

Planagumà et al. show that infusing CSF from patients with anti-NMDAR encephalitis into the mouse brain induces progressive memory and behavioural deficits that correlate with levels of brain-bound human NMDAR-antibodies and a reduction in NMDARs. The changes, which are reversible, confirm the pathogenicity of NMDAR-antibodies and have therapeutic implications.
See Honnorat (doi:10.1093/brain/awu342) for a scientific commentary on this article.
Keywords: animal model, anti-NMDAR encephalitis, antibodies, pathogenesis, mechanism
Abstract
Anti-N-methyl D-aspartate receptor (NMDAR) encephalitis is a severe neuropsychiatric disorder that associates with prominent memory and behavioural deficits. Patients’ antibodies react with the N-terminal domain of the GluN1 (previously known as NR1) subunit of NMDAR causing in cultured neurons a selective and reversible internalization of cell-surface receptors. These effects and the frequent response to immunotherapy have suggested an antibody-mediated pathogenesis, but to date there is no animal model showing that patients’ antibodies cause memory and behavioural deficits. To develop such a model, C57BL6/J mice underwent placement of ventricular catheters connected to osmotic pumps that delivered a continuous infusion of patients’ or control cerebrospinal fluid (flow rate 0.25 µl/h, 14 days). During and after the infusion period standardized tests were applied, including tasks to assess memory (novel object recognition in open field and V-maze paradigms), anhedonic behaviours (sucrose preference test), depressive-like behaviours (tail suspension, forced swimming tests), anxiety (black and white, elevated plus maze tests), aggressiveness (resident-intruder test), and locomotor activity (horizontal and vertical). Animals sacrificed at Days 5, 13, 18, 26 and 46 were examined for brain-bound antibodies and the antibody effects on total and synaptic NMDAR clusters and protein concentration using confocal microscopy and immunoblot analysis. These experiments showed that animals infused with patients’ cerebrospinal fluid, but not control cerebrospinal fluid, developed progressive memory deficits, and anhedonic and depressive-like behaviours, without affecting other behavioural or locomotor tasks. Memory deficits gradually worsened until Day 18 (4 days after the infusion stopped) and all symptoms resolved over the next week. Accompanying brain tissue studies showed progressive increase of brain-bound human antibodies, predominantly in the hippocampus (maximal on Days 13–18), that after acid extraction and characterization with GluN1-expressing human embryonic kidney cells were confirmed to be against the NMDAR. Confocal microscopy and immunoblot analysis of the hippocampus showed progressive decrease of the density of total and synaptic NMDAR clusters and total NMDAR protein concentration (maximal on Day 18), without affecting the post-synaptic density protein 95 (PSD95) and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors. These effects occurred in parallel with memory and other behavioural deficits and gradually improved after Day 18, with reversibility of symptoms accompanied by a decrease of brain-bound antibodies and restoration of NMDAR levels. Overall, these findings establish a link between memory and behavioural deficits and antibody-mediated reduction of NMDAR, provide the biological basis by which removal of antibodies and antibody-producing cells improve neurological function, and offer a model for testing experimental therapies in this and similar disorders.
Introduction
Memory, learning, and behaviour depend on the proper function of the excitatory glutamate N-methyl D-aspartate receptor (NMDAR) and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR) and underlying mechanisms of synaptic plasticity (Lau and Zukin, 2007; Shepherd and Huganir, 2007). The critical role of NMDAR in these functions has been shown in animal models in which the NMDAR are altered genetically (Mohn et al., 1999; Belforte et al., 2010) or pharmacologically (Jentsch and Roth, 1999; Mouri et al., 2007). In humans this evidence comes from more indirect observations such as studies investigating the effects of phencyclidine or ketamine (non-competitive antagonists of NMDAR that cause psychosis) (Weiner et al., 2000; Gunduz-Bruce, 2009), and brain tissue studies of patients with schizophrenia or Alzheimer’s disease in which several molecular pathways that modulate glutamate receptor trafficking or function are affected (Snyder et al., 2005; Hahn et al., 2006). In 2007 we identified a novel disorder (anti-NMDAR encephalitis) that occurs with highly specific antibodies against extracellular epitopes located at the amino terminal domain of the GluN1 subunit of NMDAR (Dalmau et al., 2007; Gleichman et al., 2012). The resulting syndrome resembles the spectrum of symptoms that occurs in genetic or pharmacologic models of NMDAR hypofunction, including memory loss and neuropsychiatric alterations ranging from psychosis to coma (Dalmau et al., 2008; Irani et al., 2010; Viaccoz et al., 2014). Regardless of the type of presentation, most patients develop severe problems forming new memories and amnesia of the disease. Symptoms are usually accompanied by systemic and intrathecal synthesis of antibodies, the latter likely produced by plasma cells contained in brain inflammatory infiltrates (Dalmau et al., 2008; Martinez-Hernandez et al., 2011). These long-lived plasma cells and persistent antibody synthesis may explain the lengthy symptoms of most patients (average hospitalization ∼3 months) (Dalmau et al., 2008). Yet, despite the severity and duration of the disease, 80% of the patients have substantial recovery after immunotherapy (accompanied by removal of an underlying tumour, usually an ovarian teratoma, when appropriate), or sometimes spontaneously (Iizuka et al., 2008; Titulaer et al., 2013).
Investigations on the potential pathogenic role of patients’ antibodies using cultured neurons showed that the antibodies caused crosslinking and selective internalization of NMDARs that correlated with the antibody titres, and these effects were reversible after removing the antibodies (Hughes et al., 2010; Mikasova et al., 2012). In contrast, patients’ antibodies did not alter the localization or expression of other synaptic proteins, number of synapses, dendritic spines, dendritic complexity, or cell survival (Hughes et al., 2010). In parallel experiments, the density of NMDAR was also significantly reduced in the hippocampus of rats infused with patients’ antibodies, a finding comparable to that observed in the hippocampus of autopsied patients (Hughes et al., 2010). Overall, these studies suggested an antibody-mediated pathogenesis, but the demonstration that patients’ antibodies caused symptoms remained pending. Modelling symptoms and showing that these correlate with antibody-mediated reduction of NMDAR would prove the pathogenicity of patients’ antibodies, support the use of treatments directed toward decreasing the levels of antibodies or antibody-producing cells, and help to investigate experimental therapies in this and similar disorders. We report here such a model using continuous 14-day cerebroventricular infusion of patients’ CSF in mice. The aims were to determine (i) if patients’ antibodies altered memory and behaviour; (ii) whether mice symptoms correlated with brain antibody-binding and reduction of NMDAR; and (iii) whether the clinical and molecular alterations recovered after stopping the antibody infusion.
Materials and methods
Animals
Male C57BL6/J mice (Charles River), 8–10 weeks old (25–30 g) were housed in cages of five until 1 week before surgery when they were housed individually. The room was maintained at a controlled temperature (21 ± 1°C) and humidity (55 ± 10%) with illumination at 12-h cycles; food and water were available ad libitum. All experiments were performed during the light phase, and animals were habituated to the experimental room for 1 week before starting the tests. All procedures were conducted in accordance with standard ethical guidelines (European Communities Directive 86/609/EU) and approved by the local ethical committees: Comitè Ètic d’Experimentació Animal, Institut Municipal d’Assistència Sanitària (Universitat Pompeu Fabra), and Institutional Animal Care and Use Committee (University of Pennsylvania).
Patients’ CSF samples
CSF from 25 patients with high titre NMDAR antibodies (all >1:320) were pooled and used for cerebroventricular infusion. CSF from 25 subjects without NMDAR antibodies (11 with normal pressure hydrocephalus and 14 with non-inflammatory CNS disorders) were similarly pooled and used as controls. Before loading the osmotic pumps (discussed below), the pooled CSF samples from patients and controls were dialyzed (Slide-A-Lyzer 7K, Thermo) against sterile phosphate-buffered saline (PBS) overnight at 4°C, and the concentration of total IgG normalized to the CSF physiologic concentration of 2 mg/dl. All mice received the same pooled CSF either from patients or controls. Studies were approved by the institutional review board of Hospital Clínic and Institut d’Investigacions Biomèdiques August Pi i Sunyer (IDIBAPS), Universitat de Barcelona.
Surgery, placement of ventricular catheters and osmotic pumps
Cerebroventricular infusion of CSF was performed using osmotic pumps (model 1002, Alzet) with the following characteristics: volume 100 µl, flow rate 0.25 µl/h, and duration 14 days. Twenty-four hours before surgery, two osmotic pumps per animal were each loaded with 100 µl of patient or control CSF. The pumps were then connected to a 0.28 mm IM (internal diameter) polyethylene tube (C314CT, PlasticsOne) and left overnight in sterile PBS at 37°C. The next day, mice were deeply anaesthetized by intraperitoneal injection of a mixture of ketamine (100 mg/kg) and xylazine (10 mg/kg) along with subcutaneous administration of the analgesic meloxicam (1 mg/kg). Mice were then placed in a stereotaxic frame, and a bilateral catheter (PlasticsOne, model 3280PD-2.0/SP) was inserted into the ventricles (0.02 mm anterior and 1.00 mm lateral from bregma, depth 0.22 mm) and secured with dental cement. Each arm of the catheter was connected to one osmotic pump, which was subcutaneously implanted on the back of the mice. Appropriate ventricular placement of the catheters was assessed in randomly selected mice injecting methylene blue through the catheters (Fig. 1A–C).
