate after immunoprecipitation of the RBP, and then identified using either microarray or sequencing technologies.35 CLIP-based assays use ultraviolet (UV) light to form permanent cross-links between RNAs and the RBP, followed by use of ribonuclease to partially digest the bound RNAs,36 leaving only small segments that are in direct contact with the RBP. Although RIP-based assays are simpler and more widely applicable, because UV-based cross-linking is difficult in some cells or tissues,37 the irreversible covalent bond introduced by cross-linking allows a more stringent washing procedure in CLIP, which reduces the number of false-positive targets during the purification step. Cross-linking also protects the target site from ribonuclease digestion, allowing a much greater resolution in determining the actual site of interaction. One method, photoactivatable-ribonucleoside-enhanced cross-linking and immunoprecipitation (PAR-CLIP), modifies CLIP by culturing living cells with a photoreactive ribonucleoside analog, such as 4-thiouridine (4-SU), to facilitate cross-linking.38 The chemical structural change of the 4-SU base upon cross-linking to the RBP causes preferential pairing of guanine (G) rather than adenine (A) with the 4-SU base, and therefore introduces a thymidine (T) to cytidine (C) transition at the cross-linked position during PCR amplification. In PAR-CLIP, the frequencies and types of mutations observed are used as indicators to pinpoint the precise RBP binding site.39 Diagnostic mutations are also observed in other CLIP approaches, though with a lower frequency.40 Although these techniques increase the resolution of these methods, they still cannot robustly achieve single-nucleotide resolution.40 Furthermore, the cross-linking step as well as the choice of RNAase can introduce nucleotide biases in the read data that, if not corrected, can mask the true sequence binding preference of the RBPs.40,41 However, when combined with computational motif finding methods that correct these biases and improve resolution, CLIP-based methods can support the definition of detailed sequence and structural RNA-binding preferences. Recent examples of combined analyses include: Lin28,34 GLD-1,42,43 FMRP,44 and HuR.45 Databases containing sets of CLIP-based target regions for RBPs include doRiNA and CLIPZ (see Table 1).
COMPUTATIONAL METHODS THAT USE PRIMARY SEQUENCE TO IDENTIFY RBP TARGET SITES
Even when experimentally defined RBP-binding sites are available, computational motif-based methods are useful to define the precise site of binding, to detect false positives and negatives, to identify degenerate motifs, to model the impact of RNA secondary structure on binding, to identify co-binding factors (e.g.,46), and to predict the likely impact of polymorphisms on RBP–RNA interactions.
Often motif models developed for DNA-binding proteins have been adapted to identify primary sequence preferences of RBPs and to scan transcripts for potential binding sites.47–52 For example, MatrixREDUCE was used to find RNA motifs associated with transcript stability in yeast48 and to recover binding preferences of RBPs from in vitro binding affinity data.53 This model represents the binding sites with a position-specific affinity matrix (PSAM) that can be used to predict the relative affinity for each potential binding site. Unlike many other motif-finding methods, MatrixREDUCE takes as input quantitative values associated with each sequence in the dataset rather than a subset predefined as ‘bound’ or ‘unbound’. MEME (multiple expectation maximization for motif elicitation)49 is another popular motif discovery algorithm originally designed to find repeated, ungapped sequence patterns in DNA or proteins. MEME has been used to predict motifs for Puf proteins in flies and yeast.54,55 Additional models, such as FIRE (finding informative regulatory elements)50 and REFINE (relative filtering by nucleotide enrichment),51 have been used to identify a group of sequence consensuses from yeast RIP-Chip datasets.1 Motif finder methods that consider the rank-order of genome-wide binding sites, like AMADEUS52 or cERMIT47, are popular for CLIP-seq data (see, e.g., Ref 56 or 45) because CLIP-seq read clusters are typically assigned a semiquantitative score (e.g., a P-value). CLIP-seq reads can be preprocessed with PARalyzer39 to score potential RNA–protein interaction sites taking into consideration the locations of the diagnostic PAR-CLIP mutations. A summary of motif discovery tools can be found in Table 2. Users who choose to run DNA-motif finders on RNA should adjust the options within these methods (e.g., searching complementary strands should be turned off).
Table 2.
Motif Finding Methods
| Software/Method | Input | Summary | Availability | References |
|---|---|---|---|---|
| AMADEUS | DNA or RNA sequences | A method for finding short sequence motifs overrepresented in the promoters or 3′UTRs of a given set of genes | Software package: http://acgt.cs.tau.ac.il/amadeus/download.html | 52 |
| Aptamotif | RNA sequences identified by SELEX | A method for finding sequence-structure motifs in SELEX-derived aptamers. RNA secondary structure is predicted with ensemble-based methods. | Software package: available upon request | 57 |
| CMfinder | RNA sequences | Extension of CM models to search for RNA motifs in a set of unaligned sequences with long flanking regions | Software package and web server: http://bio.cs.washington.edu/yzizhen/CMfinder/ | 58 |
| cERMIT | DNA or RNA sequences and associated expression or affinity measures | A rank-ordered-based method that searches for sequence motifs bested supported by the observed experimental evidence (i.e., semiquantitative genome-wide binding data). It uses the complete dataset and does not require a cutoff to define the positive set. | Software package: http://www.genome.duke.edu/labs/ohler/research/transcription/cERMIT/ | 47 |
| COVE | RNA sequences and alignment (optional) | Implementation of CMs for (1) secondary structure-based multiple sequence alignment; (2) consensus secondary structure prediction; and (iii) secondary structure-based database scanning. | Software package: ftp://selab.janelia.org/pub/software/cove/ | 59 |
| FIRE | DNA or RNA sequences | A method to detect DNA or RNA motifs that model the mutual information between sequences and gene expression measurements. | Software package: http://tavazoielab.princeton.edu/FIRE/ Web server: https://iget.princeton.edu/ |
50 |
| MatrixREDUCE | DNA or RNA sequences and associated expression or affinity measures | A biophysical model to discover sequence-specific binding affinity of the factor of interest (TF or RBP). | Software package: http://bussemaker.bio.columbia.edu/software/MatrixREDUCE/ | 48 |
| MEME | DNA or RNA sequences | A generative model for finding motifs in DNA or protein sequences. Can be used for finding sequence motifs in RNA sequences. | Software package and web server: http://meme.sdsc.edu/meme4_6_0/cgibin/meme.cgi | 49 |
| MEMERIS | RNA sequences and predicted structures | Extension of MEME for finding RNA motifs. It uses RNA structure information as a prior to guide the motif search toward single-stranded regions. | Software package (includes scripts for structure prediction): http://www.bioinf.uni-freiburg.de/∼hiller/MEMERIS/ | 60 |
| REFINE | DNA or RNA sequences | Extension of MEME, filters out regions of target sequences that are relatively devoid of discriminatory hexamers, and then applies MEME motif-finding algorithm. | Software package: http://nar.oxfordjournals.org/content/early/2010/10/18/nar.gkq920/suppl/DC1 | 51 |
| RNAalifold | RNA alignment | A method for detecting conserved RNA secondary structures in a family of related RNA sequences. | Software package: http://www.tbi.univie.ac.at/∼ivo/RNA/ Web server: http://rna.tbi.univie.ac.at/cgibin/RNAalifold.cgi |
61 |
| RNAcontext | RNA sequences, associated affinity measures and predicted structures | A discriminatory approach for finding RNA motifs that represent the sequence and structure preferences of RBPs. RNAcontext can model a wide range of structure features using a flexible alphabet. | Software package (includes scripts for structure prediction): http://morrislab.med.utoronto.ca/software.html | 53 |
| RNApromo | RNA sequences | CM-based model for finding RNA motifs. | Software package and web server: http://genie.weizmann.ac.il/pubs/rnamotifs08/rnamotifs08_predict.html | 62 |
Primary sequence motif-based models can miss important secondary structural context constraints and, in doing so, incorrectly predict the primary sequence preference of an RBP.60 For example, both REFINE and FIRE fail to identify known binding preferences of Vts1p (i.e., CNGG within a hairpin loop) from RIP-Chip data,1 whereas this primary sequence motif is easily found on the same data by motif finders that also model preferences for RNA accessibility.63
To incorporate the mRNA secondary structure information into RBP motif discovery, one must first determine the structure. This is still a very active area of research and, in the following sections, we review both computational and experimental methods used to estimate RNA secondary structure. We then review the evidence supporting a role for intrinsic RNA secondary structure in sequence-specific RBP binding and, finally, describe RBP motif discovery algorithms that incorporate secondary structure information.
COMPUTATIONAL METHODS FOR PREDICTION OF RNA STRUCTURE
The most popular computational method to fold a single RNA sequence is based on the calculation of free energy from thermodynamic parameters derived from chemical melting experiments.64–66 Often, the focus is on the structure with the minimum free energy (MFE) because it is assumed that the RNA sequence folds into the lowest free energy structure at equilibrium.65–67 However, as thermodynamic parameters have substantial uncertainties and RNA secondary structure is often dynamic,68–70 the predicted MFE structure may not accurately represent the typical base-pairing that occurs in the structure. To address these concerns, some methods consider the ensemble of all possible structures.67,71–74 One way to represent this ensemble is to use the centroid structure, which is defined as the structure with minimum total base-pair distance to all other structures in the ensemble.75
Another way is to calculate base-pair probabilities from all possible structures using the partition function, with the assumption that the frequency of any specific RNA structure obeys the Boltzmann distribution.74 However, accurately predicting the global structure of an RNA is challenging owing to the decreasing predictive power of computational methods with increasing length of the input RNA.76 For long mRNA sequences, it is, in fact, often more accurate to only estimate structure using local interactions among bases and to ignore any potential long-range pairings.77 RNAplfold is one method for predicting site accessibility by averaging across short windows of the mRNA centered on the site of interest.71,73
Other approaches predict RNA secondary structure are based on pairwise covariation in multiple alignments with the assumption that functional RNA families should have conserved patterns of base-pairing. Covariance models (CMs) are a specialized stochastic context free grammar (SCFG) (Box 1) that probabilistically model both the RNA secondary structure and the primary sequence consensus of an RNA family.59,78 CMs are fit through a procedure that iterates between aligning individual sequences to a single CM and refining the CM based on the alignment.59,78 These methods work best when a good initial alignment is available to seed the search, and are used to predict families of functional RNAs (like tRNAs); however, their ability to model RBP binding sites in general is unclear. Indeed, the main challenge for predicting consensus structure from multiple sequences is that accurate structure prediction requires an accurate multiple-sequence alignment. Not only is simultaneously folding and aligning sequences computationally challenging,79 but also this strategy may not be appropriate for modeling RBP-binding sites, as only the parts of the secondary structure that affect binding by the RBP may be conserved. We return to this issue in later sections where we introduce CM-based motif finders.
