

Abstract
Proteins frequently fold via folding intermediates that correspond to local minima on the conformational energy landscape. Probing the structure of the partially unfolded forms in equilibrium under native conditions can provide insight into the properties of folding intermediates. To elucidate the structures of folding intermediates of Escherichia coli dihydrofolate reductase (DHFR), we investigated transient partial unfolding of DHFR under native conditions. We probed the structure of a high-energy conformation susceptible to proteolysis (cleavable form) using native-state proteolysis. The free energy for unfolding to the cleavable form is clearly less than that for global unfolding. The dependence of the free energy on urea concentration (m-value) also confirmed that the cleavable form is a partially unfolded form. By assessing the effect of mutations on the stability of the partially unfolded form, we found that native contacts in a hydrophobic cluster formed by the F-G and Met-20 loops on one face of the central β-sheet are mostly lost in the partially unfolded form. Also, the folded region of the partially unfolded form is likely to have some degree of structural heterogeneity. The structure of the partially unfolded form is fully consistent with spectroscopic properties of the near-native kinetic intermediate observed in previous folding studies of DHFR. The findings suggest that the last step of the folding of DHFR involves organization in the structure of two large loops, the F-G and Met-20 loops, which is coupled with compaction of the rest of the protein.
Keywords: DHFR, partially unfolded form, folding intermediate, proteolysis, folding pathway
Introduction
Escherichia coli dihydrofolate reductase (DHFR; 5,6,7,8-tetrahydrofolate: NADP+ oxidoreductase, EC 1.5.1.3) is one of the best characterized model systems in protein folding studies as well as in enzymology and structural biology.1–4 The protein is a 159 amino acid monomeric enzyme that catalyzes the reduction of 7,8-dihydrofolate to 5,6,7,8-tetrahydrofolate using NADPH as a cofactor. Folding of DHFR has been shown to occur through two groups of distinctive kinetic intermediates, IBP and IHF5–12:
Each group is believed to include several different intermediates that share similar spectroscopic properties. Within microseconds, unfolded DHFR collapses into IBP, a group of early intermediates that are responsible for the burst phase amplitude observed by circular dichroism (CD). Formation of IBP is followed by slower formation of IHF, another group of intermediates characterized by high fluorescence (IHF) and finally by formation of a group of native conformers.6,12
Although DHFR folding and structure have been the subject of numerous studies, the structure of the folding intermediates remains elusive. Hydrogen/deuterium exchange pulse labeling during refolding showed that two hydrophobic clusters form in IBP (Fig. 1).7 The high fluorescence of IHF has been attributed to formation of a hydrophobic cluster around Trp47 and Trp74 (Fig. 1).13 However, the structure of IHF is largely unknown. Pulse labeling could not provide information on IHF because prolonged incubation at acidic pH necessary for pulse quenching causes DHFR to aggregate.7 Although several attempts have been made to computationally model the folding of DHFR, these efforts do not converge upon a single structural description of DHFR folding.14–16
Figure 1.
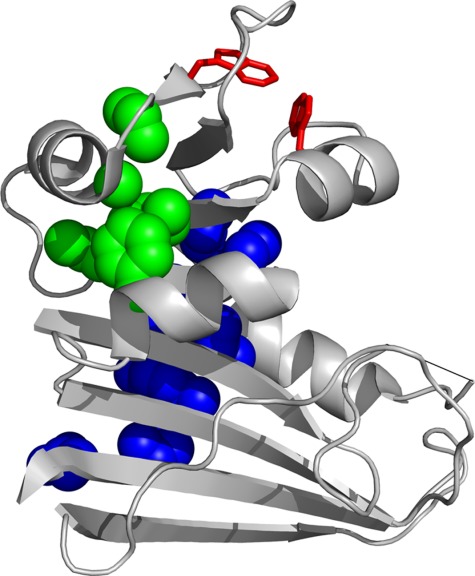
Structure of DHFR. The ribbon representation of the structure of DHFR (PDB CODE: 5DFR) is shown. The side chains of the residues in the two hydrophobic clusters that form in IBP are shown in blue and green spheres. The side chains of Trp47 and Trp74, which are responsible for high fluorescence in IHF, are shown in red sticks. The invisible part of the Met20 loop is shown in a straight line. The image was created with PyMOL.
The structures of partially unfolded forms at equilibrium with native protein provide information on protein folding intermediates.17,18 The current understanding of protein folding holds that proteins fold along the surface of funnel-like conformational energy landscapes, which contain an ensemble of partially unfolded forms in local energy minima.19–21 This view of protein folding suggests that, once folded, a natively folded protein makes transient excursions to the partially unfolded forms. Therefore, from the structures of the partially unfolded forms in equilibrium under native conditions, one can obtain valuable information on the sequence of structural acquisition along the folding pathway.22,23
To obtain structural information on the folding intermediates of DHFR, we investigated a partially unfolded form of DHFR by native-state proteolysis. Native-state proteolysis is a method to determine the energetics of partial unfolding under native conditions.24 Combined with site-directed mutagenesis, native-state proteolysis also provides information on the structure of partially unfolded forms.25 The method is based on the observation that proteolysis of folded proteins requires transient unfolding to a cleavable conformation as shown in the following scheme:
where kop and kcl are the forward and the reverse rate constant for unfolding to a cleavable form, and kint is the rate constant for proteolysis of the cleavable form. When kint is significantly smaller than kcl (EX2 condition), the apparent rate constant of the reaction (kobs) is expressed as:
| (1) |
where Kop is the equilibrium constant for unfolding to the cleavable form. By approximating kint with the rate for proteolysis of a peptide substrate, one can calculate Kop from kobs and also the free energy required for unfolding (ΔGop°) from Kop.
Native-state proteolysis is conceptually analogous to native-state hydrogen/deuterium (H/D) exchange, in which partially unfolded forms are probed by the exchange between amide protons with solvent deuterium.18,22,23 Although native-state H/D exchange may report the presence of multiple partially unfolded forms, native-state proteolysis reports only the most accessible partially unfolded forms, because unfolding to the most accessible form dominates proteolysis kinetics. Despite this limitation, native-state proteolysis is a valuable alternative to native-state H/D exchange and a suitable choice for DHFR. Because proteolysis requires unfolding of a segment with at least 8–12 residues,26 native-state proteolysis is relatively insensitive to local fluctuation, which frequently complicates detection of partial unfolding by native-state H/D exchange. Also, while the adjustment of the intrinsic exchange rate (kint) in H/D exchange requires a change in pH, the intrinsic proteolysis rate of unfolded peptides in native-state proteolysis can be controlled simply by a change in protease concentration. This aspect of native-proteolysis provides flexibility in the choice of pH. Because DHFR tends to aggregate at low pH,7 which is frequently necessary to probe partial unfolding by native-state H/D exchange, the use of a neutral pH in native-state proteolysis is especially advantageous.
