

Abstract
Channelrhodopsins (ChRs) from green flagellate algae function as light-gated ion channels when expressed heterologously in mammalian cells. Considerable interest has focused on understanding the molecular mechanisms of ChRs to bioengineer their properties for specific optogenetic applications such as elucidating the function of specific neurons in brain circuits. While most studies have used channelrhodopsin-2 from Chlamydomonas reinhardtii (CrChR2), in this work low-temperature Fourier transform infrared-difference spectroscopy is applied to study the conformational changes occurring during the primary phototransition of the red-shifted ChR1 from Chlamydomonas augustae (CaChR1). Substitution with isotope-labeled retinals or the retinal analogue A2, site-directed mutagenesis, hydrogen–deuterium exchange, and H218O exchange were used to assign bands to the retinal chromophore, protein, and internal water molecules. The primary phototransition of CaChR1 at 80 K involves, in contrast to that of CrChR2, almost exclusively an all-trans to 13-cis isomerization of the retinal chromophore, as in the primary phototransition of bacteriorhodopsin (BR). In addition, significant differences are found for structural changes of the protein and internal water(s) compared to those of CrChR2, including the response of several Asp/Glu residues to retinal isomerization. A negative amide II band is identified in the retinal ethylenic stretch region of CaChR1, which reflects along with amide I bands alterations in protein backbone structure early in the photocycle. A decrease in the hydrogen bond strength of a weakly hydrogen bonded internal water is detected in both CaChR1 and CrChR2, but the bands are much broader in CrChR2, indicating a more heterogeneous environment. Mutations involving residues Glu169 and Asp299 (homologues of the Asp85 and Asp212 Schiff base counterions, respectively, in BR) lead to the conclusion that Asp299 is protonated during P1 formation and suggest that these residues interact through a strong hydrogen bond that facilitates the transfer of a proton from Glu169.
Considerable interest has focused on understanding the molecular mechanism of channelrhodopsins (ChRs) found in green flagellate algae.1,2 ChRs expressed in mammalian cells function as optogenetic light-activated ion channels.3−9 For example, neurons expressing ChR genes in a functional form exhibit light-activated action potentials.7 Furthermore, ChRs with different wavelengths of absorption can be used to selectively activate different neurons.10 This new photonic capability has led to many incisive applications, including the spatial mapping of brain circuits,11 and holds promise for the future treatment of a variety of human neuropathologies such as Parkinson’s disease.12,13 An important goal is to understand at the molecular level key properties of ChRs such as ion gating, ion selectivity, visible absorption wavelength tuning, and channel kinetics. Such knowledge can lead to the engineering of improved light-triggered ion channels for a variety of applications.
In this work, a ChR1 from Chlamydomonas augustae (CaChR1) is studied at low temperature using Frourier transform infrared difference (FTIR-difference) spectroscopy. This approach allows us to measure conformational changes occurring in the primary phototransition of ChRs (ChR → P1) and compare the changes to those of other microbial rhodopsins, including ChR2 from Chlamydomonas reinhardtii (CrChR2). CaChR1 and other ChR1-type proteins are lower efficiency cation channels compared to CrChR2, but their red-shifted λmax values (525 nm for CaChR1 vs 470 nm for CrChR2) and slower light inactivation14 make them a candidate for some optogenetic applications. In addition, a recent resonance Raman spectroscopy (RRS) study has established that unlike CrChR2,15 light- and dark-adapted CaChR1 have an all-trans retinal structure very similar to that of the light-driven proton pump bacteriorhodopsin (BR) and sensory rhodopsin II from Natronobacterium pharonis (NpSRII).16 This feature is advantageous for spectroscopic studies because it helps minimize the possibility of parallel photocycles that can complicate analysis of structural changes.