Figure 1.
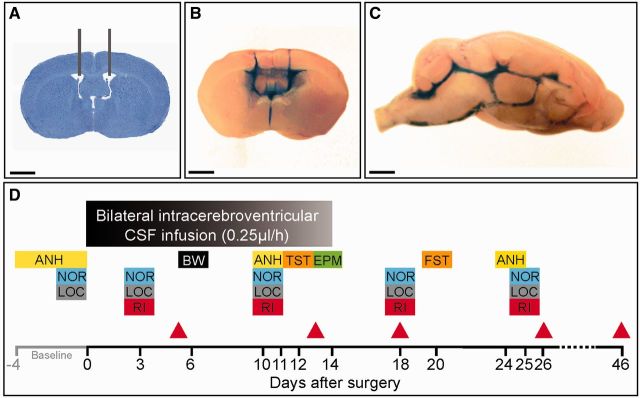
Experimental design and placement of ventricular catheters. (A) Representative coronal mouse brain section with catheter placement. Scale bar = 2 mm. (B and C) Coronal and sagittal mouse brain sections demonstrating cerebroventricular diffusion of methylene blue after ventricular infusion. Scale bars = 2 mm. (D) Schedule of cognitive testing and animal sacrifice. At Day 0, catheters and osmotic pumps were placed and bilateral ventricular infusion of patients’ or control CSF started. Infusion lasted for 14 days. Memory [novel object recognition (NOR)], anhedonia [sucrose preference test (ANH)], depressive-like behaviour [tail suspension test (TST) and forced swimming test (FST)], anxiety [black and white test (BW) and elevated plus maze test (EPM)], aggressiveness [resident intruder test (RI)] and locomotor activity (LOC) were assessed blinded to treatment at the indicated days. The novel object recognition was assessed in open field and V-maze paradigms in two different cohorts of mice. Animals were habituated for 1 to 4 days before surgery (baseline) to novel object recognition, anhedonia, and locomotor activity. Red arrowheads indicate the days of sacrifice for studies of effects of antibodies in brain.
Cognitive tasks
All behavioural tasks were performed by researchers blinded to experimental conditions using standardized tests reported by us (Maldonado et al., 1970; Filliol et al., 2000; Berrendero et al., 2005; Bura et al., 2007, 2010, 2013; Aso et al., 2008; Puighermanal et al., 2009; Burokas et al., 2012; Llorente-Berzal et al., 2013) and others (Porsolt et al., 1977; Crawley and Goodwin, 1980; Handley and Mithani, 1984; Steru et al., 1985; Konig et al., 1996; Caille et al., 1999; Strekalova et al., 2006; Taglialatela et al., 2009; Ennaceur, 2010) and following the schedule summarized in Fig. 1D. The tasks were aimed to assess memory (novel object recognition in open field and V-maze), anhedonic behaviours (sucrose preference test), depressive-like behaviours (tail suspension, and forced swimming tests), anxiety (black and white and elevated plus maze tests), aggressiveness (resident-intruder test) and locomotor activity (horizontal and vertical activity assessment). A brief description of each task is provided in the Supplementary material.
Brain tissue processing
To determine the effects of patients’ antibodies on mouse brain, animals were sacrificed at the indicated time points (Fig. 1D, Days 5, 13, 18, 26 and 46) with CO2. Brains were harvested, sagittally split, and transferred to ice-cold PBS. Half of the brain was fixed by immersion in 4% paraformaldehyde (PFA) for 1 h at 4°C, cryoprotected with 40% sucrose for 48 h at 4°C, embedded with freezing media, and snap-frozen with isopentane chilled with liquid nitrogen. The other half-brain was used for dissection of hippocampus and cerebellum for IgG and protein extraction (see below).
Immunohistochemistry and quantitative peroxidase staining
For determination of antibodies bound to brain tissue using immunoperoxidase staining, 7-µm thick tissue sections were sequentially incubated with 0.25% H2O2 for 10 min at 4°C, 5% goat serum for 15 min at room temperature, biotinylated goat anti-human IgG (1:2000, Vector labs) overnight at 4°C, and the reactivity developed using avidin-biotin-peroxidase and diaminobenzidine. Sections were mildly counterstained with haematoxylin, and results photographed under a Leica DMD108 microscope. Images were prepared creating a mask for diaminobenzidine colour, converting the mask to greyscale intensities, and inverting the pixels using Adobe Photoshop CS6 package. Hippocampal, frontal cortex, striatum and cerebellar regions were manually outlined; intensity and area were quantified in two serial sections using the public domain Fiji ImageJ software (http://fiji.sc/Fiji). Values were divided by area and normalized to the group with the highest mean (defined as 100%, patients’ CSF treated animals sacrificed at Day 18).
Immunofluorescence and confocal microscopy with brain tissue
For determination of antibodies bound to brain tissue using immunofluorescence, 5 µm-thick tissue sections were blocked with 5% goat serum and 1% bovine serum albumin for 60 min at room temperature, and incubated overnight at 4°C with Alexa Fluor® 488 goat anti-human IgG (A11013, diluted 1:1000, Molecular Probes/ Life Technologies). Slides were then mounted with ProLong® Gold (P36930, Molecular Probes) and results scanned under a LSM710 Zeiss confocal microscope. Sections from all animals were analysed in parallel. Quantification of fluorescent intensity in areas of CA1, CA3 and dentate gyrus was done using Fiji ImageJ software. Background was subtracted and intensity divided by area. Mean intensity of IgG immunostaining in animals treated with patients’ CSF and sacrificed at Day 18 was defined as 100%.
To determine the effects of patients’ antibodies on total and synaptic NMDAR clusters and PSD95, non-permeabilized 5-µm thick sections were blocked with 5% goat serum and 1% bovine serum albumin as above, incubated with human CSF antibodies for 2 h at room temperature, washed with PBS, permeabilized with Triton™ X-100 0.3% for 10 min at room temperature, and incubated with rabbit polyclonal antibody against PSD95 (diluted 1:250, Clone 18258 Abcam) overnight at 4°C. Next day, the slides were washed and incubated with the corresponding secondary antibodies, Alexa Fluor® 594 goat anti-human IgG and Alexa Fluor® 488 goat anti-rabbit IgG (A-11014, A-11008, both diluted 1:1000, Molecular Probes) for 1 h at room temperature. Slides were mounted as above and results scanned with a confocal microscope (Zeiss LSM710) with EC-Plan NEOFLUAR CS ×100/1.3 NA oil objective. Standardized z-stacks including 50 optical images were acquired from five different, equally spaced areas of CA1, CA3 and dentate gyrus of hippocampus using sequential scanning, 1024 × 1024 lateral resolution, and Nyquist optimized z-sampling frequency. Images were deconvolved with 20 iterations using theoretical point spread functions and maximum likelihood estimation algorithms of Huygens Essential software (Scientific Volume Imaging). For cluster density analysis a spot detection algorithm from Imaris suite 7.6.4 (Bitplane) was used based on automatic segmentation of the images to spots (Banovic et al., 2010). Density of clusters was expressed as spots/µm3. Three-dimensional colocalization of clusters (e.g. NMDAR and PSD95) was done using a spot co-localization algorithm implemented in Imaris suite 7.6.4. Synaptic localization was defined as co-localization of NMDAR or AMPAR with post-synaptic PSD95. Synaptic cluster density was expressed as colocalized spots/µm3. For each animal, five identical image stacks in each hippocampal area (CA1, CA2 and dentate gyrus) were acquired and the mean densities calculated for total and synaptic NMDAR and AMPAR. Densities were normalized to the mean density of control CSF treated animals (100%). For the AMPAR the antibody used was guinea pig GluA1 antibody (1:100, clone AGP-009, Alomone), and as secondary antibody Alexa Fluor® 594 goat anti-guinea pig IgG (A11076, 1:1000, Molecular Probes).
The presence of apoptosis, cellular infiltrates, and complement was assessed in the hippocampal region (CA3) in mice sacrificed on Day 18 and corresponding controls. Apoptosis was determined by standard terminal deoxynucleotidyl transferase mediated biotinylated UTP nick end labelling (TUNEL) using the TACS 2TdT-Fluor in situ apoptosis detection kit (Trevigen), and immunolabelling of cleaved caspase 3 (1:200, #9661 Cell Signalling, Technologies) using a goat anti-rabbit Alexa Fluor® 488 as secondary antibody (1:1000 Molecular Probes). The presence of complement was assessed using rabbit anti-mouse C5b-9 (1:500, Abcam) and Alexa Fluor® 488 goat anti-rabbit IgG (1:500, #A11008, Molecular Probes). Immunolabelling for T and B lymphocytes was done using rabbit anti-mouse CD3 (1:1000, #ab16669 Abcam) followed by secondary antibody goat anti-rabbit Alexa Fluor® 488 (1:1000, Molecular Probes), and rat anti-CD45R (1/10000, #ab64100) followed by goat anti-rat Alexa Fluor® 594 (1/1000, #A-11007 Molecular Probes). Results were scanned with a confocal microscope Zeiss LSM710.
