

Abstract
Purpose.
Inhibiting mechanistic target of rapamycin (mTOR) by pharmacological or genetic approaches can extend lifespan in mammals. The kinase activity of mTOR is controlled by upstream regulatory proteins and its subcellular localization. The purpose of this study was to characterize age-related alterations and functional consequences of mTOR signaling in the postmitotic RPE cells.
Methods.
Activity of mTOR complex 1 (mTORC1) was monitored by measuring phosphorylation status of its downstream effector protein S6, in either cultured human RPE cells or RPE explants prepared from mice at different ages. Subcellular distribution of mTOR was investigated by immunofluorescent staining of RPE culture or flatmount. The signaling of mTORC1 was modulated by either overexpression of a small guanosine triphosphatase, Ras homolog enriched in brain (Rheb), or disruption of the Ragulator complex with small interference RNA targeting p18. The effects of mTOR pathway on degradation of phagocytosed photoreceptor outer segments (POS) were determined by measuring the turnover rate of rhodopsin.
Results.
Aged RPE cells had more lysosome-associated mTOR and had increased response to amino acid stimulation. The lysosome distribution was essential for mTORC1 function, as disruption of the Ragulator complex abolished mTORC1 activation by amino acids. Increased mTORC1 activity caused decreased rate of degradation of internalized POS in the RPE.
Conclusions.
Aging changes the subcellular localization and function of mTOR in the RPE. Increased mTORC1 inhibits POS degradation and may further exacerbate lysosome dysfunction of aged RPE.
Keywords: mTOR, aging, eye, signaling, lysosome, retinal pigment epithelium
Aged RPE cells had increased lysosome distribution of mTOR and increased response to amino acid stimulation. Higher mTOR activity inhibited degradation of phagocytosed POS in the RPE.
Introduction
Negative regulation of longevity by TOR is firmly established in various invertebrate models.1 Similar roles have been ascribed to the mammalian counterpart, mTOR. Mice with reduced mTORC1 signaling had extended lifespan.2–4 This can be achieved either by knocking-down the immediate downstream effector, S6K1, or by administrating rapamycin, which is the prototypical inhibitor of mTORC1. The complex of mTORC1 mediates most of the known functions of mTOR, including protein translation and transcription, lipid biosynthesis, mitochondrial proliferation and function, ribosome biogenesis, and repression of autophagy.5 Age-dependent changes in those processes are associated with many late onset degenerative diseases, including AMD, which is a leading cause of blindness in elderly people. Manipulating mTORC1 signaling has been tested and proved to be a promising approach in experimental models of several types of neurodegenerative diseases.6
Nutrients, growth factors, energy, and stress regulate mTORC1 activity via distinct but interrelated signaling cascades. Except for nutrients, most of the upstream stimuli impact mTORC1 through the tuberous sclerosis complex (TSC) complex.7,8 The TSC represses mTORC1 by inhibiting the small guanosine triphosphatase (GTPase), Rheb, which binds to and activates mTORC1.9,10 Nutrients also require Rheb to activate mTORC1. However, instead of through the TSC complex, the signal is transduced to mTORC1 via Rag complex, a heterodimeric small GTPase containing RagA or B with RagC or D.11 During the process, translocation of mTOR to the lysosomal membrane is a crucial step and is mediated by Rag together with another multicomponent protein complex, Ragulator.11–13
The retinal pigment epithelium (RPE) is a monolayer of epithelial cells lying between the neural retina and choroid; and is critical for maintaining the integrity of the retina by providing structural, nutritional, and functional support such as phagocytosis of shed photoreceptor outer segments (POS) on daily base.14 The retinal pigment epithelium is particularly susceptible to age-related degeneration. Degenerating RPE not only displays decline of normal functions, such as damaged mitochondria and compromised antioxidant capacity,15,16 but may also show gain-of-function, such as autoimmune reactivity.17 These age-related changes create a microenvironment in the outer retina in favor of degeneration and inflammation, and contribute to the pathogenesis of AMD.
In the current study, we explored the subcellular localization and function of mTORC1 in our newly developed model of in vitro RPE aging. We found that aged RPE cells had increased lysosome distribution of mTOR, which resulted in enhanced activation of mTORC1. The in vitro findings were further confirmed in vivo. Nutrient-stimulated mTORC1 activation was abolished by knocking down p18, a component of the Ragulator complex. Furthermore, we demonstrated that mTORC1 negatively modulated degradation of phagocytosed POS. Collectively, the results suggest that changes in mTOR-related signaling networks can be a contributing mechanism to age-related RPE degeneration.
Methods
Materials
Antibodies were obtained from the following sources: anti-Nup93 and anti-Lamin B1 from Santa Cruz Biotechnology (Dallas, TX, USA); anti-ZO1 from Life Technologies (Grand Island, NY, USA); anti-LAMP1 and LAMP2 (developed by JT August and JEK Hildreth) from Developmental Studies Hybridoma Bank (DSHB) at University of Iowa; anti-actin from Sigma-Aldrich Corp. (St. Louis, MO, USA) and LI-COR Biosciences (Lincoln, NE, USA); anti-dimethylated histone H3 (di-Me-K9-H3), anti-histone macroH2A.1, and anticellular retinaldehyde–binding protein (CRALBP) from Abcam (Cambridge, MA, USA); anti-p16 from BD Biosciences (San Jose, CA, USA). All other antibodies were from Cell Signaling Technology (Danvers, MA, USA). Cell culture media and supplements were obtained from Mediatech (Manassas, VA, USA) unless stated otherwise.
Mice
Protocols for animal breeding, housing, and handling were approved by the Institutional Animal Care and Use Committee of University of Texas Medical Branch (UTMB). All mice were bred in-house. The rd8 mutation in the Crb1 gene, which causes early retinal degeneration,18 was not detected in the strains of mice used for experiments. All procedures were conducted in accordance with the ARVO statement for the Use of Animals in Ophthalmic and Vision Research. Knockout mice that are deficient of nuclear factor (erythroid-derived 2)-like 2 protein (Nrf2−/−) mice were described previously.19,20
Cell Culture and Treatment
Primary cultures of human fetal RPE (hfRPE) cells were established from five donor eyes (Advanced Bioscience Resources, Alameda, CA, USA).21 Cells were grown and passaged in alpha-modified Eagle's medium (α-MEM) containing 10% fetal bovine serum (FBS; Sigma-Aldrich Corp.); 10 mL/L N1 supplements (100×, Sigma-Aldrich Corp.); 10 mL/L MEM nonessential amino acid (100×); 1 mM sodium pyruvate; and 2 mM glutamine.22 Before seeding, the plates and wells were coated with collagen solution (STEMCELL Technologies, Inc., Vancouver, BC, Canada). Cells were maintained in a humidified CO2 incubator at 37°C and passaged when reaching 90% ~ 100% confluence. Cells between passages 7 and 11 were used for experiments. Cells cultured in α-MEM culture medium with either 5% or 10% FBS for 5 weeks had comparable morphology (Supplementary Fig. S1).
For amino acid deprivation and repletion, cells were first kept in Hank's balanced salt solution (HBSS) for 90 minutes and then an amino acid stock mixture (Sigma-Aldrich Corp.) was added to the final concentration of 1× and incubated for up to 30 minutes.21
Plasmid Construction and Transfection
The open reading frames of p18 and Rheb were amplified by PCR using cDNA prepared from hfRPE cells as template. Primer pairs used were: p18 forward, 5′-AAT GCT AGC ACC ATG GGG TGC TGC TAC AGC AGC-3′; p18 reverse, 5′-AAG AAT TCT CAC TTA TCA TCG TCG TCC TTG TAA TCT GGG ATC CCA AAC TGT ACA AC-3′; Rheb forward, 5′-AAT GCT AGC ACC ATG CCG CAG TCC AAG TCC CGG AAG-3′; Rheb Reverse, 5′-AAG AAT TCT CAC TTA TCA TCG TCG TCC TTG TAA TCC ATC ACC GAG CAT GAA GAC-3′. Amplified fragments were cloned into pLJM1-EGFP lentiviral vector (obtained from Addgene) between NheI and EcoRI sites and the inserts were verified by DNA sequencing. The plasmids were transfected into 293T cells along with packaging plasmids using a transfection reagent (Lipofectamine 2000; Life Technologies). Replication-deficient virus were collected 2 days after transfection and used to infect human fetal RPE cells.