FTIR-difference spectroscopy combined with site-directed mutagenesis, isotope labeling, and hydrogen–deuterium (H–D) exchange provide additional information about structural changes occurring in the protein and chromophore at the level of individual molecular groups as previously demonstrated with BR.17−19 Our results show that CaChR1 and CrChR2 undergo distinctly different protein conformational changes during the primary phototransition. Compared to BR, both appear to undergo more extensive protein changes early in the photocycle. In the case of CaChR1, a contribution from an amide II band arising from backbone peptide structural changes is detected. Spectral changes occurring in the carboxylic acid region above 1700 cm–1 reflect a distinctly different pattern of alterations of Asp and/or Glu groups for the two ChRs. Mutations at Glu169 and Asp299 in CaChR1, homologues to the counterions Asp85 and Asp212 in BR, suggest that the two residues interact together to form a strong hydrogen bond that facilitates proton transfer during the formation of the P1 intermediate. Bands caused by weakly hydrogen bonded internal water molecules that change during the primary transition are identified in both CaChR1 and CrChR2 in the 3600–3700 cm–1 region, analogous to bands previously detected in other microbial rhodopsins. In the case of CrChR2, these bands are much broader than those of CaChR1, indicating significant differences in the environment of these internal waters.
Materials and Methods
Expression, Purification, and Reconstitution of CaChR1 and Its Mutants, CrChR2, and Bacteriorhodopsin
The expression, purification, and reconstitution of wild-type and mutant CaChR1 were recently described.16 Briefly, the 7TM domain of CaChR1 was expressed from Pichia pastoris in the presence of 5 μM all-trans-retinal. Cells were grown for 2 days after induction with methanol, harvested by low-speed centrifugation, and disrupted with a bead beater. Membrane fragments were collected by centrifugation for 1 h at 38000 rpm. The protein was partially purified on a Ni-NTA agarose column (Qiagen, Hilden, Germany) after being solubilized by overnight incubation in 3% dodecyl maltoside (DDM). For membrane reconstitution, the protein was eluted in 20 mM HEPES (pH 7.4), 100 mM NaCl, 0.05% DDM, and 300 mM imidazole and mixed with Escherichia coli polar lipids (ECPL) (Avanti Polar Lipids, Alabaster, AL) at a concentration of 5 μg/mL in 10% octyl glucoside with a mass ratio of 1:10 (CaChR1:ECPL). The mixture was incubated at room temperature for 1 h. Detergent was removed using SM-2 Bio-Beads (Bio-Rad, Hercules, CA), and the reconstituted CaChR1 proteoliposomes were pelleted at 10000 rpm for 10 min. The pellet was resuspended in 10 mM phosphate buffer (pH 7.0) and 100 mM NaCl and washed twice with the same buffer.
Similar procedures were used to express and reconstitute CrChR2 as described above except the cDNA of CrChR2 encoded residues 1–309 with a C-terminal six-His tag sequence that was cloned in the pPIC9K vector (Invitrogen) within its EcoRI and NotI sites. The production of detergent-purified CrChR2 followed the methods of Bamann et al.20 and Li et al.21 For membrane reconstitution, CrChR2 in 1% decyl maltoside (DM) was mixed with DMPC (Avanti Polar Lipids) at a ratio of 1:20 (w/w). CrChR2 was also reconstituted in EPCL using procedures similar to those described here for CaChR1.
Bacteriorhodopsin in its native purple membrane was isolated from Halobacterium salinarum using standard procedures previously reported.22
Regeneration of CaChR1 and BR with Isotope-Labeled all-trans-Retinal and A2 Retinal
The synthesis of all-trans-[15-13C,15-2H]retinal and all-trans-[14,15-2H2]retinal was previously described.23 A2 retinal (3,4-dehydroretinal) was purchased from Toronto Research Chemicals (catalog no. D230075, CAS Registry No. 472-87-7). To prepare bleached CaChR1 in which the retinal chromophore is removed from the binding pocket, proteolipid prepared as described above was suspended in 25 mM hydroxylamine buffer with 50 mM K2HPO4 (pH 7.2) and exposed to 530 nm LED illumination (5 mW/cm2) for 40 min. Bleaching was monitored by UV–visible spectroscopy using a Cary 50 spectrometer (Agilent Technologies, Inc., Santa Clara, CA). After >95% conversion of the retinal chromophore to retinal oxime, the sample was then dialyzed against 300 mM NaCl and 50 mM K2HPO4 (pH 7.2) to remove excess hydroxylamine and free retinal oxime. A 2-fold stoichiometric excess of the isotope-labeled retinal or A2 analogue was then added as a 2 mM EtOH solution, and the extent of regeneration of CaChR1 was determined by measuring the 280 nm:530 nm ratio. The incorporation of the isotope-labeled retinal was verified by measuring the RRS (data not shown) using a Bruker Senterra confocal Raman microscope (Olympus BX51M) and 785 nm laser excitation similar to measurements previously reported for CaChR1.16 A similar procedure was used for the bleaching of BR where 100 mM hydroxylamine was used with a total reaction time of approximately 24 h.
