

Abstract
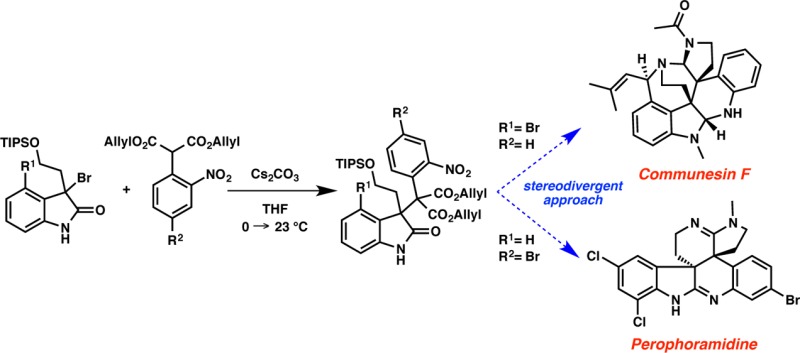
Expedient synthetic approaches to the highly functionalized polycyclic alkaloids communesin F and perophoramidine are described using a unified approach featuring a key decarboxylative allylic alkylation to access a crucial and highly congested 3,3-disubstituted oxindole. Described are two distinct, stereoselective alkylations that produce structures in divergent diastereomeric series possessing the critical vicinal all-carbon quaternary centers needed for each synthesis. Synthetic studies toward these challenging core structures have revealed a number of unanticipated modes of reactivity inherent to these complex alkaloid scaffolds. Additionally, several novel and interesting intermediates en route to the target natural products, such as an intriguing propellane hexacyclic oxindole encountered in the communesin F sequence, are disclosed. Indeed, such unanticipated structures may prove to be convenient strategic intermediates in future syntheses.
Introduction
In 1993, communesin A (1a) was isolated along with communesin B (1b) from a strain of Penicillium sp. found growing on a marine alga by the Numata group (Figure 1).1 Communesins A (1a) and B (1b) exhibit antiproliferative activity against P-388 lymphocytic leukemia cells (ED50 = 3.5 μg/mL and 0.45 μg/mL, respectively).1 In addition, communesin B (1b) disrupts actin microfilaments in cultured mammalian cells and shows cytotoxic activity against LoVo and KB cells (MIC values of 2.0 μg/mL and 4.5 μg/mL, respectively).2 Several other members of the comunesin family, communesins B–H (1b–h), were disclosed from related marine fungal strains of Penicillium sp. in the following years.3 With the exception of communesins G (1g) and H (1h), the communesins show insecticidal activity and antiproliferative activity against a variety of cancer cells, with communesin B (1b) being the most potent.1−3 These indole alkaloids contain several interesting structural features including vicinal all-carbon quaternary centers, bis-aminal functionalities, and a complex polycyclic core. The communesins are structurally unique when compared against other known microfilament-disrupting agents, which are primarily macrolides. Macrolide microfilament-disrupting agents show considerable structural similarity, and their interactions with actin have been crystallographically characterized, leading to hypotheses regarding their mechanism of action.4 The unique structure of communesin B (1b) suggests that it may exhibit a novel mechanism of action on the cytoskeleton relative to other microfilament-disrupting agents.5a The development of a unified synthetic route to the communesins would enable the understanding of their effects on the cellular cytoskeleton while addressing the scarcity of naturally occurring sources of the compounds.
Figure 1.

Communesins (1), nomofungin (2), and perophoramidine (3).
In 2001, an intriguing natural product, nomofungin (2) was isolated from an unidentified fungus found on the bark of Ficus microcarpa by the Hemscheidt group.2 Interestingly, the only structural difference between communesin B (1b) and nomofungin (2) is that communesin B has an aminal moiety instead of the N,O-acetal moiety present in nomofungin. A combination of experimental and theoretical exercises led to the independent discovery by our laboratory and the Funk group that the reported structure of nomofungin was incorrect and that it is actually that of communesin B.5a,5b Although the structure of nomofungin was erroneously assigned, its isolation and structural revision to that of an older structure can be viewed as the inception point for all synthetic efforts to the communesin family members over the past decade. Interestingly, there were no reports of synthetic efforts toward the communesins from 1993 up to our initial report in 2003.5i
A structurally related compound, perophoramidine (3) was isolated in 2002 from the ascidian Perophora namei.6 The core is comparable to the one found in the communesins, albeit in a higher oxidation state, with the alternate diastereomeric relationship between the vicinal quaternary carbons and without the azepine ring system. Perophoramidine (3) possesses modest cytotoxicity against the HCT 116 human colon carcinoma cell line (IC50 = 60 μM) and induces apoptosis.7
These complex, polycyclic, bioactive alkaloids have been the subject of intense synthetic efforts over the past decade.5 Numerous approaches have been reported in the literature, including three from our laboratory.5a,5c,5w Herein, we report the evolution of an efficient, unified approach toward the synthesis of these unique alkaloids.
Results and Discussion
1. Biosynthesis-Inspired Diels–Alder Cycloaddition Strategy to Communesin F
Our early efforts toward the communesin structure centered on the laboratory implementation of our proposed biosynthesis (Scheme 1).5a,5c,8 As the key step in the process, we envisioned a Diels–Alder cycloaddition to unite the two indole-based fragments by coupling of 5, an N-methylated derivative of the ergot alkaloid aurantioclavine (4),9,10 and an o-azaxylylene indolone 6 to generate the bridged lactam 7. We anticipated that lactam 7 would be highly reactive due to the poor alignment of the nitrogen lone pair with the carbonyl.11−13 As such, the pendant amino group would be expected to easily open the lactam, thus forming spirocycle 8. Further tailoring would produce communesin A (1a) and B (1b).
Scheme 1. Biosynthesis-Inspired Approach.

Toward this end, (±)-aurantioclavine was prepared using known methods,5a,14 and an enantioselective synthesis of (−)-aurantioclavine utilizing our oxidative kinetic resolution (OKR) technology was developed.15 We proceeded to develop an efficient cycloaddition between (±)-indole 9 as a model coupling partner and benzyl chloride 10 using conditions previously developed by Steinhagen and Corey16 that resulted in a mixture of pentacyclic diastereomers (89% yield). Removal of the tosyl group with magnesium in methanol produced a 2:1 mixture of diastereomers 11 and 12 in 80% combined yield, with the desired relative stereochemistry evident in the major diastereomer (cf. 11 and 1a) (Scheme 2).5a
Scheme 2. Model Studies for a Diels–Alder Cycloaddtion Strategy To Construct the Pentacyclic Core Structure.

Despite the success of this model system, more advanced electrophiles (e.g., mesylate 14, cyclopropane 16, or epoxide 17(17)) did not succumb to cycloaddition conditions (Scheme 3). Nor have we been successful in the oxidation of 11 and 12 at C(8), which would provide a functional handle for introduction of the second quaternary stereocenter.
Scheme 3. Attempted Diels–Alder Cycloadditions with Advanced Electrophiles.

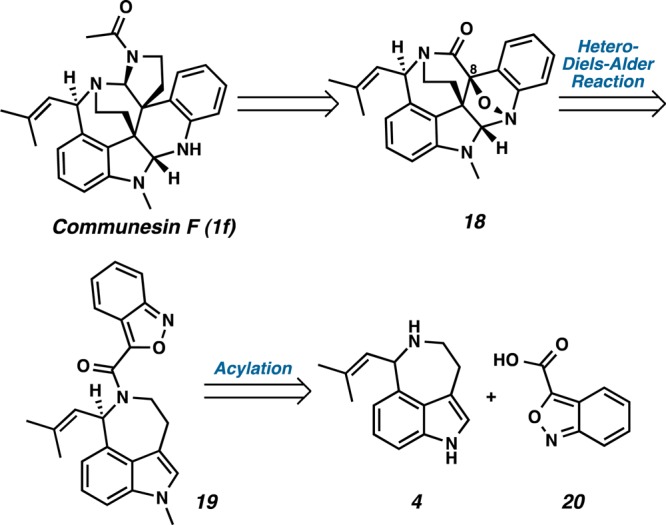
To obviate the difficulties encountered in our attempts to functionalize C(8), we next considered dienes possessing a functional handle at C(8) that could unite diene and dienophile such as benzisoxazole 19, thereby enabling an intramolecular Diels–Alder cycloaddition (Scheme 4). Thus, when coupled to aurantioclavine 4, benzisoxazole 20 would offer a stable o-methide imine that could react with the indole moiety of compound 19 in a controlled and intramolecular manner.
Scheme 4. Retrosynthetic Analysis of Communesin F by an Intramolecular Diels–Alder Cycloaddition.
Fischer esterification of commercially available carboxylic acid 21 followed by heating in neat sulfuric acid provided the benzisoxazole acid 20 in 44% yield over two steps (Scheme 5).18 Treatment of benzisoxazole acid 20 with oxalyl chloride provided the corresponding acid chloride, which was smoothly coupled with aurantioclavine 4 to furnish carboxamide 22 (91% yield, two steps). Similarly, 1-methylaurantioclavine 5 reacted with the acid chloride to afford carboxamide 19 (77% yield, two steps). Substrates 22 and 19 were subjected to an intramolecular Diels–Alder cycloaddition under acidic conditions.19 Unfortunately, the benzisoxazole reacted with the butenyl side chain of the aurantioclavine core to generate the bridged polycycles 23 and 24. Nuclear Overhauser effect NMR spectroscopy (NOESY) studies and X-ray analysis (Figure 2) demonstrated the relative stereochemistry shown for 24 and that of 23 was assigned by analogy.
Scheme 5. Intramolecular Diels–Alder Cycloaddition.

Figure 2.
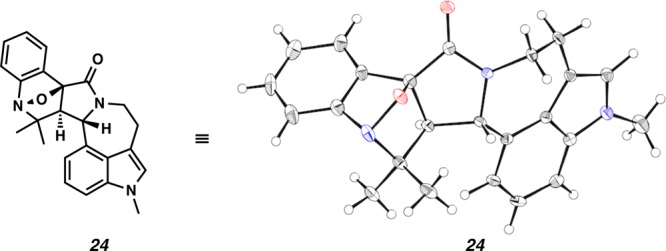
X-ray structure of bridged polycycle 24.
At this point, we turned our attention to synthesizing 3-bromooxindole 26, which would be a precursor to an o-methide imine such as reactive intermediate 6, allowing for the construction of the communesin core according to our original biosynthesis-inspired model (Scheme 1). Aurantioclavine derivative 25 was reacted with bromooxindole 26 in an effort to produce adduct 27 (Scheme 6a). Interestingly, different reactivity was observed in coordinating and noncoordinating solvents. In THF or acetonitrile, the reaction afforded indole 28 in 69% yield, wherein the oxindole was introduced to position C(2) of the indole nucleus, presumably via rearrangement of the initially formed adduct 27 at C(3) (Scheme 6b). Sulfonylation of indole 28 with o-NsCl under basic conditions was accompanied by unexpected chlorination of the indole moiety to afford chloroindolenine 29 (73% yield), the structure of which was unambiguously confirmed by X-ray crystallography (Figure 3). To the best of our knowledge, this constitutes the first use of o-NsCl for chlorination of an indole to provide the 3-chloroindolenine. On the other hand, the same coupling of derivatives 25 and 26 in benzene or dichloromethane furnished indole 28 (24% yield) and two additional undesired products 30 (32% yield) and 31 (24% yield) (Scheme 6c). Adduct 30 results from nucleophilic attack at C(6) of the aurantioclavine indole core, while double adduct 31 is produced from both C(6) and C(2) functionalization. The structure of 30 was unambiguously determined following preparation of lactam 32 (Scheme 6d). Subjecting 30 to excess sodium hydride and o-NsCl conditions functionalized both the oxindole and indole nitrogens (66% yield) and subsequent reduction of the azide allowed for cyclization to lactam 32 in 66% yield. The structure of 32 was confirmed by single-crystal X-ray diffraction (Figure 4).
Scheme 6. Reaction of Aurantioclavine Derivative 25 with Bromooxindole 26.

Figure 3.
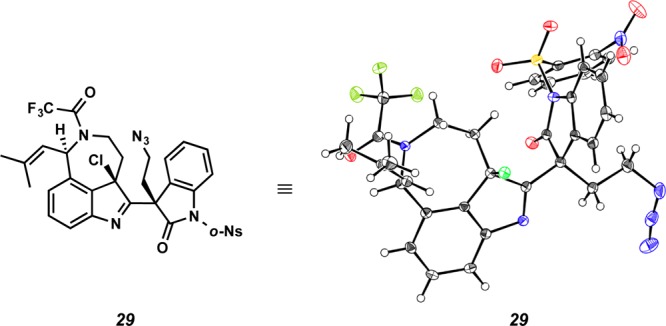
X-ray structure of chloroindoline 29.
Figure 4.
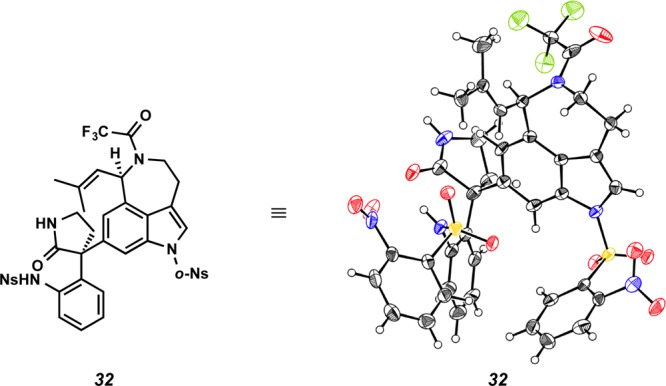
X-ray structure of lactam 32.
2. Alkylation Route to Communesin F
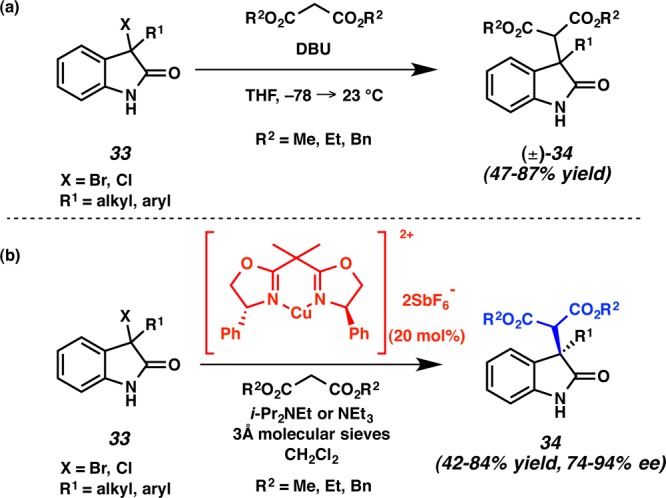
Discouraged by the unsuccessful Diels–Alder cycloaddition-based approaches to communesin F (1f), we considered an alternative strategy toward the natural product. In 2007, as a direct result of our efforts toward the communesins and perophoramidine, we developed a method to generate 3,3-disubstituted oxindoles via the base-mediated coupling of oxindole electrophiles with malonate-derived nucleophiles. (Scheme 7a).20 We also developed an asymmetric variant of this reaction utilizing copper bis(oxazoline) complexes (Scheme 7b).21
Scheme 7. Construction of 3,3-Disubstituted Oxindoles.
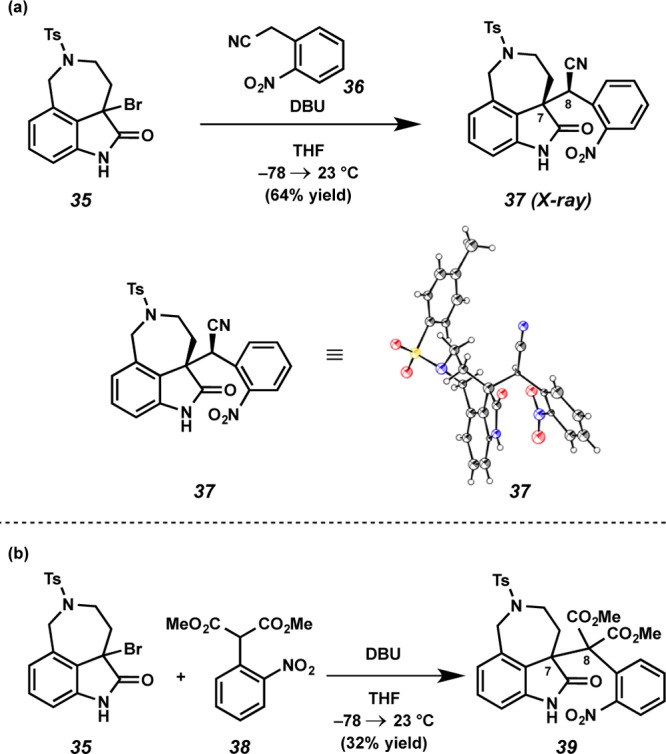
With the method shown in Scheme 7, we devised a new synthetic strategy that cast our coupling fragments in an umpolung manner, invoking an electrophilic aurantioclavine portion and a nucleophilic right-hand fragment. We first pursued this notion in the context of the model azepine 35 (Scheme 8). Treatment of 35 with DBU and a pronucleophile (e.g., 36(22) and 38) produced oxindole adducts (i.e., 37 and 39) possessing the key C(7)–C(8) linkage in modest, but encouraging yields. Importantly, adduct 37 was crystalline, and we confirmed both the new C–C bond as well as the relative stereochemistry of the sole diastereomeric isolate via X-ray analysis.
Scheme 8. Construction of C(7)–C(8) Linkage by Alkylation Strategy.
Having produced the key C(7)–C(8) linkage via an umpolung strategy, we treated aurantioclavine-derived bromooxindole 40 with malonate 41 in the presence of DBU (Scheme 9). Smooth reactivity under our standard conditions led to the isolation of a single stereoisomeric adduct 42 in 74% yield. To our delight, oxindole adduct 42 was amenable to single-crystal X-ray diffraction, however, the X-ray analysis surprisingly revealed that the alkylation occurs with high syn selectivity relative to the existing isobutenyl substituent (Figure 5). This result was intriguing, given that in the Diels–Alder cycloaddition of the corresponding indole 9 with the o-azaxylylene derived from benzyl chloride 10, the selectivity at C(7) favored the anti diastereomer 11 (cf. Schemes 9 and 2).23
Scheme 9. Alkylation of Azepine Bromooxindole 40 with Malonate 41.
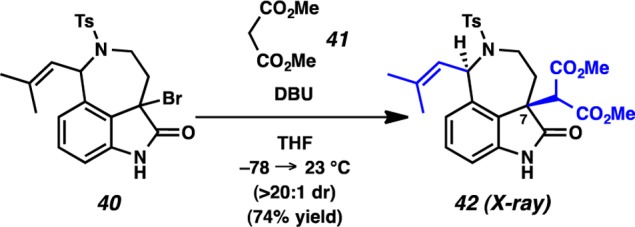
Figure 5.
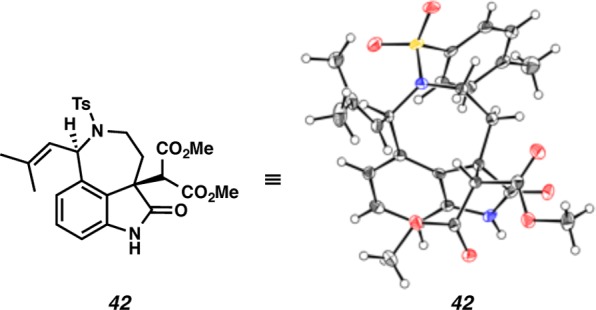
X-ray structure of oxindole adduct 42.
Since the undesired relative stereochemistry was obtained in adduct 42 from the alkylation of azepine 40 and malonate 41, we explored our strategy in a model system lacking the azepine ring of the oxindole (Scheme 10). Known silyl ether 43(21,22) was converted into malonate adducts 46 and 47 in 85% and 96% yield, respectively, under our previously reported conditions in Scheme 7. Importantly, in the nonazepine system, the efficiency of those alkylations is increased, even in these cases where vicinal quaternary centers are generated.24 Methylation of oxindoles 46 and 47 produced 48 and 49 in 99% and 92% yield, respectively.
Scheme 10. Alkylation of 3-Bromooxindole 43.