Extraction of human IgG bound to mice brain
Under a dissection microscope (Zeiss stereomicroscope, Stemi 2000), the hippocampus and cerebellum were isolated, weighed, snap-frozen, and stored at −80°C. Tissue (10 mg) was homogenized in 0.5 ml ice-cold PBS with protease inhibitors (Sigma-Aldrich) and centrifuged at 16 000g for 5 min. All steps were performed at 4°C. Washing was repeated four times to remove unbound IgG. The last wash was done in 100 µl and the supernatant saved as pre-extraction fraction. To extract the specifically bound antibodies, the pellet was solubilized for 5 min in acid (86 µl 0.1 M Na-citrate buffer pH 2.7), centrifuged at 16 000g for 5 min, and the supernatant neutralized with 14 µl 1.5 M Tris pH 8.8, and used to determine the presence of NMDAR (GluN1) antibodies (see below).
Immunofluorescence with HEK293 cells expressing GluN1
The presence of GluN1 antibodies in IgG extracts from brain was determined using a HEK293 cell-based assay expressing GluN1, as reported (Dalmau et al., 2008). After fixation with 4% paraformaldehyde and permeabilization with 0.3% Triton™ X-100, cells were blocked with 1% bovine serum albumin for 90 min, and incubated with undiluted acid-extracted IgG or pre-extraction fraction from brain of infused mice, at 4°C overnight. The next day, cells were washed and incubated with a mouse monoclonal antibody against a non-competing GluN1 epitope located at amino acid 660-811 (1:20 000; clone MAB363, Millipore) for 1 h at room temperature, followed by the corresponding Alexa Fluor® secondary antibodies (A11013, A11032, both diluted 1:1000, Molecular Probes) for 1 h at room temperature. The titre of positive samples was calculated by serial dilutions until the reactivity was no longer visible. Results were photographed under a fluorescence microscope using Zeiss Axiovision software.
Immunoblot analyses
Total protein from hippocampus and cerebellum was obtained by dissecting these regions from 20-µm thick paraformaldehyde-fixed sagittal mouse brain sections on glass slides at 4°C under a Zeiss stereomicroscope (Stemi 2000). Two consecutive sections of isolated hippocampus or cerebellum were then transferred to an Eppendorf tube in PBS supplemented with protease inhibitors. Loading buffer (RotiLoad) was added, the solubilized tissue boiled for 5 min, and the proteins separated in a 10% SDS gel electrophoresis with semi-dry blotting on PVDF membranes. Membranes were blocked in 5% non-fat skimmed milk and incubated overnight at 4°C with the following polyclonal rabbit antibodies: GluN1 (1:1000, Sigma-Aldrich), GluR2/3 (1:1000, Abcam), and PSD95 (1:1000, Synaptic Systems), or a monoclonal mouse anti-β-actin (1:20 000, Sigma-Aldrich). Membranes were incubated with secondary antibodies for 1 h at room temperature (anti-rabbit IgG HRP 1:1000, anti-mouse IgG HRP 1:10 000) and analysed by enhanced chemiluminescence (all Amersham GE Healthcare) on a LAS4000 (GE Healthcare). All studies were done in duplicate. Analysed films were in the linear range of exposure, digitally scanned, and signals quantified using Fiji ImageJ software. The signal intensity of each antigen was normalized to that of actin in the same lane. The mean intensity of signal in control CSF treated animals was defined as 100% and all other intensities expressed in per cent relative to this value.
Statistics
Behavioural tests were analysed using repeated measures two-way ANOVA for tests with multiple time points (novel object recognition, sucrose preference test, resident-intruder test, locomotor activity), independent sample t-tests for tests with single time points (forced swimming test, black and white test, elevated plus maze test) or by Mann Whitney-U for skewed distributions (tail suspension test). Non-normally distributed parameters were log-transformed (black and white test, elevated plus maze test). Significance of NMDAR antibody titre in acid-extracted IgG fractions was calculated using the Kruskal-Wallis test and Dunn’s post hoc test compared to titres at Day 46. Human IgG intensity, confocal cluster density and immunoblot data (GluN1, PSD95) from different time points or regions were analysed using two-way ANOVA with Sidak-Holm post hoc testing to calculate multiplicity-adjusted P-values. Confocal cluster density in the different hippocampal subregions (CA1, CA3, dentate gyrus) were not significantly different and were analysed pooled. All experiments were assessed visually for outliers (e.g. one animal with very different results from the other animals at the same time point), but none were identified, so measurements were pooled per time point and treatment (patient or control CSF). For confocal AMPAR cluster density measured at single time points, independent sample t-tests were used. A P-value of <0.05 was considered significant in post hoc testing after correction for multiple testing (Sidak-Holm). In the two-way ANOVA the cut-off for interaction between two factors was set at 0.10; if the P-value for interaction was <0.10, the effects of treatment were considered for the separate time points (post hoc analysis). All tests were done using GraphPad Prism (Version 6).
Results
One-hundred and eleven mice were included in the studies, 56 for cognitive and behavioural tests, and 55 for assessment of antibody binding to brain and the effects on total and synaptic NMDAR (Fig. 1).
Cerebroventricular infusion of patients’ CSF alters memory and behaviour in mice
The most robust effect during the 14-day infusion of patients’ CSF was on the novel object recognition test in both the open field and V-maze paradigms (Fig. 2A and B). Compared with animals infused with control CSF, those infused with patients’ CSF showed a progressive decrease of the object recognition index, indicative of a memory deficit (Bura et al., 2007; Puighermanal et al., 2009; Taglialatela et al., 2009). The memory deficit became significant on Day 10 and was maximal on Day 18 (4 days after the infusion of CSF had stopped). On Day 25, the object recognition index had normalized and was similar to that of animals treated with control CSF (Fig. 2A and B). For all time-points, the total time spent exploring both objects (internal control) was similar in animals infused with control or patients’ CSF (Supplementary Table 1).
Figure 2.

Infusion of CSF from patients with NMDAR antibodies causes deficits in memory, anhedonia and depressive-like behaviour. (A and B) Novel object recognition index in open field (A) or V-maze paradigms (B) in animals treated with patients’ CSF (grey circles) or control CSF (white circles). A high index indicates better object recognition memory. (C) Preference for sucrose-containing water in animals infused with patients' CSF (grey) or control CSF (white). Lower percentages indicate anhedonia. (D and E) Total time of immobility in tail-suspension test during the infusion period (D, Day 12) and in forced swimming test after the infusion period (E, Day 20). Data are presented as mean ± SEM (median ± IQR in D). Number of animals: patients’ CSF n = 18 (open field novel object recognition n = 8), control CSF n = 20 (open field novel object recognition n = 10). Significance of treatment effect was assessed by two-way ANOVA (A–C) with an α-error of 0.05 and post hoc testing with Sidak-Holm adjustment (asterisks), unpaired t-test (E) or Mann-Whitney U test (D). *P < 0.05, ***P < 0.001. See Supplementary Table 1 for detailed statistics.
The preference to drink sweetened water (sucrose preference test) was used as a measure of anhedonic behaviour. Mice infused with patients’ CSF and tested during the infusion period (Day 10) had less preference for sucrose compared with mice infused with control CSF (Fig. 2C). In contrast, the same mice tested 10 days after the infusion of CSF had stopped (Day 24) showed a preference for sucrose similar to that of the control mice. The total consumption of water with and without sucrose was similar in both groups (internal control, Supplementary Table 1). In addition, two tests of depressive-like behaviour were performed. The tail suspension test, performed on Day 12, showed that animals infused with patients’ CSF had longer periods of immobility compared with those infused with control CSF (Fig. 2D). In contrast, 6 days after the infusion of CSF had stopped (Day 20), no differences were noted with the forced swimming test (examining immobility in inescapable situations; Fig. 2E and Supplementary Table 1). Overall, these findings suggest that the infusion of NMDAR antibodies was associated with anhedonic and depressive-like behaviours.
In contrast to the prominent memory deficit, along with anhedonia and depressive behaviour, no significant differences were noted in tests of anxiety (black and white test, elevated plus maze test), aggression (resident-intruder test) and locomotor activity (Fig. 3A–D).
Figure 3.

Infusion of CSF from patients with NMDAR antibodies does not alter the tests of anxiety, aggression and locomotor activity. (A and B) Number of entries into bright/open compartments during a 5 min period in a standard black and white (A, Day 6) or elevated plus maze test (B, Day 14) in animals treated with patients’ CSF (filled circles) or control CSF (open circles). (C) Number of aggressive events over a 4-min period in a resident intruder paradigm in both treatment groups. (D) Horizontal (solid lines) and vertical (dashed lines) movement count over a 10 min period in both treatment groups. Data are presented as mean ± SEM. Number of animals: patients’ CSF n = 18, control CSF n = 20. Statistical assessment as indicated in Fig. 2 and Supplementary Table 1.
