

Abstract
Premature infants requiring supplemental oxygen are at increased risk for developing bronchopulmonary dysplasia (BPD). Rodent models involving neonatal exposure to excessive oxygen concentrations (hyperoxia) have helped to identify mechanisms of BPD-associated pathology. Genome-wide assessments of the effects of hyperoxia in neonatal mouse lungs could identify novel BPD-related genes and pathways. Newborn C57BL/6 mice were exposed to 100% oxygen for 10 days, and whole lung tissue RNA was used for high-throughput, sequencing-based transcriptomic analysis (RNA-Seq). Significance Analysis of Microarrays and Ingenuity Pathway Analysis were used to identify genes and pathways affected. Expression patterns for selected genes were validated by qPCR. Mechanistic relationships between genes were further tested in cultured mouse lung epithelial cells. We identified 300 genes significantly and substantially affected following acute neonatal hyperoxia. Canonical pathways dysregulated in hyperoxia lungs included nuclear fctor (erythryoid-derived-2)-like 2-mediated oxidative stress signaling, p53 signaling, eNOS signaling, and aryl hydrocarbon receptor (Ahr) pathways. Cluster analysis identified Ccnd1, Cdkn1a, and Ahr as critical regulatory nodes in the response to hyperoxia, with Ahr serving as the major effector node. A mechanistic role for Ahr was assessed in lung epithelial cells, and we confirmed its ability to regulate the expression of multiple hyperoxia markers, including Cdkn1a, Pdgfrb, and A2m. We conclude that a global assessment of gene regulation in the acute neonatal hyperoxia model of BPD-like pathology has identified Ahr as one driver of gene dysregulation.
Keywords: high-throughput sequencing-based transcriptional profiling, bronchopulmonary dysplasia, aryl hydrocarbon receptor
bronchopulmonary dysplasia (BPD) is a lung disease occurring in premature infants that is defined by dependence on supplemental oxygen for more than 28 days after birth and/or at 36 wk corrected gestational age (16). In the United States, every year more than 500,000 babies are born prematurely, with ∼60,000 at high risk for BPD, and 12,000 are diagnosed with the disease. The lungs of premature infants are not fully developed and often require supplemental oxygen therapy. However, because normal fetal development takes place in a hypoxic environment, exposure to high oxygen concentration can lead to lung injury, resulting in development of BPD.
Significant contributions to our current understanding of the pathogenesis of BPD have been gained from animal models of oxygen-mediated lung injury (20). Detrimental effects of neonatal hyperoxia exposure on animal lungs were first demonstrated almost half a century ago by Northway et al. using newborn guinea pigs (25). Subsequently, different animal models of BPD in primates (22), sheep (28), and rodents have helped to define the relationship between risk factors and disease pathogenesis. Recent studies, involving long-term hyperoxia exposure in neonatal rats, have identified potential therapeutic interventions for BPD (30). Mouse models may be particularly useful, since birth occurs before the alveolar stage, which corresponds to the timing of human preterm infants (18). Similar to premature infants diagnosed with BPD, neonatal mice exposed to excess oxygen at birth show persistent alveolar simplification, increased lung compliance, increased sensitivity to viral infection, and pulmonary vascular diseases (36). The transcription factor NF-κB is known to regulate vascular endothelial growth factor receptor 2, which plays an essential role in lung development by promoting alveolization. Blocking NF-κB activity in neonatal mice has been shown to induce alveolar simplification similar to that observed in BPD (14).
Over the past decade, advances in genomic technologies have helped in developing a greater understanding of the regulatory processes contributing to normal lung development, as well as aberrations leading to developmental diseases such as BPD (4). We have recently generated genome-wide expression profiling data from human early stage fetal lung tissue specimens, spanning the pseudoglandular and early canalicular stages, to identify global transcriptomic features of normal human lung development (18). However, diseases like BPD arise because of dysregulation of the developmental process at the later stages (21). Because of lack of later-stage embryonic human lung tissue, most of the current knowledge of lung development has been derived from limited molecular studies in humans and complemented by histomorphological, molecular, and genetic studies of animal models. We have previously published gene expression analysis of murine lung development beginning at embryonic day 12 and continuing to adulthood (23). Applying unsupervised analytical approaches, we identified global signatures of murine embryonic lung development (17). Similar studies by other groups have identified key temporal regulators of murine lung development (5) and also have identified potential mechanistic disruptions that may lead to pathological changes causing developmental diseases (19).