The small interfering RNA against p18 was synthesized by Integrated DNA Technologies (Coralville, IA, USA). The targeting sequence was 5′-GGA GCU GGU UGU ACA GUU UGG GAT C-3′. Scrambled siRNA was purchased from Life Technologies. Cells were transfected with 100 pmole of siRNA duplex using a transfection reagent (Lipofectamine RNAiMAX; Life Technologies) following manufacturer's instructions. Forty eight hours after transfection, cells were used for further analyses.
In Vitro Aging of Postmitotic RPE Cells
Human fetal RPE cells from each established culture were seeded in the α-MEM culture medium for a week to reach confluence. The medium was then switched to Dulbecco's modified Eagle's medium (DMEM with 1 g/L glucose, Mediatech MT10-014) supplemented with 10% FBS. The medium was refreshed every 5 ~ 7 day for up to 4 weeks. Various biochemical and morphological markers of aging were monitored with time.
Cytochemical Staining of Senescence Associated β-Galactosidase (SA-β-gal)
In situ staining for SA-β-gal in cultured RPE cells was performed using a staining kit (Cell Signaling Technology) as previously described.21 The SA-β-Gal-positive cells (blue staining) were examined under bright field illumination.
Western Blot Analyses
Cultured cells were rinsed with ice-cold PBS twice and lysed in modified RIPA buffer (50 mM Tris-HCl [pH 7.4], 1% NP-40, 150 mM NaCl, 1 mM EDTA, 1 mM Na3VO4, 1 mM NaF) supplemented with 10 mM pyrophosphate, 10 mM glycerophosphate, and protease inhibitor cocktails (Sigma-Aldrich Corp.). Mouse RPE/choroid was dissected and separated from neural retinas and sclera. The tissue was lysed in extraction buffer containing 1:1 mixture of cell lysis reagent (CelLytic M; Sigma-Aldrich Corp.) and 2× sample buffer (Laemmli; Bio-Rad, Hercules, CA, USA) plus 10 mM pyrophosphate, 10 mM glycerophosphate, 1 mM Na3VO4, 1 mM NaF and protease inhibitor cocktails, followed by brief sonication. Samples were resolved on SDS-PAGE and transferred to nitrocellulose membranes. Membranes were probed with specific antibodies followed by appropriate fluorophore-conjugated secondary antibody. The fluorescence signals were detected by an infrared imaging system (Odyssey; LI-COR, Lincoln, NE, USA).23 Detection and quantification of band intensities were performed using imaging software (Odyssey, version 3.0; LI-COR).
RNA Isolation and RT-PCR
Total RNA was extracted using an RNA extraction kit (RNeasy Mini Kit; Qiagen Valencia, CA, USA), and the yield and purity were determined using a spectrophotometer (NanoDrop ND-1000; Thermo Fisher Scientific, Waltham, MA, USA). Complementary DNA (cDNA) was synthesized from 1 μg total RNA using oligo(dT) 15 primer (Promega, Madison, WI, USA). Primers used for gene-specific PCR amplification included: RPE65: 5′-TGC AGT TGG TGC CAG AAC TC-3′ and 5′-TGG TCT CTG TGC AAG CGT AG-3′; CRALBP: 5′-AGG CAA CAT GTC AGA AGG GG-3′ and 5′-GGG TAG CCA GCT TCA ATG GT-3′; β-actin: 5′-CTG GGA CGA CAT GGA GAA AA-3′ and 5′-AAG GAA GGC TGG AAG AGT GC-3′. Polymerase chain reaction products were resolved by 1.5% agarose gel electrophoresis and imaged with a fluorescence gel analysis system (Ultralum Omega; Biovision Technologies, Claremont, CA, USA). Quantitation of band intensities was performed with ImageJ software (http://imagej.nih.gov/ij/; provided in the public domain by the National Institutes of Health [NIH], Bethesda, MD, USA).
Tissue Preparation for Ex Vivo Amino Acid Stimulation
For preparation of eyecups, eyes were enucleated from a euthanized mouse, the anterior segment was removed, and the neural retina was separated from the posterior eyecup. Both neural retina and the posterior eyecup were incubated with HBSS supplemented with 1× amino acid mixture for 25 minutes. At the end of incubation, RPE/choroid and retina were collected and lysed for Western blot analysis of S6 phosphorylation.
Indirect Immunofluorescence Staining and Confocal Microscopy
Cultured hfRPE cells were seeded on collagen-coated cover glass and stained for mTOR or marker proteins of lysosomes.24 Detection of senescence-associate heterochromatin foci (SAHF) was performed as described in the literature.25,26 Briefly cells were fixed with 4% formaldehyde and then permeabilized with 0.2% Triton X-100. After being blocked in 3% FBS-PBS buffer, cells were incubated with antibody against either dimethylated histone H3 (di-Me-K9-H3; Abcam) or variant histone MacroH2A.1 (mH2A, Abcam),25,26 followed by appropriate secondary antibody. 4,6-Diamidino-2-phenylin (DAPI) was used for nuclei counterstaining. Cryosections of mouse eyes were prepared as described previously.20 Tissues were blocked with 5% goat serum/0.1% Triton X-100/PBS overnight at 4°C. Primary antibodies were diluted with blocking buffer and incubated overnight at 4°C followed by appropriate secondary antibody. Nuclei were then counterstained with Hoechst 33342 or DAPI (Life Technologies) and the sample was mounted on positively charged glass slide with mounting medium (Electron Microscopy Service, Hatfield, PA, USA). For RPE/choroid whole mount, phalloidin (Life Technologies) was used to delineate boundaries of the RPE.27 Neighboring section or flatmount from the other eye of the same animal without staining was used to control RPE autofluorescence. Confocal microscopy was carried out with a commercial imaging system (Zeiss LSM510; Carl Zeiss Microscopy, Thornwood, NY, USA) provided by the Optical Microscopy Core of UTMB Biomedical Imaging Network. For each experiment, the confocal setting was adjusted based on unstained samples to eliminate RPE autofluorescence and the same settings were applied to all samples for comparison. For quantitation of colocalization, Pearson's correlation coefficient was calculated with the JACoP plugin within ImageJ software (NIH).28 At least 50 cells were analyzed for each experimental group.
Staining of Lysosomes by DQ Green BSA
DQ-BSA (Life Technologies) is a self-quenched fluorescent dye that emits fluorescence upon being digested by lysosomal proteases. The probe was used to visualize the lysosome mass and morphology in young and aged hfRPE cells. Cells seeded on chambered cover glass were loaded with DQ-BSA at a final concentration of 20 μg/mL, and incubated in culture medium for 5 hours. After washing with PBS, cells were kept in Eagle's balance salt solution (Sigma-Aldrich Corp.) for live cell imaging by confocal microscopy.
Phagocytosis Assays
Photoreceptor outer segments were isolated from porcine eyes (obtained from a local slaughterhouse) according to established methods published by Papermaster et al.29 Confluent RPE cells were loaded with POS at 1:10 ratio (RPE to POS) for 16 hours (pulse). Unbound POS were removed by washing cells with PBS containing 0.2 mM CaCl2 and 1 mM MgCl2 (PBS-CM) three times.30 Cells were then cultured in fresh medium without POS and incubated for the indicated time (chase). Degradation of phagocytosed POS was determined by monitoring the level of remaining opsin with Western blot analysis.
