

Background: Calpain 3 is a muscle-specific, calcium-dependent proteinase, mutations in which cause limb-girdle dystrophy 2A.
Results: Calmodulin binds the calpain 3 non-catalytic domain and promotes calpain activation.
Conclusion: Calmodulin is a positive regulator of calpain 3 activity in vivo.
Significance: Knowledge of calpain 3 activation mechanisms is crucial for understanding the etiology of limb-girdle muscular dystrophy 2A.
Keywords: Calmodulin (CaM), Calpain, Muscular Dystrophy, Proteinase, Skeletal Muscle
Abstract
Calpains are broadly distributed, calcium-dependent enzymes that induce limited proteolysis in a wide range of substrates. Mutations in the gene encoding the muscle-specific family member calpain 3 (CAPN3) underlie limb-girdle muscular dystrophy 2A. We have shown previously that CAPN3 knockout muscles exhibit attenuated calcium release, reduced calmodulin kinase (CaMKII) signaling, and impaired muscle adaptation to exercise. However, neither the precise role of CAPN3 in these processes nor the mechanisms of CAPN3 activation in vivo have been fully elucidated. In this study, we identify calmodulin (CaM), a known transducer of the calcium signal, as the first positive regulator of CAPN3 autolytic activity. CaM was shown to bind CAPN3 at two sites located in the C2L domain. Biochemical studies using muscle extracts from transgenic mice overexpressing CAPN3 or its inactive mutant revealed that CaM binding enhanced CAPN3 autolytic activation. Furthermore, CaM facilitated CAPN3-mediated cleavage of its in vivo substrate titin in tissue extracts. Therefore, these studies reveal a novel interaction between CAPN3 and CaM and identify CaM as the first positive regulator of CAPN3 activity.
Introduction
Calpain 3 (CAPN3) is the skeletal muscle-specific member of the calpain protease family. Mutations in the gene encoding CAPN3 cause the disease limb-girdle muscular dystrophy type 2A (LGMD2A).3 Although some pathogenic LGMD2A mutations interfere with the proteolytic activity of CAPN3, the consequences of many other mutations have not been explained (1–6). Mice lacking CAPN3 (C3KO) have reduced muscle mass and fiber diameter, impaired growth, and a reduction in the percentage of slow muscle fibers (7–9). These changes are in part due to insufficient activation of calcium calmodulin kinase (CaMK) signaling, and diminished adaptation to muscle loading (9). Therefore, although it is clear that impaired CaMK signaling and muscle adaptation underlie LGMD2A, the connection between CaMK and CAPN3 has not yet been clarified.
Elucidating underlying LGMD2A disease mechanisms requires an in-depth understanding of the biochemical properties of the CAPN3 enzyme. Most insights about the biochemical properties of CAPN3 are inferred from knowledge gained on the ubiquitously expressed (and more stable) “conventional” calpains (1, 10, 11). The conventional calpains (CCs), called CAPN 1 and CAPN2, exist as heterodimers, each involving a large 80-kDa catalytic subunit and a small, common 28-kDa regulatory subunit. The large subunits share structural features common to all “classical” calpains, which include two proteolytic core domains that form the active site (PC1 and PC2), a C2-like (C2L) domain, and a penta-EF-hand (PEF) domain (12). The small subunit contains a glycine-rich domain and a PEF domain that are believed to mediate association with the large subunit. This association is absolutely required for stability of the CCs. CAPN3 is similar to the CCs in that it also contains PC1, PC2, C2L, and PEF domains (Fig. 1) as well as three distinctive insertion sequences. These sequences are located at the N terminus (called NS), within PC2 (called IS1), and between the C2L and PEF domains (called IS2) (Fig. 1). The insertion sequences may offer CAPN3 some divergent characteristics from CAPN 1 and 2. For example, CAPN3 requires much lower levels of Ca2+ for activation and is much less stable. To date, no consensus cleavage site has been defined for any of the CAPNs. However, they all seem to demonstrate limited proteolysis of their substrates, and they are considered to have regulatory rather than degradative cellular functions.
FIGURE 1.

The C2L domain of CAPN3 binds to calmodulin. A, schematic of monomeric CAPN3 and its domains. PC1 is colored in orange, PC2 in yellow, C2L in green, and PEF in blue. The unique CAPN3 sequences NS, IS1, and IS2 are shown in brown, magenta, and pink, respectively. Base pair numbering is denoted above. The location of the cloned fragments is indicated. The fragments encompass the following regions of the molecule: fragment I, NS and PC1 (1–234 aa); fragment II, IS1 and PC2 (235–428 aa); fragment III, C2L and most of IS2 (429–623 aa); fragment IV, part of IS2 and PEF (624–821 aa). The inset at the bottom shows expansion of the C2L domain and the location of both sites. Amino acid numbers are indicated below each binding site. B, calpain 3 interacts with CaM through the C2L-IS2 domain in the presence of Ca2+. Top panel, Western blot analysis of the bacterially expressed, recombinant proteins that were used in the binding assays and probed with anti-GST. Lane 1, C3 = GST fused to the C129S mutant; lane 2, GST alone; lane 3, domain I; lane 4, domain II; lane 5, domain III; lane 6, domain IV. Bottom panel, results of GST pull-down experiments. Shown is a Western blot probed with anti-GST. The different constructs are indicated at the top of the blot. Also shown are GST eluates from the CaM resin blotted with anti GST. Only full-length, proteolytically inactive CAPN3 (C129S) and CAPN3 fragment III bound to CaM in the presence of Ca2+ are shown.
The CCs are activated by calcium, which triggers conformational changes necessary to properly align the active site. Calcium requirements for activation are in the micromolar (CAPN1) and millimolar (CAPN2) ranges, as measured on the basis of in vitro assays. Additional posttranslational modifications and phospholipids may further lower the calcium requirement for activity, although this aspect of calpain biology has not been not fully elucidated. It is possible that activation of the CCs occurs transiently at the sites of calcium influx, where local calcium concentrations are sufficiently high (see Ref. 13 for a review). CCs are repressed by the endogenous inhibitor calpastatin, but it is still unclear how the balance of calpain activation and inactivation is accomplished in vivo (14).
