

Significance
Mathematical models are widely used to study natural systems. They allow us to test and generate hypotheses, and help us to understand the processes underlying the observed behavior. However, such models are, by necessity, simplified representations of the true systems, so it is critical to understand the impact of assumptions made when using a particular model. Here we provide a method to assess how uncertainty about the structure of a natural system affects the conclusions we can draw from mathematical models of its dynamics. We use biological examples to illustrate the importance of considering uncertainty in both model structure and parameters. We show how solely considering the latter source of uncertainty can result in misleading conclusions and incorrect model inferences.
Keywords: robustness analysis, biological networks, network inference, dynamical systems
Abstract
Mathematical models of natural systems are abstractions of much more complicated processes. Developing informative and realistic models of such systems typically involves suitable statistical inference methods, domain expertise, and a modicum of luck. Except for cases where physical principles provide sufficient guidance, it will also be generally possible to come up with a large number of potential models that are compatible with a given natural system and any finite amount of data generated from experiments on that system. Here we develop a computational framework to systematically evaluate potentially vast sets of candidate differential equation models in light of experimental and prior knowledge about biological systems. This topological sensitivity analysis enables us to evaluate quantitatively the dependence of model inferences and predictions on the assumed model structures. Failure to consider the impact of structural uncertainty introduces biases into the analysis and potentially gives rise to misleading conclusions.
Using simple models to study complex systems has become standard practice in different fields, including systems biology, ecology, and economics. Although we know and accept that such models do not fully capture the complexity of the underlying systems, they can nevertheless provide meaningful predictions and insights (1). A successful model is one that captures the key features of the system while omitting extraneous details that hinder interpretation and understanding. Constructing such a model is usually a nontrivial task involving stages of refinement and improvement.
When dealing with models that are (necessarily and by design) gross oversimplifications of the reality they represent, it is important that we are aware of their limitations and do not seek to overinterpret them. This is particularly true when modeling complex systems for which there are only limited or incomplete observations. In such cases, we expect there to be numerous models that would be supported by the observed data, many (perhaps most) of which we may not yet have identified. The literature is awash with papers in which a single model is proposed and fitted to a dataset, and conclusions drawn without any consideration of (i) possible alternative models that might describe the observed behavior and known facts equally well (or even better); or (ii) whether the conclusions drawn from different models (still consistent with current observations) would agree with one another.
We propose an approach to assess the impact of uncertainty in model structure on our conclusions. Our approach is distinct from—and complementary to—existing methods designed to address structural uncertainty, including model selection, model averaging, and ensemble modeling (2–9). Analogous to parametric sensitivity analysis (PSA), which assesses the sensitivity of a model’s behavior to changes in parameter values, we consider the sensitivity of a model’s output to changes in its inherent structural assumptions. PSA techniques can usually be classified as (i) local analyses, in which we identify a single “optimal” vector of parameter values, and then quantify the degree to which small perturbations to these values change our conclusions or predictions; or (ii) global analyses, where we consider an ensemble of parameter vectors (e.g., samples from the posterior distribution in the Bayesian formalism) and quantify the corresponding variability in the model’s output. Although several approaches fall within these categories (10–12), all implicitly condition on a particular model architecture. Here we present a method for performing sensitivity analyses for ordinary differential equation (ODE) models where the architecture of these models is not perfectly known, which is likely to be the case for all realistic complex systems. We do this by considering network representations of our models, and assessing the sensitivity of our inferences to the network topology. We refer to our approach as topological sensitivity analysis (TSA).
Here we illustrate TSA in the context of parameter inference, but we could also apply our method to study other conclusions drawn from ODE models (e.g., model forecasts or steady-state analyses). When we use experimental data to infer parameters associated with a specific model it is critical to assess the uncertainty associated with our parameter estimates (13), particularly if we wish to relate model parameters to physical (e.g., reaction rate) constants in the real world. Too often this uncertainty is estimated only by considering the variation in a parameter estimate conditional on a particular model, while ignoring the component of uncertainty that stems from potential model misspecification. The latter can, in principle, be considered within model selection or averaging frameworks, where several distinct models are proposed and weighted according to their ability to fit the observed data (2–5). However, the models tend to be limited to a small, often diverse, group that act as exemplars for each competing hypothesis but ignore similar model structures that could represent the same hypotheses. Moreover, we know that model selection results can be sensitive to the particular experiments performed (14).
