

Abstract
Adoptive T cell therapy can target and kill widespread malignant cells thereby inducing durable clinical responses in melanoma and selected other malignances. However, many commonly targeted tumor antigens are also expressed by healthy tissues, and T cells do not distinguish between benign and malignant tissues if both express the target antigen. As such, autoimmune toxicity from T-cell-mediated destruction of normal tissue has limited the development and adoption of this otherwise promising type of cancer therapy. A review of the unique biology of T-cell therapy and of recent clinical experience compels a reassessment of target antigens that traditionally have been viewed from the perspective of weaker immunotherapeutic modalities. In selecting target antigens for adoptive T-cell therapy, expression by tumors and not by essential healthy tissues is of paramount importance. The risk of autoimmune adverse events can be further mitigated by generating antigen receptors using strategies that reduce the chance of cross-reactivity against epitopes in unintended targets. In general, a circumspect approach to target selection and thoughtful preclinical and clinical studies are pivotal to the ongoing advancement of these promising treatments.
The goal of cancer therapy is to cure the patient by killing every malignant cell while preserving vital normal tissues. Chemotherapy seeks to accomplish this by interfering with processes that are more crucial to tumors than to normal tissues. As such it is dose-limited by normal tissue toxicity and, with few exceptions, fails to cure patients with advanced disease. Although immunotherapy can specifically target tumors to the exclusion of normal tissue, it is noteworthy that thus far most targeted tumor antigens are also expressed, at least to some degree, by normal tissues1. The most frequently discussed modality for specifically targeting tumor antigens, cancer vaccines, lacks the potency to induce either regression of macroscopic tumors or injury to normal tissue2. Thus, clinical experience with cancer vaccines offers little insight into the clinical consequences of robustly targeting various antigens.
In contrast, adoptive T cell therapy (ACT)—the injection of a large number of activated tumor-specific T cells—can induce complete and durable regression of certain advanced cancers3–6. ACT can be directed against diverse target antigens by genetically engineering autologous T cells to express particular TCRs or chimeric antigen receptor (CARs). In this non-physiological scenario, virtually all injected cells bear receptors capable of recognizing—often with particularly high affinity—the target antigen. In addition, high numbers of these unusually avid T cells are typically administered to lymphoconditioned patients with no circulating leukocytes, very few regulatory cells and higher than normal amounts of cytokines that promote T cell survival7–9. As a result, the anti-tumor responses can be impressive; unfortunately, as revealed during recent clinical trials, so too can the unwanted and unanticipated autoimmune adverse events resulting from T cell recognition of antigens expressed by normal tissues (Supplemental Table 1, Figure 1)10–13.
Figure 1.

Autoimmune adverse events in ACT clinical trials. (a) Skin rash at various time points after treatment of a melanoma patient with T cells engineered to express a TCR with high affinity for MART1. (b) Cellular anterior chamber infiltrate in a melanoma patient two weeks after treatment with T cells engineered to express a TCR with high affinity for MART1 (left). Aymptomatic posterior synechiae six months after treatment of the same patient (right)10. (c) Immunohistochemical (IHC) analysis of CD8 expression in a liver biopsy obtained four days after treatment of a renal cell carcinoma patient with T cells transduced with a CAR specific for carbonic anhydrase IX. CD8 T cells line the basal side of (arrowheads) and infiltrate (arrow) the bile duct epithelium. Liver parenchyma (L), portal triangle (P), and bile duct (B) are labeled22. (d) Colonoscopy images from a colon cancer patient at various time points after administration of T cells engineered to express a TCR specific for carcinoembryonic antigen (CEA). Transient severe colitis is evident11. (e) Magnetic resonance imaging of the brain of a melanoma patient after various time points after injection of T cells expressing a receptor that recognizes MAGE-A3 but is cross-reactive with MAGE-A1212. Images show progressive white matter changes consistent with leukomalacia.
In this review, we survey the beneficial and detrimental outcomes of recent clinical trials testing genetically engineered T cells against varied tumor antigens. We discuss challenges to safe clinical testing of ACT and the implications of those challenges for selection of tumor target antigens. Finally, we look critically at commonly studied tumor antigens and propose strategies to select target antigens that are most likely to maximize the benefits and minimize the toxicities of ACT.
Lessons from clinical trials
Studies in mice and humans have illustrated the ability of adoptively transferred T cells to localize to and kill target cells expressing cognate antigen, regardless of anatomical site. For example, in experiments performed by our group, mice injected with T cells targeting gp100 (a melanocyte differentiation antigen expressed by both normal melanocytes and the B16 melanoma line), the T cells traffic indiscriminately to all tissues. However, the T cells display effector function only in tissues expressing gp100, indicating that while trafficking is ubiquitous, killing is antigen-specific14. In this mouse model, tumor regression is tightly correlated with autoimmunity in the skin and eye10,15,16. Clinical trials of T cells targeting gp100 and another melanocyte differentiation antigen, MART-1, also resulted in melanoma tumor regression and toxicity to melanocytes in the skin, eye, and ears (Supplemental Table 1, Figure 1a,b)10. Although these toxicities were mostly transient, and eye and ear inflammation could be treated with local steroids, they were common (16/20 patients treated with T cells targeting MART-1, 16/16 patients treated with T cells targeting gp100). In addition, these worrisome autoimmune manifestations were not offset by impressive tumor responses; tumor response rates were modest (6/20 patients treated with T cells targeting MART-1, 3/16 patients treated with T cells targeting gp100), and complete responses were extremely rare (0/20 patients treated with T cells targeting MART1, 1/16 patients treated with T cells targeting gp100) for these protocols. Thus, T cell targeting of two of the melanocyte differentiation antigens resulted in abundant autoimmune toxicity but limited efficacy.
The results of additional clinical trials suggest that autoimmune toxicities encountered with the melanocyte differentiation antigens are generalizable to other tissues that express antigens also expressed on tumors. Carbonic anhydrase IX (CAIX) is markedly overexpressed by some renal cell carcinomas but is also expressed by normal liver, small intestine, and gastric mucosa17–19. CAIX has been targeted with monoclonal antibodies and radioimmunotherapy without serious autoimmune toxicities but also without efficacy20. A clinical trial of T cells expressing a CAIX-specific CAR21,22 did not report objective tumor regression but did report elevated liver enzymes and cholangitis that necessitated cessation of cell infusions, administration of steroids, and reductions in cell dose. A liver biopsy from one patient revealed expression of CAIX in bile duct epithelial cells, and cholangitis with T-cell infiltration around the bile ducts (Figure 1c). Pretreatment of a cohort of patients with anti-CAIX antibody that blocks the CAR from interacting with its target prevented hepatotoxicity22. These findings suggest that CAR-expressing T cells caused the autoimmune cholangitis..
A similar phenomenon was observed in clinical trials using T cells targeting carcinoembryonic antigen (CEA). CEA is present in normal tissue throughout much of the digestive tract but is overexpressed in cancers arising from the colon23–25. It is a popular target for cancer vaccines, which can elicit immune responses against the protein but do not cause significant autoimmune toxicities or reproducible objective tumor responses26–28. However, adoptive transfer of T cells expressing a TCR having high affinity for CEA resulted in a partial tumor response in one of three patients with metastatic colorectal cancer. Unfortunately, treatment was complicated by dose-limiting diarrhea due to autoimmune colitis that occurred after infusions of relatively low numbers of T cells (2–4 x 108) in all three patients; this effect that was consistent with observations in an analogous mouse model (Supplemental Table 1, Figure 1e)11,29.
Another common target of vaccines and antibodies, the receptor tyrosine-protein kinase erbB-2 (HER2), has also been targeted with ACT. This growth receptor is amplified in a variety of cancers including breast and gastric cancer but also is expressed by normal tissues including cardiopulmonary and digestive tract organs24,25,30,31. A United States Food and Drug Administration approved antibody targeting HER2 has been used extensively and safely in patients, but with noteworthy cardiac toxicity. Cancer vaccines against HER2 have been tested in clinical trials without serious autoimmune adverse events and without robust tumor regression 32. A single patient treated with T cells expressing a HER2-targeting CAR experienced cytokine release syndrome that resulted in death13. Whether the distribution of HER2 on the patient’s heart and lung contributed to this serious adverse event cannot be determined definitively, but given the autoimmune toxicities resulting from targeting other self-antigens, it is a possibility.