Low-Temperature FTIR-Difference Spectroscopy
The protein samples for the low-temperature FTIR measurements were prepared as previously reported24−26 using approximately 50 μg of the protein for each experiment. The samples were deposited on BaF2 windows and slowly dried in a drybox. Samples were then rehydrated through the vapor phase with a small drop (∼0.5 μL) of H2O, H218O,or D2O and sealed in a sample cell with another BaF2 window, and the cell was mounted in a liquid nitrogen cryostat (Oxford Instruments, OptistatDN). For measurements of CaChR1 at 80 K, the samples were first cooled from room temperature in the dark to avoid trapping of photointermediates. Note that unlike BR, which is light-adapted before low-temperature FTIR measurements, CaChR1 is not because it does not exhibit dark adaptation.16
For CaChR1 samples, the film was allowed to equilibrate at 80 K for >1 h after which spectra are recorded using the following cycle: (1) dark, (2) illumination with a 505 nm LED (all LEDs and LED control systems from Thorlabs Inc., Newton, NJ), (3) dark, and (4) illumination with a 590 nm LED. This cycle was repeated at least 50 times, and the corresponding difference spectra (e.g., 2 – 1, 3 – 1, and 4 – 3) were calculated. In addition to the CaChR1 film, BR and CrChR2 films were measured using an identical procedure with the exception of the illumination conditions: for BR, 530 and 625 nm illumination LEDs were used; for CrChR2, 455 and 530 nm illumination LEDs were used. Each acquired spectrum consisted of 200 scans (approximately 1 min of total time) recorded at 4 cm–1 resolution using a Bio-Rad FTS-60A FTIR spectrometer (Bio-Rad, Digilab Division, Cambridge, MA) equipped with a liquid nitrogen-cooled HgCdTe detector.
Results
The Primary Phototransition of CaChR1 Involves all-trans- to 13-cis Chromophore Isomerization
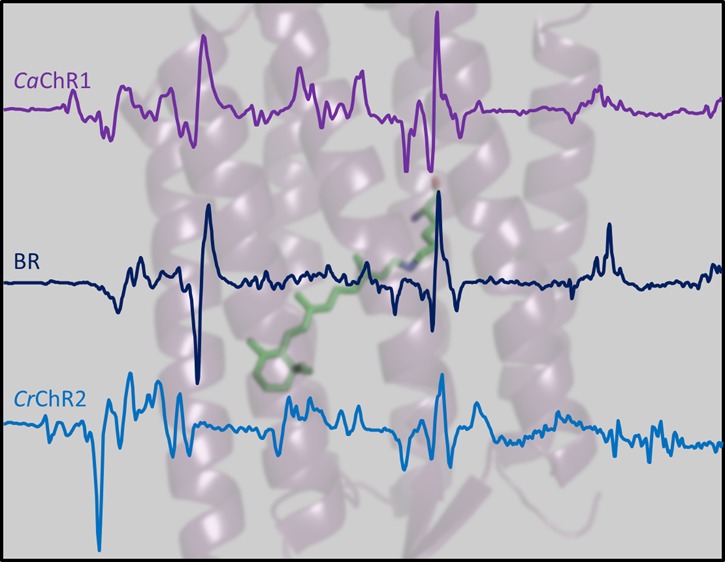
The P1 intermediate is the red-shifted primary photoproduct found in the photocycle of most ChRs and is analogous to the K intermediate in the BR photocycle (see, for example, Figure 1 of ref (27)). The difference spectrum of CaChR1 → P1 is similar to the difference spectrum of BR → K28−31 for bands assigned to the retinal chromophore as shown in Figure 1. In the ethylenic C=C stretching region from 1500 to 1600 cm–1, negative/positive bands appear at (−)1535 and (+)1521 cm–1 that are assigned to the ethylenic C=C stretching modes of the retinal chromophore for the initial dark state and P1 photointermediate, respectively (see below). The negative/positive ethylenic bands in CaChR1 are upshifted to a higher frequency compared to those of BR (1529/1514 cm–1) as expected because of CaChR1’s blue-shifted visible absorption (λmax = 523 nm)21 relative to that of light-adapted BR (λmax = 570 nm) and in agreement with the inverse linear relationship that is observed between λmax and νC=C for most microbial and animal rhodopsins.24,32,33 In addition, the negative band at 1535 cm–1 is close to the frequency assigned to the ethylenic mode of CaChR1 measured by RRS.16 The slight increase in frequency compared to that of the RRS (1532 cm–1) is expected due to splitting effects between the positive and negative ethylenic bands in the FTIR-difference spectrum.