Results
Developing a Postmitotic Aging Model of Cultured Human RPE Cells
Adult human RPE cells normally do not proliferate. To better address their age-associated changes and to explore the underlying molecular mechanisms, we developed an in vitro postmitotic aging model. Cells were first kept in nutrient-rich α- MEM culture medium22 for 1 week to reach confluence and subsequently switched to DMEM culture medium (containing 1 g/L glucose) for additional 4 weeks. Various markers of early and deep senescence were measured to monitor the RPE aging process in vitro.31 While α- MEM medium is optimized for long-term RPE culture, DMEM medium is suboptimal and promoted aging of the postmitotic RPE cells. As shown in Figure 1, early senescence markers, such as increased SA-β-Gal activity and elevated level of CDK inhibitor p16,32,33 were present in both conditions (Figs. 1A, 1B), indicating growth arrest in the postmitotic cells. Cells cultured in DMEM medium for 2 weeks had increased cell size compared with cells remained in α-MEM culture medium (Fig. 1A). After 4 weeks, cells in DMEM medium developed markers of deep senescence, including decreased level of nuclear envelope proteins Lamin B1 and Nup93 (Figs. 1B, 1C), decreased expression of RPE marker proteins RPE 65 and CRALBP (Figs. 1D–G), and appearance of SAHF which were stained by modified histone proteins (Fig. 1H).25,26,34–36 In contrast, cells maintained in α-MEM culture medium did not present those markers of deep senescence (Figs. 1B–H). Decreased Lamin B1 (Fig. 2B) and the appearance of SAHF (Fig. 2A) in the RPE were further confirmed in 16 month-old Nrf2-deficient mice, indicating that some of the aging markers can be validated in vivo. As demonstrated by our own group and others, the Nrf2−/− mice have accelerated aging and shortened lifespan.20,37 Their retinal pigment epithelium started to show age-related degenerative pathology around 12 months of the age.20 Thus, by combined morphological, biochemical, cytochemical, and immunohistochemical measurements, we demonstrated that postmitotic RPE cells cultured under less optimal conditions can develop accelerated aging. We also measured Lamin B1 in RPE of wild-type mice at different ages (Fig. 2C), and found a trend but not statistically significant decrease at 19 to 24 months.
Figure 1.

Accelerated aging in postmitotic RPE cells cultured under suboptimal condition. (A) Confluent hfRPE cells were cultured in either α-MEM or DMEM culture medium for the indicated time. Activity of SA-β-gal was stained at pH 6.0. (B) Western blot analyses of nuclear envelope proteins. Data quantitation is presented in (C). (D) Reverse transcription–PCR analyses of RPE65 and CRALBP expression in cells at different stages of aging. Data quantitation is presented in (E). (F) Western blot analyses of CRALBP. (G) Quantitation of Western data. Data are the averages of three to five independent experiments (mean ± SEM; *P < 0.05; **P < 0.01, one-way ANOVA and Dunnett's post hoc test). (H) Immunofluorescent staining of heterochromatin foci (arrows), enriched with methylated histone H3 (H3K9Me) and MacroH2A.1. Scale bar: 20 μm.
Figure 2.

Validation of RPE senescence markers in vivo. (A) Immunofluorescent staining of methylated histone H3 foci on RPE flatmount prepared from Nrf2-deficient mice at either age 4 or 16 months. (B, C) Western blot analyses of amin B1 in RPE isolated from Nrf2-deficient (B) or WT mice (C) at the indicated age (*P < 0.05, Student's t-test).
Dysregulated mTORC1 Signaling in Aged RPE
In mammalian cells mTOR-mediated signaling pathways are central regulators of aging. We measured the phosphorylation status of ribosomal protein S6 as an indicator of mTORC1 activity, before and after the in vitro aging of the RPE. As shown in Figure 3A, persistent activation of mTORC1 was detected in aged RPE cells after cultured in DMEM medium for 4 weeks. Such results are consistent with our previous finding from replicatively senescent RPE cells that showed more robust response of mTORC1 to nutrient stimuli.21 We then assessed the in vivo changes of mTORC1 signaling in the RPE. To compare basal activity of mTORC1, RPE was harvested from young (4 ~ 5 months) and old (12 ~ 16 months) Nrf2−/− mice, and S6 phosphorylation was measured by Western blot analyses. As shown in Figure 3B, the ratio of phosphorylated S6 to total S6 was significantly higher in the RPE of old mice, confirming an age-related increase of mTORC1 activity in the RPE in vivo. To further investigate the responses of mTORC1 to nutrients, we performed ex vivo treatment with RPE explants prepared from young and old Nrf2-deficient mice. Mouse eyecups were stimulated with amino acid mixture for 25 minutes and then the RPE/choroid was collected for analysis. As shown in Figure 3C, nutrient stimulation activated mTORC1 in the RPE and the S6 phosphorylation was more pronounced in RPE of older mice. Increased S6 phosphorylation was also evident in the RPE from wild-type (WT) mice at 19 to 24 months of age (Fig. 3D).
Figure 3.

Age-related changes of mTORC1 activity in the RPE, in vitro and in vivo. (A) Cultured RPE cells in DMEM medium for 4 weeks were stimulated with amino acid mixture for 15 or 30 minutes. The phosphorylation level of S6 was determined by Western blot analyses. Data presented are the averages of six independent experiments (mean ± SEM; *P < 0.05, **P < 0.01, Student's t-test). (B) Basal level of S6 phosphorylation in RPE/choroid tissues isolated from Nrf2-deficient mice at the indicated ages. (C) Ex vivo activation of mTORC1 in the RPE. Eyecups prepared from Nrf2-deficient mice at different ages were treated with amino acid mixture for 25 minutes. Activity of mTORC1 was assessed by the phosphorylation status of S6. Data presented are the averages of three separate experiments (*P < 0.05, Student's t-test). (D) Basal level of S6 phosphorylation in RPE/choroid tissues isolated from wild-type mice at the indicated ages. Data presented are the averages of four independent experiments (mean ± SEM) (*P < 0.05, one-way ANOVA, and Dunnett's post hoc test). (E) Immunostaining of phosphorylated S6 (green) on cryosections of posterior poles prepared from Nrf2-deficient mice at 6 and 14 months. Control IgG staining was presented in the lower panel. CH, choroid. Scale bar: 20 μm. (F) Activity of mTORC1 in liver or retina from young and old Nrf2-deficient mice.
We further performed immunostaining of cryosections of eyes from Nrf2-deficient mice at different ages. More intense staining of phosphorylated S6 was detected in the RPE layer from old mice, after justifying autofluorescence with unstained adjacent sections (Fig. 3E). Notably the immunofluorescence signal was uneven on cryosections and some RPE cells displayed higher S6 phosphorylation. Such data suggested that the processes of RPE aging and the related mTOR changes occurred at different pace in RPE cells of the same eye. The activity of mTORC1 in the retina and liver was not significantly affected by age (Fig. 3F). Thus, with both in vitro and in vivo systems, we found augmented mTORC1 activity in aged RPE cells.
Increased Lysosomal Distribution of mTOR in Aged RPE
The lysosome has recently emerged as an integral part of mTORC1 signaling, and the recruitment of mTORC1 to the lysosomal surface is a prerequisite for its activity.12–14,36–38 The alterations of mTORC1 activity in aged RPE promoted us to explore potential age-related changes of subcellular distribution of mTOR in the RPE. In the in vitro culture system, the lysosome content was higher in aged RPE, as indicated by more preeminent staining of lysosome-associated membrane protein 2 (LAMP2; Fig. 4A) and LAMP1 (Fig. 4C). As an alternative approach, we used DQ Green BSA, a self-quenched lysosome probe,38 for live cell imaging. Consistent with the changes in LAMP1/2 staining, aged cells harbored more fluorescent DQ-BSA–contained vesicles than young cells (Fig. 4A). For more quantitative comparisons, Western blot analyses were used to measure the protein level of LAMP1 and LAMP2 (Fig. 4B). The amounts of both proteins were considerably higher in aged cells.