Other incidences of direct T cell-mediated damage to normal tissue resulted from unanticipated cross-reactivity of TCRs with antigens other than the intended target antigen (Figure 2). For example MAGE-A3 is a cancer testis antigen (CTA), a class of antigens that in adults are normally expressed predominantly by germ cells, but that can be activated in certain cancers. MAGE-A3 is expressed by a wide variety of cancers and has been targeted with cancer vaccines without significant autoimmunity.33,34 MAGE-A3 vaccination of a HLA-A2 transgenic mouse was used to isolate a TCR recognizing an HLA-A2-restricted MAGE-A3 peptide12. Affinity of the TCR for its target peptide/HLA complex was enhanced by an amino acid substitution in the complementarity determining region 2α35. Nine patients (seven diagnosed with melanoma, and one each with synovial sarcoma and esophageal carcinoma) were treated with autologous T cells engineered to express this MAGE-A3-targeting TCR; five experienced objective tumor responses, two of which (one a complete response) were ongoing after more than 12 months. However, four patients developed severe neurological toxicities (Supplemental Table 1, Figure 1f). Careful analysis revealed TCR cross-reactivity with a non-identical epitope of MAGE-A12, and expression of MAGE-A12 in the brain. The targeted epitope was also detected within MAGE-A9, and possible low-level brain expression of MAGE-A9 was noted. A second clinical trial with T cells expressing a different MAGE-A3-targeted TCR (HLA-A1-restricted and affinity enhanced by four substitutions in the complementarity determining region 2α) also resulted in unexpected severe toxicity36. The first two patients (one diagnosed with melanoma and the other with myeloma) treated on this protocol developed fatal cardiac toxicity. These cardiac events were caused by direct cardiac muscle injury inflicted by the injected T cells, and these T cells were found to be cross-reactive with titin, a striated muscle-specific protein expressed in normal myocardium36,37.
Figure 2.

Scenarios in which TCRs and CARs can recognize and cross-react with unintended target antigens120,121. (a-d) TCRs recognize target antigen peptides bound to MHC molecules. (a) The ideal tumor antigen targeted by adoptively transferred T cells is a peptide-MHC complex that is expressed uniquely by tumor cells and is not expressed by healthy tissue. (b-f) However, even T cells engineered to target such ideal tumor antigens may recognize and damage healthy tissue in certain scenarios. (b) Healthy tissue may express distinct antigens that contain peptide epitopes identical to the one recognized by the engineered T cells. Vigilance for this type of cross-reactivity is particularly important when targeting peptides in antigens encoded by genes arising from gene duplication such as some CTA family members (e.g. MAGE). (c) Healthy tissue may express distinct antigens that contain peptide epitopes that are different in sequence than the intended target peptide, but sufficiently similar in structure to bind to the same MHC molecule and be recognized by the TCR on the engineered T cells. (d) Healthy tissue may express complexes of different MHC molecules with peptides from non-target antigens that are cross-recognized by the engineered TCR. (e-g) CARs directly engage intact target antigens, rather than antigen-derived peptides bound to MHC molecules. (e) An ideal CAR for ACT specifically recognizes a single epitope that is unique to a tumor-restricted antigen. (f-g) However, in some scenarios even T cells engineered to express such ideal CARs can recognize unintended target antigens. (f) Non-target antigens expressed by healthy tissues may contain epitopes identical to the intended CAR target and therefore be recognized by the CAR-expressing T cells. (g) Antibody specificity for an epitope can be imperfect, permitting structurally similar (but different in sequence) epitopes present on non-target antigens to be cross-recognized by a CAR targeting a tumor-specific antigen.
Taken together, these clinical trial results suggest that autoimmune toxicities result when critical normal tissues express antigens intentionally or unintentionally targeted by the injected T cells. In addition, they illustrate two important challenges to target antigen selection. First, one needs to rule out the possibility that the TCR or CAR on adoptively transferred T cells will cross-react with other self-antigens (Fig. 2). Second, one needs to detect and determine the clinical relevance of even very low levels of intended target antigen expression in normal tissues (Box 1). They also indicate that the risk of autoimmune toxicity may increase with use of TCRs that have not been subjected to thymic selection. These include TCRs obtained from immunized mice and TCRs that have been affinity-enhanced by complementarity determining region mutations; in both cases these TCRs have not been tested by normal negative selection on self-peptides expressed in the human thymus.
Box 1. Analyzing expression of candidate target antigens in healthy tissues.
When selecting candidate target antigens for T cell therapies and anticipating potential toxicities it is valuable to know if healthy tissues express the candidate target antigens. Unfortunately, current technologies are limited. cDNA microarray and RNA-seq can provide information about expression of a vast array of genes, and gene expression data for panels of healthy tissues are available through public databases such as BioGPS (www.biogps.org) and Genevestigator (www.genevestigator.com). However, these data are limited in that the minimum threshold for clinically significant expression levels is not known. Furthermore, a gene expressed by a rare but important population of cells in any given tissue may not be detected due to low overall representation of that cell’s mRNA in the tissue sample. This problem is exemplified by the expression of certain MAGE family members in the brain at marginally detectable but clinically relevant levels12. Another potential pitfall is that detection of antigen in some apparently indispensible tissues may not reflect its presence in vital cells. For example, B-cell maturation antigen (BCMA) is expressed in tissue samples from throughout the gastrointestinal tract but is restricted to plasma cells, which can be found in many tissues but are not vital47.
Immunohistochemistry (IHC)-mediated detection of protein expression can be invaluable in demonstrating which cells from particular tissues express the antigen of interest47. For some antigens, databases such as The Human Protein Atlas (http://www.proteinatlas.org) contain IHC data that clearly document tissue expression. However, the availability and reliability of antibodies able to reliably detect antigens varies and data are often unavailable or inconsistent. Furthermore, absence of detectable antigen expression does not confirm that the protein is never present in that tissue, as expression of certain genes is induced only under specific conditions36.
Adding to the challenge of ruling out candidate target antigen expression in healthy tissues is the lack of samples representing every human tissue. For some antigens, study of expression of orthologs in animal models may provide additional information. However this approach is complicated by species differences and is particularly difficult to apply to TCRs, which require species-specific MHC-dependent antigen presentation15,29. Thus, with existing technology and tissue samples, it is not possible to definitively confirm absence of a given antigen from crucial healthy tissues. As such, targeting antigens encoded by the normal human genome generally results in uncertainty about which healthy tissues are at risk of T cell-mediated attack. Carefully conducted clinical trials will be required to ultimately define the safety profile for each target antigen.
The studies described above raise serious concerns about autoimmune targeting of tissues essential for life. However, some healthy tissues or cell populations can be killed without major morbidity. For example, normal B cells and their precursors express CD19, which is also present on malignancies that arise from these cells38. Adoptive transfer of T cells expressing CARs that target CD19 can induce impressive tumor regression in patients with B-cell malignancies including follicular lymphoma, chronic lymphocytic leukemia (CLL), splenic marginal zone lymphoma, and pediatric and adult acute lymphoblastic leukemia (ALL) 5,39–43. However this tumor regression is accompanied by depletion of healthy B cells resulting in B cell cytopenia and reduced immunoglobulin levels. These effects are mostly well-tolerated (consistent with observations from therapeutic antibodies that target B cells), although immunoglobulin replacement may be required and the effects in pediatric patients are less studied (and potentially more concerning). Thus, in some scenarios engineered T cells may kill healthy cell populations or damage healthy tissues without resulting in major morbidity.
ACT can also result in a second type of toxicity, independent of the effects of healthy tissue destruction. This toxicity, which has been a serious problem in the clinic, stems from high concentrations of cytokines released by the engineered T cells. Elevated serum levels of interferon-γ, tumor necrosis factor-α (TNF-α), interleukin (IL)-2, IL-6 and a other cytokines have been detected in association with a suite of sometimes severe clinical signs and symptoms termed cytokine release syndrome (CRS)5,40–44. One clinical trial administered lymphoconditioning chemotherapy prior to infusion of T cells engineered to express a CD19-specific CAR that contained a CD28 costimulatory domain; after T cell infusion they injected high-dose bolus IL-2. This treatment resulted in consistent elevations in serum cytokine levels and frequent, severe adverse events that required protocol modifications (reducing the T-cell dose and eliminating IL-2 administration)5. In another clinical trial, three patients with CLL were treated with T cells engineered to express a CD19-specific CAR that contained a 41BB costimulatory domain; despite infusion of cells over a three-day period in an attempt to prevent sudden massive cytokine production, two patients developed what the investigators described as “… potentially dangerously high levels of cytokines,” accompanied by fever and rigors and associated with other adverse events41,42. One patient experienced dyspnea and cardiac dysfunction necessitating corticosteroids to suppress the activity of the transferred T cells. The same CAR construct was used in the treatment of two children with ALL40. Grade 3 or 4 (severe or life-threatening) febrile neutropenia, hypotension, capillary leak syndrome, acute respiratory distress syndrome, encephalopathy, and liver enzyme elevations were reported. One patient required glucocorticoids, etanercept (anti-TNF-α monoclonal antibodies), and tocilizumab (anti-IL-6 monoclonal antibodies) to ameliorate the toxicities of CRS. At another institution, two adult patients with ALL and bone marrow blasts who were treated with T cells engineered to express a CD19-targeted CAR containing a CD28 costimulatory domain suffered fevers, hypotension, and mental status changes that required high-dose lymphotoxic steroid therapy43. Thus, while loss of B cells has been well-tolerated, the indirect toxicities of CRS have been severe and dose-limiting.