We assume that an initial model, together with parameters or plausible parameter ranges, has been proposed to describe the dynamics of a given system. This model may have been constructed based on expert knowledge of the system, selected from previous studies, or (particularly in the case of large systems) proposed automatically using network inference algorithms (15–19), for example. Using TSA, we aim to identify how reliant any conclusions and inferences are on the particular set of structural assumptions made in this initial candidate model. We do this by identifying alterations to model topology that maintain consistency with the observed dynamics and test how these changes impact the conclusions we draw (Fig. 1). Analogous to PSA we may perform local or global analyses—by testing a small set of “close” models with minor structural changes, or performing large-scale searches of diverse model topologies, respectively. To do this we require efficient techniques for exploring the space of network topologies and, for each topology, inferring the parameters of the corresponding ODE models.
Fig. 1.
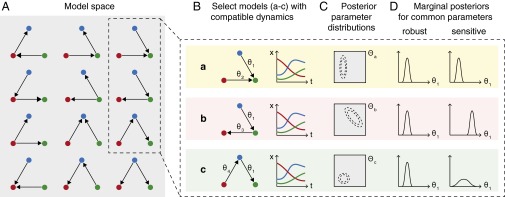
Overview of TSA applied to parameter inference. (A) Model space includes our initial candidate model and a series of altered topologies that are consistent with our chosen rules (e.g., all two-edge, three-node networks, where nodes indicate species and directed edges show interactions). One topology may correspond to one or several ODE models depending on the parametric forms we choose to represent interactions. (B) We test each ODE model to see whether it can generate dynamics consistent with our candidate model and the available experimental data. For TSA, we select a group of these compatible models and compare the conclusions we would draw using each of them. (C) Associated with each model m is a parameter space (gray); using Bayesian methods we can infer the joint posterior parameter distribution (dashed contours) for a particular model and dataset. (D) In some cases, equivalent parameters will be present in several selected models (e.g., , which is associated with the same interaction in models a–c). We can compare the marginal posterior distribution inferred using each model for a common parameter to test whether our inferences are robust to topological changes, or rely on one specific set of model assumptions (i.e., sensitive). Different models may result in marginal distributions that differ in position and/or shape for equivalent parameters, but we cannot tell from this alone which model better represents reality—this requires model selection approaches (2–4).
Even for networks with relatively few nodes (corresponding to ODE models involving few interacting entities), the number of possible topologies can be enormous. Searching this “model space” presents formidable computational challenges. We use here a gradient-matching parameter inference approach that exploits the fact that the nth node, , in our network representation is conditionally independent of all other nodes given its regulating parents, (20–26). The exploration of network topologies is then reduced to the much simpler problem of considering, independently for each n, the possible parent sets of in an approach that is straightforwardly parallelized.
We use biological examples to illustrate local and global searches of model spaces to identify alternative model structures that are consistent with available data. In some cases we find that even minor structural uncertainty in model topology can render our conclusions—here parameter inferences—unreliable and make PSA results positively misleading. However, other inferences are robust across diverse compatible model structures, allowing us to be more confident in assigning scientific meaning to the inferred parameter values.
Model Structures
We consider systems consisting of N interacting variables that can be modeled using ODEs of the form
| [1] |
where , is the value of the nth variable at time t, is the derivative of with respect to t, and θ is the vector of model parameters. The rate of change of the nth variable over time is described by the nth component of the vector ,
| [2] |
Typically, will only act on a (relatively small) subset of the variables ; we represent our systems as networks, in which nodes correspond to variables and a directed edge is drawn from to if the rate of change of depends upon . The set of parents of , , is therefore the collection of variables on which acts. The parent set, provides a complete description of the system’s network representation.
Defining the Model Search Space
To specify the space of possible ODE models we need to define (i) the set of network topologies, and (ii) the corresponding ODE model(s) that can be represented by each network topology. Exploring large model spaces is, of course, challenging due to the number of possible topologies. However, because each network is completely defined by the parent set , it is sufficient to consider (for each species n independently) the possible parents of , to fully explore the space of networks. Considering the regulation of each species independently in this way can reduce the search space sufficiently to make an exhaustive search feasible (Fig. S1A). Further simplification, such as assuming that each parent set is restricted to a relatively small size as in refs. 23–25, may also be helpful.