Figure 4.
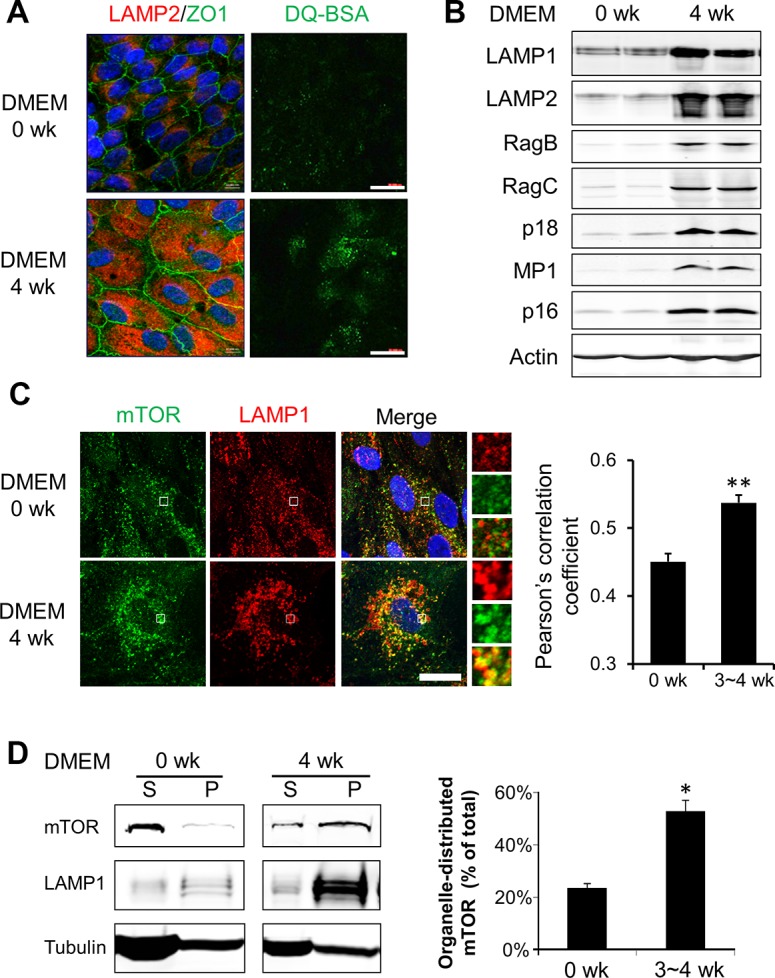
Increased lysosome distribution of mTOR in aged RPE cells in vitro. (A) Left: Immunofluorescent staining of lysosome membrane protein LAMP2 and junction-associated protein ZO-1 in young and aged hfRPE cells. Right: live cell staining with lysosome probe DQ-BSA. Scale bar: 20 μm. (B) Western blot analyses of protein components of the Ragulator-Rag complex in cultured RPE cells. (C) Colocalization of mTOR (green) and LAMP1 (red) in young and aged RPE cells (0 and 3 ~ 4 weeks in DMEM culture medium, respectively). (Insets showed an enlarged field of interest). Pearson's correlation coefficient was calculated from images from >50 cells to quantify the colocalized signals. **P < 0.01, Student's t-test. (D) Western blot analyses of soluble (S) and organelle-associated (P) mTOR in young and aged RPE cells. Cells were permeabilized by saponin and fractionated into cytosolic (S) and organelle-enriched (P) fractions. Data presented are the averages of four separate experiments. *P < 0.05, Student's t-test.
Given the importance of lysosome distribution of mTOR for its function, we next assessed the subcellular localization of mTOR in cultured RPE cells. As shown in Figure 4C, mTOR displayed a more diffused staining pattern in young cells; while with aging, more mTOR was found on the lysosomal membrane, as indicated by increased colocalization with LAMP1. Pearson's correlation coefficient, which is an indicator of the overlapping fluorescence, was significantly higher in aged RPE cells (Fig. 3C). We further used biochemical approaches to investigate the distribution of mTOR in young and aged cells. Cells were treated with a mild detergent saponin to selectively permeabilize plasma membrane and release cytosolic proteins without disrupting lysosomes.38 As shown in Figure 4D, in young RPE cells mTOR was mostly present in the soluble fraction released by saponin. In aged cells, however, there was a shift from the cytosolic pool to the saponin-resistant organelle pool, which was consistent with the results of immunostaining (Fig. 4C).
We next extended our studies to the in vivo system. Similar to cells in culture, RPE from old Nrf2-deficient mice aged 12 to 16 months showed higher amounts of lysosome marker proteins LAMP1 and LAMP2 on both Western blots (Fig. 5A) and immunostaining of RPE/choroid flatmount (Fig. 5B). Moreover, there was a significant increase of colocalization between mTOR and LAMP1 (Fig. 5B). Similar changes were observed in wild-type mice aged 19 to 24 months (Fig. 5C). Thus, in both in vitro and in vivo systems, aged RPE cells had higher lysosome content and increased distribution of mTOR to the lysosome.
Figure 5.

Increased lysosome distribution of mTOR with RPE aging in vivo. Western blot analyses of LAMP1/2 and Rag-Ragulator complex proteins in RPE isolated from Nrf2-deficient mice (A) and wild-type mice at indicated ages (C). (B) Colocalization of mTOR (green) and LAMP1 (red) on immunofluorescent staining of RPE flatmount. Insets showed the enlarged field of interest. Pearson's correlation coefficient was determined to quantify the colocalization of signals. Scale bar: 20 μm. Data presented are representative of four separate experiments. *P < 0.05, **P < 0.01, Student's t-test.
Heterodimeric small GTPase Rag proteins, in complex with lysosome-anchored protein complex Ragulator, mediate translocation of mTOR to the lysosome surface.11–13,39 We measured the protein levels of certain components of the Rag GTPase11 and Ragulator complex.12 As shown in Figure 4B, Rag B, Rag C, p18 and MP1 all had elevated levels in aged RPE cells in culture. Similar changes were detected in RPE tissue from either Nrf2-deficient (Fig. 5A) or wild-type mice at advanced age (Fig. 5C).
Among the proteins examined, p18 is the only one with lysosome anchor signal and is critical for lysosome distribution of the Ragulator complex.12 Knocking down p18 with siRNA in cultured RPE cells not only diminished p18, but also MP1, another subunit of Ragulator, suggesting p18 affected the stability of the Ragulator proteins12 (Figs. 6A, 6B). Reduction in the p18 level led to loss of lysosomal distribution of mTOR (Fig. 6A) and blunted stimulation of mTORC1 in response to amino acids in RPE cells (Fig. 6B). Putting together, these data suggested that the Rag-Ragulator complex controls the lysosome distribution of mTORC1 in RPE cells.
Figure 6.