The activation mechanism for CAPN3 has been deduced from prior biochemical studies that used a recombinant fragment of CAPN3 known to be more stable than the whole molecule. This recombinant fragment consists of the two proteolytic core domains (PC1 and PC2) and can be used to biochemically assess activation. Our current understanding of CAPN3 activation suggests that both calcium and autoproteolysis are components of the process and that autolytic activation occurs in two steps (11). The first step involves intramolecular cleavage of the N-terminal regions of NS and IS1, whereas the second step involves a slower intermolecular cleavage in IS1. The two resulting CAPN3 fragments (30 and 55 kDa) remain together by non-covalent binding to form the active enzyme. Therefore, although autolytic cleavage and calcium binding are definitive steps in the CAPN3 activation process, the specific intricacies involved in in vivo activation of CAPN3 have not yet been resolved.
Given the very low calcium requirement of CAPN3 and its high instability, it has been hypothesized that cellular activators and inhibitors of CAPN3 must exist for regulation of proper in vivo activity (15). Unlike the CCs, CAPN3 does not associate with either the small subunit or calpastatin. However, CAPN3 may homodimerize (16–18). A significant pool of CAPN3 is anchored to myofibrils, possibly through its interaction with the giant cytoskeletal protein titin, which may influence the activity and/or stability of CAPN3 (2, 7, 19–21). More recently, another protein, PLEIAD, has also been shown to reduce CAPN3 protease autolytic activity (22). Therefore, at least two regulators of CAPN3 stability and activity have been identified, but the in vivo mechanisms for how these different proteins interact are still unclear.
Although two negative regulators of CAPN3 activity have been identified, researchers have not yet identified any protein cofactors that promote CAPN3 activity. In this investigation, we show that calmodulin (CaM) binds and facilitates CAPN3 autolytic activation. CaM is a small, highly conserved protein that contains four EF-hand motifs and acts as an important calcium signal transducer. CaM regulates a variety of cellular processes, including excitation-contraction coupling, cell survival, transcription, and metabolism (23, 24). Importantly, CaM activates CaMK signaling, a pathway that is dysregulated in the absence of CAPN3. CAPN3 and CaMK colocalize at muscle triads, and, in the absence of CAPN3, CaMK and RyR1 are greatly reduced (25). Although the nature of the relationship between calcium, CaMK, CaM, CAPN3, and calpainopathy are still not fully elucidated, the identification of CaM as a cofactor of CAPN3 activity provides additional insights into the mechanisms of CAPN3 regulation in skeletal muscle.
EXPERIMENTAL PROCEDURES
Structural Modeling
The CAPN3 linear sequence was analyzed for the presence of CaM binding site(s) by exploiting the CaM Target Database and by sequence comparison of the CAPN3 sequence with the CaM binding site(s) in confirmed and predicted CaM-regulated kinases (26). The CAPN3 structure was generated by utilizing protein structure homology modeling using the SWISS-MODEL workspace (27–29) and visualized by PyMOL software.
Antibodies
Antibodies used for Western blotting included goat polyclonal anti-CAPN3 pIS2 antibody (1:1000, Cosmo Bio Co.), mouse monoclonal anti-His antibody (1:4000, GE Healthcare), mouse monoclonal anti-GST antibody (1:2000, GenScript), and mouse monoclonal anti-V5 antibody (1:3500, Invitrogen). Secondary antibodies used were anti-mouse and anti-goat peroxidase conjugates from Sigma-Aldrich (1:10000). Blots were developed using HyGLO Quick Spray chemiluminescent HRP antibody detection reagent (Denville). Signals were registered by the FluorChem FC2 digital imaging system (Alpha Innotech).
Cloning of CAPN3 into the GST Gene Fusion Vector
Fragments of CAPN3 were cloned into the pGEX-2T vector according to the recommendations of the manufacturer (Pharmacia Biotech). Boundaries of the fragments were determined in agreement with previously accepted CAPN3 domain assignments. Fragment I contains amino acids 1–234 (contains domains NS and PC1), fragment II contains amino acids 235–431 (includes IS1 and PC2), fragment III contains amino acids 429–622 (and contains C2L and most of IS2), and fragment IV contains amino acids 623–821 (and contains part of IS2 and PEF). In addition, smaller segments of fragment III were cloned in the pGEX-2T vector: 429–570 aa, 429–502 aa, 429–458 aa, 459–570 aa, 459–502 aa, and 571–623 aa. Sequences of all vectors were verified to ensure proper cloning and the absence of a frameshift.
Bacterial Expression of CAPN3 Fragments and Cell Lysate Preparation
Escherichia coli BL21(DE3)pLysS Z-competent cells were transformed with plasmid pGEX-2T (encoding glutathione S-transferase fusion) with given CAPN3 fragments/segments. Cells were grown in the presence of ampicillin (50 μg/ml) and chloramphenicol (34 μg/ml) at 37 °C until optical density reached 0.5–0.6. Expression was induced with 1 mm isopropyl 1-thio-β-d-galactopyranoside, and, after incubation for 2 h at 37 °C, cells were harvested, frozen in liquid nitrogen, and stored at −80 °C until use.
For the CaM binding assay, the cell pellet from 125-ml cultures of E. coli expressing a given CAPN3 fragment/segment was resuspended in 10 ml of buffer A containing 50 mm Tris-HCl (pH 8.0), 150 mm NaCl, 0.1% Triton X-100, and Halt protease and phosphatase inhibitor mixture, EDTA-free (Thermo Scientific). Cells were disrupted by sonication four times for 15 s on ice, and the lysate was clarified by centrifugation at 10,000 × g for 10 min at 4 °C.
Supernatant was divided into three parts and either, 5 mm EGTA or 5 mm CaCl2 was added to the experimental tubes. An equal amount of buffer A was added to the control tube. Binding to CaM affinity resin was performed according to the instructions of the manufacturer (Stratagene). In essence, 100 μl of a 50% resin slurry prewashed with Buffer A was added to each tube. After rotation for 2 h at 4 °C, beads were spun down and washed three times with either buffer A only or buffer A containing either EGTA or CaCl2. Bound material was eluted with 100 μl of 2× running sample buffer. An aliquot (30 μl) of each sample was subjected to SDS/10% PAGE and Western blot analysis with anti-GST antibody. Of note, we considered that expression levels of the fragments could vary significantly and that higher levels of expression could lead to higher nonspecific binding and affect interpretation of the binding experiment. Therefore, the expression level of each construct was initially evaluated by Western blotting with anti-GST antibody, and then the content of GST-tagged proteins was normalized with E. coli BL21(DE3)pLysS cell lysate prepared in a similar manner. This approach proved to be useful in the eradication of false positive results.