Here, we report the structure of a partially unfolded form of DHFR we probed by native-state proteolysis in combination with site-directed mutagenesis. We compare the identified partially unfolded form with known folding intermediates of DHFR, using the previously reported structural features of the folding intermediates. We also postulate the molecular events at the final stage of DHFR folding based on the structure of the partially unfolded form.
Results
Proteolysis of DHFR occurs through partial unfolding
DHFR used in this study is a cysteine-free version of DHFR (C85A/C152S DHFR), which is more resistant to oxidation than wild-type DHFR. Still, the cysteine-free DHFR has been shown to have activity, stability, and a folding mechanism comparable to wild-type DHFR.27 For simplicity, here we refer to the cysteine-free DHFR as wild type.
We probed unfolding of DHFR under native conditions by native-state proteolysis with a nonspecific protease, thermolysin. We determined the apparent first-order rate constant for proteolysis (kobs) by monitoring the change in the band intensities of remaining intact protein on a SDS-PAGE gel from a proteolysis reaction quenched at various time points. Determination of the equilibrium constant for unfolding to a cleavable form (Kop) requires EX2-like conditions (kcl >> kint). Because kint is dependent on the protease concentration, EX2-like conditions can be achieved and also confirmed by adjusting the protease concentration. To identify the optimal range of protease concentration for EX2-like conditions, we determined kobs at varying concentrations of thermolysin and confirmed that proteolysis of DHFR satisfies EX2-like conditions up to 70 µg mL−1 thermolysin (Supporting Information Fig. S1). Then, we determined Kop from kobs by estimating kint with kcat/Km for cleavage of an unstructured peptide substrate. From the Kop value, the free energy for unfolding to the cleavable form (ΔGop°) was determined to be 4.9 ± 0.1 kcal mol−1, which is significantly less than the free energy required for global unfolding (6.6 ± 0.3 kcal mol−1) as determined by equilibrium unfolding monitored by circular dichroism (Table1). The observation that unfolding to the cleavable form requires less free energy than global unfolding indicates that proteolysis occurs through a form distinct from the globally unfolded form.
Table I.
Equilibrium Unfolding Parameters
| DHFR | m-Value (kcal mol−1 M−1) | Cm (M) | ΔGU−N° (kcal mol−1) | ΔΔGU−N°a (kcal mol−1) |
|---|---|---|---|---|
| Wild type | 2.3 ± 0.1 | 2.87 ± 0.01 | 6.6 ± 0.3 | — |
| L4A | 2.0 ± 0.1 | 1.47 ±0.03 | 2.9 ± 0.1 | −3.7 ± 0.3 |
| L8A | 2.1 ± 0.1 | 2.12 ± 0.01 | 4.5 ± 0.1 | −2.1 ± 0.3 |
| I61A | 2.8 ± 0.2 | 1.77 ± 0.01 | 5.0 ± 0.4 | −1.6 ± 0.5 |
| I91A | 2.6 ± 0.1 | 2.29 ± 0.01 | 6.0 ± 0.2 | −0.6 ± 0.4 |
| L110V | 2.6 ± 0.1 | 2.14 ± 0.01 | 5.6 ± 0.2 | −1.0 ± 0.4 |
| L112V | 2.2 ± 0.1 | 2.21 ± 0.01 | 4.9 ± 0.2 | −1.7 ± 0.4 |
| V136A | 2.5 ± 0.1 | 2.26 ± 0.01 | 5.7 ± 0.1 | −0.9 ± 0.3 |
| L156A | 2.1 ± 0.1 | 1.75 ± 0.02 | 3.7 ± 0.1 | −2.9 ± 0.3 |
The m-values, Cm, and the global stability (ΔGU−N°) of each variant were determined by monitoring unfolding in urea by circular dichroism. Standard errors of m-values and Cm values are from nonlinear curve-fitting of urea unfolding to a two-state model. Standard errors of ΔGU−N° and ΔΔGU−N° are from error propagation.
ΔΔGU−N° = ΔΔGU−N°(mutant) − ΔΔGU−N°(wild type).
To assess the scale of unfolding to the cleavable form, we measured ΔGop° at varying concentrations of urea. The free energy for cooperative unfolding in proteins depends linearly on urea concentration, and the dependence (m-value) is proportional to the amount of buried surface area exposed upon unfolding.28 The plot of ΔGop° versus urea reveals two phases with distinct slopes (Fig. 2). Because the slope is proportional to the change in the solvent accessible surface area upon unfolding, the presence of two phases indicates that proteolysis occurs through two distinct cleavable forms depending on the concentration of urea. The shallow phase at lower concentrations of urea suggests proteolysis through partial unfolding, and the steep phase at higher concentrations of urea suggests proteolysis through global unfolding. At lower concentrations of urea, the partially unfolded form is energetically more accessible than the globally unfolded form. As the urea concentration increases, the globally unfolded form becomes more accessible than the partially unfolded form.
Figure 2.
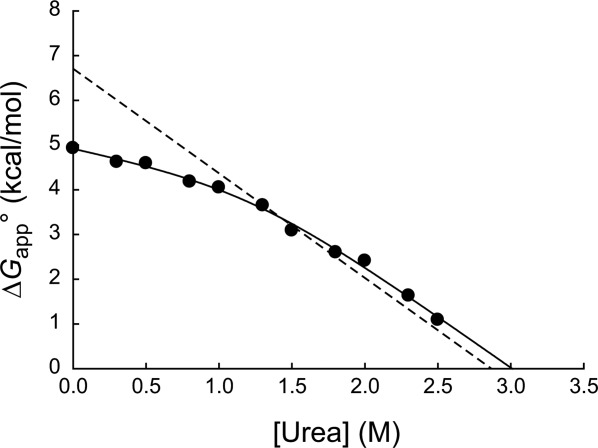
Native-state proteolysis of wild-type DHFR. The dependence of the free energy of unfolding (ΔGapp°) determined by proteolysis (•) and by equilibrium unfolding (---) is plotted against urea concentration. The plot of ΔGapp° versus urea concentration was fit to Eq. (2) (solid line) to estimate parameters for partial unfolding (mop and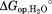 ) and global unfolding (munf and
) and global unfolding (munf and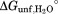 ).