BOX 1 STOCHASTIC CONTEXT-FREE GRAMMAR.
Context-free grammars (CFGs) can be used to describe valid RNA secondary structures with nested base pairs using a set of production rules generated from outside in. Stochastic context-free grammars are extensions of CFGs that assign a probability to each rule and thereby specify a probability distribution over sequences that satisfy the grammar.
EVIDENCE THAT INTRINSIC RNA SECONDARY STRUCTURE HAS AN IMPACT ON RBP BINDING
The accessibility of a potential RBP target site plays an important role in finding whether the RBP actually binds to the site. Often ‘accessibility’ is defined on the basis of predictions of RNA secondary structure and can be roughly interpreted as the proportion of transcripts in which that site is single-stranded. This calculation is based exclusively on the RNA sequence without consideration of a potential role for other binding factors. The role of mRNA accessibility in binding-site selection by microRNAs (miRNAs) and small interfering RNAs (siRNAs) is well established80–83; the role of accessibility in RBP–RNA interaction has taken longer to establish owing to the diversity and complexity of these interactions.
The effect of RNA secondary structure in recognition of target sites was first investigated for the RBP, HuR.84 A positive correlation was found between predicted site accessibility and the binding affinity of HuR to sites that matched the NNUUNUUU HuR consensus. Furthermore, it was possible to alter HuR–mRNA binding in vitro and to increase mRNA stability in cell lysates by introducing secondary structure modulators that either increased or decreased the predicted accessibility of the HuR binding site; this was accomplished by hybridization to complementary RNAs that were predicted either to ‘open’ or ‘close’ the HuR binding site within the target mRNA's secondary structure. Reduction in predicted accessibility also explained the reduction of HuR–TNFa binding upon an insertion of a sequence adjacent to the AU-rich element (ARE) in TNFa.85,86 A companion paper contained what we call the Hackermüller-Stadler model, which models the observed Ka of an RBP to a bound RNA as the product of the probability (in the RNA structure ensemble) that the site is in the preferred structural context and the Ka of the RBP for the site in this context.87
We have explored the role of accessibility in RBP–target interactions for more than a dozen RBPs from yeast, flies, and humans; these RBPs contain a range of RBDs with diverse primary sequence binding preferences.63 By analyzing RIP-Chip datasets of RBPs using these preestablished preferences, we found that, for >70% of the RBPs, when we considered target-site accessibility we significantly increased the ability to predict in vivo binding of those RBPs (Figure 2). Because this accessibility was predicted based solely on the mRNA sequence,71,73 these results suggest a greater than previously anticipated role for intrinsic mRNA secondary structure in determining RBP target preference. Furthermore, we found that more stringent methods to estimate accessibility were better predictors of RBP binding than others, suggesting that we could use this dataset as a benchmark for comparing different mRNA secondary structure estimates.88 Indeed, replacing the accessibility-based scoring system with one that considers the structural context of ssRNA further improves prediction of in vivo target selection by RBPs (Figure 3).
Figure 2.
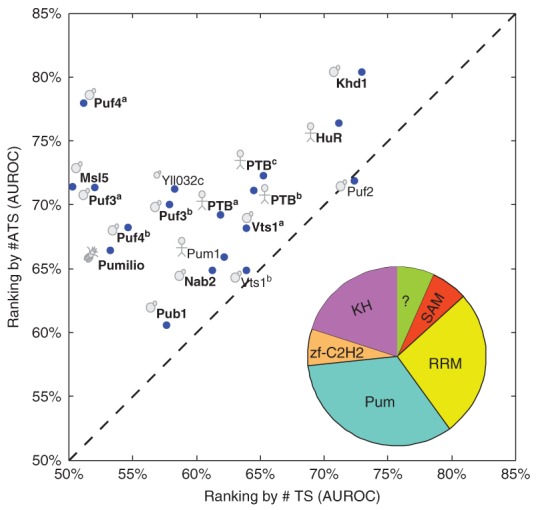
Target site accessibility predicts in vivo binding for a diverse range of RNA-binding proteins (RBPs). Comparison of accuracy in predicting bound transcripts based on a given consensus, using either #ATS (i.e., the expected number of accessible target sites, y-axis) or #TS (i.e., the number of target sites, x-axis). Each dot represents the results of an RBP coupled with its previously defined consensus sequence. If there are multiple reported consensus sequences for a protein, the result for each is shown and is distinguished from others by a superscript. Cartoons indicate the species of origin (yeast, fly, or human). RBPs in bold have significantly improved AUROC for #ATS versus #TS (P < 0.05, Delong-Delong-Clarke-Pearson test). The RBDs housed in the RBPs (using SMART domains) are summarized in the pie graph.
Figure 3.
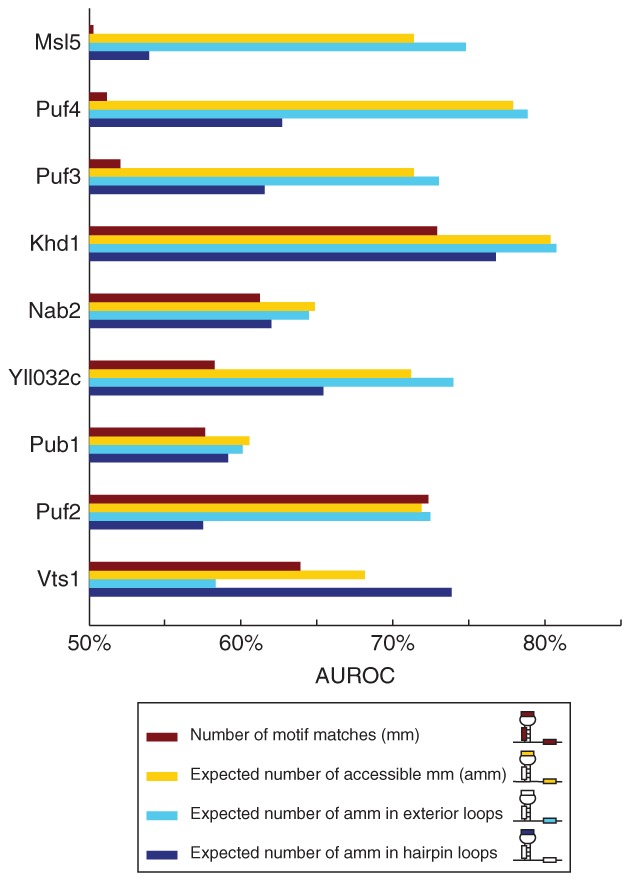
Structural context of target sites improves prediction of target mRNAs bound in vivo by RNA-binding proteins (RBPs). Bar graphs compare the accuracy of different methods that use the structural context of motif matches to predict in vivo binding of RBPs. The inset describes the different bars within the graph.
EXPERIMENTAL METHODS FOR PREDICTION OF RNA STRUCTURE
By far, the fastest, cheapest, and easiest way to estimate mRNA secondary structure is using computational prediction methods (reviewed above and summarized in Table 3). However, the accuracy of these methods is controversial and, recently, biochemical methods have been introduced that query mRNA secondary structure genome-wide. In this section we review experimental approaches used for prediction of mRNA structure.
Table 3.
Web Resource for Predicting mRNA Secondary Structure
| Software/Method | Input | Summary | Availability | References |
|---|---|---|---|---|
| Mfold | RNA sequence | It predicts the suboptimal structures within a free energy increment from the minimum free energy. | Software package and web server: http://mfold.rna.albany.edu/?q=mfold | 89 |
| RNAshapes | RNA sequence | It calculates shapes and their probabilities by analyzing the full ensemble, predicts the complete set of suboptimal structures and their probabilities | Software package and web server: http://bibiserv.techfak.uni-bielefeld.de/rnashapes/ | 90 |
| RNAstructure | RNA sequence | It includes algorithms for RNA secondary structure prediction and calculation of base-pair probabilities. | Software package with GUI: http://rna.urmc.rochester.edu/RNAstructure.html | 91 |
| SFOLD | RNA sequence | It computes base pair probabilities from a representative sample of the full ensemble | Software package and web server: http://sfold.wadsworth.org/ | 75 |
| Vienna package | RNA sequence | RNAfold: predicts MFE energy structure and base-pair probabilities RNAplfold: uses local folding to calculate base-pair probabilities |
Software package: http://www.tbi.univie.ac.at/∼ivo/RNA/Web server: http://rna.tbi.univie.ac.at/ | 72 92 |
Physical methods, including X-ray crystallography and nuclear magnetic resonance (NMR) spectroscopy, have been used to describe RNA's three-dimensional structure in great detail, but are often time-consuming and are limited to relatively short RNAs. RNA footprinting, an easier alternative, is often selected to analyze the structure of long RNAs. RNA footprinting detects RNA structure by treating the RNA of interest with a chemical or a nuclease to modify or cleave bases, respectively, that have a particular structural conformation (e.g., single-stranded, double-stranded, or solvent-exposed).93–97 RNase-cleaved products, usually radioactively end-labeled, are then detected by autoradiography, while the chemically modified bases are detected by electrophoresis of the reverse-transcribed products that have stalled at the modified bases.
RNA footprinting has been extended to a large-scale method by combining next-generation sequencing technology with traditional RNase/chemical footprinting in order to simultaneously probe a mixture of RNAs.98–100 Structure probing by chemical modification has a higher resolution than by nucleases because it is less restricted by steric hindrance; however, the read-out of the modification is much more difficult and, to date, has not been applicable to genome-wide assays of mRNA secondary structure. Selective 2′-hydroxyl acylation analyzed by primer extension (SHAPE) is the major chemical modification-based technique. Unlike other base-selective chemical reagents, the hydroxyl-selective electrophiles used in SHAPE prevent reverse transcription on flexible nucleotides (e.g., single-stranded ones) by reacting with the 2′-hydroxyl group to form a 2′-O-adduct. This method can thereby interrogate all nucleotides in an RNA molecule simultaneously without biases toward certain primary sequences.101 SHAPE-seq, which couples SHAPE chemistry with a multiplexed hierarchical barcoding and deep sequencing strategy,100 has been used to accurately and simultaneously probe structures of several in vitro-transcribed RNAs.100 However, the barcodes must be designed to target specific RNAs, thus preventing the expansion of SHAPE-seq to genome-wide assays.102
In contrast, large-scale, nuclease cleavage-based structure probing experiments have recently been developed.98,99 Parallel analysis of RNA structure (PARS) has been used to profile mRNA secondary structures in the budding yeast, S. cerevisiae.98 Purified polyadenylated transcripts were renatured in vitro and separately treated with RNase S1 (specific for single-stranded RNA) and RNase V1 (specific for double-stranded RNA). The cleaved products from these two complementary enzymes were then analyzed using deep sequencing technology to infer single- or double-strandedness at single nucleotide resolution. Related techniques have been used to probe mRNA secondary structure in Drosophila and C. elegans.103 An alternative method, fragmentation sequencing (Fragseq), has been used to provide an ‘RNA accessibility profile’ on the naked RNAs from the mouse nuclear transcriptome.99 This method differs from PARS in two ways. First, Fragseq focuses on cleavage products that are 20–100 bases long, while PARS explores all the cleavage products using random fragmentation. Fragseq, thus, primarily focuses on small RNAs.102 Second, Fragseq uses only RNase P1 to cleave single-stranded RNA and reports the log ratio between the number of sequence reads obtained from the nuclease-treated sample and the untreated sample. This is done to control the occurrence of RNA degradation in the cell or during sample preparation. PARS, however, uses both RNase V1 and RNase S1 and reports the log ratio between the number of sequence reads obtained from the RNase V1-treated sample and the RNase S1-treated sample.