Generation of C129S-V5 Mice
Transgenic mice that overexpress mutant cDNA were created using the human skeletal actin promoter to drive expression of C129S in-frame with a V5 epitope located at the C terminus of CAPN3. These mice were then crossed to the C3KO mouse (7) so that the C129S transgene was the only CAPN3 expressed. Transgenic mice that overexpress wild-type CAPN3 in skeletal muscle have been described in previous studies (2, 30). These mice were also on the C3KO background.
Muscle Sample Preparation to Assess the Effect of CaM on CAPN3 Autolysis
Saline homogenates of gastrocnemius muscles from wild-type and transgenic mice overexpressing either wild-type CAPN3 or the inactive C129S mutant with V5 epitope were prepared as described previously by Anderson et al. (31) and our prior studies (2). After homogenization, samples were immediately divided into two tubes, one of which contained 2.4 μm CaM (Millipore, catalog no. 14-368). Equal aliquots were taken after 0, 5, 15, 30, 45, 60, and 90 min of incubation at 30 °C and mixed with an equal volume of 2× running sample buffer, boiled for 2 min, and kept on ice until analysis by immunoblotting. For some experiments, autolysis of CAPN3 in the muscles of WT transgenic mice was assessed in the presence of both 2.4 μm CaM and a mixture of non-cysteine protease inhibitors: 1.5 μm aprotonin (serine), 72.5 μm bestatin (exopeptidase), 1.5 μm pepstatin (aspartic), and 200 μm AEBSF (serine) (Fig. 4B).
FIGURE 4.

Calmodulin specifically promotes CAPN3 autolysis. A, Western blot analysis of whole muscle extracts from CAPN3 transgenic mice, probed with a CAPN3-specific antibody. Muscles from CAPN3 transgenic mice were homogenized in saline and allowed to incubate over a time course to allow for CAPN3 autolysis. Extracts were incubated in the absence (left side of blot) or presence (right side of blot) of CaM. Bands of 60, 58, and 55 kDa appeared more rapidly in the presence of CaM. B, Western blot analysis of whole muscle extracts from CAPN3 transgenic mice, incubated with and without protease inhibitor mixture. The blot was probed with anti-CAPN3. The presence of inhibitors of non-cysteine proteases did not impact the ability of CaM to promote autolysis, confirming that the accumulation of cleaved CAPN3 fragments in the presence of CaM is due to CAPN3 autolysis and not the action of other cellular proteases. Right panel, rates of degradation of CAPN3 expressed as a percentage of the initial concentration of intact protein in the absence (open squares) and presence (black squares) of CaM and in the presence of both CaM and inhibitors of non-cysteine proteases (open triangles). C, Western blot analysis of whole muscle extracts expressing the proteolytically inactive CAPN3 mutant (C129S). Addition of CaM did not alter the concentration of intact CAPN3 or CAPN3 autolytic products, suggesting that accumulation of CAPN3 fragments is due to autolysis rather than the action of other cellular proteases.
To assess the effect of CaM in the soluble (membrane and cytosol) and myofibrillar fractions separately, gastrocnemius muscles of WT transgenic mice homogenized in saline (1 ml) were subjected to centrifugation at 2000 × g for 5 min at 4 °C. The pellet was resuspended in 1 ml of saline and represented the enriched myofibrillar fraction. The supernatant was subjected to further centrifugation at 15,000 × g for 5 min at 4 °C. The pellet was discarded, and the supernatant was used as the soluble fraction containing cytosolic and membrane proteins. The effect of CaM was studied as described above.
Western blots to assess the rates of CAPN3 autolysis were probed with either the IS2 antibody or (in the C129S transgenic mouse) a V5 antibody. Equal aliquots for each time point were run to SDS-PAGE and transferred to a nitrocellulose membrane (100 V, 1 h, 4 °C). Densitometry was performed utilizing ImageJ software.
Titin Gel Electrophoresis
To evaluate the effect of CaM on CAPN3 proteolytic activity toward titin, muscle homogenates were subjected to gel electrophoresis as described previously (2). Specifically, the tibialis anterior muscles from wild-type and C3KO animals (∼160 mg) were ground with a pestle and mortar in liquid nitrogen, transferred to a Dounce homogenizer, and homogenized on ice in 15 volumes of 10 mm Tris-HCl (pH 7.8), 0.25 m sucrose, and 0.2 mm EDTA (buffer B) containing a mixture of non-cysteine protease inhibitors (see above). The homogenates were centrifuged at 2000 × g for 8 min at 4 °C. The pellets containing the myofibrillar fraction were washed three times in saline containing non-cysteine protease inhibitors and resuspended in the initial volumes of the same buffer. Suspension was divided into two parts. CaCl2 (up to 2 μm) and CaM (2.4 μm) were added to the experimental tube, whereas only CaCl2 was added to the control tube. Equal aliquots (200 μl) were taken after 0, 10, 30, and 60 min of incubation at 30 °C, and myofibrils were spun down for 10 min at 10,000 × g at 4 °C. The pellet was resuspended in 240 μl of sample buffer (8 m urea, 2 m thiourea, 3% SDS, 75 mm DTT, and 50 mm Tris (pH 6.8)), vortexed, heated at 60 °C for 2 min in a water bath, vortexed, heated at 60 °C for 5 min, cooled, passed several times through a syringe needle to reduce viscosity, and centrifuged for 5 min at 13,000 × g at room temperature. The resulting supernatant was transferred to a new tube and analyzed by titin electrophoresis as described previously (2, 20). Gels were stained with Coomassie Brilliant Blue R-250 as recommended by the manufacturer (Bio-Rad). Images were captured by the FluorChem FC2 digital imaging system (Alpha Innotech), and densitometry was performed with ImageJ software.
