

ABSTRACT
Autophagy is a ubiquitous mechanism involved in the lysosomal-mediated degradation of cellular components when they are engulfed in vacuoles called autophagosomes. Autophagy is also recognized as an important regulator of the innate and adaptive immune responses against numerous pathogens, which have, therefore, developed strategies to block or use the autophagy machinery to their own benefit. Upon human immunodeficiency virus type 1 (HIV-1) infection, viral envelope (Env) glycoproteins induce autophagy-dependent apoptosis of uninfected bystander CD4+ T lymphocytes, a mechanism likely contributing to the loss of CD4+ T cells. In contrast, in productively infected CD4+ T cells, HIV-1 is able to block Env-induced autophagy in order to avoid its antiviral effect. To date, nothing is known about how autophagy restricts HIV-1 infection in CD4+ T lymphocytes. Here, we report that autophagy selectively degrades the HIV-1 transactivator Tat, a protein essential for viral transcription and virion production. We demonstrated that this selective autophagy-mediated degradation of Tat relies on its ubiquitin-independent interaction with the p62/SQSTM1 adaptor. Taken together, our results provide evidence that the anti-HIV effect of autophagy is specifically due to the degradation of the viral transactivator Tat but that this process is rapidly counteracted by the virus to favor its replication and spread.
IMPORTANCE Autophagy is recognized as one of the most ancient and conserved mechanisms of cellular defense against invading pathogens. Cross talk between HIV-1 and autophagy has been demonstrated depending on the virally challenged cell type, and HIV-1 has evolved strategies to block this process to replicate efficiently. However, the mechanisms by which autophagy restricts HIV-1 infection remain to be elucidated. Here, we report that the HIV-1 transactivator Tat, a protein essential for viral replication, is specifically degraded by autophagy in CD4+ T lymphocytes. Both Tat present in infected cells and incoming Tat secreted from infected cells are targeted for autophagy degradation through a ubiquitin-independent interaction with the autophagy receptor p62/SQSTM1. This study is the first to demonstrate that selective autophagy can be an antiviral process by degrading a viral transactivator. In addition, the results could help in the design of new therapies against HIV-1 by specifically targeting this mechanism.
INTRODUCTION
Macroautophagy, herein referred to as autophagy, is a major cellular catabolic pathway highly regulated in eukaryotes. It is involved in the degradation of cytoplasmic material after its sequestration in vacuoles called autophagosomes. The autophagosomes fuse with lysosomes to form autolysosomes in which the sequestered material is degraded and then recycled (1). Since the discovery of the Atg genes that regulate this process, autophagy has been found to be involved in a number of important cellular functions, including cellular homeostasis, development, aging, or innate and adaptive immune responses (2, 3). Autophagy is believed to be one of the most ancient defense processes against invading pathogens. Its antiviral effect has been described in many studies through different mechanisms, including a direct degradation of cytoplasmic viral components, as shown, for example for the Sindbis virus (SIN) capsids that are specifically targeted to autophagy upon interaction with p62 (4, 5). Importantly, pathogens have evolved different means to inhibit or use autophagy to their own profit (6).
At the molecular level, two signaling complexes are involved in the induction, the elongation, and the closure steps of autophagy, leading to the formation of autophagosomes. Briefly, the class III phosphatidylinositol 3-kinase (PI3K), associated with p150 and beclin 1, is responsible for the formation of the phagophore. Two ubiquitination-related conjugation systems, leading to the formation of the Atg12-Atg5-Atg16L complex and the Atg8-phosphatidylethanolamine (PE) complex, are required for the elongation and closure of the autophagosome. These two conjugates are formed upon the action of a unique E1-activating enzyme called Atg7. ATG8-PE is inserted in the autophagic vacuole membranes and is present all along the pathway, a characteristic that makes it an autophagosomal marker. As autophagy proceeds, ATG8-PE is finally degraded in autolysosomes. Six orthologs of ATG8 exist in mammals, three microtubule-associated protein 1 light chain 3 (LC3) proteins (LC3A, -B, and -C), one gamma-aminobutyrate receptor-associated protein (GABARAP), and two GABARAP-like proteins (GABARAPL1 and GATE16/GABARAPL2). All of these proteins are synthesized as precursors that are rapidly processed at their C termini, leading to the exposure of a glycine residue that can be conjugated to PE (7–9). LC3B is still the most extensively studied ATG8 protein and will be referred to hereafter as LC3.
Autophagy can be a very selective process by the action of adaptor proteins behaving as autophagy cargo receptors, themselves degraded by autophagy due to their interaction with LC3 (10). Autophagy cargo receptors share at least one domain, the LIR domain (LC3-interacting region), allowing interaction with ATG8 family members and thus targeting the cargos to autophagosomes (11). p62/SQSTM1 (sequestosome 1, hereafter called p62) is a typical autophagy receptor that interacts with ubiquitinated substrates via its UBA domain (ubiquitin-associated domain) and that is able to multimerize via its PB1 domain (NH2-terminal Phox and Bem1p domain). These two motifs are important for selective autophagic degradation of ubiquitinated substrates (10, 12–14). Of note, p62 has been involved in the lysosomal-mediated degradation of incoming pathogens, a process termed “xenophagy” (15, 16).
Human immunodeficiency virus type 1 (HIV-1) infects immune cells, mainly CD4+ T lymphocytes and macrophages. In most instances, viral entry is mediated by the interaction of the viral envelope (Env) glycoproteins (gp120 and gp41) with CD4 and a coreceptor, mainly CCR5 or CXCR4, expressed at the surface of the target cells, to initiate a membrane fusion event. The gp120 protein interacts first with CD4, triggering conformational changes leading to increased exposure of gp120 regions able to bind to the coreceptor. This interaction induces a structural rearrangement in gp41 and the insertion of its N terminus fusion peptide into the target cell membrane, leading to the fusion between viral and cell membranes and thus to viral entry (17). The RNA viral genome is then retro-transcribed in DNA by the viral reverse transcriptase and is transported to the nucleus, where it integrates the genome of the host cell by the action of the viral integrase. After the integration step, the viral transactivator Tat enables the transcription of the viral DNA, leading to the production of the viral components necessary to synthesize new infectious particles that are released from the infected cells. The HIV-1 replication cycle can thus be divided in two phases: the early phase, from viral entry to provirus integration, and the late phase, from transcription of viral genes to the release of new viral particles (18, 19).
Tat is a small HIV-1 protein of 80 to 103 amino acids, depending on the viral strain, necessary for the transcription of all the viral genes after its binding to the TAR RNA loop present in the HIV-1 long terminal repeat (LTR) promoter (20). In addition to its role in infected cells, Tat can be secreted and captured by bystander cells, where it can induce apoptosis or modulate the expression of cellular proteins, in particular, proinflammatory cytokines (21–24).
Complex links exist between HIV-1 and autophagy, depending on the cell types and on the infectious status of the target cell type, i.e., if the target cell is productively infected or not (25–27). In dendritic cells, HIV-1 is able to block autophagy, impeding viral antigen presentation (28, 29). In CD4+ T lymphocytes, we previously demonstrated that autophagy is induced by the gp41 fusogenic function and that this process leads to apoptosis of uninfected bystander CD4+ T cells, i.e., cells that have been in contact with infected cells without being productively infected (30, 31). In contrast, autophagy is blocked in productively infected CD4+ T cells (32).
In macrophages that have been challenged by the virus but not productively infected, autophagy is inhibited via the activation of Stat3 and Src-Akt by Tat and interleukin-10 (IL-10) (33). HIV-1 Tat also suppresses STAT1 activation triggered by gamma interferon (IFN-γ) in monocytes and IFN-γ-induced autophagy in primary macrophages, suggesting that inhibition of autophagy could impair the immune defenses by blocking the antigen processing required for the recognition and killing of intracellular pathogens (34). These results are consistent with the fact that macrophages do not undergo Env-mediated apoptosis and are not subjected to depletion during HIV-1 infection.
The mechanism by which autophagy exerts its restriction activity against HIV-1 infection in CD4+ T cells remains to be fully characterized. We demonstrate here that p62 interacts with Tat and targets it to lysosomal-mediated degradation via selective autophagy. This interaction is not mediated by a conventional domain of p62 since the UBA or the PB1 domain of this protein is not involved. Interestingly, both neo-synthesized Tat in infected cells and exogenous Tat internalized by uninfected cells are sensitive to autophagic degradation, indicating that all the functions of Tat could be counteracted by autophagy. In conclusion, we propose that degradation of the viral transactivator Tat by selective autophagy is a potent though unrecognized antiviral cellular mechanism against HIV-1 infection of CD4+ T lymphocytes.