Acid-catalyzed desilylation and cyclization of diester 48 proceeded smoothly to furnish lactone 50 in 85% yield as a single diastereomer (Scheme 11a).25 To our delight, lactone 50 underwent decarboxylative allylic alkylation when treated with Pd(PPh3)4, yielding 51 in 90% yield as a single diastereomer.26,27 Single-crystal X-ray analysis confirmed that lactone 51 possesses the relative stereochemistry at the vicinal quaternary carbon centers C(7) and C(8) that is needed for further elaboration to communesin F (1f). Interestingly, direct decarboxylative allylic alkylation of diester 49 again provided an alkylated product (i.e., 52) as a single diastereomer in 78% yield (Scheme 11b). Through X-ray analysis, we discovered that the relative stereochemistry at the vicinal quaternary stereocenters C(20) and C(4) of 52 was complementary to that of the lactone 51 and thus ideal for elaboration to perophoramidine (3).
Scheme 11. Model Studies for Construction of the Vicinal Quaternary Centers.
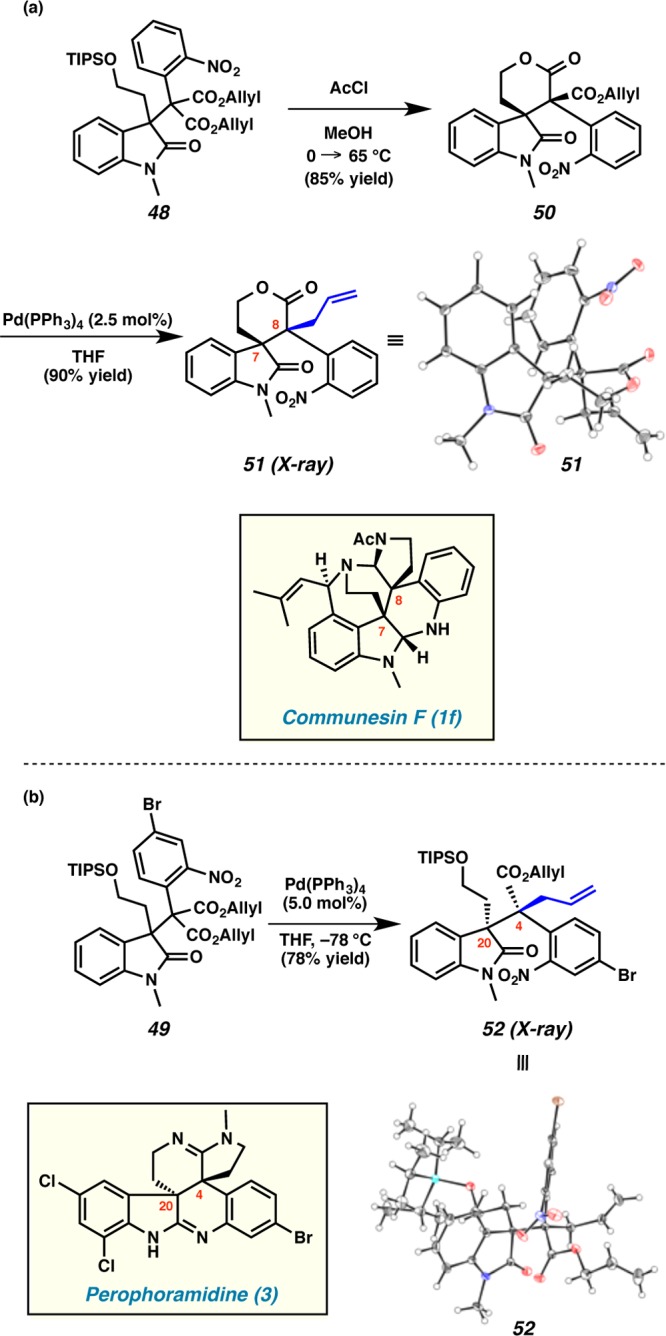
At this time, the underlying reasons for the stereochemical relationships observed in these two alkylation reactions are unclear. The fact that the reactions proceed stereodivergently with high diastereocontrol is quite remarkable. Work toward building reasonable models for stereoinduction of β-quaternary tetrasubstituted enolates in both cyclic and acyclic settings as well as the development of these interesting processes in more general cases is ongoing. Nevertheless, with the promising model systems 51 and 52 completed, we next applied our findings to expedient formal syntheses of communesin F (1f) and perophoramidine (3).
3. Formal Synthesis of Communesin F (1f)
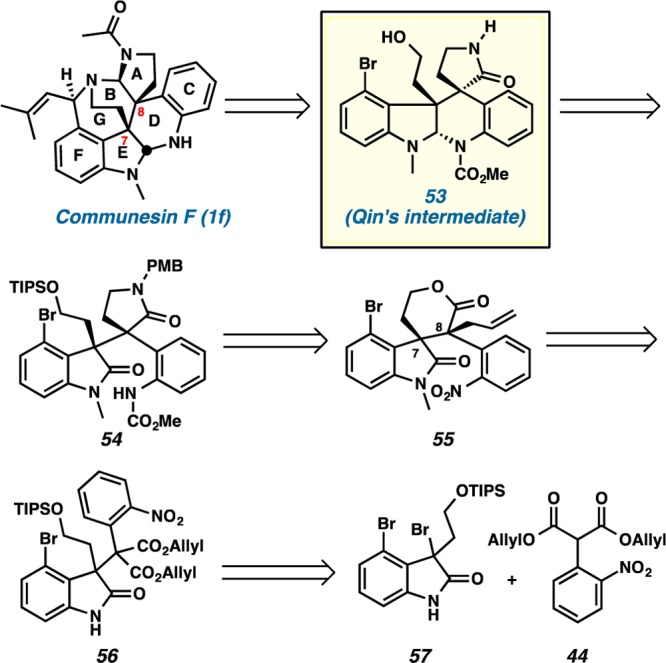
As depicted in our retrosynthetic strategy (Scheme 12), communesin F could be completed from advanced intermediate 53 in Qin’s synthesis.5g We anticipated the initial disconnection of the aminal linkage in 53, thereby revealing oxindole and aniline moieties in 54. Then, the lactam ring in 54 would be excised, affording lactone 55. We envisioned that the relative stereochemical relationship at C(7) and C(8) of lactone 55 could be established by employing our decarboxylative allylic alkylation. The quaternary center on oxindole 56 was disassembled into 3-bromooxindole 57 and diallyl malonate 44.
Scheme 12. Retrosynthesis of Communesin F (1f).
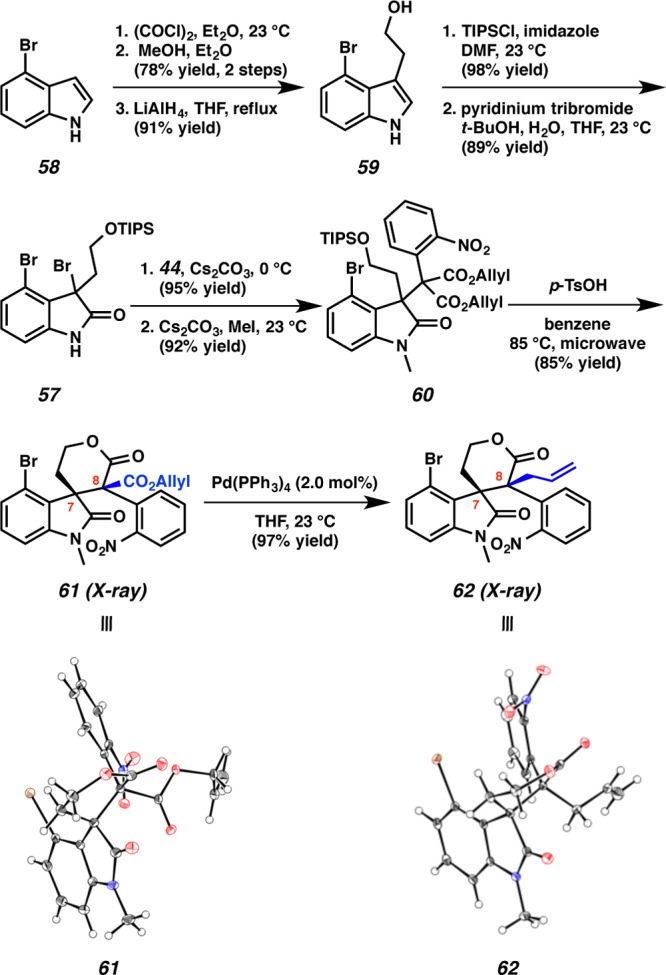
In the forward synthetic sense, our efforts toward communesin F commenced with the elaboration of 4-bromooxindole 58 to diallyl malonate 60 (Scheme 13). Treatment of 4-bromoindole 58 with oxalyl chloride and methanol provided an oxoacetate (78% yield, two steps), which was reduced to the corresponding primary alcohol 59 with LiAlH4 in 91% yield.5g Silylation of the primary alcohol with TIPSCl (98% yield) and subsequent oxidation with pyridinium tribromide afforded dibromooxindole 57 in 89% yield.28 Despite the extra steric encumbrance of C(4) substitution, we were delighted to find that smooth coupling of dibromooxindole 57 with malonate 44 produced a 3,3-disubstituted oxindole in 95% yield. Protection of the oxindole with MeI delivered adduct 60 in 92% yield. Microwave assisted lactonization of diester 60 with p-TsOH proceeded smoothly to furnish lactone 61 as a single diastereomer (85% yield). Gratifyingly, decarboxylative allylic alkylation constructed the quaternary center at C(8) of compound 62 as a single diastereomer in 97% yield under Pd(PPh3)4 catalysis. The relative stereochemistry at C(7) and C(8) of 61 and 62 was unambiguously confirmed by X-ray analysis.
Scheme 13. Development of the Vicinal Quaternary Center.
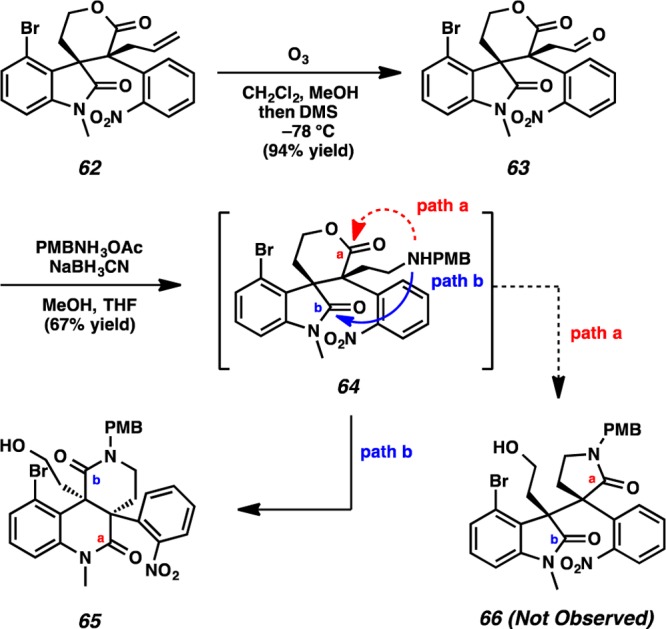
Although ozonolysis of alkene 62 delivered aldehyde 63 in 94% yield, attempted reductive amination of aldehyde 63 did not produce the desired γ–lactam 66 (Scheme 14). Upon treatment of aldehyde 63 with p-methoxybenzylammonium acetate and sodium cyanoborohydride, amine intermediate 64 was likely produced.29 Instead of opening the lactone directly (path a), nucleophilic attack by the newly generated amine at the oxindole moiety (path b), and subsequent ring-shift tautomerization delivered dihydroquinolinone 65 in 67% yield.
Scheme 14. Ozonolysis and Reductive Amination of Lactone 62.
Alternatively, we found that lactam 54 (an analogue of 66) could be obtained via the reaction sequence summarized in Scheme 15. The nitro group on compound 62 was reduced to the aniline, which resulted in concomitant lactone ring opening to furnish a bis-oxindole 67 in 80% yield. Protection of the primary alcohol with TIPSCl (90% yield) and protection of the oxindole nitrogen with methyl chloroformate afforded carbamate 68 in 98% yield. Ozonolysis of alkene 68 generated aldehyde 69 (94% yield),30 which underwent subsequent reductive amination and selective lactamization with the electron-deficient oxindole to afford γ-lactam 54 in 95% yield.
Scheme 15. Synthesis of Lactam 54.

With lactam 54 in hand, we envisioned that the piperidine D ring of 70 would be prepared by AlH3–Me2NEt mediated reductive cyclization (Scheme 16).21,31 To our disappointment, treatment of lactam 54 with AlH3–Me2NEt produced undesired pyrrolidinoindoline derivative 71 as a single diastereomer in 61% yield resulting from chemoselective reduction of the N-PMB-lactam in the presence of the oxindole. After cleavage of the TIPS group by TBAF (98% yield),32 the PMB group was removed with DDQ33 to provide alcohol 72. The structure of the pentacyclic heterocycle 72 was confirmed by X-ray analysis (Figure 6).
Scheme 16. Reductive Cyclization of Lactam 54 with AlH3-Me2NEt.

Figure 6.
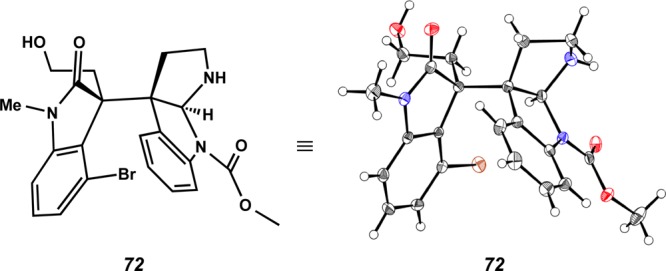
X-ray structure of pyrrolidinoindoline 72.
Having failed on our initial exploration, alternative conditions for construction of the piperidine D ring were next explored. Treatment of the lactam 54 with LiAlH434 produced debrominated compound 73 in 83% yield (Scheme 17a). X-ray analysis of compound 73 showed a hydrogen-bonding interaction between the carbonyl group of the PMB-protected amide and the NH group of the carbamate. We reasoned that the undesired pyrrolidine was formed preferentially to the piperidine due to the close proximity of the carbamate NH and the carbonyl group of the PMB-protected amide. Next, a reductive cyclization reaction was attempted by treatment of 54 with Tf2O and NaBH4 to construct the piperidine ring. To our surprise, treatment of lactam 54 with Tf2O provided the PMB-protected hexacyclic oxindole 76 in 95% yield (Scheme 17b). The PMB-protected amide of 54 was activated by Tf2O to provide 74, and nucleophilic attack by the aniline functionality furnished pyrrolidinoindoline derivative 75. After the TIPS group was removed under the reaction conditions, the resultant hydroxyl group attacked the amidinium to generate the propellane structure of hexacyclic oxindole 76. After cleavage of the PMB group using DDQ, the propellane structure of hexacyclic oxindole 77 was confirmed using X-ray analysis.
Scheme 17. Attempted Reductive Cyclization of Lactam 54.

Despite this unexpected turn of events, we envisaged that the desired aminal 81 could be accessed from the propellane compound 76 using suitable conditions, since the oxidation state at C(9) of 76 is identical to that of the desired aminal 81 (Scheme 18). Moreover, the reactive N-PMB-pyrrolidinone in 54 is now protected by the propellane structure of 76, thus leaving the oxindole as the only reducible carbonyl group. Fortunately, after extensive experimentation, we were pleased to find that reductive cyclization of hexacyclic oxindole 76 could be accomplished with DIBAL and Et2AlCl to furnish aminal 81 in 87% yield (Scheme 18). Presumably, the oxindole of 76 was reduced by DIBAL to provide 78, and rearrangement of the propellane structure generated iminium 79. After the workup, water attacked the iminium moiety of 79 to afford aniline 80, and the resultant aniline group attacked the iminium of 80 to construct aminal 81. In the last stage of the synthesis, we screened a variety of reaction conditions to remove the PMB group on the lactam 81 (e.g., DDQ, CAN, TFA, etc.), but surprisingly, removal of the PMB group failed under all conditions attempted. This unexpected turn was particularly insidious since the PMB group was easily removed from hexacyclic oxindole 76 by DDQ (Scheme 17). The cleavage of allyl or benzyl groups were also examined, but disappointingly, cleavage of these groups on the lactam was similarly unsuccessful under several conditions.35
Scheme 18. Synthesis of Aminal 81.

Given the difficulty of removal of PMB, allyl, and benzyl groups, our attention turned to exploring the o-nitrobenzyl group as a protecting group. However, subjecting the hexacyclic oxindole 77 to o-nitrobenzyl bromide under basic conditions to produce the o-nitrobenzyl-protected propellane hexacyclic oxindole turned out to be challenging. Thus, we next investigated reductive amination of aldehyde 69 and were pleased to find that treatment of 69 with o-nitrobenzylammonium acetate 82 furnished lactam 83 in 97% yield (Scheme 19). Formation of the o-nitrobenzyl-protected propellane hexacyclic oxindole using Tf2O (75% yield) was followed by reductive cyclization with DIBAL and Et2AlCl to furnish aminal 84 in 60% yield. To our delight, we found that removal of the o-nitrobenzyl group could be achieved by photolysis/irradiation at 350 nm in 40% yield.36 Surprisingly, we discovered that removal of the o-nitrobenzyl group to produce compound 53 was also accomplished using 20% aq NaOH in methanol at 75 °C in 70% yield—a previously unknown deprotection protocol.37 Aminal 53 has been advanced by the Qin group to communesin F,5g thus completing our formal synthesis of the natural product.
Scheme 19. Completion of Formal Synthesis of Communesin F.

4. Formal Synthesis of Perophoramidine (3)
Our retrosynthetic analysis of perophoramidine (3) was based on our previously established expedient synthesis of oxindole derivative 52 (Scheme 20). We speculated that the aminal and lactam ring functionalities of pentacycle 85, an intermediate in Funk’s synthesis,5o could be cleaved, thereby leading to aldehyde 86. The N–C bond of the 6-bromooxindole moiety in 86 was excised to arrive at nitroarene 52. The construction of the contiguous quaternary centers at C(20) and C(4) of allyl ester 52 with the proper relative stereochemistry was accessed by decarboxylative allylic alkylation as previously described (Scheme 11b).
Scheme 20. Retrosynthesis of Perophoramidine (3).
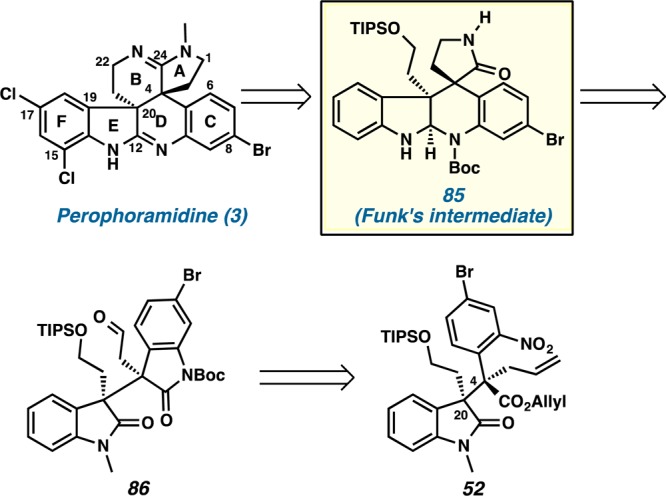
Carbamate 88 was obtained by reduction of nitroarene 52 with titanium chloride and simultaneous oxindole formation38 to furnish the bis-oxindole moiety 87 in 91% yield followed by protection with Boc anhydride in 85% yield. Ozonolysis of olefin 88 produced aldehyde 86 in 90% yield. Reductive amination of aldehyde 86 with o-nitrobenzylammonium acetate 82 resulted in an amine that underwent in situ lactam formation to afford oxindole lactam 89 in 91% yield (Scheme 21)
Scheme 21. Synthesis of Amide 89.

Initially, we attempted to generate the o-nitrobenzyl protected propellane hexacyclic oxindole 90 under analogous conditions to those used in our formal synthesis of communesin F on the pseudo-diastereomeric series (vide supra). However, treatment of lactam 89 with Tf2O yielded an unexpected azepine 91 in 70% yield (Scheme 22). Both the Boc and the TIPS groups on amide 89 were removed under the reaction conditions, and the resulting primary alcohol was presumably converted to the corresponding triflate. Finally, the aniline likely attacked the newly formed triflate to form azepine 91.
Scheme 22. Formation of Azepine 91 Using Tf2O.