Figure 1.

Comparison of FTIR-difference spectra for CaChR1, BR, and CrChR2 taken at 80 K over the 800–1800 cm–1 region. Peaks labeled in bold are discussed in the text. Y axis markers indicate 2.5 mOD for CaChR1, 5 mOD for BR, and 0.6 mOD for CrChR2.
The fingerprint region (1100–1250 cm–1), which reflects the mixed C–C stretching modes of the retinylidene chromophore, is particularly sensitive to the retinal isomeric state.34,35 An earlier RRS study of CaChR1 revealed bands near 1201, 1172, and 1163 cm–1 that are highly characteristic of an all-trans configuration of retinal in the unphotolyzed state of CaChR1.16 Negative bands appear at similar frequencies in the FTIR-difference spectrum of CaChR1 and BR (Figure 1), further indicating that the primary phototransition at low temperatures involves isomerization of an all-trans retinylidene chromophore.
The largest positive band in this region for CaChR1 appears at 1196 cm–1, close to the band at 1194 cm–1 in BR assigned to the mixed stretching vibration of the 13-cis retinylidene chromophore.35 Importantly, almost all microbial rhodopsins containing an essentially pure all-trans-retinal composition display a spectral pattern similar to that of CaChR1 and BR in this region with two negative bands near 1200 and 1165–1170 cm–1 and a positive band near 1195 cm–1, including archaerhodopsin-3 (AR3),36 green- and blue-absorbing proteorhodopsin (GPR and BPR, respectively),24,37NpSRII,38 and halorhodopsin from H. salinarum (HsHR).39
Note that the upshift in the frequency of the negative band at 1205 cm–1 compared to that of the RRS spectrum (1201 cm–1) is most likely due to overlap/splitting effects with the positive band at 1196 cm–1. Overall, the similarity of the fingerprint region between CaChR1 and BR firmly establishes that the chromophore undergoes an all-trans to 13-cis isomerization as previously established for BR on the basis of RRS studies of isotopically labeled retinals.34,35 In contrast, analysis of this region for CrChR2 reconstituted in both EPCL and DMPC (Figure S1 of the Supporting Information) along with photoreversibility measurements (Figure S2 of the Supporting Information) reveals that CrChR2 contains a higher level of 13-cis-retinal, which is consistent with earlier RRS measurements.16
Assignment of the Schiff Base C=N Stretch and Amide I Bands
An early FTIR-difference study of the primary phototransition of light-adapted BR using isotope-labeled retinals and H–D exchange led to the assignment of the Schiff base (SB) C=N stretching mode of the K intermediate to a positive band near 1609 cm–1.40 The corresponding assignment of this band for the P1 intermediate has not yet been made for any ChRs. As a first step, isotope-labeled retinals and H–D exchange were used to assign the νC=N of the dark-adapted CaChR1 that should appear as a negative band near 1646 cm–1 based on recent RRS measurements.16,41 As seen in Figure 2, a negative band at a slightly higher frequency (1651 cm–1) seen in the CaChR1 membranes and bleached CaChR1 membranes regenerated with A1 and A2 retinal (Figure 2, spectra A–C, respectively) disappears when CaChR1 is regenerated with either all-trans-[15-13C,15-2H]retinal or all-trans-[14,15-2H2]retinal and also upon H–D exchange (Figure 2D–F). The latter leads to deuteration of the protonated SB (C=NH+ → C=ND+). Because all of these substitutions (Figure 2D−F) are expected to cause a downshift of the νC=N in analogy with BR,40 this result establishes the assignment of the negative 1651 cm–1 band to the C=N stretch mode [as opposed, for example, to an amide I band (see below)]. FTIR-difference spectra measured for BR regenerated with these two isotope retinal labels also support this conclusion (see Figure S3 of the Supporting Information). The slightly higher frequency measured in the FTIR-difference spectrum at 1651 cm–1 compared to that of the spectrum in the RRS is most likely due to the overlap of hidden bands.
