

Abstract
Interspecific hybridization provides the unique opportunity for species to tap into genetic variation present in a closely related species and potentially take advantage of beneficial alleles. It has become increasingly clear that when hybridization occurs, mitochondrial DNA (mtDNA) often crosses species boundaries, raising the possibility that it could serve as a recurrent target of natural selection and source of species' adaptations. Here we report the sequences of 46 complete mitochondrial genomes of Drosophila yakuba and Drosophila santomea, two sister species known to produce hybrids in nature (∼3%). At least two independent events of mtDNA introgression are uncovered in this study, including an early invasion of the D. yakuba mitochondrial genome that fully replaced the D. santomea mtDNA native haplotypes and a more recent, ongoing event centred in the hybrid zone. Interestingly, this recent introgression event bears the signature of Darwinian natural selection, and the selective haplotype can be found at low frequency in Africa mainland populations of D. yakuba. We put forward the possibility that, because the effective population size of D. santomea is smaller than that of D. yakuba, the faster accumulation of mildly deleterious mutations associated with Muller's ratchet in the former species may have facilitated the replacement of the mutationally loaded mitochondrial genome of D.
Keywords: gene flow, positive selection, recurrent adaptation, speciation
Introduction
Shifting environments challenge species as they often throw them off their fitness optimum, and the role of adaptation is to move them to a new phenotypic optimum (Orr 2002, 2005). This can typically be achieved using standing genetic variation (i.e. variation present in the species at the time of the environmental change) or de novo mutations. Yet there is another way for species to acquire beneficial alleles: introgression via interspecific hybridization (Rieseberg & Wendel 1993; Arnold 1997; Whitney et al. 2006). Adaptive introgression was first recognized by evolutionary botanists (Anderson 1949; Stebbins 1959) and later detected in animal species (Lewontin & Birch 1966). Although the initial hybridization is frequently deleterious, introgression provides the opportunity for species to tap into additional genetic variation of a closely related species and potentially take advantage of raw material for adaptation (Seehausen 2004; Mallet 2005; Whitney et al. 2006; Arnold 2007; Baack & Rieseberg 2007; Castric et al. 2008; Rieseberg 2009; Consortium 2012). Universally beneficial alleles can spread easily across species boundaries when isolating barriers are incomplete (Coyne & Orr 2004) and produce the signature of a trans-species selective sweep (Barton & Gale 1993; Hilton et al. 1994; Stephan et al. 1998; Machado & Hey 2003; Llopart et al. 2005b; Teeter et al. 2008; Staubach et al. 2012; Brand et al. 2013).
For several reasons introgression may represent a particularly efficient way for species to adapt. First, introgression can enable populations to respond rapidly to sudden environmental changes where fast response is crucial. Cases of resistance to anthropogenic agents, such as the anticoagulant rodenticide warfarin, constitute textbook examples (Rieseberg 2011; Song et al. 2011). Interestingly, adaptation through introgression often involves a change in the fitness value of already existing alleles that were originally either neutral or weakly deleterious before the environmental shift. This change in the fitness value is an attribute also observed in adaptation through standing genetic variation within species (Orr & Betancourt 2001; Hermisson & Pennings 2005; Barrett & Schluter 2008; Peter et al. 2012; Hedrick 2013). Second, alleles lost to random genetic drift or deleterious alleles fixed in a population may be easier to replace with alleles from a closely related species that have been pretested by natural selection (Kim et al. 2008; Rieseberg 2009). This could be particularly relevant to the introgression of nonrecombining genomes with high mutation rates (e.g. mitochondrial DNA in Drosophila) in species with small effective population sizes (Ne) (e.g. endemics) as Muller's ratchet, the irreversible accumulation of mildly deleterious mutations, is expected to proceed faster in such species and thus result in mutationally loaded nonrecombining genomes (Lynch & Gabriel 1990; Charlesworth et al. 1993b; Gabriel et al. 1993; Loewe 2006). Lastly, the observation of novel traits in hybrids that are not present in either of the parental species (i.e. transgressive segregation) (Rieseberg et al. 1999, 2003) raises the interesting possibility that the placement of several genes of one species in the genetic background of another through introgression may result in new traits. The pervasive, nonadditive nature of gene regulation suggests that changes in expression could be underlying these novel phenotypes (Gibson & Weir 2005; Lai et al. 2006; Landry et al. 2007).
Traditionally viewed as fortuitous accidents of nature, hybrid zones provide the unique opportunity to study introgression (Barton & Hewitt 1985, 1989; Hewitt 1988; Harrison & Rand 1989; Harrison 1993). On the small African island of São Tomé, Drosophila yakuba and Drosophila santomea, a pair of very recently diverged sister species in the melanogaster subgroup, form a well-characterized hybrid zone where hybrids are abundant (∼3–5%) (Llopart et al. 2005a). The two species began diverging allopatrically ∼400 000 years ago (Llopart et al. 2005b), but today are both present on São Tomé due to secondary invasion by D. yakuba. (D. santomea is endemic to São Tomé.) Despite their recent divergence, D. yakuba and D. santomea show traditional forms of reproductive isolation, including sexual isolation (Lachaise et al. 2000; Coyne et al. 2002; Moehring et al. 2006b), conspecific sperm precedence (Chang 2004), prezygotic reinforcement (Matute 2010) and hybrid sterility that conforms to Haldane's rule (Cariou et al. 2001; Coyne et al. 2004; Moehring et al. 2006a; Matute & Coyne 2009).
Two previous studies in these species reported introgression of mitochondrial DNA (mtDNA) based on lack of reciprocal monophyly (Llopart et al. 2005b; Bachtrog et al. 2006). Neither study, however, had adequate statistical power to assess the selective nature of the process, rule out alternative neutral scenarios (i.e. population expansion or strong purifying selection) and obtain a detailed characterization (Llopart et al. 2005b; Bachtrog et al. 2006). To determine the evolutionary forces leading to mtDNA introgression in D. yakuba and D. santomea and understand their temporal dynamics, we sequenced 46 complete mitochondrial genomes. These sequences represent East, Central and West African populations of D. yakuba as well as populations of both species from São Tomé. We found that introgression of the mitochondrial genome is recurrent in these species, with an ancient event in which the D. yakuba mtDNA completely replaced the D. santomea native mitochondrial genome and an ongoing introgression event centred in the D. yakuba–D. santomea hybrid zone. Interestingly, the ongoing introgression shows the signature of positive natural selection and is associated with a haplotype present at low frequency on the Africa mainland. We propose that in species with very different Ne, Muller's ratchet (Lynch & Gabriel 1990; Charlesworth et al. 1993b; Gabriel et al. 1993; Loewe 2006) can have a significant influence on mitochondrial introgression resulting in the rescue of the more mutationally loaded mtDNA of the species with small Ne by the mtDNA of the species with large Ne.
Methods
Drosophila lines
A total of 46 isofemale lines, 29 Drosophila yakuba and 17 Drosophila santomea, were used in this study. To assess the direction of the mitochondrial introgression, we obtained a geographically diversified sample of D. yakuba, including 13 allopatric lines from mainland Africa (West Coast: Ivory Coast, Cameroon and Gabon; Central: Zimbabwe; East Coast: Kenya and Tanzania) and 16 lines collected in São Tomé. One of these island lines was collected in a garden outside São Tomé City, in an area where only D. yakuba is found, and the remaining 15 lines were established from females collected in the D. yakuba–D. santomea hybrid zones (Llopart et al. 2005a,b). To take into account possible population structure in D. santomea, our sampling strategy covered two different geographical areas where D. yakuba is also abundant, the hybrid zone in the Obo Natural Reserve (13 lines) and Rio Queijo in southwest São Tomé (4 lines). A detailed description of the lines is included in Table S1 (Supporting information).
DNA extraction and sequencing strategies
Sequencing was carried out using a combination of two different strategies: standard cycle sequencing preceded by PCR-based amplification (14 complete mitochondrial genomes) and next-generation sequencing (32 complete mitochondrial genomes). In the standard approach, mtDNA was isolated following Afonso et al. (1988) with minor modifications. This is a crude method for the selective isolation of covalently closed circular DNA molecules. The first step involves an alkaline denaturation followed by an acid/phenol extraction. To amplify the entire mtDNA prior to sequencing, we generated 12 overlapping PCR fragments using primers designed on a complete sequence of the D. yakuba mitochondrial genome [accession no. in GenBank X03240; (Clary & Wolstenholme 1985)]. (Primer sequences are available upon request from the authors.) PCR products were purified using the Wizard MagneSil PCR clean-up system (Promega, Madison, WI). Both strands of DNA were sequenced directly using the BigDye Terminator kit v3.1 chemistry and resolved in an ABI 3730 DNA Analyzer (Life Technologies, Carlsbad, CA). Sequences of each line were assembled using the software Sequencher 5.10 (Gene Codes, Ann Arbor, MI), and multiple alignments were obtained using ClustalX (Thompson et al. 1997).