To move from a network representation to an ODE model requires a set of rules that translate interactions (network edges) into parameterized mathematical functions. These rules are necessarily context- and model-specific and may allow a single network topology to represent multiple different ODE models. For example, we could permit several types of interaction (e.g., activation and inhibition), each represented by a different mathematical function, or several methods to model the combined effect of multiple interactions (e.g., synergistic and additive). Such rules allow us to map from each possible parent set to the possible functional forms of the corresponding differential equations,
| [3] |
To test whether a particular ODE model can generate the desired dynamic behavior, we require the associated model parameters θ. Crucially, to enable large-scale searches we need a method to infer the parameters of each ODE derivative component independently. Gradient-matching parameter inference approaches (26–28) avoid the need to simulate from the complete coupled ODE system (Eq. 1); instead, parameters are optimized using data-derived estimates of and , and minimizing the discrepancy between two gradient estimates— and —in a process which can be applied to each derivative component independently (Fig. S1B). Here we use Gaussian process (GP) regression, a nonparametric Bayesian method for nonlinear regression (28–31), to obtain data-derived estimates of and (see SI Materials and Methods for details).
Constructing and Ranking ODE Models
Given an initial parameterized candidate model, we use the following method to identify and rank realistic alternative models:
-
i)
Simulate time course data (at times ) for each species in the system using the initial candidate model.
-
ii)
Define rules to construct possible ODE models for the regulation of each species . We consider the maximum number of parents allowed per species; possible parametric forms to represent interactions; and how to model combinatorial regulation if a node has several parents. The form of the initial candidate model can guide these rules.
-
iii)
Estimate state variables and corresponding derivatives for all species in the system using GP regression and the simulated time course data (generated in step i).
-
iv)
For each species n:
-
a)
Consider all possible parent sets to construct all possible models of the form , according to the rules defined in step ii.
-
b)
Infer parameters θ for each test model using gradient-matching parameter inference and GP-derived estimates for and . We estimate θ by minimizing,
[4] -
c)
Rank models using the distance calculated in step iv(b) or an alternative metric, e.g., Akaike’s Information Criterion adapted for small samples () (2).
-
a)
-
v)
Combine the componentwise models to obtain complete ODE models .
Selected models are then used to explore how uncertainty in model structure impacts our inferences and/or predictions.
Results
We first outline how to generate alternative models which generate dynamics consistent with the initial candidate model. We then use synthetic and experimental datasets to demonstrate how we can use selected models to test the robustness of our inferred conclusions (here, parameter estimates) to altered model assumptions. We include examples using both optimization and Bayesian approaches to parameter inference. ODE models were constructed as described in Materials and Methods. SI Materials and Methods lists the parameter values and initial conditions used for simulations from the initial candidate models.
Automated Model Generation and Ranking.
To illustrate, we assume a five-species gene regulatory network, model A (Fig. 2), has been proposed to model a hypothetical system. We suppose that this network corresponds to an ODE model for which parameters (including initial conditions) have already been estimated (see SI Materials and Methods for values). We take this as our initial candidate model, and use GP regression to estimate species concentrations and corresponding derivatives from simulated trajectories (Fig. 3). For , we define 33 possible componentwise models of the form to describe the regulation of each species by allowing up to two regulating parents per gene, no self-regulation, and two types of interaction. For each putative parental set , we test all permutations of interaction types—e.g., if we consider , , , or , where the superscript indicates activation () or inhibition (). We rank these models using the distance (Eq. 4) obtained during gradient-matching parameter inference (Fig. S2A), and combine selected componentwise models to create coupled ODE models describing the network dynamics (Fig. 3). Crucially, this combination step does not require further parameter estimation so, by evaluating the possible component equations for , we can easily—and rapidly—construct and rank all complete ODE systems, . If we have information about the system dynamics under multiple experimental conditions we can consider this when ranking our complete models (SI Results and Fig. S2).
Fig. 2.
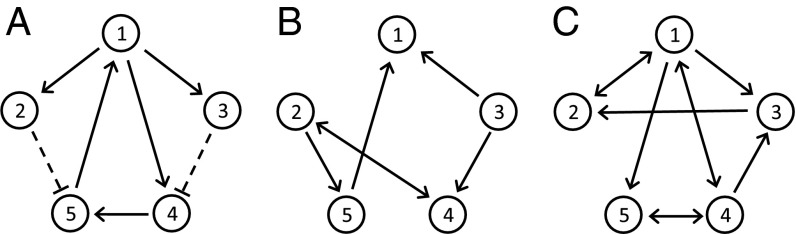
Model topologies for synthetic datasets: model A represents a gene regulatory network, and models B and C represent population dynamics models.
