

Abstract
Fitness is a central quantity in evolutionary models of viruses. However, it remains difficult to determine viral fitness experimentally, and existing in vitro assays can be poor predictors of in vivo fitness of viral populations within their hosts. Next-generation sequencing can nowadays provide snapshots of evolving virus populations, and these data offer new opportunities for inferring viral fitness. Using the equilibrium distribution of the quasispecies model, an established model of intrahost viral evolution, we linked fitness parameters to the composition of the virus population, which can be estimated by next-generation sequencing. For inference, we developed a Bayesian Markov chain Monte Carlo method to sample from the posterior distribution of fitness values. The sampler can overcome situations where no maximum-likelihood estimator exists, and it can adaptively learn the posterior distribution of highly correlated fitness landscapes without prior knowledge of their shape. We tested our approach on simulated data and applied it to clinical human immunodeficiency virus 1 samples to estimate their fitness landscapes in vivo. The posterior fitness distributions allowed for differentiating viral haplotypes from each other, for determining neutral haplotype networks, in which no haplotype is more or less credibly fit than any other, and for detecting epistasis in fitness landscapes. Our implemented approach, called QuasiFit, is available at http://www.cbg.ethz.ch/software/quasifit.
Keywords: quasi-species, fitness landscapes, epistasis, Bayesian inference, next-generation sequencing
FITNESS is a central quantity in evolutionary biology. It can be regarded as a measure of reproductive capacity of each individual. In evolving populations, individuals with higher fitness can outcompete those with lower fitness. Fitness depends on the genetic composition of the individual’s haplotype, i.e., the allelic constellation of multiple loci of its genome, and on the environment, i.e., the host conditions for viral reproduction. Determining the fitness of viruses in a population is experimentally difficult and laborious, because individual virus particles need to be isolated and analyzed separately. In vitro determination of viral fitness usually involves enzymatic, growth competition, or monoinfection assays (Quiñones-Mateu and Arts 2002). A drawback to all in vitro measurements of viral fitness is the removal of viruses from the environment to which they have adapted. Such estimates disregard effects on the fitness landscape deriving from the natural in vivo environment.
RNA viruses, such as human immunodeficiency virus (HIV), have very high mutation rates (Rezende and Prasad 2004), and the number of haplotypes that arise in the normal course of intrahost evolution can be extremely large (Steinhauer and Holland 1987). Thus, HIV populations change and explore sequence space on timescales that are much shorter than those of higher eukaryotes. As such, RNA viruses lend themselves to being studied as model systems of evolutionary theory. In clinical settings, viral fitness is also of great interest. Disease progression, the formation of escape mutants, and ultimately treatment failure depend on viral fitness (Clavel and Hance 2004; Beerenwinkel et al. 2013).
The frequencies of viral haplotypes are determined by evolutionary parameters, including mutation rate and fitness. With the advent and exponential decrease in cost of next-generation sequencing (NGS) data (Niedringhaus et al. 2011), the technical prerequisites for affordable in-depth personalized diagnostics are within reach. Acevedo et al. (2014) have shown that inference of marginal fitness effects of single nucleotide variants in viruses with NGS is possible already today. High-quality NGS data for intrahost viral populations will become ubiquitous in the near future, making in vivo fitness analysis on the basis of such data possible.
A fitness landscape is the association of a real, nonnegative fitness value to each haplotype. Fitness landscapes can be perfectly correlated, “Mount Fuji”-like with strong correlation between the fitnesses of closely related haplotypes or, on the other end of the spectrum, extremely rugged and spiky (“house of cards”), with no correlation of fitness values between related haplotypes (Gavrilets 2004). The effect of multiple alleles acting in concert to confer a fitness unexpected from the individual alleles is termed epistasis. The key factor for the ruggedness of a fitness landscape is the degree of epistasis involved in shaping it.
Several computational methods have been proposed for predicting in vitro fitness from viral sequence (Segal et al. 2004; Deforche et al. 2008; Ma et al. 2010; Hinkley et al. 2011; Ferguson et al. 2013) and for analyzing the structure of HIV in vitro fitness landscapes (Beerenwinkel et al. 2007a,b; Kouyos et al. 2012). For example, Hinkley et al. (2011) have performed large-scale in vitro fitness estimation of the HIV-1 protease and reverse transcriptase in the absence of drugs as well as in the presence of 15 antiretroviral drugs. However, neither this nor any other published study considers intrahost viral genetic diversity and hence none can account for in vivo fitness effects deriving from the host environment.
To estimate in vivo fitness landscapes and without direct observation of the growth kinetics of the viral population, an evolutionary model is required that links fitness to haplotype frequencies. Here we employ, for this purpose, the quasispecies model, an established model of intrahost viral evolution (Eigen and Schuster 1977; Burch and Chao 2000; Vignuzzi et al. 2005; Metzner et al. 2009; Domingo et al. 2012), which is mathematically tractable. One of the predictions of quasispecies theory that has stood the test of time is the existence of an error threshold in viral replication. If the mutation rate of a virus lies above this critical threshold, then mutation will cause genetic information to be lost. Anderson et al. (2004) have shown this phenomenon to exist in practice with the use of mutagenic nucleosides.
In this article, we establish a computational framework for estimating fitness from NGS data based on quasispecies theory. The Markov chain Monte Carlo (MCMC) sampler developed here infers the posterior distribution of fitness landscapes given NGS count data obtained from mixed intrahost virus populations (Figure 1). This inference scheme makes use of cross-sectional data, which are common in clinical settings, and where time series data are scarce.
Figure 1.

Schematic illustration of fitness landscapes and quasispecies. On the left is a fitness landscape on a simple biallelic two-locus genome. Here, 0 indicates a wild-type allele and 1 indicates a mutant allele and the vertical axis indicates fitness f. The fitness landscape also includes epistasis, as the fitness of the double mutant is not additive in the main fitness effects of the two mutant alleles. For this fitness landscape, at equilibrium, the quasispecies equation yields the quasispecies, i.e., the mutation–selection equilibrium (right-hand side). The vertical bars indicate the relative frequency p of each haplotype. In practice, these frequencies are not known but can be estimated from next-generation sequencing data. The stacked horizontal lines represent reads from a sequencing experiment and amount to a finite sample of the quasispecies. Solid circles and crosses indicate mutant alleles at the two loci. Due to the sampling variance inherent in the finite sample, the number of reads will not match the frequencies exactly. Note that fitness values for the haplotypes need not show a strong linear correlation with the haplotype frequencies, as mutational coupling can obscure this relationship. Given a finite sample of the population, we aim to infer properties of the fitness landscape (right-to-left arrow).