After extensive experimentation, we discovered that in contrast to the communesin system, the desired reductive cyclization in the perophoramidine diastereomer occurred directly with AlH3–Me2NEt31 to furnish cyclization product 92 in 42% yield (66% yield based on recovered starting material) (Scheme 23). The indoline methyl group was converted to a formyl group using PDC oxidation in 62% yield (93% yield based on recovered starting material).39 To our delight, an attempt to remove the formyl group with 20% aq NaOH at 75 °C resulted in removal of both the formyl group and the o-nitrobenzyl group to produce aminal 85 in 50% yield.37,40 This molecule was previously advanced by the Funk group to perophoramidine5o and constitutes an expedient formal synthesis of the natural product.
Scheme 23. Completion of Formal Synthesis of Perophoramidine.

Conclusion
In conclusion, we have conducted synthetic studies toward unique polycyclic alkaloids and completed formal syntheses of communesin F (1f) in 9% overall yield over 17 steps and perophoramidine (3) in 6% overall yield (13% overall yield, based on recovered starting material) over 10 steps using a unified stereodivergent alkylation approach. The all-carbon quaternary center on the oxindole was established via stabilized enolate alkylation of 3-bromooxindoles, a method previously developed by our laboratory and now shown to be quite versatile even in particularly sterically challenging situations. The complementary relative stereochemistry of the two contiguous quaternary stereogenic centers found in communesin F (1f) and perophoramidine (3), respectively, was established by substrate controlled diastereoselective decarboxylative allylic alkylation. A reductive amination approach furnished the A ring, and reductive cyclization produced the D ring for both communesin F (1f) and perophoramidine (3). En route to the evolution of our eventual successful strategy, we have discovered a method to convert an indole to a 3-chloroindolenine using a mild reagent such as o-NsCl during the synthesis. In addition, previously unknown, mild and efficient deprotection conditions for the o-nitrobenzyl group on the lactam were discovered. Further studies to rationalize unprecedented complementary selectivity by Pd-catalyzed allylic alkylation reactions are currently in progress.
Experimental Section
N-(2-(1-Hydroxy-2-(methoxymethoxy)ethyl)phenyl)-4-methylbenzenesulfonamide (13)
To a solution of 4-methyl-N-(2-vinylphenyl)benzenesulfonamide (SI-1) (6.11 g, 22.4 mmol, 1.00 equiv) in THF (140 mL) and water (70 mL) were added N-methylmorpholine N-oxide (5.96 g, 50.8 mmol, 2.30 equiv) and osmium tetroxide (11.6 mg, 43.9 mmol, 0.002 equiv). After addition, reaction was stirred for 3 days. The reaction was concentrated to approximately 50 mL under reduced pressure and then extracted with a mixture ether and THF (1:1) (3 × 45 mL). The organic layers were dried over sodium sulfate, and the solvent was removed under reduced pressure. Impurities were removed by washing solid with dichloromethane to afford diol SI-2 (5.43 g, 80% yield) as a white solid: Rf = 0.13 (1:1 hexane/EtOAc); 1H NMR (300 MHz, CDCl3) δ 8.47 (s, 1H), 7.71 (d, J = 8.3 Hz, 2H), 7.39 (dd, J = 8.1, 1.1 Hz, 1H), 7.25–7.18 (m, 3H), 7.15–7.02 (m, 2H), 4.82 (t, J = 6.5 Hz, 1H), 3.66–3.57 (m, 2H), 2.97 (br, s, 1H), 2.39 (s, 3H), 1.98 (br, s, 1H); 13C NMR (75 MHz, CDCl3) δ 144.1, 137.1, 136.3, 129.9, 129.8, 129.2, 128.5, 127.4, 127.0, 122.2, 74.78, 66.0, 21.8; IR (neat film NaCl) 3271, 1318, 1150 cm–1; HRMS (MM: ESI-APCI+) m/z calcd for C15H18NO4S [M + H]+ 308.0951, found 308.0967.
To a solution of diol SI-2 (500 mg, 1.63 mmol, 1.00 equiv) in toluene (70 mL) was added dibutyltin dimethoxide (410 μL, 1.79 mmol, 1.10 equiv). The flask was fitted with a short path distillation apparatus, and approximately half of the solvent was removed by distillation. To this solution were added MOMCl (136 μL, 1.79 mmol, 1.10 equiv) and tetrabutylammonium iodide (900 mg, 2.44 mmol, 1.50 equiv). After addition, the reaction was stirred for 12 h, and then brine was added to this solution. The reaction mixture was extracted with EtOAc (3 × 50 mL). The combined organic phases were dried over MgSO4 and concentrated in vacuo. The residue was purified by flash column chromatography (3:1 → 1:1 hexanes/EtOAc) to afford alcohol 13 (513 mg, 90% yield, two steps) as a white solid: Rf= 0.27 (1:1 hexane/EtOAc); 1H NMR (300 MHz, CDCl3) δ 8.76 (s, 1H), 7.72 (d, J = 8.5 Hz, 2H), 7.50 (d, J = 8.0 Hz, 1H), 7.27–7.20 (m, 3H), 7.11–7.02 (m, 2H), 4.83–4.78 (m, 1H), 4.64 (s, 2H), 3.60 (dd, J = 10.5, 3.5 Hz, 1H), 3.48–3.41 (m, 2H), 3.39 (s, 3H), 2.39 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 143.9, 137.1, 136.3, 129.7, 129.5, 128.9, 128.2, 127.2, 124.7, 122.0, 97.0, 73.3, 72.4, 55.6, 21.6; IR (neat film NaCl) 3233, 2932, 1598, 1497, 1335, 1161 cm–1; HRMS (MM: ESI-APCI+) m/z calcd for C17H22NO5S [M + H]+ 352.1213, found 352.1219.
Methyl 2′-Oxospiro[cyclopropane-1,3′-indoline]-2-carboxylate (16)
A flame-dried flask (25 mL) equipped with a Teflon stirbar was charged with sodium hydride (60% dispersion in mineral oil, 22 mg, 0.55 mmol, 1.10 equiv), which was washed 3 times with dry hexanes. Then, DMSO (5.5 mL) and trimethylsulfoxonium iodide (119 mg, 0.58 mmol, 1.20 equiv) were added. To this solution was added methyl (E)-2-(2-oxoindolin-3-ylidene)acetate (SI-3) (100 mg, 0.49 mmol, 1.00 equiv) in a solution of DMSO (2.5 mL). After addition, the reaction was stirred for 2 h, and then the temperature was raised to 50 °C. The reaction was complete after another hour. Brine was added and then the mixture was extracted with EtOAc (3 × 5 mL). The combined organic phases were dried over MgSO4 and concentrated in vacuo. The residue was purified was by flash column chromatography (3:1 → 1:1 hexanes/EtOAc) to afford oxindole 16 as two diastereomers. Diastereomer 1: (44.7 mg, 42% yield). Diastereomer 2: (28.6 mg, 27% yield). Diastereomer 1: Rf= 0.52 (1:1 hexane/EtOAc); 1H NMR (300 MHz, CDCl3) δ 8.28 (br, s, 1H), 7.34 (d, J = 7.7 Hz, 1H), 7.22 (dd, J = 7.7, 1.3 Hz, 1H), 7.02 (dd, J = 7.7, 1.1 Hz, 1H), 6.99–6.93 (m, 1H), 3.69 (s, 3H), 2.72 (dd, J = 8.6, 7.4 Hz, 1H), 2.16 (dd, J = 7.4, 4.5 Hz, 1H), 2.04 (dd, J = 8.6, 4.4 Hz, 1H); 13C NMR (75 MHz, CDCl3) δ 177.5, 169.3, 141.8, 127.9, 126.4, 123.0, 122.4, 110.3, 52.4, 34.3, 32.9, 21.1; IR (neat film NaCl) 3214, 1712, 1622, 1470, 1209 cm–1; HRMS (MM: ESI-APCI+) m/z calcd for C12H12NO3 [M + H]+ 218.0812, found 218.0825. Diastereomer 2: Rf= 0.45 (1:1 hexane/EtOAc); 1H NMR (300 MHz, CDCl3) δ 7.99 (br, s, 1H), 7.25–7.19 (m, 1H), 7.02 (td, J = 7.6, 1.0 Hz, 1H), 6.93 (d, J = 7.8 Hz, 1H), 6.85–6.80 (m, 1H), 3.75 (s, 3H), 2.66 (t, J = 8.3 Hz, 1H), 2.39 (dd, J = 5.0, 8.0 Hz, 1H), 1.84 (dd, J = 5.0, 8.6 Hz, 1H); 13C NMR (75 MHz, CDCl3) δ 176.0, 167.7, 141.1, 129.5, 188.0, 122.4, 118.9, 110.3, 52.6, 33.5, 32.9, 21.3; IR (neat film NaCl) 3256, 1739, 1710 cm–1; HRMS (MM: ESI-APCI+) m/z calcd for C12H12NO3 [M + H]+ 218.0812, found 218.0828.
Benzo[c]isoxazole-3-carboxylic Acid 20
A flame-dried flask (500 mL) equipped with a Teflon stirbar was charged with 2-nitrophenylacetic acid 21 (10.0 g, 55.2 mmol, 1.00 equiv), ethanol (60 mL), sulfuric acid (200 μL), and toluene (280 mL). The flask was fitted with a condenser, and the solution was refluxed for 14 h. The solvent was removed under reduced pressure and sulfuric acid (280 mL) was added. After addition, the reaction was heated to 110 °C and stirred for 90 min. The solution was then poured onto ice (600 g), and the mixture was extracted with ether (3 × 200 mL). The combined organic layers were dried over MgSO4 and concentrated in vacuo. Purification was performed via crystallization from water to afford acid 20 (3.94 g, 44% yield, 2 steps) as an off-white solid: 1H NMR (300 MHz, acetone-d6) δ 10.82 (br, s, 1H), 7.94 (d, J = 9.0 Hz, 1H), 7.75 (d, J = 10.0 Hz, 1H), 7.50 (dd, J = 6.5, 9.5 Hz, 1H), 7.34 (dd, J = 7.0, 8.5 Hz, 1H); 13C NMR (75 MHz, acetone-d6) δ 158.5, 158.1, 155.3, 132.4, 128.8, 121.4, 121.0, 116.7; IR (neat film NaCl) 2360, 1731, 1301, 1231, 1189, 753 cm–1; HRMS (MM: ESI-APCI+) m/z calcd for C8H6NO3 [M + H]+ 164.0342, found 164.0341.
6-Methyl-1-(2-methylprop-1-en-1-yl)-2,3,4,6-tetrahydro-1H-azepino[5,4,3-cd]indole 5
To a solution of (E)-2-methyl-4-(3-(2-nitroethyl)-1H-indol-4-yl)but-3-en-2-ol (SI-4) (386 mg, 1.41 mmol, 1.00 equiv) in THF (14 mL) was added methyl iodide (875 μL, 14.1 mmol, 10.0 equiv) at 0 °C. Sodium hydride (60% dispersion in mineral oil, 562 mg, 14.5 mmol, 10.3 equiv) was then added to the solution, and the mixture was stirred for 25 min at 23 °C. The reaction was quenched with satdrated ammonium hydroxide solution and extracted with EtOAc (3 × 10 mL). The combined organic layers were dried over MgSO4 and concentrated in vacuo. The residue was purified by flash column chromatography (3:1 → 2:1 hexanes/EtOAc) to afford (E)-2-methyl-4-(1-methyl-3-(2-nitroethyl)-1H-indol-4-yl)but-3-en-2-ol (SI-5) (363.5 mg, 90% yield) as a yellow solid.
To a solution of nitro compound SI-5 (512 mg, 1.78 mmol, 1.00 equiv) in MeOH (125 mL) and 2 N HCl (40 mL) was added amalgamated zinc, which had been formed from zinc dust (6.5 g, 98.3 mmol, 55.0 equiv) and mercuric chloride (1.10 g, 3.55 mmol, 2.00 equiv) in 2 N HCl and subsequently rinsed with MeOH. The mixture was stirred at reflux for 3 h. The reaction was then decanted from the remaining amalgam and then basified to pH >10. The solid was removed by filtration, and the resulting solution was extracted with dichloromethane (3 × 100 mL). The combined organic layers were dried over MgSO4 and concentrated in vacuo. The residue was purified by flash column chromatography (18:1 CH2Cl2/MeOH) to afford 1-methylaurantioclavine 5 (258 mg, 60% yield) as a yellow oil: Rf= 0.30 (18:1 CH2Cl2/MeOH); 1H NMR (300 MHz, CDCl3) δ 7.19–7.12 (m, 2H), 6.89–6.83 (m, 2H), 5.48 (d, J = 9.0 Hz, 1H), 4.92 (d, J = 9.0 Hz, 1H), 3.76 (s, 3H), 3.62–3.54 (m, 1H), 3.13–3.02 (m, 3H), 2.26 (br, s, 1H), 1.86 (s, 6H); 13C NMR (75 MHz, CDCl3) δ 138.5, 137.8, 133.3, 127.7, 125.9, 121.1, 117.4, 114.2, 107.3, 62.6, 48.9, 32.7, 30.8, 25.9, 18.4; IR (neat film NaCl) 3332, 2910, 1554, 1455 cm–1; HRMS (MM: ESI-APCI+) m/z calcd for C16H21N2 [M + H]+ 241.1699, found 241.1712.
Benzo[c]isoxazol-3-yl(1-(2-methylprop-1-en-1-yl)-1,3,4,6-tetrahydro-2H-azepino[5,4,3-cd]indol-2-yl)methanone (22)
To a solution of 2,1-benzisoxazole-3-carboxylic acid (20) (262 mg, 1.60 mmol, 1.25 equiv) in dichloromethane (5 mL) was added oxalyl chloride (420 μL, 4.81 mmol, 3.80 equiv) and then a small amount of DMF (∼20 μL). The reaction was stirred for 1 h, and then the solvent was removed under reduced pressure; the residue was evaporated from benzene (2 mL) to remove excess reagent. Dichloromethane (10 mL) and triethylamine (537 μL, 3.85 mmol, 3.00 equiv) were added, and to this solution was added aurantioclavine 4 (290 mg, 1.28 mmol, 1.00 equiv). After addition, the reaction was stirred for 60 min, and then brine was added. The resulting solution was extracted with EtOAc (3 × 7 mL). The combined organic layers were dried over MgSO4 and concentrated in vacuo. The residue was purified by flash column chromatography (3:1 → 1:1 hexanes/EtOAc) to afford amide 22 (435 mg, 91% yield, 2 steps) as a white solid. Rf= 0.72 (1:2 hexanes/EtOAc). (Due to the distinct presence of rotameric isomers, the 1H NMR and 13C NMR contained extra peaks. See the attached spectrum, Supporting Information): 1H NMR (300 MHz, CDCl3) δ 9.00–8.86 (m, 2H), 7.98 (dq, J = 8.9, 0.9 Hz, 1H), 7.71–7.57 (m, 3H), 7.35–7.27 (m, 2H), 7.25–7.22 (m, 1H), 7.22–7.16 (m, 3H), 7.15–7.07 (m, 2H), 7.07–7.01 (m, 2H), 7.01–6.94 (m, 2H), 6.90–6.83 (m, 2H), 6.70 (d, J = 7.5 Hz, 1H), 5.48 (ddq, J = 7.9, 2.8, 1.6 Hz, 2H), 4.82–4.66 (m, 2H), 4.07 (ddd, J = 15.4, 10.0, 5.8 Hz, 1H), 3.82 (td, J = 13.0, 2.6 Hz, 1H), 3.60–3.46 (m, 1H), 3.23–3.12 (m, 3H), 1.96 (d, J = 1.3 Hz, 3H), 1.79 (d, J = 1.5 Hz, 3H), 1.73 (d, J = 1.6 Hz, 2H), 1.64 (d, J = 1.3 Hz, 2H); 13C NMR (75 MHz, CDCl3) δ 158.5, 158.4, 157.9, 157.8, 156.9, 156.8, 137.7, 137.4, 137.2, 136.9, 135.2, 135.1, 131.5, 131.4, 126.4, 126.1, 124.7, 124.3, 123.9, 123.8, 122.1, 121.9, 121.8, 121.7, 121.1, 121.0, 119.9, 118.3, 117.5, 114.9, 113.3, 112.6, 110.1, 109.9, 61.0, 57.5, 44.5, 43.1, 29.1, 25.9, 25.8, 25.7, 18.9, 18.2; IR (neat film NaCl) 3325, 2914, 2246, 1730, 1616, 1447 cm–1; HRMS (MM: ESI-APCI+) m/z calcd for C23H22N3O2 [M + H]+ 372.1707, found 372.1721.
Benzo[c]isoxazol-3-yl(6-methyl-1-(2-methylprop-1-en-1-yl)-1,3,4,6-tetrahydro-2H-azepino[5,4,3-cd]indol-2-yl)methanone (19)