Patients’ antibodies bind to NMDAR in mouse brain
Animals infused with patients’ CSF, but not control CSF, had progressively increasing human IgG immunostaining (representing IgG bound to brain) that correlated with the duration of the infusion. The distribution of IgG immunostaining predominated in regions with high density of NMDAR, mainly the hippocampus (Fig. 4A), resembling that obtained with brain sections directly incubated with patients’ CSF or a monoclonal antibody against GluN1 (Dalmau et al., 2008). Upon quantification of immunostaining, the maximal antibody binding was identified in mice sacrificed on Day 18, which had received 14 days of CSF infusion, compared with mice sacrificed on Days 5 or 13 (Fig. 4B and C). In animals sacrificed on Days 26 and 46 the presence of IgG immunostaining progressively decreased. In frontal cortex the dynamics of IgG binding were similar to those of the hippocampus (Supplementary Fig. 1), but the amount of IgG was substantially less; in other brain regions such as the cerebellum and striatum, the IgG immunostaining was sparse and not significantly different between animals infused with patients’ CSF or control CSF (data not shown).
Figure 4.
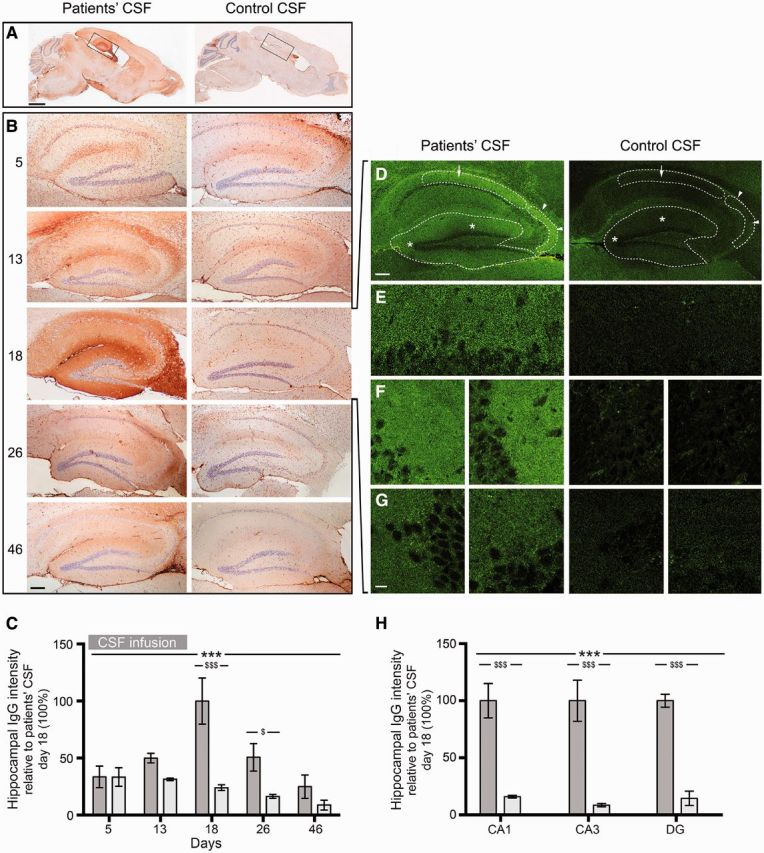
Animals infused with patient’s CSF have a progressive increase of human IgG bound to hippocampus. (A and B) Immunostaining of human IgG in sagittal brain sections (A) and hippocampus (B) of representative animals infused with patients’ CSF (left) and control CSF (right), sacrificed at the indicated experimental days. In animals infused with patients’ CSF there is a gradual increase of IgG immunostaining until Day 18, followed by decrease of immunostaining. Scale bars: A = 2 mm; B = 200 µm. (C) Quantification of intensity of human IgG immunolabelling in hippocampus of mice infused with patients’ CSF (dark grey columns) and control CSF (light grey columns) sacrificed at the indicated time points. (D–H) Confocal microscopy analysis of IgG bound to the hippocampus on Day 18. (D) Sagittal section of the hippocampus with areas examined at higher magnification in E (arrow in CA1), F (arrow heads in CA3) and G (asterisks in dentate gyrus). Note the fine punctate IgG immunolabelling surrounding neuronal bodies in mice infused with patients’ CSF; this immunolabelling is similar to that reported in brain sections directly incubated with patients’ antibodies, as in Dalmau et al. (2008). Scale bars: D = 200 µm; E–G = 10 µm. (H) Quantification of the intensity of human IgG immunofluorescence in the indicated areas in animals infused with patients’ CSF (dark grey columns) or control CSF (light grey columns). For all quantifications, mean intensity of IgG immunostaining in the group with the highest value (animals treated with patients’ CSF and sacrificed at Day 18) was defined as 100%. All data are presented as mean ± SEM. For each time point five animals infused with patients’ CSF and five with control CSF were examined. Significance of treatment effect was assessed by two-way ANOVA with an α-error of 0.05 (*) and post hoc testing with Sidak-Holm adjustment ($). ***, $$$P < 0.001; $P < 0.05. See Supplementary Table 2 for detailed statistics.
Studies with immunofluorescence and confocal microscopy showed that in animals infused with patients’ CSF the presence of hippocampal IgG was visible as a punctate immunolabelling on the surface of neurons and neuronal processes in contrast to mice infused with control CSF where minor amounts of IgG reactivity without preference for neuronal structures were noted (Fig. 4D–G). In addition, the amount of human IgG bound to all selected regions of hippocampus was significantly higher than in the control group (Fig. 4H).
To determine if the IgG immunostaining represented brain-bound NMDAR antibodies, IgG was extracted from several brain regions and examined for reactivity with HEK cells expressing GluN1. These studies showed that the IgG extracted from hippocampus of mice infused with patients’ CSF reacted specifically with GluN1 (Fig. 5A). The NMDAR antibody concentration in the extracts correlated with the duration of infusion of CSF; it increased until Day 13, reached the maximal concentration on Days 13–18, and decreased afterwards (Fig. 5A and C). NMDAR antibodies were also detected in IgG extracts from other brain regions (frontal cortex, cerebellum) but at lower concentration to that obtained from hippocampus (Fig. 5D). Demonstration that the extracted antibodies were specifically bound to the NMDAR was provided by the lack of GluN1 reactivity in the pre-extraction fractions (Fig. 5B and E). Parallel studies with tissue from animals infused with control CSF did not show NMDAR antibodies (Supplementary Fig. 2).
Figure 5.
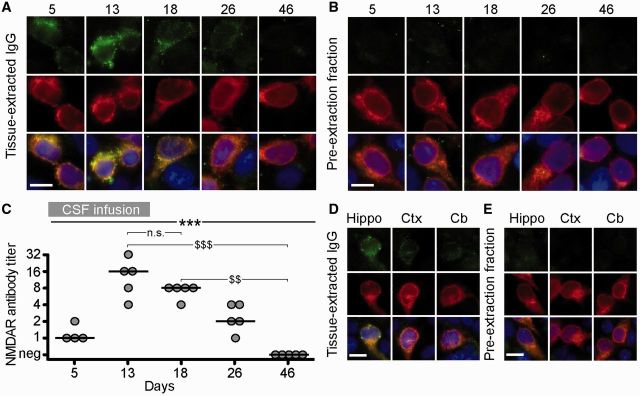
The human IgG extracted from brain of mice infused with patients’ CSF is specific for NMDARs. (A and B) HEK293 cells expressing the GluN1 subunit of the NMDAR immunolabelled with acid-extracted IgG fractions (top row in A) or pre-extraction fractions (top row in B) from hippocampus of mice infused with patients’ CSF and sacrificed on the indicated days. The maximal reactivity with GluN1-expressing cells was noted in acid-extracted IgG fractions from Days 13 and 18 (A); none of the pre-extraction fractions showed GluN1 reactivity (B) indicating that the reactivity of acid-extracted fractions corresponds to IgG antibodies that were bound to brain NMDAR receptors. The second row in A and B shows the reactivity with a monoclonal GluN1 antibody, and the third row the colocalization of immunolabelling. Scale bars = 10 µm. (C) Quantification of NMDAR antibody titre in IgG-extracted fractions from hippocampus of animals treated with patients’ CSF (n = 5 mice per each time point, except four mice for Day 5). Solid line = median. Significance was tested by Kruskal-Wallis with an α-error of 0.05 (asterisks) and post hoc testing with Dunn’s test ($). **, $$P < 0.01, ***, $$$P < 0.001. See Supplementary Table 2 for detailed statistics. (D and E) HEK293 cells expressing the GluN1 subunit of the NMDAR immunolabelled with acid-extracted IgG fractions (D) and pre-extraction fractions (E) from hippocampus (Hippo), cerebral cortex (Ctx) and cerebellum (Cb) of mice infused with patients’ CSF (Day 18). The acid-extracted IgG fraction from hippocampus showed higher level of NMDAR antibodies than those extracted from cerebral cortex (Ctx) and cerebellum (Cb). Scale bars = 10 µm. n.s = not significant.