Fig. 3.

Example of automated model generation and ranking for the five-species gene regulatory network (model A). GP regression model means (Left, solid lines) fitted to noiseless data simulated from the initial candidate model (circles). Simulated trajectories (Right) from two example alternative ODE models automatically constructed using our method (see SI Results for details)—the best models accurately capture the desired dynamics, whereas lower-ranked models deviate.
Global TSA of Optimization-Based Parameter Estimation.
Once we have identified alternative ODE models with comparable dynamics to our initial model, we can use these to explore how dependent our inferences are on the structural assumptions inherent to a particular model. We illustrate this in the context of maximum likelihood parameter estimation, using a synthetic dataset simulated from a competitive population dynamics model, model B (Fig. 2), which we also take as the initial candidate.
As in the previous example, we assume that model B corresponds to an ODE model of a hypothetical system for which parameters have previously been estimated (SI Materials and Methods). We additionally assume that we have an experimental dataset , which, for this synthetic example, is generated by adding noise to values simulated from the candidate model (SI Materials and Methods). Thus, in this illustrative example, the candidate model is also the “true” (data-generating) model. We first perform an exhaustive search to identify models with consistent dynamics to our initial candidate. We allow a maximum of 3 parent species, no self-regulation, and a single type of interaction resulting in possible complete ODE models. To illustrate results, we select for the present analysis the top 10 models, ranked by AICc values (Fig. 4A). The models ranked 2nd–10th all contain the interactions present in the true model (ranked first), but have one or two additional “incorrect” edges.
Fig. 4.

Comparison of parameter inference results for the top 10 complete ODE models (ranked by AICc) for a competitive population dynamics system. (A) Noisy data (circles) simulated from the true model (model B) are used to obtain maximum likelihood parameter estimates for each of the 10 models. Lines show the trajectories simulated using each of the resulting parameterized models (assuming the true initial conditions). (B) Comparison of parametric bootstrap distributions obtained using each model for three of the parameters present in all models; distributions for all of the parameters present in the true model are shown in Fig. S3. For each model, 1,000 replicate datasets were obtained by adding Gaussian noise to the initial fitted trajectories (Fig. 4A) and used for parameter estimation. Kernel density estimates of the resulting parameter distributions are shown by solid lines; vertical dashed gray lines indicate the true parameter values used for simulation. Incorrect edges, from species k to n, in models 2–10 are from the set .
Next we can assess the variability in the conclusions (here, maximum likelihood parameter estimates) that we obtain from the candidate models when fitting them to a noisy dataset, . We assess uncertainties in the estimated values using a parametric bootstrap (22) (Fig. 4B and Fig. S3). Most distributions are consistent across the models, but network topology has a significant impact on some parameters. For example, with seven models (including the true model) the distributions for parameter are centered close to 0.5 (which we know to be the true value for this synthetic example) with little variation; however, models containing an incorrect interaction from species 4 to 3 (models 5, 6, and 10) result in broader distributions centered around a higher value.
Thus, even models that include all of the true interactions can still lead to misleading conclusions about biophysical parameters if additional incorrect interactions are also present. If we solely rely on parameter uncertainty and sensitivity analyses, without considering the impact of potential structural misspecifications in our model, we are therefore likely to overestimate the precision of our results.
Local TSA of Bayesian Parameter Inference.
Instead of an exhaustive, global search, we may instead perform a local analysis by only considering models with minor structural modifications relative to the candidate model. This could be appropriate if we have a clear idea about key interactions, and consider large deviations from this topology to be biologically irrelevant, or for larger complex systems where exhaustive search becomes computationally infeasible. To illustrate this we use a synthetic dataset (Fig. S4), simulated from a population dynamics model, model C (Fig. 2), which we also take as the initial candidate. We then consider the set of 20 close models that differ by addition or deletion of a single network edge. For each model we obtain samples from the posterior parameter distribution using two Bayesian approaches: nested sampling (32, 33) and the Metropolis–Hastings algorithm (34, 35). As well as generating posterior samples, nested sampling provides an estimate of the evidence (marginal likelihood) for each model, allowing us to rank them (Table S1). We select the five alternative models with evidence greater than or equal to the true model and compare the estimated posterior distributions.