To a solution of 2,1-benzisoxazole-3-carboxylic acid (20) (37.5 mg, 0.229 mmol, 1.10 equiv) in dichloromethane (500 μL) was added oxalyl chloride (59 μL, 0.676 mmol, 3.30 equiv) and then a small amount of DMF (∼1 μL). The reaction was stirred for 1 h, and then the solvent was removed under reduced pressure. The residue was evaporated from benzene (1 mL) to remove excess reagent. Dichloromethane (1.1 mL) and triethylamine (30 μL, 0.215 mmol, 1.03 equiv) were added, and to this solution was added 1-methylaurantioclavine (5) (50.0 mg, 0.208 mmol, 1.00 equiv). After addition, the reaction was stirred for 60 min, and then brine was added. The resulting solution was extracted with EtOAc (3 × 1.5 mL). The combined organic layers were dried over MgSO4 and concentrated in vacuo. The residue was purified by flash column chromatography (3:1 → 2:1 hexanes/EtOAc) to afford amide 19 (61.6 mg, 77% yield, 2 steps) as a white solid: Rf= 0.79 (1:2 hexanes/EtOAc). (Due to the distinct presence of rotameric isomers, the 1H NMR and 13C NMR contained extra peaks. See the attached spectrum, Supporting Information): 1H NMR (300 MHz, CDCl3) δ 8.00 (dt, J = 8.9, 1.1 Hz, 1H), 7.71 (dt, J = 8.9, 1.1 Hz, 1H), 7.61 (ddt, J = 10.9, 9.1, 1.0 Hz, 2H), 7.34–7.26 (m, 3H), 7.24–7.16 (m, 5H), 7.12–7.04 (m, 2H), 7.00 (ddd, J = 8.9, 6.4, 0.8 Hz, 1H), 6.93–6.86 (m, 2H), 6.81 (d, J = 1.2 Hz, 1H), 6.72 (d, J = 7.5 Hz, 1H), 5.50 (ddt, J = 7.4, 2.8, 1.4 Hz, 2H), 4.83–4.64 (m, 2H), 4.13–4.00 (m, 1H), 3.81 (td, J = 13.0, 2.6 Hz, 1H), 3.69 (d, J = 8.6 Hz, 6H), 3.64–3.45 (m, 1H), 3.26–3.09 (m, 3H), 1.98 (d, J = 1.3 Hz, 3H), 1.83–1.78 (m, 3H), 1.74 (d, J = 1.4 Hz, 2H), 1.65 (d, J = 1.3 Hz, 2H); 13C NMR (75 MHz, CDCl3) δ 158.5, 158.5, 157.7, 157.6, 156.9, 156.8, 137.8, 137.7, 137.6, 136.8, 135.7, 135.6, 131.3, 131.2, 126.5, 126.3, 125.9, 124.8, 124.4, 124.3, 124.2, 122.1, 121.6, 121.5, 121.2, 121.1, 120.0, 118.1, 117.3, 115.0, 114.9, 112.3, 111.7, 107.9, 107.7, 60.9, 57.3, 44.5, 43.1, 32.6, 29.0, 25.9, 25.7, 18.9, 18.2; IR (neat film NaCl) 2913, 2245, 1615, 1455, 1410 cm–1; HRMS (MM: ESI-APCI+) m/z calcd for C24H24N3O2 [M + H]+ 386.1863, found 386.1877.
15,15-Dimethyl-4,6,7,15,15a,15b-hexahydro-9H-9a,14-epoxyindolo[4″,3″:3′,4′,5′]azepino[1′,2′:1,2]pyrrolo[3,4-c]quinolin-9-one (23)
A flame-dried vial (20 mL) equipped with a Teflon stirbar was charged with amide 22 (100 mg, 0.269 mmol, 1.00 equiv) and cooled to 0 °C. To this reaction mixture was added a 0.5 M solution of HCl in MeOH (2.7 mL, generated from addition of acetyl chloride to methanol at 0 °C) at 0 °C. The mixture was stirred for 1 h and then warmed to 23 °C over 30 min. The solvent was then removed under reduced pressure. Purification was performed by washing the solid with dichloromethane to afford indole 23 (31.1 mg, 31% yield) as a white solid: Rf= 0.22 (1:1 hexane/EtOAc); 1H NMR (300 MHz, DMSO) δ 11.05 (d, J = 2.5 Hz, 1H), 7.35–7.17 (m, 6H), 7.09 (t, J = 7.7 Hz, 1H), 6.61 (dt, J = 7.4, 1.0 Hz, 1H), 5.47 (d, J = 6.6 Hz, 1H), 4.21 (dt, J = 13.2, 3.7 Hz, 1H), 3.45 (td, J = 13.2, 12.5, 2.4 Hz, 1H), 3.15 (dt, J = 16.0, 3.0 Hz, 1H), 3.08–2.94 (m, 1H), 2.37 (dd, J = 6.6, 0.9 Hz, 1H), 1.77 (s, 3H), 1.03 (s, 3H); 13C NMR (75 MHz, DMSO) δ 164.3, 153.0, 139.6, 137.0, 134.6, 127.3, 126.6, 123.7, 122.6, 121.4, 118.6, 118.0, 115.2, 112.7, 110.2, 96.4, 70.1, 63.1, 61.2, 44.4, 26.9, 26.6, 26.0; IR (neat film NaCl) 3314, 1681, 753 cm–1; HRMS (MM: ESI-APCI+) m/z calcd for C23H22N3O2 [M + H]+ 372.1707, found 372.1710.
4,15,15-Trimethyl-4,6,7,15,15a,15b-hexahydro-9H-9a,14-epoxyindolo[4″,3″:3′,4′,5′]azepino[1′,2′:1,2]pyrrolo[3,4-c]quinolin-9-one (24)
A flame-dried vial (20 mL) equipped with a Teflon stirbar was charged with amide 19 (100 mg, 0.259 mmol, 1.00 equiv) and cooled to 0 °C. To this solution was added a 0.5 M solution of HCl in MeOH (2.6 mL, generated from addition of acetyl chloride to methanol at 0 °C) at 0 °C. The mixture was stirred for 1 h and then warmed to 23 °C over 30 min. The solvent was then removed under reduced pressure. Purification was performed via flash column chromatography (3:1 → 1:1 hexanes/EtOAc) to afford indole 24 (101 mg, 99% yield) as a white solid: Rf= 0.29 (1:1 hexane/EtOAc); 1H NMR (300 MHz, CDCl3) δ 7.33–7.32 (m, 1H), 7.24–7.14 (m, 5H), 7.00 (s, 1H), 6.76–6.73 (m, 1H), 5.51 (d, J = 6.5 Hz, 1H), 4.57–4.50 (m, 1H), 3.80 (s, 3H), 3.41 (dd, J = 12.4, 10.0 Hz, 1H), 3.33–3.24 (m, 1H), 3.19–3.12 (m, 1H), 2.57 (d, J = 6.5 Hz, 1H), 1.95 (s, 3H), 1.18 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 165.4, 153.5, 139.8, 137.9, 135.6, 127.5, 127.0, 126.9, 124.6, 121.9, 119.1, 118.4, 115.9, 113.4, 108.4, 97.2, 70.9, 64.1, 61.9, 45.2, 33.0, 27.5, 27.2, 26.6; IR (neat film NaCl) 3315, 2932, 1699, 1456, 1317, 754 cm–1; HRMS (MM: ESI-APCI+) m/z calcd for C24H24N3O2 [M + H]+ 386.1863, found 386.1867.
2,2,2-Trifluoro-1-(1-(2-methylprop-1-en-1-yl)-1,3,4,6-tetrahydro-2H-azepino[5,4,3-cd]indol-2-yl)ethan-1-one (25)
To a solution of aurantioclavine 4 (882 mg, 3.90 mmol, 1.00 equiv) in THF (14 mL) were added triethylamine (820 μL, 5.88 mmol, 1.50 equiv) and trifluoroacetic anhydride (606 μL, 4.29 mmol, 1.10 equiv) at 0 °C. The reaction was completed immediately, so it was quenched with methanol. The solvent was then removed under reduced pressure. Purification was performed via flash column chromatography (9:1 → 3:1 hexanes/EtOAc) to afford the N-trifluoroacetate-aurantioclavine 25 (1.03 g, 82% yield) as a yellow foam:Rf= 0.30 (3:1 hexanes/EtOAc). (Due to the distinct presence of rotameric isomers, the 1H NMR and 13C NMR contained extra peaks. See the attached spectrum, Supporting Information): 1H NMR (300 MHz, CDCl3) δ 8.50 (s, 2H), 7.31–7.24 (m, 2H), 7.16 (dd, J = 8.1, 7.2 Hz, 2H), 7.03–6.95 (m, 3H), 6.93–6.86 (m, 2H), 6.23 (d, J = 7.8 Hz, 1H), 5.46–5.31 (m, 2H), 4.39 (dt, J = 13.4, 3.5 Hz, 1H), 4.18–4.06 (m, 1H), 4.05–3.91 (m, 1H), 3.83 (td, J = 13.2, 2.9 Hz, 1H), 3.43 (dddd, J = 17.4, 13.1, 4.2, 1.7 Hz, 1H), 3.29–3.18 (m, 2H), 3.09 (dt, J = 16.5, 2.8 Hz, 1H), 1.91 (d, J = 1.3 Hz, 3H), 1.86 (d, J = 1.4 Hz, 3H), 1.77 (t, J = 1.9 Hz, 6H); 13C NMR (75 MHz, CDCl3) δ 157.5, 157.0, 156.8, 156.3, 138.5, 137.4, 137.2, 137.2, 135.1, 134.4, 124.3, 123.9, 123.5, 123.3, 122.1, 121.9, 121.8, 119.0, 118.8, 118.5, 117.1, 115.2, 115.0, 113.1, 112.6, 110.3, 110.1, 60.7, 60.6, 58.6, 43.7, 43.7, 28.4, 26.2, 25.7, 25.0, 18.9, 18.2; IR (neat film NaCl) 3361, 2917, 1667, 1441, 1205 cm–1; HRMS (MM: ESI-APCI+) m/z calcd for C17H18N2OF3 [M + H]+ 323.1366, found 323.1365.
3-(2-Azidoethyl)-3-bromoindolin-2-one (26
A flame-dried flask (1000 mL) equipped with a Teflon stirbar was charged with 3-(2-azidoethyl)-1H-indole (SI-6)41 (5.03 g, 30.3 mmol, 1.00 equiv), to which were subsequently added THF (150 mL), t-BuOH (150 mL), and water (3.75 mL) followed by cooling to −40 °C. A 0 °C solution of NBS (8.03 g, 45.1 mmol, 1.50 equiv) in THF (450 mL) was then added via cannula over 30 min, and the resulting solution was allowed to warm to −10 °C over 2 h. Warming continued slowly over 30 min to 0 °C. After 20 min at 0 °C, the solvent was removed under reduced pressure. Purification was performed via flash column chromatography (9:1 → 1:2 pentanes:ether) to afford bromooxindole 26 (5.46 g, 64% yield) as a yellow solid. 3-(2-azidoethyl)indolin-2-one (SI-7) (1.50 g, 25% yield) was also isolated as a light yellow solid. Bromooxindole 26: Rf= 0.46 (2:1 hexanes/EtOAc); 1H NMR (300 MHz, CDCl3) δ 1H NMR (300 MHz, CDCl3) δ 8.12 (br, s, 1H), 7.39 (ddt, J = 7.5, 1.3, 0.6 Hz, 1H), 7.31 (td, J = 7.7, 1.3 Hz, 1H), 7.12 (td, J = 7.6, 1.0 Hz, 1H), 6.93 (d, J = 7.8 Hz, 1H), 3.36 (ddd, J = 12.6, 8.1, 5.2 Hz, 1H), 3.24 (dt, J = 12.6, 7.6 Hz, 1H), 2.77 (ddd, J = 14.2, 8.1, 7.4 Hz, 1H), 2.60 (ddd, J = 14.1, 7.8, 5.2 Hz, 1H); 13C NMR (75 MHz, CDCl3) δ 180.5, 141.7, 128.4, 128.1, 123.8, 122.3, 110.2, 48.0, 43.3, 29.5; IR (neat film NaCl) 3228, 2100, 1693, 1620, 1470 cm–1; HRMS (MM: ESI-APCI+) m/z calcd for C10H10N4OBr [M + H]+ 281.0033, found 281.0040. Oxindole SI-7: Rf= 0.37 (2:1 hexanes/EtOAc); 1H NMR (300 MHz, CDCl3) δ 8.21 (br, s, 1H), 7.23 (d, J = 6.0 Hz, 2H), 7.13 (t, J = 7.5 Hz, 1H), 6.95 (d, J = 8.0 Hz, 1H), 3.41–3.20 (m, 3H), 2.84–2.57 (m, 2H); 13C NMR (75 MHz, CDCl3) δ 176.6, 139.8, 130.6, 129.1, 124.6, 123.5, 111.4, 54.5, 47.6, 38.0; IR (neat film NaCl) 3252, 2102, 1732, 1619, 1471 cm–1; HRMS (MM: ESI-APCI+) m/z calcd for C10H11N4O[M + H]+ 203.0927, found 203.0933.
3-(2-Azidoethyl)-3-(1-(2-methylprop-1-en-1-yl)-2-(2,2,2-trifluoroacetyl)-2,3,4,6-tetrahydro-1H-azepino[5,4,3-cd]indol-5-yl)indolin-2-one (28)
A flame-dried vial (20 mL) equipped with a Teflon stirbar was charged with indole 25 (120 mg, 0.372 mmol, 1.00 equiv) and bromooxindole 26 (157 mg, 0.559 mmol, 1.50 equiv), which were subsequently dissolved in THF (4 mL). Cesium carbonate (243 mg, 0.746 mmol, 2.00 equiv) was then added. After addition, the reaction was stirred for 12 h, and then water was added. The resulting solution was extracted with EtOAc (3 × 3 mL). The combined organic layers were dried over MgSO4 and concentrated in vacuo. The residue was purified by flash column chromatography (9:1 → 2:1 hexanes/EtOAc) to afford adduct 28 (134 mg, 69% yield) as a yellow foam: Rf= 0.34 (2:1 hexanes/EtOAc × 2 elutions). (Due to the distinct presence of rotameric isomers, the 1H NMR and 13C NMR contained extra peaks. See the attached spectrum, Supporting Information): 1H NMR (300 MHz, CDCl3) δ 8.35 (s, 1H), 8.26 (s, 2H), 7.41–6.95 (m, 12H), 6.91–6.86 (m, 1H), 6.81–6.74 (m, 1H), 6.09 (d, J = 7.9 Hz, 1H), 5.37 (d, J = 7.9 Hz, 1H), 5.33–5.27 (m, 1H), 4.17 (d, J = 13.8 Hz, 1H), 3.95 (d, J = 15.4 Hz, 1H), 3.81 (t, J = 13.8 Hz, 1H), 3.63 (t, J = 12.2 Hz, 1H), 3.33–3.05 (m, 5H), 3.05–2.85 (m, 3H), 2.65–2.51 (m, 2H), 1.83 (d, J = 1.4 Hz, 2H), 1.79 (d, J = 1.4 Hz, 2H), 1.73–1.67 (m, 4H), 1.63 (s, 2H); 13C NMR (75 MHz, CDCl3) δ 178.8, 178.7, 157.1, 156.7, 156.5, 156.0, 141.4, 141.3, 138.4, 137.0, 136.0, 135.6, 135.1, 134.4, 130.1, 129.9, 129.8, 129.5, 129.5, 125.5, 125.0, 124.9, 124.8, 124.4, 123.7, 123.3, 122.4, 122.2, 119.5, 119.0, 118.7, 118.1, 114.9, 111.8, 111.5, 111.2, 111.2, 110.1, 109.8, 60.5, 58.3, 52.7, 52.6, 47.3, 47.3, 43.5, 43.4, 35.0, 34.8, 28.1, 26.3, 25.8, 24.5, 19.0, 18.3; IR (neat film NaCl) 3335, 2102, 1713, 1674 cm–1; HRMS (MM: ESI-APCI+) m/z calcd for C27H26N6O2F3 [M + H]+ 523.2064, found 523.2058.
4-(3-(2-Azidoethyl)-3-(4a-chloro-1-(2-methylprop-1-en-1-yl)-2-(2,2,2-trifluoroacetyl)-2,3,4,4a-tetrahydro-1H-azepino[5,4,3-cd]indol-5-yl)-2-oxoindolin-1-yl)-3-nitrobenzenesulfonic Acid (29)
To a solution of adduct 28 (183 mg, 0.350 mmol, 1.00 equiv) in THF (4 mL) was added sodium hydride (60% dispersion in mineral oil, 42 mg, 1.05 mmol, 3.00 equiv) at 0 °C. The reaction mixture was stirred for 5 min, and o-nitrobenzylsulfonyl chloride (116 mg, 0.523 mmol, 1.50 equiv) was added at 0 °C. The reaction was stirred for 10 min, and then a satdrated solution of ammonium chloride was added. The resulting solution was extracted with EtOAc (3 × 3 mL). The combined organic layers were dried over MgSO4 and concentrated in vacuo. The residue was purified by flash column chromatography (15:1 → 2:1 hexanes/EtOAc) to afford alkyl chloride 29 (142 mg, 73% yield) as a white crystalline solid: Rf= 0.26 (2:1 hexanes/EtOAc). (Due to the distinct presence of rotameric isomers, the 1H NMR and 13C NMR contained extra peaks. See the attached spectrum, Supporting Information): 1H NMR (300 MHz, CDCl3) δ 8.64–8.59 (m, 2H), 7.95–7.84 (m, 8H), 7.55–7.47 (m, 6H), 7.37–7.26 (m, 5H), 7.15 (d, J = 7.0 Hz, 2H), 6.95 (d, J = 8.0 Hz, 1H), 6.31 (d, J = 7.5 Hz, 2H), 5.76–5.72 (m, 2H), 5.61 (d, J = 7.5 Hz, 1H), 4.20–4.08 (m, 2H), 3.94–3.67 (m, 3H), 3.31–3.19 (m, 2H), 2.99–2.74 (m, 6H), 2.29 (dd, J = 3.0, 10.5 Hz, 1H), 1.75 (s, 6H), 1.65 (s, 6H); 13C NMR (75 MHz, CDCl3) δ 175.9, 175.8, 173.8, 173.6, 156.3, 155.9, 151.7, 151.6, 148.2, 147.9, 141.6, 140.7, 139.9, 139.7, 139.2, 138.2, 137.4, 136.9, 136.4, 136.0, 135.3, 135.2, 132.6, 130.9, 130.8, 130.4, 130.4, 128.3, 126.8, 126.6, 126.4, 126.3, 125.8, 125.4, 125.3, 125.2, 124.9, 121.7, 121.6, 120.6, 119.8, 118.6, 115.4, 115.3, 114.8, 77.7, 77.4, 77.2, 76.8, 75.7, 75.5, 60.1, 57.6, 55.7, 55.6, 46.5, 46.4, 40.0, 39.1, 37.8, 37.6, 36.1, 32.6, 26.5, 26.0, 18.6, 18.1; IR (neat film NaCl) 2102, 1755, 1686, 1544, 1146 cm–1; HRMS (MM: ESI-APCI+) m/z calcd for C33H28O7N7F3SCl [M + H]+ 758.1406, found 758.1442.
4-((2-(3-(1-(2-Methylprop-1-en-1-yl)-6-(2-nitro-4-sulfophenyl)-2-(2,2,2-trifluoroacetyl)-2,3,4,6-tetrahydro-1H-azepino[5,4,3-cd]indol-8-yl)-2-oxopyrrolidin-3-yl)phenyl)amino)-3-nitrobenzenesulfonic Acid (32)
A flame-dried flask (100 mL) equipped with a Teflon stirbar was charged with indole 25 (1.03 g, 3.21 mmol, 1.00 equiv) and bromooxindole 26 (2.25 mg, 8.02 mmol, 2.50 equiv), which were subsequently dissolved in dichloromethane (32 mL). Cesium carbonate (3.14 g, 9.62 mmol, 3.00 equiv) was then added. After addition, the reaction was stirred for 3 h, and then water was added. The resulting solution was extracted with EtOAc (3 × 20 mL). The combined organic layers were dried over MgSO4 and concentrated in vacuo. The residue was purified by flash column chromatography (18:1 → 1:2 hexanes/EtOAc) to afford adduct 28 (404 mg, 24% yield), adduct 30 (538 mg, 32% yield), and adduct 31 (548 mg, 24% yield). Adduct 30: Rf= 0.18 (2:1 hexanes/EtOAc × 2 elutions). (Due to the distinct presence of rotameric isomers, the 1H NMR and 13C NMR contained extra peaks. See the attached spectrum, Supporting Information): 1H NMR (300 MHz, CDCl3) δ 8.91 (d, J = 13.0 Hz, 2H), 8.33 (d, J = 6.0 Hz, 2H), 7.28–7.03 (m, 8H), 6.95–6.86 (m, 6H), 6.75 (d, J = 7.5 Hz, 1H), 6.11 (d, J = 7.5 Hz, 1H), 5.31 (dd, J = 7.5, 29.5 Hz, 2H), 4.30 (d, J = 13.5 Hz, 1H), 4.06–4.01 (m, 1H), 3.94–3.84 (m, 1H), 3.71 (t, J = 13.0 Hz, 1H), 3.29 (t, J = 13.0 Hz, 1H), 3.17–3.13 (m, 6H), 2.98 (d, J = 16.5 Hz, 1H), 2.89–2.75 (m, 2H), 2.53–2.44 (m, 2H), 1.75–1.69 (m, 10H); 13C NMR (75 MHz, CDCl3) δ 181.1, 180.9, 157.4, 157.0, 156.7, 156.2, 141.1, 138.5, 137.5, 137.3, 137.1, 135.5, 134.8, 133.4, 133.2, 132.2, 132.1, 128.8, 128.7, 125.1, 124.0, 123.6, 123.2, 123.1, 122.8, 122.5, 119.0, 118.7, 117.0, 115.7, 115.1, 114.9, 113.3, 112.6, 110.7, 108.6, 108.5, 60.6, 58.6, 55.5, 55.4, 47.9, 43.8, 43.7, 36.6, 36.3, 28.4, 26.3, 25.8, 25.0, 18.9, 18.3; IR (neat film NaCl) 3328, 2100, 1712, 1682 cm–1. Adduct 31: Rf= 0.09 (2:1 hexanes/EtOAc tmex 2 elutions); (Due to the distinct presence of rotameric isomers, the 1H NMR and 13C NMR contained extra peaks. See the attached spectrum, Supporting Information): 1H NMR (300 MHz, acetone-d6) δ 10.43 (d, J = 11.7 Hz, 1H), 9.80 (d, J = 7.9 Hz, 1H), 9.59 (d, J = 4.9 Hz, 1H), 7.43–7.17 (m, 7H), 7.14–6.92 (m, 6H), 6.67 (dd, J = 18.0, 8.0 Hz, 1H), 6.11 (d, J = 8.1 Hz, 1H), 5.50–5.25 (m, 2H), 4.07–3.48 (m, 4H), 3.34–3.12 (m, 5H), 3.04 (s, 3H), 2.96–2.38 (m, 8H), 1.84–1.58 (m, 7H); 13C NMR (75 MHz, acetone-d6) δ 180.2, 180.1, 177.9, 177.8, 143.1, 143.0, 142.9, 138.3, 137.4, 137.2, 136.8, 135.3, 134.8, 134.6, 134.4, 133.6, 133.3, 133.2, 133.0, 131.3, 129.9, 129.3, 129.2, 126.1, 125.3, 125.2, 125.0, 124.8, 123.9, 123.6, 122.9, 118.0, 147.1, 111.0, 110.9, 110.3, 109.9, 109.4, 109.2, 61.2, 59.2, 55.8, 55.7, 52.7, 52.7, 48.6, 47.6, 44.0, 37.0, 36.9, 35.4, 28.2, 26.1, 25.7, 24.8, 18.8, 18.3; IR (neat film NaCl) 3305, 2101, 1713, 1472 cm–1; HRMS (MM: ESI-APCI+) m/z calcd for C37H34N10F3O3 [M + H]+ 723.2762, found 723.2761.