).
We fit the plot of ΔGop° versus urea using a model in which proteolysis occurs through unfolding to two cleavable forms with different m-values (Fig. 2). The m-values for the shallow phase and the steep phase were determined to be 0.7 ± 0.3 kcal mol−1 M−1 and 2.3 ± 0.2 kcal mol−1 M−1, respectively. The m-value of the steep phase agrees well with the m-value of global unfolding (2.3 ± 0.1 kcal mol−1 M−1) determined by equilibrium unfolding monitored by circular dichroism (Table1). This consistency in m-value corroborates that proteolysis occurs through global unfolding in the steep phase. The smaller m-value of the shallow phase also confirms that proteolysis occurs through partial unfolding in the shallow phase. Compared with the m-value for global unfolding (2.3 ± 0.1 kcal mol−1 M−1), the m-value of the shallow phase (0.7 ± 0.3 kcal mol−1 M−1) suggests that the partial unfolding exposes about 30% of the buried surface that is exposed upon global unfolding. The free energies of the shallow phase and the steep phase were extrapolated to be 4.9 ± 0.1 kcal mol−1 and 6.9 ± 0.6 kcal mol−1 in 0M urea, respectively. Proteolysis of the unfolded protein occurs before proline residues reach their cis–trans equilibrium. The lack of proline isomerization in the unfolded forms is expected to increase the free energy of global unfolding measured by proteolysis by 0.7 kcal mol−1 in DHFR.29 Even after this correction, the free energy of the steep phase agrees well with the free energy for global unfolding determined by equilibrium unfolding (6.6 ± 0.3 kcal mol−1). The uncertainty in the rate constant for intrinsic proteolysis (kint) that we estimate with a peptide substrate may also contribute to the slight discrepancy.
Probing the partially unfolded form by site-directed mutagenesis
To determine the region of DHFR that becomes unfolded in the partially unfolded form, we probed the structure of the partially unfolded form by site-directed mutagenesis. To destabilize the protein with only minimal structural perturbation, we limited our mutations to leucine to valine, leucine, and isoleucine to alanine, or valine to alanine.30 Specifically, we prepared L4A, L8A, I61A, I91A, L110V, L112V, V136A, and L156A DHFR. We confirmed that all variants maintain native-like structures by circular dichroism (Supporting Information Fig. S2).
From the global stability and the free energy of partial unfolding of each variant, we determined the φc value, which is the ratio of the change in the stability of the partially unfolded form (ΔΔGU–C°) to that of the native form (ΔΔGU–N°) caused by a mutation.25 If the mutated residue lies within the folded region of the partially unfolded form, the partially unfolded form and the native form will be equally destabilized, and the φc value of the variant will be close to 1. If a mutated residue lies within the unfolded region of the partially unfolded form, only the native form will be destabilized, and the φc value of the variant will be close to 0. Therefore, the distribution of φc values in a protein structure reports which part of the protein is unfolded in a partially unfolded form.25 We calculated the destabilization of the native form (ΔΔGU−N°) by comparing the global stability of each variant of DHFR to that of wild-type DHFR (Table1) after determining the global stability of each by equilibrium unfolding in urea (Supporting Information Fig. S3). We determined the destabilization of the partially unfolded form (ΔΔGU–C°) indirectly from the change in the free energy for partial unfolding (ΔΔGC–N°) determined by native-state proteolysis (Table2 and Supporting Information Fig. S4).
Table II.
Proteolysis Kinetics and φc Values
| DHFR | kpa (×10−3 s−1) | kp(mut)/kp(wt) | ΔΔGC−N°b (kcal mol−1) | φc |
|---|---|---|---|---|
| Wild type | 0.087 ± 0.003 | — | — | — |
| L4A | 2.2 ± 0.2 | 25 ± 2 | −1.91 ± 0.05 | 0.49 ± 0.04 |
| L8A | 2.4 ± 0.1 | 28 ± 2 | −1.97 ± 0.04 | 0.05 ± 0.1 |
| I61A | 0.20 ± 0.01 | 2.3 ± 0.1 | −0.49 ± 0.02 | 0.69 ± 0.09 |
| I91A | 0.140 ± 0.008 | 1.6 ± 0.1 | −0.28 ± 0.04 | 0.5 ± 0.2 |
| L110V | 1.3 ± 0.1 | 14 ± 1 | −1.56 ± 0.04 | −0.6 ± 0.5 |
| L112V | 3.3 ± 0.2 | 38 ± 2 | −2.15 ± 0.03 | −0.3 ± 0.3 |
| V136A | 0.20 ± 0.02 | 2.3 ± 0.2 | −0.49 ± 0.05 | 0.5 ± 0.2 |
| L156A | 3.3 ± 0.2 | 38 ± 2 | −2.15 ± 0.03 | 0.24 ± 0.07 |
kp is the first-order rate constant for the proteolysis of 0.10 mg mL−1 DHFR by 20 µg mL−1 thermolysin. The standard error of kp is from nonlinear curve-fitting. The standard errors of ΔΔGC−N° and φc are from error propagation.
ΔΔGC−N° = ΔGC−N°(mutant) − ΔGC−N°(wild type).
Mutations show a broad spectrum of φc values (Table2). Interestingly, no mutation has a φc value close to 1. Ile61 has the highest φc value of 0.69. Leu4, Ile91, and Val136 have φc values close to 0.5. Leu156 has a low φc value of 0.24. Finally, Leu8, Leu110, and Leu112 have φc values close to 0. When shown on the structure, the distribution of φc values clearly suggests the location of unfolding in the partially unfolded form (Fig. 3). Residues with φc values close to 0 (Leu8, Leu110, and Leu112) form a cluster on one face of the central β-sheet of the protein. This face of the β-sheet is covered by two extended loops (F-G loop and Met-20 loop). The low φc values of Leu8, Leu110, and Leu112 signify that the loops are unfolded and the face of β-sheet is exposed in the partially unfolded form. Moving away from the cluster of Leu8, Leu110, and Leu112, residues show increasing φc values. The relatively high φc values of Ile61 and Ile91 indicate that the adenosine-binding domain maintains a significant portion of its native contacts in the partially unfolded form. The relatively high φc value of Val136 also indicates the back of the central β-sheet does not lose its native contacts completely in the partially unfolded form. The gradual increase in φc values from Leu110 to Leu156 and to Val136 also indicates that the loss of the native contacts in the cluster of Leu8, Leu110, and Leu112 is mostly confined to the front face of the central β-sheet (Fig. 3).