In most cases the univariate marginal posterior distributions are broadly conserved across the selected models (Fig. S5), but in a few cases the shape and/or location of these distributions vary with a slightly different model (e.g., in Fig. S5, see parameters and in model 18, or in model 8). As well as the values of individual parameters, we may also be interested in the dependencies between parameters. In particular, the related concepts of sloppiness and identifiability in biological models have recently received much attention—in the context of possible biological significance and for optimal experimental design (12, 36–39). Fig. 5 shows bivariate scatter plots illustrating the dependencies between particular parameter pairs for each of the selected models.
Fig. 5.

Comparison of nested sampling inference results for the best close models and the true model. Data simulated from the true model (model C) assuming additive Gaussian noise are used to infer posterior parameter distributions for the true model and 20 close models (those differing from the true model by addition or deletion of a single interaction); we assumed uniform prior distributions for all parameters (). Models with estimated evidence greater than or equal to the true model are then compared. Example bivariate scatter plots are shown for three parameter pairs for these models; each circle shows a posterior sample, with color corresponding to the likelihood value. Model 18 does not include an interaction from species 5 to species 4, hence there is no plot. See Fig. S6 for additional results.
Again, whereas some dependencies are robust to varying model topology (e.g., the negative dependence between parameters and is observed in all cases), sometimes even small alterations to model topology significantly alter our conclusions about parameter dependencies—with a single edge missing from the true network, we may either infer a strong dependency (e.g., and in model 8) that is absent in the true system, or miss a true dependency (e.g., and in model 12). Of course, in this example we know the true model, but these results serve to illustrate how drawing conclusions about the underlying system based on inferences drawn from a single model may be misleading. When dealing with simplified representations of reality we believe the approach outlined here is a useful, even essential, way to determine which results strongly rely on model assumptions and which ones are robust to structural modification and may therefore more likely be biologically relevant rather than an artifact resulting from a specific single model topology.
TSA of a Model of Yeast Gene Expression Dynamics.
We use time-resolved gene expression data for cell-cycle regulated genes in Saccharomyces cerevisiae (40). Following Lu et al. (41), we used their D-NetWeaver algorithm (19) to construct an initial candidate ODE model describing gene expression dynamics (Fig. S7). The resulting network comprises 41 nodes (corresponding to gene clusters) and 148 directed edges (regulatory interactions). The system size precludes an exhaustive search of alternative topologies, so we performed a local analysis in which we sampled models with 1–30 random edges rewired (relative to the initial candidate) and identified those with comparable dynamics using gradient matching.
In total we sampled rewired models and selected 7 models for comparison with our initial candidate (see SI Results for details). We obtained maximum likelihood estimates for the 189 parameters associated with each model by fitting to the mean clustered gene expression profiles, and compared estimates for parameters common to all selected models. Fig. 6 illustrates the variety of estimated values we obtained for four parameters (results for all common parameters are shown in Fig. S8).
Fig. 6.

Comparison of parameter estimation results for eight alternative ODE models fitted to the data of ref. 40. Results for four parameters are shown; further estimates are given in Fig. S8. Estimated values are shown by circles, with error bars indicating 95% credible intervals estimated using a Laplace approximation (42); estimates for equivalent parameters in different models are joined by a line to aid comparison. The selected models are ordered by the number of rewired edges relative to the initial candidate model (at ). Estimated values for many model parameters are robust to altered model topology (e.g., p9 and p78), but some are more sensitive (e.g., estimates for p29 are consistent in sign but not magnitude, whereas p22 estimates span positive and negative values in the selected models).
Many estimated parameter values are consistent across these models (i.e., robust to topological changes). However, Fig. 6 shows one example where the sign of an edge-associated parameter (p22) varies with model choice, suggesting we cannot reliably infer the nature of this regulatory interaction (activating or inhibitory). Relying solely on estimates of parameter uncertainty that condition on a chosen model structure (e.g., the confidence intervals estimated here) but ignoring the potential impact of structural uncertainty could lead us to draw unreliable conclusions about the true network. TSA provides us with a way to assess the latter component of uncertainty, and thus gain confidence in results which are consistent among the selected models while identifying less reliable inferences.
Discussion
For mechanistic mathematical modeling to be useful, we must assess the robustness of conclusions drawn from our models. This is particularly important in fields such as systems biology, where we usually rely on (knowingly) oversimplified representations of the true complex systems (7, 43, 44). To gain meaningful insights into the real underlying processes, we must acknowledge that our conclusions are conditional on the chosen model architecture, and understand the impact of changing these model assumptions. Frequently, once a model has been selected, researchers only consider uncertainty at the level of model parameters but ignore the contribution of potential (and likely) structural misspecifications. One reason for this is that it has been difficult to explore a diverse range of models in a computationally efficient manner.