A flame-dried vial (4 mL) equipped with a Teflon stirbar was charged with adduct 30 (43.7 mg, 0.0836 mmol, 1.00 equiv), which was subsequently dissolved in THF (800 μL). The solution was cooled to 0 °C, and then sodium hydride (60% dispersion in mineral oil, 17 mg, 0.425 mmol, 5.00 equiv) was added. Five minutes after the sodium hydride addition, o-nitrobenzylsulfonyl chloride (93 mg, 0.420 mmol, 5.02 equiv) was added. The reaction was stirred for 10 min, and then a satdrated solution of ammonium chloride was added. The resulting solution was extracted 3 times with ether, the organic layers were combined and dried over magnesium sulfate, and the solvent was removed under reduced pressure. Purification was performed via flash column chromatography (9:1 →1:1 hexanes/EtOAc) to afford the 4-((R)-8-((R)-3-(2-azidoethyl)-1-(2-nitro-4-sulfophenyl)-2-oxoindolin-3-yl)-1-(2-methylprop-1-en-1-yl)-2-(2,2,2-trifluoroacetyl)-1,2,3,4-tetrahydro-6H-azepino[5,4,3-cd]indol-6-yl)-3-nitrobenzenesulfonic acid (SI-8) (49.0 mg, 66% yield) as a yellow solid. Rf= 0.42 (1:1 hexanes/EtOAc). (Due to the distinct presence of rotameric isomers, the 1H NMR and 13C NMR contained extra peaks. See the attached spectrum, Supporting Information): 1H NMR (300 MHz, CDCl3) δ 8.54–8.51 (m, 2H), 8.03–8.00 (m, 2H), 7.88–7.56 (m, 16H), 7.50 (s, 1H), 7.42–7.35 (m, 5H), 7.18–7.12 (m, 3H), 6.95 (s, 1H), 6.61 (d, J = 7.5 Hz, 1H), 5.97 (d, J = 7.5 Hz, 1H), 5.23–5.16 (m, 2H), 4.27 (d, J = 13.5 Hz, 1H), 4.02 (d, J = 15.0 Hz, 1H), 3.89–3.79 (m, 1H), 3.64 (t, J = 11.0 Hz, 1H), 3.32–2.80 (m, 11H), 2.48–2.33 (m, 2H), 1.73 (s, 12H); 13C NMR (75 MHz, CDCl3) δ 176.2, 176.0, 157.3, 156.9, 156.5, 148.0, 148.0, 140.2, 140.1, 140.0, 138.6, 137.2, 136.6, 136.2, 136.0, 135.7, 135.5, 134.8, 134.7, 132.8, 132.8, 132.4, 131.2, 131.2, 131.1, 130.9, 130.6, 129.9, 129.8, 129.5, 129.2, 127.4, 127.0, 125.7, 125.7, 125.4, 125.4, 125.2, 125.1, 124.9, 122.8, 121.9, 121.1, 120.0, 118.8, 118.5, 117.9, 115.5, 114.9, 110.6, 60.2, 58.1, 55.0, 55.0, 47.5, 42.8, 42.7, 36.6, 36.6, 39.9, 28.0, 26.3, 25.8, 25.0, 18.9, 18.3; IR (neat film NaCl) 2930, 2101, 1754, 1684, 1544 cm–1.
To a solution of nosylate SI-8 (43.1 mg, 0.0483 mmol, 1.00 equiv) in THF (1 mL) and water (250 μL) was added triphenylphosphine (25 mg, 0.0953 mmol, 2.00 equiv). The reaction was stirred for 3 h at 50 °C, and then the solvent was removed under reduced pressure. The residue was purified by flash column chromatography (3:1 → 1:1 hexanes/EtOAc) to afford the lactam 32 (49.0 mg, 66% yield) as a yellow crystalline solid: Rf= 0.14 (1:1 hexanes/EtOAc). (Due to the distinct presence of rotameric isomers, the 1H NMR and 13C NMR contained extra peaks. See the attached spectrum, Supporting Information): 1H NMR (300 MHz, CDCl3) δ 10.82 (s, 2H), 7.75–7.50 (m, 17H), 7.41 (td, J = 7.8, 1.4 Hz, 2H), 7.36–7.23 (m, 9H), 7.13 (dd, J = 7.5, 1.9 Hz, 1H), 6.80 (dd, J = 6.8, 1.6 Hz, 2H), 6.54 (d, J = 7.6 Hz, 1H), 5.99 (d, J = 7.7 Hz, 1H), 5.34–5.16 (m, 2H), 4.26 (d, J = 13.4 Hz, 1H), 4.00 (d, J = 15.6 Hz, 1H), 3.80 (dd, J = 14.3, 10.2 Hz, 1H), 3.68–3.48 (m, 3H), 3.32–2.97 (m, 9H), 2.29 (q, J = 7.8, 6.7 Hz, 2H), 1.66 (d, J = 1.3 Hz, 6H), 1.60 (s, 6H); 13C NMR (75 MHz, CDCl3) δ 180.0, 157.3, 156.8, 156.7, 156.2, 147.9, 147.8, 147.7, 139.5, 139.4, 137.8, 137.5, 137.4, 136.9, 136.5, 136.4, 136.2, 136.1, 135.6, 135.3, 135.1, 133.6, 132.9, 132.5, 132.2, 132.0, 131.0, 130.9, 130.5, 129.0, 127.9, 127.8, 126.3, 126.2, 125.2, 125.1, 125.0, 124.9, 124.8, 124.5, 124.4, 124.3, 123.3, 122.4, 121.4, 120.5, 119.2, 118.9, 118.5, 118.4, 115.1, 110.2, 109.7, 77.7, 77.4, 77.2, 76.8, 60.1, 58.2, 57.7, 43.0, 42.8, 39.8, 38.8, 28.5, 26.1, 25.7, 25.3, 18.9, 18.3; IR (neat film NaCl) 3098, 2916, 1682, 1545, 1368, 1170, 732 cm–1; HRMS (MM: ESI-APCI+) m/z calcd for C39H34N6O10F3S2 [M + H]+ 867.1729, found 867.1735.
4a-Bromo-2-tosyl-1,2,3,4,4a,6-hexahydro-5H-azepino[5,4,3-cd]indol-5-one (35)
To a solution of 2,3,4,6-tetrahydro-1H-azepino[5,4,3-cd]indole (SI-9) (106 mg, 0.614 mmol, 1.00 equiv) and Et3N (0.17 mL, 1.23 mmol, 2.00 equiv) in CH2Cl2 (4 mL) cooled to 0 °C was added a solution of TsCl (117 mg, 0.614 mmol, 1.00 equiv) in CH2Cl2 (3 mL) dropwise. The ice bath was removed, and the reaction mixture was stirred for 5 h, diluted with EtOAc (160 mL), and washed with 0.5 N HCl (2 × 30 mL) and brine. The organic layers were combined, dried over MgSO4, and concentrated in vacuo. The residue was purified by column chromatography (2:1 hexanes/EtOAc) to afford 2-tosyl-2,3,4,6-tetrahydro-1H-azepino[5,4,3-cd]indole (SI-10) (164 mg, 82% yield).
Indole SI-10 was dissolved in THF (10 mL), t-BuOH (10 mL), and water (1 mL). The solution was cooled to 0 °C, and pyridinium tribromide (504 mg, 1.54 mmol, 1.02 equiv) was added. The reaction mixture was stirred at 0 °C for 45 min and then allowed to warm to ambient temperature. The reaction was quenched by addition of 10 mL of 1:1 v/v 1 M Na2S2O3/satd NaHCO3. The reaction mixture was diluted with brine (50 mL) and extracted with EtOAc (3 × 50 mL). The combined organic extracts were dried with MgSO4 and concentrated in vacuo. The residue was purified by silica gel chromatography (2:1 hexanes/acetone) to afford 2-tosyl-1,2,3,4,4a,6-hexahydro-5H-azepino[5,4,3-cd]indol-5-one (SI-11) (397 mg, 75% yield) as a white solid: Rf= 0.15 (1:1 hexanes/EtOAc); 1H NMR (300 MHz, CDCl3) δ 8.77 (br, s, 1H), 7.56 (d, J = 8.2 Hz, 2H), 7.21–7.14 (m, 3H), 6.94 (d, J = 7.7 Hz, 1H), 6.80 (d, J = 7.7 Hz, 1H), 4.80 (d, J = 15.5 Hz, 1H), 4.30 (d, J = 13.6 Hz, 1H), 4.15 (d, J = 15.5 Hz, 1H), 3.52 (dd, J = 12.5, 3.5 Hz, 1H), 3.24 (t, J = 12.6 Hz, 1H), 2.38 (s, 3H), 2.30 (m, 1H), 1.53 (m, 1H); 13C (75 MHz, CDCl3) δ 178.4, 143.3, 140.5, 137.1, 135.9, 129.6, 128.4, 128.3, 127.0, 121.5, 109.1, 53.3, 51.6, 46.2, 28.6, 21.5; IR (neat film NaCl) 3276, 2925, 2853, 1698, 1618, 1463, 1326, 1153 cm–1; HRMS (MM: ESI-APCI+) m/z calcd for C18H19N2O3S [M + H]+ 343.1111, found 343.1104.
LiHMDS (429 mg, 2.57 mmol, 2.50 equiv) was dissolved in THF (5 mL). The solution was cooled to −78 °C, and a solution of oxindole SI-11 (352 mg, 1.03 mmol, 1.00 equiv) in THF (20 mL) was added dropwise over 20 min. The reaction mixture was stirred at −78 °C for 20 min and transferred to a precooled solution of N-bromosuccinimide (457 mg, 2.57 mmol, 2.50 equiv) in THF (10 mL) that was protected from light. The resulting reaction mixture was placed in a −40 °C bath for 1 h, while being protected from light, and then quenched with satd NH4Cl. The reaction mixture was allowed to warm to ambient temperature, diluted with brine (100 mL), and extracted with EtOAc (3 × 70 mL). The combined organic extracts were dried over MgSO4 and concentrated to afford a yellow oil, which was purified by silica gel chromatography (2:1 hexanes/EtOAc) to afford bromooxindole 35 (334 mg, 76% yield) as a yellow solid: Rf = 0.40 (1:1 hexane/EtOAc); 1H NMR (300 MHz, CDCl3) δ 8.49 (br, s, 1H), 7.58 (d, J = 8.2 Hz, 2H), 7.24–7.19 (m, 3H), 6.96 (d, J = 7.7 Hz, 1H), 6.83 (d, J = 7.8 Hz, 1H), 4.76 (d, J = 15.4 Hz, 1H), 4.32 (m, 2H), 3.80 (t, J = 13.2 Hz, 1H), 2.39 (s, 3H), 2.33 (m, 1H), 1.90 (m, 1H); 13C NMR (75 MHz, CDCl3) δ 175.1, 143.5, 139.0, 137.7, 136.9, 130.7, 129.7, 128.7, 126.9, 122.6, 110.1, 59.2, 51.6, 48.3, 35.2, 21.5; IR (neat film NaCl) 3313, 2930, 1734, 1615, 1460, 1334, 1155, 1096, 727 cm–1; HRMS (MM: ESI-APCI+) m/z calcd for C18H18BrN2O3S [M + H]+ 421.0216, found 421.0213.
2-(2-Nitrophenyl)-2-(5-oxo-2-tosyl-1,2,3,4,5,6-hexahydro-4aH-azepino[5,4,3-cd]indol-4a-yl)acetonitrile (37)
A solution of bromooxindole 35 (44.9 mg, 0.107 mmol, 1.00 equiv) and 2-nitrophenylacetonitrile 36 (34.6 mg, 0.213 mmol, 2.00 equiv) in THF (3 mL) was cooled to −78 °C. DBU (64 mL, 0.426 mmol, 4.00 equiv) was added dropwise. The reaction mixture was then allowed to gradually warm to ambient temperature. After 8 h, the reaction mixture was quenched with satd NH4Cl (10 mL) and the mixture was extracted with EtOAc (4 × 5 mL). The combined organic extracts were dried over MgSO4 and concentrated under reduced pressure to afford a brown oil, which was purified by preparatory thin-layer chromatography on silica gel (1:1 hexane/EtOAc x2 elutions) to afford nitrile 37 (34.8 mg, 64% yield) as an orange-yellow solid: Rf = 0.28 (1:1 hexane/EtOAc); 1H NMR (300 MHz, CDCl3) δ7.92 (m, 1H), 7.63 (m, 2H), 7.55 (m, 2H), 7.43 (m, 1H), 7.29 (d, J = 8.0 H, 2H), 7.18 (t, J = 7.8 Hz, 1H), 6.97 (d, J = 7.7 Hz, 1H), 6.48 (d, J = 7.4 Hz, 1H), 6.07 (s, 1H), 4.80 (d, J = 16.2 Hz, 1H), 4.29 (m, 2H), 3.45 (app. t, J = 13.2 Hz, 1H), 2.74 (app. dd, J = 15.0, 2.6 Hz, 1H), 2.43 (s, 3H), 1.94 (m, 1H); 13C NMR (75 MHz, CDCl3) δ 176.2, 147.9, 143.8, 140.3, 137.9, 136.5, 133.5, 132.5, 131.0, 130.3, 130.0, 127.0, 124.8, 124.5, 124.5, 123.7, 116.2, 109.4, 54.4, 52.6, 46.8, 34.2, 32.1, 21.6; IR (neat film NaCl) 3302, 2920, 1724, 1527, 1155, 724 cm–1; HRMS (MM: ESI-APCI+) m/z calcd for C26H23N4O5S [M + H]+ 503.1384, found 503.1411.
Dimethyl 2-(2-Nitrophenyl)-2-(5-oxo-2-tosyl-1,2,3,4,5,6-hexahydro-4aH-azepino[5,4,3-cd]indol-4a-yl)malonate (39)
To a solution of bromooxindole 35 (50.0 mg, 0.119 mmol, 1.00 equiv) and malonate 38 (90.1 mg, 0.356 mmol, 3.00 equiv) in THF (0.6 mL) was added DBU (54.2 mg, 0.356 mmol, 3.00 equiv) at −78 °C. The reaction solution was slowly warmed to 23 °C. The reaction solution was stirred for 12 h and quenched with satd NH4Cl. The mixture was extracted with EtOAc (3 × 1 mL) and brine. The combined organic extracts were dried over MgSO4 and concentrated under reduced pressure, and then the residue was purified by preparatory thin-layer chromatography on silica gel (1:1 hexane/EtOAc) to afford nitrile 39 (23 mg, 32% yield): Rf = 0.15 (1:1 hexanes/EtOAc); 1H NMR (300 MHz, CDCl3) δ 8.85 (br, s, 1H), 8.23 (dd, J = 8.0, 1.5 Hz, 1H), 7.58–7.51 (m, 5H), 7.24 (d, J = 8.0 Hz, 2H), 6.96 (dd, J = 7.5, 1.5 Hz, 1H), 6.76 (d, J = 1.5 Hz, 1H), 6.66 (d, J = 1.0 Hz, 1H), 4.44 (d, J = 15.4 Hz, 1H), 4.15 (d, J = 15.4 Hz, 1H), 3.95 (d, J = 14.6 Hz, 1H), 3.82 (s, 3H), 3.75 (s, 3H), 3.69 (m, 1H), 2.40 (s, 3H), 1.95 (d, J = 14.6 Hz, 1H), 1.66 (m, 1H); 13C NMR (75 MHz, CDCl3) δ 179.1, 168.8, 148.1, 143.4, 141.2, 138.7, 137.0, 134.7, 133.9, 132.0, 129.7, 129.1, 128.3, 126.8, 125.7, 122.8, 110.7, 74.8, 68.7, 53.7, 53.5, 51.3, 46.0, 33.4, 21.5; IR (neat film NaCl) 3313, 1737, 1623, 1530, 1450, 1347, 1241, 1153, 1091, 895, 729 cm–1; HRMS (FAB) m/z calcd for C29H26N3O9S [M – H]− 592.1395, found 592.1382.
4a-Bromo-1-(2-methylprop-1-en-1-yl)-2-tosyl-1,2,3,4,4a,6-hexahydro-5H-azepino[5,4,3-cd]indol-5-one (40)
To a solution of aurantioclavine 4 (300 mg, 0.00133 mol, 1.00 equiv) and Et3N (0.37 mL, 0.00265 mol, 2.00 equiv) in CH2Cl2 (4 mL) cooled to 0 °C was added a solution of TsCl (254 mg, 0.00133 mmol, 1.00 equiv) in CH2Cl2 (3 mL) dropwise. The ice bath was removed, and the reaction mixture was stirred for 5 h, then diluted with EtOAc (200 mL) and washed with 0.5 N HCl (2 × 35 mL) and brine. The organic layers were combined, dried over MgSO4 and concentrated in vacuo. The residue was purified by column chromatography (2:1 hexanes/EtOAc) to afford 1-(2-methylprop-1-en-1-yl)-2-tosyl-2,3,4,6-tetrahydro-1H-azepino[5,4,3-cd]indole (SI-12) (405 mg, 80% yield).
A solution of indole SI-12 (902.2 mg, 2.371 mmol, 1.00 equiv) in THF/t-BuOH/H2O (10:10:1 v/v/v, 52.5 mL) was cooled to 0 °C, and pyridinium tribromide (834.2 mg, 2.608 mmol, 1.10 equiv) was added in small portions over 5 min. The reaction mixture was stirred at 0 °C for 15 min, and then allowed to warm to ambient temperature. After 5 min at ambient temperature, the reaction mixture was quenched by addition of 1:1 v/v satd NaHCO3/1 M aq Na2S2O3 (15 mL), poured into brine (150 mL), and extracted with EtOAc (3 × 100 mL). The combined extracts were dried over Na2SO4 and concentrated under reduced pressure to afford a brown solid, which was purified by silica gel chromatography (2:1 → 1:1 hexanes/EtOAc) to afford 1-(2-methylprop-1-en-1-yl)-2-tosyl-1,2,3,4,4a,6-hexahydro-5H-azepino[5,4,3-cd]indol-5-one (SI-13) (747.5 mg, 80% yield): Rf = 0.22 (5:1 benzene/MeCN); 1H NMR (300 MHz, CDCl3) δ 8.12 (br, s, 1H), 7.33 (dd, J = 8.5, 2.1 Hz, 2H), 7.03 (m, 1H), 6.96 (d, J = 7.2 Hz, 2H), 6.82 (d, J = 7.7 Hz, 1H), 6.64 (d, J = 8.0 Hz, 1H), 5.76 (d, J = 8.0 Hz, 1H), 5.31 (m, 1H), 4.00 (dt, J = 15.7, 2.8 Hz, 1H), 3.64–3.50 (m, 2H), 2.22 (s, 3H), 2.00 (m, 1H), 1.67 (s, 3H), 1.65 (s, 3H), 1.25 (m, 1H); 13C NMR (75 MHz, CDCl3) δ 178.4, 142.9, 140.9, 140.5, 138.2, 129.2, 128.3, 128.3, 127.0, 126.7, 121.3, 119.0, 108.7, 59.0, 46.0, 43.8, 27.8, 26.0, 21.4, 18.5; IR (neat film NaCl) 3246, 2925, 1713, 1615, 1460, 1326, 1155, 732 cm–1; HRMS (MM: ESI-APCI+) m/z calcd for C22H25N2O3S [M + H]+ 397.1580, found 397.1586.