These methodologies are still undergoing development. Currently, PARS requires multiple manipulations on cellular RNA including heating and refolding—it is unclear how the resulting product reflects the in vivo mRNA secondary structure. Although recent reports suggest that SHAPE-like methodologies can be applied in vivo,104 to date, genome-wide SHAPE has not been reported.
COMPARISON OF EXPERIMENTAL AND COMPUTATIONAL METHODS FOR PREDICTION OF RNA STRUCTURE
To assess the relative accuracy of experimentally assayed versus computationally determined mRNA secondary structure, we applied a slightly modified version of our benchmark63 to compare how well each set of structures supports the prediction of in vivo RBP binding. Specifically, we compared the ability of PARS98 and RNAplfold71,73 to recover RBP binding sites. RIP-Chip data was used to define sets of bound transcripts (i.e., positives) and co-expressed but unbound transcripts (i.e., negatives) for nine yeast RBPs with defined consensus single-stranded binding motifs that are predictive of in vivo binding.63 For each RBP, we scored every bound or unbound transcript according to the structural accessibility of all sites in that mRNA that matched the RBP's previously described consensus motif (as described in Ref 63). Briefly, for each RBP, the accessibility score for an entire transcript was set to be the maximum of the RNAplfold-predicted accessibility scores for each match to the RBP consensus motif in that transcript. The accessibility score for a match was set to be the minimum of the accessibilities of all nucleotides in the match. For many sites, PARS scores were unavailable for every nucleotide, so this minimum was calculated over all nucleotides for which PARS data were available. For PARS, we used the inverse of the PARS score as a measure of single-nucleotide accessibility and, for RNAplfold, we used the predicted probability that the nucleotide was single-stranded. We have previously reported that the minimum is the best single-nucleotide predictor of the accessibility of the entire binding site and the maximum is nearly as good as the sum at consolidating estimates from multiple sites.63 This slight modification of our original methodology allows us compare PARS and RNAplfold fairly without requiring us to make arbitrary choices in order to calibrate the PARS scores. For each RBP, we then ranked transcripts according to their accessibility scores and evaluated how well that ranking distinguished positive and negative transcripts using the area under the receiver operating characteristic (AUROC), a standard metric commonly used for this purpose.
Using these methods, we found that RNAplfold-based calculations of site accessibility are significantly better predictors of in vivo binding than those provided by PARS (P = 0.004, two-tailed sign test; Figure 4(a)). Note that only 58% of nucleotides have a defined PARS score, possibly due to the non-uniform ability of V1 and S1 nucleases to cleave different parts of an mRNA and/or insufficient sequencing depth. RNAplfold remains a better predictor than PARS even when only those nucleotides with PARS scores are considered (P = 0.04, two-tailed sign test; Figure 4(b)).
Figure 4.
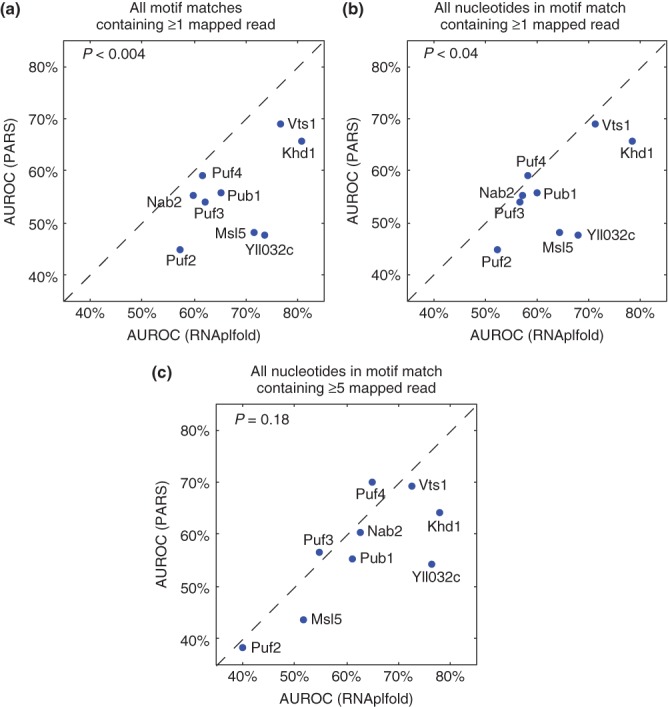
Comparison of prediction accuracy for in vivo binding of nine yeast RNA-binding proteins (RBPs) using parallel analysis of RNA structure (PARS) and RNAplfold to estimate the secondary structure of bound versus unbound transcripts. The results using PARS are shown on the y-axis, those using RNAplfold on the x-axis. (a) The analysis was performed on all consensus sites containing at least one nucleotide with a nonzero PARS score. (b) The analysis was performed only considering nucleotides with nonzero PARS score. (c) As for (b) but with the additional constraint that the transcript load (i.e., reads/nucleotide) was at least five. P-values were calculated using the two-tailed sign test.
As mentioned above, one factor that contributes to the unexpectedly poor performance of PARS is that in vitro refolding may not capture the in vivo structure.105 Sequencing errors are likely to lead to additional inaccuracies; reliable quantification of structure profiles may require a higher read count than the one read/nucleotide that was used to define the PARS scores. Indeed, when we restricted our analysis to transcripts with an average of five reads/nucleotide or higher, the performance difference between PARS and RNAplfold was no longer statistically significant (P = 0.18, two-tailed sign test; Figure 4(c)). Unfortunately, very few transcripts have any PARS data at this restrictive threshold.
In summary, while PARS provides a useful empirical tool to assess mRNA secondary structure on a genome-wide basis, at present data can be collected for only a subset of nucleotides and coverage is strongly biased toward highly expressed transcripts. On the other hand, computational methods such as those based on RNAplfold provide information on every nucleotide and are not sensitive to transcript abundance.
COMBINING EXPERIMENTAL AND COMPUTATIONAL METHODS
In the previous section, we directly compared the performance of experimentally and computationally predicted secondary structure. However, it is possible to incorporate experimentally derived RNA structure profiling data as a guide to computational prediction of RNA secondary structures. For example, chemical/RNase-probe-based measurements of nucleotide structural conformation can be used as additional energy potentials to guide folding.55,56,88 In this case, the folding computation is biased toward RNA secondary structures that are consistent with the experimental data by assigning a large positive free-energy penalty to all possible alternatives.65,66,106 The resulting algorithm has a similar time and space complexity as default secondary structure prediction.106 It is also possible to include an additional term that reflects the inverse correlation between the SHAPE score and the base-pairing probability, and to integrate this term into the RNAstructure91 software. An alternative approach, named ‘sample and select’, uses experimental data to identify the correct structure among the Boltzmann ensemble of structures.107 SeqFold88 modified this approach so that only centroids of structure clusters (identified by Sfold) are considered as candidate structures. Seqfold is less sensitive to noise in experimental data than RNAstructure and ‘sample and select’. Binding site accessibility assessed by SeqFold has been shown to be a better predictor of in vivo binding than RNAfold for at least some yeast RBPs; however, not all available RBPs have been assessed. However, initial results suggest that incorporating current experimental data does, indeed, improve secondary structure prediction by computational methods.88
RBP MOTIF DISCOVERY ALGORITHMS THAT INCORPORATE SECONDARY STRUCTURE INFORMATION
Because mRNA secondary structure can either be predicted or measured, and these estimates improve the ability of computational methods to predict in vivo binding, it makes sense to incorporate secondary structure preferences into motif models used to scan for RBP target sites.
There are two main approaches to incorporating structural information: (1) methods that model the preferred structural context of the primary sequence motif bound by the RBP53,60,63,108 and (2) methods that explicitly model the secondary structure recognized by the RBP using stochastic context-free grammars.59,58,62 Table 2 summarizes the motif models described below.
Structural Context-Based Methods
The first structural context-based method, MEMERIS, incorporates the Hackermüller-Stadler model into the popular DNA motif finding program, MEME, by annotating nucleotides according to their predicted RNA secondary structure. MEMERIS precomputes for each word (i.e., k-mer) the probability that the word is in single-stranded context (as predicted by RNAfold), and then uses these values as priors on possible motif start positions. This adaptation changes the search so that motifs that are enriched in single-stranded regions are preferentially found. Compared with MEME, MEMERIS is more accurate at recovering RNA motifs in both artificial and in vitro datasets.60 As expected, MEMERIS is able to identify the correct motifs in single-stranded regions even with the existence of a stronger sequence motif embedded in a double-stranded region. MEMERIS could, in principle, easily be modified to incorporate the probability that each word is in another secondary structure context as a way of identifying, say, motifs for an RBP that binds hairpin loops; to date, such a modification has not been tested. MEMERIS is a ‘generative’ motif finding algorithm in that it tries to find motifs enriched among a set of bound transcripts.