Toxicities relating to cytokine release and tumor destruction may be an inevitable consequence of effective therapies mediated by engineered T cells, but it might be possible to mitigate these effects. For example, investigators are exploring the influence of particular signaling domains of CAR constructs (e.g. CD28 or 4-1BB) on T cell cytokine production. Also, the risks and benefits of lymphoconditioning regimens are not fully understood in this setting and protocols with and without pretreatment chemotherapy are ongoing. Finally, although B-cell loss is well-tolerated and suggests that tumor antigens shared by certain normal tissues can be reasonable targets, targeting antigens that are restricted to malignant cells and not expressed on healthy B-cell populations might circumvent both B-cell depletion and cytokine-related toxicities and is an important area of ongoing research45–47.
Increasing the safety of ACT
Given that it is difficult to exclude the possibility that some tumor antigens targeted by ACT may be expressed, at least to some degree, on healthy tissues (Box 1) methods of safely testing the potential toxicity of ACT are all the more important. However, intrinsic differences between traditional drugs (e.g. chemotherapeutics, small molecules and monoclonal antibodies) and T-cell therapies have important implications for phase I clinical trials aimed at testing safety and determining maximum tolerated dose (Fig. 3). In contrast to chemotherapeutics, which are produced in a standardized way and might be subjected to batch-to-batch variation but not patient-to-patient variation, T-cell therapies, because they are produced from patient cells, are inherently variable from patient-to-patient. These differences are compounded by the unpredictable but massive proliferation and long-term persistence of the T cells following infusion. As a result, drug exposure for T-cell therapy is highly variable and virtually limitless. Because of this variability, it is not clear that the maximum tolerated dose for one cohort of patients can be broadly applied to others. Further complicating phase 1 clinical testing of T-cell therapies, particularly those targeted to antigens expressed by vital tissues as well as tumors, is the inability to reliably terminate patient exposure to the transferred T cells. With traditional drugs, when adverse events occur drug dosing is stopped and drugs are eliminated predictably, allowing recovery of the injured tissue. In contrast, T cell-driven autoimmune toxicity may actually worsen as target antigen-expressing healthy tissue cells trigger a self-amplifying T cell response and ignite a feed forward loop of mutual T cell and antigen presenting cell activation that is not reliably extinguished.
Figure 3.
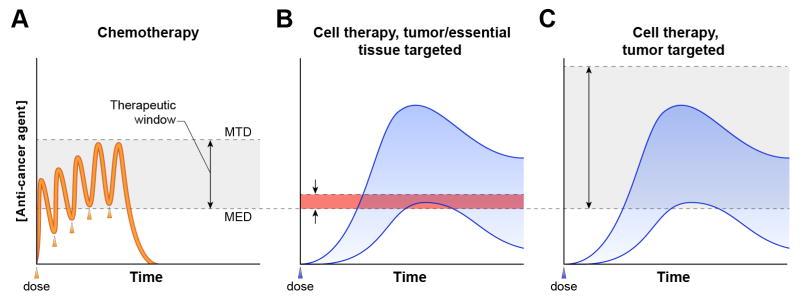
Differences in the pharmacokinetics and mechanisms of action between cytotoxic chemotherapy and adoptive T cell therapies have important implications for phase I clinical trials and determination of therapeutic range. (a) Patient-to-patient variation in drug levels (orange line) is small for chemotherapeutics. Drug exposure (the area under the curve) is predictable and, in the case of toxicity, can be terminated by stopping drug dosing (each dose is indicated by an arrowhead). The efficacy of chemotherapy derives from the greater sensitivity of tumors than vital normal tissues to the cytotoxic actions of the drug. This difference in tissue sensitivity creates a therapeutic window (gray shaded area) between the minimum effective dose (MED) (the dose at which tumor regression occurs) and the maximum tolerated dose (MTD) (the dose at which intolerable toxicities occur) of the drug. (b-c) Because engineered T cells used in ACT are produced on a patient-to-patient basis their pharmacokinetics fluctuate substantially (blue shaded area). Given this variability, it is not clear that the MTD from one cohort of patients can be broadly applied to others. Furthermore, safe phase I testing is challenging because infused T cells do not degrade predictably but rather – unlike any other type of cancer treatment – increase in quantity (due to T cell proliferation) and can persist indefinitely (due to formation of long-lived memory T cell populations) following infusion. Therefore, in the case of adverse events, exposure to T cell therapies cannot be terminated reliably by the standard approach of stopping drug dosing 13,90. (b) A therapeutic window (red shaded area) in which durable regression of high-tumor-burden disease occurs without debilitating autoimmunity has thus far not been identified for any cellular therapy directed against antigens expressed by tumors and essential healthy tissues10–12,21,22. (c) Targeting an antigen that is not expressed by healthy tissue opens the therapeutic window (gray shaded area, by increasing the MTD) for ACT because direct cytotoxicity to normal tissues does not occur, even at high doses of cells4.
Several approaches to controlling T-cell toxicities have been used or are in development. Immunosuppressive drug such as glucocorticoids and antibodies specific for TNF-α or IL-6 can diminish toxicities, and tumor responses have been observed despite their use; however these drugs cannot always halt the T cell response in time to prevent irreversible tissue damage12,13,40,43,48. One evolving tactic is to engineer T cells to express inducible suicide genes in tandem with TCR or CAR genes49. Another is to engineer T cells to express two TCRs or CARs, each targeting a different tumor antigen; crucially, although each targeted antigen is also expressed on healthy tissue no single healthy tissue expresses both antigens. Because each of the two TCRs or CARs provides only a portion of the signal required for T-cell activation and effector function, engineered T cells that encounter healthy tissue—which will express one or none of the antigens—will not receive sufficient signal to induce effector function50,51. This strategy, in theory, permits targeting of a tumor based on coexpression of two antigens (e.g. prostate stem cell antigen and prostate-specific membrane antigen in prostate cancer) and may provide the means to exploit shared tumor/self-antigens that would otherwise incur autoimmune toxicity50,51. These approaches might broaden the application and increase the safety of cellular therapies, but they have the drawback of adding complexity to the T cell engineering process. In addition, experience with these approaches in patients is only beginning to accumulate. With limited ability to safely escalate T cell dose, the selection of target antigens with expression restricted to dispensable tissues and/or tumors has become increasingly important.
Determining the therapeutic range of ACT
Also driving the focus on tumor-specific antigens is uncertainty about the therapeutic window for ACT targeting antigens shared with vital health tissues (Fig. 3). Chemotherapy and newer ‘molecularly targeted’ agents (e.g., tyrosine kinase inhibitors, mammalian target of rapamycin (mTOR) inhibitors, antibodies against growth receptors) act on both healthy tissues and tumors but are effective by virtue of a range of doses at which tumor regression occurs but toxicities due to effects on healthy tissue are tolerable. This dosing ‘window’ is due to the greater sensitivity of tumors than healthy tissues to the drugs. However, the same rationale may not apply to cancer immunotherapy.
In particular, antigen processing and presentation defects in tumors and immune inhibitory/immunosuppressive factors in the tumor microenvironment may actually render tumors less susceptible than healthy tissue to T-cell-mediated killing (Fig. 4). That this is indeed the case is suggested by the results of clinical trials of T cells targeting CAIX and CEA; these trials reported consistent healthy tissue injury but only a single partial tumor response11,21. Thus, although it is appealing to propose that a therapeutic window exists for antigens that are expressed more abundantly on tumors than on healthy tissue, clinical experience does not (at least thus far) support this concept. Furthermore, if such a therapeutic window does exist, the need to dose T cells within a limited range would cap the potential potency of the T cell-mediated therapy and preclude implementation of discoveries that are aimed at boosting the potency of adoptively transferred T cells. Recent preclinical studies from our group and others have revealed the potential to enhance the efficacy of T cells for adoptive immunotherapy through generation of effector T cells directly from naïve precursors, by polarization to a type 17 phenotype, or by ectopic T cell production of IL-12. These advances cannot be exploited if the strength of the T cell response is restricted by a ceiling of autoimmunity52–57. Another method to reduce T cell potency, the use of TCRs and CARs with lower affinity for tumor target antigens, may prevent or reduce toxicity to healthy tissues but may at the same time lessen or eliminate altogether the anti-tumor effects of the treatment50,58. In contrast to strategies that diminish the treatment in an effort to stay within a therapeutic window, targeting tumor-specific antigens completely circumvents T-cell-mediated cytolysis of healthy tissues, thereby avoiding autoimmune toxicity, creating a broader therapeutic range, and enabling a treatment in which a stronger T cell response can be brought to bear against the tumor (Fig. 3).
Figure 4.