A solution of oxindole SI-13 (172.0 mg, 0.434 mmol, 1.00 equiv) in THF (5 mL) was added dropwise to a freshly prepared solution of LiHMDS (217.8 mg, 1.301 mmol, 3.00 equiv) in THF (5 mL) that had been precooled to −78 °C. After 20 min at −78 °C, the resulting solution was transferred via cannula to a solution of N-bromosuccinimide (231.6 mg, 1.301 mmol, 3.00 equiv) in THF (5 mL) that had been precooled to −78 °C. The resulting yellow reaction mixture was allowed to warm to −15 °C (the reaction flask was transferred to a bath composed of ethylene glycol and dry ice) and maintained at this temperature for 2 h. The reaction mixture was then cooled to −78 °C and quenched by addition of satd NH4Cl (5 mL). The yellow reaction mixture was allowed to warm to ambient temperature and diluted with H2O (80 mL), then extracted with EtOAc (3 × 70 mL). The combined organic extracts were washed with brine (100 mL), dried over MgSO4, and concentrated under reduced pressure to afford a yellow oil, which was purified immediately by silica gel chromatography (2:1 hexanes/EtOAc) to afford a 3:1 mixture of bromooxindole 40 (>20:1 dr) and dehydrobromination product, 1-(2-methylprop-1-en-1-yl)-2-tosyl-1,2,3,6-tetrahydro-5H-azepino[5,4,3-cd]indol-5-one (SI-14) (131.1 mg, 64% combined yield, 50% yield of bromooxindole 40). Bromooxindole 40 was stored frozen in benzene and used without further purification. The relative configuration of this bromooxindole was assigned based on the stereochemistry of the malonate adduct obtained (see below). Quenching the reaction prior to completion afforded the bromooxindole 40 as a single diastereomer, which showed greater stability, and could be fully characterized: Rf = 0.50 (1:1 hexane/EtOAc); 1H NMR (300 MHz, CDCl3) δ 8.62 (br, s, 1H), 7.49 (d, J = 8.0 Hz, 2H), 7.21 (t, J = 8.0, 1H), 7.13 (d, J = 8.0 Hz, 2H), 6.90 (d, J = 7.5 Hz, 1H), 6.79 (dd, J = 8.0, 1.0 Hz, 1H), 5.96 (td, J = 8.5, 1.5 Hz, 1H), 5.88 (d, J = 8.5 Hz, 1H), 4.18–4.02 (m, 2H), 2.35 (s, 3H), 2.27 (m, 1H), 1.97 (m, 1H), 1.76 (d, J = 1.0 Hz, 3H), 1.75 (d, J = 1.5 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 174.8, 143.2, 142.4, 140.2, 139.2, 137.8, 130.8, 129.4, 126.9, 126.1, 122.7, 120.1, 109.7, 66.8, 59.5, 39.8, 34.4, 26.1, 21.4, 18.4; IR (neat film NaCl) 3291, 1734, 1617, 1602, 1457, 1326, 1156, 1092, 738 cm–1; HRMS (MM: ESI-APCI+) m/z calcd for C22H24BrN2O3S [M + H]+ 475.0686, found 475.0668.
Dimethyl 2-(1-(2-Methylprop-1-en-1-yl)-5-oxo-2-tosyl-1,2,3,4,5,6-hexahydro-4aH-azepino[5,4,3-cd]indol-4a-yl)malonate (42)
Bromooxindole 40 (>20:1 dr, 51.3 mg, 0.108 mmol, 1.00 equiv) was dissolved in THF (2 mL). Dimethyl malonate 41 (37 mL, 0.324 mmol, 3.00 equiv) was added, and the reaction mixture was cooled to −78 °C. DBU (48 mL, 0.324 mmol, 3.00 equiv) was added dropwise. The reaction mixture was stirred at −78 °C for 15 min and then warmed to 23 °C. After the reaction mixture was maintained at 23 °C for 6 h, satd NH4Cl (2 mL) was added and the mixture was warmed to ambient temperature. The reaction mixture was diluted with EtOAc (50 mL) and satd NH4Cl (50 mL). The phases were separated, and the aqueous phase was extracted with EtOAc (2 × 50 mL). The combined organic extracts were dried over Na2SO4 and concentrated to afford colorless oil. Analysis of the crude oil by 1H NMR indicated >20:1 dr of the malonate adduct 42. The residue was purified by silica gel chromatography (1:1 hexane/EtOAc) to afford malonate 42 (42 mg, 74% yield): Rf = 0.20 (1:1 hexane/EtOAc); 1H NMR (300 MHz, CDCl3) δ 7.88 (br, s, 1H), 7.26 (d, J = 8.0 Hz, 2H), 7.14 (t, J = 7.5 Hz, 1H), 7.00 (d, J = 8.0 Hz, 2H), 6.91, (d, J = 7.5 Hz, 1H), 6.72 (d, J = 8.0 Hz, 1H), 5.92 (d, J = 4.5 Hz, 1H), 5.38 (d, J = 4.5 Hz, 1H), 4.62 (s, 1H), 4.06 (m, 1H), 3.89 (m, 1H), 3.78 (s, 3H), 3.46 (s, 3H), 2.40 (m, 1H), 2.30 (s, 3H), 1.82 (s, 3H), 1.79 (s, 3H), 1.07 (m, 1H); 13C NMR (75 MHz, CDCl3) δ 177.8, 166.8, 166.8, 143.0, 141.9, 141.4, 141.0, 137.9, 129.2, 128.9, 127.6, 127.0, 123.0, 124.0, 109.0, 59.6, 53.9, 52.9, 52.3, 51.3, 39.8, 28.9, 26.4, 21.4, 18.6; IR (neat film NaCl) 3338, 2953, 1733, 1618, 1597, 1458, 1327, 1158 cm–1; HRMS (MM: ESI-APCI+) m/z calcd for C27H31N2O7S [M + H]+ 527.1846, found 527.1848.
Diallyl 2-(2-Nitrophenyl)-2-(2-oxo-3-(2-((triisopropylsilyl)oxy)ethyl)indolin-3-yl)malonate (46)
To a suspension of Cs2CO3 (5.37 g, 16.5 mmol, 3.00 equiv) and bromooxindole 43 (2.27 g, 5.50 mmol, 1.00 equiv) in THF (100 mL) was added malonate 44 (5.04 g, 16.5 mmol, 3.00 equiv) at 0 °C. The reaction mixture was then allowed to slowly warm to 23 °C and stirred for 16 h. Solids were removed via filtration through a Celite plug (rinsed with EtOAc), and the resulting purple solution was concentrated under reduced pressure. The residue was purified by column chromatography using a Teledyne Isco CombiFlash (SiO2, 120 g column, 100:0 → 3:1 hexanes/EtOAc) to provide alkylation product 46 (8.9 g, 85% yield) as a colorless oil: Rf = 0.18 (3:1 hexanes/EtOAc); 1H NMR (500 MHz, CDCl3) δ 7.97 (d, J = 8.0 Hz, 1H), 7.94 (s, 1H), 7.73 (dd, J = 7.9, 1.6 Hz, 1H), 7.41 (dt, J = 8.0, 1.6 Hz, 1H), 7.38–7.33 (m, 2H), 7.13 (dt, J = 7.7, 1.1 Hz, 1H), 6.89 (dt, J = 7.7, 1.0 Hz, 1H), 6.72 (d, J = 7.6 Hz, 1H), 5.88–5.73 (m, 2H), 5.25 (ddd, J = 17.2, 2.8, 1.4 Hz, 1H), 5.19–5.11 (m, 3H), 4.69 (tdd, J = 13.3, 5.7, 1.4 Hz, 1H), 4.64 (tdd, J = 13.3, 5.7, 1.3 Hz, 1H), 4.57 (tdd, J = 13.4, 5.9, 1.4 Hz, 1H), 4.48 (tdd, J = 13.1, 5.9, 1.1 Hz, 1H), 3.35 (ddd, J = 9.5, 8.5, 6.9 Hz, 1H), 3.07 (dt, J = 9.5, 4.5 Hz, 1H), 2.95 (ddd, J = 12.9, 8.7, 7.0 Hz, 1H), 2.63 (ddd, J = 12.9, 8.5, 4.5 Hz, 1H), 0.97–0.77 (m, 21H); 13C NMR (125 MHz, CDCl3) δ 177.8, 166.7, 166.3, 150.2, 140.7, 132.4, 131.3, 131.0, 130.8, 129.5, 129.2, 128.5, 126.9, 125.3, 122.4, 119.2, 118.4, 109.1, 66.8, 66.7, 59.5, 56.7, 38.3, 17.8, 11.8; IR (neat film NaCl) 3332, 2942, 1714, 1649, 1618, 1538, 1471, 1356, 1230, 1114, 995, 933, 885, 850, 752, 683 cm–1; HRMS (MM: ESI-APCI+) m/z calcd for C34H45N2O8Si [M + H]+ 637.2940, found 637.2945.
Diallyl 2-(1-Methyl-2-oxo-3-(2-((triisopropylsilyl)oxy)ethyl)indolin-3-yl)-2-(2-nitrophenyl)malonate (48)
To a suspension of oxindole 46 (0.50 g, 0.79 mmol, 1.00 equiv) and Cs2CO3 (0.77 g, 2.37 mmol, 3.00 equiv) in THF (4.0 mL) was added methyl iodide (0.3 mL, 4.7 mmol, 6.00 equiv) at 0 °C. The reaction mixture was stirred for 12 h at 23 °C. After the reaction was complete, satd NH4Cl was added. The aqueous phase was extracted with EtOAc (3 × 3 mL). The combined organic phases were washed with brine, dried over MgSO4, and concentrated in vacuo. The residue was purified by flash column chromatography on silica gel (3:1 hexanes/EtOAc) to give methylated oxindole 48 (0.51g, 99% yield): Rf = 0.33 (3:1 hexanes/EtOAc); 1H NMR (500 MHz, CDCl3) δ 7.82 (d, J = 8.0 Hz, 1H), 7.54 (dd, J = 7.9, 1.5 Hz, 1H), 7.42 (d, J = 7.6 Hz, 1H), 7.31 (dt, J = 7.7, 1.6 Hz, 1H), 7.25 (dt, J = 7.8, 1.2 Hz, 1H), 7.10 (dt, J = 7.7, 0.9 Hz, 1H), 6.85 (dt, J = 7.8, 0.8 Hz, 1H), 6.57 (d, J = 7.7 Hz, 1H), 5.80 (tdd, J = 16.3, 10.7, 5.8 Hz, 1H), 5.71 (tdd, J = 16.4, 10.5, 5.9 Hz, 1H), 5.22–5.02 (m, 4H), 4.69–4.56 (m, 2H), 4.52 (tdd, J = 13.1, 5.8, 1.3 Hz, 1H), 4.36 (tdd, J = 13.3, 5.9, 1.3 Hz, 1H), 3.15–3.02 (m, 4H), 2.96 (ddd, J = 9.7, 8.3, 4.7 Hz, 1H), 2.85 (td, J = 13.2, 7.4 Hz, 1H), 2.67 (ddd, J = 12.8, 8.0, 4.7 Hz, 1H), 0.86–0.71 (m, 21H); 13C NMR (125 MHz, CDCl3) δ 176.3, 166.5, 166.5, 150.3, 143.6, 132.7, 131.3, 130.9, 130.3, 128.8, 128.5, 128.5, 128.3, 126.9, 125.1, 122.3, 119.1, 118.5, 107.3, 66.7, 66.7, 59.6, 56.8, 37.9, 26.1, 17.8, 11.7; IR (neat film NaCl) 3421, 3054, 2944, 2866, 1723, 1613, 1539, 1473, 1356, 1253, 1180, 1104, 1068, 935, 862, 840, 752, 690 cm–1; HRMS (MM: ESI-APCI+) m/z calcd for C35H47N2O8Si [M + H]+ 651.3096, found 651.3092.
Allyl 1-Methyl-3′-(2-nitrophenyl)-2,2′-dioxo-2′,3′,5′,6′-tetrahydrospiro[indoline-3,4′-pyran]-3′-carboxylate (50)
Acetyl chloride (46.0 μL, 650 μmol, 10.0 equiv) was added to MeOH (1.0 mL) and cooled to 0 °C. The resulting mixture was stirred for 30 min, and then the solution of oxindole 48 (21.0 mg, 32.0 μmol, 1.00 equiv) in MeOH (2.0 mL) was added. The reaction was stirred for 2 h at 23 °C and then heated to 65 °C. The reaction mixture was stirred for 16 h at that temperature. The colorless solution was cooled to ambient temperature, concentrated under reduced pressure, and subjected to column chromatography (4:1 hexanes/EtOAc) to afford the desired lactone 50 (12 mg, 85% yield) as a colorless solid: Rf = 0.29 (50% EtOAc in hexanes); 1H NMR (400 MHz, CDCl3) δ 7.80 (d, J = 7.4 Hz, 1H), 7.39–7.31 (m, 3H), 7.23–7.15 (m, 2H), 6.82–6.75 (m, 2H), 5.92 (tdd, J = 17.1, 10.6, 5.8 Hz, 1H), 5.30 (ddd, J = 17.2, 2.7, 1.3 Hz, 1H), 5.21 (ddd, J = 10.4, 2.4, 1.2 Hz, 1H), 4.98–4.90 (m, 2H), 4.74 (tdd, J = 13.1, 5.8, 1.3 Hz, 1H), 4.71–4.64 (m, 1H), 3.32 (s, 3H), 2.84–2.70 (m, 1H), 2.03 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 175.8, 166.5, 165.7, 149.2, 142.2, 131.7, 131.4, 131.1, 128.8, 129.7, 129.3, 129.0, 128.9, 125.6, 124.8, 122.5, 118.9, 108.5, 67.5, 65.4, 53.7, 30.2, 26.7; IR (neat film NaCl) 2096, 1718, 1637, 1533, 1475, 1358, 1232, 1184, 760 cm–1; HRMS (MM: ESI-APCI+) m/z calcd for C23H21N2O7 [M + H]+ 437.1343, found 437.1298.
3′-Allyl-1-methyl-3′-(2-nitrophenyl)-5′,6′-dihydrospiro[indoline-3,4′-pyran]-2,2′(3′H)-dione (51)
An oven-dried flask was charged with ester 50 (1.3 g, 3.05 mmol, 1.00 equiv), sealed with a rubber stopper, and evacuated. The flask was brought in a glovebox, and Pd(PPh3)4 (84 mg, 75.0 μmol, 0.025 equiv) was added. The flask was brought out of the drybox and THF (60 mL) was added. The reaction mixture was stirred for 5 min and concentrated under reduced pressure. Column chromatography using a Teledyne Isco CombiFlash Rf (SiO2, 80 g column, 25 → 50% EtOAc in hexanes) afforded the desired protected alkylation product 51 (1.1 g, 90% yield) as a colorless solid: Rf = 0.40 (1:1 hexane/EtOAc); 1H NMR (400 MHz, CDCl3) δ 7.63 (dd, J = 7.9, 1.6 Hz, 1H), 7.18 (dt, J = 7.8, 1.4 Hz, 1H), 7.12 (dt, J = 7.5, 1.7 Hz, 1H), 7.07 (dt, J = 7.7, 1.1 Hz, 1H), 7.00 (d, J = 7.7 Hz, 1H), 6.85 (dd, J = 8.0, 1.3 Hz, 1H), 6.68 (d, J = 7.7 Hz, 1H), 6.64 (dt, J = 7.7, 1.0 Hz, 1H), 5.55 (tdd, J = 17.0, 10.2, 6.7 Hz, 1H), 5.33 (ddd, J = 11.6, 10.0, 3.4 Hz, 1H), 5.01 (ddd, J = 17.1, 3.0, 1.5 Hz, 1H), 4.93 (ddd, J = 10.3, 2.7, 1.2 Hz, 1H), 4.68 (td, J = 11.6, 4.7 Hz, 1H), 3.45 (tdd, J = 15.6, 6.6, 1.3 Hz, 1H), 3.26 (s, 3H), 2.99 (tdd, J = 9.0, 6.9, 1.3 Hz, 1H), 2.87 (ddd, J = 14.6, 10.0, 4.6 Hz, 1H), 2.21 (ddd, J = 14.7, 4.8, 3.5 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 177.3, 168.5, 149.8, 142.1, 133.9, 133.1, 131.1, 130.6, 130.4, 128.6, 127.8, 125.7, 124.8, 122.2, 119.0, 107.6, 64.8, 54.2, 54.0, 43.7, 30.7, 26.5; IR (neat film NaCl) 1701, 1614, 1531, 1473, 1356, 1300, 1259, 1202, 1105, 929, 739 cm–1; HRMS (MM: ESI-APCI+) m/z calcd for C22H21N2O5 [M + H]+ 393.1445, found 393.1458.
2-(4-Bromo-1-methyl-3′-(2-nitrophenyl)-2,2′-dioxo-2′,3′,5′,6′-tetrahydrospiro[indoline-3,4′-pyran]-3′-yl)acetaldehyde (63)
A solution of alkene 62 (47.1 mg, 100 μmol, 1.00 equiv) in a mixture of CH2Cl2 (2.5 mL) and MeOH (2.5 mL) in a Schlenk flask hooked up to an ozone generator was purged with oxygen gas at −78 °C (5 min, flow 0.25). Then the ozone generator was turned on (low–medium setting), and an ozone/oxygen gas mixture was bubbled through the reaction. The progress of the reaction was checked via TLC (9:1 hexanes/CH2Cl2) in short time intervals (1–2 min). Upon completion of the reaction, the mixture was purged with oxygen gas for 5 min, and DMS (36.0 μg, 500 μmol, 5.00 equiv) was added. The reaction mixture was slowly warmed to ambient temperature, and stirred for 16 h. The residue was purified by column chromatography (9:1 CH2Cl2:EtOAc) on silica gel to afford aldehyde 63 (44 mg, 94% yield): Rf = 0.28 (9:1 CH2Cl2/EtOAc); 1H NMR (500 MHz, DMSO-d6) δ 8.64 (d, J = 3.4 Hz, 1H), 7.83–7.79 (m, 1H), 7.65–7.59 (m, 1H), 7.43–7.39 (m, 1H), 7.31 (t, J = 8.0 Hz, 1H), 7.21 (d, J = 7.9 Hz, 1H), 7.05 (d, J = 8.1 Hz, 1H), 6.92 (d, J = 8.3 Hz, 1H), 5.15 (ddd, J = 12.5, 10.8, 4.7 Hz, 1H), 4.59 (dd, J = 11.1, 6.9 Hz, 1H), 3.94 (ddd, J = 14.5, 12.6, 7.1 Hz, 1H), 3.22 (s, 3H), 3.08 (d, J = 17.0 Hz, 1H), 2.67 (dd, J = 17.0, 3.5 Hz, 1H), 1.96 (dd, J = 14.7, 4.6 Hz, 1H); 13C NMR (125 MHz, DMSO) δ 198.0, 174.8, 171.0, 151.5, 146.3, 133.3, 131.9, 131.3, 129.9, 129.7, 127.6, 125.5, 124.6, 122.4, 109.3, 65.4, 55.6, 52.6, 49.8, 26.4, 22.3; IR (neat film NaCl) 1695, 1600, 1528, 1458, 1354, 1294, 1222, 1118, 850, 787 cm–1; HRMS (MM: ESI-APCI+) m/z calcd for C21H18BrN2O6 [M + H]+ 473.0343, found 473.0346.
10-Bromo-10b-(2-hydroxyethyl)-2-(4-methoxybenzyl)-6-methyl-4a-(2-nitrophenyl)-2,3,4,4a,6,10b-hexahydrobenzo[c][2,6]naphthyridine-1,5-dione (65)