Figure 2.

FTIR differences of CaChR1 recorded at 80 K over the 1600–1700 cm–1 region for an untreated sample and samples subjected to bleaching and retinal regeneration as described in Materials and Methods: (A) CaChR1 in H2O, (B) CaChR1 in H2O regenerated with A1 retinal, (C) CaChR1 in H2O regenerated with A2 retinal, (D) CaChR1 in H2O regenerated with [15-13C,15-2H]retinal, (E) CaChR1 in H2O regenerated with [14,15-2H2]retinal, and (F) CaChR1 in D2O. Peaks labeled in bold are discussed in the text. Y axis markers are approximately 1.5 mOD for all spectra. For detailed bleaching and regeneration procedures as well as spectral acquisition parameters, see Materials and Methods.
As a control, CaChR1 regenerated with normal all-trans-A1 retinal was also measured. As shown in Figure 2B, no spectral changes are observed because of the regeneration procedures, as expected. Interestingly, no changes are seen in this region for CaChR1 regenerated with A2 retinal (Figure 2C), which, unlike the isotope labels, causes a substantial red-shift in the λmax from 525 nm with A1 retinal to 550 nm with A2 (data not shown; see ref (42)). This result indicates that the extra double bond in the A2 retinal located in the β-ionone ring while causing an increased level of electron delocalization in the polyene chain, does not significantly affect the charge distribution over the C=N bond.
On a similar basis, the positive band near 1635 cm–1 can be assigned to the C=N stretch of the P1 intermediate that is tentatively downshifted to 1610, 1617, and 1606 cm–1 for all-trans-[15-13C,15-2H]retinal, all-trans-[14,15-2H2]retinal, and deuterated SB of the all-trans-retinal, respectively (Figure 2D–F). Note that in comparison to the 1635 cm–1 band, the 1624 cm–1 band is insensitive to these retinal isotope alterations and most likely arises from a protein vibration (see below). One exception is the case of H–D exchange in which the 1651 cm–1 negative band is expected to downshift ∼26 cm–1 based on RRS measurements,16 thus explaining the drop in intensity near 1624 cm–1. Because the overall downshift of the C=N vibration from 1651 to 1635 cm–1 during the CaChR1 → P1 transition is much smaller than that during the BR → K transition (from 1639 to 1608 cm–1),40 this may indicate that the change in microenvironment near the SB occurring during this transition is smaller in CaChR1 than in BR (see Discussion and Conclusions). Note also that no significant shift was found for the 1635 cm–1 band for A2 retinal (3,4-dehydroretinal) (Figure 2C), a further indication that the electron density for the SB C=N bond is unaltered in the P1 intermediate even though the λmax of the P1 intermediate is red-shifted (data not shown).
The 1600–1700 cm–1 region is also expected to exhibit bands due to the amide I mode that consists predominantly of the C=O stretch of peptide backbone groups. In the case of CaChR1, several bands in this region may be due to the amide I mode, including the negative bands at 1661 and 1687 cm–1 and the positive band at 1667 cm–1 (Figure 1). Note, however, that the intensity of these bands does not compare to the intensity of the negative band at 1667 cm–1 observed in CrChR2 (see Discussion and Conclusions).
Evidence of Changes in the Amide II Protein Mode in the Primary Phototransition of CaChR1
In addition to the negative/positive bands at 1535 and 1521 cm–1 assigned to the ethylenic mode of the CaChR1 dark-adapted state and P1 intermediate, respectively, as discussed above, a second prominent negative band appears in the ethylenic region at 1553 cm–1 (Figures 1 and 3). This band may indicate the existence of a second blue-shifted species or additional ethylenic vibration from the 523 nm absorbing species that contributes to the CaChR1 photocycle and gives rise to additional ethylenic bands. A second possibility, however, is that this peak arises from alterations in the protein backbone structure. In fact, the amide II band that consists of a coupled C–N stretch and NH bending motion of the peptide group of the protein backbone also falls in this region.43
Figure 3.