Often, it is more accurate to identify ‘discriminative’ motifs, which distinguish between sets of bound and unbound transcripts.53,109,110 This approach removes the necessity to define a ‘background model’ because it uses the unbound set. We have described a discriminative motif-finding method called #ATS (i.e., expected number of accessible target sites)63 that incorporates accessibility. #ATS also differs from MEMERIS in that it fits a degenerate consensus sequence motif model (e.g., CNGG, where N could be any base). #ATS uses a greedy heuristic to build its model: it starts from the five hexamers with the largest predictive power and iteratively refines them (by shortening, lengthening, or introducing degenerate bases) until it can no longer improve the discriminative power of the motif. Applying the #ATS model to several RIP-Chip datasets has successfully recovered the previously identified motifs indicating its ability to identify in vivo RBP binding sites.63
The methods mentioned above allow query of single-strandedness only. However, some RBPs may have more complex structural-context preferences. StructRED108 extends MatrixREDUCE48 to find RNA cis-regulatory elements that are located in hairpin loops. Briefly, StructRED pre-filters all k-mers for those that are flanked by at least three bases that can pair (e.g., A-U, G-C, and G-U) and applies MatrixREDUCE to these k-mers. Unlike the two methods described above, no consideration is given to the thermodynamic stability of the stem in the naked mRNA; however, it is known that RBPs such as Vts1p can stabilize otherwise unstable loop structures.16–18 StructRED correctly recovered the known binding preferences of Vts1p in yeast and its ortholog, Smaug, in flies, and discovered a number of RNA-regulatory elements in humans and flies; however, its limited representation of secondary structure elements makes it difficult to apply it to RBPs other than stem-loop binders.
Some RBPs can bind their target site in a variety of structural contexts; for example, SNRPA (aka U1A) binds AUUGCAC when it is at the 5′ end of a hairpin or internal loop111 but can bind the same sequence with lower affinity if it is single-stranded but not in a loop.112 RNAcontext53 is the first motif-finding algorithm that is designed to detect the relative preferences of an RBP for multiple structural contexts. Like #ATS, it is a discriminative motif finding method that outputs an RNA sequence motif but, unlike either #ATS or MEMERIS, it also outputs a vector indicating the relative preferences for a nonoverlapping set of structural contexts (e.g., paired, hairpin loop, etc.). When applied to RNAcompete data from nine RBPs,9 RNAcontext recovered known structural context preferences, as well as, showing an improved ability to predict in vitro binding to sequences not used to train the motif model.53 RNAcontext uses represent the structural context of a base by annotating it with probability distribution over an alphabet representing the possible contexts (Figure 5). The input into RNAcontext consists of a set of RNA sequences, their associated structure profiles as computed by RNAplfold or Sfold, and estimates of binding affinities of the RBP of interest. Each input RNA sequence is scored by using the sequence and structure context parameters. RNAcontext has been applied to in vitro binding-affinity data. The RBPmotif webserver implements RNAcontext.113
Figure 5.
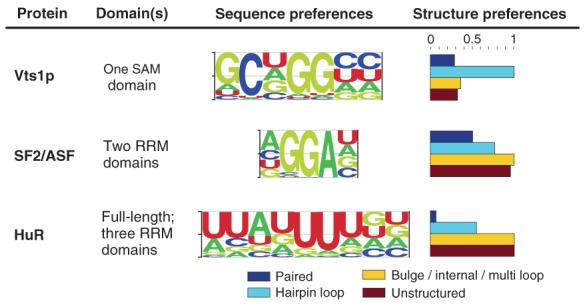
RNAcontext-predicted motifs. The figure shows motifs and their structural contexts predicted by RNAcontext using RNAcompete binding data.9 (Reprinted with permission from Ref 53. Copyright 2010, PLoS Computational Biology Creative Commons Attribution License.)
Stochastic Context-Free Grammar (SCFG)-Based Methods
There are two main methods in this category: CMfinder58 and RNApromo.62 Both fit CM-based motif models similar to those used to define RNA families.58,62,114 However, unlike many RNA families, sets of RBP target sites from different transcripts rarely have conserved sequence in paired regions, making it difficult to establish the initial alignment required for the CM iterations. CMfinder and RNApromo both use strategies based on thermodynamic stability to establish an initial structural alignment of putative RBP binding sites. CMfinder identifies (and aligns) shared secondary structures among the minimum free energy structures of the input sequences. RNApromo replaces this initialization with nonredundant substructures that are overrepresented in the positive set versus the background set. Methods like these are well-suited to modeling complex primary and secondary structure preferences such as those recently reported for LIN28A34 and ADAR2.15,115 Although neither method has yet been used for these specific RBPs, CMfinder has successfully identified complex structures such as riboswitches.116
CMfinder focuses on large secondary structures with extensive base-pairing. Recently, two CM-based methods have been described, Aptamotif57 and TEISER,117 which can detect motifs that contain shorter stems. Aptamotif57 adapts the iterative learning procedure of CMs to find sequence-structure motifs in SELEX-derived aptamers. As a first step, Aptamotif parses both optimal and suboptimal structures of input sequences to generate a set of loop substructures. Next, a set of seed motifs is randomly selected from the set of all loop substructures. Input sequences are scanned with these seed motifs and matching regions are aligned. The motifs with the best alignment score are retained for the next iteration. Aptamotif has been able to recover the reported binding preference of L22, a ribosomal protein that binds a long primary-sequence motif within a hairpin loop; although MEMERIS correctly identified the motif, it was not able to capture permitted gaps or the requirement for the hairpin structural context. Aptamotif's search procedure ignores single-stranded motifs outside of loops, and the use of suboptimal structures may limit its use with longer RNA sequences as the number of such structures increases exponentially with sequence length. This suggests that Aptamotif will be most useful for RBPs that have a strong requirement for specific secondary structural contexts. TEISER is a method specifically designed to identify short stem-loop structures with primary sequence preferences in either the loop or the stem. It represents these using a non-stochastic context-free grammar and does a combinatorial search over all possible stem-loops up to a given size to detect those with highest mutual information with RBP binding or mRNA stability.117 To date, it has not been validated on RBPs with known stem-loop binding preferences, and its predicted binding preference for HNRNPA2B1 differs from previously reported ones for close homologs118,119 and from an in vitro binding assay,26 all of which are in agreement with one another.
One must exercise care when using SCFG-based methods to model the binding preferences of sequence-specific RBPs. Because these methods explicitly search for secondary structures, while ignoring the impact that sequence flanking the structure might have on its folding, these methods are unsuitable for use with RBPs that simply bind an accessible mRNA sequence. For these RBPs, the ‘structural context’ of their binding site cannot be detected by SCFG methods because it is defined by the absence of nearby flanking sequence that can pair with the binding site. This, coupled with the tendency of CMs to over-predict pairing,63 makes it hard to interpret motif models generated by these methods for HuR120 and Puf3p62 which place their target sites partially within the stem of a hairpin loop. Both RBPs had been previously reported to bind ssRNA84,121 and to prefer ATS.63 Subsequent reanalysis of the HuR stem-loop binding model on different data122 suggested that the binding model was simply capturing biases in dinucleotide frequencies rather than secondary structure. Over-prediction of secondary structure may also account for differences between TEISER's motif model for HNRNPA2B1 and those of others.
USING SEQUENCE AND STRUCTURE CONSERVATION TO FIND RBP BINDING SITES
Another strategy for identification of likely RBP binding sites is to search for motifs in the 5′UTRs or 3′UTRs (untranslated region) that are surprisingly highly conserved, which display a bias toward conservation when they are in the sense strand and do not correspond to miRNA seeds. This approach was first used within the context of genome-wide discovery of regulatory motifs.123 On the basis of distinct patterns of genome-wide conservation of known motifs versus random sequences across four yeast species (S. paradoxus, S. mikatae, S. bayanus, and S. cerevisiae), conservation criteria were used to discover regulatory motifs. The algorithm uses an enumeration approach to select strongly conserved motif cores and then extend or collapse these motifs to produce candidate regulatory motifs. A similar comparative genomics analysis approach has been applied to the genomes of 12 Drosophila species.124 This particular method used the total branch length over which a motif is conserved, to estimate the conservation level of a motif instance. Such a scoring system is robust to comparative genomic analysis because it does not explicitly penalize missing instances, but instead rewards the motif instances in distantly related species more than ones in closely related species in order to capture neutral divergence of the motifs.124 Motifs describing primary and secondary structure preferences have been detected using a comparative method called EvoFam, which uses phylogenetic, stochastic context-free grammars125 to identify conserved, potentially regulatory, RNA structures in a 41-way genomic vertebrate alignment.126
FUTURE CHALLENGES AND DIRECTIONS
To this point, we have described experimental methods to query RBP–RNA interactions and also how computational models can be used to infer binding preferences from experimental data. The following sections summarize the existing challenges, and point to possible improvements that might be made in several areas.
Combinatorial Interactions among RBPs, miRNAs, and mRNAs
There is increasing evidence for widespread nonadditive interactions among trans-factors in post-transcriptional regulation. A number of such interactions have already been described among specific RBPs1,41,127,128 or between RBPs and miRNAs.45,46,129–133 Other interactions are suggested by computational analyses: for example, miRNA sites are significantly enriched in the human PUM1 and PUM2 targets defined by RIP-Chip experiments, and the PUM-binding motifs (UGUANAUA) is enriched in the vicinity of the predicted miRNA sites.130 Furthermore, hundreds of short k-mer sequences have recently been reported to have significant correlation with increases or decreases in steady-state mRNA abundance following transfection of small RNAs; a number of these k-mers match the known sequence-binding preference of RBPs including U-rich sequences (bound by ELAVL1/HuR and HuD) and AREs bound by a number of ARE-binding proteins.132
These combinatorial interactions can be mediated by the mRNA sequence itself. Competitive interactions can occur as a result of overlapping binding sites. For example, Dead end 1 (Dnd1) positively regulates its targets by counteracting miRNA-mediated repression through binding to U-rich regions in the 3′UTR of the target, thereby physically blocking access to overlapping miRNA target sites.131,134 On the other hand, the secondary structure of the target mRNAs may induce the cooperative binding of trans-acting factors. For instance, the binding of RBPs, PUM1 and PUM2, induce a conformational change in the 3′UTR of P27 mRNA, thus making a target sequence accessible to an miRNA.46 Currently, little computational methodology exists to detect and model these phenomena.
Gapped Motif Finders for RBPs
Many RBPs have multiple RBDs: either repeated copies of the same domain or a mixture of distinct domains.135 For instance, the human PUM1 protein has eight repeats of the Puf domain where each domain recognizes a single nucleotide.14 The poly(A) binding protein (PABP) has four RRM domains and each pairwise combination of these domains has a different RNA-binding activity.136 RBD3 and RBD4 of polypyrimidine-tract binding (PTB) protein bind RNA with a fixed orientation relative to each other such that a single RNA cannot be bound simultaneously by these two RBDs unless the two cis-elements are separated by a linker sequence (Figure 6(a)).137 The two dsRBDs in ADAR2 bind to distinct locations in the GluR-2 R/G RNA and both are essential for R/G editing15,115(Figure 6(b)).
Figure 6.