MATERIALS AND METHODS
Ethics statement.
Primary CD4+ T cells were purified from blood obtained from the Etablissement Français du Sang (EFS). Procedures using human cells were approved by the Human Experimentation and Ethics Committee of the CNRS Institute. The HIV-1 infection experiments were performed in a biological safety level 3 laboratory. Cell culture was performed in a biological safety level 2 laboratory.
Cell culture.
The original HEK.293T cell line, HEK cells expressing wild-type (WT) CD4 and wild-type CXCR4 (HEK/CD4/CXCR4), and HEK cells stably expressing Env at the their surface were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with antibiotics and 10% fetal calf serum (FCS). MAGIC5B cells (cell line expressing CXCR4 and CCR5 and βgalactosidase under the control of the HIV long-terminal repeat [LTR]) were obtained from T. Masashi (Tokyo, Japan) and cultured in DMEM supplemented with antibiotics and 10% FCS. MOLT cells are nonadherent human acute lymphoblastic leukemia cells. The chronically HIV-1-infected MOLT cells (MOLT-X4) and the uninfected parental cell line (MOLT) were provided by J. Blanco (Barcelona, Spain). The 8.E5 cell line is a CEM-derived T cell line containing a single integrated copy of HIV-1 and unable to produce infectious virions. T cell lines were cultured in RPMI 1640 medium supplemented with antibiotics and 10% FCS. Primary CD4+ T cells were isolated from peripheral blood mononuclear cells and purified by negative selection using the CD4+ Rosette separation technique (Stem Cell Technologies). Cells were phenotypically analyzed, stimulated with 2 μg of phytohemagglutinin (PHA) for 24 h, and maintained in culture by the addition of 100 U/ml of interleukin 2 (IL-2) (Boehringer Mannheim) every 2 to 3 days.
Reagents and Abs.
Torin 1 (To) was purchased from Tocris, and 3-methyladenine (3-MA), PHA, E64d, and pepstatin A (anti-protease [AP]) were purchased from Sigma-Aldrich. T20 was obtained from the NIH AIDS Reagent Program. Anti-LC3 (catalog no. L7543), anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH; catalog no. G9545), anti-ATG7 (catalog no. A2856), and anti-FLAG-horseradish peroxidase (HRP) (catalog no. A8592) were obtained from Sigma-Aldrich; anti-Tat (catalog no. sc65912) and anti-beclin 1 (catalog no. sc11427) were purchased from Santa-Cruz Biotechnologies. The monoclonal anti-p24 antibody (Ab) was from the NIH AIDS Reagent program.
Plasmids.
The pNL4-3 molecular clone was obtained from the NIH AIDS Reagent Program. The different p62 constructs, cloned in a pENTR plasmid, were a kind gift from Terje Johansen (University of Tromsø, Tromsø, Norway). After an LR Clonase reaction, we cloned the different p62 cDNAs in pDEST vectors in order to obtain glutathione S-transferase (GST)-fused proteins, using Gateway cloning technology (Life Technologies). The LC3B cDNA was first cloned in a pENTR vector and then cloned in a pDEST vector using Gateway cloning technology (Life Technologies). The GST-LC3 F52A mutant was obtained by mutagenesis using a QuickChange kit from Stratagene. The FLAG-tagged Tat (86 residues; BH10 isolate) was cloned in the pBi-GL expression vector under the control of a tetracycline response element (TRE) (the FLAG tag is placed at the C-terminal part of Tat). To express the gene of interest, the plasmid needs to be cotransfected with a pUHD-neo vector responsible for the expression of the tTA regulatory proteins needed to transactivate the TRE. A pBi-Tat K50/51A mutant expressing the FLAG-Tat K50/51A mutant was generated from pBi-Tat using a QuickChange kit from Stratagene (35). The FLAG-Tat K71R expression vector was a kind gift from Monsef Benkirane (as with pBi-GL-Tat, the FLAG tag is located at the C terminus of Tat) (36).
The different constructs expressing Vif, Nef, and Vpr were cloned in a pENTR plasmid and transferred in a pCI-neo3×FLAG expression vector after an LR Clonase reaction using Gateway cloning technology (Life Technologies). The expression vector coding for GFP-LC3 (where GFP is green fluorescent protein) was kindly provided by T. Yoshimori (Osaka University, Japan) and R. Willey (NIAID, NIH). The expression vector coding for Gag was kindly provided by Nathalie Chazal (Centre d'Études d'Agents Pathogènes et Biotechnologies pour la Santé [CPBS], Montpellier, France) (37).
TEM analysis.
Cells were fixed in situ with 2.5% glutaraldehyde in cacodylate buffer (pH 7.4) for 60 min at 4°C, postfixed with 2% osmium tetroxide, and then washed in cacodylate buffer containing 0.5% tannic acid. After extensive washes in 0.1 M Sorensen phosphate buffer (pH 7.2), cells were included in a fibrin clot as described by Charret and Fauré-Fremiet (38). Cells were then postfixed with 2% osmium tetroxide and 0.5% tannic acid, dehydrated, and embedded in Epon (Embed-812; Electron Microscopy Sciences, Inc.). Sections were counterstained with uranyl acetate and lead citrate and examined with a Hitachi H7100 transmission electron microscope (TEM).
HIV-1 infection and production.
To produce HIV virions, HEK cells were transfected with pNL4-3, and supernatants were collected at 2 days posttransfection. Viral particles were concentrated by ultracentrifugation. Jurkat cells were then challenged for 24 h with dilutions of the concentrated viruses. Flow cytometry analysis of Gag+ cells was performed in order to quantify the multiplicity of infection (MOI). Quantification of viral particle production was performed with a p24 enzyme-linked immunosorbent assay (ELISA) from Innogenetics (Innotest HIV antigen monoclonal Ab [MAb]) according to the manufacturer's instructions. CD4+ T cells were infected with concentrated viruses at an MOI between 0.1 and 1. Coculture experiments were done with chronically HIV-infected (MOLT-X4) or uninfected (MOLT) T cells coincubated for indicated times (see the figure legends) with HEK/CD4/CXCR4 or MAGIC5B cells. Quantification of HIV infection of target cells was measured by flow cytometry (for HEK cells) or β-galactosidase activity assay (for MAGIC5B cells).
GST pulldown experiments.
Constructs expressing GST proteins fused to wild-type LC3 (GST-LC3), mutated LC3 (GST-LC3 F52A), wild-type p62 (GST-p62), or mutated p62 (GST-p62ΔLIR, GST-p62ΔUBA, and GST-p62ΔPB1, with deletions of the LIR, UBA, and PB1 domains, respectively) were cotransfected in HEK cells with the pBi-FLAG-Tat and pUHD vectors. Corresponding empty vectors were used as controls. Anti-proteases (E64d plus pepstatin A) were added to the transfected cells in order to optimize the expression of Tat. Cell lysis was performed at 24 h posttransfection in 150 mM NaCl, 20 mM Tris, pH 7, and 0.5% NP-40 at 4°C. After centrifugation for 20 min at 4°C, the lysates were incubated with glutathione-conjugated Sepharose beads for 2 h at 4°C. Unbound proteins were removed by four washes with cold lysis buffer. Bound proteins were then eluted by boiling for 10 min in Laemmli buffer and separated by gel electrophoresis, followed by immunoblotting with specific antibodies.
Flow cytometry.
The percentage of HIV-1-infected cells was determined by quantifying the cellular levels of p24Gag by flow cytometry using an HIV KC57 fluorescein isothiocyanate (FITC) kit according to the manufacturer's instructions (Beckman Coulter). Briefly, cells were fixed and permeabilized, and the anti-p24 antibody was added to the cells. After staining for 30 min, cells were washed with phosphate-buffered saline (PBS), and fluorescence intensity was measured on a Coulter Epics XL Flow Cytometer (Beckman Coulter).
β-Galactosidase assay.
A chemiluminescent reporter assay system, Galacto-Star β-Galactosidase Reporter Gene Assay System for Mammalian Cells, from Life Technologies, was used to quantitate ß-galactosidase activity according to the manufacturer's instructions. Briefly, 0.5 × 106 MAGIC5B cells were lysed in 200 μl of lysis buffer provided by the manufacturer. Fifty microliters of cell lysate was used in each assay, in duplicates. The results were normalized by quantifying the total protein concentrations using a Bradford assay from Sigma-Aldrich.
Transfections.