T cells may target healthy tissues more efficiently than tumors, independent of the relative abundance of target antigen on each tissue. This phenomenon may preclude identification of a therapeutic window based on enhanced sensitivity of tumor compared to healthy tissue to T cell-mediated killing. Because high-avidity T cells have exquisite sensitivity for their target antigens and can recognize even single complexes of target peptide-MHC122, healthy tissues expressing even small quantities of antigen but possessing intact antigen processing and presentation machinery might be highly vulnerable to T cell-mediated killing (left panel). Once initiated, such T cell killing can be amplified by the proliferation of T cells and by the mutual activation of T cells and antigen presenting cells. Despite expressing potentially large quantities of target antigen, tumors are prone to genomic instability, defects in antigen processing and presentation (e.g. defects in transporter associated with antigen processing function (TAP), proteosomal subunits, β-2 microglobulin, and MHC molecules) (right panel). As such they may not present target antigen to engineered T cells. Tumors may be further protected by their production of cell surface and soluble molecules that inhibit T cell activation.
Previously top-ranked target antigens
Nonetheless, there continues to be strong support for targeting antigens that are expressed on healthy tissues as well as tumors. A National Cancer Institute (NCI; Bethesda, MD) pilot project gathered experts in the field in July 2007 (recommendations published in August 2009) to prioritize tumor antigens as therapeutic targets for cancer vaccines and T-cell therapy52. Antigens were ranked using nine weighted criteria with specificity the fourth priority. The distribution of healthy tissue expression of the 25 top-ranked antigens, excluding those not encoded by normal human genes (such as mutated gene products and viral antigens, which by definition are not expressed by normal tissues), is displayed in Table 1.
Table 1.
Healthy tissue distribution of antigens previously ranked highly by NCI as candidate ACT target antigens.
| Antigen | NCI priority rank | Gene | Healthy tissue expression that may cause major morbiditya |
|---|---|---|---|
| WT1 | 1 | WT1 | kidney, hematopoetic cells25,62,63,93 |
| MUC1 | 2 | MUC1 | lung, liver, pancreas, esophagus, stomach, small bowel, colon, rectum, kidney, bone marrow, lymph node, peripheral nerve, skin, parathyroid gland, adrenal gland24,25,66 |
| HER-2/neu | 6 | ERBB2 | heart, lung, esophagus, stomach, small bowel, colon, rectum, kidney, urinary bladder24,25 |
| MAGE-A3 | 8 | MAGEA3 | None25,94 |
| p53 | 9 | TP53 | bone marrow, spleen, stomach, esophagus, small bowel, colon, rectum, skin24,25,59 |
| NY-ESO-1 | 10 | CTAG1B | None25,94 |
| PSMA | 11 | FOLH1 | brain, kidney, liver, spinal cord, nervous tissue, skin24,25,95 |
| GD2 | 12 | N/A | brain, connective tissue from colon and kidney, skin, peripheral nerve, posterior pituitary72,74,96,97 |
| CEA | 13 | CEACAM5 | bone marrow, liver, lung, esophagus, stomach, small bowel, colon, rectum23–25 |
| MelanA/MART1 | 14 | MLANA | melanocytes including skin, eye, ear10,24 |
| gp100 | 16 | PMEL | melanocytes including skin, eye, ear10 |
| Proteinase 3 (PR1) | 18 | PRTN3 | hematopoetic stem cells25,98 |
| Tyrosinase | 20 | TYR | melanocytes including skin, eye, and ear24,99 |
| Survivin | 21 | BIRC5 | bone marrow, esophagus, stomach, small bowel, colon, rectum, heart, urinary bladder24,25,100 |
| PSA | 22 | KLK3 | pancreas, salivary gland101,102 |
| hTERT | 23 | TERT | hematopoetic cells, lymphocytes, skin, intestine103–107 |
| EphA2 | 25 | EPHA2 | skeletal muscle, liver, colon, lung, esophagus25,108 |
Tissues that might be associated with tolerable toxicities, such as reproductive organs, were not included.
N/A Not applicable.
Gene and protein expression data suggest that these antigens, with the exception of the cancer testis antigens cancer/testis antigen 1B (CTAG1B/NY-ESO-1) and melanoma antigen family A3 (MAGEA3), are expressed in potentially important normal tissues. Three of these antigens (CEA, melan-A (MART1) and glycoprotein 100 (gp100)) have been targeted using TCR-engineered T cells, and as discussed above the resulting severe autoimmune toxicities are consistent with their predicted tissue distribution. One antigen, HER 2/neu(erbB2), has been targeted using CAR-engineered T cells and, as discussed above, resulted in severe toxicity13. Tyrosinase has the same tissue distribution as MART1 and gp100 and likely carries the same risks as a target. P53 is expressed by activated healthy T cells so targeting it would likely lead to T-cell fratricide59. Thus, clinical experience suggests that six of the 25 top-ranked target antigens are suboptimal, if not untenable, choices.
Two of the very highest ranked antigens, Wilm’s tumor 1 (WT1) and mucin 1 (MUC1), may also be suboptimal choices. Microarray data indicate expression of WT1 and MUC1 by a variety of healthy tissues25,60. Some of these tissues, such as reproductive organs and breast tissue are not necessary for life, and related toxicities may be tolerable; however others have important, non-redundant functions. WT1 has been detected in glomerular podocytes of normal adult kidney, which are essential to kidney function25,61–63, and in CD34+ bone marrow cells, which if depleted might lead to bone marrow aplasia64. Treatment of patients with leukemia with repeated infusions of WT-1 specific T cell clones has been reported65. One of three patients with detectable leukemic blasts experienced a transient reduction in peripheral blood blasts (lasting less than a week). That patient also developed unspecified toxicity, attributed to prior therapy, which prevented additional cell infusions. Whether a potent T cell transfer therapy can be safely directed against the WT1 antigen remains untested. MUC1 is expressed in the cytoplasm and membranes of glandular cells of hepatobiliary and gastrointestinal tissues as well as in airway epithelial cells, lymph node germinal centers, and the kidney24,66. The consequences of injury to these tissues might include respiratory failure, gastroenteritis and colitis, and renal failure. An abnormally glycosylated form of MUC1 was reported to be preferentially expressed by tumor cells, but evidence for absence of this antigen on vital healthy tissues is lacking67.
Rational targets for potent ACT
The considerations detailed above compel a reassessment of strategies used to select target antigens for ACT. Rather than the current emphasis on perceived clinical benefit in vaccine trials, immunogenicity, and oncogenic function, tumor specificity may need to be the top priority, with expression by vital cell types weighing heavily against a candidate antigen1. With this in mind, four classes of tumor-specific or highly tissue-restricted antigens could be targeted: CTA (testis-restricted and tissue-restricted), tissue-restricted antigens, mutated gene products and viral proteins (Supplemental Table 2).
CTA are normally expressed predominantly by germ cells, but can be expressed in certain cancers. However, because they do not express major histocompatibility complex (MHC) molecules, germ cells are not recognized by TCRs. Thus, CT antigens, if truly restricted in expression to the testis and not expressed on other healthy tissues, function as essentially tumor-specific T-cell target antigens. However, CT antigens can be sub-categorized as testis-restricted, testis/brain-restricted, or testis-selective (predominantly expressed in testis but also present in other tissues) based on their expression patterns. Among these, testis-restricted and certain testis-selective CT antigens may be rational targets for T-cell therapies (Supplemental Table 2). One advantage of this class of antigens is that they are often expressed by various cancers, some of which are quite common. In addition, the proteins encoded by CTA genes are large enough to generate multiple peptides, and this increases the chances of identifying an epitope presented by common HLA alleles. However, CTAs have some disadvantages. First, with current technology it is impossible to prospectively rule out their expression in all vital healthy tissues (Box 1). Second, they are mostly intracellular and therefore require MHC-restricted recognition by a TCR, rather than a CAR, which can only bind to surface proteins. Nevertheless, HLA-A2-restricted epitopes have been described for many testis-restricted CTAs4,31,45,68–70. And injection of T cells expressing TCRs having high affinity for an HLA-A2-restricted epitope of NY-ESO-1 resulted in tumor responses in four of six patients with synovial cell sarcoma and five of 11 patients with melanoma4. Two of the melanoma responses were complete and ongoing after more than a year. No autoimmune adverse events occurred. This experience seems to indicate that testis-restricted CT antigens can be targeted safely and effectively. The challenge may be in identifying CT antigens that are truly restricted to testis, or perhaps to non-essential tissues, and in isolating TCRs that bind them with high-affinity but do not cross-react with other epitopes.
The second type of tumor antigen that may be safely targeted is those with well-defined expression restricted to non-vital cell lineages and/or tissues (Supplemental Table 2). CD19, which as discussed above is expressed exclusively on the surface of malignant and healthy B cells, has been targeted by CAR-expressing T cells and toxicities related to depletion of healthy B cells have been relatively well tolerated5,5,39–43. B-cell maturation antigen (BCMA), consistently detected on multiple myeloma cells, is also expressed on B cells and plasma cells; because it is a surface protein it may be targeted with a CAR and is therefore a promising ACT candidate target antigen47. CD22 is another potential B-cell lineage-specific surface antigen that might be targeted with CARs71. However, for solid tumors, tissue-specific cell surface antigens have been difficult to identify. Disialoganglioside (GD2) (ranked 12th in Table 2) is an acidic glycosphingolipid that is expressed by neuroectodermic tumors such as neuroblastoma but is also present in normal neurons, skin, melanocytes, and peripheral nerves72–74. Its expression is restricted but not necessarily to dispensable tissues. GD2 was targeted with CAR-expressing T cells without severe toxicity, but with limited evidence of strong anti-tumor activity (one partial response in seven patients with bulky disease)75. There may be additional antigens that are restricted to more dispensable tissue lineages, such as T-cell receptor gamma alternant reading frame protein (TARP), a purportedly prostate-specific intracellular antigen76,77. The primary drawback of targeting tissue-restricted antigens is uncertainty about tissue distribution. The truest test of tissue expression of some candidate target antigens may be clinical trials of T cells expressing receptors having high affinity for these antigens; this approach obviously carries the risk of unanticipated toxicities but may be necessary for continued advancement of the field.