Animals
All experimental protocols were conducted in accordance with the National Institutes of Health Guide for Care and Use of Laboratory Animals and approved by the UCLA Institutional Animal Care and Use Committee.
RESULTS
CAPN3 Interacts with CaM through Predicted CaM-binding Sites Located in the C2L Domain
To ascertain information about potential binding partners that might serve as modulators of CAPN3 activity, its sequence and structure were examined for conserved domains. Using two different approaches, consensus CaM binding sites were identified in the CAPN3 sequence. The first binding site in the C2L domain of CAPN3 (amino acids 487–499) was predicted by the Calmodulin Target Database. A second CaM binding site was identified in the same C2L domain (amino acids 436–453) through comparison with known CaM binding kinases (Fig. 1A). Both areas lie in close proximity to each other in the C2L domain and share surprisingly similar secondary and tertiary structure on the basis of the structural model of CAPN3 (Fig. 2A, inset).
FIGURE 2.

Mapping the calmodulin binding sites within the C2L domain of CAPN3. A, left, structural model of CAPN3 representing the location of the C2L/IS2 domain with the predicted CaM-binding sites, indicated as red or green mesh. Center, model of the C2L/IS2 fragment, which includes amino acids 429–623. Right, a different view of the structural arrangement of the C2L/IS2 fragment and putative CaM binding sites. B, graphic representation of cloned segments used in GST pull-down assays shown in Fig. 3B. CaM binding sites are shown in red and green.
To biochemically test the interaction between CAPN3 and CaM, we carried out binding assays using recombinant proteins. For the assays we used either full-length proteolytically inactive CAPN3 (C129S) or four different CAPN3 fragments representing four non-overlapping pieces of CAPN3 (from the N to C terminus, labeled I-IV) (Fig. 1A). All recombinant proteins were tested for their ability to bind to a CaM resin in the absence and presence of calcium or EGTA. Recombinant GST was used as a negative control. The data demonstrate that CAPN3 binds to CaM in a calcium-dependent manner (Fig. 1B). Furthermore, CaM binding was mapped to fragment III, which includes the C2L domain.
To further narrow down the CaM binding site(s), we generated a set of deletions in fragment III containing the C2L domain on the basis of the expected structural features of the protein and the location of the predicted CaM binding sites (Fig. 2, A and B). Binding assays were carried out with the CaM resin using either GST alone or the segments indicated in Fig. 2B. As shown in Fig. 3B, each recombinant peptide that contained at least one of the predicted CaM binding sites was able to bind to CaM. Interestingly, the ability of CaM binding site 2 to bind CaM is higher than that of CaM binding site 1, but only when the region that extended beyond CaM binding site 2 was present in the fragment. The efficiency of the binding was increased significantly when both sites were included, suggesting a cooperative effect of the two CaM binding sites. We assessed the binding affinity of various segments of the C2L domain using a technique described by Pollard (33). This analysis validated our initial conclusion that the affinity of the binding to CaM of the C2L domain was increased when both binding sites were present (data not shown). These data demonstrate the presence of two distinct, functional CaM binding sites located in the C2L domain of CAPN3 and validate cooperative binding to CaM in vitro in the presence of calcium.
FIGURE 3.
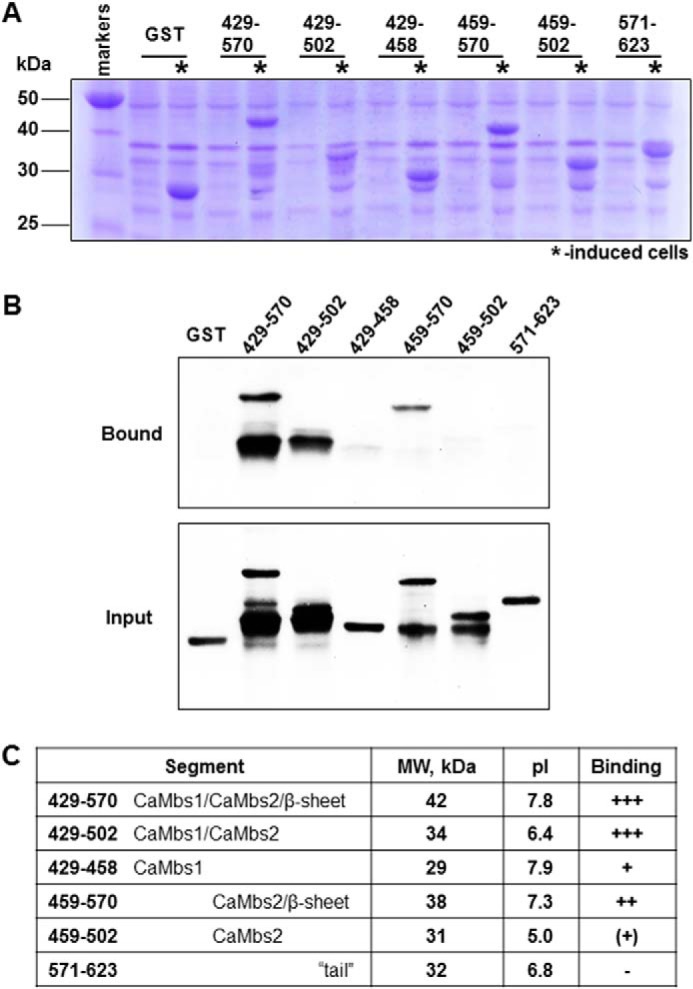
A, expression of the segments cloned into the pGEX-2T vector after 2 h of induction with 1 mm IPTG (asterisks). B, results of GST pull-down experiments. Shown is a Western blot of fragments bound to CaM (Bound) or lysates used in assay (Input). Blots were probed using anti-GST antibody. C, size, structural elements, molecular weights, pI, and binding ability of the tested segments. Estimation of pI values was accomplished by using a web tool.
Calmodulin Specifically Facilitates CAPN3 Autolytic Activity
The observation of CaM binding to CAPN3 led us to hypothesize that CaM might modulate CAPN3 autolytic activity. Because CAPN3 autolytically cleaves as part of the activation mechanism, we examined the rate of CAPN3 autolysis in the presence and absence of CaM by Western blotting. Autolytic cleavages occur in three places in CAPN3, leading to loss of the 94-kDa band (representing full-length CAPN3) and progressive accumulation of proteolytic fragments migrating at 55–60 kDa.