Effects of patients’ antibodies on NMDAR
To determine the effects of patients’ antibodies on NMDAR, we focused on the hippocampus, which was the region with maximal concentration of NMDAR-bound antibodies. Compared with animals infused with control CSF, those infused with patients' CSF had on Days 13 and 18 a significant decrease of the density of total and synaptic hippocampal NMDAR clusters followed by a gradual recovery after Day 18 (pooled analysis of CA1, CA3 and dentate gyrus; Fig. 6A–D). No significant differences in between hippocampal subregions (CA1, CA3, dentate gyrus) were observed (not shown). In contrast, patients’ antibodies did not alter the density of PSD95 or AMPAR clusters (Fig. 6E and F).
Figure 6.
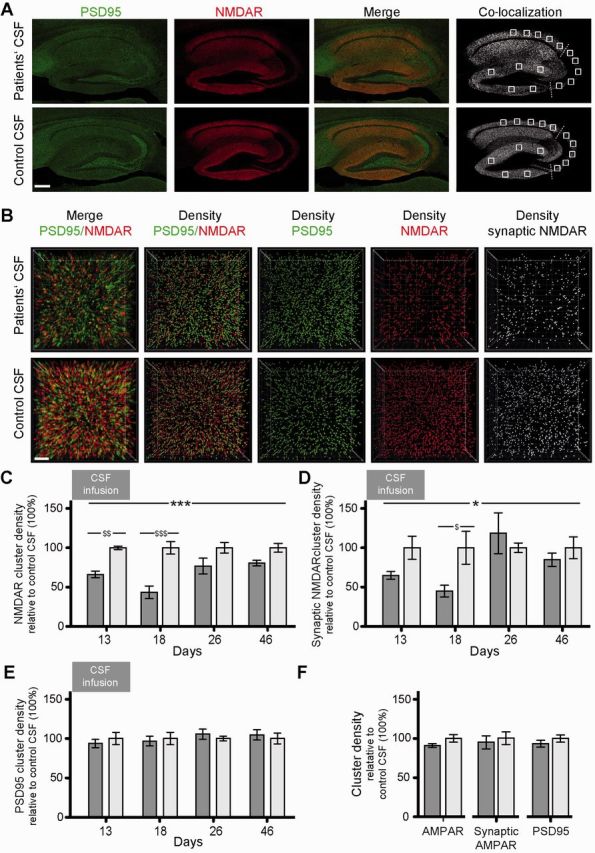
Patients’ NMDAR antibodies selectively reduce the density of total and synaptic NMDAR clusters in hippocampus of mice. (A) Hippocampus of mice infused for 14 days (Day 18) with patients’ CSF (upper row) or control CSF (lower row) immunolabelled for PSD95 and NMDAR. Images were merged (merge) and post-processed to demonstrate co-localizing clusters (co-localization). Squares in ‘co-localization’ indicate the analysed areas in CA1, CA3 and dentate gyrus. Scale bar = 200 µm. (B) Three-dimensional projection and analysis of the density of total clusters of PSD95 and NMDAR, and synaptic clusters of NMDAR (defined as NMDAR clusters colocalizing with PSD95) in a representative CA3 region (square in A ‘co-localization’). Merged images (merge, PSD95 green, NMDAR red) were post-processed and used to calculate the density of clusters (density = spots/µm3). Scale bar = 2 µm. (C–F) Quantification of the density of total (C) and synaptic (D) NMDAR clusters, PSD95 clusters (E), and total/synaptic AMPAR and PSD95 clusters (Day 18 only, F) in a pooled analysis of hippocampal subregions (CA1, CA3, dentate gyrus) in animals treated with patients’ CSF (dark grey) or control CSF (light grey) on the indicated days. Mean density of clusters in control CSF treated animals was defined as 100%. Data are presented as mean ± SEM. For each time point five animals infused with patients’ CSF and five with control CSF were examined. Significance of treatment effect was assessed by two-way ANOVA with an α-error of 0.05 (asterisks) and post hoc testing with Sidak-Holm adjustment ($) (C–E) or unpaired t-test (F). *, $P < 0.05; **, $$P < 0.01; ***, $$$P < 0.001. See Supplementary Table 2 for detailed statistics.
Immunoblot analysis of total protein extracted from hippocampus showed that on Days 13 and 18, mice infused with patients’ CSF had a significant decrease of total NMDAR protein concentration compared with mice infused with control CSF (Fig. 7A and B). The magnitude of this effect was greater in animals with higher concentration of IgG bound to hippocampus (Fig. 7C). Parallel studies examining the effect on the protein concentrations of PSD95 (Fig. 7A and E) and AMPAR (Fig. 7D) demonstrated no significant differences between mice infused with patients’ CSF or control CSF.
Figure 7.

Patients’ NMDAR antibodies selectively reduce the protein concentration of NMDAR in hippocampus of mice. (A) Representative immunoblots of proteins extracted from hippocampus of animals infused with patients’ CSF (P) or control CSF (C) sacrificed at the indicated time points and probed for expression of GluN1 (NMDAR), PSD95 and β-actin (loading control). Note that there is less visible GluN1 expression on Days 13 and 18. (B, D and E) Quantification of total NMDAR (B), AMPAR (D) or PSD95 (E) protein in animals treated with patients’ CSF (filled columns) or control CSF (open columns) sacrificed at the indicated time points (AMPAR Day 18 only, D). Results were normalized to β-actin (loading control). Mean band density of animals treated with control CSF was defined as 100%. Data are presented as mean ± SEM. For each time point six animals infused with patients’ CSF and six with control CSF were examined (for Days 26 and 46, only five animals treated with patient’s CSF were available). Significance of treatment effect was assessed by two-way ANOVA with an α-error of 0.05 (asterisks) and post hoc testing with Sidak-Holm adjustment ($). $$P < 0.01; ***P < 0.001. See Supplementary Table 2 for detailed statistics. (C) Correlation between concentration of human IgG bound to hippocampus (x-axis, highest hippocampal IgG intensity was defined as 100%) and hippocampal NMDAR protein concentration in mice sacrificed on Day 18 (R2 = 0.69, P = 0.003). Filled circles: mice infused with patients’ CSF (n = 5), open circles: mice infused with control CSF (n = 5).
In cerebellum, no significant effects on the cluster density or total protein concentration of NMDAR, PSD95 and AMPAR were noted in animals infused with patients’ CSF compared to those infused with control CSF (data not shown).
Immunohistochemical studies for neuronal apoptosis, infiltrates of T or B cells, and deposits of complement in hippocampus of animals infused with patients’ or control CSF, examined on Day 18, showed no abnormalities (Fig. 8).
Figure 8.

Absence of neuronal apoptosis, deposits of complement, and lymphocytic infiltrates in the hippocampus of mice infused with patients’ CSF. (A and B) TUNEL and cleaved caspase 3 immunolabelling of a representative area of CA3 (area with maximal IgG binding and lower NMDAR concentration) of an animal infused with patients’ CSF, showing lack of apoptotic cells. A section of the same region in an animal with transient middle cerebral artery occlusion (stroke model) shows apoptotic cells in the penumbra (left). (C) Same CA3 region as in (A) immunostained for C5b-9 showing lack of deposit of complement. A section of the same region in the indicated stroke model shows presence of complement in the penumbra (left). (D and E) Same CA3 region as in (A) immunostained for T (CD3) and B (CD45R) lymphocytes showing absence of inflammatory infiltrates. A section of spleen was used as control tissue showing the presence of CD3 (green) and CD45R (red) cells. Scale bar = 10 µm. Total number of animals examined: patients’ CSF n = 5; control CSF n = 5. Scale bars = 20 µm.
Discussion
We report that passive transfer of NMDAR antibodies by continuous ventricular infusion of CSF from patients with anti-NMDAR encephalitis causes memory and behavioural deficits in mice, and that the effects are likely mediated by the binding of antibodies to NMDAR resulting in a specific decrease of the density of these receptors. Data from earlier reports showing that despite the severity and duration of symptoms, most patients with anti-NMDAR encephalitis respond to immunotherapy (Gresa-Arribas et al., 2014), and findings at the cellular level demonstrating that patients’ antibodies cause a titre-dependent decrease of synaptic NMDAR receptors fulfilled most of the Witebsky’s criteria for an antibody-mediated disease (Rose and Bona, 1993), but the transfer of symptoms to animals was pending. In the current study, four sets of experiments satisfy this postulate: (i) the development of symptoms in animals infused with patients’ CSF, but not control CSF; (ii) the demonstration that the infused antibodies reacted predominantly with brain regions with high density of NMDAR (e.g. hippocampus) and specifically recognized these receptors; (iii) the identification of a selective decrease of the density of total and synaptic NMDAR clusters and total NMDAR protein concentration without affecting PSD95, and that these effects correlated with the concentration of brain-bound antibodies; and (iv) the correlation noted between the intensity of the abovementioned findings and time-course of patients’ antibody infusion, as well as between the reversibility of symptoms and restoration of NMDAR levels after stopping the infusion of CSF antibodies.