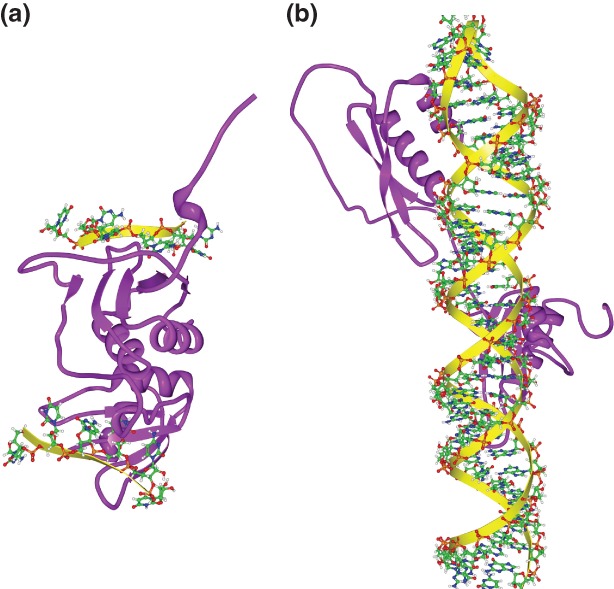
Three-dimensional structures of multiple RNA-binding domains (RBDs) in complex with RNA. (a) Solution structure of polypyrimidine tract binding (PTB), RBD3, and RBD4 in complex with CUCUCU RNA [Protein Data Bank (PDB): 2ADC]. RBD3 and RBD4 have different binding specificity: RBD3 binds YCUNN and RBD4 binds YCN (Y, pyrimidine; N, any nucleotide). RBD3 and RBD4 interact extensively, resulting in an antiparallel orientation of their bound RNAs, suggesting that the only way to make these two RBDs bind to a single RNA is to separate their sites by a linker sequence.137 (b) Solution structure of ADAR2 dsRBD1 and dsRBD2 in complex with GluR-2 R/G RNA (PDB: 2L3J). The dsRBDs recognize their targets by the shape and by the primary sequence in the minor groove. Sequence-specific recognition is achieved through a hydrogen bond to the amino group of G (in the GG mismatch for dsRBD1; in the GC pair for dsRBD2) via a β1-β2 loop and via a hydrophobic contact to adenine H2 (in the AU pair for dsRBD1; in the AC mismatch for dsRBD2) via helix α1. The two dsRBDs bind one face of the RNA and cover about 120° of the turn of the RNA helix.15
The unique modular structure of each RBP is crucial for definition of its mode of target recognition, especially for those RBPs equipped with multiple copies of the same RBD.137 Modeling the modular structure of RBPs in identification of their binding sites is crucial. Some work has, for example, modeled gapped DNA motifs, such as GLAM2 (gapped local alignment of motifs).138 However, finding gapped RNA motifs is much more difficult than finding gapped DNA motifs as one must take into account primary sequence, secondary and even tertiary, structural elements.
Motif Finding for dsRNA Binding Proteins
How do dsRNA binding proteins achieve specificity? Possibly through base-specific interactions with the minor groove of the dsRNA helix139 or subtle differences in dsRNA structures140,141 or a combination of the two.15 To date, few motif finding methods are available for modeling the preferences of dsRNA binding proteins. Early efforts in this area include those that model preferences for length and pairedness of dsRNA stems arising from inter-molecular140 or intra-molecular interactions,141 but more work is needed in this area to develop general methodology.
CONCLUSION
New technologies have rapidly increased the quantity of in vivo and in vitro binding data available for RBPs. At the same time, new methods are being developed to measure or model RNA secondary structure on a genome-wide scale. Motif-finding methods that capture RBP-binding preferences are still in their infancy and have been derived largely from DNA motif finding methods. Motif finding for RBPs is poised to become a rapidly expanding field.
Acknowledgments
This work was funded by the Canadian Institutes of Health Research grants (MOP-125894 to Q.D.M. and MOP-14409 to H.D.L.).
REFERENCES
- 1.Hogan DJ, Riordan DP, Gerber AP, Herschlag D, Brown PO. Diverse RNA-binding proteins interact with functionally related sets of RNAs, suggesting an extensive regulatory system. PLoS Biol. 2008;6:e255. doi: 10.1371/journal.pbio.0060255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Blanchette M, Green RE, MacArthur S, Brooks AN, Brenner SE, Eisen MB, Rio DC. Genome-wide analysis of alternative pre-mRNA splicing and RNA-binding specificities of the Drosophila hnRNP A/B family members. Mol Cell. 2009;33:438–449. doi: 10.1016/j.molcel.2009.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lukong KE, Chang KW, Khandjian EW, Richard S. RNA-binding proteins in human genetic disease. Trends Genet. 2008;24:416–425. doi: 10.1016/j.tig.2008.05.004. [DOI] [PubMed] [Google Scholar]
- 4.Keene JD. RNA regulons: coordination of post-transcriptional events. Nat Rev Genet. 2007;8:533–543. doi: 10.1038/nrg2111. [DOI] [PubMed] [Google Scholar]
- 5.Seeman NC, Rosenberg JM, Suddath FL, Kim JJ, Rich A. RNA double-helical fragments at atomic resolution. I. The crystal and molecular structure of sodium adenylyl-3′,5′-uridine hexahydrate. J Mol Biol. 1976;104:109–144. doi: 10.1016/0022-2836(76)90005-x. [DOI] [PubMed] [Google Scholar]
- 6.Alden CJ, Kim SH. Solvent-accessible surfaces of nucleic acids. J Mol Biol. 1979;132:411–434. doi: 10.1016/0022-2836(79)90268-7. [DOI] [PubMed] [Google Scholar]
- 7.Nagai K, Mattaj IW. RNA-Protein Interactions. Oxford: Oxford University Press; 1994. [Google Scholar]
- 8.Messias AC, Sattler M. Structural basis of single-stranded RNA recognition. Acc Chem Res. 2004;37:279–287. doi: 10.1021/ar030034m. [DOI] [PubMed] [Google Scholar]
- 9.Ray D, Kazan H, Chan ET, Pena Castillo L, Chaudhry S, Talukder S, Blencowe BJ, Morris Q, Hughes TR. Rapid and systematic analysis of the RNA recognition specificities of RNA-binding proteins. Nat Biotechnol. 2009;27:667–670. doi: 10.1038/nbt.1550. [DOI] [PubMed] [Google Scholar]
- 10.Draper DE. Themes in RNA-protein recognition. J Mol Biol. 1999;293:255–270. doi: 10.1006/jmbi.1999.2991. [DOI] [PubMed] [Google Scholar]
- 11.Allers J, Shamoo Y. Structure-based analysis of protein-RNA interactions using the program ENTANGLE. J Mol Biol. 2001;311:75–86. doi: 10.1006/jmbi.2001.4857. [DOI] [PubMed] [Google Scholar]
- 12.Jones S, Daley DT, Luscombe NM, Berman HM, Thornton JM. Protein-RNA interactions: a structural analysis. Nucleic Acids Res. 2001;29:943–954. doi: 10.1093/nar/29.4.943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maris C, Dominguez C, Allain FH. The RNA recognition motif: a plastic RNA-binding platform to regulate post-transcriptional gene expression. FEBS J. 2005;272:2118–2131. doi: 10.1111/j.1742-4658.2005.04653.x. [DOI] [PubMed] [Google Scholar]
- 14.Wang X, McLachlan J, Zamore PD, Hall TM. Modular recognition of RNA by a human pumilio-homology domain. Cell. 2002;110:501–512. doi: 10.1016/s0092-8674(02)00873-5. [DOI] [PubMed] [Google Scholar]
- 15.Stefl R, Oberstrass FC, Hood JL, Jourdan M, Zimmermann M, Skrisovska L, Maris C, Peng L, Hofr C, Emeson RB, et al. The solution structure of the ADAR2 dsRBM-RNA complex reveals a sequence-specific readout of the minor groove. Cell. 2010;143:225–237. doi: 10.1016/j.cell.2010.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Oberstrass FC, Lee A, Stefl R, Janis M, Chanfreau G, Allain FH. Shape-specific recognition in the structure of the Vts1p SAM domain with RNA. Nat Struct Mol Biol. 2006;13:160–167. doi: 10.1038/nsmb1038. [DOI] [PubMed] [Google Scholar]
- 17.Aviv T, Lin Z, Ben-Ari G, Smibert CA, Sicheri F. Sequence-specific recognition of RNA hairpins by the SAM domain of Vts1p. Nat Struct Mol Biol. 2006;13:168–176. doi: 10.1038/nsmb1053. [DOI] [PubMed] [Google Scholar]
- 18.Johnson PE, Donaldson LW. RNA recognition by the Vts1p SAM domain. Nat Struct Mol Biol. 2006;13:177–178. doi: 10.1038/nsmb1039. [DOI] [PubMed] [Google Scholar]
- 19.Wu H, Henras A, Chanfreau G, Feigon J. Structural basis for recognition of the AGNN tetraloop RNA fold by the double-stranded RNA-binding domain of Rnt1p RNase III. Proc Natl Acad Sci U S A. 2004;101:8307–8312. doi: 10.1073/pnas.0402627101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ryter JM, Schultz SC. Molecular basis of double-stranded RNA-protein interactions: structure of a dsRNA-binding domain complexed with dsRNA. EMBO J. 1998;17:7505–7513. doi: 10.1093/emboj/17.24.7505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ramos A, Grunert S, Adams J, Micklem DR, Proctor MR, Freund S, Bycroft M, St Johnston D, Varani G. RNA recognition by a Staufen double-stranded RNA-binding domain. EMBO J. 2000;19:997–1009. doi: 10.1093/emboj/19.5.997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ellington AD, Szostak JW. In vitro selection of RNA molecules that bind specific ligands. Nature. 1990;346:818–822. doi: 10.1038/346818a0. [DOI] [PubMed] [Google Scholar]
- 23.Zhao Y, Granas D, Stormo GD. Inferring binding energies from selected binding sites. PLoS Comput Biol. 2009;5:e1000590. doi: 10.1371/journal.pcbi.1000590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jolma A, Kivioja T, Toivonen J, Cheng L, Wei G, Enge M, Taipale M, Vaquerizas JM, Yan J, Sillanpaa MJ, et al. Multiplexed massively parallel SELEX for characterization of human transcription factor binding specificities. Genome Res. 2010;20:861–873. doi: 10.1101/gr.100552.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Philippakis AA, Qureshi AM, Berger MF, Bulyk ML. Design of compact, universal DNA microarrays for protein binding microarray experiments. J Comput Biol. 2008;15:655–665. doi: 10.1089/cmb.2007.0114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ray D, Kazan H, Cook KB, Weirauch MT, Najafabadi HS, Li X, Gueroussov S, Albu M, Zheng H, Yang A, et al. A compendium of RNA-binding motifs for decoding gene regulation. Nature. 2013;499:172–177. doi: 10.1038/nature12311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Available at: http://cisbp-rna.ccbr.utoronto.ca/ (Accessed 19 October 2013)