Figure 3.
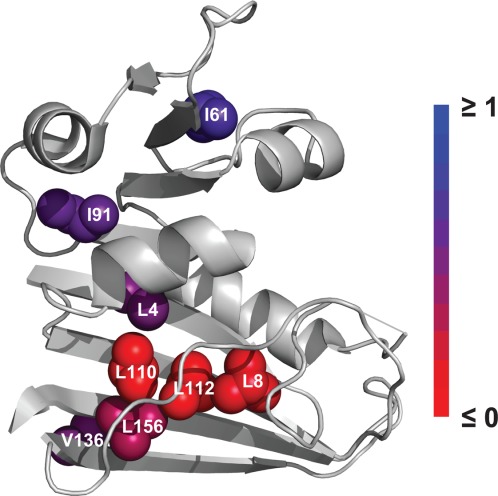
φc values of the mutated residues. Residues probed by mutation are shown in spheres in the native structure of DHFR (PDB CODE: 5DFR), colored by φc value from 0 (red) to 1 (blue). For clarity, φc values less than 0 are shown as 0. The invisible part of the Met20 loop is shown in a straight line. The image was created with PyMOL.
Discussion
The structure of the partially unfolded form
The pattern of φc values reveals the location of unfolding in the partially unfolded form of DHFR (Fig. 3). Leu8, Leu110, and Leu112 have φc values close to 0 and form a continuous cluster, suggesting that the region experiences significant structural disruption in the partially unfolded form. However, none of the mutated residues has a φc value close to 1. The observation that a majority of the mutated residues have fractional φc values is distinct from the observation we made with E. coli maltose binding protein in which the majority of the mutated residues from an initial survey show φc values close to 1.25 We interpret the fractional φc values similarly to the way that fractional transition-state φ values have been interpreted.31 Fractional φc values could mean that some but not all of a residue's native contacts are lost, or an ensemble exists with forms that are close in energy but possess somewhat different structural features. One of the folding intermediates of DHFR, IHF, has been described as a group of four species with similar properties.6,32 The fractional φc values may have resulted from a different contribution of each species in a group of partially unfolded forms. Although the partially unfolded form we probed may be a group of structurally distinct forms with similar energetic properties, the residues with φc values close to 0 allow us to determine the common unfolded region of the partially unfolded forms in the group. Another source of the fractional φc values is underestimation of ΔGC−N° due to proteolysis through global unfolding. The free energy for unfolding of the partially unfolded form (ΔGU–C°) is just 1.8 ± 0.3 kcal mol−1. If a mutation destabilizes the partially unfolded form by more than 1.8 kcal mol−1, proteolysis would occur through the globally unfolded form rather than the partially unfolded form even in 0M urea. In this case, ΔGC–N° determined by native-state proteolysis is not the free energy for partial unfolding but the global stability of the mutant. Underestimated ΔGC−N° results in underestimation of ΔΔGU−C° and subsequent underestimation of φc. We found one mutant that may have this scenario. The global stability of L4A is only 2.9 kcal mol−1, and its ΔGC–N° determined by proteolysis is 3.0 kcal mol−1. This similarity between the global stability and ΔGC–N° suggests that proteolysis of L4A may occur through global unfolding and the φc value of 0.49 may be an underestimated value. Even if L4A has a φc value of 1, underestimation of ΔΔGU−C° to 1.8 kcal mol−1 would result in φc value of ∼0.5.
A significant portion of the native contacts of I61 and I91 seems to be retained in the partially unfolded form (Fig. 3). This result suggests that the adenosine-binding domain mostly remains in the native-like conformation in the partially unfolded form. From m-values for partial and global unfolding, we estimate that partial unfolding exposes only ∼30% of the buried surface that is exposed upon global unfolding. The relatively small m-value of partial unfolding is also consistent with intact adenosine-binding domain in the partially unfolded form. Still, the fractional φc values of I61 and I91 may arise from structural heterogeneity or partial loss of the native contacts within the adenosine-binding domain in the partially unfolded form.
The native contacts of Leu8, Leu110, and Leu112 seem to be mostly lost in the partially unfolded form (Fig. 3). These residues belong to the discontinuous loop domain, the large domain containing the N- and C-termini, and contact quite a few residues in the central β-sheet, the F-G loop, and the Met-20 loop (Fig. 4). The low φc values of Leu8, Leu110, and Leu112, therefore, indicate that the partial unfolding involves a concerted loss of structure in the F-G and Met-20 loops and exposure of the residues on the β-sheet that the two loops cover. The Met-20 loop is known to undergo conformational changes between the “occluded” and “closed” conformations along the catalytic steps of DHFR.33,34 Moreover, residues 16–20 of the Met-20 loop are invisible in the structure of the apo form of DHFR determined by X-ray crystallography. This apparent disorder of the Met-20 loop was suggested to result from an interconversion between the occluded and closed conformations.35 The conformations that the two loops experience during the catalytic cycle seem to be distinct from their conformations in the partially unfolded form. The significant free energy (4.9 ± 0.1 kcal mol−1) required for partial unfolding indicates that the partially unfolded form is rarely populated under native conditions (1 out of 4000 molecules), while the conformations accessible during the catalytic cycle are significantly populated under native condition.34 Although the partially unfolded form is energetically distinct from the conformations accessed during the catalytic cycle, the partial unfolding in the F-G and Met-20 loops seems to be the consequence of the conformational energy landscape of DHFR optimized for its function. To achieve the dynamic nature of the catalytic site, the protein structure may have sacrified the local stability of the loop regions, which results in transient partial unfolding of the region under native conditions.
Figure 4.
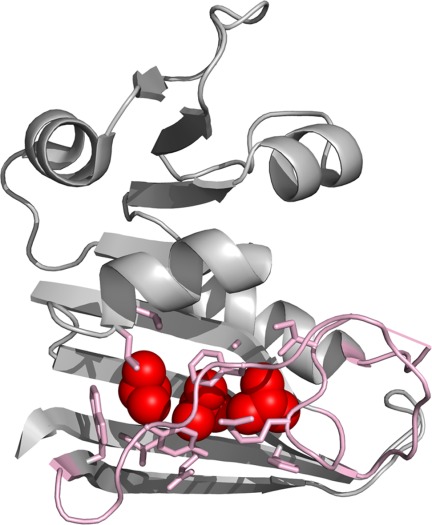
Unfolded region in the partially unfolded form. Leu8, Leu110, and Leu112, which have low φc values, are shown in red spheres. Side chains in contact with the three residues are shown in pink sticks. Contacts are defined as residues within 5 Å of any carbon atom lost upon mutation. The F-G loop and the Met-20 loops are also shown in pink. The invisible part of the Met20 loop is shown in a straight line. The image was created with PyMOL.