Genome-wide expression profiling using DNA microarrays has been previously used to identify the lung response to hyperoxia in mice (24, 27). We recently used this approach to study genome-wide expression changes in lungs of patients with BPD (2). High-throughput sequencing-based transcriptional profiling, otherwise known as RNA-seq, provides improved sensitivity, specificity, and dynamic range. In this study, we used RNA-seq to generate transcriptome-level gene expression data for neonatal lung tissue from hyperoxia-exposed neonatal mice and age-matched controls. Using standard statistical approaches, we identified genes and pathways dysregulated in murine lungs because of exposure to neonatal hyperoxia. These data identified critical regulatory factors and suggested potential mechanisms involved in disease pathogenesis.
METHODS
Animal Exposure
Mice were housed in sterile microisolator cages in a specified pathogen-free environment and exposed to superphysiological levels of oxygen as previously described (36), according to a protocol approved by the University Committee on Animal Resources at the University of Rochester. Briefly, newborn C57BL/6J mice from several litters were mixed within 6 h of birth and randomly separated into two groups. Each group was then exposed to room air (21% oxygen) or 100% oxygen until postnatal day 10. The mixed gas stream was humidified to 40–70% by passage through deionized water-jacketed Nafion membrane tubing and delivered thorough a 0.22-μm filter before passage into a sealed Lexan polycarbonate chamber. Dams were cycled between litters exposed to room air and 100% oxygen every 24 h to protect them from acute oxygen toxicity and to ensure that maternal nutrition provided to the pups exposed to 100% oxygen was similar to that provided to the pups exposed to room air.
RNA Isolation
Lung tissue collection.
Mice were anesthetized with pentobarbital sodium (100 mg/kg). Whole lung lobes were removed from hyperoxia-challenged (n = 6) and normoxic, age-matched controls (n = 8), flash-frozen in liquid nitrogen, and stored at −80°C. Frozen tissue was homogenized using TRIzol (Invitrogen), and total RNA was isolated. Isolated RNA was repurified and rendered DNA-free by an on-column DNase I treatment according to the Agilent MiniPrep protocol (Agilent Technologies, Santa Clara, CA). The quality of isolated RNA was assessed using microcapillary electrophoresis, and concentration was assessed by measuring ultraviolet absorbance. Only RNA samples with an RNA concentration >100 ng/μl and a RNA integrity number >6 were used for RNA-Seq.
RNA-Seq
Total RNA was pooled from two animals of the same treatment group for the generation of each cDNA library. Each animal contributed to a single pool. RNA samples were pooled in an effort to reduce the effects of biological variation. cDNA libraries (n = 4 for hyperoxia, n = 3 for control) were generated, and high-throughput sequencing was completed by the University of Rochester Functional Genomics Center using an Illumina Genome Analyzer II. Approximately 20 million reads were generated for each sample. Sequences were aligned to the mouse reference genome (mm10) containing 21,718 annotated genes. Count estimation was performed using ELAND (version 1.7) as implemented in CASAVA (Illumina, San Diego, CA).
Expression estimate data obtained from CASAVA were normalized using trimmed mean (TM; Eq. 1) or reads per million bases (RPM; Eq. 2), independently.
| (1) |
| (2) |
The normalized datasets were filtered to remove values indistinguishable from noise. Expression values, derived using each normalization algorithm, were assessed for undesirable and unanticipated structure or associations among the samples by unsupervised clustering with the nonparametric bootstrap (3). Differential expression of genes was assessed using Significance Analysis of Microarrays (SAM) (33) on each version of normalized data, independently. SAM, a permutation-based modification of t-test, was performed using MeV 3.0 (www.tm4.org/mev.html).
Pathway Analysis
Genes identified as differentially expressed were used for pathway analysis. Ingenuity Pathway Analysis (IPA) software was applied in an effort to identify significantly overrepresented canonical pathways, defined by Fisher's exact test at a P value of <0.05. Gene symbols for the selected genes were used as the input list to explore direct interactions among those genes in both in vivo and in vitro studies.
In Vitro Experiments
MLE15 cells were cultured under standard conditions, using DMEM medium supplemented with 10% FBS, 1% penicillin/streptomycin, 1% nonessential amino acids, 1% Fungizone, sodium pyruvate, and l-glutamine. Subconfluent cultures of MLE15 cells were exposed to 95% oxygen (5% CO2) for 48 h or grown in room air (as control). In addition, confluent cultures of MLE15 cells were treated for 48 h with either 2 nM of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in DMSO or DMSO alone (as a vehicle control). RNA samples were isolated using the Absolutely RNA Microprep Kit (Agilent).
Quantitative Reverse-Transcriptase PCR
cDNA were synthesized from 1,000 ng RNA using the iScript Reverse Transcription Kit (Bio-Rad, Hercules, CA). Quantitative reverse-transcriptase PCR (qPCR) was performed on a Viia7 (Applied Biosytems) using SYBR green chemistry (2). Primer sequences for individual genes were obtained from the MGH Primer Bank (http://www.pga.mgh.harvard.edu/primerbank). Gene expression levels (dCT) were calculated relative to the measured CT value of peptidyl prolyl isomerase A (or cyclophilin A) as an internal endogenous control and are reported as relative expression according to the ddCT method.