Here we describe a rapid, parallelizable method to automatically generate models consistent with the observed dynamics of a biological system. Constraints on the parametric forms of these models are selected for the system of interest, to ensure that proposed models are plausible given the types of interactions believed to be possible. Within computational limitations we can consider any number of possible parametric forms to describe the dynamics and interactions within a network, and investigate different rules for combinatorial regulation. This permits an extensive search of local or global model space for alternative, dynamically consistent models. These can then be used to test the sensitivity of inferences to the structural assumptions inherent to a specific model.
We demonstrate how our approach, TSA, may be used to explore the impact of changes to our model upon the conclusions that we draw, in the particular context of parameter inference. Whereas Bayesian analysis and bootstrapping approaches are often used to assess parameter variability (and identifiability), it is important to remember that these assessments condition upon a single, specific model. Even minor changes in the model structure can significantly alter the conclusions that we draw (in our case, the values and dependencies of the inferred parameters). Even if, for one particular model, the parameter values are very tightly constrained (and thus, for that model, we feel we have “high confidence” in the parameter values), it does not follow that the same will be true for other feasible models. If our parameters have a real, biophysical interpretation, we therefore need to be very careful not to assert that we know the true values of these quantities in the underlying system, just because––for a given model––we can pin them down with relative certainty. Considering the sensitivity of model behavior to both parameter and structural variation enables us to identify inferences that are robust to possible errors in our model. We can therefore be more confident about using these inferences to draw scientifically meaningful conclusions.
Materials and Methods
Models for Synthetic Gene Regulatory Networks.
We use the model A structure (Fig. 2) and ODE modeling format described in ref. 45. Transcription regulation is modeled using equations of the form , where is species n mRNA concentration, and are basal synthesis and degradation rates, respectively, and the sum accounts for regulatory interactions influencing gene n (assuming additive combinatorial regulation) (45). An interaction from species k to species n is modeled using a strength parameter and a Hill function (with associated parameters and ). The latter takes the form for activating interactions or for inhibitory regulation.
Models for Competitive Population Dynamics.
Population sizes of N species competing for finite resources are modeled by (46) , where is the inherent growth rate of species n and represents the influence of species k on species n. Here , , and for all n.
Models for Yeast Gene Expression Data.
We used D-NetWeaver (19) to construct an initial candidate model for the 613 complete α-factor synchronized gene-expression profiles in ref. 40. As in ref. 41, genes were clustered into 41 groups based on expression profile similarities, and a linear ODE model inferred to describe regulatory interactions between these clusters with the form , where , is the mean expression level of genes in cluster n at time t, A is a connectivity matrix with entry indicating a directed interaction from cluster j to cluster i (with and indicating activation or inhibition, respectively), is a vector of constant terms, and N is the total number of clusters.
Supplementary Material
Acknowledgments
This work was supported by Human Frontier Science Program Grant RG0061/2011 and Biotechnology and Biological Sciences Research Council Grant BB/K017284/1. M.P.H.S. is a Royal Society Wolfson Research Merit Award Holder.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1414026112/-/DCSupplemental.