The relatively high φc values of Leu4 and Val136 could indicate the boundary of the unfolded region in the partially unfolded form (Fig. 3). Again, the fractional φc values may arise from structural heterogeneity or a partial loss of the contacts around Leu4 and Val136. Still, the significant difference in the φc values of the two residues from those of Leu8, Leu110, and Leu112 implies that the degree of unfolding is much less severe outside of the cluster of Leu8, Leu110, and Leu112. The relatively high φc value of Val136 supports that the central β-sheet is largely intact in the partially unfolded form, which is consistent with the observation that many of the amide hydrogens in the central β-sheet are protected quite early in the folding pathway of DHFR.7
The partially unfolded form and folding intermediates
The structure and stability of the partially unfolded form are consistent with the folding intermediate IHF. The folding of DHFR has been shown to involve multiple intermediates.5,6,10 Matthews and coworkers describe DHFR folding as occurring through an early intermediate (IBP) and a group of four later intermediates (IHF) that lead to four native conformers via parallel folding channels.6 The parallel channels may form even earlier,7 and the native β-sheet topology begins to form before the formation of IBP.11,12
Pulse labeling H/D exchange and the effect of mutations on the burst-phase CD amplitude suggest that IBP contains native-like secondary structure and limited hydrophobic packing.7,9 The burst phase amplitude is affected by mutations at Ile91, Ile94, and Ile155 but is not substantially affected by mutations at Ile2, Ile61, and Leu112, indicating that the later group does not participate in hydrophobic packing in IBP.9 The φc values of Ile91 (0.5 ± 0.2) and Leu122 (−0.3 ± 0.3) are in line with this result. However, the relatively high φc value of Ile61 (0.69 ± 0.09) clearly indicates that IBP is different from the partially unfolded form we observed. IBP buries about 20% of the surface buried in the native form10 and is 1.5 ± 0.5 kcal mol−1 more stable than globally unfolded DHFR.9 Our partially unfolded form has stability similar to IBP (1.8 ± 0.3 kcal mol−1) but buries a significantly larger fraction of surface (∼70%) than IBP. Therefore, the partially unfolded form is distinct from IBP.
The later intermediate IHF forms within the range of hundreds of milliseconds and is an essential, on-pathway intermediate.10 IHF has a higher fluorescence than IBP, native, or unfolded DHFR. The high fluorescence is attributed to the formation of a hydrophobic cluster in the adenosine-binding domain in which Trp47 and Trp74 form their native interaction and become protected from solvent.10,13,36 Ile61 forms a hydrophobic cluster with Trp47 and Trp74. The high φc value of Ile61 strongly supports that the hydrophobic cluster including Trp47 and Trp74 is intact in the partially unfolded form. Furthermore, IHF was previously found to bury about 65% of the buried surface of the native form,14 which agrees well with our finding that ∼70% of the buried surface of the native form remains buried in the partially unfolded form. The structure and fraction of buried surface suggest that the partially unfolded form and IHF are likely to be the same species.
Frieden and coworkers explain the fluorescence change upon DHFR refolding with a linear model containing three intermediates (I1–I3).5,8 I1 forms with an increase in fluorescence intensity and occurs on the timescale of hundreds of milliseconds.5,8 I1 and IHF are the species with the highest fluorescence in their respective models, and both form on the millisecond timescale. Because the structure of our partially unfolded form is similar to IHF, it is possible that the partially unfolded form corresponds to I1. I2 and I3 form with a decrease in fluorescence. I3 has been shown to be only 1–2 kcal mol−1 less stable than the native form.8 Because our partially unfolded form is closer in energy to the unfolded form, I3 is clearly distinct from the partially unfolded form. However, we cannot rule out the possibility that the partially unfolded form corresponds to I2.
The partially unfolded form is also similar to an equilibrium intermediate of DHFR, which was discovered in an extensive analysis of the dependence of spectroscopic signal on temperature and urea concentration.37 The equilibrium intermediate has a compact structure and, like IHF, has higher tryptophan fluorescence than native or unfolded DHFR. The m-value for the equilibrium between the native form and the equilibrium intermediate was determined to be 0.7 ± 0.2 kcal mol−1 M−1, which agrees well with the m-value (0.7 ± 0.3 kcal mol−1 M−1) for unfolding to our partially unfolded form. The equilibrium intermediate, the kinetic intermediate IHF, and the partially unfolded form detected by native-state proteolysis all appear to be the same species that is the most accessible non-native form of DHFR.
Comparison with computational studies
Computational approaches to DHFR unfolding have previously suggested the structures of folding intermediates. Sham et al. investigated thermal unfolding of DHFR by molecular dynamics simulation.16 The first step of unfolding was the loss of the contacts between β-strands 1 and 2, which include Ile61 and Trp74. The effect of mutations on the burst-phase CD intensity showed that this region is folded in IHF but not IBP.9 Disruption of the interaction between β-strands 1 and 2 captured by the molecular dynamics simulation seems to indicate that the unfolding simulation quickly reaches a conformation that resembles IBP. The unfolding to IHF or a conformation equivalent to our partially unfolded form may be of too small a scale to be detected in this simulation because the analysis only takes into account contacts with >70% occupancy during a control simulation.
Pan et al. investigated unfolding of DHFR using the COREX algorithm.15 This approach estimates the energetic impact of iteratively unfolding small segments of a protein to identify residues that have a high probability of unfolding.21 Their study found that residues 60–90 have the highest probability to unfold in DHFR. Interestingly, the region includes Ile61 and Trp74, which are believed to be folded in our partially unfolded form. The high-energy conformation that COREX predicts is clearly different from the partially unfolded form we observed in this study. A possible explanation for this discrepancy is the difference in the choice of the native state. The structure used for the COREX study was the crystallographic structure of DHFR:folate:NADP+ ternary complex with the two ligand removed, which is clearly different from the structure of the apo-DHFR under our experimental conditions.