We used muscle extracts from transgenic mice for those experiments because CAPN3 is more stable when expressed in muscle than when it is expressed as a recombinant protein in vitro. We assayed CAPN3 activity by homogenizing muscle from C3Tg mice in saline and allowing the homogenate to sit at 30 °C. Samples were collected at defined points along a time course to assess autolysis by Western blotting. Fig. 4A shows that addition of supplemental CaM increased the rate of CAPN3 cleavage, as revealed by more rapid loss of the 94-kDa band. Therefore, these data demonstrate that CaM facilitates CAPN3 cleavage in muscle homogenates.
To ensure that the enhanced cleavage we observed in the presence of CaM was solely attributable to the autolytic activity of CAPN3 and not because of proteolysis by other cellular proteases, we assayed CAPN3 autolysis in the presence of both CaM and proteinase inhibitors. In prior investigations, we identified LGMD2A pathogenic mutations that increase susceptibility of mutant CAPN3 to proteolysis by non-cysteine, cellular proteases (2). The protease inhibitor mixture that was used had a wide inhibitor spectrum but did not contain cysteine protease inhibitors, therefore preserving the autolytic activity of CAPN3. The data showed that addition of inhibitors did not alter the effect of CaM on CAPN3 autolysis (Fig. 4B), which confirms that the increased CAPN3 cleavage that appeared in the presence of CaM was due to autolysis and not the action of cellular proteases.
To provide additional evidence that CaM facilitates CAPN3 autolysis and to further demonstrate that the observed cleavage products derive from autolysis and not other cellular proteases, we assayed CAPN3 cleavage in muscle homogenates derived from transgenic mice that express the proteolytically inactive mutant of CAPN3, C129SV5. This mouse was crossed to the C3KO background so that the inactive mutant was the only CAPN3 expressed. As shown in Fig. 4C (top panel), the full-length C129S protein was stable in the presence of CaM. Therefore, these data show that the cleavage products that occur in the presence of CaM derive purely from CAPN3 autolysis.
Calmodulin Facilitates CAPN3 Autolysis in Both Soluble (Membrane and Cytosol) and Insoluble (Myofibrillar) Fractions
Previous investigations revealed that CAPN3 is present in different fractions of skeletal muscle obtained by differential centrifugation, including the myofibrillar, cytosolic, and membrane fractions (2, 25). In the myofibrillar fraction, CAPN3 is bound to titin, and this association is hypothesized to stabilize CAPN3 and/or inhibit its autolytic activity (21, 34). To determine whether CAPN3 anchorage to myofibrils impairs its ability to be activated by CaM, we probed the effect of CaM on CAPN3 autolysis in different fractions from muscle. Homogenates were subjected to a two-step centrifugation protocol to obtain either the myofibrillar-enriched fraction (Fig. 5B) or the soluble fraction (Fig. 5A). The latter contains both cytosolic and membrane pools of CAPN3. Autolysis assays of CAPN3 demonstrated that CaM enhanced CAPN3 autolysis in both the cytosolic/membrane and myofibrillar fractions. This analysis also showed that CAPN3 is more stable in the myofibrillar fraction compared with the soluble fraction (Fig. 5, compare left top and bottom panels), consistent with the notion that CAPN3 is stabilized by association with myofibrils. Therefore, these data show that CAPN3 is more stable in the myofibrillar fraction but that anchorage to myofibrils does not appear to prevent the ability of CaM to enhance CAPN3 autolysis.
FIGURE 5.

Calmodulin promotes CAPN3 autolysis in both the soluble and myofibrillar fractions. Whole muscle extracts from CAPN3 transgenic mice were subjected to differential centrifugation to obtain the cytosolic and membrane fraction (top panel) or myofibrillar fraction (bottom panel). Fractions were incubated in the presence or absence of CaM and subjected to autolysis assays, followed by Western blot analysis using CAPN3-specific antibodies. Samples on the left side of the blot did not receive supplemental CaM, whereas samples on the right side of the blot were supplied with exogenous CaM following differential centrifugation. Despite the increased stability of CAPN3 in the myofibrillar fraction, the effect of CaM on CAPN3 autolysis was similar. Right panels, rates of degradation of CAPN3 expressed as a percentage of the initial concentration of intact protein in the absence (open squares) and presence (black squares) of CaM.
Calmodulin Facilitates CAPN3 Proteolytic Activity toward Its in Vivo Substrate, Titin
Given the findings of increased CAPN3 autolysis in the presence of CaM, we next tested whether CaM could facilitate CAPN3 cleavage of its substrates. As we showed previously, titin is an in vivo substrate of CAPN3 (2). Titin is a 3-MDa protein that runs as a doublet on specialized polyacrylamide/agarose gels that are optimized to detect large titin fragments. In muscles lacking CAPN3, the upper band of the titin doublet is the most pronounced band, whereas in muscles overexpressing CAPN3, the cleaved, lower molecular weight band of the titin doublet is most prevalent (2) (Fig. 6A). In this study, we utilized the same technical approach to evaluate titin cleavage by CAPN3 in the presence or absence of exogenous CaM. C3KO mice and CAPN3 transgenic mice served as negative and positive controls for CAPN3 cleavage of titin, respectively. Examination of titin gels revealed differing titin banding patterns between WT (Fig. 6A, left) and C3KO samples (Fig. 6A, right). Addition of CaM to the WT muscle extracts increased the rate of loss of the high molecular weight titin band (Fig. 6, A, left, and B). At the same time, no significant accumulation of cleaved titin was observed in C3KO muscles with the addition of CaM, confirming that titin cleavage is specific to CAPN3. Similar results were obtained using a recombinant fragment of M-line titin (data not shown). Taken together, these observations strongly support CaM as a positive regulator of CAPN3 proteolytic activity against its substrate titin.
FIGURE 6.