- 28.Gruber AR, Fallmann J, Kratochvill F, Kovarik P, Hofacker IL. AREsite: a database for the comprehensive investigation of AU-rich elements. Nucleic Acids Res. 2011;39:D66–69. doi: 10.1093/nar/gkq990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Khorshid M, Rodak C, Zavolan M. CLIPZ: a database and analysis environment for experimentally determined binding sites of RNA-binding proteins. Nucleic Acids Res. 2011;39:D245–252. doi: 10.1093/nar/gkq940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Anders G, Mackowiak SD, Jens M, Maaskola J, Kuntzagk A, Rajewsky N, Landthaler M, Dieterich C. doRiNA: a database of RNA interactions in post-transcriptional regulation. Nucleic Acids Res. 2012;40:D180–186. doi: 10.1093/nar/gkr1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cook KB, Kazan H, Zuberi K, Morris Q, Hughes TR. RBPDB: a database of RNA-binding specificities. Nucleic Acids Res. 2011;39:D301–D308. doi: 10.1093/nar/gkq1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Burge SW, Daub J, Eberhardt R, Tate J, Barquist L, Nawrocki EP, Eddy SR, Gardner PP, Bateman A. Rfam 11.0: 10 years of RNA families. Nucleic Acids Res. 2013;41:D226–232. doi: 10.1093/nar/gks1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Grillo G, Turi A, Licciulli F, Mignone F, Liuni S, Banfi S, Gennarino VA, Horner DS, Pavesi G, Picardi E, et al. UTRdb and UTRsite (RELEASE 2010): a collection of sequences and regulatory motifs of the untranslated regions of eukaryotic mRNAs. Nucleic Acids Res. 2010;38:D75–80. doi: 10.1093/nar/gkp902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wilbert ML, Huelga SC, Kapeli K, Stark TJ, Liang TY, Chen SX, Yan BY, Nathanson JL, Hutt KR, Lovci MT, et al. LIN28 binds messenger RNAs at GGAGA motifs and regulates splicing factor abundance. Mol Cell. 2012;48:195–206. doi: 10.1016/j.molcel.2012.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tenenbaum SA, Carson CC, Lager PJ, Keene JD. Identifying mRNA subsets in messenger ribonucleoprotein complexes by using cDNA arrays. Proc Natl Acad Sci U S A. 2000;97:14085–14090. doi: 10.1073/pnas.97.26.14085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ule J, Jensen KB, Ruggiu M, Mele A, Ule A, Darnell RB. CLIP identifies Nova-regulated RNA networks in the brain. Science. 2003;302:1212–1215. doi: 10.1126/science.1090095. [DOI] [PubMed] [Google Scholar]
- 37.Kishore S, Luber S, Zavolan M. Deciphering the role of RNA-binding proteins in the post-transcriptional control of gene expression. Brief Funct Genomics. 2010;9:391–404. doi: 10.1093/bfgp/elq028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hafner M, Landthaler M, Burger L, Khorshid M, Hausser J, Berninger P, Rothballer A, Ascano M, Jr, Jungkamp AC, Munschauer M, et al. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell. 2010;141:129–141. doi: 10.1016/j.cell.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Corcoran DL, Georgiev S, Mukherjee N, Gottwein E, Skalsky RL, Keene JD, Ohler U. PARalyzer: definition of RNA binding sites from PAR-CLIP short-read sequence data. Genome Biol. 2011;12:R79. doi: 10.1186/gb-2011-12-8-r79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kishore S, Jaskiewicz L, Burger L, Hausser J, Khorshid M, Zavolan M. A quantitative analysis of CLIP methods for identifying binding sites of RNA-binding proteins. Nat Methods. 2011;8:559–564. doi: 10.1038/nmeth.1608. [DOI] [PubMed] [Google Scholar]
- 41.Klass DM, Scheibe M, Butter F, Hogan GJ, Mann M, Brown PO. Quantitative proteomic analysis reveals concurrent RNA-protein interactions and identifies new RNA-binding proteins in Saccharomyces cerevisiae. Genome Res. 2013;23:1028–1038. doi: 10.1101/gr.153031.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wright JE, Gaidatzis D, Senften M, Farley BM, Westhof E, Ryder SP, Ciosk R. A quantitative RNA code for mRNA target selection by the germline fate determinant GLD-1. EMBO J. 2011;30:533–545. doi: 10.1038/emboj.2010.334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brummer A, Kishore S, Subasic D, Hengartner M, Zavolan M. Modeling the binding specificity of the RNA-binding protein GLD-1 suggests a function of coding region-located sites in translational repression. RNA. 2013;19:1317–1326. doi: 10.1261/rna.037531.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ascano M, Jr, Mukherjee N, Bandaru P, Miller JB, Nusbaum JD, Corcoran DL, Langlois C, Munschauer M, Dewell S, Hafner M, et al. FMRP targets distinct mRNA sequence elements to regulate protein expression. Nature. 2012;492:382–386. doi: 10.1038/nature11737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mukherjee N, Corcoran DL, Nusbaum JD, Reid DW, Georgiev S, Hafner M, Ascano M, Jr, Tuschl T, Ohler U, Keene JD. Integrative regulatory mapping indicates that the RNA-binding protein HuR couples pre-mRNA processing and mRNA stability. Mol Cell. 2011;43:327–339. doi: 10.1016/j.molcel.2011.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kedde M, van Kouwenhove M, Zwart W, Oude Vrielink JA, Elkon R, Agami R. A Pumilio-induced RNA structure switch in p27-3′ UTR controls miR-221 and miR-222 accessibility. Nat Cell Biol. 2010;12:1014–1020. doi: 10.1038/ncb2105. [DOI] [PubMed] [Google Scholar]
- 47.Georgiev S, Boyle AP, Jayasurya K, Ding X, Mukherjee S, Ohler U. Evidence-ranked motif identification. Genome Biol. 2010;11:R19. doi: 10.1186/gb-2010-11-2-r19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Foat BC, Houshmandi SS, Olivas WM, Bussemaker HJ. Profiling condition-specific, genome-wide regulation of mRNA stability in yeast. Proc Natl Acad Sci U S A. 2005;102:17675–17680. doi: 10.1073/pnas.0503803102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bailey TL, Elkan C. The value of prior knowledge in discovering motifs with MEME. Proc Int Conf Intell Syst Mol Biol. 1995;3:21–29. [PubMed] [Google Scholar]
- 50.Elemento O, Slonim N, Tavazoie S. A universal framework for regulatory element discovery across all genomes and data types. Mol Cell. 2007;28:337–350. doi: 10.1016/j.molcel.2007.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Riordan DP, Herschlag D, Brown PO. Identification of RNA recognition elements in the Saccharomyces cerevisiae transcriptome. Nucleic Acids Res. 2011;39:1501–1509. doi: 10.1093/nar/gkq920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Linhart C, Halperin Y, Shamir R. Transcription factor and microRNA motif discovery: the Amadeus platform and a compendium of metazoan target sets. Genome Res. 2008;18:1180–1189. doi: 10.1101/gr.076117.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kazan H, Ray D, Chan ET, Hughes TR, Morris Q. RNAcontext: a new method for learning the sequence and structure binding preferences of RNA-binding proteins. PLoS Comput Biol. 2010;6:e1000832. doi: 10.1371/journal.pcbi.1000832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gerber AP, Herschlag D, Brown PO. Extensive association of functionally and cytotopically related mRNAs with Puf family RNA-binding proteins in yeast. PLoS Biol. 2004;2:E79. doi: 10.1371/journal.pbio.0020079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gerber AP, Luschnig S, Krasnow MA, Brown PO, Herschlag D. Genome-wide identification of mRNAs associated with the translational regulator PUMILIO in Drosophila melanogaster. Proc Natl Acad Sci U S A. 2006;103:4487–4492. doi: 10.1073/pnas.0509260103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Leveille N, Elkon R, Davalos V, Manoharan V, Hollingworth D, Oude Vrielink J, le Sage C, Melo CA, Horlings HM, Wesseling J, et al. Selective inhibition of microRNA accessibility by RBM38 is required for p53 activity. Nat Commun. 2011;2:513. doi: 10.1038/ncomms1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hoinka J, Zotenko E, Friedman A, Sauna ZE, Przytycka TM. Identification of sequence-structure RNA binding motifs for SELEX-derived aptamers. Bioinformatics. 2012;28:i215–223. doi: 10.1093/bioinformatics/bts210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yao Z, Weinberg Z, Ruzzo WL. CMfinder--a covariance model based RNA motif finding algorithm. Bioinformatics. 2006;22:445–452. doi: 10.1093/bioinformatics/btk008. [DOI] [PubMed] [Google Scholar]
- 59.Eddy SR, Durbin R. RNA sequence analysis using covariance models. Nucleic Acids Res. 1994;22:2079–2088. doi: 10.1093/nar/22.11.2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hiller M, Pudimat R, Busch A, Backofen R. Using RNA secondary structures to guide sequence motif finding towards single-stranded regions. Nucleic Acids Res. 2006;34:e117. doi: 10.1093/nar/gkl544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hofacker IL, Fekete M, Stadler PF. Secondary structure prediction for aligned RNA sequences. J Mol Biol. 2002;319:1059–1066. doi: 10.1016/S0022-2836(02)00308-X. [DOI] [PubMed] [Google Scholar]
- 62.Rabani M, Kertesz M, Segal E. Computational prediction of RNA structural motifs involved in post-transcriptional regulatory processes. Proc Natl Acad Sci U S A. 2008;105:14885–14890. doi: 10.1073/pnas.0803169105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Li X, Quon G, Lipshitz HD, Morris Q. Predicting in vivo binding sites of RNA-binding proteins using mRNA secondary structure. RNA. 2010;16:1096–1107. doi: 10.1261/rna.2017210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Xia T, SantaLucia J, Jr, Burkard ME, Kierzek R, Schroeder SJ, Jiao X, Cox C, Turner DH. Thermodynamic parameters for an expanded nearest-neighbor model for formation of RNA duplexes with Watson-Crick base pairs. Biochemistry. 1998;37:14719–14735. doi: 10.1021/bi9809425. [DOI] [PubMed] [Google Scholar]
- 65.Mathews DH, Sabina J, Zuker M, Turner DH. Expanded sequence dependence of thermodynamic parameters improves prediction of RNA secondary structure. J Mol Biol. 1999;288:911–940. doi: 10.1006/jmbi.1999.2700. [DOI] [PubMed] [Google Scholar]