Clementi et al. simulated DHFR folding only explicitly accounting for the native topology of the protein.14 Their simulation predicted that the adenosine binding domain is mostly folded but the discontinuous loop domain is mostly unfolded in IHF. This structure is clearly distinct from the model of the partially unfolded form we describe here in which the central β-sheet of the discontinuous loop domain is mostly intact. The extensive protection of the central β-sheet in the hydrogen/deuterium exchange pulse chase experiment7 is rather more in line with our model of IHF than the model proposed from the simulation. It is possible that the intermediate observed in the simulation occurs much earlier in the folding pathway than IHF and is not observable in our native-state proteolysis experiment. Still, their calculation shows that contacts between the Met-20 and F-G loops have some of the lowest probabilities of contacts at the intermediate stage of folding, which is consistent with the concerted unfolding of the loops in the partially unfolded form.
Conclusion
We report here the structure of a partially unfolded form of DHFR that transiently forms under native conditions. Our comparison with previously observed folding intermediates of DHFR suggests that the identified partially unfolded form is likely the same species as the folding intermediate IHF. The partially unfolded form shows a similar degree of unfolding to that of IHF. Also, the spectroscopic nature of IHF is consistent with the structure of the partially unfolded form. The structure of the partially unfolded form provides much greater details of the structure of IHF; the adenosine-binding domain is mostly folded, and the hydrophobic core formed by the F-G and Met-20 loops and one face of the central β-sheet is largely unfolded. However, even the folded region seems to have some degree of structural heterogeneity. The final and also the slowest step of DHFR folding is then the docking of the two loops to the central β-sheet as observed in the structure of the native form. The completion of the folding of the two loops may also cause the rest of the protein to achieve more compact packing and reduce the structural heterogeneity. As shown by previous native-state H/D exchange studies,22,23 our study clearly demonstrates that investigating partially unfolded forms under native conditions is a valuable means to elucidate structures of folding intermediates. Also, partial unfolding of the F-G and Met-20 loops whose dynamics have functional importance for catalysis by DHFR implies that optimization of the conformational energy landscape of a protein for its function may influence how the protein achieves its native structure along the folding pathway.
Materials and Methods
Preparation of proteins
We expressed DHFR in E. coli BL21(DE3)pLysS cells grown to OD600 of 0.6 and induced with 0.50M isopropyl-β-d-thiogalactopyranoside (IPTG). Plasmids with mutated DHFR genes were prepared by thermal cycling of the plasmid carrying the DHFR gene sequence and mutagenic oligonucleotide primers according to the protocol for Quikchange Site-Directed Mutagenesis (Agilent Technologies, Santa Clara, CA). All the DHFR proteins we used in this study have C85A/C152S mutations. We purified DHFR by DEAE Sepharose Fast Flow (GE Healthcare Life Sciences; Piscataway, NJ) anion exchange chromatography and Superdex 200 (GE Healthcare Life Sciences) size exclusion chromatography. Thermolysin was prepared by dissolving lyophilized thermolysin (Type X; Sigma-Aldrich; St. Louis, MO) in 2.5M NaCl and 10 mM CaCl2.38 Concentrations of all proteins were determined by absorbance at 280 nm using extinction coefficients determined according to their amino acid composition.39 We confirmed that less than 10% of purified DHFR was bound to the cofactor NADPH by monitoring change in fluorescence upon NADPH titration.
Native-state proteolysis
For wild-type DHFR, we determined proteolysis kinetics at varying concentrations of thermolysin (0.6–70 µg mL−1) in 0–2.5M urea. We initiated proteolysis by adding concentrated thermolysin to DHFR in a buffer to achieve the final condition of 0.10 mg mL−1 DHFR, 20 mM Tris–HCl buffer (pH 8.0), 100 mM NaCl, 10 mM CaCl2, and 0–2.5M urea. At desired time points, 15-µL aliquots were quenched with 5 µL of 50 mM EDTA. The low protein concentration (0.10 mg mL−1 DHFR) was chosen to minimize the inhibition by cleavage products (Kasper and Park, unpublished result).
We analyzed quenched reaction samples by 15% SDS-PAGE gels. Mark12 Protein Standard (Life Technologies; Grand Island, NY) was used as a molecular weight marker. Gels were stained with SYPRO Red Protein Gel Stain (Life Technologies), and fluorescent images were taken with a Typhoon scanner (GE Healthcare Life Sciences). Intact protein gel bands were quantified from images with ImageJ software. Apparent rates of proteolysis (kp) were calculated from fitting the change in band intensity over time to a first-order rate equation in OriginPro 8.5.1 (OriginLab; Northampton, MA). We confirmed proteolysis through the EX2-like kinetics by examining the linear dependence of kp on thermolysin concentration at each urea concentration. Thermolysin maintains its structure under our experimental condition, but the activity decreases as urea concentration is increased.24,40 We approximated the kint value at each urea concentration using the kcat/Km value for proteolysis of a generic peptide substrate for thermolysin, 2-aminobenzoyl-Ala-Gly-Leu-Ala-4-nitrobenzylamide by thermolysin.25 Because proteolysis of DHFR by thermolysin does not generate any cleavage products observable by SDS PAGE, the initial cleavage site is not known, and a generic peptide substrate is used instead of a peptide substrate with the actual sequence of the initial cleavage site. We determined the equilibrium constant for unfolding to the cleavable form (Kop) from the slope of the plot of kp versus thermolysin concentration and the kcat/Km value for proteolysis of the peptide substrate.24 Then, we calculated the free energy for unfolding to the cleavable form (ΔGapp°) from the Kop value at each urea concentration.
We fit the plot of ΔGapp° versus urea (Fig. 2) to
| (2) |
in which the free energies ΔGop° and ΔGunf° are expressed as functions of urea by the linear-extrapolation method:
| (3) |
| (4) |
From this fitting, we determined the m-value and free energy of unfolding for both partial unfolding (mop,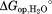 ) and global unfolding (munf,
) and global unfolding (munf,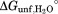 ). The use of the approximate kint value from the generic substrate may introduce a systematic error in Kop and ΔGop. However, the m-values are independent of kint and can be determined reliably regardless of the uncertainty in kint.
). The use of the approximate kint value from the generic substrate may introduce a systematic error in Kop and ΔGop. However, the m-values are independent of kint and can be determined reliably regardless of the uncertainty in kint.