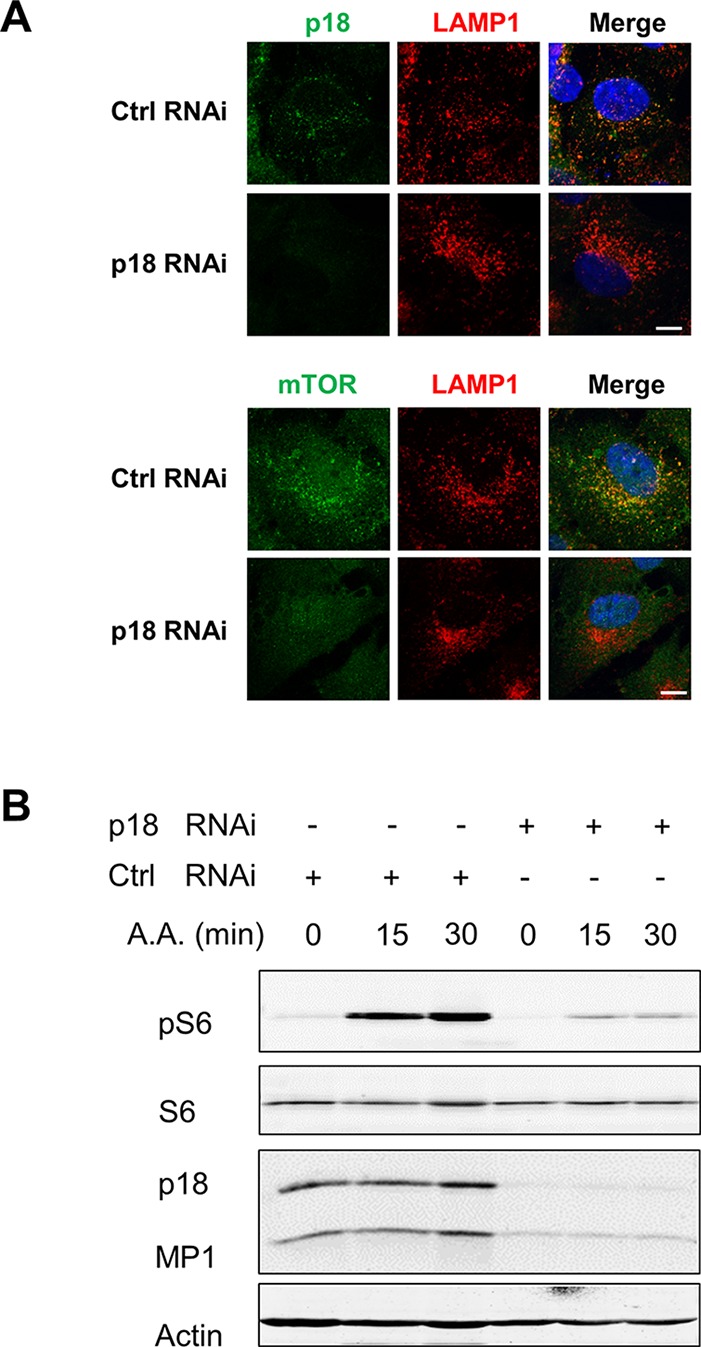
Requirement of p18 for lysosomal distribution and activity of mTOR. (A) Immunofluorescent staining of p18 and LAMP1 in RPE cells transfected with either control scrambled siRNA or siRNA targeting p18. Scale bar: 10 μm. (B) Amino acid-induced mTORC1 activation in RPE cells with p18 knockdown. Cells transfected with control or p18 siRNA were treated with amino acids for 15 and 30 minutes. The protein levels of p18 and MP1, and S6 phosphorylation were measured by Western blot analyses. Data presented are representative of three separate experiments.
mTORC1 as a Negative Regulator of POS Turnover in the RPE
To explore potential functional consequences of elevated mTOR activity in the RPE, we overexpressed p18 or/and Rheb, both of which are mTORC1 activators. As anticipated, cells transduced with either Rheb or p18 showed higher S6 phosphorylation and coexpression of Rheb and p18 led to more pronounced mTORC1 activation (Fig. 7A). We then looked at the degradation rate of internalized POS in those cells. Cells were first loaded with POS at 10:1 ratio (POS/RPE). After removing unbound POS by stringent washes, cells were incubated in media without POS and allowed to chase for 8 hours, when at least 80% of internalized POS were degraded. As shown in Figure 7B, there was a remarkable blockade in digestion of POS in cells transduced with Rheb, p18 or Rheb plus p18, as measured by the amount of remaining rhodopsin. The efficiency of POS degradation was negatively associated with mTORC1 activity. Cells overexpressed with both p18 and Rheb had the highest mTORC1 activity and the slowest rate of POS degradation. We further measured the POS turnover rates in cultured young and aged RPE cells (Fig. 7C). Consistent with their higher mTORC1 activity, aged RPE cells were much less efficient in degrading internalized POS and retained nearly 60% of rhodopsin at 8 hours after the initial loading.
Figure 7.

Mechanistic target of rapamycin complex 1 negatively regulated degradation of phagocytosed POS. Retinal pigment epithelium cells were transduced with vector, p18, Rheb, or p18 with Rheb. Overexpression of exogenous proteins and activation of mTOR were confirmed by Western blot analyses (A). (B) Rate of POS degradation in RPE cells with elevated mTORC1 activity. Cells loaded with POS were chased for 8 hours and the amount of remaining rhodopsin was determined by Western blot analyses. Data presented are the averages of three independent experiments (mean ± SEM; *P < 0.05, **P < 0.01, ***P < 0.001, one-way ANOVA, and Dunnett's post hoc test). (C) Rate of POS degradation in young and aged RPE cells in culture. After being fed with POS, young and aged RPE cells (0 and 3 ~ 4 weeks after being switched to DMEM culture medium) were allowed to chase for 8 hours. The amount of remaining rhodopsin was determined by Western blot analyses. Data presented are the averages of three independent experiments (mean ± SEM; *P < 0.05, Student's t-test).
Discussion
Cultured RPE cells have been an invaluable tool for characterizing their unique physiological functions such as tight junction,40,41 phagocytosis and autophagy42,43 and immune regulation.44,45 One of the goals of our current study was to develop an in vitro culture model that can be used to probe mechanisms of RPE aging. We found that by replacing α-MEM with the less optimal DMEM culture medium, confluent RPE cells gradually developed markers of aging and lost their functionality in a relatively short period of time (Fig. 1). The aging markers appeared around 2 weeks in DMEM culture medium and continued to progress. After 4 weeks the majority of cells lost proliferative potential and could not be replated.
Oncogene-induced senescence is the most commonly used approach to study cellular aging processes in vitro46; and literature studies have identified a number of established markers of early and deep senescence.47,48 Currently, there is no single marker that can be used to conclusively prove senescence of cultured cells47; the consensus is to use a combination of various biochemical and morphological approaches to define cell remodeling and functional alterations caused by aging. Retinal pigment epithelium cells maintained in DMEM medium for 4 weeks had decreased Nup93 and Lamin B1 (Fig. 1C), indicating disassembly of the nuclear pore complex as a marker of deep senescence.34–36,49 The levels of RPE65 and CRALBP decreased in aged RPE cells in culture (Figs. 1D–G). We also detected an increase in H3K9Me-enriched heterochromatic foci (Fig. 1H). The accumulation of heterochromatin has been well documented in aging mouse tissues and postmitotic neuron.50,51 Using criteria of (1) increased cell size; (2) increased lysosome content; (3) appearance of SA-β-Gal and increased p16; (4) loss of nuclear envelope protein Nup93 and Lamin B1; (5) formation of SAHF; (6) loss of RPE-specific markers RPE65 and CRALBP; and (7) permanent loss of proliferative potential, we conclude that RPE cells in our model developed deep senescence in vitro. Although loss of epithelial markers and cell function can happen in both dedifferentiation and aging, the cell fate is distinct between the two processes. Numerous literature reports have demonstrated that dedifferentiation in mammalian cells is a reprogramming of differentiated cells to pluripotency for proliferation and regeneration.52–54 Aging, on the other hand, is defined by the loss of proliferative potential and eventually cell death. In our culture system, confluent RPE cells maintained in DMEM medium were unable to proliferate after 4 weeks, even after replating. They did not express genes associated with early development (Supplementary Fig. S2). Thereby, the observed changes in cell phenotype were caused by aging instead of dedifferentiation.