Methods
The quasispecies model
The quasispecies model describes the evolution of an infinite population of DNA (or RNA) sequences (Eigen and Schuster 1977). We define the DNA alphabet and DNA sequence space of a genomic region of length L as the Cartesian product The elements of are synonymously referred to as viral haplotypes, genotypes, or strains. They are indexed by
The quasispecies equation is a first-order coupled nonlinear differential equation describing the temporal dynamics of a population subject to mutation and selection. Selection results from increased replication due to higher fitness, and coupling between haplotypes is maintained by mutation. A quasispecies is a cloud of closely connected haplotypes that evolve according to
| (1) |
where pi(t) denotes the relative frequency at time t ≥ 0, fi the fitness of haplotype i, and qji the probability of haplotype j mutating into haplotype i upon replication. The term pj(t) fjqji in the first sum denotes the flux, i.e., the approximate amount of haplotype j mutating into haplotype i per unit time. The second sum is a normalization constant and ensures that the frequencies of all haplotypes sum to 1 for all time points. The fitness landscape is static and does not change with time. In matrix notation, the quasispecies Equation 1 becomes
| (2) |
with the (m − 1)-dimensional probability simplex, average fitness φ(p(t), f) = p ⋅ f, and mutation probability matrix Q = (qij). In the literature, the mutation–selection matrix QTdiag(f) is often denoted W (Eigen et al. 1988; Wilke 2005).
Equilibrium distribution:
The equilibrium distribution p* of the quasispecies Equation 2 is well known (Eigen et al. 1988). It is obtained by setting such that
| (3) |
If Q has only positive entries, then every haplotype has a nonzero probability of mutating into any other haplotype, and the transition matrix Q is irreducible and a Perron matrix. If all fitness values are also positive, then the matrix QTdiag(f), which is a column-wise reweighting of QT, is still a Perron matrix. As a consequence of the Perron–Frobenius theorem, there exists a unique real eigenvalue φ larger than the absolute value of the real part of any other eigenvalue. To determine the equilibrium distribution (Equation 3), we first calculate the largest real eigenvalue φ of QTdiag(f) and its associated eigenvector possessing only positive components. Normalizing this eigenvector by dividing it by the sum of its components yields the global equilibrium distribution of the quasispecies equation (Eigen et al. 1988; Nowak and May 2000).
The mutation–selection equilibrium p* is referred to as the quasispecies in quasispecies theory. The quasispecies model can be formulated for finite populations, where it is very similar to the Wright–Fisher model (Park et al. 2010; Musso 2012). When the effective population size Ne and the mutation rate μ are such that Neμ > 1, then the quasispecies is closely related to classical mutation–selection equilibrium from population genetics as exemplified in Wright’s equation (Wilke 2005).
The single globally stable mutation–selection equilibrium is one appealing feature of the quasispecies equation, making it more tractable than other models of evolution. For fixed Q, we denote by the set of all stationary distributions p* arising under the quasispecies model for positive fitness landscapes
Haplotype space and mutation probabilities:
Working with the full combinatorial DNA sequence space is infeasible, because its dimension grows exponentially in L. To employ this model for real data, we work on a reduced haplotype subset that is sufficiently small to allow for computational analysis but large enough to account for HIV’s large heterogeneity. In practice, the haplotype space ℋ contains all haplotypes observed in the sequencing data, plus additional unobserved ones, such that it is sufficiently connected, as detailed below. Working on a reduced haplotype space also reflects biological reality, where most DNA sequences of length L do not encode viable viruses and hence are extremely unlikely to arise in the course of HIV evolution and can safely be ignored.
To define the mutation probabilities (supporting information, File S1, section 1), we assume an identical per-site mutation probability μ > 0. This constant reflects the fidelity of reverse transcription and is ∼3 × 10−5 per replication for HIV. We denote by d(i, j) the Hamming distance between haplotypes i and j, i.e., the number of loci at which they differ. For a haplotype subset ℋ of cardinality n, we define Q = (qij) by setting
| (4) |
for all i ≠ j, and for all i = 1, … , n. In the case that the uniform mutation probabilities are considered too restricted, a more general two-rate model can be used for setting up the mutation matrix Q (File S1, section 1A).
Either way, the matrix Q is positive, symmetric, and stochastic. Furthermore, as Q is strictly diagonally dominant, it is also regular due to the Levy–Desplanques theorem (Horn and Johnson 1985). We fix Q for the rest of this article and focus on estimating the fitness landscape. In this setting, the quasispecies model is identifiable. By contrast, it has been shown that even with time series data, Q and f jointly are structurally nonidentifiable, because the quasispecies equation is overparameterized (Falugi and Giarré 2009).
While Q is mathematically irreducible, i.e., every haplotype can mutate into every other haplotype with positive probability, technical precision limits imposed by machine precision can lead to situations where Q is not numerically irreducible any longer. Let be the undirected graph with vertices ℋ and edges whenever For a given k that depends on machine precision and on the mutation rate μ, Q will be numerically irreducible if the haplotype graph Gk is connected, i.e., if a path exists between any pair of haplotypes. For HIV, we require k ≤ 1 when using standard precision and k ≤ 2 when using quadruple precision, for three reasons. First, biologically, any three mutations in one replication cycle occur with a probability of ∼10−15, which is orders of magnitude lower than the inverse of the number of virions. Second, the approximate transition rate of 10−15 is numerically bordering on machine ε = 2.22 × 10−16, leaving little overhead for precise calculations (see File S1, section 5A). Finally, most haplotypes will be generated by mutation from haplotypes closely related to them, such that in the asymptotic limit of an infinitely large population size, the great majority of the influx by mutation will still originate from very closely related haplotypes.
In practice, we construct ℋ from observed data as follows (see Figure 2 for an illustration). For a given k (1 in the case of standard precision), we first construct Gk from the observed haplotypes and determine its connected components. If the number of connected components is >1, we proceed to augment ℋ by including additional unobserved haplotypes to increase connectivity. We iterate over all pairs of connected components and determine the haplotype in each connected component closest to the haplotypes in the other connected component. We draw an edge between these two haplotypes with weight equal to their Hamming distance. We then build the minimum weight spanning tree between connected components. Finally, we replace all of the edges of the minimum weight spanning tree by linear chains of k mutational steps by inserting unobserved haplotypes. The stability of this procedure is detailed in File S1, section 5B.
Figure 2.

Illustration of the procedure to make the haplotype graph G1 connected. Solid circles indicate observed haplotypes from sequencing in sequence space. Observed haplotypes are grouped in three connected components denoted with uppercase letters in boldface type, A, B, and C. The shortest mutational path between any pair of connected components is indicated with lines and labeled with lowercase italic letters a, b, and c. Blue solid circles indicate those haplotypes forming the endpoints of the shortest path between any pair of connected components. The aim is to make the whole graph connected while inserting the least number of unobserved haplotypes. In this case, the two paths a and b require inserting only two unobserved haplotypes each, while path c requires inserting eight haplotypes and is not used. The edges a and b form the minimum spanning tree of connected components and the haplotypes representing the solid circles are inserted.