A total of 0.5 × 106 HEK or HEK/CD4/CXCR4 cells were cultured in six-well plates 24 h prior to transfection. Plasmids were then transfected using TurboFect reagent (Fermentas) according to the manufacturer's instructions. Smartpool p62 small interfering RNA (siRNA) and control siRNA (siCT) were from ABgene. Beclin 1 siRNA and Atg7 siRNA were synthesized and annealed by Eurogentec. The mRNA sequences to be targeted for beclin 1 and Atg7 siRNAs were 5′-CAGUUUGGCACAAUCAAUA-3′ and 5′-GCAUCAUCUUCGAAGUGAA-3′, respectively. Transfection of siRNA into HEK/CD4/CXCR4 or MAGIC5B cells (0.5 × 106 cells maintained in six-well plates) was performed with LipoRNAiMax reagent (Invitrogen) according to the manufacturer's instructions. In the case of siRNA and plasmid cotransfections, 0.25 × 106 HEK cells were first transfected with siRNA using LipoRNAiMax reagent, and, 24 h later, siRNA-transfected cells were transfected with the plasmid using TurboFect reagent.
Western blotting.
Cell lysates were loaded in 12% Prosieve 50 gels (Lonza) and transferred to polyvinylidene (PVDF) membranes. After a blocking step for 1 h at room temperature in PBS containing 0.5% casein, membranes were incubated overnight at 4°C with the primary antibody (Ab) in the blocking buffer. After three washes with PBS supplemented with 0.05% Tween, the membranes were incubated for 1 h at room temperature with peroxidase-coupled secondary Ab. Upon extensive washes, membranes were incubated with Luminata Western HRP Substrate (Millipore). Proteins were detected by chemiluminescence and imaged with a G-box camera (Syngene imaging system). The expression level of GAPDH was used as a loading control in all of the Western blotting experiments and for quantification. Quantification of protein expression was done with Genesys software (Syngene).
The form of LC3 conjugated to PE, named LC3-II, presents a higher electrophoretic mobility in gels and was, therefore, used to quantify autophagy.
Fluorescence analysis of autophagy.
HEK/CD4/CXCR4 cells were cultured in six-well plates and transfected with pEGFP-LC3 using TurboFect reagent (Fermentas) according to the manufacturer's instructions. Transfected cells were then left to adhere on coverslips before coculture with MOLT cells (negative control) or chronically infected cells (MOLT-X4) for the indicated times (from 2 h to 48 h) (see Fig. 4A). Human primary CD4+ T cells were transfected with pEGFP-LC3 using a Nucleofector apparatus (Amaxa Biosystems) according to the manufacturer's instructions. Transfected primary T cells were then cocultured on coverslips for 4 h with effector cells either expressing or not expressing the viral envelope at their surface (HEK.Env or HEK cells, respectively). Coverslips were washed with PBS, and cells were fixed in a 3.7% paraformaldehyde-PBS solution for 10 min at room temperature and analyzed by epifluorescence using a Leica microscope. Autophagic cells were defined as cells containing at least five GFP-LC3 puncta. More than 100 transfected cells were analyzed by two different investigators. Results are from at least three independent experiments.
FIG 1.
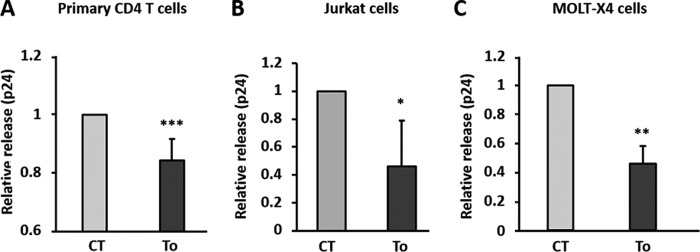
Autophagy has an anti-HIV-1 effect in CD4+ T lymphocytes. Primary CD4+ T cells (A) or Jurkat cells (B) were infected with HIV-1 at an MOI of 1 for 3 days. Cells were then extensively washed to eliminate excess free viruses and cultured for 24 h in the presence of 2 μM torin 1 to induce autophagy. HIV Gag-p24 levels were quantified by ELISA in the culture supernatants. (C) Chronically infected MOLT-X4 cells were treated for 24 h with 2 μM torin 1, and the HIV Gag-p24 concentration was quantified in the culture supernatants by ELISA. Data are representative of triplicates from at least three independent experiments. CT, control. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Statistics.
Analysis of the variance of the results was performed after arc sine transformation of the data when percentages were compared (39).
RESULTS
Induction of autophagy decreases viral production in HIV-1-infected CD4+ T lymphocytes.
We, along with others, have previously shown that autophagy is blocked in productively HIV-1-infected CD4+ T lymphocytes, suggesting that autophagy is an anti-HIV process counteracted by this virus (32, 40). To examine this hypothesis, activated primary CD4+ T cells were infected by HIV-1 for 3 days (∼20% infection). Then, autophagy was induced by adding torin 1, an inhibitor of the mTOR kinase, for 24 h. The level of viral particle production was measured by quantifying the viral capsid protein p24 by ELISA. As expected, we detected a significant decrease of HIV-1 p24 levels in the supernatants of cells treated with torin 1 (Fig. 1A). The same results were obtained when torin 1 was added to infected Jurkat cells (Fig. 1B) or chronically infected CD4+ T cells (MOLT-X4) (Fig. 1C). Thus, addition of torin 1 decreases viral production, supporting the anti-HIV role of autophagy.
Tat interacts with p62 and is specifically degraded by torin 1-induced autophagy.
We performed a two-hybrid screen to analyze the interactions between several HIV-1 proteins (Tat, Vif, Vpr, and Nef) and a bank of ATG proteins. Among the viral proteins tested, the transactivator Tat was the only viral protein able to bind p62 (data not shown), which is highly suggestive of a role for selective autophagy. This interaction was further confirmed in vitro using a GST pulldown assay in which Tat was found to interact with GST-p62 but not GST alone (Fig. 2A). As p62 is an autophagy receptor involved in the specific degradation of proteins or organelles by autophagy, we then analyzed whether induction of autophagy could affect the expression level of Tat. To this end, we transfected human embryonic kidney (HEK) cells with a plasmid expressing a FLAG-Tat construct, and autophagy was induced upon addition of torin 1 for 3 h. We observed a significant decrease in Tat levels upon torin 1 treatment (Fig. 2B). Importantly, this effect was reversed in cells pretreated with a cocktail of anti-proteases ([APs] E64d plus pepstatin A) previously described to block lysosomal-mediated degradation (Fig. 2B). Furthermore, inhibition of lysosomal proteases, in the absence of autophagy induction, led to increased levels of Tat expression in cells, suggesting that the viral transactivator can be degraded by lysosomes (Fig. 2B and D). The levels of the ectopically expressed viral proteins Gag, Vif, and Nef were not affected by the addition of torin 1 (Fig. 2C). These data strongly suggest that the torin 1-induced decrease in Tat level is selective and relies on an autophagy-dependent lysosomal-mediated degradation. The results were confirmed in a context of viral infection using HIV-1 chronically infected T cells (MOLT-X4 cells) which constitutively produce the viral Tat protein. As shown in Fig. 2D, Tat levels were also decreased in MOLT-X4 cells upon torin 1 treatment for 3 h, and the autophagy-inducing drug effect could be reversed by AP treatment. Since Tat is essential for the HIV-1 replication cycle, these results extend further those presented in Fig. 1, in which drug-mediated induction of autophagy strongly decreased viral release.
FIG 2.