To a suspension of aldehyde 63 (47.3 mg, 100 μmol, 1.00 equiv) and the acetic acid ammonium salt of p-methoxybenzylamine (59.2 mg, 300 μmol, 3.00 equiv) in MeOH (4 mL) was added NaBH3CN (2.60 mg, 300 μmol, 3.00 equiv) in THF (2 mL). The reaction mixture was stirred at ambient temperature for 16 h (conversion of the suspension to a clear, colorless solution usually indicated the completion of the reaction) and then concentrated under reduced pressure. Column chromatography using a Teledyne Isco CombiFlash Rf (SiO2, 12 g column, 1. 1:1 → 1:4 hexanes/EtOAc) yielded lactam 65 (39.7 mg, 67% yield) as a colorless solid: Rf = 0.12 (19:1 CH2Cl2/MeOH); 1H NMR (500 MHz, CDCl3) δ 9.10 (dd, J = 8.3, 1.4 Hz, 1H), 7.59–7.55 (m, 1H), 7.43 (ddd, J = 8.4, 7.3, 1.3 Hz, 1H), 7.36 (dd, J = 8.0, 1.6 Hz, 1H), 7.17 (t, J = 7.9 Hz, 1H), 7.10 (dd, J = 8.2, 1.0 Hz, 1H), 7.06–7.02 (m, 2H), 6.81–6.76 (m, 3H), 4.71 (d, J = 14.6 Hz, 1H), 3.98 (d, J = 14.6 Hz, 1H), 3.76 (s, 3H), 3.53–3.49 (m, 1H), 3.18 (s, 3H), 3.16–3.08 (m, 3H), 2.90 (ddd, J = 9.6, 8.5, 3.2 Hz, 1H), 2.78 (dt, J = 9.6, 7.3 Hz, 1H), 2.25 (ddd, J = 14.1, 7.1, 3.1 Hz, 1H), 2.14 (ddd, J = 14.0, 8.6, 7.6 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ 177.7, 171.7, 159.2, 152.6, 147.2, 134.4, 130.9, 130.4, 130.3, 129.9, 129.0, 128.0, 127.9, 126.3, 125.5, 122.4, 114.1, 107.3, 60.1, 58.5, 55.7, 55.4, 47.4, 44.0, 32.1, 27.5, 26.8; IR (neat film NaCl) 3459, 2931, 1682, 1601, 1574, 1530, 1457, 1360, 1249, 1176, 1037, 910, 849, 783, 731 cm–1; HRMS (MM: ESI-APCI+) m/z calcd for C29H29BrN3O6 [M + H]+ 594.1234, found 594.1230.
Methyl (2-(3-(4-Bromo-1-methyl-2-oxo-3-(2-((triisopropylsilyl)oxy)ethyl)indolin-3-yl)-1-(4-methoxybenzyl)-2-oxopyrrolidin-3-yl)phenyl)carbamate (54)
To a solution of aldehyde 69 (100 mg, 0.15 mmol, 1.00 equiv) and (p-methoxybenzyl)ammonium acetate (90 mg, 0.46 mmol, 3.00 equiv) in methanol (7.6 mL) was added NaBH3CN (21 mg, 0.30 mmol, 2.00 equiv) in THF (3.8 mL) at 0 °C. The reaction mixture was slowly warmed to 23 °C and stirred overnight. The reaction mixture was washed with EtOAc (3 × 10 mL), and brine. The combined organic phases were dried over MgSO4 and concentrated in vacuo. The residue was purified by column chromatography (4:1 hexanes/EtOAc) on silica gel to afford lactam 54 (112 mg, 95% yield): Rf = 0.20 (4:1 hexanes/EtOAc); 1H NMR (500 MHz, CDCl3) δ 12.14 (s, 1H), 8.00 (d, J = 8.3 Hz, 1H), 7.18 (d, J = 8.3 Hz, 2H), 7.15–7.10 (m, 2H), 7.05 (q, J = 8.5, 7.9 Hz, 2H), 6.88 (t, J = 7.7 Hz, 1H), 6.85–6.80 (m, 2H), 6.49 (d, J = 7.7 Hz, 1H), 4.80 (d, J = 14.4 Hz, 1H), 4.36 (d, J = 14.4 Hz, 1H), 3.78 (s, 3H), 3.70 (s, 1H), 3.63 (s, 3H), 3.52 (td, J = 6.9, 3.6 Hz, 1H), 3.43–3.36 (m, 1H), 3.27 (td, J = 9.3, 5.7 Hz, 1H), 3.17 (t, J = 9.6 Hz, 1H), 2.87 (ddd, J = 13.3, 6.1, 3.5 Hz, 1H), 2.66–2.60 (m, 1H), 2.56 (s, 3H), 2.54–2.49 (m, 1H), 0.85 (q, J = 3.8 Hz, 21H); 13C NMR (125 MHz, CDCl3) δ 175.94, 173.92, 159.34, 154.32, 146.89, 139.65, 132.30, 130.98, 130.07, 129.75, 128.88, 128.50, 127.57, 127.42, 126.71, 126.45, 121.74, 120.99, 120.75, 114.18, 107.15, 68.21, 61.31, 60.86, 60.43, 55.34, 51.42, 47.61, 44.98, 31.20, 25.78, 17.86, 11.88; IR (neat film NaCl) 2944, 2865, 2073, 1716, 1667, 1604, 1513, 1455, 1247, 1227, 1109, 1069, 1034, 883, 761 cm–1; HRMS (MM: ESI-APCI+) m/z calcd for C40H53BrN3O6Si [M + H]+ 778.2882, found 778.2879.
Methyl 3a-(4-Bromo-1-methyl-2-oxo-3-(2-((triisopropylsilyl)oxy)ethyl)indolin-3-yl)-1-(4-methoxybenzyl)-2,3,3a,8a-tetrahydropyrrolo[2,3-b]indole-8(1H)-carboxylate (71)
To a solution of amide 54 (0.32 g, 0.41 mmol, 1.00 equiv) in THF (41.1 mL) was added AlH3–Me2NEt (0.5 M in toluene; 1.64 mL, 0.82 mmol, 2.00 equiv) dropwise at 0 °C. The reaction was stirred for 2 h and quenched with MeOH. The reaction solution was concentrated in vacuo and purified by column chromatography (4:1 hexanes/EtOAc) to afford aminal 71 (0.19 g, 61% yield): Rf = 0.20 (4:1 hexanes/EtOAc); 1H NMR (300 MHz, CDCl3) δ 7.58–7.47 (m, 1H), 7.21 (d, J = 8.2 Hz, 2H), 7.04–6.93 (m, 3H), 6.80 (dq, J = 14.8, 7.4 Hz, 4H), 6.39–6.24 (m, 2H), 4.30 (m, 1H), 3.92–3.79 (m, 4H), 3.78 (s, 3H), 3.68 (dd, J = 10.2, 5.0 Hz, 1H), 3.57–3.43 (m, 1H), 3.03 (s, 3H), 2.65 (m, 4H), 2.26 (dt, J = 15.0, 7.2 Hz, 1H), 2.09 (dd, J = 11.9, 4.6 Hz, 1H), 0.87 (d, J = 3.2 Hz, 21H); 13C NMR (125 MHz, CDCl3) δ 176.0, 175.7, 160.6, 146.3, 141.7, 133.9, 132.2, 130.3, 130.1, 126.8, 123.6, 123.2, 123.1, 115.1, 114.6, 113.9, 107.3, 107.1, 106.6, 82.4, 60.7, 60.1, 57.2, 55.3, 52.6, 51.0, 31.9, 31.1, 30.7, 29.7, 26.1, 17.7, 11.8; IR (neat film NaCl) 2943, 1722, 1604, 1464, 1386, 1344, 1254, 1107, 1033, 885, 760 cm–1; HRMS (MM: ESI-APCI+) m/z calcd for C40H53BrN3O5Si [M + H]+ 762.2932, found 762.2936.
Methyl 3a-(4-Bromo-3-(2-hydroxyethyl)-1-methyl-2-oxoindolin-3-yl)-2,3,3a,8a-tetrahydropyrrolo[2,3-b]indole-8(1H)-carboxylate (72)
To a solution of silyl ether 71 (98 mg, 0.13 mmol, 1.00 equiv) in THF (1.28 mL) was added TBAF (0.15 mL, 1.0 M solution in THF) at 0 °C. The reaction mixture was stirred for 3 h at 23 °C and quenched with satd NH4Cl. The reaction mixture was washed with EtOAc (3 × 1.5 mL) and brine. The combined organic phases were dried over MgSO4 and concentrated in vacuo. The residue was purified by column chromatography (2:1 hexanes/EtOAc) on silica gel to afford PMB-protected aminal compound (78 mg, 98% yield).
To a solution of PMB-protected aminal compound (96 mg, 0.16 mmol, 1.00 equiv) in CH2Cl2 (3.1 mL) and H2O (0.8 mL) was added DDQ (53 mg, 0.23 mmol, 1.50 equiv) in portions over 30 min at 0 °C. The reaction mixture was stirred for 2 h at 23 °C. The solution was diluted with CH2Cl2 and quenched with satd NaHCO3. The reaction mixture was washed with CH2Cl2 (3 × 2.5 mL) and brine. The combined organic phases were dried over MgSO4, and concentrated in vacuo. The residue was purified by column chromatography (4:1 CH2Cl2/acetone) on silica gel to afford aminal 72 (64 mg, 85% yield): Rf = 0.31 (4:1 CH2Cl2/acetone); 1H NMR (600 MHz, CDCl3) δ 7.50 (d, J = 8.1 Hz, 1H), 7.01–6.96 (m, 2H), 6.84 (p, J = 8.6 Hz, 1H), 6.78 (d, J = 7.6 Hz, 1H), 6.73 (t, J = 7.5 Hz, 1H), 6.48 (s, 1H), 6.35 (d, J = 7.8 Hz, 1H), 3.87 (d, J = 17.2 Hz, 3H), 3.66 (ddd, J = 10.8, 6.8, 3.7 Hz, 1H), 3.35 (dt, J = 10.9, 5.3 Hz, 1H), 3.11 (s, 3H), 3.01 (dd, J = 10.0, 6.5 Hz, 1H), 2.80–2.76 (m, 1H), 2.76–2.62 (m, 2H), 2.50 (td, J = 10.3, 4.7 Hz, 1H), 2.24 (br, s, 1H), 2.20 (dd, J = 12.7, 4.8 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ 177.6, 152.8, 146.1, 142.7, 130.4, 129.7, 128.9, 127.4, 127.2, 122.8, 121.6, 118.5, 113.8, 106.8, 80.5, 63.4, 60.0, 57.2, 52.6, 44.6, 35.1, 31.5, 26.3; IR (neat film NaCl) 3417, 2925, 1710, 1604, 1487, 1458, 1392, 1362, 1272, 1093, 756 cm–1; HRMS (MM: ESI-APCI+) m/z calcd for C23H25BrN3O4 [M + H]+ 486.1023, found 486.1014.
Methyl (2-(1-(4-Methoxybenzyl)-3-(1-methyl-2-oxo-3-(2-((triisopropylsilyl)oxy)ethyl)indolin-3-yl)-2-oxopyrrolidin-3-yl)phenyl)carbamate (73)
To a solution of lactam 54 (10.6 mg, 0.0136 mmol, 1.00 equiv) in THF (1.36 mL) was added LiAlH4 (5.2 mg, 0.136 mmol, 10.0 equiv) in portions at 0 °C. The reaction was stirred for 1 h and then quenched with satd NaCl. The reaction mixture was washed with EtOAc (3 × 2 mL) and brine. The combined organic phases were dried over MgSO4 and concentrated in vacuo. The residue was purified by column chromatography (4:1 hexanes/EtOAc) to afford oxindole 73 (7.9 mg, 83% yield): Rf = 0.25 (4:1 hexanes/EtOAc); 1H NMR (600 MHz, CDCl3) δ 9.64 (br, s, 1H), 8.02 (d, J = 7.5 Hz, 1H), 7.60 (br, s, 1H), 7.13 (d, J = 8.4 Hz, 3H), 7.09 (t, J = 7.6 Hz, 1H), 7.04 (t, J = 7.7 Hz, 1H), 6.97 (s, 1H), 6.83 (d, J = 8.3 Hz, 3H), 6.38 (d, J = 7.7 Hz, 1H), 4.55 (d, J = 14.5 Hz, 1H), 4.44 (d, J = 14.5 Hz, 1H), 3.77 (s, 3H), 3.71 (s, 3H), 3.31–3.27 (m, 1H), 3.24 (br, s, 2H), 3.14 (br, s, 1H), 3.05–3.00 (m, 1H), 2.85 (s, 3H), 2.72 (br, s, 1H), 2.52 (m, 1H), 2.41 (br, s, 1H), 0.86 (q, J = 7.5, 6.2 Hz, 21H); 13C NMR (125 MHz, CDCl3) δ 177.5, 159.4, 154.7, 143.7, 129.6, 129.4, 129.3, 129.2, 128.6, 128.4, 127.7, 127.5, 127.4, 122.2, 122.0, 121.8, 114.3, 107.3, 60.0, 56.6, 55.4, 52.1, 47.1, 44.0, 34.3, 29.9, 26.0, 18.0, 12.0; IR (neat film NaCl) 2941, 1732, 1711, 1610, 1515, 1442, 1375, 1248, 1225, 1105, 1070, 1036, 750 cm–1; HRMS (MM: ESI-APCI+) m/z calcd for C40H54N3O6Si [M + H]+ 700.3776, found 700.3776.
Methyl 4-Bromo-10′-(4-methoxybenzyl)-1-methyl-2-oxo-2′,3′-dihydro-9′H-spiro[indoline-3,4′-[9a,4a](epiminoethano)pyrano[2,3-b]indole]-9′-carboxylate (76)
To a solution of lactam 54 (70 mg, 0.090 mmol, 1.00 equiv) in CH2Cl2 (9 mL) was added Tf2O (45 mL, 0.27 mmol, 3.00 equiv) dropwise at 0 °C. The reaction mixture was slowly warmed to 23 °C and stirred for 2 h. The solution was neutralized by adding satd NaHCO3. The reaction mixture was washed with CH2Cl2 (3 × 5 mL) and brine. The combined organic phases were dried over MgSO4 and concentrated in vacuo. The residue was purified by column chromatography (2:1 hexanes/EtOAc) on silica gel to afford propellane hexacycle 76 (52 mg, 95% yield): Rf = 0.25 (3:1 hexanes/EtOAc); 1H NMR (500 MHz, CDCl3) δ 7.67 (s, 1H), 7.16–7.11 (m, 3H), 6.95–6.93 (m, 2H), 6.78 (d, J = 8.6 Hz, 2H), 6.73 (td, J = 7.6, 1.1 Hz, 1H), 6.60 (t, J = 4.4 Hz, 1H), 6.36 (dd, J = 7.6, 1.5 Hz, 1H), 4.53 (dt, J = 11.5, 5.8 Hz, 1H), 4.47 (d, J = 14.9 Hz, 1H), 4.23–4.18 (m, 1H), 3.87 (s, 3H), 3.76 (s, 3H), 3.72 (d, J = 13.7 Hz, 1H), 3.21 (s, 3H), 3.01 (ddd, J = 14.4, 11.7, 8.4 Hz, 1H), 2.77–2.73 (m, 1H), 2.54 (q, J = 9.3 Hz, 1H), 2.13 (td, J = 8.8, 5.3 Hz, 1H), 1.93 (dd, J = 14.2, 6.3 Hz, 1H), 1.80–1.75 (m, 1H); 13C NMR (125 MHz, CDCl3) δ 178.0, 158.5, 153.7, 145.8, 142.5, 132.0, 130.9, 130.1, 129.5, 129.4, 128.9, 128.1, 124.6, 122.9, 121.5, 116.1, 113.6, 112.1, 106.6, 58.2, 57.0, 55.4, 54.4, 52.7, 50.2, 47.9, 33.7, 26.6, 23.2; IR (neat film NaCl) 1718, 1601, 1575, 1513, 1484, 1455, 1365, 1245, 1134, 1099, 1037, 912, 764, 731 cm–1; HRMS (MM: ESI-APCI+) m/z calcd for C31H31BrN3O5 [M + H]+ 604.1442, found 604.1433.
Methyl 4-Bromo-1-methyl-2-oxo-2′,3′-dihydro-9′H-spiro[indoline-3,4′-[9a,4a](epiminoethano)pyrano[2,3-b]indole]-9′-carboxylate (77)
To a solution of oxindole 76 (46 mg, 0.075 mmol, 1.00 equiv) in CH2Cl2 (3.8 mL) and H2O (0.94 mL) was added DDQ (34 mg, 0.15 mmol, 2.00 equiv) at 0 °C. The reaction mixture was slowly warmed to 23 °C and stirred for 2 h. The solution was quenched with satd NaHCO3. The reaction mixture was washed with CH2Cl2 (3 × 3.0 mL) and brine. The combined organic phases were dried over MgSO4 and concentrated in vacuo. The residue was purified by column chromatography (4:1 CH2Cl2/acetone) on silica gel to afford propellane hexacycle 77 (33 mg, 92% yield): Rf = 0.1 (1:1 hexane/EtOAc); 1H NMR (500 MHz, CDCl3) δ 7.70 (d, J = 8.2 Hz, 1H), 7.16 (ddd, J = 8.3, 7.4, 1.4 Hz, 1H), 7.03 (dd, J = 8.2, 7.6 Hz, 1H), 6.95 (dd, J = 8.2, 1.1 Hz, 1H), 6.72 (ddd, J = 14.7, 7.6, 1.1 Hz, 2H), 6.26 (ddd, J = 7.6, 1.4, 0.5 Hz, 1H), 4.47 (td, J = 11.2, 6.6 Hz, 1H), 4.12–4.07 (m, 1H), 3.93 (s, 3H), 3.23 (s, 3H), 3.14 (dd, J = 9.4, 6.0 Hz, 1H), 3.00 (ddd, J = 14.4, 11.2, 8.2 Hz, 1H), 2.68–2.57 (m, 2H), 1.89–1.85 (m, 1H), 1.82 (ddd, J = 14.4, 6.6, 1.6 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ 177.9, 153.6, 145.8, 142.4, 133.3, 130.4, 129.7, 129.2, 128.1, 125.7, 123.2, 122.5, 114.3, 111.6, 106.9, 58.5, 55.7, 53.8, 53.0, 43.3, 36.5, 26.6, 23.5; IR (neat film NaCl) 2958, 1713, 1602, 1485, 1446, 1373, 1242, 1095, 754 cm–1; HRMS (MM: ESI-APCI+) m/z calcd for C23H23BrN3O4 [M + H]+ 484.0866, found 484.0874.
Methyl 10-Bromo-10b-(2-hydroxyethyl)-1′-(4-methoxybenzyl)-6-methyl-2′-oxo-6,10b-dihydrospiro[indolo[2,3-b]quinoline-11,3′-pyrrolidine]-5(5aH)-carboxylate (81)
To a solution of propellane hexacycle 76 (13 mg, 0.021 mmol, 1.00 equiv) in CH2Cl2 (2.14 mL) was added DIBAL (1.0 M in THF; 0.11 mL, 0.11 mmol, 5.00 equiv) dropwise at −78 °C. The reaction mixture was stirred for 1 h and warmed to 0 °C. DIBAL (1.0 M in THF; 21.4 mL, 21.4 mmol, 1.00 equiv) was added dropwise at 0 °C and the mixture stirred for 1 h. Then, DIBAL (1.0 M in THF; 21.4 mL, 21.4 mmol, 1.00 equiv) was added one more time dropwise at 0 °C and the mixture stirred for another 1 h. The reaction mixture was warmed to 23 °C, and Et2AlCl (1.0 M in hexane; 42.8 mL, 42.8 mmol, 2.00 equiv) was added dropwise. The mixture was stirred for 30 min and quenched with satd NH4Cl and satd potassium sodium tartrate. The reaction mixture was washed with CH2Cl2 (3 × 2.0 mL) and brine. The combined organic phases were dried over MgSO4 and concentrated in vacuo. The residue was purified by column chromatography (1:1 hexane/EtOAc) on silica gel to afford aminal 81 (10.5 mg, 87% yield): Rf = 0.25 (1:1 hexane/EtOAc); 1H NMR (500 MHz, CDCl3) δ 7.39 (d, J = 7.7 Hz, 1H), 7.24–7.22 (m, 2H), 7.17 (m, 1H), 7.08–7.03 (m, 1H), 6.87–6.84 (m, 2H), 6.82 (t, J = 7.9 Hz, 1H), 6.71 (dd, J = 8.0, 1.0 Hz, 1H), 6.23 (br, s, 1H), 6.01 (dd, J = 7.9, 1.0 Hz, 1H), 5.15 (d, J = 14.5 Hz, 1H), 3.94 (d, J = 14.5 Hz, 1H), 3.84 (s, 3H), 3.79 (s, 3H), 3.72–3.63 (m, 2H), 3.40 (dd, J = 9.5, 7.7 Hz, 1H), 3.12 (td, J = 9.7, 1.5 Hz, 1H), 3.10–3.04 (m, 1H), 2.89–2.82 (m, 1H), 2.48 (s, 3H), 2.32 (dt, J = 14.2, 7.2 Hz, 1H), 1.65 (dt, J = 14.7, 9.7 Hz, 1H), 1.59 (br, s, 1H); 13C NMR (125 MHz, CDCl3) δ 170.5, 159.2, 152.7, 139.2, 136.3, 134.0, 130.4, 129.8, 128.8, 126.8, 126.7, 126.1, 125.2, 125.0, 122.9, 122.4, 114.2, 104.7, 83.3, 61.0, 60.5, 55.4, 53.6, 53.4, 47.1, 44.3, 35.3, 33.0, 31.1; IR (neat film NaCl) 2922, 1689, 1597, 1512, 1444, 1334, 1249, 1178, 1032, 754 cm–1; HRMS (MM: ESI-APCI+) m/z calcd for C31H33BrN3O5 [M + H]+ 606.1598, found 606.1592.
8′-Bromo-1-methyl-1″-(2-nitrobenzyl)-2′,3′-dihydro-1′H-dispiro[indoline-3,4′-benzo[b]azepine-5′,3″-pyrrolidine]-2,2″-dione (91)
