

Abstract
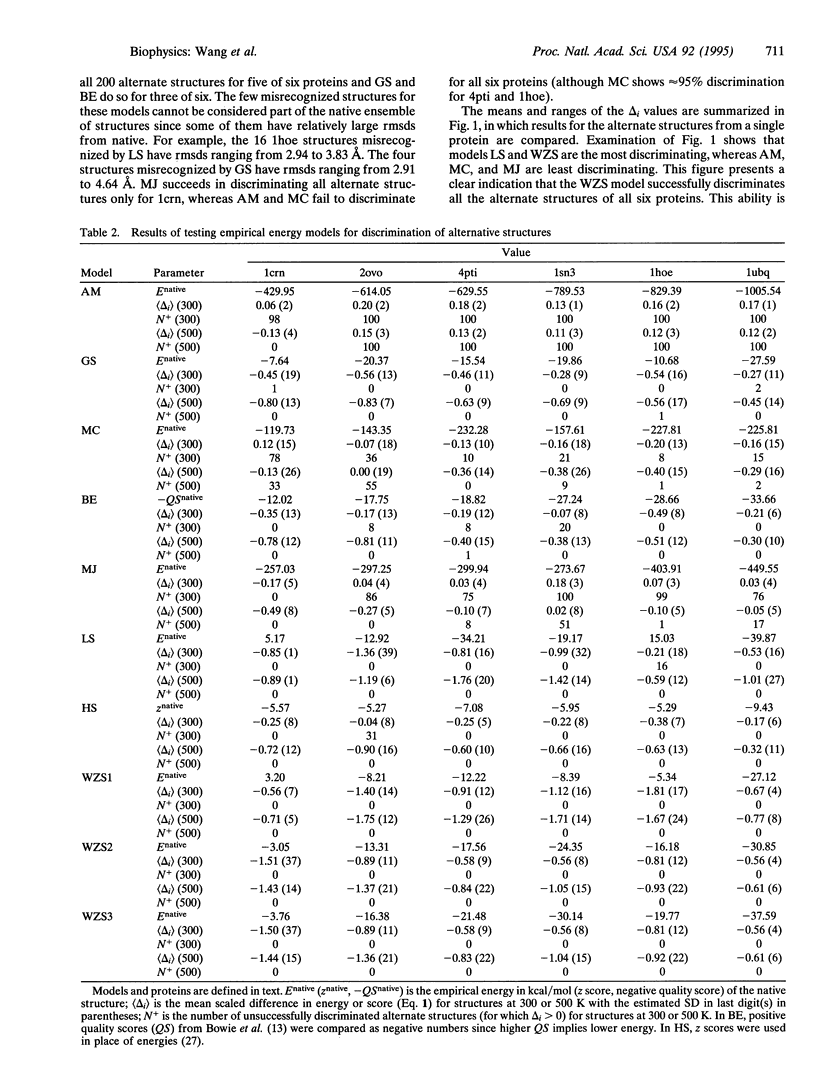
Prediction of the native tertiary structure of a globular protein from the primary sequence will require a potential energy model that can discriminate all nonnative structures from the native structure(s). A successful model must distinguish not only alternate structures that are very nonnative but also alternate structures that are compact and near-native. We describe here a method, based on molecular dynamics simulation, that allows generation of hundreds of compact alternate structures that are arbitrarily close to the native structure. In this way, a significant amount of conformational space in the neighborhood of the native structure can be sampled and these alternate structures can be used as a stringent test of protein folding models. We have used two sets of these alternate structures generated for six crystallographically characterized small globular proteins (1200 alternate structures in all) to test eight empirical energy models for their ability to discriminate alternate from native structures. Seven of the models fail to correctly identify at least some of the alternate structures as nonnative. An atomic solvation model is presented that succeeds in discriminating all 1200 alternate structures from native.
Full text
PDF



Images in this article

Selected References
These references are in PubMed. This may not be the complete list of references from this article.