Next-generation technology was also used to obtain the mtDNA sequence of 32 lines. RNA-free DNA extracted from single flies using the Qiagen DNeasy Blood and Tissue Kit (Qiagen, Valencia, CA) was fragmented in a Bioruptor UCD-200 (Diagenode, Denville, NJ). We prepared genomic DNA libraries following the Illumina protocol (‘Preparing Samples for Sequencing Genomic DNA’, Illumina, San Diego, CA). We used custom-designed adapters that have unique 7-nucleotide tags, which were added to standard Illumina adapters, and allowed multiplexing of several samples prior to sequencing. The sequences and a detailed description of these custom-designed adapters can be found in Comeron et al. (2012). Prior to multiplexing for sequencing, library quantitation was performed using the Quant-iT PicoGreen kit (Invitrogen, Carlsbad, CA). Cluster generation and sequencing (75 bp) were carried out at the Iowa State University DNA Facility (Ames, IA) using a GAII instrument. Filtering of reads, mapping and generation of consensus sequences were carried out using the FASTX toolkit (http://hannonlab.cshl.edu/fastx_toolkit), BWA (Li & Durbin 2009), SAMtools v1.4 (Li et al. 2009) and custom scripts. After mapping, the average coverage and consensus quality per nucleotide of the mitochondrial genomes were 130.2 × (median 115.1×) and 222.1 (median 242.8), respectively. SNPs were called with quality Q40 or greater.
Analysis of introgression
To determine whether there has been introgression of the mitochondrial genome between D. yakuba and D. santomea and explore posterior probabilities of demographic parameters under an Isolation-with-Migration model (IM) (Nielsen & Wakeley 2001), we performed simulations using the program IMa2 (Hey & Nielsen 2004, 2007). Our IMa2 analysis included GenBank sequence X03240 and excluded the control region of the mitochondrial genome (positions 14943–16019 in X03240, positions 14990–16127 in the multiple sequence alignment). We assumed a simple demographic model in which ancestral populations do not experience migration and there is one migration parameter between each pair of sampled populations. Log-likelihood-ratio (LLR) tests were used to evaluate how two different models, with and without gene flow between populations, fit the data (Nielsen & Wakeley 2001; Hey 2010). Several preliminary runs were conducted to assess the mixing of the Markov chain. We tried different numbers of Metropolis-coupled chains and heating terms, always using a geometric scheme. We determined that 100–180 chains with a = 0.99 and b = 0.75 produce high swapping rates between adjacent chains and lead to good mixing (i.e. low autocorrelations, high effective sample sizes and no trends in the plots over the course of a run). Upper bounds of prior distributions were originally set following recommendations in the IMa2 manual and adjusted afterwards, as needed, based on the results of a complete first run. The burn-in period was carried out until the stationary phase was reached, which typically took between 1–4 million steps depending on the specific population model (e.g. two populations vs. three populations). To ensure convergence, we performed two well-mixed independent runs using different random number seeds, each with its own burn-in period, and confirmed that they produced similar estimates for all parameters. Results from two well-mixed runs based on no less than 100 000 genealogies each, saved after the burn-in period, were combined to obtain parameter estimates. In all analyses, we used the HKY mutational model (Hasegawa et al. 1985), which is adequate for Drosophila mitochondrial sequences. To determine the role of selection on mitochondrial introgression, we performed coalescent simulations with population growth using the program ms and obtained expectations under strict neutrality (Hudson 2002).
Dating introgression events
To estimate the time to most recent common ancestor (i.e. TMRCA), we used the beast v.1.7.4 software package (Drummond & Rambaut 2007; Drummond et al. 2012). BEAST, with its interface program BEAUti, performs Bayesian MCMC analysis of molecular sequences related by a tree that allows estimates of divergence times (Drummond & Rambaut 2007; Drummond et al. 2012). To this end, the models implemented in BEAST fix the external nodes of the tree to a specified date or rate and then sample the time of the internal nodes from their posterior probability distribution using MCMC. We assumed a strict molecular clock with no rate variation among lineages, which is reasonable with very closely related species such as D. yakuba and D. santomea, a gamma heterogeneity site model with a fraction of invariant sites (initial pinv. = 0.64) and a coalescent-based framework. The initial value of the fraction of invariable sites was determined identifying the model that best fit the data using Akaike information criterion tests in the software jModeltest (Guindon & Gascuel 2003; Darriba et al. 2012). To calibrate the molecular clock, we considered that D. yakuba–D. santomea split from Drosophila erecta 10.4 Mya (Tamura et al. 2004), which correspond to 1.4446 synonymous changes per synonymous site as estimated using the program PAML (Yang 1997, 2007). We conducted two independent runs each of 100 million steps with 10 million steps of burn-in. In each run, we sampled genealogies every 1000 steps to produce 90 000 trees that were analysed with the program Tracer v.1.5 (http://beast.bio.ed.ac.uk/Tracer). This resulted in effective sample sizes greater than 2000 for all the parameters and in all analyses.
Gene genealogy reconstruction
Phylogenetic analyses were conducted using the program MEGA5 (Tamura et al. 2011) and the available sequence of D. erecta as the outgroup (Clark et al. 2007). We obtained gene genealogies using (1) the neighbour-joining (NJ) algorithm (Saitou & Nei 1987) with evolutionary distances corrected for multiple hits at a site (Kimura 1980) and (2) maximum-likelihood and maximum-parsimony methods. Statistical confidence in the nodes of the reconstructed gene tree was assessed using bootstrap (Felsenstein 1985) with 1000 replicates.
Results
Drosophila yakuba and Drosophila santomea mitochondrial genomes are not genetically different
We sequenced the complete mitochondrial genomes of 29 Drosophila yakuba (13 from mainland Africa and 16 from São Tomé) and 17 Drosophila santomea lines. Levels of polymorphism across the different genes are similar in D. yakuba and D. santomea (Table1), in agreement with previous observations in the same species pair (Llopart et al. 2005b; Bachtrog et al. 2006). We detect population structure within D. yakuba, for there is significant genetic differentiation between the Africa mainland and São Tomé populations of this species (nearest-neighbour statistic Snn = 0.79, permutation test P < 0.001) (Hudson 2000). However, this differentiation is based on differences in SNP frequencies rather than on the presence of fixed differences between populations. In striking contrast, mitochondrial haplotypes of D. santomea are not genetically distinct from those of the São Tomé population of D. yakuba (Snn = 0.54, P = 0.24), the first hint of mtDNA introgression. This becomes particularly informative when we consider that genes on the Y chromosome, the other nonrecombining, uni-parentally inherited genomic element, do show significant genetic differentiation between D. yakuba and D. santomea (Snn = 1.0, P < 0.0001), with numerous fixed differences between species (Llopart et al. 2005b). Our analysis of population differentiation strongly suggests that mitochondrial gene flow has indeed occurred from D. yakuba to D. santomea.
Table 1.
Polymorphism data summary
| Locus | Species | n* | S† | ΠS†(×10+3) | ΠN† (×10+3) | Fixed§ | Shared§ | L (bp)¶ |
|---|---|---|---|---|---|---|---|---|
| ATPase 6 | yak | 30 | 8 | 7.97 | 0.51 | 0 | 2 | 672 |
| san | 17 | 6 | 9.11 | 0.23 | ||||
| ATPase 8 | yak | 30 | 1 | 2.1 | 0 | 0 | 0 | 159 |
| san | 17 | 1 | 0 | 0.93 | ||||
| COI | yak | 30 | 14 | 3.82 | 0.11 | 0 | 3 | 1533 |
| san | 17 | 9 | 5.57 | 0.10 | ||||
| COII | yak | 30 | 7 | 4.03 | 0 | 0 | 1 | 684 |
| san | 17 | 3 | 5.11 | 0 | ||||
| COIII | yak | 30 | 6 | 0.74 | 0.44 | 0 | 0 | 786 |
| san | 17 | 3 | 1.31 | 0.19 | ||||
| cyt_b | yak | 30 | 10 | 4.77 | 0.22 | 0 | 3 | 1134 |
| san | 17 | 6 | 3.03 | 0.40 | ||||
| ND1 | yak | 30 | 5 | 2.40 | 0 | 0 | 0 | 972 |
| san | 17 | 2 | 0.55 | 0.58 | ||||
| ND2 | yak | 30 | 6 | 3.82 | 0.41 | 0 | 1 | 1023 |
| san | 17 | 3 | 3.10 | 0.29 | ||||
| ND3 | yak | 30 | 3 | 1.88 | 0.47 | 0 | 0 | 351 |
| san | 17 | 1 | 1.66 | 0 | ||||
| ND4 | yak | 30 | 14 | 3.19 | 0.38 | 0 | 3 | 1338 |
| san | 17 | 6 | 3.35 | 0.23 | ||||
| ND4L | yak | 30 | 1 | 0 | 0.29 | 0 | 0 | 288 |
| san | 17 | 0 | 0 | 0 | ||||
| ND5 | yak | 30 | 27 | 6.62 | 0.58 | 0 | 10 | 1719 |
| san | 17 | 23 | 10.59 | 0.75 | ||||
| ND6 | yak | 30 | 5 | 5.96 | 0.32 | 0 | 1 | 522 |
| san | 17 | 2 | 6.0 | 0 | ||||
| Total coding | yak | 30 | 107 | 4.13 | 0.32 | 0 | 24 | 11181 |
| san | 17 | 65 | 4.76 | 0.31 | ||||
| Total noncoding | yak | 30 | 31 | 0.81 | – | 0 | 11 | 4949 |
| san | 17 | 25 | 1.13 | – |
yak, Drosophila yakuba and san, Drosophila santomea.