RESULTS
Dataset Filtering and Normalization
The dataset consisted of complete RNA-Seq transcriptome data from seven samples, three hyperoxia-challenged and four age-matched controls. To reduce noise and avoid spurious results, only data from genes that were detected as expressed in all samples were used for analysis. After filtering, the dataset contained expression values for a total of 16,079 genes. The dataset was normalized using two separate approaches (TM and RPM). We performed hierarchical clustering analysis on the raw and normalized datasets. A comparison of cluster analysis confirmed normalization leads to improvement of the data structure, whereby samples segregated according to exposure (Fig. 1). Based upon these results, we performed gene selection using normalized data only.
Fig. 1.
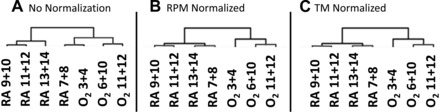
Normalization methods improved sample classification. Shown are the results of hierarchical clustering of all samples using 16,079 genes using Euclidean distance and average linkage. Samples were clustered using raw (A), reads per million bases (RPM, B)-, or trimmed mean (TM, C)-normalized data.
Gene Selection
Both normalized datasets were tested separately using SAM to identify genes differentially expressed in response to acute neonatal hyperoxia. We identified 1,019 genes that were significantly different using SAM analysis on RPM-normalized data, whereas 1,017 genes were different in TM-normalized data. A total of 798 genes were common in both analyses. Of the 798 genes identified as differentially expressed by SAM, we focused our studies on the set of genes (n = 300) that also had a magnitude of change greater than twofold (Fig. 2). A list of all 300 genes has been provided in Supplemental Table 1.
Fig. 2.
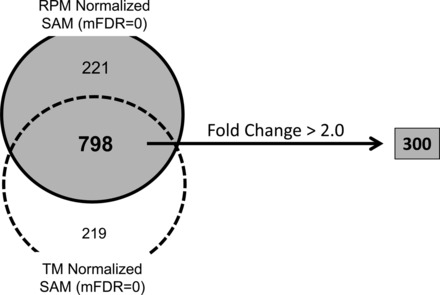
Gene selection results. A total of 798 genes were identified as significantly affected by hyperoxia exposure using Significance Analysis of Microarrays (SAM) (at median false discovery rate or mFDR = 0) on both TM- and RPM-normalized data. Of these 708 genes, 300 had a magnitude of change >2-fold.
RNA-Seq Validation
We attempted validation of differential expression for 31 of the 300 genes by qPCR. Fourteen of these genes appeared to have increased expression in response to hyperoxia, whereas 17 were predicted to have reduced expression. These genes were selected based on prior knowledge of their relevance to the disease process or lung biology. Evidence for significant differences in gene expression was confirmed in 24 of the 31 (77%) genes tested (Fig. 3). There was a clear association between the magnitude change identified by RNA-Seq and subsequent qPCR validation. Essentially all genes showing a magnitude of change greater than threefold by RNA-seq were validated by qPCR.
Fig. 3.
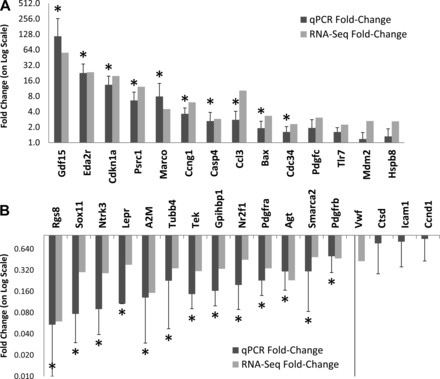
Quantitative reverse-transcriptase PCR (qPCR) validation. We tested differential expression using qPCR for 31 selected genes identified as significantly affected in hyperoxia by high-throughput sequencing-based transcriptional profiling (RNA-Seq). Significant changes in expression were confirmed for 24 out of 31 of these genes, including 14 of 17 downregulated genes (A) and 10 of 14 upregulated genes (B). Dark bars indicate relative fold change by qPCR in hyperoxia-exposed mice compared with age-matched controls, and gray bars indicate their corresponding fold change from RNA-Seq data. *Significance at P < 0.05 by t-test.