A third class of antigens suitable for investigation is mutated gene products, which by definition are not expressed by normal tissues. As our understanding of the molecular basis of cancer has evolved, mutated signaling proteins in particular have emerged as promising candidate ACT target antigens. These constitutively activated intracellular proteins induce and sustain malignant transformation and drive development and progression of many common cancers. Targeting them with small molecule inhibitors has resulted in some of the most notable advances in oncology78–81. The mutations that cause activation of these signaling proteins can indirectly create non-self ‘neoepitopes’ that can be potentially recognized by T cells. The main downside of targeting these neoepitopes is that few peptides span the mutated region, and thus the chances of finding a peptide suitable for ACT are lower than with other classes of target antigens; in addition, the mutations are rarely present in the most common types of cancer (Supplemental Table 2). Nevertheless, high-throughput in silico methods based on MHC binding predictions and mutations collected in the Catalogue of Somatic Mutations in Cancer (COSMIC) database have been used to predict mutational epitopes suitable for ACT 82. Mutations at residues 12 and 13 of KRAS result in mutated KRAS peptides that are predicted to bind to HLA-A282. Although the frequency of these mutations in any given type of cancer is generally low, the incidence of and death rate from cancers that bear them can be high; therefore considerable numbers of patients might be treated82–84. Even so, it remains to be determined if the MHC-binding algorithm predictions are correct and if T cells having high affinity for the predicted epitopes can be generated. In general, while theoretically interesting, the strategy of targeting mutated antigens is mostly untested. Another way that mutated gene products might be targeted in the future is through rapid identification of all mutations in the expressed genes from a particular tumor85,86. TCRs against mutated epitopes might be rapidly generated and novel gene transfer technologies used to express them in T cells for therapy85,86. However, while the technologies required for this strategy, such as whole exome sequencing, MHC/peptide elution and peptide sequencing systems, and gene transfer methods are evolving rapidly, at this time such an approach is not possible.
Viral antigens, in particular those encoded by viral oncogenes, form a final class of candidate tumor target antigens worthy of investigation. In contrast to the limited epitopes within and generally low frequencies of mutated signaling proteins, viral oncogenes encode large antigens containing multiple epitopes, and they are expressed in most patients with certain types of cancer. They are ideal therapeutic targets in that they are constitutively and exclusively expressed by malignant cells, and they are functionally important. For example, Epstein-Barr virus (EBV) is associated with diverse cancers including lymphoproliferative disease, undifferentiated nasopharyngeal carcinoma, and Hodgkin’s lymphoma. T cells bearing receptors targeting EBV antigens have shown promise, particularly for lymphoproliferative disorders for which these therapies often induce prolonged complete remission6,85,86. Another example is antigens encoded by the high-risk human papillomaviruses (HPV) that cause cervical cancer and are commonly associated with oropharyneal, anal, vulvar, vaginal, and penile cancers. The HPV 16 E6 and E7 oncoproteins have been targeted in premalignant disease with peptide vaccines without major toxicity and with signs of efficacy in a single arm clinical study87. HLA-A2 restricted epitopes of E6 and E7 have been described and eluted from the surface of HPV-16 positive cell lines confirming that they are naturally processed and presented88. T cells bearing receptors specific for these epitopes but lacking the avidity to recognize unmanipulated tumor lines have been described89. However, surprisingly, unambiguous direct recognition of HPV-positive tumors by any T cell specific for E6 or E7 is yet to be demonstrated, raising the question of whether high-affinity receptors can be generated against these targets. Nevertheless, E6 and E7 are particularly attractive ACT targets if high-affinity TCRs for can be isolated.
None of the antigen classes described above is ideal in that none are present with high frequency in common cancers, constitutively expressed exclusively by malignant cells, functionally important for tumor growth and progression, and targetable with MHC-independent systems (CARs) (Supplemental Table 2). Nevertheless, mutated gene products and viral antigens provide added safety in clinical testing because of their unequivocal restriction to malignant (and, for viral proteins, infected) cells. For some cancers, such as ovarian cancer, non-small cell lung cancer, hepatocellular carcinoma, and melanoma, CTAs are well-characterized, tumor/testis-specific, and shared by substantial cohorts of patients; thus they are conspicuous targets. CTAs must, however, be approached circumspectly because of uncertainties about their tissue restriction and the potential for antigen receptor cross-reactivity against related proteins. For certain types of cancers, particularly lymphoid malignancies, lineage-specific antigens (e.g., CD19, CD20, CD22 and B-cell maturation protein (BCMA/Tnfrsf17)) appear to be viable targets as is suggested by the encouraging early results of clinical ACT protocols targeting CD19. However, the indirect toxicities, especially CRS, associated with this type of therapy may signal caution about using CARs to target antigens expressed by healthy cells widespread throughout the body. For some of the most common and deadly cancers, currently studied candidate target antigens are limited; for example, in pancreatic and colon cancer, no high-affinity receptors specific for commonly expressed tumor antigens restricted by common HLA alleles have been described. Identification of appropriate targets and isolation of antigen receptors with appropriate affinity and specificity for these targets is an important goal of ongoing research.
A path forward
The community of clinicians and researchers developing adoptive T cell therapies can take steps to support ongoing progress in the field. The first is to take a circumspect approach to target selection, giving higher priority to antigens with less chance of expression by vital normal tissues. The second is to minimize the chances of unexpected toxicities to normal tissue by conducting careful preclinical studies. When antigens encoded by normal unmutated human genes are targeted these studies should include extensive testing for target expression by healthy tissue panels from multiple donors. Techniques employed currently rely primarily on analysis of whole genome expression (microarrays), RNA sequencing, and immunohistochemistry, although other technologies are evolving, including newer forms of high throughput mass spectrometry proteomics that enable sensitive detection of potential targets. These data, especially unanticipated expression by important tissues, should be published in journals and/or shared in public online databases such as those organized by the National Library of Medicine. Receptors should be also assessed for cross-reactivity to antigens with similar protein sequences. Finally the risk of cross-reactivity to self antigens might be further reduced by preferentially using unmanipulated TCRs that are isolated from humans rather than HLA-transgenic mice, because receptors isolated from humans have undergone thymic negative selection against human proteins, and therefore have reduced autoreactivity. Similarly, affinity-enhancement through mutagenesis of TCR CDRs, which has resulted in severe autoimmune toxicities, should be undertaken with great caution12,90.
Advances in the field will come with risk, but a thoughtful approach that avoids past mistakes might help expedite innovation and development of the most promising therapies. In particular, exploitation of the full potential of T-cell based therapies may require a more incremental approach in which a smaller population of patients is treated with T cells designed to target truly tumor-specific antigens. Although the potency of ACT may restrict its safe application to tumor-specific or highly tissue-restricted antigens, the flexibility to target both intracellular and cell-surface antigens broadens its potential use. ACT is a promising treatment modality, and we are increasingly learning how to direct it against diverse cancers. Judicious target antigen selection is crucial to the continued advancement of this approach91.