Sample size.
Number of polymorphic sites.
Nucleotide diversity at synonymous, or silent in noncoding regions (⊓S), and nonsynonymous (⊓N) sites calculated with DnaSP v.5 (Librado & Rozas 2009).
Differences fixed between D. yakuba and D. santomea and shared polymorphisms.
Size of the sequenced region (including alignment gaps).
Significant post-split mitochondrial gene flow
To formally investigate whether gene flow between D. yakuba and D. santomea occurred after the colonization of São Tomé by the ancestral species (i.e. post-split gene flow), we fit an IM model to our data of complete mtDNA sequences using the software IMa2 (Nielsen & Wakeley 2001; Hey & Nielsen 2004, 2007). As polymorphism levels are low in the Drosophila mitochondrial genome due to strong functional constraints (Nabholz et al. 2013), maternal inheritance, and the effects of hitchhiking (Maynard Smith & Haigh 1974; Kaplan et al. 1989; Sella et al. 2009) and/or background selection (Charlesworth et al. 1993a; Charlesworth 2012) associated with the lack of recombination, there is not enough polymorphism information in the data to obtain precise maximum-likelihood estimates of population mutation rate parameters (i.e. θ). To assess whether our results are robust to different θ values, we tested the null hypothesis of no gene flow under three different models using θ set to 1, 2.5 and 5× the value observed in D. yakuba mtDNA (θ per sequence = 29.79). In all conditions investigated, LLR tests indicated that a model in which post-split gene flow between D. yakuba and D. santomea had occurred was significantly more likely than a scenario with 0 migration (LLR test statistic ≥14.08 in all cases, P < 0.001). In addition, marginal posterior probability distributions of the migration rate parameter (m) excluded the smallest value significantly, producing the unequivocal signal of gene flow (posterior P < 1.2 × 10−8, P < 3.8 × 10−6 and P < 9.9 × 10−5 for m = 0.004 in the 1, 2.5 and 5× models, respectively; Fig.1). Note that these posterior probabilities suggest that ancestral populations with large Ne, which are expected to have more ancestral polymorphism, make the test to zero migration conservative. The lack of fixed differences between the island populations (D. yakuba and D. santomea combined) and mainland D. yakuba strongly indicates that some, if not all, D. santomea mitochondrial genomes have been replaced by haplotypes from its sister species D. yakuba.
Fig 1.
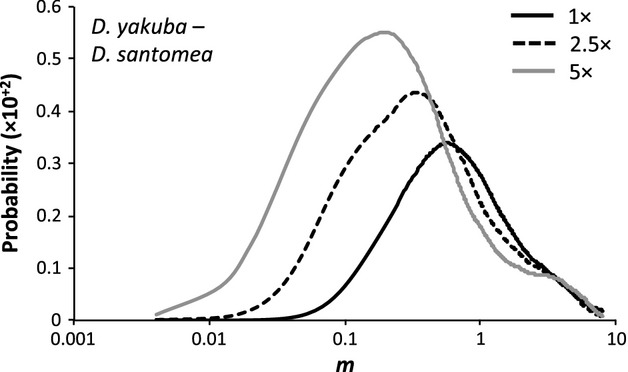
Marginal density for the migration rate parameter (m) obtained by fitting the isolation-with-migration model to a data set with two descendant populations, Drosophila yakuba and Drosophila santomea. 1, 2.5 and 5× correspond to θ per sequence of ∼30, ∼75 and ∼150, respectively (θ per sequence observed in D. yakuba and D. santomea are 29.79 and 21, respectively).
Although we were able to rule out isolation between D. yakuba and D. santomea, a model in which each species is represented by a single population is not very realistic considering that there is population structure within D. yakuba (see above). A more realistic scenario can be modelled using three populations, two D. yakuba populations (one mainland and one island) and one population of D. santomea. This scenario is also expected to be more sensitive to recent events of introgression. We fit a three-population IM model to our data set and investigated the possibility of recent introgression on São Tomé. LLR tests produced the same results across replicates and population sizes. There is significant post-split gene flow between D. santomea and the island population of D. yakuba (LLR test statistics = 3.38–4.84, P < 0.05; Fig.2a) but not between the two D. yakuba populations, or the mainland D. yakuba population and D. santomea (LLR test statistics = 0–0.5, P > 0.05; Fig.2b, c). In summary, IMa2 results are consistent with previous findings that suggested gene flow of mtDNA between D. yakuba and D. santomea (Llopart et al. 2005b; Bachtrog et al. 2006).
Fig 2.
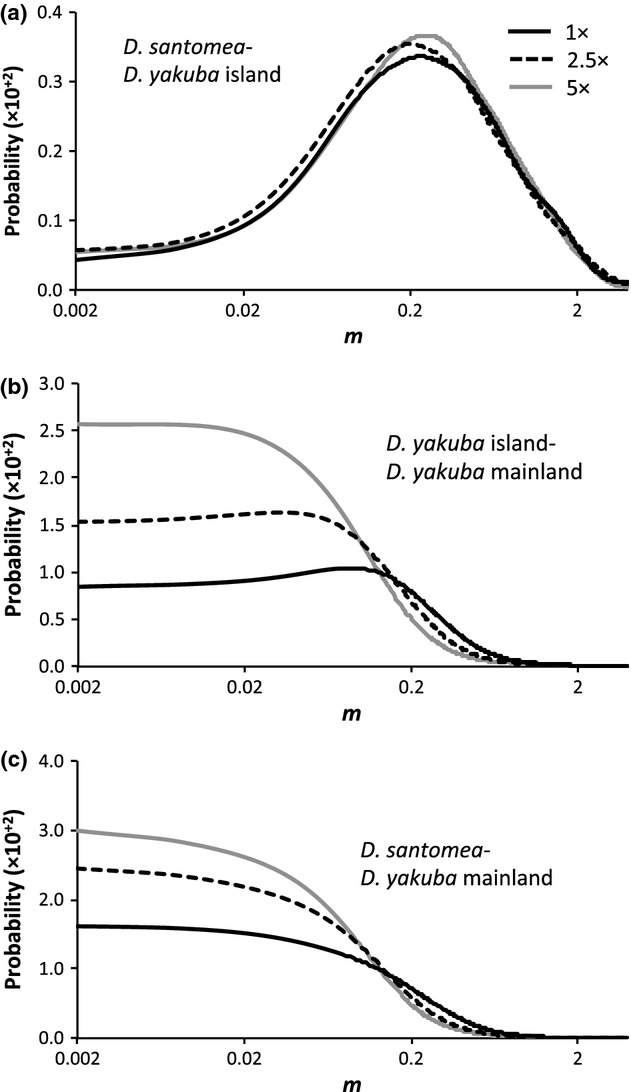
Marginal density for the migration rate parameter (m) obtained by fitting the isolation-with-migration model to a data set with three descendant populations. Marginal density for m between (a) Drosophila santomea–Drosophila yakuba island populations, (b) D. yakuba island–D. yakuba mainland populations and (c) D. santomea –D. yakuba mainland populations. 1, 2.5 and 5× correspond to θ per sequence of ∼30, ∼75 and ∼150, respectively (θ per sequence observed in the D. yakuba mainland, D. yakuba island and D. santomea populations are 23.27, 18.68 and 21, respectively).
Ongoing adaptive introgression in the mitochondrial genome
To investigate the temporal dynamics of the mitochondrial introgression on São Tomé, we applied a phylogenetic approach. Gene trees obtained using different methods (i.e. distance, parsimony and maximum likelihood) revealed the same pattern: a distinct, starlike clade strongly supported by high bootstrap values (>90%) clusters together 13 D. yakuba and 8 D. santomea sequences (21 of 47 total sequences; 46 newly reported here and X03240 from GenBank) (Fig.3). This clade is significantly enriched in sequences derived from the classic hybrid zone (HZ) of these two species (Llopart et al. 2005a) (16/21 in the HZ clade vs. 10/26 outside the HZ clade; FET, P = 0.017). The starlike topology is consistent with a rapid increase in frequency of a single haplotype (i.e. HZ haplotype) in the population due to either population growth or positive selection. The presence of both D. yakuba and D. santomea sequences in this clade indicates that the HZ haplotype spread through the population after it transferred from one species to the other. This suggests that the HZ clade potentially represents an ongoing adaptive introgression event of the mitochondrial genome.