There are certain limitations in our in vitro aging system of the RPE. We used 10% FBS to support long-term RPE culture. The serum in the culture medium can influence RPE tight junction55 and gene expression.56 Future studies will be needed to identify serum replacement57 with defined supplements for the postmitotic culture system. Early senescence markers, such as SA-β-Gal staining and increased p16 level, are controlled by status of cell proliferation and growth, and should be interpreted together with markers of deep senescence. In fact, a number of previous studies reported that SA-β-Gal activity would increase substantially in cells cultured under confluent condition and was not required for senescence.58–62 Identifying definitive marker(s) of RPE aging, both in vitro and in vivo, will greatly facilitate the research in this field. Also signaling pathways of the RPE cells can be distinct from oncogene-transformed cancer cells. For instance, histone 3 K9 methylation requires phosphorylation and activation of retinoblastoma protein,63,64 and its appearance in SAHF can be different between cancer cells and non-transformed RPE cells. Some previous studies suggested that the heterochromatin foci may represent pericentromeric heterochromatin.51 Regardless of those limitations, further improvement of this in vitro aging system can potentially develop it into a novel tool for studying the biology of RPE aging.
We found that, with aging, there was increased mTORC1 activity (Fig. 3), along with increases in lysosome mass and lysosomal localization of mTOR (Figs. 4, 5). Recently, the lysosome distribution has been shown to be required for mTORC1 activity.12,65 Similar to previously reported findings from other types of cells, the RPE used Rag-Ragulator complex to target mTORC1 to lysosomes (Fig. 6). Moreover, key components of the Rag-Ragulator complex were upregulated at protein levels in both aged RPE cells in culture (Fig. 4) and RPE tissue from old animals (Fig. 5). The increased lysosome anchoring machinery likely accounted for the accumulation of lysosome-distributed mTOR and augmented mTOR activity in old RPE.
In both in vitro and in vivo systems of postmitotic RPE aging, we detected an increase in lysosome content and accumulation of lysosomal mTOR (Figs. 4, 5). Although the causative relationship between lysosome expansion and mTOR redistribution and activation remains to be established, the two events in unison could be of great significance in development of phenotypic changes in aging cells. Expansion of lysosomal compartment has long been known to occur in replicative senescent cells and several aging tissues in vivo.58,66–69 Recently, Narita et al.70 showed an induction of lysosome biogenesis in oncogene-induced senescent cells, as well as spatial association of mTOR with lysosomes. The association was thought to couple protein degradation with new protein synthesis, and lead to active protein turnover demanded by the drastic phenotypic transitions during aging. In our current study we did not measure the activities of lysosomal hydrolases, such as cathepsin D, in aged mice tissue. Thereby, it is unclear whether the increased lysosome content was due to expansion of functional lysosomes or accumulation of nonfunctional secondary lysosomes.
Mechanistic target of rapamycin is a ubiquitously expressed kinase, but its regulation is tissue- and cell-type specific. We only observed the age-associated changes of mTORC1 in the RPE, but not in the neural retina or liver from the same animal (Fig. 3F). The activity of mTORC1 can be stimulated by other upstream signaling such as growth factors and cellular energy status. Our current study mainly focused on amino acids which are the prerequisite for other pathways. The retinal pigment epithelium is part of the blood-retina barrier and is highly active in metabolism and nutrient transport. Age-dependent increase of mTORC1 activity may reflect metabolic needs to maintain the balance between catabolism and anabolism. Thickening of Bruch's membrane occurs with age, accompanied with changes in its components such as accumulation of the advanced glycation end products.71 The efficiency of amino acid transport across the Bruch's membrane tends to decrease with aging.72 The retinal pigment epithelium may compensate for such changes by increasing the amino acid sensing via mTOR signaling, and the resulted downstream effects can play causative roles that eventually lead to RPE degeneration. Such notion is supported by a previous report which showed that administration of rapamycin attenuates progressive RPE and retinal degeneration in mice with RPE-specific deletion of mitochondrial transcription factor A.27
The activity of mTORC1 is inversely related to longevity. When the cell cycle is blocked, mTOR drives senescence instead of growth.5 How downstream effector proteins of mTOR regulate aging and age-related degeneration of the RPE remains elusive. One of the novel findings from our present study was that mTORC1 activity negatively regulated degradation of phagocytosed POS in the RPE (Fig. 7). Upregulation of mTORC1 activity was achieved by overexpressing either its activator Rheb or the trafficking protein p18 to force its lysosome distribution. Both approaches induced mTORC1 activation and delayed digestion of POS (Fig. 7). When the cells coexpressed Rheb and p18, they had higher mTORC1 activation and slower degradation of POS compared with cells overexpressing individual plasmid. The results demonstrated an inverse relationship between mTORC1 activity and the ability of RPE to degrade POS. Our finding is consistent with results from a recent study by Sinha's group that showed mTORC1 pathway was involved in regulating lysosomal clearance in RPE of Cryba1 conditional knockout mice.43
Aging is controlled by multiple levels of genetic and epigenetic mechanisms.73 Particularly to the RPE cells, oxidative stress is an important contributing factor to their aging. Oxidants can decrease RPE-specific gene expression74 and advanced glycation end products, generated by oxidative damage, accumulate in Bruch's membrane with age.71 Our results (Figs. 3B, 3D) showed that increased mTORC1 activity occurred earlier in Nrf2-deficient RPE than in wild-type RPE. Nrf2 mediates the transcription of antioxidant genes. Chronic oxidative stress in Nr2−/− RPE can cause mitochondrial dysfunction and proteolytic stress,75,76 both of which may influence the mTOR signaling. Future studies can be needed to further decipher the interactions between oxidative stress and mTOR pathways in the RPE.
The beneficial effects of rapamycin in extending lifespan and ameliorating age-related degenerative changes have been well acknowledged; however, there is emerging evidence that administration of rapamycin may not be a universal strategy in treating age-related diseases.4,6,77 A recent clinical trial conducted by the National Eye Institute showed that administration of rapamycin to patients with advanced form of atrophic AMD had no beneficial outcomes in preventing the progression of the disease. Instead, it may be associated with the adverse effects on visual acuity.77 The unfavorable effects may be resulted from the well-known fact that long-term treatment of rapamycin causes aberrant mTORC2/Akt signaling in certain tissues. The disrupted mTORC2/Akt signaling may lead to negative effects independent of longevity.78,79 For treatment of chronic diseases like AMD, agents selectively targeting mTORC1 pathway will be better solutions. On the other hand, persistent mTOR activation in degenerated RPE cells may be a compensatory response to metabolic stress.80 Because rapamycin can slow the aging process, it could be more effective for prevention rather than treatment of advanced AMD.
In summary, we have identified aging-associated changes in mTORC1 location and activity in the RPE. We also showed that mTOR participated in the regulation of phagocytosis, a unique and major function of the RPE. Future studies are warranted to further decipher mTOR signaling and pinpoint downstream effectors linking mTOR to phagocytosis and other important physiological functions of the RPE.
Acknowledgments
Supported by NIH Grants EY019706 (YC), EY021937 (JC) and EY07892 (PS); the International Retina Research Foundation; BrightFocus Foundation; Ted Nash Long Life Foundation; and Research to Prevent Blindness, Inc.