Pathway Analysis
To better identify the global effects of acute hyperoxia challenge in neonatal mice, the 300 genes (identified by SAM with a fold change greater than 2) were used for pathway analysis. IPA was used to test for canonical pathways overrepresented in these 300 genes (Fig. 4). IPA discovered biological processes previously known to be associated with acute hyperoxia, including nuclear fctor (erythryoid-derived-2)-like 2 (NRF2) activity (9) and p53 signaling (26). IPA also identified “Cellular Effects of Sildenafil”/eNOS pathways, which represents a therapeutic target in BPD (12). In addition, IPA identified pathways previously underappreciated in this model that were significantly affected in response to acute neonatal hyperoxia, including dendritic cell pathways and aryl hydrocarbon receptor (Ahr) signaling.
Fig. 4.
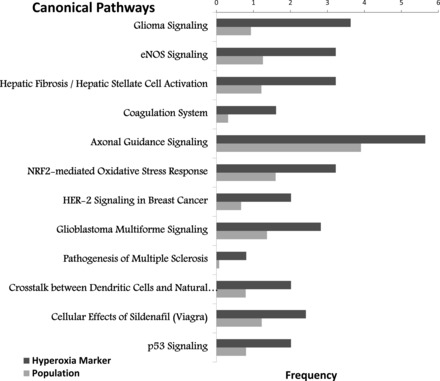
Canonical pathways affected. Pathway analysis was performed using Ingenuity Pathway Analysis (IPA) software, as described in methods. Shown are “canonical” pathways that were identified as significant by Fisher's exact t-test (P ≤ 0.01), listing the percentage of the hyperoxia-associated genes in each pathway (dark gray bar) vs. the percentage of all genes in the genome that correspond to each listed pathway (light gray bar).
Cluster analysis was used to explore the interrelationships among the differentially expressed genes. IPA generated a network including 81 of the 300 genes based on regulatory and interaction information (available in their curated database). This network contained three main centroids, or central regulatory nodes (Fig. 5). These central regulatory nodes were defined by Ccnd1, Cdkn1a, and Ahr, each of which is linked directly with 10 or more genes. Of these three central regulatory nodes, Ccnd1 and Cdkn1a appeared to be targets of regulation. Alternately, Ahr appeared to have predominantly effector functions, where it was predicted to play an instructive regulatory role. In particular, Ahr was predicted as a regulator of other connected genes, including Ccnd1, Cdkn1a, Pdgfrb, and A2M.
Fig. 5.
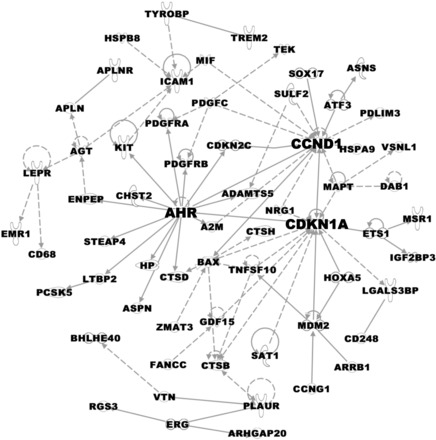
A hyperoxia-regulated gene expression network. IPA identified a network including 81 of the 300 genes (represented as nodes with Gene IDs) significantly affected in hyperoxia, based on experimental evidence of regulatory or physical interactions. This network contained three centroids, or central regulatory nodes [aryl hydrocarbon receptor (Ahr), Ccnd1, and Cdkn1a], interconnecting most of the genes regulated by hyperoxia. A solid line denotes a direct relationship, whereas a broken line denotes an indirect relationship between two genes, and the arrows indicate the direction of the interaction.
In Vitro Mechanistic Studies of Ahr
We tested the direct response of mouse lung epithelial cells to high ambient oxygen concentrations in vitro. We exposed subconfluent cultures of MLE15 cells to 95% oxygen (5% CO2) for 48 h and tested genes regulated by hyperoxia in vivo for differential expression by qPCR. Direct exposure of MLE15 cells resulted in significant changes in the expression of six genes we identified in vivo (Fig. 6). Five of these genes (Cdkn1a, Eda2r, Gdf15, Casp4, and Agt) demonstrated a robust response (>10-fold), consistent with the one observed in vivo, whereas one gene demonstrated a small response (<2-fold) in the opposite direction (Tek). These data suggest that MLE15 cells can serve as a relevant in vitro model system for the investigation of mechanistic responses to hyperoxia exposure in the neonatal murine lung.
Fig. 6.
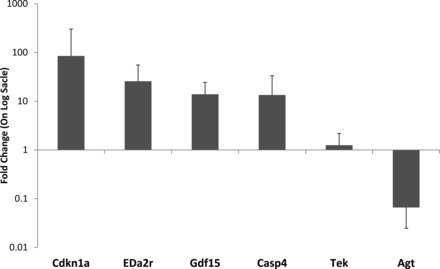
Epithelial cell model of direct response to hyperoxia. The MLE15 mouse lung epithelial cell line was treated for 48 h with 95% oxygen. Cells were harvested immediately following exposure, and gene expression was assessed by qPCR. Shown is the relative mean gene expression level (+SD) for 6 genes that were significantly different either by t-test (P < 0.05) or Mann-Whitmey U-test (MWU) (P < 0.05).