Fitness landscape space:
The quasispecies equation describes the dynamics of an evolving population of haplotypes, but in clinical practice, time series data are difficult and expensive to produce and thus scarce. Hence, we apply the model to cross-sectional data by analyzing the quasispecies in mutation–selection equilibrium (Equation 3). With this assumption, we cannot determine the timescale of approaching the equilibrium, which is reflected in the magnitude of the fitness landscape f. We therefore constrain the average fitness to φ = 1, removing 1 d.f. The constraint fitness space is denoted
| (5) |
We can now ask, for a given equilibrium distribution p, what is the corresponding fitness landscape f? This amounts to solving Equation 3 for f. The solution defines the mapping
| (6) |
where is the quasispecies space defined above (File S1, section 2B). The mapping h is a bijection with inverse denoted by (Figure 3; File S1, sections 2 and 2A). This property is critical, as it allows for estimating fitness parameters in from haplotype frequencies in
Figure 3.
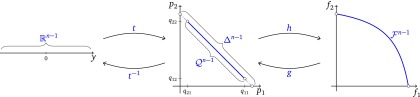
Sample space (left), quasispecies space (center), and fitness space (right). Sample space is mapped to the population distribution space by t with inverse The quasispecies space is mapped to fitness space by h with inverse g. Note the lower and upper bounds of the marginal relative frequencies in the two-haplotype model q21 < p1 < q11 and q12 < p2 < q22.
Inference of the fitness landscape
To estimate fitness in the equilibrium quasispecies model, we require a sample of the viral population. The data vector records the count Xi of each haplotype i among reads sampled in total. Classical methods such as limiting dilution assays with Sanger sequencing can be used to produce such data. Nowadays, NGS is far less laborious and produces data with ever increasing depth. As NGS data are generally noisy, i.e., they include erroneously incorporated bases with respect to the true template, several methods have been developed to address this problem. Probabilistic and combinatorial approaches can be used for preprocessing raw NGS data to reduce errors and to infer the composition of the viral population X (Beerenwinkel et al. 2012).
If we assume that haplotypes have been inferred from raw sequencing data, then the read counts from one NGS experiment represent a sample from the multinomial distribution Mult(X|p). Furthermore, if we assume p = g(f) as the quasispecies equilibrium distribution, this narrows the parameter space. Whereas usually Δn−1 is the parameter space of the multinomial distribution, we have to work with quasispecies space as only elements from this set can represent true underlying quasispecies distributions. For certain data sets where a maximum-likelihood estimator will not exist, because is an open set and is therefore not compact. Such situations can arise, for example, when Xi = 0 for some haplotype This property makes resampling techniques like bootstrapping intractable for realistic data sets due to the increasingly large number of bootstrap samples not having a maximum-likelihood estimator (MLE).
We address this statistical problem in a Bayesian fashion. This approach not only gives us the full posterior p(f|X) but also circumvents the aforementioned shortcomings of a likelihood-based approach. We employ a noninformative, maximum-entropy uniform prior on by setting It remains to compute the posterior distribution
| (7) |
where P(X|g(f)) is the multinomial likelihood. As with most practical Bayesian inference problems, the posterior distribution cannot be derived in closed form. This is due to the eigenvalue constraint in Equation 5, which is a nonlinear constraint and thus yields the nonlinear bounded space
MCMC sampler:
We have developed a Metropolis–Hastings MCMC sampler that can draw samples from this posterior distribution. This task is daunting, because working directly on the fitness space would require knowledge of the neighborhood structure of this nonlinear space. Furthermore, working on is also difficult as, in general, its boundary is not analytically known. Finally, the most common distributions on the simplex, such as the Dirichlet distribution, have strong conditional independence properties that do not allow for flexible covariance structures (Aitchison and Shen 1980).
We use as our sampling space and map samples to the fitness manifold via using the composed mapping h ˚ t, where is the logistic transformation defined by for i = 1, … , n − 1, and (Figure 3; File S1, section 4). The approach of mapping samples from a simpler space to a manifold has been demonstrated by Diaconis et al. (2013). Working in Euclidean space is much easier and we have more distributions at our disposal for constructing proposal distributions. For the functional form of the posterior we have, up to a normalization constant,
| (8) |
where d(y) = |det(J[h ˚ t])| and J denotes the Jacobian (see File S1, section 3).
One widely characterized class of fitness landscapes is Stuart Kauffman’s LK fitness landscapes (Kauffman and Weinberger 1989), where L denotes the number of genomic loci and K denotes the number of interacting loci of each locus. Campos et al. (2002) have shown that the only LK fitness landscapes that lack any correlation between closely related haplotypes are the house of cards fitness landscapes, i.e., fitness landscapes where all genes interact with all other genes concurrently. Thus, it is natural to assume at least some degree of correlation inherent in real fitness landscapes.
Capturing any potential correlation present in fitness landscapes is key to an efficient Bayesian estimation of the posterior. To this end, we employ a differential evolution MCMC as a sampling algorithm. This sampler is of the globally adaptive type and can estimate the covariance structure efficiently. It has the advantage of requiring no a priori specification of the posterior’s covariance structure, which is generally not known. Differential evolution MCMC (Ter Braak 2006) is very similar in nature to parallel tempering (Earl and Deem 2005) for estimating equilibrium distributions of energy states. The basic idea is that the difference of two chains of the current population optimally captures the correlation structure. An advantage of our implementation over other globally adaptive MCMC schemes is the ease of parallelizing this otherwise iterative procedure. Furthermore, due to crossover of chains, the inference scheme involves minimal overhead computation.
In practice, for an n-dimensional problem, we run ∼2n independent chains. At every generation, we cycle through all chains and update the current chain by generating a new proposal. For this, we calculate the proposal for chain i with where R1 ≠ R2 denote two randomly chosen indexes of vectors of chains other than i in each trial, and is responsible for detailed balance to hold. This proposal sample is retained only if all its components are positive by inspection of Equation 6. If the sample has not been rejected in the previous step, then we calculate the log posterior (Equation 8) and determine by the usual MCMC acceptance probability min{1, p(yp|X)/p(yi|X)} whether to accept this sample as a draw from the posterior. We start the parallel chains at the MLE if it exists; otherwise we start near the boundary where the MLE would be if the parameter space was closed.
Implementation
We implemented our MCMC inference scheme, called QuasiFit, in C++. For the linear algebra we employed the Eigen suite (Guennebaud and Jacob 2010), which is a flexible framework for calculating Lower Upper (LU) decompositions that are crucial for matrix inversion and for calculating the determinant of a matrix.
Convergence of all MCMC runs was assessed with the coda package in R (Plummer et al. 2006). For the clinical data sets, we plotted the Gelman and Rubin scale reduction factor vs. trial number and the autocorrelation, and we tested for equality of distribution in the purported stationary distribution samples (File S1, section 6).
The major computational bottleneck in our inference scheme is the calculation of the determinant log d(y) in each step. The computational complexity is therefore for every trial due to the LU decomposition. In total this makes for a complexity of We conducted simulations to investigate the required central processing unit (CPU) time per MCMC trial (File S1, section 9). In practice, the asymptotic regime is reached for n > 64. If all considered haplotypes have at least one observation, i.e., Xi > 0 for all then we have the best-case decline in efficiency of in Metropolis–Hastings schemes (Hanson and Cunningham 1998).