Spectra from Figure 2 over the 1500–1600 cm–1 region: (A) CaChR1 in H2O, (B) CaChR1 in H2O regenerated with A1 retinal, (C) CaChR1 in H2O regenerated with A2 retinal, (D) CaChR1 in H2O regenerated with [15-13C,15-2H]retinal, (E) CaChR1 in H2O regenerated with [14,15-2H2]retinal, and (F) CaChR1 in D2O. Peaks labeled in bold are discussed in the text. Y axis markers are approximately 1.75 mOD for all spectra.
To distinguish between the two possibilities of chromophore versus protein structural changes, the two retinal isotope substitutions and substitution with A2 retinal were analyzed in this region. As expected, the negative/positive ethylenic bands at 1535 and 1521 cm–1 are all altered by the isotope substitutions near the SB (see spectra D and E of Figure 3). As established previously, the ethylenic normal modes consist of a mixture of C=C stretches involving various double bonds, including the C13=C14 bond44,45 in all-trans and 13-cis protonated SB (PSB) retinals. Hence, it is not surprising that the all-trans-[14,15-2H2]retinal has the largest effect with an apparent shift of the negative/positive bands from 1535/1521 to 1527/1508 cm–1. In the case of all-trans-[15-13C,15-2H]retinal, the shift is smaller to 1530/1515 cm–1. As expected, the ethylenic bands also downshift in the A2 retinal substitution (1525/1513 cm–1) in agreement with the red-shift of the visible absorption.14 Similar qualitative effects for bands assigned to the ethylenic mode in BR are also observed for the same isotope labels as shown in Figure S3 of the Supporting Information.
In contrast to the behavior of the assigned ethylenic bands, the negative 1553 cm–1 band is not affected by any of isotope labels or A2 retinal, strongly indicating it arises from a protein vibration. The one exception is its near disappearance upon H–D exchange. One likely explanation is that H–D exchange of peptide NH groups causes a shift of the amide II band to the 1430–1460 cm–1 range (often termed the amide II′ band) as is observed in the case of BR.46 Consistent with this explanation, a decrease in positive intensity is observed near 1440 cm–1 in the CaChR1 → P1 spectrum recorded in D2O but not in the other spectra (see Figure 4). This could be explained by the downshift of the 1553 cm–1 negative band to near this frequency. However, complete exchange of all the peptide groups in a protein backbone is not expected because many are resistant due to strong hydrogen bonding and inaccessibility to the external bulk water, especially in the case of a membrane protein. For example, BR undergoes only a partial exchange of its NH peptide groups, unless it is fully delipidated and then reconstituted and/or regenerated in the presence of D2O.46 Under these conditions, a negative band appears in the BR → M difference spectrum at 1439 cm–1 that was assigned to the amide II′ mode.46
Figure 4.

Spectra from Figure 2 over the 1400–1500 cm–1 region: (A) CaChR1 in H2O, (B) CaChR1 in H2O regenerated with A1 retinal, (C) CaChR1 in H2O regenerated with A2 retinal, (D) CaChR1 in H2O regenerated with [15-13C,15-2H]retinal, (E) CaChR1 in H2O regenerated with [14,15-2H2]retinal, and (F) CaChR1 in D2O. Peaks labeled in bold are discussed in the text. Y axis markers are approximately 0.5 mOD for all spectra.
Detection of Weakly Hydrogen Bonded Internal Water Molecules
The OH stretching mode of weakly hydrogen bonded waters is normally found in the region from 3550 to 3700 cm–1.47 In the case of BR, negative/positive bands appear in the BR → K FTIR-difference spectrum at 3642/3636 cm–1 and a second positive band at 3625 cm–1, all of which downshift approximately 10–13 cm–1 when H218O is substituted for H2O.48 A similar pattern is also observed in this region, although at shifted frequencies, for GPR and BPR.37
In the case of CaChR1, a different pattern is found in this region with positive/negative bands appearing at 3632/3626 cm–1 and an additional positive band at 3595 cm–1 (Figure 5). All of these bands can be assigned to one or more weakly hydrogen bonded internal waters on the basis of a H218O-induced downshift in frequency (Figure 5). In contrast, none of the retinal substitutions, including the A2 retinal analogue, affects these bands (Figure S5 of the Supporting Information). As expected, H–D exchange causes a complete disappearance of these bands (Figure S5 of the Supporting Information, spectrum F) with their downshift and appearance in the OD stretching region at (+)2682 and (−)2677 cm–1 (Figure 6F).
Figure 5.