With AHR being identified as a central regulator in the hyperoxia gene network, we tested the ability of Ahr agonists to regulate the expression of selected genes in MLE15 cells by qPCR. MLE15 cells were grown under standard conditions and treated with TCDD, a specific Ahr-activating ligand, for 48 h. TCDD treatment did not affect steady-state mRNA levels for Ahr. However, treatment of MLE15 cells with TCDD resulted in modest but significant changes in expression for four of six hyperoxia-dysrelated genes tested. Treatment with 2 nM TCDD significantly increased the expression of Cdkn1a and Bax and significantly suppressed the expression of both Pdgfrb and A2m (Fig. 7).
Fig. 7.
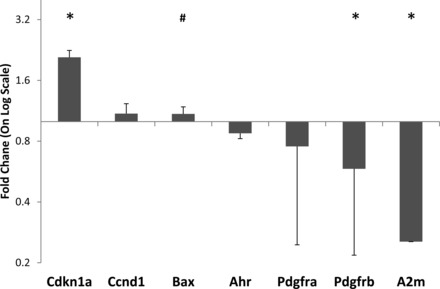
Ahr activation regulates hyperoxia-associated gene expression in lung epithelial cells. Mouse lung epithelial cells were treated for 48 h with the Ahr-activating ligand 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD, 2 mM). Shown is the relative mean gene expression level (+SD) for 6 genes tested. *Significance at P < 0.05 by t-test. #Significance at P < 0.05 by MWU.
DISCUSSION
A number of animal models, in different species, have been developed to mimic the pathophysiology of BPD. These models emulate the phenotype of chronic lung disease of the newborn either through genetic or environmental manipulation. Our group recently described a mouse model of BPD-like lung pathology resulting from compound mutation of fibroblast growth factor receptor 3 and 4, resulting in deficiencies in alveolar formation and extracellular matrix accumulation (32). Murine models of newborn chronic lung disorders include wild-type mice exposed to persistent hypoxia, which recapitulates some pathological processes observed in BPD, such as abnormal vascular remodeling (10, 24). High level of oxygen has been shown to suppress proliferation of alveolar epithelial type II cells (11). Hyperoxia exposure also impairs endothelial proliferation (13), increases expression of β-catenin and plasminogen activator inhibitor-1 (29), and reduces the number of endothelial progenitor cells in lungs of newborn mice, but not in adults (1). We have previously shown that neonatal exposure to hyperoxia alters the epithelial and vascular progenitor cells required for normal lung development and maintenance, including increasing their susceptibility to viral infections (6, 7, 35, 36). Here, applying the latest genome-wide expression profiling methodology, we attempt to improve our understanding of how neonatal oxygen exposure adversely affects lung development in children born prematurely.
We identified over 1,000 genes with some evidence for abnormal expression following acute neonatal oxygen exposure. We focused our analysis on the subset of 300 genes that were both consistently identified by multiple analytical approaches and display a substantial magnitude of change in expression. This strategy is supported by qPCR-based validation results, which demonstrate a clear relationship between the magnitude of expression changes identified using RNA-seq and reliability to confirm these expression differences as statistically significant. Interestingly, qPCR validation data suggest that our RNA-Seq data analysis methods overestimated the magnitude of gene expression induction and underestimated the magnitude of gene expression reduction. The basis for this bias in estimation is not clear.
Although a number of the genes and pathways we identified were previously described, the results of our analyses include the identification of many gene expression changes that occur in this model of BPD-like neonatal lung pathology, which have not been previously reported. We assessed the representation of canonical pathways for the genes we found to be responsive to acute neonatal hyperoxia. Our results confirmed both the efficacy of our methods and the relevance of this model, by identifying pathways previously known to be involved in this animal model, as well as in human BPD pathology. For instance, we recently reported a transcriptomic analysis of human BPD lung tissue (2) that revealed DNA damage-related pathways are highly affected in BPD. However, although murine models of neonatal exposure to hyperoxia may replicate some aspects of BPD-like pathophysiology, their relevance is yet to be completely established, since most use oxygen concentrations higher than that provided to the preterm infant. Furthermore, the experimental design of the different animal models of hyperoxia lack one critical component of therapeutic intervention, namely the fluctuations in oxygen concentrations occurring in preterm infants. These variations could potentially create alterations in gene expression and pathways not observed in the experimental settings. Regardless, the RNA-Seq data presented here helped to identify additional novel pathways affected in this model (including dendritic cell, complement/coagulation, and Ahr pathways) that may warrant further investigation.