Fig 3.
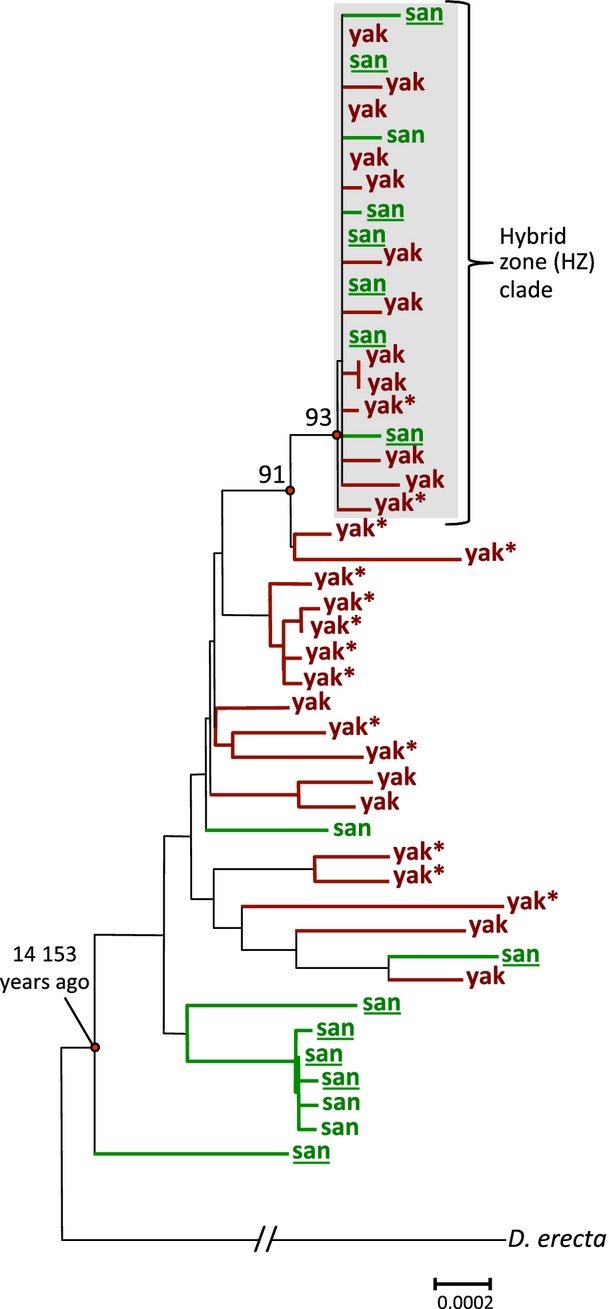
Neighbour-joining trees reconstructed using complete sequences of 48 mitochondrial genomes from Drosophila erecta, Drosophila yakuba (yak) and Drosophila santomea (san). D. santomea sequences from the classic hybrid zone in the Obo Natural Reserve are underlined, and asterisks indicate Africa mainland lines of D. yakuba. Bootstrap values were obtained after 1000 replicates.
To formally assess the adaptive nature of the ongoing introgression, we performed neutral coalescent simulations using the program ms (Hudson 2002). We simulated population growth and evaluated whether the severe reduction in nucleotide variation in the HZ clade could be explained by demographic factors. Our simulations are thus conditional on the observed number of total polymorphisms and the frequency spectrum, which is skewed towards rare variants, as captured by the summary statistic Tajima's D (Tajima 1989) and in agreement with population growth. We analysed a subset of 10 000 genealogies showing an average D = −2.20 ± 1 SD. (Note that this value in our simulations closely resembles our observation in the entire sample of D. yakuba and D. santomea mitochondrial genomes, D = −2.24.) For each simulation, we tracked the number of polymorphisms present in a cluster formed by the 21 most closely related sequences. Our results indicate that there is a significant deficit of polymorphisms in the HZ clade given the overall distribution of variation in the entire gene tree (23 or fewer polymorphisms in a cluster containing 21 of 47 sequences, P < 0.0001). Similar results were obtained when we analysed D. yakuba (13 sequences with 15 or fewer polymorphisms in a clade of 21 sequences, P < 0.0001) and D. santomea (eight sequences with eight or fewer polymorphisms in a clade of 21 sequences, P = 0.008) separately. We conclude that the presence of a haplotype at a frequency of >40% cannot be explained by population expansion and is thus consistent with positive selection. The HZ clade constitutes a case of ‘caught-in-the-act’ mitochondrial adaptive introgression.
Sequential introgression events
To determine whether we could formally rule out the possibility of a single introgression event, we obtained estimates of the time to the most recent common ancestor (TMRCA) of all D. yakuba and D. santomea sequences, as well as those in the HZ clade. We assumed a neutral mutation rate of 0.06945 mutations per site and million year based on synonymous divergence between Drosophila erecta and D. yakuba/D. santomea in the 13 mitochondrial genes (dS = 1.4446; see materials and methods) and a time since split of 10.4 My (Tamura et al. 2004). The MRCA of the entire data set of D. yakuba and D. santomea mitochondrial sequences was estimated to have lived 14 153 years ago [upper and lower 95% highest posterior density (HPD): 17 888–10 792]. The estimate appears unusually recent and therefore contradicts divergence times between D. yakuba and D. santomea based on nuclear genes (Llopart et al. 2005b). One could argue, however, that the comparison of times based on nuclear and mitochondrial genomes is unfair as the latter do not recombine, are maternally inherited and are thus susceptible to shorter coalescent times due to smaller Ne (Hudson & Coyne 2002; Hudson & Turelli 2003; Ballard & Whitlock 2004). The Y chromosome, instead, provides the optimal comparison and, in addition, is more resilient to introgression in the D. yakuba and D. santomea system, for it has a large effect on hybrid male sterility (Coyne et al. 2004; Llopart et al. 2005b). Divergence times estimated for the Y chromosome using BEAST are significantly older than those obtained using mtDNA [116 100 years with 95% HPD: 187 900–51 730; nuclear mutation rate assumed to be 5× lower than the mitochondrial mutation rate (Moriyama & Powell 1997; Haag-Liautard et al. 2007, 2008)] uncovering an old event of mtDNA introgression that drove the D. santomea mtDNA to extinction. The inclusion of the Y chromosome data set in our mitochondrial IMa2 simulations results in a more than 8-fold increase in the maximum-likelihood estimate of m between D. yakuba and D. santomea (m = 0.19 for mtDNA alone and m = 1.6 for mtDNA and Y chromosome data sets combined, respectively, both for the θ ∼5× model; Fig.4). This is expected as the power to detect older introgression events is hampered by the analysis of a single, albeit lengthy, locus that has experienced introgression. The incorporation of Y chromosome data provides information on the speciation time of D. yakuba and D. santomea, allowing IMa2 to estimate m more accurately.
Fig 4.
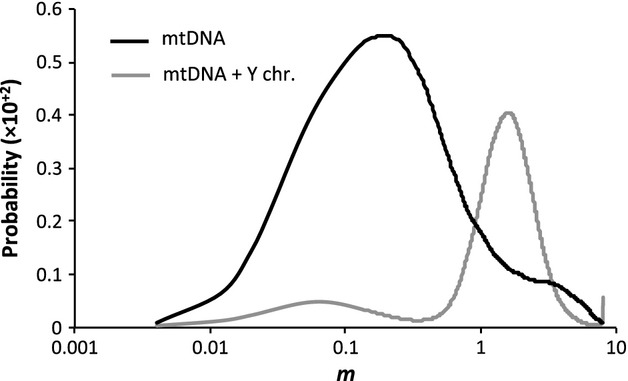
Marginal density for the migration rate parameter (m) obtained by fitting a two-population isolation-with-migration model to a data set of mtDNA sequences only (mtDNA) or mtDNA and Y chromosome sequences combined (mtDNA + Y chr.).
The TMRCA for the HZ clade was estimated to be 2539 years ago (95% HPD: 3624–1557). Similar results were obtained when population expansion was allowed [13 360 years for all sequences (HPD: 16 936–10 008) and 2905 years for the HZ clade (95% HPD: 4010–1893)]. Although precise dating of the different clades depends on the particular assumptions of the underlying molecular model, nonoverlapping HPDs clearly indicate that there have been at least two distinct waves of mitochondrial gene flow between D. yakuba and D. santomea (Fig.5).
Fig 5.