Equilibrium unfolding
We conducted equilibrium unfolding experiments with 1.0 µM DHFR in 20 mM Tris–HCl buffer (pH 8.0) containing 100 mM NaCl, 10 mM CaCl2, and varying concentrations of urea. We incubated all samples overnight at 25°C before measurement. We measured ellipticity for each sample at 222 nm by a JASCO J-815 CD spectrophotometer (JASCO, Easton, MD). We determined Cm and m-value by fitting the dependence of ellipticity on urea concentration to a two-state unfolding model using OriginPro 8.5.1 (OriginLab, Northampton, MA).41 ΔGU–N° was calculated as the product of Cm and m, and ΔΔGU–N° was calculated by subtracting ΔGU–N° of wild type DHFR from that of a mutant.
Determination of φc values
We calculated φc by
| (5) |
using the relationship of ΔΔGU−C° = ΔΔGU−N° − ΔΔGC−N°.25 ΔGU−C° is the free energy for unfolding of the partially unfolded form, ΔGU−N° is the free energy for global unfolding, and ΔGC−N° is the free energy for partial unfolding. We determined ΔΔGC−N° by native-state proteolysis with the assumption that proteolysis occurs with EX2-like kinetics for all DHFR variants under our experimental condition, using
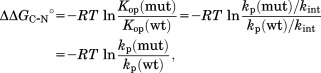 |
(6) |
in which Kop is the equilibrium constant for unfolding to the cleavable form, kp is the apparent rate constant for proteolysis, and kint is the intrinsic rate of proteolysis for the cleavable form.25 The kp value of each variant was determined at a single concentration of thermolysin (20 µg mL−1).
Acknowledgments
The authors thank Youngil Chang for generation of the cysteine-free version of DHFR and Chen Chen and Nathan Gardner for helpful comments on this manuscript.
Glossary
- DHFR
C85A/C152S E. coli dihydrofolate reductase
- EDTA
ethylenediaminetetraacetic acid
- PAGE
polyacrylamide gel electrophoresis
- SDS
sodium dodecyl sulfate.
Supporting Information
Additional Supporting Information may be found in the online version of this article.
Supplementary Information
References
- Fierke CA, Johnson KA, Benkovic SJ. Construction and evaluation of the kinetic scheme associated with dihydrofolate reductase from Escherichia coli. Biochemistry. 1987;26:4085–4092. doi: 10.1021/bi00387a052. [DOI] [PubMed] [Google Scholar]
- Bolin JT, Filman DJ, Matthews DA, Hamlin RC, Kraut J. Crystal structures of Escherichia coli and Lactobacillus casei dihydrofolate reductase refined at 1.7 Å resolution. I. General features and binding of methotrexate. J Biol Chem. 1982;257:13650–13662. [PubMed] [Google Scholar]
- Touchette NA, Perry KM, Matthews CR. Folding of dihydrofolate reductase from Escherichia coli. Biochemistry. 1986;25:5445–5452. doi: 10.1021/bi00367a015. [DOI] [PubMed] [Google Scholar]
- Schnell JR, Dyson HJ, Wright PE. Structure, dynamics, and catalytic function of dihydrofolate reductase. Ann Rev Biophys Biomol Struct. 2004;33:119–140. doi: 10.1146/annurev.biophys.33.110502.133613. [DOI] [PubMed] [Google Scholar]
- Frieden C. Refolding of Escherichia coli dihydrofolate reductase: sequential formation of substrate binding sites. Proc Natl Acad Sci USA. 1990;87:4413–4416. doi: 10.1073/pnas.87.12.4413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jennings PA, Finn BE, Jones BE, Matthews CR. A reexamination of the folding mechanism of dihydrofolate reductase from Escherichia coli: verification and refinement of a four-channel model. Biochemistry. 1993;32:3783–3789. doi: 10.1021/bi00065a034. [DOI] [PubMed] [Google Scholar]
- Jones BE, Matthews CR. Early intermediates in the folding of dihydrofolate reductase from Escherichia coli detected by hydrogen exchange and NMR. Protein Sci. 1995;4:167–177. doi: 10.1002/pro.5560040204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark AC, Frieden C. Native Escherichia coli and murine dihydrofolate reductases contain late-folding non-native structures. J Mol Biol. 1999;285:1765–1776. doi: 10.1006/jmbi.1998.2402. [DOI] [PubMed] [Google Scholar]
- O'Neill JC, Jr, Matthews CR. Localized, stereochemically sensitive hydrophobic packing in an early folding intermediate of dihydrofolate reductase from Escherichia coli. J Mol Biol. 2000;295:737–744. doi: 10.1006/jmbi.1999.3403. [DOI] [PubMed] [Google Scholar]
- Heidary DK, O'Neill JC, Jr, Roy M, Jennings PA. An essential intermediate in the folding of dihydrofolate reductase. Proc Natl Acad Sci USA. 2000;97:5866–5870. doi: 10.1073/pnas.100547697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arai M, Kondrashkina E, Kayatekin C, Matthews CR, Iwakura M, Bilsel O. Microsecond hydrophobic collapse in the folding of Escherichia coli dihydrofolate reductase, an a/b-type protein. J Mol Biol. 2007;368:219–229. doi: 10.1016/j.jmb.2007.01.085. [DOI] [PubMed] [Google Scholar]
- Arai M, Iwakura M, Matthews CR, Bilsel O. Microsecond subdomain folding in dihydrofolate reductase. J Mol Biol. 2011;410:329–342. doi: 10.1016/j.jmb.2011.04.057. [DOI] [PubMed] [Google Scholar]
- Kuwajima K, Garvey EP, Finn BE, Matthews CR, Sugai S. Transient intermediates in the folding of dihydrofolate reductase as detected by far-ultraviolet circular dichroism spectroscopy. Biochemistry. 1991;30:7693–7703. doi: 10.1021/bi00245a005. [DOI] [PubMed] [Google Scholar]
- Clementi C, Jennings PA, Onuchic JN. How native-state topology affects the folding of dihydrofolate reductase and interleukin-1b. Proc Natl Acad Sci USA. 2000;97:5871–5876. doi: 10.1073/pnas.100547897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan H, Lee JC, Hilser VJ. Binding sites in Escherichia coli dihydrofolate reductase communicate by modulating the conformational ensemble. Proc Natl Acad Sci USA. 2000;97:12020–12025. doi: 10.1073/pnas.220240297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sham YY, Ma B, Tsai CJ, Nussinov R. Thermal unfolding molecular dynamics simulation of Escherichia coli dihydrofolate reductase: thermal stability of protein domains and unfolding pathway. Proteins. 2002;46:308–320. doi: 10.1002/prot.10040. [DOI] [PubMed] [Google Scholar]