The effects of hyperoxia exposure on lung gene expression have previously been assessed through transcriptomic analyses using DNA microarrays. Perkowski et al. used customized microarrays to identify molecular events occurring during the early response to hyperoxia exposure. Consistent with our data, they observed significant increase in p21, Bax, and reduction in thrombomodulin expression (27). McGrath-Morrow et al. used microarray analysis to demonstrate that Nrf2, a major canonical pathway identified in our data, is essential for inflammatory gene expression and inflammation, but not cell cycle-associated gene regulation or alveolar growth arrest, in neonates exposed to hyperoxia (24). Zhu et al. studied hyperoxia-induced photoreceptor degeneration in the retina of C57BL/6J mice using Affymetrix Mouse Gene ST 1.0 arrays and identified differentially expressed genes and pathways (37). Wollen et al. used the same platform to assess transcriptional changes in lung tissue from newborn mice exposed to hypoxia followed by graded during reoxygenation (through FiO2 supplementation) and observed that hyperoxic reoxygenation affects pathways regulating cell growth and survival (34). Chambellan et al. studied transcriptomic changes in human airway epithelial cells derived from subjects under ambient oxygen conditions and after breathing 100% oxygen for 12 h and identified induction of genes in the chaperone and proteasome-ubiquitin-conjugation pathways that together comprise an integrated cellular response to manage and degrade damaged proteins (8).
Aryl Hydrocarbon Receptor
In a regulatory network created using 300 hyperoxia-affected genes, the Ahr was one of three genes (Ccnd1 and Cdkn1a being the other two) with a large number of interactions. Even though both Ccnd1 and Cdkn1a had greater number of interactions, Ahr uniquely appeared to be playing an effector role. Interestingly, recent reports have implicated Ahr as a regulatory factor mediating the response for hyperoxia (15, 31). Our network model suggested that Ahr may be able to regulate many of the hyperoxia-related gene expression responses. We tested this hypothesis in vitro, treating mouse lung epithelial cells with the Ahr-activating ligand TCDD. TCDD effectively regulated three of six target genes tested, including Cdkn1a, another of the three genes with the largest number of interactions in the gene regulation network. Our data support those previous studies, expand upon them by identifying specific regulatory target genes for Ahr in the hyperoxia response, and support additional studies to assess Ahr as a therapeutic target in preclinical models of BPD-like lung pathology.
DISCLOSURES
No conflicts of interest, financial or otherwise are declared by the authors.
AUTHOR CONTRIBUTIONS
S.B., Z.Z., M.Y., A.M.L., V.A.L., S.K.S., E.R., and B.W.B. performed experiments; S.B., Z.Z., and C.-Y.C. analyzed data; S.B., C.-Y.C., S.K.S., T.J.M., and M.A.O. interpreted results of experiments; S.B. prepared figures; S.B. drafted manuscript; T.J.M. and M.A.O. conception and design of research; T.J.M. and M.A.O. edited and revised manuscript; T.J.M. and M.A.O. approved final version of manuscript.
Supplementary Material
ACKNOWLEDGMENTS
We thank Stephen L. Welle and Michelle Zanche of the Functional Genomics Center of the University of Rochester Medical Center for generating and sequencing the cDNA libraries.