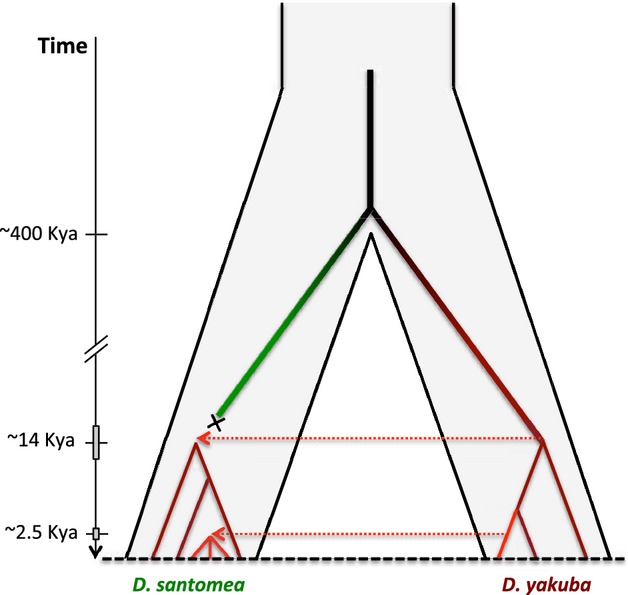
Schematic representation of the temporal dynamics of mitochondrial introgression in the Drosophila yakuba–Drosophila santomea system. Grey rectangles represent upper and lower 95% highest posterior density of the time to the most recent common ancestor.
Discussion
Although Drosophila yakuba and Drosophila santomea began diverging ∼400 Kya (Cariou et al. 2001; Llopart et al. 2002, 2005b), today they share the same mitochondrial genome. This observation stands out when we consider several facts. First, multiple nuclear loci investigated in these same species, including some on the Y chromosome, do show extensive genetic differentiation between D. yakuba and D. santomea (Llopart et al. 2005b; Bachtrog et al. 2006). Second, mutations rates of mtDNA in Drosophila are ∼5–10 times higher than those of nuclear genes (Moriyama & Powell 1997; Haag-Liautard et al. 2007, 2008). Thus, the observed genetic similarity can only be explained by mitochondrial exchange through interspecific hybridization.
Our gene tree reconstruction and molecular dating indicate two independent mitochondrial introgression events (Fig.5). Estimates of the TMRCA for all D. yakuba and D. santomea mitochondrial genomes are more than 8-fold smaller than estimates of speciation times based on the Y chromosome and indicate a common ancestor as recent as ∼14 000 years ago. This is not only evidence of an early mitochondrial introgression event in D. yakuba and D. santomea, but it also suggests that only the mitochondrial genome of one of the two species survived this early introgression. Because the sequences of the mainland and island populations of D. yakuba do not form distinct clades, we infer that the direction of the gene flow was from D. yakuba to D. santomea. (The alternative possibility requires introgression from D. santomea into island populations of D. yakuba and then a species-wide selective sweep of the D. santomea haplotype among mainland populations of D. yakuba.) The basal sequences of D. santomea shown in the gene tree of Fig.3 may represent D. yakuba-like remnants of this early introgression event. The replacement of the mtDNA of one species by that of another has been also reported in D. pseudoobscura and D. persimilis (Powell 1983; Machado et al. 2002) but contrasts with the D. simulans–D. mauritiana case, wherein the mtDNA of both species is still present today in D. mauritiana (Ballard 2000; Nunes et al. 2010; Garrigan et al. 2012). It is tempting to speculate that the early replacement of the D. santomea mitochondrial genome was driven by positive selection. However, as the signature of positive selection is typically short-lived (Przeworski 2002), it is not likely to detect its fingerprints. Indeed, we find no evidence in our data to support the possibility that the original D. santomea mtDNA replacement was adaptive. We detected a second, ongoing, mitochondrial exchange between D. yakuba and D. santomea centred in the classic hybrid zone (Llopart et al. 2005a). Contrary to the early introgression event, in this case, we were able to detect the fingerprints of natural selection using coalescent simulations. This event represents a prime example of adaptive introgression or the acquisition of beneficial alleles from closely related species (Rieseberg 2011; Song et al. 2011).
Several explanations could potentially account for the exchange of mitochondrial genomes among species. Theoretical population genetic models show that very small amounts of gene flow between two species are sufficient to homogenize neutral mtDNA, even when hybrids experience substantial fitness loss caused by nuclear incompatibilities (Takahata & Slatkin 1984). Recurrent hybridization between D. yakuba females and D. santomea males and selection against hybrid nuclear genotypes, but not against mtDNA variants, could lead to some of the observed patterns. Selectively advantageous factors also need to be considered. mtDNA could hitchhike with the spread of the endosymbiont Wolbachia, as reported in D. simulans (Turelli & Hoffmann 1991; Turelli et al. 1992), because they are both maternally transmitted, and infected females experience a selective advantage relative to uninfected ones due to cytoplasmic incompatibility (Hoffmann et al. 1986; Nigro & Prout 1990). Although to date D. yakuba and close relatives lack cytoplasmic incompatibility associated with Wolbachia (Charlat et al. 2004; Zabalou et al. 2004), the endosymbiont may induce other potentially beneficial effects, such as resistance to virus infections (Hedges et al. 2008; Teixeira et al. 2008; Kambris et al. 2009) or increased fecundity (Fast et al. 2011). Another possible selective scenario is that mitochondrial introgression could be mediated by cytonuclear interactions. Experimental populations show that mitochondrial alleles can experience significant changes in frequency that are not necessarily associated with mitochondrial fitness differences directly, but depend on the nuclear genetic background (Clark 1985; Clark & Lyckegaard 1988; MacRae & Anderson 1988; Scribner & Avise 1994; Hutter & Rand 1995; Kilpatrick & Rand 1995; Cruzan & Arnold 1999). This could explain the recent introgression event. The presence of the mitochondrial haplotype at low frequency on the Africa mainland indicates that it is not universally beneficial and raises the possibility that it became advantageous only in the nuclear genetic background and/or environment of the hybrid zone.
Lastly, we propose a third selective explanation for mitochondrial introgression and replacement that is based on Muller's ratchet (Muller 1964; Felsenstein 1974; Maynard Smith 1978; Lynch & Gabriel 1990; Charlesworth et al. 1993b). While Muller's ratchet has often been proposed to explain the irreversible eventual extinction of asexual species (or populations) (Gabriel et al. 1993; Lynch et al. 1993; Gordo & Charlesworth 2000), it can also operate in nonrecombining genetic elements, such as mtDNA and the Y chromosome (Gabriel et al. 1993; Charlesworth & Charlesworth 2000; Filatov et al. 2000; Loewe 2006; Kaiser & Charlesworth 2010) as well as in nuclear regions with severely reduced recombination rates (Dolgin & Charlesworth 2008). The larger Ne of D. yakuba relative to that of D. santomea likely resulted in a decreased rate of accumulation of mildly deleterious mutations (i.e. slower Muller's ratchet) in the mtDNA. Interspecific hybridization after the secondary colonization of São Tomé by D. yakuba may have allowed D. santomea to tap into additional genetic variation. The lower mutational load of the D. yakuba mitochondrial genome may have driven the early replacement of the D. santomea mtDNA. Rieseberg (2009) proposed that introgression could serve as an efficient way to replace damaged or lost alleles using those of a closely related species. The early mtDNA replacement in D. yakuba and D. santomea, and perhaps even the ongoing introgression event, could well constitute examples of this scenario.
Acknowledgments
We thank all the members of the ‘Isolation with Migration Discussion Group’ (https://groups.google.com/forum/?fromgroups#!forum/Isolation-with-Migration) for tireless and helpful advice. We are also grateful to Josep Comeron and four anonymous reviewers for comments on the manuscript. This work was funded by University of Iowa funds to A.L. D.H. was partially supported by NIH Predoctoral Training Grant in Genetics T32 GM008629014.
A.L. conceived and supervised the project and also performed the analyses. A.L., D.H., E.B. and Z.S. carried out laboratory experiments and obtained DNA sequences. A.L. wrote the manuscript with comments from all the other authors.
Data accessibility
DNA sequences: GenBank/EMBL accession numbers KF824856–KF824901. IMa2 and MEGA input files: Dryad doi:10.5061/dryad.9kh1b.
Supporting Information
Additional supporting information may be found in the online version of this article.
Detailed description of the lines used in this study.