Tat interaction with p62 leads to its selective degradation by autophagy. (A) HEK cells were cotransfected with the FLAG-Tat vector and the GST or GST-p62 plasmid. At 24 h posttransfection, cells were lysed, and lysates were subjected to GST pulldown before immunoblotting with anti-FLAG or anti-GST Ab. A fraction of lysates was preserved and used as an input control and immunoblotted with the anti-FLAG Ab. The result is representative of at least five different experiments. (B) HEK cells were transfected with plasmids expressing FLAG-Tat. After 24 h, the transfected cells were either left untreated or treated with 2 μM torin 1 in the presence or absence of anti-proteases (APs; E64d plus pepstatin A; 10 μg/ml each) for 3 h. The expression level of FLAG-Tat was revealed by Western blotting using anti-FLAG Ab. Levels of expression were quantified by densitometry and normalized to GAPDH expression levels. Data are representative of at least eight independent experiments for FLAG-Tat. (C) HEK cells were transfected with plasmids expressing Flag-Tat, FLAG-Nef, Flag-Vif, or Gag. After 24 h, the transfected cells were either left untreated or treated with 2 μM torin for 3 h. The expression level of the FLAG-tagged proteins or Gag was revealed by Western blotting using anti-FLAG or anti-p24Gag Ab, respectively. Levels of expression were quantified by densitometry and normalized to GAPDH expression levels. Data are representative of at least three independent experiments. (D) Chronically infected MOLT-X4 cells were either left untreated or treated for 3 h with 2 μM torin 1 in the presence or absence of AP. Cell lysates were then subjected to immunoblotting with anti-Tat Ab and anti-GAPDH Ab as a loading control. Tat expression levels were quantified by densitometry and normalized to GAPDH protein levels. Data are representative of at least three independent experiments. (E) HEK cells, previously transfected with unspecific (siCT) or ATG7-specific (siAtg7) siRNA, were transfected with the FLAG-Tat vector for 24 h. Cells were then either left untreated or treated with 2 μM torin 1 for 3 h before lysis. Cell lysates were immunoblotted with anti-Atg7 Ab, anti-FLAG Ab, or anti-GAPDH Ab as a loading control. Expression levels of Atg7 (left panels) and Tat (right panels) were quantified by densitometry and normalized to GAPDH protein levels. Data are representative of at least three independent experiments. (F) HEK cells were transfected with a p62-specific siRNA (sip62) or control siRNA (siCT) and then transfected with the plasmid expressing FLAG-Tat. The following day, cells were lysed, and p62 and Tat levels were assessed in lysates by Western blotting using Abs against p62 and FLAG. Sample loading was controlled by immunoblotting with anti-GAPDH Ab. Expression levels of p62 (left panels) and Tat (right panels) were quantified by densitometry and normalized to GAPDH protein levels. Data are representative of four independent experiments. AU, arbitrary units. *, P < 0.05; ***, P < 0.001.
To confirm that the autophagic process is responsible for the lysosomal-mediated degradation of Tat, we downregulated the expression of the essential autophagy-related protein ATG7 using siRNA. As shown in Fig. 2E, while the control siRNA did not prevent torin-1-induced Tat degradation, the viral transactivator expression levels were significantly increased in cells for which ATG7 expression was reduced. Notably, even if siRNA-mediated ATG7 decreased expression was only 30%, Tat levels became totally insensitive to torin 1 treatment, confirming the contribution of the autophagy pathway in Tat degradation. This result also indicates that this process is finely tuned and relies on a fully functional autophagy process.
Since we observed that Tat was interacting with p62 (Fig. 2A), we aimed at analyzing Tat levels when p62 expression was decreased. Remarkably, we noted that the siRNA-mediated decrease of p62 expression correlated with a significant increase in Tat protein signal compared to levels obtained in lysates from siCT-treated cells (Fig. 2F). Overall, these results demonstrated that the autophagy pathway significantly contributes to Tat degradation and that the autophagy receptor p62, shown to interact with Tat, could confer selectivity in this process.
The interaction between Tat and p62 is ubiquitin independent.
As we demonstrated that Tat interacts with p62 and since p62 is known to induce the selective autophagic degradation of substrates by interacting with LC3, we further analyzed whether Tat could be present in complexes with LC3. We thus performed GST pulldown experiments after cotransfection of plasmids expressing GST-LC3 and FLAG-Tat in HEK cells. As shown in Fig. 3A, Tat coprecipitates with GST-LC3. To assess whether this interaction was p62 dependent, we cotransfected cells with constructs expressing FLAG-Tat and a GST-tagged mutant of LC3 unable to interact with p62 (GST-LC3 F52A) (36, 41). We observed that Tat did not coprecipitate with LC3 F52A (Fig. 3A), indicating that the interaction between Tat and LC3 was indirect and seemingly regulated by p62. To confirm this hypothesis, we tested the interaction between Tat and a mutated form of p62 that does not interact with LC3 (GST-p62ΔLIR). As expected, this mutant still interacted with Tat with an apparently increased efficacy, which could be explained by the lack of targeting to autophagy-mediated degradation and thus a prolonged protein complex stability (Fig. 3A, lane 6).
FIG 3.

p62-dependent interaction of Tat with LC3. (A) HEK cells were cotransfected with the FLAG-Tat vector and plasmids expressing either GST, GST-LC3, GST-LC3 F52A, GST-p62, or GST-p62ΔLIR, a indicated. At 24 h posttransfection, cells were lysed, and lysates subjected to GST pulldown before immunoblotting with anti-FLAG Ab and anti-GST Ab. In each case, a fraction of lysates was preserved and used as an input control and immunoblotted with the anti-FLAG Ab. (B) HEK cells were cotransfected with plasmids expressing FLAG-Tat and either GST alone, GST-p62, GST-p62ΔUBA, or GST-p62ΔPB1 before GST pulldown. Results are representative of at least three independent experiments. (C) HEK cells transfected with plasmids expressing wild-type FLAG-Tat (Tat WT) or mutated forms of Tat (Tat K71R or Tat K50/51A) were either left untreated or treated or with 2 μM torin 1 for 3 h. Lysates obtained from each condition were used to analyze the expression levels of Tat and GAPDH as previously described. FLAG-Tat levels were quantified by densitometry upon normalization with GAPDH expression levels in the corresponding samples. AU, arbitrary units.
The autophagy receptor p62 has been reported to selectively direct ubiquitinated proteins or ubiquitinated organelles to autophagic degradation via specific recognition of ubiquitin moieties through its UBA domain, and the PB1 domain contributes to this process. We thus analyzed the interaction between Tat and mutated forms of p62 in which the UBA (GST-p62ΔUBA) or the PB1 (GST-p62ΔPB1) domain was deleted. As shown in Fig. 3B (lanes 3 and 4), Tat still interacted with these two p62 mutants, indicating that ubiquitination might not influence the interaction between p62 and Tat in vitro. In accordance with this result, Tat mutants on known ubiquitinated lysines, previously reported to regulate Tat activity and stability (K71 and K50/51, respectively) (36, 42, 43) are still degraded upon induction of autophagy by torin 1 (Fig. 3C).
Autophagy is induced by HIV-1 envelope glycoproteins (Env) at the very first steps of HIV-1 infection.
To further decipher whether Tat degradation by autophagy could be relevant in vivo, we analyzed its occurrence in the context of HIV-1 infection. Indeed, we already demonstrated that autophagy is induced in uninfected bystander CD4+ T lymphocytes after 48 h of coculture with Env-expressing cells (31). We extended this finding with experiments showing that the fusogenic function of gp41 is responsible for the observed Env-induced autophagy (30). Given that the gp41-mediated fusion process is an early HIV-1 entry event, we investigated whether autophagy could be induced very rapidly after contact of Env with its receptors on target cells before productive infection. To this aim, HEK cells expressing HIV receptors (HEK/CD4/CXCR4) and transfected with a plasmid expressing GFP-LC3 were cocultured with chronically infected T cells (MOLT-X4) or with uninfected T cells (MOLT) as a negative control. The level of autophagy and the percentage of infected cells were analyzed at different time points of coculture. As shown in Fig. 4A, autophagy was rapidly induced in read-out cells upon coculture with MOLT-X4, with a peak around 4 h corresponding to the early phase of HIV-1 infection (19). Autophagy was then negatively regulated and finally completely inhibited after 24 h of coculture, a step corresponding to the late phases of HIV-1 infection. Importantly, induction of autophagy is inversely correlated with the level of infection. Of note, the same pattern was obtained when GFP-LC3-transfected cells were challenged with cell-free HIV-1 (data not shown). We next analyzed whether the viral envelope was contributing to this early autophagy induction during early events of HIV-1 infection of CD4+ T cells. Primary CD4+ T cells, expressing GFP-LC3, were cocultured with effector cells stably expressing Env, or not, at their surface (HEK.Env or HEK cells, respectively). As shown in Fig. 4B, Env alone was able to rapidly and significantly induce autophagy in target cells. To confirm and reinforce these data, we took advantage of another autophagy read-out by analyzing the phenotype of primary CD4+ T cells by transmission electron microscopy upon 4 h of coculture with HEK cells stably expressing Env or not. We detected the appearance of numerous autophagic vacuoles in the cytoplasm of target CD4+ T cells only in the presence of Env (Fig. 4C). Thus, Env is able to induce autophagy in target cells upon early contact with infected cells. It is interesting that, as shown for Env-mediated autophagy in longer kinetics of coculture (30), this early autophagy induction was dependent on the gp41 fusogenic function. Indeed, the addition of T20 to the coculture, a gp41-mediated fusion peptide inhibitor, completely abolished the Env-induced autophagy (Fig. 4D).