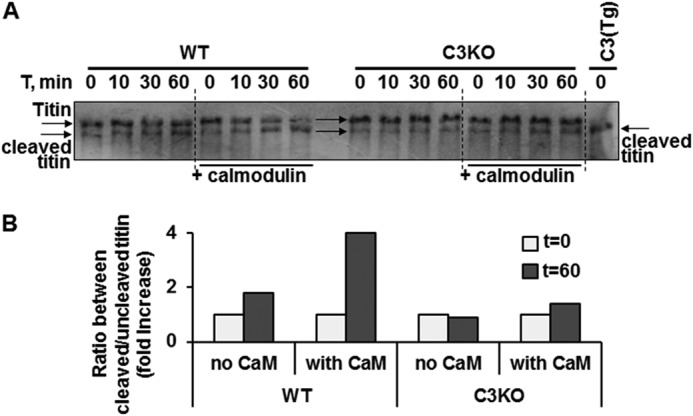
Calmodulin enhances CAPN3-mediated cleavage of titin in vivo. The effect of CaM on CAPN3-mediated proteolytic activity against titin was accessed in the myofibrillar fraction of muscle lysates. In specialized Coomassie-stained gels optimized for detection of titin, WT skeletal muscle extracts display titin as a doublet that runs at the top of the gel (2). A, left, WT muscle extracts were run on gels after incubation for selected time points in the absence and presence of CaM. In the absence of CaM, the upper band stays constant over the time course. In the presence of CaM, there is gradual loss of the upper band and an increase in the amount of the lower band representing cleaved titin. In contrast, when C3KO muscles are run to the gel (right), the upper band is more prominent and does not change with the addition of CaM. CAPN3 transgenic muscle extract was run at the far right lane of the gel as a positive control. In CAPN3 transgenic extracts, only the lower band of the titin doublet is present (2). B, accumulation of cleaved titin after 1 h of incubation in the absence and presence of CaM. Results are presented as -fold increase in the ratio between cleaved and uncleaved titin.
DISCUSSION
Understanding the biochemical nature of CAPN3 has been the focus of numerous studies since 1995, when it was established that mutations in this enzyme caused LGMD2A. LGMD2A is one of the most frequently occurring forms of LGMD2A and one of the most perplexing. From the earliest investigations, researchers recognized that defining mechanisms of disease would require an in-depth understanding of the biochemical nature of CAPN3. However, despite numerous efforts to explain its biochemical properties, specific details of the regulation of CAPN3 are still very poorly understood. One reason for this is that it has not been possible to purify full-length CAPN3 because of its tremendous instability. Therefore, our knowledge of the biochemical nature of CAPN3 derives from in vitro studies of the proteolytic core domains and from extrapolation of data generated from CCs. There are, however, some fundamental differences between CAPN3 and CCs. Therefore, not all data on the CCs can be extrapolated to CAPN3. The insertion domains (NS, IS1, and IS2) change its calcium requirement, and, importantly, neither the small regulatory subunit nor the inhibitor calpastatin impact CAPN3 activity.
Given the low calcium requirement and instability of CAPN3, it has been hypothesized that positive and negative regulators of CAPN3 must exist. In this study, we explored the possibility that the calcium sensor CaM could be a positive modulator of CAPN3 activity because two putative CaM binding motifs were identified in the C2L domain of CAPN3. Using a set of recombinant proteins, we confirmed that CAPN3 binds CaM in the presence of calcium and that this interaction indeed occurred at the predicted CaM binding sites. Although CAPN3 fragments containing either CaM binding site demonstrated the ability to bind to CaM to some extent, binding was significantly higher when both of these sites were present, suggesting cooperative binding. Interestingly, structural analysis of CaM binding sites revealed that they are strikingly similar (Fig. 2A, inset), providing further support for the functional importance of both sites.
As mentioned above, CAPN3 must first undergo intermolecular autolytic cleavage to become proteolytically active. This initial cleavage occurs at known sites within the CAPN3 molecule and produces fragments with molecular weights of 60, 58, and 55 kDa. Monitoring the appearance of these fragments serves as a good indicator of CAPN3 activity. We used this approach to evaluate the effect of CaM on CAPN3 autolytic activity and demonstrated that the addition of exogenous CaM to muscle extracts facilitated accumulation of CAPN3 autolytic fragments. This effect was absent in the proteolytically inactive C129S mutant of CAPN3 and was not affected by the presence of inhibitors of non-cysteine proteases. Taken together, these observations suggest that CaM specifically facilitates CAPN3 activation by promoting its autolytic cleavage.
Interestingly the CaM-induced increase in the autocatalytic activity of CAPN3 was observed in both the soluble (cytosolic plus membrane) and insoluble (myofibrillar) fractions of muscle lysates. It is widely accepted that myofibril-associated CAPN3 remains inactive because of its interaction with the sarcomeric protein titin. Although the stabilizing effect of titin has never been shown directly, this relationship has been inferred because of secondary reductions of CAPN3 in mice carrying titin deletions that eliminate CAPN3 binding sites (19, 34). It has been suggested that this reduction in CAPN3 was caused by loss of association with myofibrils and subsequent degradation. Our data showed that even though the rate of autoproteolysis was indeed lower in the myofibrillar fraction compared with the cytosolic plus membrane fraction, CaM was still able to facilitate autolysis in both fractions. Moreover, increased autoproteolytic activation of CAPN3 resulted in an increased rate of proteolytic cleavage of titin, an in vivo substrate of CAPN3, as we have demonstrated earlier (2). Therefore, although myofibril anchorage seems to stabilize CAPN3, it does not abolish the effect of CaM on CAPN3 activation.
CAPN3 can be detected in various cellular compartments, including the myofibrillar, cytosolic, and membrane fractions. In prior studies, we showed that CAPN3 appeared to be much more stable in the cytosolic fraction than in the membrane fraction (25). In recent work by Ono et al. (22), a novel protein called PLEIAD has been shown to suppress CAPN3 autolytic activity in nonmuscle cell cultures, suggesting that PLEIAD might serve as a stabilizing factor for the cytosolic pool of CAPN3. Our data provide a possible explanation for the increased activity of CAPN3 in the membrane fractions, attributable to increased local Ca2+ concentrations and CaM activation. We have found previously that CAPN3 is concentrated at the triad, where it colocalizes with a complex of proteins that include RyR1, CaMK, and aldolase (25). Although our previous data suggest a structural role for CAPN3 at this complex, we cannot exclude the possibility that there may also be a proteolytic role important for maintenance of the RyR complex. In this scenario, CaM may serve as a transducer of the local calcium signal and lead to CAPN3 activation. Further study of the triad fraction is necessary to reveal the proteolytic role of CAPN3 at this site.