References
- Afonso JM, Pestano J, Hernandez M. Rapid isolation of mitochondrial DNA from Drosophila adults. Biochemical Genetics. 1988;26:381–386. doi: 10.1007/BF02401791. [DOI] [PubMed] [Google Scholar]
- Anderson E. Introgressive Hybridization. New York: John Wiley & Sons; 1949. [Google Scholar]
- Arnold ML. Natural Hybridization and Evolution. Oxford: Oxford University Press; 1997. [Google Scholar]
- Arnold ML. Evolution through Genetic Exchange. Oxford: Oxford University Press; 2007. [Google Scholar]
- Baack EJ, Rieseberg LH. A genomic view of introgression and hybrid speciation. Current Opinion in Genetics & Development. 2007;17:513–518. doi: 10.1016/j.gde.2007.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachtrog D, Thornton K, Clark A, Andolfatto P. Extensive introgression of mitochondrial DNA relative to nuclear genes in the Drosophila yakuba species group. Evolution. 2006;60:292–302. [PubMed] [Google Scholar]
- Ballard JW. When one is not enough: introgression of mitochondrial DNA in Drosophila. Molecular Biology and Evolution. 2000;17:1126–1130. doi: 10.1093/oxfordjournals.molbev.a026394. [DOI] [PubMed] [Google Scholar]
- Ballard JWO, Whitlock MC. The incomplete natural history of mitochondria. Molecular Ecology. 2004;13:729–744. doi: 10.1046/j.1365-294x.2003.02063.x. [DOI] [PubMed] [Google Scholar]
- Barrett RD, Schluter D. Adaptation from standing genetic variation. Trends in Ecology & Evolution. 2008;23:38–44. doi: 10.1016/j.tree.2007.09.008. [DOI] [PubMed] [Google Scholar]
- Barton NH, Gale KS. Genetic analysis of hybrid zones. In: Harrison RG, editor. Hybrid zones and the Evolutionary Process. New York and Oxford: Oxford University Press; 1993. pp. 13–45. [Google Scholar]
- Barton NH, Hewitt GM. Analysis of hybrid zones. Annual Review of Ecology, Evolution and Systematics. 1985;16:113–148. [Google Scholar]
- Barton NH, Hewitt GM. Adaptation, speciation and hybrid zones. Nature. 1989;341:497–503. doi: 10.1038/341497a0. [DOI] [PubMed] [Google Scholar]
- Brand CL, Kingan SB, Wu L, Garrigan D. A selective sweep across species boundaries in Drosophila. Molecular Biology and Evolution. 2013;30:2177–2186. doi: 10.1093/molbev/mst123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cariou ML, Silvain JF, Daubin V, Da Lage JL, Lachaise D. Divergence between Drosophila santomea and allopatric or sympatric populations of D. yakuba using paralogous amylase genes and migration scenarios along the Cameroon volcanic line. Molecular Ecology. 2001;10:649–660. doi: 10.1046/j.1365-294x.2001.01225.x. [DOI] [PubMed] [Google Scholar]
- Castric V, Bechsgaard J, Schierup MH, Vekemans X. Repeated adaptive introgression at a gene under multiallelic balancing selection. PLoS Genetics. 2008;4:e1000168. doi: 10.1371/journal.pgen.1000168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang AS. Conspecific sperm precedence in sister species of Drosophila with overlapping ranges. Evolution. 2004;58:781–789. doi: 10.1111/j.0014-3820.2004.tb00411.x. [DOI] [PubMed] [Google Scholar]
- Charlat S, Ballard JW, Mercot H. What maintains noncytoplasmic incompatibility inducing Wolbachia in their hosts: a case study from a natural Drosophila yakuba population. Journal of Evolutionary Biology. 2004;17:322–330. doi: 10.1046/j.1420-9101.2003.00676.x. [DOI] [PubMed] [Google Scholar]
- Charlesworth B. The effects of deleterious mutations on evolution at linked sites. Genetics. 2012;190:5–22. doi: 10.1534/genetics.111.134288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth B, Charlesworth D. The degeneration of Y chromosomes. Philosophical Transactions of the Royal Society B. 2000;355:1563–1572. doi: 10.1098/rstb.2000.0717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth B, Morgan MT, Charlesworth D. The effect of deleterious mutations on neutral molecular variation. Genetics. 1993a;134:1289–1303. doi: 10.1093/genetics/134.4.1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth D, Morgan MT, Charlesworth B. Mutation accumulation in finite outbreeding and inbreeding populations. Genetical Research. 1993b;61:39–56. [Google Scholar]
- Clark AG. Natural selection with nuclear and cytoplasmic transmission. II. Tests with Drosophila from diverse populations. Genetics. 1985;111:97–112. doi: 10.1093/genetics/111.1.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark AG, Lyckegaard EM. Natural selection with nuclear and cytoplasmic transmission. III. Joint analysis of segregation and mtDNA in Drosophila melanogaster. Genetics. 1988;118:471–481. doi: 10.1093/genetics/118.3.471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark AG, Eisen MB, Smith DR, et al. Evolution of genes and genomes on the Drosophila phylogeny. Nature. 2007;450:203–218. doi: 10.1038/nature06341. [DOI] [PubMed] [Google Scholar]
- Clary DO, Wolstenholme DR. The ribosomal RNA genes of Drosophila mitochondrial DNA. Nucleic Acids Research. 1985;13:4029–4045. doi: 10.1093/nar/13.11.4029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comeron JM, Ratnappan R, Bailin S. The many landscapes of recombination in Drosophila melanogaster. PLoS Genetics. 2012;8:e1002905. doi: 10.1371/journal.pgen.1002905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyne JA, Orr HA. Speciation. Sunderland, MA: Sinauer Associates; 2004. [Google Scholar]
- Coyne JA, Kim SY, Chang AS, Lachaise D, Elwyn S. Sexual isolation between sibling species with overlapping ranges: Drosophila santomea and Drosophila yakuba. Evolution. 2002;56:2424–2434. doi: 10.1111/j.0014-3820.2002.tb00168.x. [DOI] [PubMed] [Google Scholar]
- Coyne JA, Elwyn S, Kim SY, Llopart A. Genetic studies of two sister species in the Drosophila melanogaster subgroup, D. yakuba and D. santomea. Genetical Research. 2004;84:11–26. doi: 10.1017/s0016672304007013. [DOI] [PubMed] [Google Scholar]
- Cruzan MB, Arnold ML. Consequences of cytonuclear epistasis and assortative mating for the genetic structure of hybrid populations. Heredity. 1999;82:36–45. [Google Scholar]
- Darriba D, Taboada GL, Doallo R, Posada D. jModelTest 2: more models, new heuristics and parallel computing. Nature Methods. 2012;9:772. doi: 10.1038/nmeth.2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolgin ES, Charlesworth B. The effects of recombination rate on the distribution and abundance of transposable elements. Genetics. 2008;178:2169–2177. doi: 10.1534/genetics.107.082743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond AJ, Rambaut A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evolutionary Biology. 2007;7:214. doi: 10.1186/1471-2148-7-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond AJ, Suchard MA, Xie D, Rambaut A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Molecular Biology and Evolution. 2012;29:1969–1973. doi: 10.1093/molbev/mss075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fast EM, Toomey ME, Panaram K. Wolbachia enhance Drosophila stem cell proliferation and target the germline stem cell niche. Science. 2011;334:990–992. doi: 10.1126/science.1209609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenstein J. The evolutionary advantage of recombination. Genetics. 1974;78:737–756. doi: 10.1093/genetics/78.2.737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenstein J. Confidence limits on phylogenies: an approach using the bootstrap. Evolution. 1985;39:783–791. doi: 10.1111/j.1558-5646.1985.tb00420.x. [DOI] [PubMed] [Google Scholar]
- Filatov DA, Moneger F, Negrutiu I, Charlesworth D. Low variability in a Y-linked plant gene and its implications for Y-chromosome evolution. Nature. 2000;404:388–390. doi: 10.1038/35006057. [DOI] [PubMed] [Google Scholar]
- Gabriel W, Lynch M, Burger R. Muller's Ratchet and mutational meltdowns. Evolution. 1993;47:1744–1757. doi: 10.1111/j.1558-5646.1993.tb01266.x. [DOI] [PubMed] [Google Scholar]
- Garrigan D, Kingan SB, Geneva AJ, et al. Genome sequencing reveals complex speciation in the Drosophila simulans clade. Genome Research. 2012;22:1499–1511. doi: 10.1101/gr.130922.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson G, Weir B. The quantitative genetics of transcription. Trends in Genetics. 2005;21:616–623. doi: 10.1016/j.tig.2005.08.010. [DOI] [PubMed] [Google Scholar]
- Gordo I, Charlesworth B. The degeneration of asexual haploid populations and the speed of Muller's ratchet. Genetics. 2000;154:1379–1387. doi: 10.1093/genetics/154.3.1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon S, Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Systematic Biology. 2003;52:696–704. doi: 10.1080/10635150390235520. [DOI] [PubMed] [Google Scholar]
- Haag-Liautard C, Dorris M, Maside X, et al. Direct estimation of per nucleotide and genomic deleterious mutation rates in Drosophila. Nature. 2007;445:82–85. doi: 10.1038/nature05388. [DOI] [PubMed] [Google Scholar]