FIG 4.

Autophagy is induced at early steps of HIV-1 infection. (A) HEK/CD4/CXCR4 cells, previously transfected with a plasmid expressing GFP-LC3, were cocultured with either MOLT cells (CT, negative control) or chronically infected MOLT-X4 cells (HIV) for the indicated times. The number of autophagic HEK/CD4/CXCR4 cells was counted by three different experimenters, and the result is the mean of three independent experiments. The percentage of autophagic cells obtained after coculture with MOLT-X4 cells was normalized with the percentage of autophagic cells obtained after coculture with MOLT cells. A representative pattern of GFP-LC3 puncta in HEK/CD4/CXCR4 cells upon 4 h of coculture with MOLT-X4 cells is presented on the left-hand side and compared to the fluorescence pattern obtained upon 4 h of coculture with control MOLT cells. In parallel, the percentage of HIV-infected cells was quantified by flow cytometry using the anti-p24 Ab. (B) Human primary CD4+ T cells were transfected with a plasmid expressing GFP-LC3 and cocultured with HEK cells (negative control) or Env-transfected HEK cells for 2 h or 4 h. Quantification was performed as described above, and the results are presented as mean fold value of each condition from three independent experiments. A representative pattern of GFP-LC3 puncta in primary CD4+ T cells upon 4 h of coculture with HEK-Env cells is presented and compared to the fluorescence pattern obtained upon 4 h of coculture with control HEK cells. (C) Human primary CD4+ T cells were cocultured with HEK cells expressing Env, or not, for 4 h, and autophagic vacuoles were observed by transmission electron microscopy. The percentage of autophagic cells is presented. (D) Human primary CD4+ T cells were cocultured with HEK cells expressing Env, or not, for 4 h in the presence or absence of AP. CD4+ T cells were harvested, and lysates were immunoblotted with anti-LC3 Ab and anti-GAPDH Ab as a loading control. LC3-II levels were quantified by densitometry and normalized with GAPDH expression levels. (E) HEK/CD4/CXCR4 cells were transfected with the FLAG-Tat vector and then cocultured with MOLT cells (negative control) or with MOLT-X4 cells for 4 h and 48 h. Suspension cells were removed by washing, and autophagy induction was monitored in the lysates of target cells upon immunoblotting with anti-LC3 Ab (left panel). Tat levels were assessed with the anti-FLAG Ab (right panel), while sample loading was controlled with the anti-GAPDH Ab for each condition. LC3-II and Tat levels were quantified by densitometry upon normalization with GAPDH expression levels in the corresponding samples. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
We then analyzed whether later steps of autophagy could be induced or blocked when cellular lysosomal proteases were inhibited by AP. As evidenced in Fig. 4E, the increased level of LC3-II obtained upon 4 h of contact with Env-expressing HEK cells was further enhanced in lysates from cells also pretreated with AP, confirming that the induced autophagy flux was not blocked during the early time points of contact between the viral envelope and CD4+ T cells.
To confirm that Tat could be a selective target for autophagic degradation in the context of productive infection, we analyzed its expression levels upon Env-induced autophagy during HIV-1 infection. For this purpose, we transfected a plasmid expressing FLAG-Tat in HEK/CD4/CXCR4 cells and cocultured these cells with MOLT-X4 cells for 4 h, a time previously shown to correspond to the peak of Env-mediated autophagy induction, or 48 h, a time for which autophagy was seemingly inhibited (Fig. 4A). As expected, the level of LC3-II was increased up to 70% after 4 h of coculture while being decreased by 40% upon 48 h of coculture with MOLT-X4 cells, strongly supporting an early induction of autophagy followed by its blockade during HIV-1 infection. Remarkably, Tat expression levels were inversely correlated to those of LC3-II, meaning that Tat is depleted when autophagy is induced, while this effect was not observed under conditions where autophagy is inhibited by the virus (Fig. 4F).
Env-induced autophagy triggers Tat degradation and consequently represses HIV-1 LTR activation.
In order to assess the contribution of Env-mediated autophagy induction in Tat degradation, we transfected a plasmid expressing FLAG-Tat in HEK/CD4/CXCR4 cells and treated these cells with the autophagy initiation inhibitor 3-methyladenine (3-MA) before coculture for 4 h with uninfected or chronically HIV-infected T cells. As shown in Fig. 5A, Tat levels markedly decreased upon coculture with MOLT-X4 cells, while they were fully rescued in the presence of 3-MA. This result confirms that Env-mediated autophagy flux induction is responsible for Tat degradation. We reproduced the experiments using 8.E5 cells as donor cells. These CD4+ T cells are productively infected, thus expressing Env at the surface, but because they do not express the reverse transcriptase, they do not produce infectious viral particles. With this model, the Env-mediated autophagy induction was even more pronounced, as evidenced by the increased and sustained Tat degradation observed in target cells (Fig. 5B).
FIG 5.

Autophagy-mediated Tat degradation inhibits HIV-1 LTR activation and viral replication. (A) HEK/CD4/CXCR4 cells transfected with the FLAG-Tat vector were cocultured with uninfected (MOLT) or chronically HIV-1-infected cells (MOLT-X4) cells for 4 h in the presence or absence of 10 mM 3-MA. Tat expression was monitored by Western blotting using the anti-FLAG Ab and quantified upon normalization with GAPDH levels. (B) HEK/CD4/CXCR4 cells, transfected as above, were cocultured for 4 h with effector control cells (Env negative) or cells expressing HIV-1 envelope (8.E5 cells). Tat detection and quantification were done as previously described. (C) MAGIC5B cells were cocultured with MOLT-X4 cells for 4 h or 8 h in the presence or absence of 3-MA. After cell lysis, β-galactosidase activity was quantified by luminescence and normalized to the condition in the absence of 3-MA for each time of coculture. Data represent the means ± standard errors of at least three independent experiments. (D) MAGIC5B cells were transfected with control (CT) or beclin 1 (Bec1)- or Atg7-specific siRNA for 16 h. Transfected cells were then cocultured with MOLT-X4 cells for 4 h. The cells were washed, harvested, and then lysed. A fraction of lysates was kept to control beclin 1, ATG7, and GAPDH levels by immunoblotting (left panels) while remaining lysates were used to measure β-galactosidase activity, normalized to the activity obtained from cells transfected with CT siRNAs for each time point. Data are means ± standard errors of four independent experiments. (E) MAGIC5B cells cocultured with MOLT-X4 cells were incubated or not with 2 μM torin 1 for 16 h in the presence or absence of AP. Fold induction of β-galactosidase activity was calculated as the enzymatic activity obtained in treated cells compared to that in untreated cells. Data are representative of at least three independent experiments. (F) MAGIC5B cells were incubated for 48 h with 200 nM recombinant wild-type Tat (Tat), mutant Tat (W11Y), or medium alone (Ct) before β-galactosidase activity was assayed. (G) MAGIC5B cells, treated as in described for panel D, were incubated for 48 h with 200 nM recombinant Tat or W11Y. A fraction of lysates was kept to for beclin 1 and GAPDH levels by immunoblotting while remaining lysates were used to measure β-galactosidase activity. Data are representative of at least five independent experiments. RLU, relative light units. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
While viral challenge could rapidly and transiently induce autophagy, productive infection appeared to block this defense mechanism, thus potentially protecting the viral transactivator Tat from degradation. Interestingly, Tat can exert multiple effects on bystander cells when secreted from productively infected cells. For instance, secreted Tat was reported to transactivate the expression of genes under the control of the HIV LTR in bystander cells. To examine whether incoming Tat could be degraded by autophagy, we used HeLa cells expressing CD4 and CXCR4 together with β-galactosidase under the control of the HIV LTR promoter (MAGIC5B cells) (44). MAGIC5B cells were cocultured for 4 h and 8 h with MOLT-X4 cells in the presence or absence of 3-MA, and β-galactosidase activity was measured. Enzymatic activity could be detected in reporter target cells, supporting the bystander effect of secreted Tat. Transactivation was significantly increased in the presence of 3-MA, strongly suggesting that autophagy controls Tat-mediated transactivation of the HIV LTR (Fig. 5C). Because 3-MA could have some side effects possibly affecting Tat secretion from infected cells, we aimed at confirming these results by directly downregulating the expression of essential ATG proteins in target cells. Therefore, MAGIC5B cells were transfected with siRNA directed against beclin 1 or Atg7 or unrelated siRNA before coculture with MOLT-X4 cells. As expected, the level of β-galactosidase activity was increased at both time points of coculture when beclin 1 or Atg7 expression was silenced (Fig. 5D). Inversely, autophagy induction should inhibit HIV LTR transactivation due to Tat degradation (Fig. 2). Indeed, when MAGIC5B cells were cocultured with MOLT-X4 cells in the presence of torin 1, we observed a significant decrease of β-galactosidase activity, inversely correlated with the increased enzymatic activity obtained upon autophagy flux blockade with AP (Fig. 5E). Hence, the effect of drug-mediated autophagy induction on transactivation by Tat appeared to be sustained, as evidenced by the decreased transactivation upon 16 h of coculture (Fig. 5E). These data thus show that the autophagy pathway can be a major regulator of Tat-mediated transactivation and viral replication. This conclusion is valid whether Tat is produced intracellularly (i.e., by infected cells) or delivered from the outside (i.e., endocytosed).