The major factor for convergence, well-mixing and statistical efficiency is determined by the number of haplotypes with no observations (Xi = 0). In practice, this condition is identical to asking for the existence of a MLE. In the case where some Xi = 0, an MLE does not exist. This in turn will lead to a situation where the posterior on will be located on the boundary. The more haplotypes are considered with Xi = 0, the longer it will take for the initial burn-in phase to approach the boundary and the less efficient the overall procedure becomes. Furthermore, once in the stationary distribution at the boundary, with increasing number of haplotypes without observations an increasing number of rejections in the Metropolis–Hastings schemes result not from the general curse of dimensionality of MCMC schemes but from proposal samples that are not elements of In practice, data sets of up to 300 haplotypes can currently be analyzed on a 48-core system.
Results
Simulation studies
As there are no known in vivo fitness values of viruses from the same host, we resorted to simulations to assess the goodness of our fitness landscape estimates.
Five-haplotype simulation model:
We first devised a small five-haplotype example to illustrate the intricate relationship between haplotype fitness and frequency. Figure 4 shows the haplotype network G1 and parameters used for the simulation. We drew multinomial samples with different read coverages N ∈ {200, 2000, 20000} and applied our MCMC approach to obtain the marginal posterior fitness distributions shown in Figure 5. As coverage N increases, so does the confidence in the estimated fitness values, which manifests itself in smaller credibility intervals. The most fit haplotype is only the second least frequent and the haplotype with the highest frequency is the second least fit. This weak correlation between the ranks of relative abundances and fitness is an important consequence of mutational coupling. It highlights the potential pitfalls of simply relying on abundance as a measure of fitness.
Figure 4.

Five-haplotype simulation model. The haplotype graph G1, where haplotypes can mutate into all other haplotypes taking only steps of one mutation at a time, is a chain. The true fitness ranks are indicated by the size of the nodes and the equilibrium relative abundance by the height of the vertical line above each haplotype node.
Figure 5.
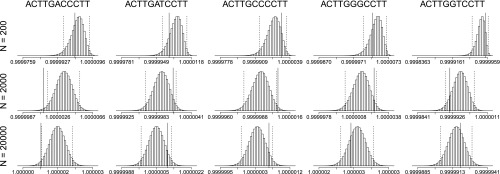
Posterior fitness distributions of each of the five haplotypes in the simulations. Each histogram displays the marginal posterior distributions of a simulation run with a particular coverage N indicated to the left of each row. The true parameter fi of each haplotype from Figure 4 is indicated by a solid vertical line. The 95% highest posterior density intervals are demarcated with dashed vertical lines. The true parameter is not included in the 95% highest posterior density intervals in some cases, as is to be expected from a finite sample.
LK fitness landscape simulations:
To validate more realistic fitness landscapes, where n is at least on the order of the expected number of viral haplotypes in patient samples, as presented below, we employed the LK model for simulating fitness landscapes. Stuart Kauffman’s LK model (originally called the NK model) is a widely used scheme for generating random fitness landscapes of tunable ruggedness (Kauffman and Weinberger 1989; Szendro et al. 2013).
The LK model is defined such that L determines the total number of loci and K < L determines the number of other loci affecting the fitness of any one allele at some locus. Let be a DNA sequence of length L and be the interaction structure of locus i, i.e., the K other loci affecting locus i. The fitness landscape is then
| (9) |
where bi(⋅) denotes the fitness contribution of allele ai. To generate a random fitness landscape according to the LK model with given L and K, we proceed by first generating random interaction sites. For every locus i, we randomly select K elements from {1, …, i − 1, i + 1, …, N} (without replacement) as the loci ei on which the fitness at locus i depends. Second, we generate the mapping by randomly sampling values from where the parameters η and v determine the mean and, respectively, the standard deviation of the log-normal distribution. We set η = 10−2 and v = 2 × 10−6 to produce fitness values with small differences between each other such that the quasispecies shows significant diversity in equilibrium.
For every pair of L ∈ {2, 3, 4} and K ∈ {0, …, L − 1}, we generated random fitness landscapes and calculated the stationary distribution, sampled one multinomial sample with simulated read coverage of N = 100,000, and repeated this procedure until we had 100 samples possessing a fitness MLE. It should be emphasized here that our inference scheme does not require an MLE (File S1, section 5B). Requiring all samples to have an MLE was solely done to facilitate convergence for high-dimensional haplotype sets with L = 4. Finally, we ran QuasiFit on all multinomial samples and took the mean of the posterior as an estimator of the underlying fitness landscape.
To compare our model-based predictions to those of merely using the ranks of the estimated frequencies as a proxy for the ranks of the fitness landscape, we used Kendall’s τ as a measure of agreement in the ranks of different methods of estimating fitness landscapes (Figure 6). The case K = 0 represents fitness landscapes possessing only main/additive effects; i.e., there is no epistasis and as such we can envision a Mount Fuji-like fitness landscape, where mutations will cause the population to ultimately climb to the maximum fitness, as there is only one local optimum that is also the global optimum. The ranks of the fitness landscape and its equilibrium population distribution closely agree in this case (Figure 6). For K > 0, our fitness landscape estimates recover the ranks of the true fitness landscape significantly better than the naive count-based estimator as our model accounts for mutational neighborhood structure (Figure 6).
Figure 6.
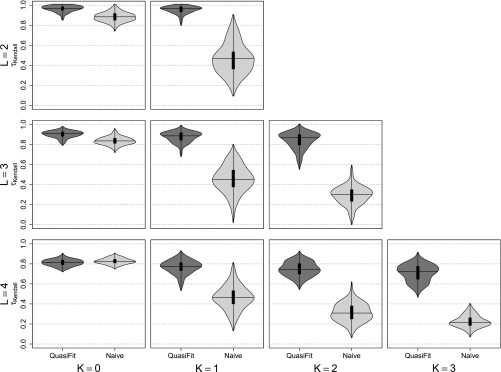
Rank correlation coefficient τKendall for different L and K. The rows depict results for increasingly high-dimensional DNA spaces, where n = 4L denotes the number of haplotypes. The columns depict the density estimators for the rank correlation coefficients between an estimator and the true fitness landscape with increasing K. Densities with dark shading represent τKendall for the QuasiFit-based estimator and densities with light shading represent τKendall for the count-based estimator.
Next, we assessed the ability of our model to estimate the rank fitness landscape as a function of the magnitude of epistatic relative to additive effects. We decomposed the LK fitness landscape model into its parametric interaction terms and simulated random fitness landscapes with prescribed epistatic strengths, using interactions of order at most K + 1. We found that our model becomes superior to the naive ranking method as soon as epistatic effects are on the order of 10% of the additive effects, which is well within a biologically plausible range of epistatic effects in HIV (File S1, section 5E).