- Haag-Liautard C, Coffey N, Houle D, et al. Direct estimation of the mitochondrial DNA mutation rate in Drosophila melanogaster. PLoS Biology. 2008;6:e204. doi: 10.1371/journal.pbio.0060204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison RG. Hybrid Zones and the Evolutionary Process. New York: Oxford University Press; 1993. [Google Scholar]
- Harrison RG, Rand D. Mosaic hybrid zones. In: Endler J, editor. Speciation and its Consequences. 1989. [Google Scholar]; Otte D, editor. Sunderland, MA: Sinauer Associates; pp. 111–133. [Google Scholar]
- Hasegawa M, Kishino H, Yano T. Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. Journal of Molecular Evolution. 1985;22:160–174. doi: 10.1007/BF02101694. [DOI] [PubMed] [Google Scholar]
- Hedges LM, Brownlie JC, O'Neill SL, Johnson KN. Wolbachia and virus protection in insects. Science. 2008;322:702. doi: 10.1126/science.1162418. [DOI] [PubMed] [Google Scholar]
- Hedrick PW. Adaptive introgression in animals: examples and comparison to new mutation and standing variation as sources of adaptive variation. Molecular Ecology. 2013;22:4606–4618. doi: 10.1111/mec.12415. [DOI] [PubMed] [Google Scholar]
- Hermisson J, Pennings PS. Soft sweeps: molecular population genetics of adaptation from standing genetic variation. Genetics. 2005;169:2335–2352. doi: 10.1534/genetics.104.036947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hewitt GM. Hybrid zones-Natural laboratories for evolutionary studies. Trends in Ecology & Evolution. 1988;3:158–167. doi: 10.1016/0169-5347(88)90033-X. [DOI] [PubMed] [Google Scholar]
- Hey J. Isolation with migration models for more than two populations. Molecular Biology and Evolution. 2010;27:905–920. doi: 10.1093/molbev/msp296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hey J, Nielsen R. Multilocus methods for estimating population sizes, migration rates and divergence time, with applications to the divergence of Drosophila pseudoobscura and D. persimilis. Genetics. 2004;167:747–760. doi: 10.1534/genetics.103.024182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hey J, Nielsen R. Integration within the Felsenstein equation for improved Markov chain Monte Carlo methods in population genetics. Proceedings of the National Academy of Sciences USA. 2007;104:2785–2790. doi: 10.1073/pnas.0611164104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilton H, Kliman RM, Hey J. Using hitchhiking genes to study adaptation and divergence during speciation within the Drosophila melanogaster species complex. Evolution. 1994;48:1900–1913. doi: 10.1111/j.1558-5646.1994.tb02222.x. [DOI] [PubMed] [Google Scholar]
- Hoffmann AA, Turelli M, Simmons GM. Unidirectional incompatibility between populations of Drosophila simulans. Evolution. 1986;40:692–701. doi: 10.1111/j.1558-5646.1986.tb00531.x. [DOI] [PubMed] [Google Scholar]
- Hudson RR. A new statistic for detecting genetic differentiation. Genetics. 2000;155:2011–2014. doi: 10.1093/genetics/155.4.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson RR. Generating samples under a Wright-Fisher neutral model of genetic variation. Bioinformatics. 2002;18:337–338. doi: 10.1093/bioinformatics/18.2.337. [DOI] [PubMed] [Google Scholar]
- Hudson RR, Coyne JA. Mathematical consequences of the genealogical species concept. Evolution. 2002;56:1557–1565. doi: 10.1111/j.0014-3820.2002.tb01467.x. [DOI] [PubMed] [Google Scholar]
- Hudson RR, Turelli M. Stochasticity overrules the “three-times rule”: genetic drift, genetic draft, and coalescence times for nuclear loci versus mitochondrial DNA. Evolution. 2003;57:182–190. doi: 10.1111/j.0014-3820.2003.tb00229.x. [DOI] [PubMed] [Google Scholar]
- Hutter CM, Rand DM. Competition between mitochondrial haplotypes in distinct nuclear genetic environments: Drosophila pseudoobscura vs. D. persimilis. Genetics. 1995;140:537–548. doi: 10.1093/genetics/140.2.537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser VB, Charlesworth B. Muller's ratchet and the degeneration of the Drosophila miranda neo-Y chromosome. Genetics. 2010;185:339–348. doi: 10.1534/genetics.109.112789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kambris Z, Cook PE, Phuc HK, Sinkins SP. Immune activation by life-shortening Wolbachia and reduced filarial competence in mosquitoes. Science. 2009;326:134–136. doi: 10.1126/science.1177531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan NL, Hudson RR, Langley CH. The “hitchhiking effect” revisited. Genetics. 1989;123:887–899. doi: 10.1093/genetics/123.4.887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilpatrick ST, Rand DM. Conditional hitchhiking of mitochondrial DNA: frequency shifts of Drosophila melanogaster mtDNA variants depend on nuclear genetic background. Genetics. 1995;141:1113–1124. doi: 10.1093/genetics/141.3.1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M, Cui ML, Cubas P, et al. Regulatory genes control a key morphological and ecological trait transferred between species. Science. 2008;322:1116–1119. doi: 10.1126/science.1164371. [DOI] [PubMed] [Google Scholar]
- Kimura M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. Journal of Molecular Evolution. 1980;16:111–120. doi: 10.1007/BF01731581. [DOI] [PubMed] [Google Scholar]
- Lachaise D, Harry M, Solignac M. Evolutionary novelties in islands: Drosophila santomea, a new melanogaster sister species from Sao Tome. Proceedings of the Royal Society B-Biological Sciences. 2000;267:1487–1495. doi: 10.1098/rspb.2000.1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai Z, Gross BL, Zou Y, Andrews J, Rieseberg LH. Microarray analysis reveals differential gene expression in hybrid sunflower species. Molecular Ecology. 2006;15:1213–1227. doi: 10.1111/j.1365-294X.2006.02775.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landry CR, Hartl DL, Ranz JM. Genome clashes in hybrids: insights from gene expression. Heredity. 2007;99:483–493. doi: 10.1038/sj.hdy.6801045. [DOI] [PubMed] [Google Scholar]
- Lewontin RC, Birch LC. Hybridization as a source of variation for adaptation to new environments. Evolution. 1966;20:315–336. doi: 10.1111/j.1558-5646.1966.tb03369.x. [DOI] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25:1451–1452. doi: 10.1093/bioinformatics/btp187. [DOI] [PubMed] [Google Scholar]
- Llopart A, Elwyn S, Lachaise D, Coyne JA. Genetics of a difference in pigmentation between Drosophila yakuba and Drosophila santomea. Evolution. 2002;56:2262–2277. doi: 10.1111/j.0014-3820.2002.tb00150.x. [DOI] [PubMed] [Google Scholar]
- Llopart A, Lachaise D, Coyne JA. An anomalous hybrid zone in Drosophila. Evolution. 2005a;59:2602–2607. [PubMed] [Google Scholar]
- Llopart A, Lachaise D, Coyne JA. Multilocus analysis of introgression between two sympatric sister species of Drosophila Drosophila yakuba and D. santomea. Genetics. 2005b;171:197–210. doi: 10.1534/genetics.104.033597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loewe L. Quantifying the genomic decay paradox due to Muller's ratchet in human mitochondrial DNA. Genetical Research. 2006;87:133–159. doi: 10.1017/S0016672306008123. [DOI] [PubMed] [Google Scholar]
- Lynch M, Gabriel W. Mutation load and the survival of small populations. Evolution. 1990;44:1725–1737. doi: 10.1111/j.1558-5646.1990.tb05244.x. [DOI] [PubMed] [Google Scholar]
- Lynch M, Burger R, Butcher D, Gabriel W. The mutational meltdown in asexual populations. Journal of Heredity. 1993;84:339–344. doi: 10.1093/oxfordjournals.jhered.a111354. [DOI] [PubMed] [Google Scholar]
- Machado CA, Hey J. The causes of phylogenetic conflict in a classic Drosophila species group. Proceedings of the Royal Society B-Biological Sciences. 2003;270:1193–1202. doi: 10.1098/rspb.2003.2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machado CA, Kliman RM, Markert JA, Hey J. Inferring the history of speciation from multilocus DNA sequence data: the case of Drosophila pseudoobscura and close relatives. Molecular Biology and Evolution. 2002;19:472–488. doi: 10.1093/oxfordjournals.molbev.a004103. [DOI] [PubMed] [Google Scholar]
- MacRae AF, Anderson WW. Evidence for non-neutrality of mitochondrial DNA haplotypes in Drosophila pseudoobscura. Genetics. 1988;120:485–494. doi: 10.1093/genetics/120.2.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallet J. Hybridization as an invasion of the genome. Trends in Ecology & Evolution. 2005;20:229–237. doi: 10.1016/j.tree.2005.02.010. [DOI] [PubMed] [Google Scholar]
- Matute DR. Reinforcement of gametic isolation in Drosophila. PLoS Biology. 2010;8:e1000341. doi: 10.1371/journal.pbio.1000341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matute DR, Coyne JA. Intrinsic reproductive isolation between two sister species of Drosophila. Evolution. 2009;64:903–920. doi: 10.1111/j.1558-5646.2009.00879.x. [DOI] [PubMed] [Google Scholar]
- Maynard Smith J. The Evolution of Sex. Cambridge, UK: Cambridge University Press; 1978. [Google Scholar]
- Maynard Smith J, Haigh J. The hitch-hiking effect of a favorable gene. Genetical Research. 1974;23:23–35. [PubMed] [Google Scholar]