Indeed, to confirm that the detected viral promoter transactivation was exclusively Tat dependent and controlled by autophagy, we took advantage of a previously described Tat mutant that lacks its single tryptophan residue (Tat-W11Y). This mutant retains transactivation activity but is unable to enter bystander cells (35) (Fig. 5F). MAGIC5B cells were transfected with a beclin 1-specific siRNA or a control siRNA and then treated with 200 nM recombinant wild-type Tat (Tat-WT) or Tat-W11Y, and β-galactosidase activity was measured after 48 h. We observed that, in contrast to cells treated with Tat-WT, transactivation was not significant when Tat-W11Y was used (Fig. 5G). These results confirm that transactivation by incoming Tat is targeted by selective autophagy.
DISCUSSION
HIV-1, like other viruses, has to deal with cellular defenses in order to replicate efficiently. Although several cellular HIV-1 restriction factors have been identified, most of them were shown to be affected by viral countermeasures (45). We previously demonstrated that HIV-1 Env induces autophagy in uninfected CD4+ T lymphocytes via the fusogenic function of gp41, leading to their apoptotic cell death (30, 31). In this study, we demonstrate that induction of autophagy is readily detectable early after viral challenge in an Env-dependent manner, while being compromised at later times. Autophagic activity is inversely correlated with HIV-1 production. We further provide evidence that autophagy, when pharmacologically induced in productively infected CD4+ T cells, leads to a significant decrease in viral production, thus identifying this cellular pathway as an HIV-1 restriction mechanism. This observation led us to decipher the molecular mechanism of the autophagy-mediated antiviral effect. A two-hybrid screen revealed a strong and specific interaction between p62 (also called SQSTM1), an autophagy receptor, and the viral transactivator Tat. We first confirmed the interaction between the proteins at the cellular level in pulldown assays and confirmed that Tat expression levels were modulated in cells upon autophagy activation or inhibition. We then demonstrated that Tat expression was significantly increased in p62-deficient cells, suggesting that p62 could be a mediator of Tat degradation by selective autophagy. We therefore analyzed the domains of p62 possibly involved in the interaction with Tat. Surprisingly, the interaction between Tat and p62 did not rely on a functional UBA domain of p62, suggesting that ubiquitination would not regulate p62-mediated Tat degradation, in contrast to the general paradigm reported for other substrates (46). Consistently, Tat mutations at the level of lysines, which were previously shown to regulate Tat stability and activity, did not prevent its degradation upon autophagy induction. Although we cannot exclude modification of other residues or the involvement of unidentified cellular proteins belonging to the complex Tat/p62, our data represent the first demonstration of selective autophagic degradation of a viral transactivator. Indeed, in a context of viral infection, a selective p62-mediated degradation has been demonstrated only for two structural viral proteins, the capsid protein of the chikungunya virus (CHIKV) and the capsid protein of the Sindbis virus (SIN) (4, 47). In the case of CHIKV, p62 binds to ubiquitinated capsid and targets it to autophagic degradation, while in the case of the SIN, and reminiscent of our data for Tat, the interaction between p62 and the SIN capsid does not require the p62 UBA domain, highlighting a potential original mechanism by which p62 recognizes autophagic substrates. As p62 could frequently interact with viral proteins (48), it would be interesting to decipher the molecular mechanism behind this ubiquitin-independent p62-mediated autophagic degradation, with a particular emphasis on the identification of other putative cellular partners linking p62 to its substrates. One potential candidate is the histone deacetylase HDAC6, whose deacetylase activity was shown to be regulated upon interaction with p62 (49). Furthermore, HDAC6 was also reported to deacetylate Tat, leading to a decrease in Tat transactivation activity (50).
Although cellular factors are involved in HIV-1 transcription, Tat is absolutely required for HIV-1 replication since it is needed for the elongation of viral transcripts (20). Tat is thus a perfect target to shut down viral production. Interestingly, Tat was shown to be actively secreted from infected cells and captured by several cell types (21, 42). Our results suggest that incoming Tat is also a target for autophagic degradation following internalization by bystander cells. The consequences of this degradation on the different known effects of Tat and, in particular, on the homeostasis of target cells warrant further investigation.
We discovered that autophagy is induced by HIV-1 Env at the very first steps of infection. This Env-induced autophagy is time regulated and initially triggers the degradation of any incoming Tat, avoiding the death of the target cells long enough to allow the virus to initiate its replication. If productive infection of the target cells proceeds, the virus blocks autophagy, and Tat, thereby protected from degradation, can fulfill its transcriptional role, enabling robust viral production.
Intracellular Tat concentration is tightly regulated. Indeed, we demonstrated here that Tat can be degraded by basal constitutive autophagy and upon Env-induced autophagy. Tat was also reported to inhibit the autophagy process in uninfected cells (33, 34), underlining the complex relationship between autophagy and this viral protein. This also seems to be the case for another retroviral transactivator, the human T-cell leukemia virus type 1 (HTLV-1) Tax protein. Indeed, it has been demonstrated that Tax is a substrate for autophagy degradation, but the underlying mechanism is still unknown (51). They also demonstrated that Tax is able to block the degradative phases of autophagy, suggesting that this viral protein could control its own stability and turnover (51).
Highly active antiretroviral therapy (HAART) can efficiently block viral replication and reduce the levels of circulating virus below the detection limit. Despite its efficacy, the virus is not eradicated due to the establishment of latent but replication-competent proviruses in reservoir cells probably located in peripheral blood and tissues. Viral latency is characterized by HIV-1 LTR transcriptional repression, and, interestingly, a hallmark of this process is the low level of Tat. Thus, specific degradation of Tat by autophagy could adversely contribute to the establishment of HIV-1 latency, which should be taken into account in the context of future antiviral strategies.
Since Tat is absolutely required for viral transcription, many therapeutic strategies targeting this viral protein have been considered, such as Tat antagonists (52), dominant negative Tat (53, 54), Tat-inhibitory peptides (55–58), or siRNAs against Tat (59–61). However, none of these strategies has been successful to date. The autophagy-mediated degradation of Tat presented here could represent a new way to target this viral protein in infected cells, blocking viral transcription and thus viral replication. Indeed, while HAART acts directly on viral enzymatic proteins, the induced degradation of Tat by autophagy could represent a new strategy based on a cellular response. Interestingly, some sirolimus-derived compounds (rapamycin and rapamune), which are strong inducers of autophagy and already used in therapeutics, were shown to possess anti-HIV-1 properties (62). Recently, Campbell and Spector demonstrated that vitamin D3 inhibits HIV replication through induction of autophagy (63). It would be interesting, therefore, to examine the autophagy-induced degradation of Tat in these contexts and its possible outcome on inhibition of HIV replication.
The results obtained in our study provide new evidence of the antiviral action of autophagy and highlight why the viruses have evolved strategies to block this process.
ACKNOWLEDGMENTS
The work was supported by institutional funds from the Centre National de la Recherche Scientifique (CNRS) and the Montpellier University I and II (UMI and UMII) and by grants from Sidaction and the Agence Nationale de Recherche sur le SIDA (ANRS). S.S. was the recipient of a fellowship from ANRS and Sidaction.
We thank M. Benkirane for providing the Tat K71R construct, T. Johansen for the GST-p62 plasmids, T. Yoshimori for the GFP-LC3 construct, and N. Chazal for the Gag expression vector. The anti-p24 antibody was obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH.