Robustness of mutation parameter μ:
To assess the robustness of our inference scheme with respect to the presumed mutation rate, we performed a large-scale analysis over a range of mutation rates, 10−6 ≤ μ ≤ 10−3. To be realistic in view of the clinical data and rudimentary current knowledge of fitness landscapes, we simulated one fitness landscape on a DNA space of L = 3 (64 haplotypes) and with some epistasis K = 1, using the procedure outlined in the previous section. The choice of the inclusion of first-order epistatic interactions is motivated by the analysis in Hinkley et al. (2011), where such pairwise interactions (equivalent to K = 1) have been found to be an important feature of HIV-1 fitness landscapes.
We generated the fitness landscape with parameters where η = 10−2 and v = 5 × 10−7. The latter parameter is one-fourth of the corresponding v in the previous LK simulations such that the average selective advantages produced in the fitness landscape are not much larger than the mutation rate at the lower bound of 10−6; otherwise the coupling between haplotypes is too weak and no diversity will be present at equilibrium. We have iterated over 100 log-uniformly spaced μ-values in the interval [10−6, 10−3]. For each value of μ, we calculated the equilibrium distribution given our fitness landscape and the mutation matrix (Equation 4). For each equilibrium distribution, we simulated a read coverage of N = 100,000 by drawing from a multinomial distribution with p = g(f) and then applying our sampler with fixed μ = 3 × 10−5. We sampled a total of Ntrials = 43.2 × 106 with 144 chains and a thinning interval of 100, giving us 432,000 samples after each run for every μ. We calculated the mean marginal fitnesses of the last 100,000 samples and determined the rank correlation coefficient τKendall with respect to the initially fixed true fitness landscape for the QuasiFit-based estimator and the naive count-based estimator. The actual mutation rates vs. τKendall for the back-inference and the naive estimator are shown in Figure 7.
Figure 7.

Rank correlation coefficient τKendall between the true fitness landscape and one of the two estimators plotted against different actual simulated mutation rates. The dotted vertical line marks the mutation rate that we assume in our model.
We found our model to be very robust within half an order of magnitude below and above our presumed mutation rate of μ = 3 × 10−5. Our estimates also reproduce the ranks of the true fitness landscape better than the naive estimator over a wide range of μ-values. In general, reproducing a perfect agreement (i.e., τ = 1) between ranks of the true and reinferred fitness landscapes is not possible, due to the introduced sampling variance of the finite multinomial draw for each μ.
We also conducted simulations to assess the robustness toward violations of the transition/transversion rate κ (File S1, section 5D). These simulations suggest that our inference scheme still predicts better ranks of the fitness landscape when κ shows large deviations from 1.
Sensitivity analysis:
In addition, we performed a sensitivity analysis that shows our method to be significantly better at recovering the ranks of the underlying fitness landscape up to 500 time units away from equilibrium (File S1, section 5C).
Fitness landscapes of clinical p7 quasispecies
To apply our model to clinical data, we selected two patients from the Swiss HIV Cohort Study (Schoeni-Affolter et al. 2010). We analyzed parts of the spacer peptide 1 (p2) and the nucleocapsid protein (p7) comprising a total of 207 bases or 69 amino acids. We aligned the 2 × 250-bp Illumina MiSeq reads with bwa (Li and Durbin 2010) to the reference sequence HXB2 and focused on the p2–p7 reading frame. The compositions of the viral populations had been inferred with the probabilistic viral haplotype reconstruction tool QuasiRecomb (Töpfer et al. 2013). In both cases, QuasiRecomb was employed only for error correction, as the raw reads contain too many errors, but read assembly was not necessary on these short segments. After inference, the observed quasispecies distribution for patient 1 and patient 2 consisted of n1 = 86 and, respectively, n2 = 123 haplotypes. Both data sets possess a connected haplotype graph G1, such that no further unobserved haplotypes needed to be included.
We determined the fitness landscape of the quasispecies by running QuasiFit on the estimated composition of the viral population. Default parameters were used. In total, 288 chains were run in parallel, with a total of 500,000 trials per chain for both patient samples. The burn-in phase for both MCMC processes was ∼30,000 and 60,000 (Figure 8) and every chain was thinned by retaining every 1150th sample (File S1, Figure S6 and section 6). In Figure 8, the observed drop from the initial value is due to the general curse of dimensionality of sampling in high dimensions when starting at the single highest point of the posterior.
Figure 8.
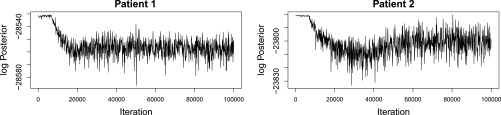
Log-posterior of the first 100,000 MCMC trials. The x-axis denotes the trial number and the y-axis denotes the logarithm of the posterior distribution, Equation 8.
We determined neutral haplotype networks by 95% highest posterior density (HPD) intervals of marginal fitness differences. A 95% HPD region is the smallest region that has 95% probability mass. We determine these for the marginals of the posterior by minimizing over all 95% credibility intervals. Neutrality between two haplotypes is called when 0 is an element of the 95% HPD of fitness differences between two such haplotypes. On the other hand, a ranking of haplotypes by fitness can be established when there is a credible difference in fitnesses, i.e., when 0 is not an element of the pairwise fitness difference (Figure 9; haplotype sequences in File S1, section 7). Visualizing fitness landscapes by drawing a directed edge between haplotypes of differing fitness is a popular and intuitive way of visualizing these high-dimensional mathematical objects (Crona et al. 2013). Both graphs are transitively reduced; i.e., all directed paths between haplotypes represent credible fitness differences. In patient 1, the fitness landscape is dominated by a few highly fit haplotypes and a large fraction of unfit haplotypes. In contrast, patient 2 shows a fitness landscape where the two highly fit haplotypes are surrounded by a cloud of haplotypes of intermediate, average fitness. It also shows a stronger star-like topology in comparison to that of patient 1.
Figure 9.
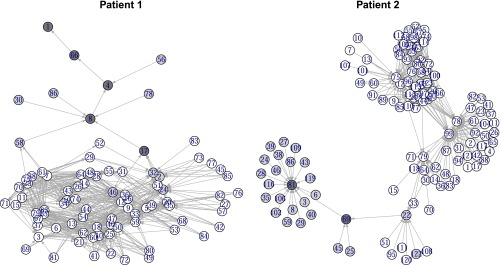
Rank fitness landscapes of the haplotypes in each of two patients. A directed edge i → j exists between haplotypes i and j if the posterior fitness difference fj − fi can credibly be inferred to be >0, i.e., if, given the model, there is evidence for haplotype j being fitter than haplotype i. Both graphs possess the transitive property; i.e., if j is fitter than i (indicated by an edge i → j) and k is fitter than j, then k is also fitter than i and a directed path exists from i to k. Dark gray vertices possess credibly larger than average, light gray vertices possess average, and colorless vertices possess lower than average fitness φ = 1.