- Almassy R. J., Fontecilla-Camps J. C., Suddath F. L., Bugg C. E. Structure of variant-3 scorpion neurotoxin from Centruroides sculpturatus Ewing, refined at 1.8 A resolution. J Mol Biol. 1983 Oct 25;170(2):497–527. doi: 10.1016/s0022-2836(83)80159-4. [DOI] [PubMed] [Google Scholar]
- Bode W., Epp O., Huber R., Laskowski M., Jr, Ardelt W. The crystal and molecular structure of the third domain of silver pheasant ovomucoid (OMSVP3). Eur J Biochem. 1985 Mar 1;147(2):387–395. doi: 10.1111/j.1432-1033.1985.tb08762.x. [DOI] [PubMed] [Google Scholar]
- Bowie J. U., Lüthy R., Eisenberg D. A method to identify protein sequences that fold into a known three-dimensional structure. Science. 1991 Jul 12;253(5016):164–170. doi: 10.1126/science.1853201. [DOI] [PubMed] [Google Scholar]
- Bryant S. H., Lawrence C. E. An empirical energy function for threading protein sequence through the folding motif. Proteins. 1993 May;16(1):92–112. doi: 10.1002/prot.340160110. [DOI] [PubMed] [Google Scholar]
- Covell D. G. Lattice model simulations of polypeptide chain folding. J Mol Biol. 1994 Jan 21;235(3):1032–1043. doi: 10.1006/jmbi.1994.1055. [DOI] [PubMed] [Google Scholar]
- Eisenberg D., McLachlan A. D. Solvation energy in protein folding and binding. Nature. 1986 Jan 16;319(6050):199–203. doi: 10.1038/319199a0. [DOI] [PubMed] [Google Scholar]
- Fetrow J. S., Bryant S. H. New programs for protein tertiary structure prediction. Biotechnology (N Y) 1993 Apr;11(4):479–484. doi: 10.1038/nbt0493-479. [DOI] [PubMed] [Google Scholar]
- Godzik A., Kolinski A., Skolnick J. Topology fingerprint approach to the inverse protein folding problem. J Mol Biol. 1992 Sep 5;227(1):227–238. doi: 10.1016/0022-2836(92)90693-e. [DOI] [PubMed] [Google Scholar]
- Goldstein R. A., Luthey-Schulten Z. A., Wolynes P. G. Optimal protein-folding codes from spin-glass theory. Proc Natl Acad Sci U S A. 1992 Jun 1;89(11):4918–4922. doi: 10.1073/pnas.89.11.4918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein R. A., Luthey-Schulten Z. A., Wolynes P. G. Protein tertiary structure recognition using optimized Hamiltonians with local interactions. Proc Natl Acad Sci U S A. 1992 Oct 1;89(19):9029–9033. doi: 10.1073/pnas.89.19.9029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendlich M., Lackner P., Weitckus S., Floeckner H., Froschauer R., Gottsbacher K., Casari G., Sippl M. J. Identification of native protein folds amongst a large number of incorrect models. The calculation of low energy conformations from potentials of mean force. J Mol Biol. 1990 Nov 5;216(1):167–180. doi: 10.1016/S0022-2836(05)80068-3. [DOI] [PubMed] [Google Scholar]
- Hinds D. A., Levitt M. A lattice model for protein structure prediction at low resolution. Proc Natl Acad Sci U S A. 1992 Apr 1;89(7):2536–2540. doi: 10.1073/pnas.89.7.2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm L., Sander C. Evaluation of protein models by atomic solvation preference. J Mol Biol. 1992 May 5;225(1):93–105. doi: 10.1016/0022-2836(92)91028-n. [DOI] [PubMed] [Google Scholar]
- Jones D. T., Taylor W. R., Thornton J. M. A new approach to protein fold recognition. Nature. 1992 Jul 2;358(6381):86–89. doi: 10.1038/358086a0. [DOI] [PubMed] [Google Scholar]
- Kang H. S., Kurochkina N. A., Lee B. Estimation and use of protein backbone angle probabilities. J Mol Biol. 1993 Jan 20;229(2):448–460. doi: 10.1006/jmbi.1993.1045. [DOI] [PubMed] [Google Scholar]
- Kocher J. P., Rooman M. J., Wodak S. J. Factors influencing the ability of knowledge-based potentials to identify native sequence-structure matches. J Mol Biol. 1994 Feb 4;235(5):1598–1613. doi: 10.1006/jmbi.1994.1109. [DOI] [PubMed] [Google Scholar]
- Lüthy R., Bowie J. U., Eisenberg D. Assessment of protein models with three-dimensional profiles. Nature. 1992 Mar 5;356(6364):83–85. doi: 10.1038/356083a0. [DOI] [PubMed] [Google Scholar]
- Maiorov V. N., Crippen G. M. Contact potential that recognizes the correct folding of globular proteins. J Mol Biol. 1992 Oct 5;227(3):876–888. doi: 10.1016/0022-2836(92)90228-c. [DOI] [PubMed] [Google Scholar]
- Novotný J., Bruccoleri R., Karplus M. An analysis of incorrectly folded protein models. Implications for structure predictions. J Mol Biol. 1984 Aug 25;177(4):787–818. doi: 10.1016/0022-2836(84)90049-4. [DOI] [PubMed] [Google Scholar]
- Novotný J., Rashin A. A., Bruccoleri R. E. Criteria that discriminate between native proteins and incorrectly folded models. Proteins. 1988;4(1):19–30. doi: 10.1002/prot.340040105. [DOI] [PubMed] [Google Scholar]