To a solution of amide 89 (20 mg, 0.0239 mmol, 1.00 equiv) in CH2Cl2 (2.39 mL), was added Tf2O (0.0121 mL, 0.0718 mmol, 3.00 equiv) dropwise at 0 °C. The reaction mixture was slowly warmed to 23 °C and the mixture stirred for 2 h. After the reaction was complete, the solution was brought to pH 10.5–11.0 by addition of satd NaHCO3. The reaction mixture was extracted with EtOAc (3 × 3 mL) and washed with brine. The combined organic phases were dried over MgSO4 and concentrated in vacuo. The residue was purified by column chromatography (2:1 hexanes/EtOAc) on silica gel to afford tetrahydroazepine 91 (9.4 mg, 70% yield): Rf = 0.33 (2:1 hexanes/EtOAc); 1H NMR (600 MHz, CDCl3) δ 8.04 (d, J = 8.1 Hz, 1H), 7.69 (t, J = 7.6 Hz, 1H), 7.53 (d, J = 7.8 Hz, 1H), 7.43 (t, J = 7.8 Hz, 1H), 7.19 (t, J = 7.7 Hz, 1H), 7.09 (d, J = 2.1 Hz, 1H), 6.98 (dd, J = 8.3, 2.0 Hz, 1H), 6.82 (d, J = 7.8 Hz, 1H), 6.73 (t, J = 7.7 Hz, 1H), 6.70 (dd, J = 8.4, 1.3 Hz, 1H), 6.18 (d, J = 7.6 Hz, 1H), 5.08 (d, J = 17.0 Hz, 1H), 4.47 (d, J = 17.0 Hz, 1H), 3.89 (br, s, 1H), 3.78 (q, J = 13.0, 12.0 Hz, 1H), 3.31–3.26 (m, 2H), 3.23 (td, J = 9.3, 8.5, 4.1 Hz, 1H), 3.18 (s, 3H), 2.96–2.89 (m, 2H), 2.70 (dt, J = 13.5, 8.5 Hz, 1H), 1.42 (d, J = 13.3 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ 180.4, 175.1, 150.3, 148.3, 143.8, 134.2, 132.5, 131.1, 131.1, 129.4, 129.2, 128.3, 127.7, 126.0, 125.3, 125.0, 124.6, 121.7, 121.3, 108.0, 58.8, 50.3, 44.6, 44.1, 43.4, 36.1, 27.6, 26.4; IR (neat film NaCl) 3343, 2942, 1703, 1611, 1588, 1524, 1471, 1357, 1285, 1137, 1106, 1065, 984, 858, 732 cm–1; HRMS (MM: ESI-APCI+) m/z calcd for C28H26BrN4O4 [M + H]+ 561.1132, found 561.1165.
Acknowledgments
The authors wish to thank NIH-NIGMS (R01GM080269), Amgen, the Gordon and Betty Moore Foundation, and Caltech for financial support. S.-J.H. thanks Fulbright (Foreign Student Program, No. 15111120) and the Ilju Foundation of Education & Culture (Predoctoral Research Fellowship) for financial support. F.V. thanks the German Academic Exchange Service (DAAD) for a postdoctoral fellowship. J.A.M. is grateful for a fellowship by Bristol-Myers Squibb and Amgen. S.K. thanks California TRDRP for financial support (postdoctoral fellowship 14FT-0002). M.G. is grateful to the Swiss National Science Foundation for financial support (postdoctoral fellowship). Dr. David VanderVelde (Caltech) is gratefully acknowledged for assistance with the characterization of compounds by NMR spectroscopy. Lawrence Henling (Caltech) is acknowledged for X-ray crystallographic structural determination. The authors are thankful to Dr. Ryan Zeidan, Dr. Sandy Ma, and Mr. Chung Whan Lee for experimental assistance.
Supporting Information Available
1H and 13NMR spectral data of all compounds and single-crystal X-ray analysis. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
† BASF SE. GVA/HC-B009, 67056 Ludwigshafen, Germany.
Author Present Address
‡ University of Houston, Department of Chemistry, 112 Fleming Building, Houston, TX 77204.
Author Present Address
§ Howard Hughes Medical Institute, University of California, San Francisco, Genentech Hall, 600, 16th St, MC 2280, San Francisco, CA 94158.
Author Present Address
# BASF SE. GMU/A-B001, 67056 Ludwigshafen, Germany.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Numata A.; Takahashi C.; Ito Y.; Takada T.; Kawai K.; Usami Y.; Matsumura E.; Imachi M.; Ito T.; Hasegawa T. Tetrahedron Lett. 1993, 34, 2355. [Google Scholar]
- a Ratnayake A. S.; Yoshida W. Y.; Mooberry S. L.; Hemscheidt T. K. J. Org. Chem. 2001, 66, 8717. [DOI] [PubMed] [Google Scholar]; b Retraction of this article was prompted by a revision of structure; see ref (5a) andRatnayake A. S.; Yoshida W. Y.; Mooberry S. L.; Hemscheidt T. K. J. Org. Chem. 2003, 68, 1640. [DOI] [PubMed] [Google Scholar]; However, the biological activity reported in this manuscript was not specifically called into question and is distinct relative to previous reports.1
- a Jadulco R.; Edrada R. A.; Ebel R.; Berg A.; Schaumann K.; Wray V.; Steube K.; Proksch P. J. Nat. Prod. 2004, 67, 78. [DOI] [PubMed] [Google Scholar]; b Hayashi H.; Matsumoto H.; Akiyama K. Biosci. Biotechnol. Biochem. 2004, 68, 753. [DOI] [PubMed] [Google Scholar]; c Andersen B.; Smedsgaard J.; Frisvad J. C. J. Agric. Food Chem. 2004, 52, 2421. [DOI] [PubMed] [Google Scholar]; d Dalsgaard P. W.; Blunt J. W.; Munro M. H. G.; Frisvad J. C.; Christophersen C. J. Nat. Prod. 2005, 68, 258. [DOI] [PubMed] [Google Scholar]; e Blunt J. W.; Copp B. R.; Munro M. H. G.; Northcote P. T.; Prinsep M. R. Nat. Prod. Rep. 2006, 23, 26. [DOI] [PubMed] [Google Scholar]; f Wigley L. J.; Mantle P. G.; Perry D. A. Phytochemistry 2006, 67, 561. [DOI] [PubMed] [Google Scholar]
- a Tanaka J.; Yan Y.; Choi J.; Bai J.; Klenchin V. A.; Rayment I.; Marriott G. Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 13851. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Klenchin V. A.; Allingham J. S.; King R.; Tanaka J.; Marriott G.; Rayment I. Nat. Struct. Biol. 2003, 10, 1058. [DOI] [PubMed] [Google Scholar]
- a May J. A.; Zeidan R. K.; Stoltz B. M. Tetrahedron Lett. 2003, 44, 1203. [Google Scholar]; b Crawley S. L.; Funk R. L. Org. Lett. 2003, 5, 3169. [DOI] [PubMed] [Google Scholar]; c May J. A.; Stoltz B. Tetrahedron 2006, 62, 5262. [Google Scholar]; d Yang J.; Song H.; Xiao X.; Wang J.; Qin Y. Org. Lett. 2006, 8, 2187. [DOI] [PubMed] [Google Scholar]; e Crawley S. L.; Funk R. L. Org. Lett. 2006, 8, 3995. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Seo J. H.; Artman G. D. III; Weinreb S. M. J. Org. Chem. 2006, 71, 8891. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Yang J.; Wu H.; Shen L.; Qin Y. J. Am. Chem. Soc. 2007, 129, 13794. [DOI] [PubMed] [Google Scholar]; h George J. H.; Adlington R. M. Synlett 2008, 2093. [Google Scholar]; i Siengalewicz P.; Gaich T.; Mulzer J. Angew. Chem., Int. Ed. 2008, 47, 8170. [DOI] [PubMed] [Google Scholar]; j Liu P.; Seo J. H.; Weinreb S. M. Angew. Chem., Int. Ed. 2010, 49, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]; k Zuo Z.; Xie W.; Ma D. J. Am. Chem. Soc. 2010, 132, 13226. [DOI] [PubMed] [Google Scholar]; l Zuo Z.; Ma D. Angew. Chem., Int. Ed. 2011, 50, 12008. [DOI] [PubMed] [Google Scholar]; m Belmar J.; Funk R. L. J. Am. Chem. Soc. 2012, 134, 16941. [DOI] [PubMed] [Google Scholar]; n Artman G. D. III; Weinreb S. M. Org. Lett. 2003, 5, 1523. [DOI] [PubMed] [Google Scholar]; o Fuchs J. R.; Funk R. L. J. Am. Chem. Soc. 2004, 126, 5068. [DOI] [PubMed] [Google Scholar]; p Sabahi A.; Novikov A.; Rainier J. D. Angew. Chem., Int. Ed. 2006, 45, 4317. [DOI] [PubMed] [Google Scholar]; q Evans M. A.; Sacher J. R.; Weinreb S. M. Tetrahedron 2009, 65, 6712. [Google Scholar]; r Wu H.; Xue F.; Xiao X.; Qin Y. J. Am. Chem. Soc. 2010, 132, 14052. [DOI] [PubMed] [Google Scholar]; s Schammel A. W.; Chiou G.; Garg N. K. Org. Lett. 2012, 14, 4556. [DOI] [PMC free article] [PubMed] [Google Scholar]; t Ishida T.; Takemoto Y. Tetrahedron 2013, 69, 4517. [Google Scholar]; u Ishida T.; Ikota H.; Kurahashi K.; Tsukano C.; Takemoto Y. Angew. Chem., Int. Ed. 2013, 52, 10204. [DOI] [PubMed] [Google Scholar]; v Zhang H.; Hong L.; Kang H.; Wang R. J. Am. Chem. Soc. 2013, 135, 14098. [DOI] [PubMed] [Google Scholar]; w Han S.-J.; Vogt F.; Krishnan S.; May J. A.; Gatti M.; Virgil S. C.; Stoltz B. M. Org. Lett. 2014, 16, 3316. [DOI] [PMC free article] [PubMed] [Google Scholar]; x Trost B. M.; Osipov M.; Krüger S.; Zhang Y. Chem. Sci. 2014, 10.1039/C4SC01826E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verbitski S. M.; Mayne C. L.; Davis R. A.; Concepcion G. P.; Ireland C. M. J. Org. Chem. 2002, 67, 7124. [DOI] [PubMed] [Google Scholar]
- Denault J.-B.; Salvesen G. S. Chem. Rev. 2002, 102, 4489. [DOI] [PubMed] [Google Scholar]
- Wigley L. J.; Perry D. A.; Mantle P. G. Mycol. Res. 2008, 112, 131. [DOI] [PubMed] [Google Scholar]
- a Kozlovskii A. G.; Solov’eva T. F.; Sahkarovskii V. G.; Adanin V. M. Dokl. Akad. Nauk SSSR 1981, 260, 230. [PubMed] [Google Scholar]; b Sahkarovskii V. G.; Aripovskii A. V.; Baru M. B.; Kozlovskii A. G. Khim. Prir. Soedin. 1983, 656. [Google Scholar]
- a Hegedus L. S.; Toro J. L.; Miles W. H.; Harrington P. J. J. Org. Chem. 1987, 52, 3319. [Google Scholar]; b Yokoyama Y.; Matsumoto T.; Murakami Y. J. Org. Chem. 1995, 60, 1486. [Google Scholar]; c Yokoyama Y.; Hikawa H.; Mitsuhashi M.; Uyama A.; Hiroki Y.; Murakami Y. Eur. J. Org. Chem. 2004, 1244. [Google Scholar]
- Greenberg A. Mol. Struct. Enegetics 1988, 7, 139. [Google Scholar]
- Indeed, it was the recognition that an anti-Bredt, twisted amide might be involved in a potentially biogenetic or laboratory synthesis of alkaloids 1a–h that inspired our fundamental synthetic study in this area.Tani K.; Stoltz B. M. Nature 2006, 441, 731. [DOI] [PubMed] [Google Scholar]
- a Szostak M.; Aubé J. Org. Biomol. Chem. 2011, 9, 27. [DOI] [PubMed] [Google Scholar]; b Szostak M.; Aubé J. Chem. Rev. 2013, 113, 5701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Yamada F.; Makita Y.; Suzuki T.; Somei M. Chem. Pharm. Bull. 1985, 33, 2162. [Google Scholar]; b Somei M.; Yamada F. Jpn Patent JP 85–47044 19850308, 1986.; c Iwao M.; Motoi O. Tetrahedron Lett. 1995, 36, 5929. [Google Scholar]
- Krishnan S.; Bagdanoff J. T.; Ebner D. C.; Ramtohul Y. K.; Tambar U. K.; Stoltz B. M. J. Am. Chem. Soc. 2008, 130, 13745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinhagen H.; Corey E. J. Angew. Chem., Int. Ed. 1999, 38, 1928. [DOI] [PubMed] [Google Scholar]
- Kennewell P. D.; Miller D. J.; Scrowston R. M.; Westwood R. J. Chem. Res. Miniprint 1995, 10, 2380. [Google Scholar]
- a Hrib N. J.; Jurcak J. G.; Burgher K. L.; Conway P. G.; Hartman H. B.; Kerman L. L.; Roehr J. E.; Woods A. T. J. Med. Chem. 1994, 37, 2308. [DOI] [PubMed] [Google Scholar]; b Spence T. W. M.; Tennant G. J. Chem. Soc. C 1971, 3712. [Google Scholar]; c Eckroth D. R.; Cochran T. G. J. Chem. Soc. C 1970, 2660. [Google Scholar]
- He L.; Bekkaye M.; Retailleau P.; Masson G. Org. Lett. 2012, 14, 3158. [DOI] [PubMed] [Google Scholar]
- Krishnan S.; Stoltz B. M. Tetrahedron Lett. 2007, 48, 7571. [Google Scholar]
- a Ma S.; Han X.; Krishnan S.; Virgil S. C.; Stoltz B. M. Angew. Chem., Int. Ed. 2009, 48, 8037. [DOI] [PubMed] [Google Scholar]; b Dou X.; Yao W.; Zhou B.; Lu Y. Chem. Commun. 2013, 49, 9224. [DOI] [PubMed] [Google Scholar]
- Liu B.; Hu L. Bioorg. Med. Chem. 2003, 11, 3889. [DOI] [PubMed] [Google Scholar]
- We thought that the diastereoselectivity
might be related to protecting groups and attempted the Diels–Alder
cycloaddition with tosyl-protected aurantioclavine derivative and o-azaxylylene derivative. However, a change of the protecting
groups did not significantly affect the diastereoselectivity of the
cycloaddition.
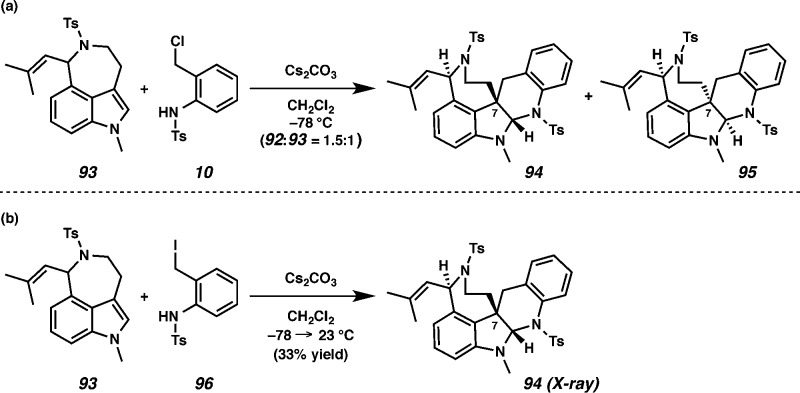
- Lee C. W.; Han S.-J.; Virgil S. C.; Stoltz B. M. Tetrahedron 2014, 10.1016/j.tet.2014.10.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath A.; Grassberger M. A.; Schulz G.; Haidl E.; Sperner H.; Steck A. Tetrahedron 2000, 56, 7469. [Google Scholar]
- a Tsuda T.; Chujo Y.; Nishi S.; Tawara K.; Saegusa T. J. Am. Chem. Soc. 1980, 102, 6381. [Google Scholar]; b Tsuji J.; Minami I.; Shimizu I. Tetrahedron Lett. 1983, 24, 1793. [Google Scholar]; c Tsuji J.; Minami I. Acc. Chem. Res. 1987, 20, 140. [Google Scholar]; d Trost B. M.; Xu J.; Schmidt T. J. Am. Chem. Soc. 2009, 131, 18343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For reviews on allylic alkylation, see:; a Trost B. M.; Van Vranken D. L. Chem. Rev. 1996, 96, 395. [DOI] [PubMed] [Google Scholar]; b Tunge J. A.; Burger E. C. Eur. J. Org. Chem. 2005, 1715. [Google Scholar]; c Mohr J. T.; Stoltz B. M. Chem.—Asian J. 2007, 2, 1476. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Weaver J. D.; Recio A. III; Grenning A. J.; Tunge J. A. Chem. Rev. 2011, 111, 1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marfat A.; Carta M. P. Tetrahedron Lett. 1987, 28, 4027. [Google Scholar]
- Abdel-Magid A. F.; Carson K. G.; Harris B. D.; Maryanoff C. A.; Shah R. D. J. Org. Chem. 1996, 61, 3849. [DOI] [PubMed] [Google Scholar]
- Lu H.; Li C. Org. Lett. 2006, 8, 5365. [DOI] [PubMed] [Google Scholar]
- a Trost B. M.; Malhotra S.; Chan W. H. J. Am. Chem. Soc. 2011, 133, 7328. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Li P.; Buchwald S. L. Angew. Chem., Int. Ed. 2011, 50, 6396. [DOI] [PubMed] [Google Scholar]
- Kotoku N.; Sumii Y.; Kobayashi M. Org. Lett. 2011, 13, 3514. [DOI] [PubMed] [Google Scholar]
- Fernandes R. A.; Yamamoto Y. J. Org. Chem. 2004, 69, 3562. [DOI] [PubMed] [Google Scholar]
- Rege P. D.; Johnson F. J. Org. Chem. 2003, 68, 6133. [DOI] [PubMed] [Google Scholar]
- Treatment
of hexacyclic oxindole 77 with allyl iodide and benzyl
bromide under basic conditions provided 97 and 98 in 85% and 80% yield, respectively.
Although smooth reductive cyclization of the propellane hexacyclic
oxindole 97 and 98 led to aminal 99 and 100, respectively, removal of both allyl and benzyl
groups on lactam 99 and 100 turned out to
be challenging.
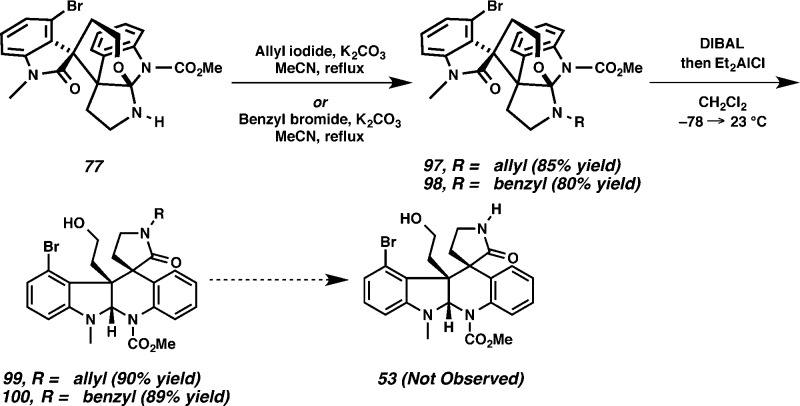
- a Snider B. B.; Busuyek M. V. Tetrahedron 2001, 57, 3301. [Google Scholar]; b Gareau Y.; Zamboni R.; Wong A. W. J. Org. Chem. 1993, 58, 1582. [Google Scholar]; c Voelker T.; Ewell T.; Joo J.; Edstrom E. D. Tetrahedron Lett. 1998, 39, 359. [Google Scholar]
- Han S.-J.; Fernando de Melo G.; Stoltz B. M. Tetrahedron Lett. 2014, 55, 6467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuyama T.; Liu G. J. Am. Chem. Soc. 1996, 118, 7426. [Google Scholar]
- Wang Y.; Kong C.; Du Y.; Song H.; Zhang D.; Qin Y. Org. Biomol. Chem. 2012, 10, 2793. [DOI] [PubMed] [Google Scholar]
- Yamada Y.; Arima S.; Okada C.; Akiba A.; Kai T.; Harigaya Y. Chem. Pharm. Bull. 2006, 54, 788. [DOI] [PubMed] [Google Scholar]
- Suzuki T.; Ota Y.; Ri M; Bando M.; Gotoh A.; Itoh Y.; Tsumoto H.; Tatum P. R.; Mizukami T.; Nakagawa H.; Iida S.; Ueda R.; Shirahige K.; Miyata N. J. Med. Chem. 2012, 55, 9562. [DOI] [PubMed] [Google Scholar]


Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.