Supplementary Material
References
- 1.Cheever MA, et al. The prioritization of cancer antigens: a National Cancer Institute pilot project for the acceleration of translational research. Clin Cancer Res. 2009;15:5323–5337. doi: 10.1158/1078-0432.CCR-09-0737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nat Med. 2004;10:909–915. doi: 10.1038/nm1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rosenberg SA, et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res. 2011;17:4550–4557. doi: 10.1158/1078-0432.CCR-11-0116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Robbins PF, et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J Clin Oncol. 2011;29:917–924. doi: 10.1200/JCO.2010.32.2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kochenderfer JN, et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood. 2012;119:2709–2720. doi: 10.1182/blood-2011-10-384388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Heslop HE, et al. Long-term outcome of EBV-specific T-cell infusions to prevent or treat EBV-related lymphoproliferative disease in transplant recipients. Blood. 2010;115:925–935. doi: 10.1182/blood-2009-08-239186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Paulos CM, et al. Microbial translocation augments the function of adoptively transferred self/tumor-specific CD8+ T cells via TLR4 signaling. J Clin Invest. 2007;117:2197–2204. doi: 10.1172/JCI32205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Antony PA, et al. CD8+ T cell immunity against a tumor/self-antigen is augmented by CD4+ T helper cells and hindered by naturally occurring T regulatory cells. J Immunol. 2005;174:2591–2601. doi: 10.4049/jimmunol.174.5.2591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gattinoni L, et al. Removal of homeostatic cytokine sinks by lymphodepletion enhances the efficacy of adoptively transferred tumor-specific CD8+ T cells. J Exp Med. 2005;202:907–912. doi: 10.1084/jem.20050732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Johnson LA, et al. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood. 2009;114:535–546. doi: 10.1182/blood-2009-03-211714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Parkhurst MR, et al. T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Mol Ther. 2010;19:620–626. doi: 10.1038/mt.2010.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Morgan RA, et al. Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J Immunother. 2013;36:133–151. doi: 10.1097/CJI.0b013e3182829903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Morgan RA, et al. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther. 2010;18:843–851. doi: 10.1038/mt.2010.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Palmer DC, et al. Vaccine-stimulated, adoptively transferred CD8+ T cells traffic indiscriminately and ubiquitously while mediating specific tumor destruction. J Immunol. 2004;173:7209–7216. doi: 10.4049/jimmunol.173.12.7209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Palmer DC, et al. Effective tumor treatment targeting a melanoma/melanocyte-associated antigen triggers severe ocular autoimmunity. Proc Natl Acad Sci. 2008;105:8061–8066. doi: 10.1073/pnas.0710929105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Overwijk WW, et al. Tumor regression and autoimmunity after reversal of a functionally tolerant state of self-reactive CD8+ T cells. J Exp Med. 2003;198:569–580. doi: 10.1084/jem.20030590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Leibovich BC, et al. Carbonic anhydrase IX is not an independent predictor of outcome for patients with clear cell renal cell carcinoma. J Clin Oncol. 2007;25:4757–4764. doi: 10.1200/JCO.2007.12.1087. [DOI] [PubMed] [Google Scholar]
- 18.Brouwers AH, Mulders PFA, Oyen WJG. Carbonic anhydrase IX expression in clear cell renal cell carcinoma and normal tissues: experiences from (radio) immunotherapy. J Clin Oncol. 2008;26:3808–3809. doi: 10.1200/JCO.2008.17.6073. author reply 3811–3812. [DOI] [PubMed] [Google Scholar]
- 19.Ivanov S, et al. Expression of hypoxia-inducible cell-surface transmembrane carbonic anhydrases in human cancer. Am J Pathol. 2001;158:905–919. doi: 10.1016/S0002-9440(10)64038-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brouwers AH, et al. Lack of efficacy of two consecutive treatments of radioimmunotherapy with 131I-cG250 in patients with metastasized clear cell renal cell carcinoma. J Clin Oncol Off J Am Soc Clin Oncol. 2005;23:6540–6548. doi: 10.1200/JCO.2005.07.732. [DOI] [PubMed] [Google Scholar]
- 21.Lamers CHJ, et al. Treatment of metastatic renal cell carcinoma with autologous T-lymphocytes genetically retargeted against carbonic anhydrase IX: first clinical experience. J Clin Oncol. 2006;24:e20–e22. doi: 10.1200/JCO.2006.05.9964. [DOI] [PubMed] [Google Scholar]
- 22.Lamers CH, et al. Treatment of metastatic renal cell carcinoma with CAIX CAR-engineered T cells: clinical evaluation and management of on-target toxicity. Mol Ther. 2013;21:904–912. doi: 10.1038/mt.2013.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hammarström S. The carcinoembryonic antigen (CEA) family: structures, suggested functions and expression in normal and malignant tissues. Semin Cancer Biol. 1999;9:67–81. doi: 10.1006/scbi.1998.0119. [DOI] [PubMed] [Google Scholar]
- 24.Uhlen M, et al. Towards a knowledge-based Human Protein Atlas. Nat Biotechnol. 2010;28:1248–1250. doi: 10.1038/nbt1210-1248. [DOI] [PubMed] [Google Scholar]
- 25.McCall MN, Uppal K, Jaffee HA, Zilliox MJ, Irizarry RA. The Gene Expression Barcode: leveraging public data repositories to begin cataloging the human and murine transcriptomes. Nucleic Acids Res. 2011;39:D1011–D1015. doi: 10.1093/nar/gkq1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gulley JL, et al. Pilot study of vaccination with recombinant CEA-MUC-1-TRICOM poxviral-based vaccines in patients with metastatic carcinoma. Clin Cancer Res. 2008;14:3060–3069. doi: 10.1158/1078-0432.CCR-08-0126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Marshall JL, et al. Phase I study of sequential vaccinations with fowlpox-CEA(6D)-TRICOM alone and sequentially with vaccinia-CEA(6D)-TRICOM, with and without granulocyte-macrophage colony-stimulating factor, in patients with carcinoembryonic antigen-expressing carcinomas. J Clin Oncol. 2005;23:720–731. doi: 10.1200/JCO.2005.10.206. [DOI] [PubMed] [Google Scholar]
- 28.Ullenhag GJ, et al. Durable carcinoembryonic antigen (CEA)-specific humoral and cellular immune responses in colorectal carcinoma patients vaccinated with recombinant CEA and granulocyte/macrophage colony-stimulating factor. Clin Cancer Res. 2004;10:3273–3281. doi: 10.1158/1078-0432.CCR-03-0706. [DOI] [PubMed] [Google Scholar]
- 29.Bos R, et al. Balancing between antitumor efficacy and autoimmune pathology in T-cell-mediated targeting of carcinoembryonic antigen. Cancer Res. 2008;68:8446–8455. doi: 10.1158/0008-5472.CAN-08-1864. [DOI] [PubMed] [Google Scholar]
- 30.Natali PG, et al. Expression of the p185 encoded by HER2 oncogene in normal and transformed human tissues. Int J Cancer J Int Cancer. 1990;45:457–461. doi: 10.1002/ijc.2910450314. [DOI] [PubMed] [Google Scholar]
- 31.Kawashima I, et al. The multi-epitope approach for immunotherapy for Cancer: Identification of several CTL epitopes from various tumor-associated antigens expressed on solid epithelial tumors. Hum Immunol. 1998;59:1–14. doi: 10.1016/s0198-8859(97)00255-3. [DOI] [PubMed] [Google Scholar]
- 32.Jones KL, Buzdar AU. Evolving novel anti-HER2 strategies. Lancet Oncol. 2009;10:1179–1187. doi: 10.1016/S1470-2045(09)70315-8. [DOI] [PubMed] [Google Scholar]
- 33.Atanackovic D, et al. Booster vaccination of cancer patients with MAGE-A3 protein reveals long-term immunological memory or tolerance depending on priming. Proc Natl Acad Sci. 2008;105:1650–1655. doi: 10.1073/pnas.0707140104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Van Baren N, et al. Tumoral and immunologic response after vaccination of melanoma patients with an ALVAC virus encoding MAGE antigens recognized by T cells. J Clin Oncol. 2005;23:9008–9021. doi: 10.1200/JCO.2005.08.375. [DOI] [PubMed] [Google Scholar]
- 35.Nachimuthu Chinnasamy JAW. A TCR targeting the HLA-A*0201-restricted epitope of MAGE-A3 recognizes multiple epitopes of the MAGE-A antigen superfamily in several types of cancer. J Immunol Baltim Md 1950. 2011;186:685–96. doi: 10.4049/jimmunol.1001775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Linette GP, et al. Cardiovascular toxicity and titin cross-reactivity of affinity enhanced T cells in myeloma and melanoma. Blood. 2013 doi: 10.1182/blood-2013-03-490565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cameron BJ, et al. Identification of a titin-derived HLA-A1-presented peptide as a cross-reactive target for engineered MAGE A3-directed T cells. Sci Transl Med. 2013;5:197ra103. doi: 10.1126/scitranslmed.3006034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Uckun FM, et al. Detailed studies on expression and function of CD19 surface determinant by using B43 monoclonal antibody and the clinical potential of anti-CD19 immunotoxins. Blood. 1988;71:13–29. [PubMed] [Google Scholar]