References
- 1.Box GEP, Draper NR. Empirical Model-Building and Response Surfaces. John Wiley & Sons; New York: 1987. [Google Scholar]
- 2.Burnham KP, Anderson DR. Model Selection and Multimodel Inference. A Practical Information-Theoretic Approach. Springer; New York: 2002. [Google Scholar]
- 3.Kirk P, Thorne T, Stumpf MPH. Model selection in systems and synthetic biology. Curr Opin Biotechnol. 2013;24(4):767–774. doi: 10.1016/j.copbio.2013.03.012. [DOI] [PubMed] [Google Scholar]
- 4.Toni T, Stumpf MPH. Simulation-based model selection for dynamical systems in systems and population biology. Bioinformatics. 2010;26(1):104–110. doi: 10.1093/bioinformatics/btp619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hoeting JA, Madigan D, Raftery AE, Volinsky CT. Bayesian model averaging: A tutorial. Stat Sci. 1999;14(4):382–417. [Google Scholar]
- 6.Kuepfer L, Peter M, Sauer U, Stelling J. Ensemble modeling for analysis of cell signaling dynamics. Nat Biotechnol. 2007;25(9):1001–1006. doi: 10.1038/nbt1330. [DOI] [PubMed] [Google Scholar]
- 7.Kaltenbach H-M, Dimopoulos S, Stelling J. Systems analysis of cellular networks under uncertainty. FEBS Lett. 2009;583(24):3923–3930. doi: 10.1016/j.febslet.2009.10.074. [DOI] [PubMed] [Google Scholar]
- 8.Sunnåker M, et al. Automatic generation of predictive dynamic models reveals nuclear phosphorylation as the key Msn2 control mechanism. Sci Signal. 2013;6(277):ra41. doi: 10.1126/scisignal.2003621. [DOI] [PubMed] [Google Scholar]
- 9.Sunnåker M, et al. Topological augmentation to infer hidden processes in biological systems. Bioinformatics. 2014;30(2):221–227. doi: 10.1093/bioinformatics/btt638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rabitz H. Systems analysis at the molecular scale. Science. 1989;246(4927):221–226. doi: 10.1126/science.246.4927.221. [DOI] [PubMed] [Google Scholar]
- 11.Saltelli A, et al. Global Sensitivity Analysis. The Primer. John Wiley & Sons; Chichester, UK: 2008. [Google Scholar]
- 12.Erguler K, Stumpf MPH. Practical limits for reverse engineering of dynamical systems: A statistical analysis of sensitivity and parameter inferability in systems biology models. Mol Biosyst. 2011;7(5):1593–1602. doi: 10.1039/c0mb00107d. [DOI] [PubMed] [Google Scholar]
- 13.Vanlier J, Tiemann CA, Hilbers PAJ, van Riel NAW. Parameter uncertainty in biochemical models described by ordinary differential equations. Math Biosci. 2013;246(2):305–314. doi: 10.1016/j.mbs.2013.03.006. [DOI] [PubMed] [Google Scholar]
- 14.Silk D, Kirk PDW, Barnes CP, Toni T, Stumpf MPH. Model selection in systems biology depends on experimental design. PLOS Comput Biol. 2014;10(6):e1003650. doi: 10.1371/journal.pcbi.1003650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bonneau R, et al. The Inferelator: An algorithm for learning parsimonious regulatory networks from systems-biology data sets de novo. Genome Biol. 2006;7(5):R36. doi: 10.1186/gb-2006-7-5-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Greenfield A, Hafemeister C, Bonneau R. Robust data-driven incorporation of prior knowledge into the inference of dynamic regulatory networks. Bioinformatics. 2013;29(8):1060–1067. doi: 10.1093/bioinformatics/btt099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thorne T, Stumpf MPH. Inference of temporally varying Bayesian networks. Bioinformatics. 2012;28(24):3298–3305. doi: 10.1093/bioinformatics/bts614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Oates CJ, et al. Causal network inference using biochemical kinetics. Bioinformatics. 2014;30(17):i468–i474. doi: 10.1093/bioinformatics/btu452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu S, Liu Z-P, Qiu X, Wu H. Modeling genome-wide dynamic regulatory network in mouse lungs with influenza infection using high-dimensional ordinary differential equations. PLoS ONE. 2014;9(5):e95276. doi: 10.1371/journal.pone.0095276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Friedman N, Geiger D, Goldszmidt M. Bayesian network classifiers. Mach Learn. 1997;29(2-3):131–163. [Google Scholar]
- 21.Friedman N, Linial M, Nachman I, Pe’er D. Using Bayesian networks to analyze expression data. J Comput Biol. 2000;7(3-4):601–620. doi: 10.1089/106652700750050961. [DOI] [PubMed] [Google Scholar]
- 22.Murphy KP. Machine Learning. A Probabilistic Perspective. MIT Press; Cambridge, MA: 2012. [Google Scholar]
- 23.Aijö T, Lähdesmäki H. Learning gene regulatory networks from gene expression measurements using non-parametric molecular kinetics. Bioinformatics. 2009;25(22):2937–2944. doi: 10.1093/bioinformatics/btp511. [DOI] [PubMed] [Google Scholar]
- 24.Penfold CA, Wild DL. How to infer gene networks from expression profiles, revisited. Interface Focus. 2011;1(6):857–870. doi: 10.1098/rsfs.2011.0053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Penfold CA, Buchanan-Wollaston V, Denby KJ, Wild DL. Nonparametric Bayesian inference for perturbed and orthologous gene regulatory networks. Bioinformatics. 2012;28(12):i233–i241. doi: 10.1093/bioinformatics/bts222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dondelinger F, Husmeier D, Rogers S, Filippone M. 2013. ODE parameter inference using adaptive gradient matching with Gaussian processes. JMLR Workshop Conf Proc 31:216–228.