REFERENCES
- 1.Tanida I. 2011. Autophagy basics. Microbiol Immunol 55:1–11. doi: 10.1111/j.1348-0421.2010.00271.x. [DOI] [PubMed] [Google Scholar]
- 2.Suzuki K, Noda T, Ohsumi Y. 2004. Interrelationships among Atg proteins during autophagy in Saccharomyces cerevisiae. Yeast 21:1057–1065. doi: 10.1002/yea.1152. [DOI] [PubMed] [Google Scholar]
- 3.Uchiyama Y, Shibata M, Koike M, Yoshimura K, Sasaki M. 2008. Autophagy-physiology and pathophysiology. Histochem Cell Biol 129:407–420. doi: 10.1007/s00418-008-0406-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Orvedahl A, MacPherson S, Sumpter R Jr, Talloczy Z, Zou Z, Levine B. 2010. Autophagy protects against Sindbis virus infection of the central nervous system. Cell Host Microbe 7:115–127. doi: 10.1016/j.chom.2010.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sumpter R Jr, Levine B. 2011. Selective autophagy and viruses. Autophagy 7:260–265. doi: 10.4161/auto.7.3.14281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Deretic V, Saitoh T, Akira S. 2013. Autophagy in infection, inflammation and immunity. Nat Rev Immunol 13:722–737. doi: 10.1038/nri3532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pyo JO, Nah J, Jung YK. 2012. Molecules and their functions in autophagy. Exp Mol Med 44:73–80. doi: 10.3858/emm.2012.44.2.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Weidberg H, Shpilka T, Shvets E, Elazar Z. 2010. Mammalian Atg8s: one is simply not enough. Autophagy 6:808–809. doi: 10.4161/auto.6.6.12579. [DOI] [PubMed] [Google Scholar]
- 9.Weidberg H, Shvets E, Shpilka T, Shimron F, Shinder V, Elazar Z. 2010. LC3 and GATE-16/GABARAP subfamilies are both essential yet act differently in autophagosome biogenesis. EMBO J 29:1792–1802. doi: 10.1038/emboj.2010.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Johansen T, Lamark T. 2011. Selective autophagy mediated by autophagic adapter proteins. Autophagy 7:279–296. doi: 10.4161/auto.7.3.14487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Birgisdottir AB, Lamark T, Johansen T. 2013. The LIR motif—crucial for selective autophagy. J Cell Sci 126:3237–3247. doi: 10.1242/jcs.126128. [DOI] [PubMed] [Google Scholar]
- 12.Bjorkoy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, Stenmark H, Johansen T. 2005. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol 171:603–614. doi: 10.1083/jcb.200507002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bjorkoy G, Lamark T, Johansen T. 2006. p62/SQSTM1: a missing link between protein aggregates and the autophagy machinery. Autophagy 2:138–139. doi: 10.4161/auto.2.2.2405. [DOI] [PubMed] [Google Scholar]
- 14.Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Overvatn A, Bjorkoy G, Johansen T. 2007. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 282:24131–24145. doi: 10.1074/jbc.M702824200. [DOI] [PubMed] [Google Scholar]
- 15.Ishimura R, Tanaka K, Komatsu M. 2014. Dissection of the role of p62/Sqstm1 in activation of Nrf2 during xenophagy. FEBS Lett 588:822–828. doi: 10.1016/j.febslet.2014.01.045. [DOI] [PubMed] [Google Scholar]
- 16.Knodler LA, Celli J. 2011. Eating the strangers within: host control of intracellular bacteria via xenophagy. Cell Microbiol 13:1319–1327. doi: 10.1111/j.1462-5822.2011.01632.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Arrildt KT, Joseph SB, Swanstrom R. 2012. The HIV-1 env protein: a coat of many colors. Curr HIV/AIDS Rep 9:52–63. doi: 10.1007/s11904-011-0107-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Freed EO, Mouland AJ. 2006. The cell biology of HIV-1 and other retroviruses. Retrovirology 3:77. doi: 10.1186/1742-4690-3-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mohammadi P, Desfarges S, Bartha I, Joos B, Zangger N, Munoz M, Gunthard HF, Beerenwinkel N, Telenti A, Ciuffi A. 2013. 24 hours in the life of HIV-1 in a T cell line. PLoS Pathog 9:e1003161. doi: 10.1371/journal.ppat.1003161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ott M, Geyer M, Zhou Q. 2011. The control of HIV transcription: keeping RNA polymerase II on track. Cell Host Microbe 10:426–435. doi: 10.1016/j.chom.2011.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Debaisieux S, Rayne F, Yezid H, Beaumelle B. 2012. The ins and outs of HIV-1 Tat. Traffic 13:355–363. doi: 10.1111/j.1600-0854.2011.01286.x. [DOI] [PubMed] [Google Scholar]
- 22.Huigen MC, Kamp W, Nottet HS. 2004. Multiple effects of HIV-1 trans-activator protein on the pathogenesis of HIV-1 infection. Eur J Clin Invest 34:57–66. doi: 10.1111/j.1365-2362.2004.01282.x. [DOI] [PubMed] [Google Scholar]
- 23.Rayne F, Vendeville A, Bonhoure A, Beaumelle B. 2004. The ability of chloroquine to prevent tat-induced cytokine secretion by monocytes is implicated in its in vivo anti-human immunodeficiency virus type 1 activity. J Virol 78:12054–12057. doi: 10.1128/JVI.78.21.12054-12057.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vendeville A, Rayne F, Bonhoure A, Bettache N, Montcourrier P, Beaumelle B. 2004. HIV-1 Tat enters T cells using coated pits before translocating from acidified endosomes and eliciting biological responses. Mol Biol Cell 15:2347–2360. doi: 10.1091/mbc.E03-12-0921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Biard-Piechaczyk M, Borel S, Espert L, de Bettignies G, Coux O. 2012. HIV-1, ubiquitin and ubiquitin-like proteins: the dialectic interactions of a virus with a sophisticated network of post-translational modifications. Biol Cell 104:165–187. doi: 10.1111/boc.201100112. [DOI] [PubMed] [Google Scholar]
- 26.Borel S, Espert L, Biard-Piechaczyk M. 2012. Macroautophagy regulation during HIV-1 infection of CD4+ T cells and macrophages. Front Immunol 3:97. doi: 10.3389/fimmu.2012.00097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Espert L, Biard-Piechaczyk M. 2009. Autophagy in HIV-induced T cell death. Curr Top Microbiol Immunol 335:307–321. doi: 10.1007/978-3-642-00302-8_15. [DOI] [PubMed] [Google Scholar]
- 28.Blanchet FP, Moris A, Nikolic DS, Lehmann M, Cardinaud S, Stalder R, Garcia E, Dinkins C, Leuba F, Wu L, Schwartz O, Deretic V, Piguet V. 2010. Human immunodeficiency virus-1 inhibition of immunoamphisomes in dendritic cells impairs early innate and adaptive immune responses. Immunity 32:654–669. doi: 10.1016/j.immuni.2010.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Blanchet FP, Piguet V. 2010. Immunoamphisomes in dendritic cells amplify TLR signaling and enhance exogenous antigen presentation on MHC-II. Autophagy 6:816–818. doi: 10.4161/auto.6.6.12623. [DOI] [PubMed] [Google Scholar]
- 30.Denizot M, Varbanov M, Espert L, Robert-Hebmann V, Sagnier S, Garcia E, Curriu M, Mamoun R, Blanco J, Biard-Piechaczyk M. 2008. HIV-1 gp41 fusogenic function triggers autophagy in uninfected cells. Autophagy 4:998–1008. doi: 10.4161/auto.6880. [DOI] [PubMed] [Google Scholar]
- 31.Espert L, Denizot M, Grimaldi M, Robert-Hebmann V, Gay B, Varbanov M, Codogno P, Biard-Piechaczyk M. 2006. Autophagy is involved in T cell death after binding of HIV-1 envelope proteins to CXCR4. J Clin Invest 116:2161–2172. doi: 10.1172/JCI26185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Espert L, Varbanov M, Robert-Hebmann V, Sagnier S, Robbins I, Sanchez F, Lafont V, Biard-Piechaczyk M. 2009. Differential role of autophagy in CD4 T cells and macrophages during X4 and R5 HIV-1 infection. PLoS One 4:e5787. doi: 10.1371/journal.pone.0005787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Van Grol J, Subauste C, Andrade RM, Fujinaga K, Nelson J, Subauste CS. 2010. HIV-1 inhibits autophagy in bystander macrophage/monocytic cells through Src-Akt and STAT3. PLoS One 5:e11733. doi: 10.1371/journal.pone.0011733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li JC, Au KY, Fang JW, Yim HC, Chow KH, Ho PL, Lau AS. 2011. HIV-1 trans-activator protein dysregulates IFN-gamma signaling and contributes to the suppression of autophagy induction. AIDS 25:15–25. doi: 10.1097/QAD.0b013e328340fd61. [DOI] [PubMed] [Google Scholar]