Disclosure: B. Yu, None; P. Xu, None; Z. Zhao, None; J. Cai, None; P. Sternberg, None; Y. Chen, None
References
- 1. Kenyon CJ. The genetics of ageing. Nature. 2010; 464: 504–512. [DOI] [PubMed] [Google Scholar]
- 2. Harrison DE, Strong R, Sharp ZD, et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. 2009; 460: 392–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Selman C, Tullet JM, Wieser D, et al. Ribosomal protein S6 kinase 1 signaling regulates mammalian life span. Science. 2009; 326: 140–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wilkinson JE, Burmeister L, Brooks SV, et al. Rapamycin slows aging in mice. Aging Cell. 2012; 11: 675–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011; 12: 21–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bove J, Martinez-Vicente M, Vila M. Fighting neurodegeneration with rapamycin: mechanistic insights. Nat Rev Neurosci. 2011; 12: 437–452. [DOI] [PubMed] [Google Scholar]
- 7. Manning BD, Tee AR, Logsdon MN, Blenis J, Cantley LC. Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/Akt pathway. Mol Cell. 2002; 10: 151–162. [DOI] [PubMed] [Google Scholar]
- 8. Inoki K, Li Y, Zhu T, Wu J, Guan KL. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol. 2002; 4: 648–657. [DOI] [PubMed] [Google Scholar]
- 9. Garami A, Zwartkruis FJ, Nobukuni T, et al. Insulin activation of Rheb, a mediator of mTOR/S6K/4E-BP signaling, is inhibited by TSC1 and 2. Mol Cell. 2003; 11: 1457–1466. [DOI] [PubMed] [Google Scholar]
- 10. Castro AF, Rebhun JF, Clark GJ, Quilliam LA. Rheb binds tuberous sclerosis complex 2 (TSC2) and promotes S6 kinase activation in a rapamycin- and farnesylation-dependent manner. J Biol Chem. 2003; 278: 32493–32496. [DOI] [PubMed] [Google Scholar]
- 11. Sancak Y, Peterson TR, Shaul YD, et al. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science. 2008; 320: 1496–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sancak Y, Bar-Peled L, Zoncu R, Markhard AL, Nada S, Sabatini DM. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell. 2010; 141: 290–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kim E, Goraksha-Hicks P, Li L, Neufeld TP, Guan KL. Regulation of TORC1 by Rag GTPases in nutrient response. Nat Cell Biol. 2008; 10: 935–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Strauss O. The retinal pigment epithelium in visual function. Physiol Rev. 2005; 85: 845–881. [DOI] [PubMed] [Google Scholar]
- 15. Feher J, Kovacs I, Artico M, Cavallotti C, Papale A. Balacco Gabrieli C. Mitochondrial alterations of retinal pigment epithelium in age-related macular degeneration. Neurobiol Aging. 2006; 27: 983–993. [DOI] [PubMed] [Google Scholar]
- 16. Davies S, Elliott MH, Floor E, et al. Photocytotoxicity of lipofuscin in human retinal pigment epithelial cells. Free Radic Biol Med. 2001; 31: 256–265. [DOI] [PubMed] [Google Scholar]
- 17. Chen H, Liu B, Lukas TJ, Neufeld AH. The aged retinal pigment epithelium/choroid: a potential substratum for the pathogenesis of age-related macular degeneration. PLoS One. 2008; 3: e2339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mattapallil MJ, Wawrousek EF, Chan CC, et al. The Rd8 mutation of the Crb1 gene is present in vendor lines of C57BL/6N mice and embryonic stem cells, and confounds ocular induced mutant phenotypes. Invest Ophthalmol Vis Sci. 2012; 53: 2921–2927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chan K, Lu R, Chang JC, Kan YW. NRF2, a member of the NFE2 family of transcription factors, is not essential for murine erythropoiesis, growth, and development. Proc Natl Acad Sci U S A. 1996; 93: 13943–13948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhao Z, Chen Y, Wang J, et al. Age-related retinopathy in NRF2-deficient mice. PLoS One. 2011; 6: e19456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chen Y, Wang J, Cai J, Sternberg P. Altered mTOR signaling in senescent retinal pigment epithelium. Invest Ophthalmol Vis Sci. 2010; 51: 5314–5319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Maminishkis A, Chen S, Jalickee S, et al. Confluent monolayers of cultured human fetal retinal pigment epithelium exhibit morphology and physiology of native tissue. Invest Ophthalmol Vis Sci. 2006; 47: 3612–3624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chen Y, Cai J, Jones DP. Mitochondrial thioredoxin in regulation of oxidant-induced cell death. FEBS Lett. 2006; 580: 6596–6602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chen Y, Sternberg P, Cai J. Characterization of a Bcl-XL-interacting protein FKBP8 and its splice variant in human RPE cells. Invest Ophthalmol Vis Sci. 2008; 49: 1721–1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Narita M, Nunez S, Heard E, et al. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell. 2003; 113: 703–716. [DOI] [PubMed] [Google Scholar]
- 26. Zhang R, Poustovoitov MV, Ye X, et al. Formation of MacroH2A-containing senescence-associated heterochromatin foci and senescence driven by ASF1a and HIRA. Dev Cell. 2005; 8: 19–30. [DOI] [PubMed] [Google Scholar]
- 27. Zhao C, Yasumura D, Li X, et al. mTOR-mediated dedifferentiation of the retinal pigment epithelium initiates photoreceptor degeneration in mice. J Clin Invest. 2011; 121: 369–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gordiyenko NV, Fariss RN, Zhi C, MacDonald IM. Silencing of the CHM gene alters phagocytic and secretory pathways in the retinal pigment epithelium. Invest Ophthalmol Vis Sci. 2010; 51: 1143–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Papermaster DS, Dreyer WJ. Rhodopsin content in the outer segment membranes of bovine and frog retinal rods. Biochemistry. 1974; 13: 2438–2444. [DOI] [PubMed] [Google Scholar]
- 30. Mao Y, Finnemann SC. Analysis of photoreceptor outer segment phagocytosis by RPE cells in culture. Methods Mol Biol. 2013; 935: 285–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Baker DJ, Sedivy JM. Probing the depths of cellular senescence. J Cell Biol. 2013; 202: 11–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Dimri GP, Lee X, Basile G, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci U S A. 1995; 92: 9363–9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013; 153: 1194–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. D'Angelo MA, Raices M, Panowski SH, Hetzer MW. Age-dependent deterioration of nuclear pore complexes causes a loss of nuclear integrity in postmitotic cells. Cell. 2009; 136: 284–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dreesen O, Chojnowski A, Ong PF, et al. Lamin B1 fluctuations have differential effects on cellular proliferation and senescence. J Cell Biol. 2013; 200: 605–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Freund A, Laberge RM, Demaria M, Campisi J. Lamin B1 loss is a senescence-associated biomarker. Mol Biol Cell. 2012; 23: 2066–2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yoh K, Itoh K, Enomoto A, et al. Nrf2-deficient female mice develop lupus-like autoimmune nephritis. Kidney Int. 2001; 60: 1343–1353. [DOI] [PubMed] [Google Scholar]
- 38. Roczniak-Ferguson A, Petit CS, Froehlich F, et al. The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci Signal. 2012; 5: ra42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bar-Peled L, Schweitzer LD, Zoncu R, Sabatini DM. Ragulator is a GEF for the rag GTPases that signal amino acid levels to mTORC1. Cell. 2012; 150: 1196–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rizzolo LJ, Peng S, Luo Y, Xiao W. Integration of tight junctions and claudins with the barrier functions of the retinal pigment epithelium. Prog Retin Eye Res. 2011; 30: 296–323. [DOI] [PubMed] [Google Scholar]
- 41. Chang CW, Roque RS, Defoe DM, Caldwell RB. An improved method for isolation and culture of pigment epithelial cells from rat retina. Curr Eye Res. 1991; 10: 1081–1086. [DOI] [PubMed] [Google Scholar]