REFERENCES
- 1.Balasubramaniam V, Mervis CF, Maxey AM, Markham NE, Abman SH. Hyperoxia reduces bone marrow, circulating, and lung endothelial progenitor cells in the developing lung: implications for the pathogenesis of bronchopulmonary dysplasia. Am J Physiol Lung Cell Mol Physiol 292: L1073–L1084, 2007. [DOI] [PubMed] [Google Scholar]
- 2.Bhattacharya S, Go D, Krenitsky DL, Huyck HL, Solleti SK, Lunger VA, Metlay L, Srisuma S, Wert SE, Mariani TJ, Pryhuber GS. Genome-wide transcriptional profiling reveals connective tissue mast cell accumulation in bronchopulmonary dysplasia. Am J Respir Crit Care Med 186: 349–358, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bhattacharya S, Long D, Lyons-Weiler J. Overcoming confounded controls in the analysis of gene expression data from microarray experiments. Appl Bioinformatics 2: 197–208, 2003. [PubMed] [Google Scholar]
- 4.Bhattacharya S, Mariani TJ. Systems biology approaches to identify developmental bases for lung diseases. Pediatr Res 73: 514–522, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bonner AE, Lemon WJ, You M. Gene expression signatures identify novel regulatory pathways during murine lung development: implications for lung tumorigenesis. J Med Genet 40: 408–417, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Buczynski BW, Yee M, Martin KC, Lawrence BP, O'Reilly MA. Neonatal hyperoxia alters the host response to influenza A virus infection in adult mice through multiple pathways. Am J Physiol Lung Cell Mol Physiol 305: L282–L290, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Buczynski BW, Yee M, Paige Lawrence B, O'Reilly MA. Lung development and the host response to influenza A virus are altered by different doses of neonatal oxygen in mice. Am J Physiol Lung Cell Mol Physiol 302: L1078–L1087, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chambellan A, Cruickshank PJ, McKenzie P, Cannady SB, Szabo K, Comhair SA, Erzurum SC. Gene expression profile of human airway epithelium induced by hyperoxia in vivo. Am J Respir Cell Mol Biol 35: 424–435, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cho HY, Jedlicka AE, Reddy SP, Kensler TW, Yamamoto M, Zhang LY, Kleeberger SR. Role of NRF2 in protection against hyperoxic lung injury in mice. Am J Respir Cell Mol Biol 26: 175–182, 2002. [DOI] [PubMed] [Google Scholar]
- 10.Ciuclan L, Bonneau O, Hussey M, Duggan N, Holmes AM, Good R, Stringer R, Jones P, Morrell NW, Jarai G, Walker C, Westwick J, Thomas M. A novel murine model of severe pulmonary arterial hypertension. Am J Respir Crit Care Med 184: 1171–1182, 2011. [DOI] [PubMed] [Google Scholar]
- 11.Clement A, Edeas M, Chadelat K, Brody JS. Inhibition of lung epithelial cell proliferation by hyperoxia. Posttranscriptional regulation of proliferation-related genes. J Clin Invest 90: 1812–1818, 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Donohue PK, Gilmore MM, Cristofalo E, Wilson RF, Weiner JZ, Lau BD, Robinson KA, Allen MC. Inhaled nitric oxide in preterm infants: a systematic review. Pediatrics 127: e414–e422, 2011. [DOI] [PubMed] [Google Scholar]
- 13.Grant MB, Khaw PT, Schultz GS, Adams JL, Shimizu RW. Effects of epidermal growth factor, fibroblast growth factor, and transforming growth factor-beta on corneal cell chemotaxis. Invest Ophthalmol Visual Sci 33: 3292–3301, 1992. [PubMed] [Google Scholar]
- 14.Iosef C, Alastalo TP, Hou Y, Chen C, Adams ES, Lyu SC, Cornfield DN, Alvira CM. Inhibiting NF-kappaB in the developing lung disrupts angiogenesis and alveolarization. Am J Physiol Lung Cell Mol Physiol 302: L1023–L1036, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jiang W, Welty SE, Couroucli XI, Barrios R, Kondraganti SR, Muthiah K, Yu L, Avery SE, Moorthy B. Disruption of the Ah receptor gene alters the susceptibility of mice to oxygen-mediated regulation of pulmonary and hepatic cytochromes P4501A expression and exacerbates hyperoxic lung injury. J Pharmacol Exp Ther 310: 512–519, 2004. [DOI] [PubMed] [Google Scholar]
- 16.Jobe AH, Bancalari E. Bronchopulmonary dysplasia. Am J Respir Crit Care Med 163: 1723–1729, 2001. [DOI] [PubMed] [Google Scholar]
- 17.Kho AT, Bhattacharya S, Mecham BH, Hong J, Kohane IS, Mariani TJ. Expression profiles of the mouse lung identify a molecular signature of time-to-birth. Am J Respir Cell Mol Biol 40: 47–57, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kho AT, Bhattacharya S, Tantisira KG, Carey VJ, Gaedigk R, Leeder JS, Kohane IS, Weiss ST, Mariani TJ. Transcriptomic analysis of human lung development. Am J Respir Crit Care Med 181: 54–63, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kramer EL, Deutsch GH, Sartor MA, Hardie WD, Ikegami M, Korfhagen TR, Le Cras TD. Perinatal increases in TGF-α disrupt the saccular phase of lung morphogenesis and cause remodeling: microarray analysis. Am J Physiol Lung Cell Mol Physiol 293: L314–L327, 2007. [DOI] [PubMed] [Google Scholar]