Approximately 75% of patients with anti-NMDAR encephalitis present with mood and psychiatric alterations ranging from manic or depressive behaviour to psychosis, often followed by stereotyped movements, seizures, or decreased level of consciousness (Kayser et al., 2013; Titulaer et al., 2013). Regardless of the presentation, most patients develop severe problems forming new memories and have amnesia of the disease. Close examination during the phase of recovery shows, in some patients, impairment in the visual recognition of objects or faces (e.g. physicians, nurses) (Frechette et al., 2011). Owing to the wide range of symptoms of the disease and lack of previous studies examining the distribution of brain tissue NMDAR-antibody binding when these antibodies are infused intraventricularly, we used standardized memory and behavioural tests. The most notable effects were observed in the tests of memory (novel object recognition) using different groups of animals in two different paradigms (open field and V-maze). While the first depends predominantly on normal hippocampal function, the second is dependent of perirhinal-hippocampal structures (Winters et al., 2004). Compared with animals infused with control CSF, those infused with patients’ CSF developed progressive memory deficits, which were maximal on Days 13–18 when the highest concentration of brain-bound NMDAR antibodies and lowest density of NMDAR occurred. Other paradigms affected were related to depressive-like behaviours (tail suspension test) and anhedonic behaviours (sucrose preference test). We did not find significant abnormalities in the tests of aggression and anxiety, which are often present in the human disease, or in locomotor activity (an expected finding given that paralysis rarely occurs in patients).
The high levels of brain-bound NMDAR antibodies between Days 13–18 suggests that after stopping the infusion of patients’ CSF on Day 14, the NMDAR antibodies continued being distributed from mice cerebroventricular system to parenchyma. This distribution occurred slowly; for example, 5 days after starting the infusion of patients’ CSF the amount of NMDAR antibodies that had reached the hippocampus was very limited compared to that seen on Days 13–18 (shown in Fig. 4B). Moreover, previous studies using cultured neurons treated with patients’ CSF showed that once the antibodies bound to the NMDARs, the reduction of receptors was microscopically visible in 2 h but it took 12 h to result in the lowest receptor density. Subsequently, there was a steady state of low NMDAR density for as long as the neurons were exposed to patients’ antibodies (Moscato et al., 2014). Together, these findings explain the progressive worsening of symptoms along with continued antibody binding and decrease of NMDAR for at least 4 days after the ventricular infusion stops and the subsequent recovery starts.
Although the hippocampus was the region with the highest concentration of brain-bound NMDAR antibodies, these antibodies were also extracted from cerebral cortex or cerebellum though at much lower levels. The higher concentration of antibodies and predominant decrease of NMDAR in the hippocampus are consistent with the predominant binding of human antibodies to this brain region when sections of rodent brain are directly incubated with patients’ antibodies (Dalmau et al., 2007; Moscato et al., 2014). Additionally, because of the close spatial relationship to the ventricles, the intraventricular infusion of human CSF antibodies might have contributed to the preferential binding to the hippocampus.
The correlation between the concentration of brain-bound antibodies and selective reduction of NMDAR cluster density and protein concentration was similar to that reported using in vitro studies with cultured rat hippocampal neurons (Hughes et al., 2010; Moscato et al., 2014). Moreover, autopsies of patients with anti-NMDAR encephalitis showed that the hippocampal regions with highest concentration of brain-bound antibodies were also the areas with lower expression of NMDAR (Dalmau et al., 2007). In the current model, patients’ antibodies did not alter AMPAR cluster density or protein concentration; these findings are in line with those reported with cultured neurons where the clusters of AMPAR and AMPAR-mediated currents were not directly affected (Hughes et al., 2010). These experiments, however, did not explore whether paradigms that normally induce long-term potentiation, and therefore increase the number of synaptic AMPAR, were altered by patients’ antibodies. Mikasova et al. (2012) showed that neurons exposed to patients’ NMDAR antibodies failed to show an increase in cell surface AMPAR after induction of chemical long-term potentiation. Another study examining the acute metabolic effects of patients’ antibodies after injection into rat brain showed impairment of NMDA and AMPA-mediated synaptic function (Manto et al., 2010). In the present model, we did not perform electrophysiological studies on acute slices of brain (a goal of future studies); however, there is reported evidence that patients’ NMDAR antibodies suppress induction of long-term potentiation when directly applied to mouse hippocampal slices (Zhang et al., 2012). Work with cultured neurons indicates that the decrease of synaptic NMDAR currents is likely a result of the antibody-mediated low receptor levels, as no direct antibody blockade was detected (Moscato et al., 2014).
Our study has limitations related to the type of disease and symptoms to model. For example, different from other models of antibody-mediated CNS disorders where the antibodies result in characteristic symptoms (e.g. amphiphysin antibodies and visible muscle spasms) (Sommer et al., 2005) or focal deficits with visible tissue changes (e.g. AQP4 antibodies and neuromyelitis optica) (Hinson et al., 2012; Bradl and Lassmann, 2014), anti-NMDAR encephalitis results in a broader spectrum of symptoms where memory and behavioural deficits occur early, and the structural alterations are not visible unless the NMDAR clusters or protein concentration are measured. It is not surprising that in the current model the full spectrum of symptoms, such as seizures, dyskinesias or coma, did not occur. Studies with NMDAR antagonists have shown that the progression of symptoms (from behavioural and memory deficits to unresponsiveness with catatonic features and coma) correlated with the intensity of the decrease of receptor function (Javitt and Zukin, 1991). Therefore, it is likely that prolonged infusion or higher concentration of patients’ antibodies would cause additional symptoms. This is supported by the current model, in which the time course of symptom development, brain-bound antibody concentration, and decrease of synaptic NMDAR correlated well with each other. Future experiments using prolonged infusion or higher concentration of patients’ antibodies may also result in symptoms beyond hippocampal-parahippocampal regions. Compared with the hippocampus, other brain regions normally have lower density of NMDAR, and appeared to be less accessible to the ventricularly infused antibodies. Direct injection of antibodies into those brain regions can be considered, but we previously tried bilateral hippocampal infusion using the same osmotic pump approach, resulting in more limited antibody diffusion and no symptoms (data not published). Moreover, the phenotype of the current model is likely influenced by the strain of mice. In this study we used C57BL6/J mice because we were interested in the effects on memory and behaviour, but this strain is one of the most resistant to develop seizures (Ferraro et al., 2002).
The antibody-induced depletion of synaptic NMDAR along with the similarities between the human disease and the phenotypes induced by NMDAR antagonists (phencyclidine, ketamine or MK801) have suggested points of convergence with one of the most influential theories of schizophrenia, the NMDA-hypofunction model (Olney and Farber, 1995; Kehrer et al., 2008). The presence of positive (hallucinations, delusions, hyperactivity) and negative (decreased motivation, flat affect, deficit of memory and learning) symptoms is, however, not identical among the drug-induced phenotypes and also varies among animal species (Javitt and Zukin, 1991). It has been suggested that NMDAR-bearing parvalbumin-positive GABAergic interneurons are disproportionally more sensitive to NMDAR antagonists than other neurons (Li et al., 2002). Interestingly, a genetic model of partial ablation of the GluN1 subunit of NMDAR in corticolimbic GABAergic interneurons resulted in symptoms partially resembling our GluN1 immunological model of receptor depletion, including memory deficits and anhedonic behaviours (Belforte et al., 2010). Differences related to the underlying mechanisms (pharmacologic blockade, genetic or immunologic NMDAR depletion) and regions where the NMDAR function is depleted (general, corticolimbic, or hippocampal-parahippocampal) likely influence the clinical phenotypes.
Overall, the current findings provide robust evidence that antibodies from patients with anti-NMDAR encephalitis alter memory and behaviour through reduction of cell-surface and synaptic NMDAR, and therefore support the use of treatments directed at decreasing the levels of antibodies or antibody-producing cells. This approach can now be adapted to (i) model other aspects of the disease by changing the duration and dosing of antibody infusion, or strain of mice; (ii) investigate other disorders of memory and behaviour that occur in association with antibodies against other cell surface or synaptic proteins, such as AMPAR or GABA(B)R (Lai et al., 2009; Lancaster et al., 2010); and (iii) determine whether compounds such as Ephrin-B2 ligand that has been shown to prevent the destabilizing NMDAR crosslinking effects of patients’ antibodies improve or alter the course of the disease (Mikasova et al., 2012).
Acknowledgements
We thank Anna Planas and Vanessa Brait for providing stroke brain tissue and Jordi Andilla for technical support.