- 39.Kochenderfer JN, et al. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood. 2010;116:4099–4102. doi: 10.1182/blood-2010-04-281931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Grupp SA, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. 2013;368:1509–1518. doi: 10.1056/NEJMoa1215134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011;365:725–733. doi: 10.1056/NEJMoa1103849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kalos M, et al. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med. 2011;3:95ra73. doi: 10.1126/scitranslmed.3002842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brentjens RJ, et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med. 2013;5:177ra38. doi: 10.1126/scitranslmed.3005930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brentjens RJ, et al. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood. 2011;118:4817–4828. doi: 10.1182/blood-2011-04-348540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.LDA, et al. Identification of MAGE-C1 (CT-7) epitopes for T-cell therapy of multiple myeloma. Cancer Immunol Immunother. 2011;60:985–997. doi: 10.1007/s00262-011-1009-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Maus MV, June CH. Zoom Zoom: Racing CARs for Multiple Myeloma. Clin Cancer Res. 2013;19:1917–1919. doi: 10.1158/1078-0432.CCR-13-0168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Carpenter RO, et al. B-cell maturation antigen is a promising target for adoptive T-cell therapy of multiple myeloma. Clin Cancer Res. 2013 doi: 10.1158/1078-0432.CCR-12-2422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hinrichs CS, Palmer DC, Rosenberg SA, Restifo NP. Glucocorticoids do not inhibit antitumor activity of activated CD8+ T cells. J Immunother. 2005;28:517–524. doi: 10.1097/01.cji.0000177999.95831.7b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Di Stasi A, et al. Inducible apoptosis as a safety switch for adoptive cell therapy. N Engl J Med. 2011;365:1673–1683. doi: 10.1056/NEJMoa1106152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kloss CC, Condomines M, Cartellieri M, Bachmann M, Sadelain M. Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat Biotechnol. 2013;31:71–75. doi: 10.1038/nbt.2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hanada K, Restifo NP. Double or nothing on cancer immunotherapy. Nat Biotechnol. 2013;31:33–34. doi: 10.1038/nbt.2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hinrichs CS, et al. Adoptively transferred effector cells derived from naive rather than central memory CD8+ T cells mediate superior antitumor immunity. Proc Natl Acad Sci. 2009;106:17469–17474. doi: 10.1073/pnas.0907448106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hinrichs CS, et al. Type 17 CD8+ T cells display enhanced antitumor immunity. Blood. 2009;114:596–599. doi: 10.1182/blood-2009-02-203935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hinrichs CS, et al. Human effector CD8+ T cells derived from naive rather than memory subsets possess superior traits for adoptive immunotherapy. Blood. 2011;117:808–814. doi: 10.1182/blood-2010-05-286286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kerkar SP, et al. IL-12 triggers a programmatic change in dysfunctional myeloid-derived cells within mouse tumors. J Clin Invest. 2011;121:4746–4757. doi: 10.1172/JCI58814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Muranski P, et al. Th17 cells are long lived and retain a stem cell-like molecular signature. Immunity. 2011;35:972–985. doi: 10.1016/j.immuni.2011.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gattinoni L, et al. A human memory T cell subset with stem cell-like properties. Nat Med. 2011;17:1290–1297. doi: 10.1038/nm.2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhong S, et al. T-cell receptor affinity and avidity defines antitumor response and autoimmunity in T-cell immunotherapy. Proc Natl Acad Sci U S A. 2013;110:6973–6978. doi: 10.1073/pnas.1221609110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Theoret MR, et al. Relationship of p53 Overexpression on Cancers and Recognition by Anti-p53 T Cell Receptor-Transduced T Cells. Hum Gene Ther. 2008;19:1219–1231. doi: 10.1089/hum.2008.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wu C, et al. BioGPS: an extensible and customizable portal for querying and organizing gene annotation resources. Genome Biol. 2009;10:R130. doi: 10.1186/gb-2009-10-11-r130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Grubb GR, Yun K, Williams BR, Eccles MR, Reeve AE. Expression of WT1 protein in fetal kidneys and Wilms tumors. Lab Investig J Tech Methods Pathol. 1994;71:472–479. [PubMed] [Google Scholar]
- 62.Mundlos S, et al. Nuclear localization of the protein encoded by the Wilms’ tumor gene WT1 in embryonic and adult tissues. Dev Camb Engl. 1993;119:1329–1341. doi: 10.1242/dev.119.4.1329. [DOI] [PubMed] [Google Scholar]
- 63.Ramani P, Cowell JK. The expression pattern of Wilms’ tumour gene (WT1) product in normal tissues and paediatric renal tumours. J Pathol. 1996;179:162–168. doi: 10.1002/(SICI)1096-9896(199606)179:2<162::AID-PATH545>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 64.Inoue K, et al. Aberrant Overexpression of the Wilms Tumor Gene (WT1) in Human Leukemia. Blood. 1997;89:1405–1412. [PubMed] [Google Scholar]
- 65.Chapuis AG, et al. Transferred WT1-reactive CD8+ T cells can mediate antileukemic activity and persist in post-transplant patients. Sci Transl Med. 2013;5:174ra27. doi: 10.1126/scitranslmed.3004916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Seregni E, et al. Structure, function and gene expression of epithelial mucins. Tumori. 1997;83:625–632. doi: 10.1177/030089169708300301. [DOI] [PubMed] [Google Scholar]
- 67.Cao Y, et al. Mucins (MUC1 and MUC3) of Gastrointestinal and Breast Epithelia Reveal Different and Heterogeneous Tumor-associated Aberrations in Glycosylation. J Histochem Cytochem. 1997;45:1547–1557. doi: 10.1177/002215549704501111. [DOI] [PubMed] [Google Scholar]
- 68.Ayyoub M, et al. Proteasome-Assisted Identification of a SSX-2-Derived Epitope Recognized by Tumor-Reactive CTL Infiltrating Metastatic Melanoma. J Immunol. 2002;168:1717–1722. doi: 10.4049/jimmunol.168.4.1717. [DOI] [PubMed] [Google Scholar]
- 69.Zhao Y, et al. Primary Human Lymphocytes Transduced with NY-ESO-1 Antigen-Specific TCR Genes Recognize and Kill Diverse Human Tumor Cell Lines. J Immunol. 2005;174:4415–4423. doi: 10.4049/jimmunol.174.7.4415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ottaviani S, Zhang Y, Boon T, van der Bruggen P. A MAGE-1 antigenic peptide recognized by human cytolytic T lymphocytes on HLA-A2 tumor cells. Cancer Immunol Immunother. 2005;54:1214–1220. doi: 10.1007/s00262-005-0705-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Haso W, et al. Anti-CD22-chimeric antigen receptors targeting B-cell precursor acute lymphoblastic leukemia. Blood. 2013;121:1165–1174. doi: 10.1182/blood-2012-06-438002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hersey P, Jamal O, Henderson C, Zardawi I, D’Alessandro G. Expression of the gangliosides GM3, GD3 and GD2 in tissue sections of normal skin, naevi, primary and metastatic melanoma. Int J Cancer. 1988;41:336–343. doi: 10.1002/ijc.2910410303. [DOI] [PubMed] [Google Scholar]
- 73.Alvarez-Rueda N, et al. A Monoclonal Antibody to O-Acetyl-GD2 Ganglioside and Not to GD2 Shows Potent Anti-Tumor Activity without Peripheral Nervous System Cross- Reactivity. PLoS ONE. 2011;6:e25220. doi: 10.1371/journal.pone.0025220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mennel HD, et al. Expression of GD2-epitopes in human intracranial tumors and normal brain. Exp Toxicol Pathol. 1992;44:317–324. doi: 10.1016/S0940-2993(11)80218-6. [DOI] [PubMed] [Google Scholar]
- 75.Louis CU, et al. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood. 2011;118:6050–6056. doi: 10.1182/blood-2011-05-354449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wolfgang CD, Essand M, Vincent JJ, Lee B, Pastan I. TARP: a nuclear protein expressed in prostate and breast cancer cells derived from an alternate reading frame of the T cell receptor gamma chain locus. Proc Natl Acad Sci U S A. 2000;97:9437–9442. doi: 10.1073/pnas.160270597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hillerdal V, Nilsson B, Carlsson B, Eriksson F, Essand M. T cells engineered with a T cell receptor against the prostate antigen TARP specifically kill HLA-A2+ prostate and breast cancer cells. Proc Natl Acad Sci U S A. 2012;109:15877–15881. doi: 10.1073/pnas.1209042109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kris MGNR. Efficacy of gefitinib, an inhibitor of the epidermal growth factor receptor tyrosine kinase, in symptomatic patients with non–small cell lung cancer: A randomized trial. JAMA. 2003;290:2149–2158. doi: 10.1001/jama.290.16.2149. [DOI] [PubMed] [Google Scholar]
- 79.Savage DG, Antman KH. Imatinib Mesylate — A New Oral Targeted Therapy. N Engl J Med. 2002;346:683–693. doi: 10.1056/NEJMra013339. [DOI] [PubMed] [Google Scholar]
- 80.Kwak EL, et al. Anaplastic Lymphoma Kinase Inhibition in Non–Small-Cell Lung Cancer. N Engl J Med. 2010;363:1693–1703. doi: 10.1056/NEJMoa1006448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Flaherty KT, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010;363:809–819. doi: 10.1056/NEJMoa1002011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Warren RL, Holt RA. A census of predicted mutational epitopes suitable for immunologic cancer control. Hum Immunol. 2010;71:245–254. doi: 10.1016/j.humimm.2009.12.007. [DOI] [PubMed] [Google Scholar]
- 83.Cancer Facts & Figures. 2013 acspc-036845.pdf. at < http://www.cancer.org/acs/groups/content/@epidemiologysurveilance/documents/document/acspc-036845.pdf>.