To summarize the haplotype networks of size n1 = 86 and n2 = 123, we translated their DNA sequences into peptides and calculated the joint posterior fitness distribution of each peptide i as where the sums run over all DNA sequences j that code for peptide i. We discarded all loci with conserved residues and denote peptides with their alleles subscripted by their loci. Figure 10 illustrates the fitness landscapes for the four peptides in patients 1 and 2. This analysis shows again the importance of working with fitness values instead of ranking frequencies. In patient 1, while K25T64 and R25N64 show no credible differences when comparing their posterior frequencies, i.e., zero is an element of the 95% HPD region, they do show a credible difference in their fitness values, i.e., zero is not an element of the 95% HPD region (Figure 11).
Figure 10.

Rank fitness landscapes of four peptides in each of two patients. Each peptide is denoted by amino acids subscripted with their loci. A directed edge i → j exists between peptides i and j if the posterior fitness difference Fj − Fi can credibly be inferred to be >0, i.e., if, given the model, there is evidence for peptide j being fitter than peptide i. Vertices with dark shading possess larger than average and open vertices possess lower than average fitness φ = 1.
Figure 11.
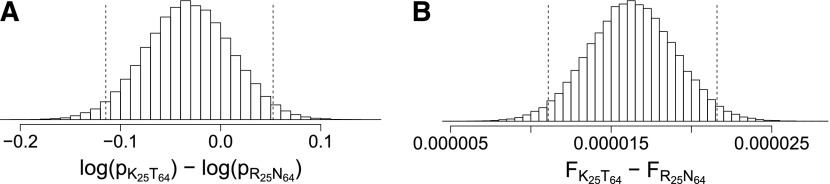
(A and B) Histograms of the difference of log-marginal frequencies (A) and of marginal fitness values (B). Differences in fitness values need not necessarily correspond to differences in marginal frequencies. The differences here are based on peptides from patient 1. The 95% highest posterior density intervals are demarcated with dashed vertical lines.
In addition to pairwise fitness differences, we also used our method for detecting epistasis, i.e., nonadditive effects of multiple alleles on fitness. We denote with 0 the wild-type or major allele and with 1 the mutant or minor allele. In patient 1, K25 and N64 are the major alleles and in patient 2, R25 and R48 are the major alleles. To determine whether a nonlinear interaction exists between alleles at two loci, we considered the random variable δ = F11 + F00 − F10 − F01 and tested whether δ is credibly different from 0. In the clinical data, we found a credible, nonzero epistatic effect between loci 25 and 64 in patient 1, but not between loci 25 and 48 in patient 2 (Figure 12).
Figure 12.
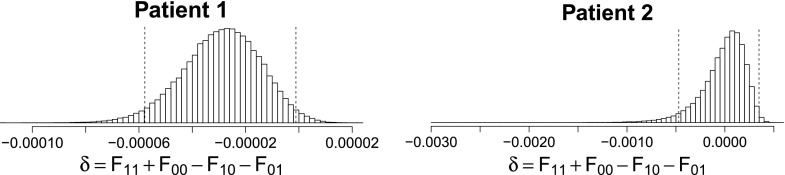
Posterior distributions of the epistasis term δ in both patients. A credible interaction term was detected in patient 1, but not in patient 2. The 95% highest posterior density intervals are demarcated with dashed vertical lines.
Finally, we also analyzed codon usage effects. We found that the marginal fitness differences between major and minor codons at synonymous amino acid residues were all credible, mainly due to the large difference in their frequencies (File S1, section 8).
Discussion
We have developed a computational framework for inferring fitness landscapes from NGS samples of HIV-1 patient-derived viruses, using quasispecies theory. Our inference scheme represents a novel approach to derive a measure of fitness from cross-sectional data. We obtained unimodal posterior distributions because of the existence of a single global stable mutation–selection equilibrium in the quasispecies model. This makes the inference scheme particularly efficient, as multimodality and suboptimal exploration of fitness space are not a problem. Our inference scheme is strongly parallelizable. For instance, for 300 haplotypes, we can run 624 chains on a 48-core server. Then each core needs to update only 13 chains by itself. This efficient partitioning scheme for sampling cannot be attained with ordinary globally adaptive MCMC schemes, where much CPU power cannot be utilized due to inherent lack of concurrency.
Every evolutionary model includes assumptions to make analysis possible. While established in the field of virology with many ubiquitous applications, the quasispecies framework nonetheless has limitations. One central weakness of the quasispecies model is at the same time its greatest strength, namely the assumption of a constant fitness landscape. Due to the inherent feedback loop of the host immune system, the quasispecies model will likely fail in genomic regions that can experience strong immunological pressure such as env, where fitness is more likely to be time dependent. While adaptations exist of the quasispecies model to time-varying, frequency-dependent fitness landscapes, such approaches necessarily include more involved mathematical machinery. The replicator–mutator equation is one such extension. In light of the minimum n2-dimensional parameter space and the highly complicated nonlinear trajectories of the replicator equation coupled with mutation (Pais and Leonard 2011), such a model would require prohibitively large amounts of time series, rather than cross-sectional, data to be useful for inference.
The centerpiece of quasispecies theory is the quasispecies, i.e., the population in mutation–selection equilibrium. Whether in reality such an equilibrium can ever exist remains an open question in the field of virology. One early study by Domingo et al. (1978) supported such a dynamic equilibrium for a multiply passaged Qβ bacteriophage. Ramratnam et al. (1999) highlight the existence of a dynamic equilibrium of production and clearance of HIV particles in vivo. Quasispecies theory likely fails to account for the acute phase of HIV infection, which is characterized by very strong initial immune responses that will show strong dynamics and where coupling between haplotypes is less of a driving force than immune escape.
Due to the high rates of mutation in RNA viruses and their large population sizes, quasispecies theory makes the implicit assumption that the expected number of produced mutants per replication cycle Neμ is large and hence can be modeled quasi-deterministically (Rodrigo 1999). It should be noted that the precise value of Ne in viral populations is an open question. In particular, if Ne < μ−1, the evolutionary process is dominated by stochastic effects, such that genetic drift trumps deterministic forces like selection. In this stochastic regime, the informative value of one sample of a viral population diminishes rapidly with decreasing Ne. Given these large random fluctuations in the stochastic regime, inferring selection requires multiple replicates of time series data, to disentangle deterministic effects from random fluctuations. At least one study of linkage disequilibrium in HIV-1 suggests Ne to be >μ−1 (Rouzine and Coffin 1999). The Ne limitation does not just affect the quasispecies model, but all deterministic models of virus evolution, such as those based on ordinary differential equations.
In addition to the presumed quasispecies, we do not take recombination into account. While extensions of the quasispecies model exist that account for this phenomenon (Boerlijst et al. 1996; Jacobi and Nordahl 2006), they are exceedingly complicated by nonlinear dynamics arising from bimolecular production reactions. This generally leads to bistability, such that a unique global quasispecies is not guaranteed anymore. Here, we analyzed genomic regions for which we assume recombination within the region to be negligible but recombination outside of the region may be somewhat larger, such that genetic variation that exists outside of the region of interest does not confound the analysis. This is reasonable, as the genomic ranges analyzed here are <200 bp. In general though, the recombination rate will affect epistasis and above a certain threshold, allelic selection takes over, where the selective advantage of alleles depends only on their own intrinsic fitness contribution and not on combinations with other alleles (Neher and Shraiman 2009).