Glossary
Abbreviations
- AMPAR
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor
- NMDAR
N-methyl D-aspartate receptor
- PSD95
post-synaptic density protein 95
Funding
This work was supported by the National Institutes of Health RO1NS077851 (J.D.), RO1MH094741 (R.B-G. and J.D.), Fundació La Marató de TV3 #101530 (J.D.), Fondo de Investigaciones Sanitarias/Instituto Carlos III (FIS PI11/01780 J.D.), ErasmusMC fellowship (M.T.), and Forschungsförderungsfonds Hamburg-Eppendorf FL).
Conflict of interest
Dr Dalmau holds a patent for the use of NMDA receptor as an autoantibody test. Dr Dalmau has received a research grant from Euroimmun Inc.
Supplementary material
Supplementary material is available at Brain online.
References
- Aso E, Ozaita A, Valdizan EM, Ledent C, Pazos A, Maldonado R, et al. BDNF impairment in the hippocampus is related to enhanced despair behavior in CB1 knockout mice. J Neurochem. 2008;105:565–72. doi: 10.1111/j.1471-4159.2007.05149.x. [DOI] [PubMed] [Google Scholar]
- Banovic D, Khorramshahi O, Owald D, Wichmann C, Riedt T, Fouquet W, et al. Drosophila neuroligin 1 promotes growth and postsynaptic differentiation at glutamatergic neuromuscular junctions. Neuron. 2010;66:724–38. doi: 10.1016/j.neuron.2010.05.020. [DOI] [PubMed] [Google Scholar]
- Belforte JE, Zsiros V, Sklar ER, Jiang Z, Yu G, Li Y, et al. Postnatal NMDA receptor ablation in corticolimbic interneurons confers schizophrenia-like phenotypes. Nat Neurosci. 2010;13:76–83. doi: 10.1038/nn.2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berrendero F, Mendizabal V, Robledo P, et al. Nicotine-induced antinociception, rewarding effects, and physical dependence are decreased in mice lacking the preproenkephalin gene. J Neurosci. 2005;25:1103–12. doi: 10.1523/JNEUROSCI.3008-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradl M, Lassmann H. Experimental models of neuromyelitis optica. Brain Pathol. 2014;24:74–82. doi: 10.1111/bpa.12098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bura AS, Guegan T, Zamanillo D, Vela JM, Maldonado R. Operant self-administration of a sigma ligand improves nociceptive and emotional manifestations of neuropathic pain. Eur J Pain. 2013;17:832–43. doi: 10.1002/j.1532-2149.2012.00251.x. [DOI] [PubMed] [Google Scholar]
- Bura SA, Burokas A, Martin-Garcia E, Maldonado R. Effects of chronic nicotine on food intake and anxiety-like behaviour in CB(1) knockout mice. Eur Neuropsychopharmacol. 2010;20:369–78. doi: 10.1016/j.euroneuro.2010.02.003. [DOI] [PubMed] [Google Scholar]
- Bura SA, Castane A, Ledent C, Valverde O, Maldonado R. Genetic and pharmacological approaches to evaluate the interaction between the cannabinoid and cholinergic systems in cognitive processes. Br J Pharmacol. 2007;150:758–65. doi: 10.1038/sj.bjp.0707152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burokas A, Gutierrez-Cuesta J, Martin-Garcia E, Maldonado R. Operant model of frustrated expected reward in mice. Addict Biol. 2012;17:770–82. doi: 10.1111/j.1369-1600.2011.00423.x. [DOI] [PubMed] [Google Scholar]
- Caille S, Espejo EF, Reneric JP, Cador M, Koob GF, Stinus L. Total neurochemical lesion of noradrenergic neurons of the locus ceruleus does not alter either naloxone-precipitated or spontaneous opiate withdrawal nor does it influence ability of clonidine to reverse opiate withdrawal. J Pharmacol Exp Ther. 1999;290:881–92. [PubMed] [Google Scholar]
- Crawley J, Goodwin FK. Preliminary report of a simple animal behavior model for the anxiolytic effects of benzodiazepines. Pharmacol Biochem Behav. 1980;13:167–70. doi: 10.1016/0091-3057(80)90067-2. [DOI] [PubMed] [Google Scholar]
- Dalmau J, Gleichman AJ, Hughes EG, Rossi JE, Peng X, Lai M, et al. Anti-NMDA-receptor encephalitis: case series and analysis of the effects of antibodies. Lancet Neurol. 2008;7:1091–8. doi: 10.1016/S1474-4422(08)70224-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalmau J, Tuzun E, Wu HY, Masjuan J, Rossi JE, Voloschin A, et al. Paraneoplastic anti-N-methyl-D-aspartate receptor encephalitis associated with ovarian teratoma. Ann Neurol. 2007;61:25–36. doi: 10.1002/ana.21050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ennaceur A. One-trial object recognition in rats and mice: methodological and theoretical issues. Behav Brain Res. 2010;215:244–54. doi: 10.1016/j.bbr.2009.12.036. [DOI] [PubMed] [Google Scholar]
- Ferraro TN, Golden GT, Smith GG, DeMuth D, Buono RJ, Berrettini WH. Mouse strain variation in maximal electroshock seizure threshold. Brain Res. 2002;936:82–6. doi: 10.1016/s0006-8993(02)02565-9. [DOI] [PubMed] [Google Scholar]
- Filliol D, Ghozland S, Chluba J, et al. Mice deficient for delta- and mu-opioid receptors exhibit opposing alterations of emotional responses. Nat Genet. 2000;25:195–200. doi: 10.1038/76061. [DOI] [PubMed] [Google Scholar]
- Frechette ES, Zhou L, Galetta SL, Chen L, Dalmau J. Prolonged follow-up and CSF antibody titers in a patient with anti-NMDA receptor encephalitis. Neurology. 2011;76:S64–6. doi: 10.1212/WNL.0b013e31820c34de. [DOI] [PubMed] [Google Scholar]
- Gleichman AJ, Spruce LA, Dalmau J, Seeholzer SH, Lynch DR. Anti-NMDA receptor encephalitis antibody binding is dependent on amino acid identity of a small region within the GluN1 amino terminal domain. J Neurosci. 2012;32:11082–94. doi: 10.1523/JNEUROSCI.0064-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gresa-Arribas N, Titulaer MJ, Torrents A, Aguilar E, McCracken L, Leypoldt F, et al. Antibody titres at diagnosis and during follow-up of anti-NMDA receptor encephalitis: a retrospective study. Lancet Neurol. 2014;13:167–77. doi: 10.1016/S1474-4422(13)70282-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunduz-Bruce H. The acute effects of NMDA antagonism: from the rodent to the human brain. Brain Res Rev. 2009;60:279–86. doi: 10.1016/j.brainresrev.2008.07.006. [DOI] [PubMed] [Google Scholar]
- Hahn CG, Wang HY, Cho DS, Talbot K, Gur RE, Berrettini WH, et al. Altered neuregulin 1-erbB4 signaling contributes to NMDA receptor hypofunction in schizophrenia. Nat Med. 2006;12:824–8. doi: 10.1038/nm1418. [DOI] [PubMed] [Google Scholar]
- Handley SL, Mithani S. Effects of alpha-adrenoceptor agonists and antagonists in a maze-exploration model of ‘fear’-motivated behaviour. Naunyn Schmiedebergs Arch Pharmacol. 1984;327:1–5. doi: 10.1007/BF00504983. [DOI] [PubMed] [Google Scholar]
- Hinson SR, Romero MF, Popescu BF, Lucchinetti CF, Fryer JP, Wolburg H, et al. Molecular outcomes of neuromyelitis optica (NMO)-IgG binding to aquaporin-4 in astrocytes. Proc Natl Acad Sci USA. 2012;109:1245–50. doi: 10.1073/pnas.1109980108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes EG, Peng X, Gleichman AJ, Lai M, Zhou L, Tsou R, et al. Cellular and synaptic mechanisms of anti-NMDA receptor encephalitis. J Neurosci. 2010;30:5866–75. doi: 10.1523/JNEUROSCI.0167-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iizuka T, Sakai F, Ide T, Monzen T, Yoshii S, Iigaya M, et al. Anti-NMDA receptor encephalitis in Japan: long-term outcome without tumor removal. Neurology. 2008;70:504–11. doi: 10.1212/01.wnl.0000278388.90370.c3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irani SR, Bera K, Waters P, et al. N-methyl-D-aspartate antibody encephalitis: temporal progression of clinical and paraclinical observations in a predominantly non-paraneoplastic disorder of both sexes. Brain. 2010;133:1655–67. doi: 10.1093/brain/awq113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javitt DC, Zukin SR. Recent advances in the phencyclidine model of schizophrenia. Am J Psychiatry. 1991;148:1301–8. doi: 10.1176/ajp.148.10.1301. [DOI] [PubMed] [Google Scholar]