- 27.Brunel NJ-B. Parameter estimation of ODEs via nonparametric estimators. Electron J Stat. 2008;2(0):1242–1267. [Google Scholar]
- 28.Calderhead B, Girolami M. Accelerating Bayesian inference over nonlinear differential equations with Gaussian processes. Adv Neural Inf Process Syst. 2008;21:217–224. [Google Scholar]
- 29.Kirk PDW, Stumpf MPH. Gaussian process regression bootstrapping: Exploring the effects of uncertainty in time course data. Bioinformatics. 2009;25(10):1300–1306. doi: 10.1093/bioinformatics/btp139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rasmussen CE, Williams CKI. Gaussian Processes for Machine Learning. MIT Press; Cambridge, MA: 2006. [Google Scholar]
- 31.Rasmussen CE, Nickisch H. Gaussian processes for machine learning (GPML) toolbox. J Mach Learn Res. 2010;11:3011–3015. [Google Scholar]
- 32.Skilling J. Nested sampling for general Bayesian computation. Bayesian Anal. 2006;1(4):833–860. [Google Scholar]
- 33.Johnson R, Kirk P, Stumpf MPH. SYSBIONS: Nested sampling for systems biology. Bioinformatics. 2014 doi: 10.1093/bioinformatics/btu675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hastings WK. Monte Carlo sampling methods using Markov chains and their applications. Biometrika. 1970;57:97–109. [Google Scholar]
- 35.Wilkinson DJ. Stochastic Modelling for Systems Biology. 2nd Ed CRC Press; Boca Raton, FL: 2011. [Google Scholar]
- 36.Daniels BC, Chen Y-J, Sethna JP, Gutenkunst RN, Myers CR. Sloppiness, robustness, and evolvability in systems biology. Curr Opin Biotechnol. 2008;19(4):389–395. doi: 10.1016/j.copbio.2008.06.008. [DOI] [PubMed] [Google Scholar]
- 37.Gutenkunst RN, et al. Universally sloppy parameter sensitivities in systems biology models. PLOS Comput Biol. 2007;3(10):1871–1878. doi: 10.1371/journal.pcbi.0030189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Apgar JF, Witmer DK, White FM, Tidor B. Sloppy models, parameter uncertainty, and the role of experimental design. Mol Biosyst. 2010;6(10):1890–1900. doi: 10.1039/b918098b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Villaverde AF, Banga JR. Reverse engineering and identification in systems biology: Strategies, perspectives and challenges. J R Soc Interface. 2014;11(91):20130505. doi: 10.1098/rsif.2013.0505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Spellman PT, et al. Comprehensive identification of cell cycle-regulated genes of the yeast Saccharomyces cerevisiae by microarray hybridization. Mol Biol Cell. 1998;9(12):3273–3297. doi: 10.1091/mbc.9.12.3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lu T, Liang H, Li H, Wu H. High dimensional ODEs coupled with mixed-effects modelling techniques for dynamic gene regulatory network identification. J Am Stat Assoc. 2011;106(496):1242–1258. doi: 10.1198/jasa.2011.ap10194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Statisticat LLC. 2014 LaplacesDemon: Complete Environment for Bayesian Inference. www.bayesian-inference.com/software (R package Version 14.06.23)
- 43.Schaber J, Klipp E. Model-based inference of biochemical parameters and dynamic properties of microbial signal transduction networks. Curr Opin Biotechnol. 2011;22(1):109–116. doi: 10.1016/j.copbio.2010.09.014. [DOI] [PubMed] [Google Scholar]
- 44.Gunawardena J. Models in biology: ‘Accurate descriptions of our pathetic thinking’. BMC Biol. 2014;12:29. doi: 10.1186/1741-7007-12-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mazur J, Ritter D, Reinelt G, Kaderali L. Reconstructing nonlinear dynamic models of gene regulation using stochastic sampling. BMC Bioinformatics. 2009;10(1):448. doi: 10.1186/1471-2105-10-448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vano JA, Wildenberg JC, Anderson MB, Noel JK, Sprott JC. Chaos in low-dimensional Lotka–Volterra models of competition. Nonlinearity. 2006;19(10):2391–2404. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.