- Englander SW. Protein folding intermediates and pathways studied by hydrogen exchange. Ann Rev Biophys Biomol Struct. 2000;29:213–238. doi: 10.1146/annurev.biophys.29.1.213. [DOI] [PubMed] [Google Scholar]
- Englander SW, Mayne L, Krishna MM. Protein folding and misfolding: mechanism and principles. Q Rev Biophys. 2007;40:287–326. doi: 10.1017/S0033583508004654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dill KA, Chan HS. From Levinthal to pathways to funnels. Nat Struct Biol. 1997;4:10–19. doi: 10.1038/nsb0197-10. [DOI] [PubMed] [Google Scholar]
- Street TO, Barrick D. Predicting repeat protein folding kinetics from an experimentally determined folding energy landscape. Protein Sci. 2009;18:58–68. doi: 10.1002/pro.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilser VJ, Garcia-Moreno EB, Oas TG, Kapp G, Whitten ST. A statistical thermodynamic model of the protein ensemble. Chem Rev. 2006;106:1545–1558. doi: 10.1021/cr040423+. [DOI] [PubMed] [Google Scholar]
- Bai Y, Sosnick TR, Mayne L, Englander SW. Protein folding intermediates: native-state hydrogen exchange. Science. 1995;269:192–197. doi: 10.1126/science.7618079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamberlain AK, Handel TM, Marqusee S. Detection of rare partially folded molecules in equilibrium with the native conformation of RNase H. Nat Struct Biol. 1996;3:782–787. doi: 10.1038/nsb0996-782. [DOI] [PubMed] [Google Scholar]
- Park C, Marqusee S. Probing the high energy states in proteins by proteolysis. J Mol Biol. 2004;343:1467–1476. doi: 10.1016/j.jmb.2004.08.085. [DOI] [PubMed] [Google Scholar]
- Chang Y, Park C. Mapping transient partial unfolding by protein engineering and native-state proteolysis. J Mol Biol. 2009;393:543–556. doi: 10.1016/j.jmb.2009.08.006. [DOI] [PubMed] [Google Scholar]
- Hubbard SJ, Eisenmenger F, Thornton JM. Modeling studies of the change in conformation required for cleavage of limited proteolytic sites. Protein Sci. 1994;3:757–768. doi: 10.1002/pro.5560030505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwakura M, Jones BE, Luo J, Matthews CR. A strategy for testing the suitability of cysteine replacements in dihydrofolate reductase from Escherichia coli. J Biochem. 1995;117:480–488. doi: 10.1093/oxfordjournals.jbchem.a124733. [DOI] [PubMed] [Google Scholar]
- Myers JK, Pace CN, Scholtz JM. Denaturant m values and heat capacity changes: relation to changes in accessible surface areas of protein unfolding. Protein Sci. 1995;4:2138–2148. doi: 10.1002/pro.5560041020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huyghues-Despointes BM, Scholtz JM, Pace CN. Protein conformational stabilities can be determined from hydrogen exchange rates. Nat Struct Biol. 1999;6:910–912. doi: 10.1038/13273. [DOI] [PubMed] [Google Scholar]
- Fersht A. Structure and mechanism in protein science: a guide to enzyme catalysis and protein folding. New York: W.H. Freeman; 1999. [Google Scholar]
- Fersht AR, Matouschek A, Serrano L. The folding of an enzyme. I. Theory of protein engineering analysis of stability and pathway of protein folding. J Mol Biol. 1992;224:771–782. doi: 10.1016/0022-2836(92)90561-w. [DOI] [PubMed] [Google Scholar]
- Jones B, Jennings P, Pierre R, Matthews C. Development of nonpolar surfaces in the folding of Escherichia coli dihydrofolate reductase detected by 1-anilinonaphthalene-8-sulfonate binding. Biochemistry. 1994;33:15250–15258. doi: 10.1021/bi00255a005. [DOI] [PubMed] [Google Scholar]
- Bystroff C, Oatley SJ, Kraut J. Crystal structures of Escherichia coli dihydrofolate reductase: the NADP+ holoenzyme and the folate·NADP+ ternary complex. Substrate binding and a model for the transition state. Biochemistry. 1990;29:3263–3277. doi: 10.1021/bi00465a018. [DOI] [PubMed] [Google Scholar]
- Boehr DD, McElheny D, Dyson HJ, Wright PE. The dynamic energy landscape of dihydrofolate reductase catalysis. Science. 2006;313:1638–1642. doi: 10.1126/science.1130258. [DOI] [PubMed] [Google Scholar]
- Sawaya MR, Kraut J. Loop and subdomain movements in the mechanism of Escherichia coli dihydrofolate reductase: crystallographic evidence. Biochemistry. 1997;36:586–603. doi: 10.1021/bi962337c. [DOI] [PubMed] [Google Scholar]
- Garvey EP, Swank J, Matthews CR. A hydrophobic cluster forms early in the folding of dihydrofolate reductase. Protein. 1989;6:259–266. doi: 10.1002/prot.340060308. [DOI] [PubMed] [Google Scholar]
- Ionescu RM, Smith VF, O'Neill JC, Jr, Matthews CR. Multistate equilibrium unfolding of Escherichia coli dihydrofolate reductase: thermodynamic and spectroscopic description of the native, intermediate, and unfolded ensembles. Biochemistry. 2000;39:9540–9550. doi: 10.1021/bi000511y. [DOI] [PubMed] [Google Scholar]
- Inouye K, Kuzuya K, Tonomura B. Sodium chloride enhances markedly the thermal stability of thermolysin as well as its catalytic activity. Biochim Biophys Acta. 1998;1388:209–214. doi: 10.1016/s0167-4838(98)00189-7. [DOI] [PubMed] [Google Scholar]
- Pace CN, Vajdos F, Fee L, Grimsley G, Gray T. How to measure and predict the molar absorption coefficient of a protein. Protein Sci. 1995;4:2411–2423. doi: 10.1002/pro.5560041120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park C, Marqusee S. Pulse proteolysis: a simple method for quantitative determination of protein stability and ligand binding. Nat Methods. 2005;2:207–212. doi: 10.1038/nmeth740. [DOI] [PubMed] [Google Scholar]
- Pace CN. Determination and analysis of urea and guanidine hydrochloride denaturation curves. Methods Enzymol. 1986;131:266–280. doi: 10.1016/0076-6879(86)31045-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information