- 35.Rayne F, Debaisieux S, Yezid H, Lin YL, Mettling C, Konate K, Chazal N, Arold ST, Pugniere M, Sanchez F, Bonhoure A, Briant L, Loret E, Roy C, Beaumelle B. 2010. Phosphatidylinositol-(4,5)-bisphosphate enables efficient secretion of HIV-1 Tat by infected T-cells. EMBO J 29:1348–1362. doi: 10.1038/emboj.2010.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bres V, Kiernan RE, Linares LK, Chable-Bessia C, Plechakova O, Treand C, Emiliani S, Peloponese JM, Jeang KT, Coux O, Scheffner M, Benkirane M. 2003. A non-proteolytic role for ubiquitin in Tat-mediated transactivation of the HIV-1 promoter. Nat Cell Biol 5:754–761. doi: 10.1038/ncb1023. [DOI] [PubMed] [Google Scholar]
- 37.Schneider R, Campbell M, Nasioulas G, Felber BK, Pavlakis GN. 1997. Inactivation of the human immunodeficiency virus type 1 inhibitory elements allows Rev-independent expression of Gag and Gag/protease and particle formation. J Virol 71:4892–4903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Charret R, Fauré-Fremiet E. 1967. Technique de rassemblement de microorganismes: preinclusion dans un caillot de fibrine. J Microsc 6:1063–1066. [Google Scholar]
- 39.Zar JH. 1996. Biostatistical analysis, 3rd ed. Prentice-Hall, Upper Saddle River, NJ. [Google Scholar]
- 40.Zhou D, Spector SA. 2008. Human immunodeficiency virus type-1 infection inhibits autophagy. AIDS 22:695–699. doi: 10.1097/QAD.0b013e3282f4a836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shvets E, Fass E, Scherz-Shouval R, Elazar Z. 2008. The N terminus and Phe52 residue of LC3 recruit p62/SQSTM1 into autophagosomes. J Cell Sci 121:2685–2695. doi: 10.1242/jcs.026005. [DOI] [PubMed] [Google Scholar]
- 42.Johri MK, Mishra R, Chhatbar C, Unni SK, Singh SK. 2011. Tits and bits of HIV Tat protein. Expert Opin Biol Ther 11:269–283. doi: 10.1517/14712598.2011.546339. [DOI] [PubMed] [Google Scholar]
- 43.Kiernan RE, Vanhulle C, Schiltz L, Adam E, Xiao H, Maudoux F, Calomme C, Burny A, Nakatani Y, Jeang KT, Benkirane M, Van Lint C. 1999. HIV-1 tat transcriptional activity is regulated by acetylation. EMBO J 18:6106–6118. doi: 10.1093/emboj/18.21.6106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kimpton J, Emerman M. 1992. Detection of replication-competent and pseudotyped human immunodeficiency virus with a sensitive cell line on the basis of activation of an integrated beta-galactosidase gene. J Virol 66:2232–2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Strebel K. 2013. HIV accessory proteins versus host restriction factors. Curr Opin Virol 3:692–699. doi: 10.1016/j.coviro.2013.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rogov V, Dotsch V, Johansen T, Kirkin V. 2014. Interactions between autophagy receptors and ubiquitin-like proteins form the molecular basis for selective autophagy. Mol Cell 53:167–178. doi: 10.1016/j.molcel.2013.12.014. [DOI] [PubMed] [Google Scholar]
- 47.Judith D, Mostowy S, Bourai M, Gangneux N, Lelek M, Lucas-Hourani M, Cayet N, Jacob Y, Prevost MC, Pierre P, Tangy F, Zimmer C, Vidalain PO, Couderc T, Lecuit M. 2013. Species-specific impact of the autophagy machinery on Chikungunya virus infection. EMBO Rep 14:534–544. doi: 10.1038/embor.2013.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gregoire IP, Rabourdin-Combe C, Faure M. 2012. Autophagy and RNA virus interactomes reveal IRGM as a common target. Autophagy 8:1136–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yan J, Seibenhener ML, Calderilla-Barbosa L, Diaz-Meco MT, Moscat J, Jiang J, Wooten MW, Wooten MC. 2013. SQSTM1/p62 interacts with HDAC6 and regulates deacetylase activity. PLoS One 8:e76016. doi: 10.1371/journal.pone.0076016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Huo L, Li D, Sun X, Shi X, Karna P, Yang W, Liu M, Qiao W, Aneja R, Zhou J. 2011. Regulation of Tat acetylation and transactivation activity by the microtubule-associated deacetylase HDAC6. J Biol Chem 286:9280–9286. doi: 10.1074/jbc.M110.208884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tang SW, Chen CY, Klase Z, Zane L, Jeang KT. 2013. The cellular autophagy pathway modulates human T-cell leukemia virus type 1 replication. J Virol 87:1699–1707. doi: 10.1128/JVI.02147-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hsu MC, Schutt AD, Holly M, Slice LW, Sherman MI, Richman DD, Potash MJ, Volsky DJ. 1991. Inhibition of HIV replication in acute and chronic infections in vitro by a Tat antagonist. Science 254:1799–1802. doi: 10.1126/science.1763331. [DOI] [PubMed] [Google Scholar]
- 53.Green M, Ishino M, Loewenstein PM. 1989. Mutational analysis of HIV-1 Tat minimal domain peptides: identification of trans-dominant mutants that suppress HIV-LTR-driven gene expression. Cell 58:215–223. doi: 10.1016/0092-8674(89)90417-0. [DOI] [PubMed] [Google Scholar]
- 54.Pearson L, Garcia J, Wu F, Modesti N, Nelson J, Gaynor R. 1990. A transdominant Tat mutant that inhibits tat-induced gene expression from the human immunodeficiency virus long terminal repeat. Proc Natl Acad Sci U S A 87:5079–5083. doi: 10.1073/pnas.87.13.5079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Agbottah E, Zhang N, Dadgar S, Pumfery A, Wade JD, Zeng C, Kashanchi F. 2006. Inhibition of HIV-1 virus replication using small soluble Tat peptides. Virology 345:373–389. doi: 10.1016/j.virol.2005.09.062. [DOI] [PubMed] [Google Scholar]
- 56.D'Orso I, Grunwell JR, Nakamura RL, Das C, Frankel AD. 2008. Targeting tat inhibitors in the assembly of human immunodeficiency virus type 1 transcription complexes. J Virol 82:9492–9504. doi: 10.1128/JVI.00763-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hamy F, Felder ER, Heizmann G, Lazdins J, Aboul-ela F, Varani G, Karn J, Klimkait T. 1997. An inhibitor of the Tat/TAR RNA interaction that effectively suppresses HIV-1 replication. Proc Natl Acad Sci U S A 94:3548–3553. doi: 10.1073/pnas.94.8.3548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Okamoto H, Cujec TP, Peterlin BM, Okamoto T. 2000. HIV-1 replication is inhibited by a pseudo-substrate peptide that blocks Tat transactivation. Virology 270:337–344. doi: 10.1006/viro.2000.0311. [DOI] [PubMed] [Google Scholar]
- 59.Coburn GA, Cullen BR. 2002. Potent and specific inhibition of human immunodeficiency virus type 1 replication by RNA interference. J Virol 76:9225–9231. doi: 10.1128/JVI.76.18.9225-9231.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lee MT, Coburn GA, McClure MO, Cullen BR. 2003. Inhibition of human immunodeficiency virus type 1 replication in primary macrophages by using Tat- or CCR5-specific small interfering RNAs expressed from a lentivirus vector. J Virol 77:11964–11972. doi: 10.1128/JVI.77.22.11964-11972.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Surabhi RM, Gaynor RB. 2002. RNA interference directed against viral and cellular targets inhibits human immunodeficiency virus type 1 replication. J Virol 76:12963–12973. doi: 10.1128/JVI.76.24.12963-12973.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Donia M, McCubrey JA, Bendtzen K, Nicoletti F. 2010. Potential use of rapamycin in HIV infection. Br J Clin Pharmacol 70:784–793. doi: 10.1111/j.1365-2125.2010.03735.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Campbell GR, Spector SA. 2012. Toll-like receptor 8 ligands activate a vitamin D mediated autophagic response that inhibits human immunodeficiency virus type 1. PLoS Pathog 8:e1003017. doi: 10.1371/journal.ppat.1003017. [DOI] [PMC free article] [PubMed] [Google Scholar]