- Moehring AJ, Llopart A, Elwyn S, Coyne JA, Mackay TF. The genetic basis of postzygotic reproductive isolation between Drosophila santomea and D. yakuba due to hybrid male sterility. Genetics. 2006a;173:225–233. doi: 10.1534/genetics.105.052985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moehring AJ, Llopart A, Elwyn S, Coyne JA, Mackay TF. The genetic basis of prezygotic reproductive isolation between Drosophila santomea and D. yakuba due to mating preference. Genetics. 2006b;173:215–223. doi: 10.1534/genetics.105.052993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriyama EN, Powell JR. Synonymous substitution rates in Drosophila: mitochondrial versus nuclear genes. Journal of Molecular Evolution. 1997;45:378–391. doi: 10.1007/pl00006243. [DOI] [PubMed] [Google Scholar]
- Muller HJ. The relation of recombination to mutational advance. Mutation Research. 1964;106:2–9. doi: 10.1016/0027-5107(64)90047-8. [DOI] [PubMed] [Google Scholar]
- Nabholz B, Ellegren H, Wolf JB. High levels of gene expression explain the strong evolutionary constraint of mitochondrial protein-coding genes. Molecular Biology and Evolution. 2013;30:272–284. doi: 10.1093/molbev/mss238. [DOI] [PubMed] [Google Scholar]
- Nielsen R, Wakeley J. Distinguishing migration from isolation: a Markov chain Monte Carlo approach. Genetics. 2001;158:885–896. doi: 10.1093/genetics/158.2.885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nigro L, Prout T. Is there selection on RFLP differences in mitochondrial DNA? Genetics. 1990;125:551–555. doi: 10.1093/genetics/125.3.551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunes MD, Wengel PO, Kreissl M, Schlotterer C. Multiple hybridization events between Drosophila simulans and Drosophila mauritiana are supported by mtDNA introgression. Molecular Ecology. 2010;19:4695–4707. doi: 10.1111/j.1365-294X.2010.04838.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr HA. The population genetics of adaptation: the adaptation of DNA sequences. Evolution. 2002;56:1317–1330. doi: 10.1111/j.0014-3820.2002.tb01446.x. [DOI] [PubMed] [Google Scholar]
- Orr HA. The genetic theory of adaptation: a brief history. Nature Reviews Genetics. 2005;6:119–127. doi: 10.1038/nrg1523. [DOI] [PubMed] [Google Scholar]
- Orr HA, Betancourt AJ. Haldane's sieve and adaptation from the standing genetic variation. Genetics. 2001;157:875–884. doi: 10.1093/genetics/157.2.875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peter BM, Huerta-Sanchez E, Nielsen R. Distinguishing between selective sweeps from standing variation and from a de novo mutation. PLoS Genetics. 2012;8:e1003011. doi: 10.1371/journal.pgen.1003011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell JR. Interspecific cytoplasmic gene flow in the absence of nuclear gene flow: evidence from Drosophila. Proceedings of the National Academy of Sciences USA. 1983;80:492–495. doi: 10.1073/pnas.80.2.492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Przeworski M. The signature of positive selection at randomly chosen loci. Genetics. 2002;160:1179–1189. doi: 10.1093/genetics/160.3.1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieseberg LH. Evolution: replacing genes and traits through hybridization. Current Biology. 2009;19:R119–R122. doi: 10.1016/j.cub.2008.12.016. [DOI] [PubMed] [Google Scholar]
- Rieseberg L. Adaptive introgression: the seeds of resistance. Current Biology. 2011;21:R581–R583. doi: 10.1016/j.cub.2011.06.038. [DOI] [PubMed] [Google Scholar]
- Rieseberg LH, Wendel JF. Introgression and its consequences in plants. In: Harrison RG, editor. Hybrid Zones and the Evolutionary Process. New York: Oxford University Press; 1993. pp. 70–109. [Google Scholar]
- Rieseberg LH, Archer MA, Wayne RK. Transgressive segregation, adaptation and speciation. Heredity. 1999;83(Pt 4):363–372. doi: 10.1038/sj.hdy.6886170. [DOI] [PubMed] [Google Scholar]
- Rieseberg LH, Widmer A, Arntz AM, Burke JM. The genetic architecture necessary for transgressive segregation is common in both natural and domesticated populations. Philosophical Transactions of the Royal Society B. 2003;358:1141–1147. doi: 10.1098/rstb.2003.1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitou N, Nei M. The Neighbor-joining method: a new method for reconstructing phylogenetic trees. Molecular Biology and Evolution. 1987;4:404–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- Scribner KT, Avise JC. Cytonuclear genetics of experimental fish hybrid zones inside Biosphere 2. Proceedings of the National Academy of Sciences USA. 1994;91:5066–5069. doi: 10.1073/pnas.91.11.5066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seehausen O. Hybridization and adaptive radiation. Trends in Ecology & Evolution. 2004;19:198–207. doi: 10.1016/j.tree.2004.01.003. [DOI] [PubMed] [Google Scholar]
- Sella G, Petrov DA, Przeworski M, Andolfatto P. Pervasive natural selection in the Drosophila genome? PLoS Genetics. 2009;5:e1000495. doi: 10.1371/journal.pgen.1000495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y, Endepols S, Klemann N, et al. Adaptive introgression of anticoagulant rodent poison resistance by hybridization between old world mice. Current Biology. 2011;21:1296–1301. doi: 10.1016/j.cub.2011.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staubach F, Lorenc A, Messer PW, et al. Genome patterns of selection and introgression of haplotypes in natural populations of the house mouse (Mus musculus. PLoS Genetics. 2012;8:e1002891. doi: 10.1371/journal.pgen.1002891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stebbins GL. The role of hybridization in evolution. Proceedings of the American Philosophical Society. 1959;103:231–251. [Google Scholar]
- Stephan W, Xing L, Kirby DA, Braverman JM. A test of the background selection hypothesis based on nucleotide data from Drosophila ananassae. Proceedings of the National Academy of Sciences USA. 1998;95:5649–5654. doi: 10.1073/pnas.95.10.5649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123:585–595. doi: 10.1093/genetics/123.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahata N, Slatkin M. Mitochondrial gene flow. Proceedings of the National Academy of Sciences USA. 1984;81:1764–1767. doi: 10.1073/pnas.81.6.1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Subramanian S, Kumar S. Temporal patterns of fruit fly (Drosophila) evolution revealed by mutation clocks. Molecular Biology and Evolution. 2004;21:36–44. doi: 10.1093/molbev/msg236. [DOI] [PubMed] [Google Scholar]
- Tamura K, Peterson D, Peterson N, et al. MEGA5: Molecular Evolutionary Genetics Analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Molecular Biology and Evolution. 2011;28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teeter KC, Payseur BA, Harris LW, et al. Genome-wide patterns of gene flow across a house mouse hybrid zone. Genome Research. 2008;18:67–76. doi: 10.1101/gr.6757907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teixeira L, Ferreira A, Ashburner M. The bacterial symbiont Wolbachia induces resistance to RNA viral infections in Drosophila melanogaster. PLoS Biology. 2008;6:e2. doi: 10.1371/journal.pbio.1000002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Heliconius. Butterfly genome reveals promiscuous exchange of mimicry adaptations among species. Nature. 2012;487:94–98. doi: 10.1038/nature11041. Genome Consortium. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Research. 1997;25:4876–4882. doi: 10.1093/nar/25.24.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turelli M, Hoffmann AA. Rapid spread of an inherited incompatibility factor in California Drosophila. Nature. 1991;353:440–442. doi: 10.1038/353440a0. [DOI] [PubMed] [Google Scholar]
- Turelli M, Hoffmann AA, McKechnie SW. Dynamics of cytoplasmic incompatibility and mtDNA variation in natural Drosophila simulans populations. Genetics. 1992;132:713–723. doi: 10.1093/genetics/132.3.713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitney KD, Randell RA, Rieseberg LH. Adaptive introgression of herbivore resistance traits in the weedy sunflower Helianthus annuus. American Naturalist. 2006;167:794–807. doi: 10.1086/504606. [DOI] [PubMed] [Google Scholar]
- Yang Z. PAML: a program package for phylogenetic analysis by maximum likelihood. Computer Applications in Biosciences. 1997;13:555–556. doi: 10.1093/bioinformatics/13.5.555. [DOI] [PubMed] [Google Scholar]
- Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Molecular Biology and Evolution. 2007;24:1586–1591. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- Zabalou S, Charlat S, Nirgianaki A, et al. Natural Wolbachia infections in the Drosophila yakuba species complex do not induce cytoplasmic incompatibility but fully rescue the wRi modification. Genetics. 2004;167:827–834. doi: 10.1534/genetics.103.015990. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Detailed description of the lines used in this study.
Data Availability Statement
DNA sequences: GenBank/EMBL accession numbers KF824856–KF824901. IMa2 and MEGA input files: Dryad doi:10.5061/dryad.9kh1b.