- 20.Lindner U, Tutdibi E, Binot S, Monz D, Hilgendorff A, Gortner L. Levels of cytokines in umbilical cord blood in small for gestational age preterm infants. Klin Padiatr 225: 70–74, 2013. [DOI] [PubMed] [Google Scholar]
- 21.Madurga A, Mizikova I, Ruiz-Camp J, Morty RE. Recent advances in late lung development and the pathogenesis of bronchopulmonary dysplasia. Am J Physiol Lung Cell Mol Physiol 305: L893–L905, 2013. [DOI] [PubMed] [Google Scholar]
- 22.Maniscalco WM, Watkins RH, Pryhuber GS, Bhatt A, Shea C, Huyck H. Angiogenic factors and alveolar vasculature: development and alterations by injury in very premature baboons. Am J Physiol Lung Cell Mol Physiol 282: L811–L823, 2002. [DOI] [PubMed] [Google Scholar]
- 23.Mariani TJ, Budhraja V, Mecham BH, Gu CC, Watson MA, Sadovsky Y. A variable fold change threshold determines significance for expression microarrays. FASEB J 17: 321–323, 2003. [DOI] [PubMed] [Google Scholar]
- 24.McGrath-Morrow SA, Lauer T, Collaco JM, Lopez A, Malhotra D, Alekseyev YO, Neptune E, Wise R, Biswal S. Transcriptional responses of neonatal mouse lung to hyperoxia by Nrf2 status. Cytokine 65: 4–9, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Northway WH, Jr, Rosan RC, Shahinian L, Jr, Castellino RA, Gyepes MT, Durbridge T. Radiologic and histologic investigation of pulmonary oxygen toxicity in newborn guinea pigs. Invest Radiol 4: 148–155, 1969. [DOI] [PubMed] [Google Scholar]
- 26.O'Reilly MA, Staversky RJ, Stripp BR, Finkelstein JN. Exposure to hyperoxia induces p53 expression in mouse lung epithelium. Am J Respir Cell Mol Biol 18: 43–50, 1998. [DOI] [PubMed] [Google Scholar]
- 27.Perkowski S, Sun J, Singhal S, Santiago J, Leikauf GD, Albelda SM. Gene expression profiling of the early pulmonary response to hyperoxia in mice. Am J Respir Cell Mol Biol 28: 682–696, 2003. [DOI] [PubMed] [Google Scholar]
- 28.Pierce RA, Albertine KH, Starcher BC, Bohnsack JF, Carlton DP, Bland RD. Chronic lung injury in preterm lambs: disordered pulmonary elastin deposition. Am J Physiol Lung Cell Mol Physiol 272: L452–L460, 1997. [DOI] [PubMed] [Google Scholar]
- 29.Popova AP, Bentley JK, Anyanwu AC, Richardson MN, Linn MJ, Lei J, Wong EJ, Goldsmith AM, Pryhuber GS, Hershenson MB. Glycogen synthase kinase-3β/β-catenin signaling regulates neonatal lung mesenchymal stromal cell myofibroblastic differentiation. Am J Physiol Lung Cell Mol Physiol 303: L439–L448, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sakurai R, Villarreal P, Husain S, Liu J, Sakurai T, Tou E, Torday JS, Rehan VK. Curcumin protects the developing lung against long-term hyperoxic injury. Am J Physiol Lung Cell Mol Physiol 305: L301–L311, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shivanna B, Zhang W, Jiang W, Welty SE, Couroucli XI, Wang L, Moorthy B. Functional deficiency of aryl hydrocarbon receptor augments oxygen toxicity-induced alveolar simplification in newborn mice. Toxicol Appl Pharmacol 267: 209–217, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Srisuma S, Bhattacharya S, Simon DM, Solleti SK, Tyagi S, Starcher B, Mariani TJ. Fibroblast growth factor receptors control epithelial-mesenchymal interactions necessary for alveolar elastogenesis. Am J Respir Crit Care Med 181: 838–850, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci USA 98: 5116–5121, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wollen EJ, Sejersted Y, Wright MS, Bik-Multanowski M, Madetko-Talowska A, Gunther CC, Nygard S, Kwinta P, Pietrzyk JJ, Saugstad OD. Transcriptome profiling of the newborn mouse lung after hypoxia and reoxygenation: hyperoxic reoxygenation affects mTOR signaling pathway, DNA repair, and JNK-pathway regulation. Pediatr Res 74: 536–544, 2013. [DOI] [PubMed] [Google Scholar]
- 35.Yee M, Buczynski BW, O'Reilly MA. Neonatal hyperoxia stimulates the expansion of alveolar epithelial type II cells. Am J Respir Cell Mol Biol 50: 757–766, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yee M, White RJ, Awad HA, Bates WA, McGrath-Morrow SA, O'Reilly MA. Neonatal hyperoxia causes pulmonary vascular disease and shortens life span in aging mice. Am J Pathol 178: 2601–2610, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhu Y, Natoli R, Valter K, Stone J. Microarray analysis of hyperoxia stressed mouse retina: differential gene expression in the inferior and superior region. Adv Exp Med Biol 664: 217–222, 2010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.