Every computational approach to inference of high-dimensional data includes certain assumptions and approximations necessary for making practical analysis possible. Our approach to analyzing NGS data in the form of an MCMC sampler is no different in this regard. Our inference scheme does not yet capture overdispersion, the effect of the data having more variance than postulated by the statistical model. It is well known that NGS data involve a number of experimental steps for sample preparation. Sequencing has practical limitations and is error prone. All these steps will eventually yield a population sample that is overdispersed with respect to the multinomial distribution we employ. However, without replicates, technical overdispersion cannot be estimated jointly with the fitness landscape. An alternative would be to specify experimental overdispersion upfront as additional model parameters.
Inferring in vivo fitness landscapes for comparative analysis becomes possible with our inference approach. It could prove to be fruitful with regard to fitness landscapes of, for example, genomic loci that experience negligible immunological pressure and are not subject to drug pressure. One example is HIV-1’s gag gene, parts of which we have analyzed here. We can determine reliably the number of distinct fitness classes. In the analyzed patient data, we deduced a total order of peptides with increasing fitness in patient 1 and a partial order in patient 2. The reason for not being able to infer a total order in the case of patient 2 lies in the neutral network formed by the peptides K25R48 and K25K48. Additionally, we can also analyze properties of fitness landscapes, such as epistatic interactions.
In clinical settings, the vast majority of data will be cross-sectional and not time series. To go beyond the current standard of practice of equating fitness ranks to frequency ranks, any fitness inference method based on cross-sectional data will need to make strong assumptions on the population dynamics By accounting for mutational neighborhood structure, an important factor of intrahost viral evolution (Burch and Chao 2000), our model performs significantly better at inferring the rank fitness landscape than equating fitness ranks to frequency ranks. This is important in light of the observation that fitness need not necessarily show a strong connection to relative haplotype abundance (De la Torre and Holland 1990).
In summary, we have devised a mathematical framework based on the quasispecies model and an efficient sampling scheme for estimating in vivo viral fitness from intrahost NGS data. It will help in analyzing viral populations and understanding their evolutionary dynamics and eventually their clinical consequences.
Supplementary Material
Acknowledgments
This work was supported by ETH research grant ETH-33 13-1 (to N.B.), by the University of Zurich’s Clinical Research Priority Program “Viral infectious diseases: Zurich Primary HIV Infection Study” (H.F.G.), and by the Swiss National Science Foundation under grant CR32I2_146331 (to N.B., K.J.M., and H.F.G.).
Footnotes
Supporting information is available online at http://www.genetics.org/lookup/suppl/doi:10.1534/genetics.114.172312/-/DC1.
Available freely online through the author-supported open access option.
Communicating editor: J. Hermisson
Literature Cited
- Acevedo A., Brodsky L., Andino R., 2014. Mutational and fitness landscapes of an RNA virus revealed through population sequencing. Nature 505(7485): 686–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aitchison J., Shen S. M., 1980. Logistic-normal distributions: some properties and uses. Biometrika 67(2): 261–272. [Google Scholar]
- Anderson J. P., Daifuku R., Loeb L. A., 2004. Viral error catastrophe by mutagenic nucleosides. Annu. Rev. Microbiol. 58: 183–205. [DOI] [PubMed] [Google Scholar]
- Beerenwinkel N., Pachter L., Sturmfels B., 2007a Epistasis and shapes of fitness landscapes. Stat. Sin. 17: 1317–1342. [Google Scholar]
- Beerenwinkel N., Pachter L., Sturmfels B., Elena S. F., Lenski R. E., 2007b Analysis of epistatic interactions and fitness landscapes using a new geometric approach. BMC Evol. Biol. 7(1): 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beerenwinkel N., Günthard H. F., Roth V., Metzner K. J., 2012. Challenges and opportunities in estimating viral genetic diversity from next-generation sequencing data. Front. Microbiol. 3: 329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beerenwinkel N., Montazeri H., Schuhmacher H., Knupfer P., von Wyl V., et al. , 2013. The individualized genetic barrier predicts treatment response in a large cohort of HIV-1 infected patients. PLoS Comput. Biol. 9(8): e1003203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boerlijst M. C., Bonhoeffer S., Nowak M. A., 1996. Viral quasi-species and recombination. Proc. R. Soc. Lond. B Biol. Sci. 263(1376): 1577–1584. [Google Scholar]
- Burch C. L., Chao L., 2000. Evolvability of an RNA virus is determined by its mutational neighbourhood. Nature 406(6796): 625–628. [DOI] [PubMed] [Google Scholar]
- Campos, P. R., C. Adami, and C. O. Wilke, 2002 Optimal adaptive performance and delocalization in NK fitness landscapes. Physica A 304(3): 495–506.
- Clavel F., Hance A. J., 2004. HIV drug resistance. N. Engl. J. Med. 350(10): 1023–1035. [DOI] [PubMed] [Google Scholar]
- Crona K., Greene D., Barlow M., 2013. The peaks and geometry of fitness landscapes. J. Theor. Biol. 317: 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De la Torre J., Holland J., 1990. RNA virus quasispecies populations can suppress vastly superior mutant progeny. J. Virol. 64(12): 6278–6281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deforche K., Camacho R., Van Laethem K., Lemey P., Rambaut A., et al. , 2008. Estimation of an in vivo fitness landscape experienced by HIV-1 under drug selective pressure useful for prediction of drug resistance evolution during treatment. Bioinformatics 24(1): 34–41. [DOI] [PubMed] [Google Scholar]
- Diaconis, P., S. Holmes, M. Shahshahani, 2013 Sampling from a manifold, edited by T. Seppäläinen, K. Burdzy, S. Fienberg, P. Hall, R. Li, P. Green, A. DasGupta, and T.N. Sriram, pp. 102–125 in Advances in Modern Statistical Theory and Applications: A Festschrift in Honor of Morris L. Eaton. Institute of Mathematical Statistics. Beachwood, OH. [Google Scholar]
- Domingo E., Sabo D., Taniguchi T., Weissmann C., 1978. Nucleotide sequence heterogeneity of an RNA phage population. Cell 13(4): 735–744. [DOI] [PubMed] [Google Scholar]
- Domingo E., Sheldon J., Perales C., 2012. Viral quasispecies evolution. Microbiol. Mol. Biol. Rev. 76(2): 159–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earl D. J., Deem M. W., 2005. Parallel tempering: theory, applications, and new perspectives. Phys. Chem. Chem. Phys. 7(23): 3910–3916. [DOI] [PubMed] [Google Scholar]
- Eigen M., Schuster P., 1977. A principle of natural self-organization. Naturwissenschaften 64(11): 541–565. [DOI] [PubMed] [Google Scholar]
- Eigen M., McCaskill J., Schuster P., 1988. Molecular quasi-species. J. Phys. Chem. 92(24): 6881–6891. [Google Scholar]
- Falugi P., Giarré L., 2009. Identification and validation of quasispecies models for biological systems. Syst. Control Lett. 58(7): 529–539. [Google Scholar]
- Ferguson A. L., Mann J. K., Omarjee S., Ndung’u T., Walker B. D., et al. , 2013. Translating HIV sequences into quantitative fitness landscapes predicts viral vulnerabilities for rational immunogen design. Immunity 38(3): 606–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavrilets, S., 2004 Fitness Landscapes and the Origin of Species. Princeton University Press, Princeton, NJ. [Google Scholar]
- Guennebaud, G., and B. Jacob, 2010 Eigen v3. Available at: http://eigen.tuxfamily.org.