- Ooi T., Oobatake M., Némethy G., Scheraga H. A. Accessible surface areas as a measure of the thermodynamic parameters of hydration of peptides. Proc Natl Acad Sci U S A. 1987 May;84(10):3086–3090. doi: 10.1073/pnas.84.10.3086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouzounis C., Sander C., Scharf M., Schneider R. Prediction of protein structure by evaluation of sequence-structure fitness. Aligning sequences to contact profiles derived from three-dimensional structures. J Mol Biol. 1993 Aug 5;232(3):805–825. doi: 10.1006/jmbi.1993.1433. [DOI] [PubMed] [Google Scholar]
- Pflugrath J. W., Wiegand G., Huber R., Vértesy L. Crystal structure determination, refinement and the molecular model of the alpha-amylase inhibitor Hoe-467A. J Mol Biol. 1986 May 20;189(2):383–386. doi: 10.1016/0022-2836(86)90520-6. [DOI] [PubMed] [Google Scholar]
- Ramachandran G. N., Sasisekharan V. Conformation of polypeptides and proteins. Adv Protein Chem. 1968;23:283–438. doi: 10.1016/s0065-3233(08)60402-7. [DOI] [PubMed] [Google Scholar]
- Rooman M. J., Kocher J. P., Wodak S. J. Extracting information on folding from the amino acid sequence: accurate predictions for protein regions with preferred conformation in the absence of tertiary interactions. Biochemistry. 1992 Oct 27;31(42):10226–10238. doi: 10.1021/bi00157a009. [DOI] [PubMed] [Google Scholar]
- Rooman M. J., Kocher J. P., Wodak S. J. Prediction of protein backbone conformation based on seven structure assignments. Influence of local interactions. J Mol Biol. 1991 Oct 5;221(3):961–979. doi: 10.1016/0022-2836(91)80186-x. [DOI] [PubMed] [Google Scholar]
- Sali A., Shakhnovich E., Karplus M. Kinetics of protein folding. A lattice model study of the requirements for folding to the native state. J Mol Biol. 1994 Feb 4;235(5):1614–1636. doi: 10.1006/jmbi.1994.1110. [DOI] [PubMed] [Google Scholar]
- Shakhnovich E, Farztdinov G, Gutin AM, Karplus M. Protein folding bottlenecks: A lattice Monte Carlo simulation. Phys Rev Lett. 1991 Sep 16;67(12):1665–1668. doi: 10.1103/PhysRevLett.67.1665. [DOI] [PubMed] [Google Scholar]
- Sikorski A., Skolnick J. Monte Carlo simulation of equilibrium globular protein folding: alpha-helical bundles with long loops. Proc Natl Acad Sci U S A. 1989 Apr;86(8):2668–2672. doi: 10.1073/pnas.86.8.2668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sippl M. J. Calculation of conformational ensembles from potentials of mean force. An approach to the knowledge-based prediction of local structures in globular proteins. J Mol Biol. 1990 Jun 20;213(4):859–883. doi: 10.1016/s0022-2836(05)80269-4. [DOI] [PubMed] [Google Scholar]
- Sippl M. J. Recognition of errors in three-dimensional structures of proteins. Proteins. 1993 Dec;17(4):355–362. doi: 10.1002/prot.340170404. [DOI] [PubMed] [Google Scholar]
- Sippl M. J., Weitckus S. Detection of native-like models for amino acid sequences of unknown three-dimensional structure in a data base of known protein conformations. Proteins. 1992 Jul;13(3):258–271. doi: 10.1002/prot.340130308. [DOI] [PubMed] [Google Scholar]
- Skolnick J., Kolinski A. Dynamic Monte Carlo simulations of a new lattice model of globular protein folding, structure and dynamics. J Mol Biol. 1991 Sep 20;221(2):499–531. doi: 10.1016/0022-2836(91)80070-b. [DOI] [PubMed] [Google Scholar]
- Skolnick J., Kolinski A. Simulations of the folding of a globular protein. Science. 1990 Nov 23;250(4984):1121–1125. doi: 10.1126/science.250.4984.1121. [DOI] [PubMed] [Google Scholar]
- Tanaka S., Scheraga H. A. Medium- and long-range interaction parameters between amino acids for predicting three-dimensional structures of proteins. Macromolecules. 1976 Nov-Dec;9(6):945–950. doi: 10.1021/ma60054a013. [DOI] [PubMed] [Google Scholar]
- Teeter M. M. Water structure of a hydrophobic protein at atomic resolution: Pentagon rings of water molecules in crystals of crambin. Proc Natl Acad Sci U S A. 1984 Oct;81(19):6014–6018. doi: 10.1073/pnas.81.19.6014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijay-Kumar S., Bugg C. E., Cook W. J. Structure of ubiquitin refined at 1.8 A resolution. J Mol Biol. 1987 Apr 5;194(3):531–544. doi: 10.1016/0022-2836(87)90679-6. [DOI] [PubMed] [Google Scholar]
- Wesson L., Eisenberg D. Atomic solvation parameters applied to molecular dynamics of proteins in solution. Protein Sci. 1992 Feb;1(2):227–235. doi: 10.1002/pro.5560010204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilmanns M., Eisenberg D. Three-dimensional profiles from residue-pair preferences: identification of sequences with beta/alpha-barrel fold. Proc Natl Acad Sci U S A. 1993 Feb 15;90(4):1379–1383. doi: 10.1073/pnas.90.4.1379. [DOI] [PMC free article] [PubMed] [Google Scholar]