- Jentsch JD, Roth RH. The neuropsychopharmacology of phencyclidine: from NMDA receptor hypofunction to the dopamine hypothesis of schizophrenia. Neuropsychopharmacology. 1999;20:201–25. doi: 10.1016/S0893-133X(98)00060-8. [DOI] [PubMed] [Google Scholar]
- Kayser MS, Titulaer MJ, Gresa-Arribas N, Dalmau J. Frequency and characteristics of isolated psychiatric episodes in anti-N-methyl-d-aspartate receptor encephalitis. JAMA Neurol. 2013;70:1133–9. doi: 10.1001/jamaneurol.2013.3216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kehrer C, Maziashvili N, Dugladze T, Gloveli T. Altered excitatory-inhibitory balance in the NMDA-hypofunction model of schizophrenia. Front Mol Neurosci. 2008;1:6. doi: 10.3389/neuro.02.006.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konig M, Zimmer AM, Steiner H, Holmes PV, Crawley JN, Brownstein MJ, et al. Pain responses, anxiety and aggression in mice deficient in pre-proenkephalin. Nature. 1996;383:535–8. doi: 10.1038/383535a0. [DOI] [PubMed] [Google Scholar]
- Lai M, Hughes EG, Peng X, Zhou L, Gleichman AJ, Shu H, et al. AMPA receptor antibodies in limbic encephalitis alter synaptic receptor location. Ann Neurol. 2009;65:424–34. doi: 10.1002/ana.21589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancaster E, Lai M, Peng X, Hughes E, Constantinescu R, Raizer J, et al. Antibodies to the GABA(B) receptor in limbic encephalitis with seizures: case series and characterisation of the antigen. Lancet Neurol. 2010;9:67–76. doi: 10.1016/S1474-4422(09)70324-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau CG, Zukin RS. NMDA receptor trafficking in synaptic plasticity and neuropsychiatric disorders. Nat Rev Neurosci. 2007;8:413–26. doi: 10.1038/nrn2153. [DOI] [PubMed] [Google Scholar]
- Li Q, Clark S, Lewis DV, Wilson WA. NMDA receptor antagonists disinhibit rat posterior cingulate and retrosplenial cortices: a potential mechanism of neurotoxicity. J Neurosci. 2002;22:3070–80. doi: 10.1523/JNEUROSCI.22-08-03070.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llorente-Berzal A, Puighermanal E, Burokas A, Ozaita A, Maldonado R, Marco EM, et al. Sex-dependent psychoneuroendocrine effects of THC and MDMA in an animal model of adolescent drug consumption. PLoS One. 2013;8:e78386. doi: 10.1371/journal.pone.0078386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maldonado JE, Kyle RA, Ludwig J. Meningeal myeloma. Arch Intern Med. 1970;126:660–3. [PubMed] [Google Scholar]
- Manto M, Dalmau J, Didelot A, Rogemond V, Honnorat J. In vivo effects of antibodies from patients with anti-NMDA receptor encephalitis: further evidence of synaptic glutamatergic dysfunction. Orphanet J Rare Dis. 2010;5:31. doi: 10.1186/1750-1172-5-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Hernandez E, Horvath J, Shiloh-Malawsky Y, Sangha N, Martinez-Lage M, Dalmau J. Analysis of complement and plasma cells in the brain of patients with anti-NMDAR encephalitis. Neurology. 2011;77:589–93. doi: 10.1212/WNL.0b013e318228c136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikasova L, De RP, Bouchet D, Georges F, Rogemond V, Didelot A, et al. Disrupted surface cross-talk between NMDA and Ephrin-B2 receptors in anti-NMDA encephalitis. Brain. 2012;135:1606–21. doi: 10.1093/brain/aws092. [DOI] [PubMed] [Google Scholar]
- Mohn AR, Gainetdinov RR, Caron MG, Koller BH. Mice with reduced NMDA receptor expression display behaviors related to schizophrenia. Cell. 1999;98:427–36. doi: 10.1016/s0092-8674(00)81972-8. [DOI] [PubMed] [Google Scholar]
- Moscato EH, Peng X, Jain A, Parsons TD, Dalmau J, Balice-Gordon RJ. Acute mechanisms underlying antibody effects in anti-N-methyl-D-aspartate receptor encephalitis. Ann Neurol. 2014;76:108–90. doi: 10.1002/ana.24195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouri A, Noda Y, Noda A, Nakamura T, Tokura T, Yura Y, et al. Involvement of a dysfunctional dopamine-D1/N-methyl-d-aspartate-NR1 and Ca2+/calmodulin-dependent protein kinase II pathway in the impairment of latent learning in a model of schizophrenia induced by phencyclidine. Mol Pharmacol. 2007;71:1598–609. doi: 10.1124/mol.106.032961. [DOI] [PubMed] [Google Scholar]
- Olney JW, Farber NB. Glutamate receptor dysfunction and schizophrenia. Arch Gen Psychiatry. 1995;52:998–1007. doi: 10.1001/archpsyc.1995.03950240016004. [DOI] [PubMed] [Google Scholar]
- Porsolt RD, Bertin A, Jalfre M. Behavioral despair in mice: a primary screening test for antidepressants. Arch Int Pharmacodyn Ther. 1977;229:327–36. [PubMed] [Google Scholar]
- Puighermanal E, Marsicano G, Busquets-Garcia A, Lutz B, Maldonado R, Ozaita A. Cannabinoid modulation of hippocampal long-term memory is mediated by mTOR signaling. Nat Neurosci. 2009;12:1152–8. doi: 10.1038/nn.2369. [DOI] [PubMed] [Google Scholar]
- Rose NR, Bona C. Defining criteria for autoimmune diseases (Witebsky's postulates revisited) Immunol Today. 1993;14:426–30. doi: 10.1016/0167-5699(93)90244-F. [DOI] [PubMed] [Google Scholar]
- Shepherd JD, Huganir RL. The cell biology of synaptic plasticity: AMPA receptor trafficking. Annu Rev Cell Dev Biol. 2007;23:613–43. doi: 10.1146/annurev.cellbio.23.090506.123516. [DOI] [PubMed] [Google Scholar]
- Snyder EM, Nong Y, Almeida CG, Paul S, Moran T, Choi EY, et al. Regulation of NMDA receptor trafficking by amyloid-beta. Nat Neurosci. 2005;8:1051–8. doi: 10.1038/nn1503. [DOI] [PubMed] [Google Scholar]
- Sommer C, Weishaupt A, Brinkhoff J, Biko L, Wessig C, Gold R, et al. Paraneoplastic stiff-person syndrome: passive transfer to rats by means of IgG antibodies to amphiphysin. Lancet. 2005;365:1406–11. doi: 10.1016/S0140-6736(05)66376-3. [DOI] [PubMed] [Google Scholar]
- Steru L, Chermat R, Thierry B, Simon P. The tail suspension test: a new method for screening antidepressants in mice. Psychopharmacology (Berl) 1985;85:367–70. doi: 10.1007/BF00428203. [DOI] [PubMed] [Google Scholar]
- Strekalova T, Gorenkova N, Schunk E, Dolgov O, Bartsch D. Selective effects of citalopram in a mouse model of stress-induced anhedonia with a control for chronic stress. Behav Pharmacol. 2006;17:271–87. doi: 10.1097/00008877-200605000-00008. [DOI] [PubMed] [Google Scholar]
- Taglialatela G, Hogan D, Zhang WR, Dineley KT. Intermediate- and long-term recognition memory deficits in Tg2576 mice are reversed with acute calcineurin inhibition. Behav Brain Res. 2009;200:95–9. doi: 10.1016/j.bbr.2008.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Titulaer MJ, McCracken L, Gabilondo I, Armangué T, Glaser C, Iizuka T, et al. Treatment and prognostic factors for long-term outcome in patients with anti-NMDA receptor encephalitis: an observational cohort study. Lancet Neurol. 2013;12:157–65. doi: 10.1016/S1474-4422(12)70310-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viaccoz A, Desestret V, Ducray F, Picard G, Cavillon G, Rogemond V, et al. Clinical specificities of adult male patients with NMDA receptor antibodies encephalitis. Neurology. 2014;82:556–63. doi: 10.1212/WNL.0000000000000126. [DOI] [PubMed] [Google Scholar]
- Weiner AL, Vieira L, McKay CA, Bayer MJ. Ketamine abusers presenting to the emergency department: a case series. J Emerg Med. 2000;18:447–51. doi: 10.1016/s0736-4679(00)00162-1. [DOI] [PubMed] [Google Scholar]
- Winters BD, Forwood SE, Cowell RA, Saksida LM, Bussey TJ. Double dissociation between the effects of peri-postrhinal cortex and hippocampal lesions on tests of object recognition and spatial memory: heterogeneity of function within the temporal lobe. J Neurosci. 2004;24:5901–8. doi: 10.1523/JNEUROSCI.1346-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Tanaka K, Sun P, Nakata M, Yamamoto R, Sakimura K, et al. Suppression of synaptic plasticity by cerebrospinal fluid from anti-NMDA receptor encephalitis patients. Neurobiol Dis. 2012;45:610–15. doi: 10.1016/j.nbd.2011.09.019. [DOI] [PubMed] [Google Scholar]