- 66.Mathews DH, Disney MD, Childs JL, Schroeder SJ, Zuker M, Turner DH. Incorporating chemical modification constraints into a dynamic programming algorithm for prediction of RNA secondary structure. Proc Natl Acad Sci U S A. 2004;101:7287–7292. doi: 10.1073/pnas.0401799101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zuker M, Stiegler P. Optimal computer folding of large RNA sequences using thermodynamics and auxiliary information. Nucleic Acids Res. 1981;9:133–148. doi: 10.1093/nar/9.1.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mandal M, Lee M, Barrick JE, Weinberg Z, Emilsson GM, Ruzzo WL, Breaker RR. A glycine-dependent riboswitch that uses cooperative binding to control gene expression. Science. 2004;306:275–279. doi: 10.1126/science.1100829. [DOI] [PubMed] [Google Scholar]
- 69.Solomatin SV, Greenfeld M, Chu S, Herschlag D. Multiple native states reveal persistent ruggedness of an RNA folding landscape. Nature. 2010;463:681–684. doi: 10.1038/nature08717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Woodson SA. Compact intermediates in RNA folding. Annu Rev Biophys. 2010;39:61–77. doi: 10.1146/annurev.biophys.093008.131334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bompfunewerer AF, Backofen R, Bernhart SH, Hertel J, Hofacker IL, Stadler PF, Will S. Variations on RNA folding and alignment: lessons from Benasque. J Math Biol. 2008;56:129–144. doi: 10.1007/s00285-007-0107-5. [DOI] [PubMed] [Google Scholar]
- 72.Hofacker ILF W, Stadler PF, Bonhoeffer S, Tacker M, Schuster P. Fast folding and comparison of RNA secondary structures. Monatsh Chem. 1994:167–188. [Google Scholar]
- 73.Bernhart SH, Hofacker IL, Stadler PF. Local RNA base-pairing probabilities in large sequences. Bioinformatics. 2006;22:614–615. doi: 10.1093/bioinformatics/btk014. [DOI] [PubMed] [Google Scholar]
- 74.McCaskill JS. The equilibrium partition function and base pair binding probabilities for RNA secondary structure. Biopolymers. 1990;29:1105–1119. doi: 10.1002/bip.360290621. [DOI] [PubMed] [Google Scholar]
- 75.Ding Y, Chan CY, Lawrence CE. RNA secondary structure prediction by centroids in a Boltzmann weighted ensemble. RNA. 2005;11:1157–1166. doi: 10.1261/rna.2500605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Doshi KJ, Cannone JJ, Cobaugh CW, Gutell RR. Evaluation of the suitability of free-energy minimization using nearest-neighbor energy parameters for RNA secondary structure prediction. BMC Bioinformatics. 2004;5:105. doi: 10.1186/1471-2105-5-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lange SJ, Maticzka D, Mohl M, Gagnon JN, Brown CM, Backofen R. Global or local? Predicting secondary structure and accessibility in mRNAs. Nucleic Acids Res. 2012;40:5215–5226. doi: 10.1093/nar/gks181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sakakibara Y, Brown M, Hughey R, Mian IS, Sjolander K, Underwood RC, Haussler D. Stochastic context-free grammars for tRNA modeling. Nucleic Acids Res. 1994;22:5112–5120. doi: 10.1093/nar/22.23.5112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sankoff D. Simultaneous solution of the RNA folding, alignment and proto sequence problems. SIAM J Appl Math. 1984;45:810–825. [Google Scholar]
- 80.Robins H, Li Y, Padgett RW. Incorporating structure to predict microRNA targets. Proc Natl Acad Sci U S A. 2005;102:4006–4009. doi: 10.1073/pnas.0500775102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kertesz M, Iovino N, Unnerstall U, Gaul U, Segal E. The role of site accessibility in microRNA target recognition. Nat Genet. 2007;39:1278–1284. doi: 10.1038/ng2135. [DOI] [PubMed] [Google Scholar]
- 82.Long D, Lee R, Williams P, Chan CY, Ambros V, Ding Y. Potent effect of target structure on microRNA function. Nat Struct Mol Biol. 2007;14:287–294. doi: 10.1038/nsmb1226. [DOI] [PubMed] [Google Scholar]
- 83.Tafer H, Ameres SL, Obernosterer G, Gebeshuber CA, Schroeder R, Martinez J, Hofacker IL. The impact of target site accessibility on the design of effective siRNAs. Nat Biotechnol. 2008;26:578–583. doi: 10.1038/nbt1404. [DOI] [PubMed] [Google Scholar]
- 84.Meisner NC, Hackermuller J, Uhl V, Aszodi A, Jaritz M, Auer M. mRNA openers and closers: modulating AU-rich element-controlled mRNA stability by a molecular switch in mRNA secondary structure. Chembiochem. 2004;5:1432–1447. doi: 10.1002/cbic.200400219. [DOI] [PubMed] [Google Scholar]
- 85.Jacob CO, Lee SK, Strassmann G. Mutational analysis of TNF-alpha gene reveals a regulatory role for the 3′-untranslated region in the genetic predisposition to lupus-like autoimmune disease. J Immunol. 1996;156:3043–3050. [PubMed] [Google Scholar]
- 86.Di Marco S, Hel Z, Lachance C, Furneaux H, Radzioch D. Polymorphism in the 3'-untranslated region of TNFalpha mRNA impairs binding of the post-transcriptional regulatory protein HuR to TNFalpha mRNA. Nucleic Acids Res. 2001;29:863–871. doi: 10.1093/nar/29.4.863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hackermuller J, Meisner NC, Auer M, Jaritz M, Stadler PF. The effect of RNA secondary structures on RNA-ligand binding and the modifier RNA mechanism: a quantitative model. Gene. 2005;345:3–12. doi: 10.1016/j.gene.2004.11.043. [DOI] [PubMed] [Google Scholar]
- 88.Ouyang Z, Snyder MP, Chang HY. SeqFold: genome-scale reconstruction of RNA secondary structure integrating high-throughput sequencing data. Genome Res. 2013;23:377–387. doi: 10.1101/gr.138545.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Steffen P, Voss B, Rehmsmeier M, Reeder J, Giegerich R. RNAshapes: an integrated RNA analysis package based on abstract shapes. Bioinformatics. 2006;22:500–503. doi: 10.1093/bioinformatics/btk010. [DOI] [PubMed] [Google Scholar]
- 91.Deigan KE, Li TW, Mathews DH, Weeks KM. Accurate SHAPE-directed RNA structure determination. Proc Natl Acad Sci U S A. 2009;106:97–102. doi: 10.1073/pnas.0806929106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lorenz R, Bernhart SH, Honer Zu Siederdissen C, Tafer H, Flamm C, Stadler PF, Hofacker IL. ViennaRNA Package 2.0. Algorithms Mol Biol. 2011;6:26. doi: 10.1186/1748-7188-6-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Clarke PA. RNA footprinting and modification interference analysis. Methods Mol Biol. 1999;118:73–91. doi: 10.1385/1-59259-676-2:73. [DOI] [PubMed] [Google Scholar]
- 94.Wurst RM, Vournakis JN, Maxam AM. Structure mapping of 5′-32P-labeled RNA with S1 nuclease. Biochemistry. 1978;17:4493–4499. doi: 10.1021/bi00614a021. [DOI] [PubMed] [Google Scholar]
- 95.Gornicki P, Baudin F, Romby P, Wiewiorowski M, Kryzosiak W, Ebel JP, Ehresmann C, Ehresmann B. Use of lead(II) to probe the structure of large RNA's. Conformation of the 3′ terminal domain of E. coli 16S rRNA and its involvement in building the tRNA binding sites. J Biomol Struct Dyn. 1989;6:971–984. doi: 10.1080/07391102.1989.10506525. [DOI] [PubMed] [Google Scholar]
- 96.Wells SE, Hughes JM, Igel AH, Ares M., Jr Use of dimethyl sulfate to probe RNA structure in vivo. Methods Enzymol. 2000;318:479–493. doi: 10.1016/s0076-6879(00)18071-1. [DOI] [PubMed] [Google Scholar]
- 97.Merino EJ, Wilkinson KA, Coughlan JL, Weeks KM. RNA structure analysis at single nucleotide resolution by selective 2′-hydroxyl acylation and primer extension (SHAPE) J Am Chem Soc. 2005;127:4223–4231. doi: 10.1021/ja043822v. [DOI] [PubMed] [Google Scholar]
- 98.Kertesz M, Wan Y, Mazor E, Rinn JL, Nutter RC, Chang HY, Segal E. Genome-wide measurement of RNA secondary structure in yeast. Nature. 2010;467:103–107. doi: 10.1038/nature09322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Underwood JG, Uzilov AV, Katzman S, Onodera CS, Mainzer JE, Mathews DH, Lowe TM, Salama SR, Haussler D. FragSeq: transcriptome-wide RNA structure probing using high-throughput sequencing. Nat Methods. 2010;7:995–1001. doi: 10.1038/nmeth.1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lucks JB, Mortimer SA, Trapnell C, Luo S, Aviran S, Schroth GP, Pachter L, Doudna JA, Arkin AP. Multiplexed RNA structure characterization with selective 2′-hydroxyl acylation analyzed by primer extension sequencing (SHAPE-Seq) Proc Natl Acad Sci U S A. 2011;108:11063–11068. doi: 10.1073/pnas.1106501108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Mortimer SA, Weeks KM. Time-resolved RNA SHAPE chemistry: quantitative RNA structure analysis in one-second snapshots and at single-nucleotide resolution. Nat Protoc. 2009;4:1413–1421. doi: 10.1038/nprot.2009.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wan Y, Kertesz M, Spitale RC, Segal E, Chang HY. Understanding the transcriptome through RNA structure. Nat Rev Genet. 2011;12:641–655. doi: 10.1038/nrg3049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Li F, Zheng Q, Ryvkin P, Dragomir I, Desai Y, Aiyer S, Valladares O, Yang J, Bambina S, Sabin LR, et al. Global analysis of RNA secondary structure in two metazoans. Cell Rep. 2012;1:69–82. doi: 10.1016/j.celrep.2011.10.002. [DOI] [PubMed] [Google Scholar]
- 104.Spitale RC, Crisalli P, Flynn RA, Torre EA, Kool ET, Chang HY. RNA SHAPE analysis in living cells. Nat Chem Biol. 2013;9:18–20. doi: 10.1038/nchembio.1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Mauger DM, Weeks KM. Toward global RNA structure analysis. Nat Biotechnol. 2010;28:1178–1179. doi: 10.1038/nbt1110-1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Mathews DH. Using an RNA secondary structure partition function to determine confidence in base pairs predicted by free energy minimization. RNA. 2004;10:1178–1190. doi: 10.1261/rna.7650904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Quarrier S, Martin JS, Davis-Neulander L, Beauregard A, Laederach A. Evaluation of the information content of RNA structure mapping data for secondary structure prediction. RNA. 2010;16:1108–1117. doi: 10.1261/rna.1988510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Foat BC, Stormo GD. Discovering structural cis-regulatory elements by modeling the behaviors of mRNAs. Mol Syst Biol. 2009;5:268. doi: 10.1038/msb.2009.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Eden E, Lipson D, Yogev S, Yakhini Z. Discovering motifs in ranked lists of DNA sequences. PLoS Comput Biol. 2007;3:e39. doi: 10.1371/journal.pcbi.0030039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Redhead E, Bailey TL. Discriminative motif discovery in DNA and protein sequences using the DEME algorithm. BMC Bioinformatics. 2007;8:385. doi: 10.1186/1471-2105-8-385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Oubridge C, Ito N, Evans PR, Teo CH, Nagai K. Crystal structure at 1.92 A resolution of the RNA-binding domain of the U1A spliceosomal protein complexed with an RNA hairpin. Nature. 1994;372:432–438. doi: 10.1038/372432a0. [DOI] [PubMed] [Google Scholar]