- Hanson K. M., Cunningham G. S., 1998. Posterior sampling with improved efficiency, edited by K. M. Hanson, pp. 371–382 in Medical Imaging’98. International Society for Optics and Photonics; Bellingham. WA. [Google Scholar]
- Hinkley T., Martins J., Chappey C., Haddad M., Stawiski E., et al. , 2011. A systems analysis of mutational effects in HIV-1 protease and reverse transcriptase. Nat. Genet. 43(5): 487–489. [DOI] [PubMed] [Google Scholar]
- Horn, R. A., and C. R. Johnson, 1985 Matrix Analysis. Cambridge University Press, Cambridge, UK. [Google Scholar]
- Jacobi M. N., Nordahl M., 2006. Quasispecies and recombination. Theor. Popul. Biol. 70(4): 479–485. [DOI] [PubMed] [Google Scholar]
- Kauffman S. A., Weinberger E. D., 1989. The NK model of rugged fitness landscapes and its application to maturation of the immune response. J. Theor. Biol. 141(2): 211–245. [DOI] [PubMed] [Google Scholar]
- Kouyos R. D., Leventhal G. E., Hinkley T., Haddad M., Whitcomb J. M., et al. , 2012. Exploring the complexity of the HIV-1 fitness landscape. PLoS Genet. 8(3): e1002551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Durbin R., 2010. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 26(5): 589–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J., Dykes C., Wu T., Huang Y., Demeter L., et al. , 2010. vFitness: a web-based computing tool for improving estimation of in vitro HIV-1 fitness experiments. BMC Bioinformatics 11(1): 261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzner K. J., Giulieri S. G., Knoepfel S. A., Rauch P., Burgisser P., et al. , 2009. Minority quasispecies of drug-resistant HIV-1 that lead to early therapy failure in treatment-naive and -adherent patients. Clin. Infect. Dis. 48(2): 239–247. [DOI] [PubMed] [Google Scholar]
- Musso F., 2012. On the relation between the Eigen model and the asexual Wright–Fisher model. Bull. Math. Biol. 74(1): 103–115. [DOI] [PubMed] [Google Scholar]
- Neher R. A., Shraiman B. I., 2009. Competition between recombination and epistasis can cause a transition from allele to genotype selection. Proc. Natl. Acad. Sci. USA 106(16): 6866–6871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niedringhaus T., Milanova D., Kerby M., Snyder M., Barron A., et al. , 2011. Landscape of next-generation sequencing technologies. Anal. Chem. 83(12): 4327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowak, M., and R. M. May, 2000 Virus Dynamics: Mathematical Principles of Immunology and Virology. Oxford University Press, London/New York/Oxford. [Google Scholar]
- Pais, D., and N. E. Leonard, 2011 Limit cycles in replicator-mutator network dynamics, edited by E. K. P. Chong, M. M. Polycarpou, J. A. Farrell, and E. F. Camacho, pp. 3922–3927 in 50th IEEE Conference on Decision and Control and European Control Conference (CDC-ECC). Institute of Electrical and Electronic Engineers. Piscataway, NJ. [Google Scholar]
- Park J.-M., Munoz E., Deem M. W., 2010. Quasispecies theory for finite populations. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 81(1): 011902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plummer, M., N. Best, K. Cowles, and K. Vines, 2006 CODA: convergence diagnosis and output analysis for MCMC. R News 6(1): 7–11.
- Quiñones-Mateu M. E., Arts E. J., 2002. Fitness of drug resistant HIV-1: methodology and clinical implications. Drug Resist. Updat. 5(6): 224–233. [DOI] [PubMed] [Google Scholar]
- Ramratnam B., Bonhoeffer S., Binley J., Hurley A., Zhang L., et al. , 1999. Rapid production and clearance of HIV-1 and hepatitis C virus assessed by large volume plasma apheresis. Lancet 354(9192): 1782–1785. [DOI] [PubMed] [Google Scholar]
- Rezende L. F., Prasad V. R., 2004. Nucleoside-analog resistance mutations in HIV-1 reverse transcriptase and their influence on polymerase fidelity and viral mutation rates. Int. J. Biochem. Cell Biol. 36(9): 1716–1734. [DOI] [PubMed] [Google Scholar]
- Rodrigo A. G., 1999. HIV evolutionary genetics. Proc. Natl. Acad. Sci. USA 96(19): 10559–10561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouzine I., Coffin J., 1999. Linkage disequilibrium test implies a large effective population number for HIV in vivo. Proc. Natl. Acad. Sci. USA 96(19): 10758–10763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoeni-Affolter F., Ledergerber B., Rickenbach M., Rudin C., Günthard H. F., et al. , 2010. Cohort profile: the Swiss HIV Cohort study. Int. J. Epidemiol. 39: 1179–1189. [DOI] [PubMed] [Google Scholar]
- Segal M. R., Barbour J. D., Grant R. M., 2004. Relating HIV-1 sequence variation to replication capacity via trees and forests. Stat. Appl. Genet. Mol. Biol. 3(1): 1031. [DOI] [PubMed] [Google Scholar]
- Steinhauer D., Holland J., 1987. Rapid evolution of RNA viruses. Annu. Rev. Microbiol. 41(1): 409–431. [DOI] [PubMed] [Google Scholar]
- Szendro I. G., Schenk M. F., Franke J., Krug J., de Visser J. A. G., 2013. Quantitative analyses of empirical fitness landscapes. J. Stat. Mech. 2013(01): P01005. [Google Scholar]
- Ter Braak C. J., 2006. A Markov Chain Monte Carlo version of the genetic algorithm Differential Evolution: easy Bayesian computing for real parameter spaces. Stat. Comput. 16(3): 239–249. [Google Scholar]
- Töpfer A., Zagordi O., Prabhakaran S., Roth V., Halperin E., et al. , 2013. Probabilistic inference of viral quasispecies subject to recombination. J. Comput. Biol. 20(2): 113–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vignuzzi M., Stone J. K., Arnold J. J., Cameron C. E., Andino R., 2005. Quasispecies diversity determines pathogenesis through cooperative interactions in a viral population. Nature 439(7074): 344–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilke C. O., 2005. Quasispecies theory in the context of population genetics. BMC Evol. Biol. 5(1): 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.