Currently over 400 mutations have been identified in the CAPN3 gene, many of which are missense mutations (Leiden Muscular Dystrophy Database). Interestingly, pathogenic mutations are spread along the length of the CAPN3 molecule and can be found in regions located far away from the catalytic domains. The question of genotype-phenotype correlation remains unanswered for the majority of mutations. It has been proposed that some of these mutations cause mislocalization and/or decreased stability of the enzyme (2, 35). The results of this investigation suggest that some pathogenic mutations might impair CAPN3 interaction with CaM, therefore affecting CAPN3 activity. Examination of the Leiden database reveals a hot spot of LGMD2A mutations in the CaM binding sites described here (32). Further support for the hypothesis that disrupted CaM-CAPN3 binding might lead to impaired in vivo activation of CAPN3 derives from biochemical studies of LGMD2A biopsies, where four of six missense mutations in this region caused reduced autolytic activity (6). Therefore, although further studies are necessary, the observations suggest that disruption of CaM-CAPN3 interaction and impaired autolytic activation may be an underlying pathogenic mechanism for some LGMD2A mutations.
Acknowledgments
We thank Jane Wen for technical assistance.
This study was supported, in whole or in part, by NIAMS/National Institutes of Health Grants U54AR052646, 2RO1AR048177, and 2 P30 AR057230-06 (to M. S.). This work was also supported by Muscular Dystrophy Association of America Grant MDA241489 (to M. S.), by the Coalition to Cure Calpain 3, by the Crystal Ball Fund, and by My Directives.
- LDMD2A
- limb-girdle muscle dystrophy type 2A
- CaMK
- calmodulin kinase
- CC
- conventional calpain
- C2L
- C2-like
- PEF
- penta-EF-hand
- CAPN
- calpain
- CaM
- calmodulin
- PLEIAD
- platform element for inhibition of autolytic degradation
- AEBSF
- 4-(2-aminoethyl)benzenesulfonylfluoride.
REFERENCES
- 1. Jia Z., Petrounevitch V., Wong A., Moldoveanu T., Davies P. L., Elce J. S., Beckmann J. S. (2001) Mutations in calpain 3 associated with limb girdle muscular dystrophy: analysis by molecular modeling and by mutation in m-calpain. Biophys. J. 80, 2590–2596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ermolova N., Kudryashova E., DiFranco M., Vergara J., Kramerova I., Spencer M. J. (2011) Pathogenity of some limb girdle muscular dystrophy mutations can result from reduced anchorage to myofibrils and altered stability of calpain 3. Hum. Mol. Genet. 20, 3331–3345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Richard I., Broux O., Allamand V., Fougerousse F., Chiannilkulchai N., Bourg N., Brenguier L., Devaud C., Pasturaud P., Roudaut C. (1995) Mutations in the proteolytic enzyme calpain 3 cause limb-girdle muscular dystrophy type 2A. Cell 81, 27–40 [DOI] [PubMed] [Google Scholar]
- 4. Ono Y., Shimada H., Sorimachi H., Richard I., Saido T. C., Beckmann J. S., Ishiura S., Suzuki K. (1998) Functional defects of a muscle-specific calpain, p94, caused by mutations associated with limb-girdle muscular dystrophy type 2A. J. Biol. Chem. 273, 17073–17078 [DOI] [PubMed] [Google Scholar]
- 5. Richard I., Roudaut C., Saenz A., Pogue R., Grimbergen J. E., Anderson L. V., Beley C., Cobo A. M., de Diego C., Eymard B., Gallano P., Ginjaar H. B., Lasa A., Pollitt C., Topaloglu H., Urtizberea J. A., de Visser M., van der Kooi A., Bushby K., Bakker E., Lopez de Munain A., Fardeau M., Beckmann J. S. (1999) Calpainopathy: a survey of mutations and polymorphisms. Am. J. Hum. Genet. 64, 1524–1540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fanin M., Nascimbeni A. C., Fulizio L., Trevisan C. P., Meznaric-Petrusa M., Angelini C. (2003) Loss of calpain-3 autocatalytic activity in LGMD2A patients with normal protein expression. Am. J. Pathol. 163, 1929–1936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kramerova I., Kudryashova E., Tidball J. G., Spencer M. J. (2004) Null mutation of calpain 3 (p94) in mice causes abnormal sarcomere formation in vivo and in vitro. Hum. Mol. Genet. 13, 1373–1388 [DOI] [PubMed] [Google Scholar]
- 8. Kramerova I., Kudryashova E., Venkatraman G., Spencer M. J. (2005) Calpain 3 participates in sarcomere remodeling by acting upstream of the ubiquitin-proteasome pathway. Hum. Mol. Genet. 14, 2125–2134; Correction (2007) Hum. Mol. Genet. 16, 1006 [DOI] [PubMed] [Google Scholar]
- 9. Kramerova I., Kudryashova E., Ermolova N., Saenz A., Jaka O., López de Munain A., Spencer M. J. (2012) Impaired calcium calmodulin kinase signaling and muscle adaptation response in the absence of calpain 3. Hum. Mol. Genet. 21, 3193–3204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jia Z., Hosfield C. M., Davies P. L., Elce J. S. (2002) Crystal structure of calpain and insights into Ca2+-dependent activation. Methods Mol. Biol. 172, 51–67 [DOI] [PubMed] [Google Scholar]
- 11. García Díaz B. E., Gauthier S., Davies P. L. (2006) Ca2+ dependency of calpain 3 (p94) activation. Biochemistry 45, 3714–3722 [DOI] [PubMed] [Google Scholar]
- 12. Ono Y., Sorimachi H. (2012) Calpains: an elaborate proteolytic system. Biochim. Biophys. Acta 1824, 224–236 [DOI] [PubMed] [Google Scholar]
- 13. Campbell R. L., Davies P. L. (2012) Structure-function relationships in calpains. Biochem. J. 447, 335–351 [DOI] [PubMed] [Google Scholar]