- 112.Kishimoto T, Sato T. Two cases of malignant mesothelioma controlled by pleurectomy. Nihon Kyobu Shikkan Gakkai Zasshi. 1992;30:1996–2001. [PubMed] [Google Scholar]
- 113.Kazan H, Morris Q. RBPmotif: a web server for the discovery of sequence and structure preferences of RNA−binding proteins. Nucleic Acids Res. 2013;41:180–186. doi: 10.1093/nar/gkt463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Nawrocki EP, Kolbe DL, Eddy SR. Infernal 1.0: inference of RNA alignments. Bioinformatics. 2009;25:1335–1337. doi: 10.1093/bioinformatics/btp157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Stefl R, Xu M, Skrisovska L, Emeson RB, Allain FH. Structure and specific RNA binding of ADAR2 double-stranded RNA binding motifs. Structure. 2006;14:345–355. doi: 10.1016/j.str.2005.11.013. [DOI] [PubMed] [Google Scholar]
- 116.Weinberg Z, Barrick JE, Yao Z, Roth A, Kim JN, Gore J, Wang JX, Lee ER, Block KF, Sudarsan N, et al. Identification of 22 candidate structured RNAs in bacteria using the CMfinder comparative genomics pipeline. Nucleic Acids Res. 2007;35:4809–4819. doi: 10.1093/nar/gkm487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Goodarzi H, Najafabadi HS, Oikonomou P, Greco TM, Fish L, Salavati R, Cristea IM, Tavazoie S. Systematic discovery of structural elements governing stability of mammalian messenger RNAs. Nature. 2012;485:264–268. doi: 10.1038/nature11013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Burd CG, Dreyfuss G. RNA binding specificity of hnRNP A1: significance of hnRNP A1 high-affinity binding sites in pre-mRNA splicing. EMBO J. 1994;13:1197–1204. doi: 10.1002/j.1460-2075.1994.tb06369.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Eiring AM, Neviani P, Santhanam R, Oaks JJ, Chang JS, Notari M, Willis W, Gambacorti-Passerini C, Volinia S, Marcucci G, et al. Identification of novel post-transcriptional targets of the BCR/ABL oncoprotein by ribonomics: requirement of E2F3 for BCR/ABL leukemogenesis. Blood. 2008;111:816–828. doi: 10.1182/blood-2007-05-090472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Lopez de Silanes I, Zhan M, Lal A, Yang X, Gorospe M. Identification of a target RNA motif for RNA-binding protein HuR. Proc Natl Acad Sci U S A. 2004;101:2987–2992. doi: 10.1073/pnas.0306453101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Zhu D, Stumpf CR, Krahn JM, Wickens M, Hall TM. A 5′ cytosine binding pocket in Puf3p specifies regulation of mitochondrial mRNAs. Proc Natl Acad Sci U S A. 2009;106:20192–20197. doi: 10.1073/pnas.0812079106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Mukherjee N, Lager PJ, Friedersdorf MB, Thompson MA, Keene JD. Coordinated post-transcriptional mRNA population dynamics during T-cell activation. Mol Syst Biol. 2009;5:288. doi: 10.1038/msb.2009.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Kellis M, Patterson N, Birren B, Berger B, Lander ES. Methods in comparative genomics: genome correspondence, gene identification and regulatory motif discovery. J Comput Biol. 2004;11:319–355. doi: 10.1089/1066527041410319. [DOI] [PubMed] [Google Scholar]
- 124.Stark A, Lin MF, Kheradpour P, Pedersen JS, Parts L, Carlson JW, Crosby MA, Rasmussen MD, Roy S, Deoras AN, et al. Discovery of functional elements in 12 Drosophila genomes using evolutionary signatures. Nature. 2007;450:219–232. doi: 10.1038/nature06340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Pedersen JS, Bejerano G, Siepel A, Rosenbloom K, Lindblad-Toh K, Lander ES, Kent J, Miller W, Haussler D. Identification and classification of conserved RNA secondary structures in the human genome. PLoS Comput Biol. 2006;2:e33. doi: 10.1371/journal.pcbi.0020033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Parker BJ, Moltke I, Roth A, Washietl S, Wen J, Kellis M, Breaker R, Pedersen JS. New families of human regulatory RNA structures identified by comparative analysis of vertebrate genomes. Genome Res. 2011;21:1929–1943. doi: 10.1101/gr.112516.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Zarnack K, Konig J, Tajnik M, Martincorena I, Eustermann S, Stevant I, Reyes A, Anders S, Luscombe NM, Ule J. Direct competition between hnRNP C and U2AF65 protects the transcriptome from the exonization of Alu elements. Cell. 2013;152:453–466. doi: 10.1016/j.cell.2012.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Zhang C, Frias MA, Mele A, Ruggiu M, Eom T, Marney CB, Wang H, Licatalosi DD, Fak JJ, Darnell RB. Integrative modeling defines the Nova splicing-regulatory network and its combinatorial controls. Science. 2010;329:439–443. doi: 10.1126/science.1191150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.van Kouwenhove M, Kedde M, Agami R. MicroRNA regulation by RNA-binding proteins and its implications for cancer. Nat Rev Cancer. 2011;11:644–656. doi: 10.1038/nrc3107. [DOI] [PubMed] [Google Scholar]
- 130.Galgano A, Forrer M, Jaskiewicz L, Kanitz A, Zavolan M, Gerber AP. Comparative analysis of mRNA targets for human PUF-family proteins suggests extensive interaction with the miRNA regulatory system. PLoS One. 2008;3:e3164. doi: 10.1371/journal.pone.0003164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Kedde M, Strasser MJ, Boldajipour B, Oude Vrielink JA, Slanchev K, le Sage C, Nagel R, Voorhoeve PM, van Duijse J, Orom UA, et al. RNA-binding protein Dnd1 inhibits microRNA access to target mRNA. Cell. 2007;131:1273–1286. doi: 10.1016/j.cell.2007.11.034. [DOI] [PubMed] [Google Scholar]
- 132.Jacobsen A, Wen J, Marks DS, Krogh A. Signatures of RNA binding proteins globally coupled to effective microRNA target sites. Genome Res. 2010;20:1010–1019. doi: 10.1101/gr.103259.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Jiang P, Singh M, Coller HA. Computational assessment of the cooperativity between RNA binding proteins and MicroRNAs in Transcript Decay. PLoS Comput Biol. 2013;9:e1003075. doi: 10.1371/journal.pcbi.1003075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Ketting RF. A dead end for microRNAs. Cell. 2007;131:1226–1227. doi: 10.1016/j.cell.2007.12.004. [DOI] [PubMed] [Google Scholar]
- 135.Lunde BM, Moore C, Varani G. RNA-binding proteins: modular design for efficient function. Nat Rev Mol Cell Biol. 2007;8:479–490. doi: 10.1038/nrm2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Burd CG, Matunis EL, Dreyfuss G. The multiple RNA-binding domains of the mRNA poly(A)-binding protein have different RNA-binding activities. Mol Cell Biol. 1991;11:3419–3424. doi: 10.1128/mcb.11.7.3419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Oberstrass FC, Auweter SD, Erat M, Hargous Y, Henning A, Wenter P, Reymond L, Amir-Ahmady B, Pitsch S, Black DL, et al. Structure of PTB bound to RNA: specific binding and implications for splicing regulation. Science. 2005;309:2054–2057. doi: 10.1126/science.1114066. [DOI] [PubMed] [Google Scholar]
- 138.Frith MC, Saunders NF, Kobe B, Bailey TL. Discovering sequence motifs with arbitrary insertions and deletions. PLoS Comput Biol. 2008;4:e1000071. doi: 10.1371/journal.pcbi.1000071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Masliah G, Barraud P, Allain FH. RNA recognition by double-stranded RNA binding domains: a matter of shape and sequence. Cell Mol Life Sci. 2013;70:1875–1895. doi: 10.1007/s00018-012-1119-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Khorshid M, Hausser J, Zavolan M, van Nimwegen E. A biophysical miRNA-mRNA interaction model infers canonical and noncanonical targets. Nat Methods. 2013;10:253–255. doi: 10.1038/nmeth.2341. [DOI] [PubMed] [Google Scholar]
- 141.Laver JD, Li X, Ancevicius K, Westwood JT, Smibert CA, Morris QD, Lipshitz HD. Genome-wide analysis of Staufen-associated mRNAs identifies secondary structures that confer target specificity. Nucleic Acids Res. 2013;41:9438–9460. doi: 10.1093/nar/gkt702. [DOI] [PMC free article] [PubMed] [Google Scholar]
FURTHER READING
- Messias AC, Sattler M. Structural basis of single-stranded RNA recognition. Acc Chem Res. 2004;37:279–287. doi: 10.1021/ar030034m. [DOI] [PubMed] [Google Scholar]
- Chang KY, Ramos A. The double-stranded RNA-binding motif a versatile macromolecular docking platform. FEBS J. 2005;272:2109–2117. doi: 10.1111/j.1742-4658.2005.04652.x. [DOI] [PubMed] [Google Scholar]
- Auweter SD, Oberstrass FC, Allain FH. Sequence-specific binding of single-stranded RNA: is there a code for recognition? Nucleic Acids Res. 2006;34:4943–4959. doi: 10.1093/nar/gkl620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fierro-Monti I, Mathews MB. Proteins binding to duplexed RNA: one motif, multiple functions. Trends Biochem Sci. 2000;25:241–246. doi: 10.1016/s0968-0004(00)01580-2. [DOI] [PubMed] [Google Scholar]