- 84.Forbes SA, et al. COSMIC (the Catalogue of Somatic Mutations in Cancer): a resource to investigate acquired mutations in human cancer. Nucleic Acids Res. 2009;38:D652–D657. doi: 10.1093/nar/gkp995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Robbins PF, et al. Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nat Med. 2013 doi: 10.1038/nm.3161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Restifo NP, Dudley ME, Rosenberg SA. Adoptive immunotherapy for cancer: harnessing the T cell response. Nat Rev Immunol. 2012;12:269–281. doi: 10.1038/nri3191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kenter GG, et al. Vaccination against HPV-16 Oncoproteins for Vulvar Intraepithelial Neoplasia. N Engl J Med. 2009;361:1838–1847. doi: 10.1056/NEJMoa0810097. [DOI] [PubMed] [Google Scholar]
- 88.Riemer AB, et al. A Conserved E7-derived Cytotoxic T Lymphocyte Epitope Expressed on Human Papillomavirus 16-transformed HLA-A2+ Epithelial Cancers. J Biol Chem. 2010;285:29608–29622. doi: 10.1074/jbc.M110.126722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Evans M, et al. Antigen processing defects in cervical carcinomas limit the presentation of a CTL epitope from human papillomavirus 16 E6. J Immunol. 2001;167:5420–5428. doi: 10.4049/jimmunol.167.9.5420. [DOI] [PubMed] [Google Scholar]
- 90.NIH VideoCasting. 129th Recombinant DNA Advisory Meeting; June 2012; at < http://videocast.nih.gov/summary.asp?Live=11393&bhcp=1>. [Google Scholar]
- 91.Do no harm. Nat Biotechnol. 2013;31:365–365. doi: 10.1038/nbt.2587. [DOI] [PubMed] [Google Scholar]
- 92.Pule MA, et al. Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nat Med. 2008;14:1264–1270. doi: 10.1038/nm.1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Inoue K, et al. Aberrant Overexpression of the Wilms Tumor Gene (WT1) in Human Leukemia. Blood. 1997;89:1405–1412. [PubMed] [Google Scholar]
- 94.Hofmann O, et al. Genome-wide analysis of cancer/testis gene expression. Proc Natl Acad Sci. 2008;105:20422–20427. doi: 10.1073/pnas.0810777105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Silver DA, Pellicer I, Fair WR, Heston WD, Cordon-Cardo C. Prostate-specific membrane antigen expression in normal and malignant human tissues. Clin Cancer Res. 1997;3:81–85. [PubMed] [Google Scholar]
- 96.Zhang S, et al. Selection of tumor antigens as targets for immune attack using immunohistochemistry: I. Focus on gangliosides. Int J Cancer. 1997;73:42–49. doi: 10.1002/(sici)1097-0215(19970926)73:1<42::aid-ijc8>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 97.Yuki N, Yamada M, Tagawa Y, Takahashi H, Handa S. Pathogenesis of the neurotoxicity caused by anti-GD2 antibody therapy. J Neurol Sci. 1997;149:127–130. doi: 10.1016/s0022-510x(97)05390-2. [DOI] [PubMed] [Google Scholar]
- 98.Sturrock AB, et al. Structure, chromosomal assignment, and expression of the gene for proteinase-3. The Wegener’s granulomatosis autoantigen. J Biol Chem. 1992;267:21193–21199. [PubMed] [Google Scholar]
- 99.Battyani Z, Xerri L, Hassoun J, Bonerandi JJ, Grob JJ. Tyrosinase gene expression in human tissues. Pigment Cell Res. 1993;6:400–405. doi: 10.1111/j.1600-0749.1993.tb00622.x. [DOI] [PubMed] [Google Scholar]
- 100.Fukuda S, Pelus LM. Survivin, a cancer target with an emerging role in normal adult tissues. Mol Cancer Ther. 2006;5:1087–1098. doi: 10.1158/1535-7163.MCT-05-0375. [DOI] [PubMed] [Google Scholar]
- 101.Elgamal AA, et al. Detection of prostate specific antigen in pancreas and salivary glands: a potential impact on prostate cancer overestimation. J Urol. 1996;156:464–468. doi: 10.1097/00005392-199608000-00040. [DOI] [PubMed] [Google Scholar]
- 102.Ishikawa T, et al. Expression of alpha-fetoprotein and prostate-specific antigen genes in several tissues and detection of mRNAs in normal circulating blood by reverse transcriptase-polymerase chain reaction. Jpn J Clin Oncol. 1998;28:723–728. doi: 10.1093/jjco/28.12.723. [DOI] [PubMed] [Google Scholar]
- 103.Hiyama E, Hiyama K, Yokoyama T, Shay JW. Immunohistochemical detection of telomerase (hTERT) protein in human cancer tissues and a subset of cells in normal tissues. Neoplasia. 2001;3:17–26. doi: 10.1038/sj.neo.7900134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hiyama K, et al. Activation of telomerase in human lymphocytes and hematopoietic progenitor cells. J Immunol. 1995;155:3711–3715. [PubMed] [Google Scholar]
- 105.Hiyama E, et al. Telomerase activity in human intestine. Int J Oncol. 1996;9:453–458. doi: 10.3892/ijo.9.3.453. [DOI] [PubMed] [Google Scholar]
- 106.Shay JW, Bacchetti S. A survey of telomerase activity in human cancer. Eur J Cancer. 1997;33:787–791. doi: 10.1016/S0959-8049(97)00062-2. [DOI] [PubMed] [Google Scholar]
- 107.Taylor RS, et al. Detection of telomerase activity in malignant and nonmalignant skin conditions. J Invest Dermatol. 1996;106:759–765. doi: 10.1111/1523-1747.ep12345811. [DOI] [PubMed] [Google Scholar]
- 108.Hirai H, Maru Y, Hagiwara K, Nishida J, Takaku F. A novel putative tyrosine kinase receptor encoded by the eph gene. Science. 1987;238:1717–1720. doi: 10.1126/science.2825356. [DOI] [PubMed] [Google Scholar]
- 109.Roselli M, et al. Brachyury, a driver of the epithelial-mesenchymal transition, is overexpressed in human lung tumors: an opportunity for novel interventions against lung cancer. Clin Cancer Res. 2012;18:3868–3879. doi: 10.1158/1078-0432.CCR-11-3211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Palena C, et al. The human T-box mesodermal transcription factor Brachyury is a candidate target for T-cell-mediated cancer immunotherapy. Clin Cancer Res. 2007;13:2471–2478. doi: 10.1158/1078-0432.CCR-06-2353. [DOI] [PubMed] [Google Scholar]
- 111.Jäger D, et al. Identification of a Tissue-specific Putative Transcription Factor in Breast Tissue by Serological Screening of a Breast Cancer Library. Cancer Res. 2001;61:2055–2061. [PubMed] [Google Scholar]
- 112.Humphrey PA, et al. Anti-synthetic peptide antibody reacting at the fusion junction of deletion-mutant epidermal growth factor receptors in human glioblastoma. Proc Natl Acad Sci. 1990;87:4207–4211. doi: 10.1073/pnas.87.11.4207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Wong AJ, et al. Structural alterations of the epidermal growth factor receptor gene in human gliomas. Proc Natl Acad Sci. 1992;89:2965–2969. doi: 10.1073/pnas.89.7.2965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Sok JC, et al. Mutant Epidermal Growth Factor Receptor (EGFRvIII) Contributes to Head and Neck Cancer Growth and Resistance to EGFR Targeting. Clin Cancer Res. 2006;12:5064–5073. doi: 10.1158/1078-0432.CCR-06-0913. [DOI] [PubMed] [Google Scholar]
- 115.Ang KK, et al. Human Papillomavirus and Survival of Patients with Oropharyngeal Cancer. N Engl J Med. 2010;363:24–35. doi: 10.1056/NEJMoa0912217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Wheeler CM, et al. Human papillomavirus genotype distributions: implications for vaccination and cancer screening in the United States. J Natl Cancer Inst. 2009;101:475–487. doi: 10.1093/jnci/djn510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.De Vuyst H, Clifford GM, Nascimento MC, Madeleine MM, Franceschi S. Prevalence and type distribution of human papillomavirus in carcinoma and intraepithelial neoplasia of the vulva, vagina and anus: a meta-analysis. Int J Cancer. 2009;124:1626–1636. doi: 10.1002/ijc.24116. [DOI] [PubMed] [Google Scholar]
- 118.Heideman DAM, et al. Human papillomavirus-16 is the predominant type etiologically involved in penile squamous cell carcinoma. J Clin Oncol. 2007;25:4550–4556. doi: 10.1200/JCO.2007.12.3182. [DOI] [PubMed] [Google Scholar]
- 119.CTpedia. at < http://www.cta.lncc.br/index.php>.
- 120.Sewell AK. Why must T cells be cross-reactive? Nat Rev Immunol. 2012;12:669–677. doi: 10.1038/nri3279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Oldstone MBA. Molecular mimicry and immune-mediated diseases. FASEB J. 1998;12:1255–1265. doi: 10.1096/fasebj.12.13.1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Irvine DJ, Purbhoo MA, Krogsgaard M, Davis MM. Direct observation of ligand recognition by T cells. Nature. 2002;419:845–849. doi: 10.1038/nature01076. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.