- 14. Hanna R. A., Garcia-Diaz B. E., Davies P. L. (2007) Calpastatin simultaneously binds four calpains with different kinetic constants. FEBS Lett. 581, 2894–2898 [DOI] [PubMed] [Google Scholar]
- 15. Beckmann J. S., Spencer M. (2008) Calpain 3, the “gatekeeper” of proper sarcomere assembly, turnover and maintenance. Neuromuscul. Disord. 18, 913–921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kinbara K., Ishiura S., Tomioka S., Sorimachi H., Jeong S. Y., Amano S., Kawasaki H., Kolmerer B., Kimura S., Labeit S., Suzuki K. (1998) Purification of native p94, a muscle-specific calpain, and characterization of its autolysis. Biochem. J. 335, 589–596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ravulapalli R., Diaz B. G., Campbell R. L., Davies P. L. (2005) Homodimerization of calpain 3 penta-EF-hand domain. Biochem. J. 388, 585–591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Partha S. K., Ravulapalli R., Allingham J. S., Campbell R. L., Davies P. L. (2014) Crystal structure of calpain-3 penta-EF-hand domain: a homodimerized PEF family member with calcium bound at the fifth EF-hand. FEBS J. 281, 3138–3149 [DOI] [PubMed] [Google Scholar]
- 19. Haravuori H., Vihola A., Straub V., Auranen M., Richard I., Marchand S., Voit T., Labeit S., Somer H., Peltonen L., Beckmann J. S., Udd B. (2001) Secondary calpain3 deficiency in 2q-linked muscular dystrophy: titin is the candidate gene. Neurology 56, 869–877 [DOI] [PubMed] [Google Scholar]
- 20. Huebsch K. A., Kudryashova E., Wooley C. M., Sher R. B., Seburn K. L., Spencer M. J., Cox G. A. (2005) Mdm muscular dystrophy: interactions with calpain 3 and a novel functional role for titin's N2A domain. Hum. Mol. Genet. 14, 2801–2811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sorimachi H., Kinbara K., Kimura S., Takahashi M., Ishiura S., Sasagawa N., Sorimachi N., Shimada H., Tagawa K., Maruyama K. (1995) Muscle-specific calpain, p94, responsible for limb girdle muscular dystrophy type 2A, associates with connectin through IS2, a p94-specific sequence. J. Biol. Chem. 270, 31158–31162 [DOI] [PubMed] [Google Scholar]
- 22. Ono Y., Iemura S., Novak S. M., Doi N., Kitamura F., Natsume T., Gregorio C. C., Sorimachi H. (2013) PLEIAD/SIMC1/C5orf25, a novel autolysis regulator for a skeletal-muscle-specific calpain, CAPN3, scaffolds a CAPN3 substrate, CTBP1. J. Mol. Biol. 425, 2955–2972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bossuyt J., Bers D. M. (2013) Visualizing CaMKII and CaM activity: a paradigm of compartmentalized signaling. J. Mol. Med. 91, 907–916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Persechini A., Stemmer P. M. (2002) Calmodulin is a limiting factor in the cell. Trends Cardiovasc. Med. 12, 32–37 [DOI] [PubMed] [Google Scholar]
- 25. Kramerova I., Kudryashova E., Wu B., Ottenheijm C., Granzier H., Spencer M. J. (2008) Novel role of calpain-3 in the triad-associated protein complex regulating calcium release in skeletal muscle. Hum. Mol. Genet. 17, 3271–3280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gautel M. (2011) Cytoskeletal protein kinases: titin and its relations in mechanosensing. Pfluegers Arch. Eur. J. Physiol. 462, 119–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Arnold K., Bordoli L., Kopp J., Schwede T. (2006) The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics 22, 195–201 [DOI] [PubMed] [Google Scholar]
- 28. Kiefer F., Arnold K., Künzli M., Bordoli L., Schwede T. (2009) The SWISS-MODEL repository and associated resources. Nucleic Acids Res. 37, D387–D392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Peitsch M. C., Wells T. N., Stampf D. R., Sussman J. L. (1995) The Swiss-3DImage collection and PDB-Browser on the World-Wide Web. Trends Biochem. Sci. 20, 82–84 [DOI] [PubMed] [Google Scholar]
- 30. Spencer M. J., Guyon J. R., Sorimachi H., Potts A., Richard I., Herasse M., Chamberlain J., Dalkilic I., Kunkel L. M., Beckmann J. S. (2002) Stable expression of calpain 3 from a muscle transgene in vivo: immature muscle in transgenic mice suggests a role for calpain 3 in muscle maturation. Proc. Natl. Acad. Sci. U.S.A. 99, 8874–8879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Anderson L. V., Davison K., Moss J. A., Richard I., Fardeau M., Tomé F. M., Hübner C., Lasa A., Colomer J., Beckmann J. S. (1998) Characterization of monoclonal antibodies to calpain 3 and protein expression in muscle from patients with limb-girdle muscular dystrophy type 2A. Am. J. Pathol. 153, 1169–1179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kramerova I., Beckmann J. S., Spencer M. J. (2007) Molecular and cellular basis of calpainopathy (limb girdle muscular dystrophy type 2A). Biochim. Biophys. Acta 1772, 128–144 [DOI] [PubMed] [Google Scholar]
- 33. Pollard T. D. (2010) A guide to simple and informative binding assays. Mol. Biol. Cell 21, 4061–4067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Garvey S. M., Rajan C., Lerner A. P., Frankel W. N., Cox G. A. (2002) The muscular dystrophy with myositis (mdm) mouse mutation disrupts a skeletal muscle-specific domain of titin. Genomics 79, 146–149 [DOI] [PubMed] [Google Scholar]
- 35. Garnham C. P., Hanna R. A., Chou J. S., Low K. E., Gourlay K., Campbell R. L., Beckmann J. S., Davies P. L. (2009) Limb-girdle muscular dystrophy type 2A can result from accelerated autoproteolytic inactivation of calpain 3. Biochemistry 48, 3457–3467 [DOI] [PubMed] [Google Scholar]