- 42. Finnemann SC, Bonilha VL, Marmorstein AD, Rodriguez-Boulan E. Phagocytosis of rod outer segments by retinal pigment epithelial cells requires alpha(v)beta5 integrin for binding but not for internalization. Proc Natl Acad Sci U S A. 1997; 94: 12932–12937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Valapala M, Wilson C, Hose S, et al. Lysosomal-mediated waste clearance in retinal pigment epithelial cells is regulated by CRYBA1/betaA3/A1-crystallin via V-ATPase-MTORC1 signaling. Autophagy. 2014; 10: 480–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Jorgensen A, Wiencke AK, la Cour M, et al. Human retinal pigment epithelial cell-induced apoptosis in activated T cells. Invest Ophthalmol Vis Sci. 1998; 39: 1590–1599. [PubMed] [Google Scholar]
- 45. Johnson LV, Forest DL, Banna CD, et al. Cell culture model that mimics drusen formation and triggers complement activation associated with age-related macular degeneration. Proc Natl Acad Sci U S A. 2011; 108: 18277–18282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997; 88: 593–602. [DOI] [PubMed] [Google Scholar]
- 47. Kuilman T, Michaloglou C, Mooi WJ, Peeper DS. The essence of senescence. Genes Dev. 2010; 24: 2463–2479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Collado M, Serrano M. The power and the promise of oncogene-induced senescence markers. Nat Rev Cancer. 2006; 6: 472–476. [DOI] [PubMed] [Google Scholar]
- 49. Shimi T, Butin-Israeli V, Adam SA, et al. The role of nuclear lamin B1 in cell proliferation and senescence. Genes Dev. 2011; 25: 2579–2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Jurk D, Wang C, Miwa S, et al. Postmitotic neurons develop a p21-dependent senescence-like phenotype driven by a DNA damage response. Aging Cell. 2012; 11: 996–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kreiling JA, Tamamori-Adachi M, Sexton AN, et al. Age-associated increase in heterochromatic marks in murine and primate tissues. Aging Cell. 2011; 10: 292–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Tata PR, Mou H, Pardo-Saganta A, et al. Dedifferentiation of committed epithelial cells into stem cells in vivo. Nature. 2013; 503: 218–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Friedmann-Morvinski D, Bushong EA, Ke E, et al. Dedifferentiation of neurons and astrocytes by oncogenes can induce gliomas in mice. Science. 2012; 338: 1080–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Jopling C, Boue S. Izpisua Belmonte JC. Dedifferentiation, transdifferentiation and reprogramming: three routes to regeneration. Nat Rev Mol Cell Biol. 2011; 12: 79–89. [DOI] [PubMed] [Google Scholar]
- 55. Chang C, Wang X, Caldwell RB. Serum opens tight junctions and reduces ZO-1 protein in retinal epithelial cells. J Neurochem. 1997; 69: 859–867. [DOI] [PubMed] [Google Scholar]
- 56. Tian J, Ishibashi K, Honda S, Boylan SA, Hjelmeland LM, Handa JT. The expression of native and cultured human retinal pigment epithelial cells grown in different culture conditions. Br J Ophthalmol. 2005; 89: 1510–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Chang CW, Defoe DM, Caldwell RB. Retinal pigment epithelial cells from dystrophic rats form normal tight junctions in vitro. Invest Ophthalmol Vis Sci. 1997; 38: 188–195. [PubMed] [Google Scholar]
- 58. Lee BY, Han JA, Im JS, et al. Senescence-associated beta-galactosidase is lysosomal beta-galactosidase. Aging Cell. 2006; 5: 187–195. [DOI] [PubMed] [Google Scholar]
- 59. Krishna DR, Sperker B, Fritz P, Klotz U. Does pH 6 beta-galactosidase activity indicate cell senescence? Mech Ageing Dev. 1999; 109: 113–123. [DOI] [PubMed] [Google Scholar]
- 60. Severino J, Allen RG, Balin S, Balin A, Cristofalo VJ. Is beta-galactosidase staining a marker of senescence in vitro and in vivo? Exp Cell Res. 2000; 257: 162–171. [DOI] [PubMed] [Google Scholar]
- 61. Wei W, Sedivy JM. Differentiation between senescence (M1) and crisis (M2) in human fibroblast cultures. Exp Cell Res. 1999; 253: 519–522. [DOI] [PubMed] [Google Scholar]
- 62. Yegorov YE, Akimov SS, Hass R, Zelenin AV, Prudovsky IA. Endogenous beta-galactosidase activity in continuously nonproliferating cells. Exp Cell Res. 1998; 243: 207–211. [DOI] [PubMed] [Google Scholar]
- 63. Nielsen SJ, Schneider R, Bauer UM, et al. Rb targets histone H3 methylation and HP1 to promoters. Nature. 2001; 412: 561–565. [DOI] [PubMed] [Google Scholar]
- 64. Chandra T, Kirschner K, Thuret JY, et al. Independence of repressive histone marks and chromatin compaction during senescent heterochromatic layer formation. Mol Cell. 2012; 47: 203–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Zoncu R, Bar-Peled L, Efeyan A, Wang S, Sancak Y, Sabatini DM. mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H(+)-ATPase. Science. 2011; 334: 678–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. De Priester W, Van Manen R, Knook DL. Lysosomal activity in the aging rat liver: II. Morphometry of acid phosphatase positive dense bodies. Mech Ageing Dev. 1984; 26: 205–216. [DOI] [PubMed] [Google Scholar]
- 67. Schmucker DL, Sachs HG. Age-dependent alterations in rat ventricular myocardium: a quantitative analysis. Mech Ageing Dev. 1985; 31: 89–101. [DOI] [PubMed] [Google Scholar]
- 68. Meihuizen SP, Blansjaar N. Stereological analysis of liver parenchymal cells from young and old rats. Mech Ageing Dev. 1980; 13: 111–118. [DOI] [PubMed] [Google Scholar]
- 69. Mishima H, Hasebe H. Some observations in the fine structure of age changes of the mouse retinal pigment epithelium. Albrecht Von Graefes Arch Klin Exp Ophthalmol. 1978; 209: 1–9. [DOI] [PubMed] [Google Scholar]
- 70. Narita M, Young ARJ, Arakawa S, et al. Spatial coupling of mTOR and autophagy augments secretory phenotypes. Science. 2011; 332: 966–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Handa JT, Verzijl N, Matsunaga H, et al. Increase in the advanced glycation end product pentosidine in Bruch's membrane with age. Invest Ophthalmol Vis Sci. 1999; 40: 775–779. [PubMed] [Google Scholar]
- 72. Hussain AA, Rowe L, Marshall J. Age-related alterations in the diffusional transport of amino acids across the human Bruch's-choroid complex. J Opt Soc Am A Opt Image Sci Vis. 2002; 19: 166–172. [DOI] [PubMed] [Google Scholar]
- 73. Campisi J. Aging, cellular senescence, and cancer. Annu Rev Physiol. 2013; 75: 685–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Alizadeh M, Wada M, Gelfman CM, Handa JT, Hjelmeland LM. Downregulation of differentiation specific gene expression by oxidative stress in ARPE-19 cells. Invest Ophthalmol Vis Sci. 2001; 42: 2706–2713. [PubMed] [Google Scholar]
- 75. Cano M, Thimmalappula R, Fujihara M, et al. Cigarette smoking, oxidative stress, the anti-oxidant response through Nrf2 signaling, and age-related macular degeneration. Vision Res. 2010; 50: 652–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Rangasamy T, Cho CY, Thimmulappa RK, et al. Genetic ablation of Nrf2 enhances susceptibility to cigarette smoke-induced emphysema in mice. J Clin Invest. 2004; 114: 1248–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Wong WT, Dresner S, Forooghian F, et al. Treatment of geographic atrophy with subconjunctival sirolimus: results of a phase I/II clinical trial. Invest Ophthalmol Vis Sci. 2013; 54: 2941–2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Sarbassov DD, Ali SM, Sengupta S, et al. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell. 2006; 22: 159–168. [DOI] [PubMed] [Google Scholar]
- 79. Lamming DW, Ye L, Katajisto P, et al. Rapamycin-induced insulin resistance is mediated by mTORC2 loss and uncoupled from longevity. Science. 2012; 335: 1638–1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Punzo C, Kornacker K, Cepko CL. Stimulation of the insulin/mTOR pathway delays cone death in a mouse model of retinitis pigmentosa. Nat Neurosci. 2009; 12: 44–52. [DOI] [PMC free article] [PubMed] [Google Scholar]