Comparison of the weak hydrogen bonding region from 3550 to 3700 cm–1 for CaChR1 and CrChR2 in H2O and H218O at 80 K. Peaks labeled in bold are discussed in the text. Y axis markers are approximately 0.1 mOD for both CaChR1 spectra and 0.05 mOD for both CrChR2 spectra. For detailed H218O exchange procedures as well as spectral acquisition parameters, see Materials and Methods.
Figure 6.

Spectra from Figure 2 over the 2500–2700 cm–1 region with the addition of the bottom trace: (A) CaChR1 in H2O, (B) CaChR1 in H2O regenerated with A1 retinal, (C) CaChR1 in H2O regenerated with A2 retinal, (D) CaChR1 in H2O regenerated with [15-13C,15-2H]retinal, (E) CaChR1 in H2O regenerated with [14,15-2H2]retinal, and (F) CaChR1 in D2O. Peaks labeled in bold are discussed in the text. Y axis markers are approximately 0.25 mOD for all spectra.
A similar spectral pattern is also observed in this region for CrChR2. However, the bands appear much broader, and the negative band at 3617 cm–1 is at a lower frequency and the positive band at 3602 cm –1 at a higher frequency relative to the corresponding bands in CaChR1 (Figure 5). The much broader bands in CrChR2 indicate these waters are located in a more heterogeneous environment and have more disordered structure. Overall, the band pattern observed in both ChRs indicates that at least one weakly hydrogen bonded water undergoes a further decrease in hydrogen bonding strength during the primary phototransition. In contrast, in BR it has been shown that a weakly hydrogen bonded water (W401) that is part of a hydrogen-bonded pentagonal cluster located near the retinal Schiff base49−51 undergoes an increase in its strength of hydrogen bonding. Furthermore, as discussed below and in contrast with BR, in CaChR1 this water does not appear to be significantly affected by substitutions in the putative counterion residues near the SB. We also note that recent FTIR-difference measurements of the unphotolyzed state to P2 intermediate reveal bands in the OH stretching region of weakly hydrogen bonded waters (J. I. Ogren et al., unpublished observations).52
Detection of Cysteine Vibrations
The SH stretching mode of cysteine residues is typically found in the spectral region from 2500 to 2600 cm–1.53−55 For example, bands in this region have been assigned to cysteine in the FTIR-difference spectrum of bovine rhodopsin,56 sensory rhodopsin from the fungus Neurospora crassa (NOP-1),32 and sensory rhodopsin from cyanobacterium Anabaena (ASR).57 In contrast, bands do not appear in this region of BR or sensory rhodopsin II from Natronomonas pharaonis, which have no cysteines in their primary sequences.
In the case of CaChR1, two positive bands appear in this region at 2563 and 2543 cm–1 (Figure 6A), which are highly reproducible in CaChR1 regenerated with the various isotope substitutions that are not expected to cause a shift in this vibration (Figure 6B–E). The higher-frequency bands disappear as expected because of H–D exchange of the cysteine SH group (Figure 6F). However, the lower-frequency band at 2543 cm–1 could not be definitively assigned to cysteine because there is still positive intensity near 2543 cm–1 in D2O. Instead, this band may arise from positive/negative bands appearing in this region from the OD stretch mode of internal D2O molecules. Alternatively, it is possible that this particular cysteine SH group is inaccessible to H–D exchange as seen, for example, for some NH peptide groups in BR.46 We also note that recent FTIR measurements of the unphotolyzed state to P2 intermediate differences reveal bands in the SH stretching region (J. I. Ogren et al., unpublished observations).52
Effects of Mutations of Glu169 and Asp299 in the Carboxylic Acid C=O Stretch Region
A number of positive and negative bands appear in the 1700–1800 cm–1 region in the CaChR1 → P1 difference spectrum (Figure 7A) that could arise from the C=O stretch of carboxylic acid groups from Asp and Glu residues or from the carboxamide C=O stretch mode of Asn and Gln residues that are generally found near or below 1700 cm–1.37,58 As described below and in the Supporting Information, all of these bands are tentatively assigned to carboxylic acid stretch vibrations. We also note that there is a significant difference in this region compared to the case for CrChR2, where two prominent positive/negative bands appear at 1741/1736 cm–1 but most of the bands appearing in CaChR1 are absent (see Figure 1 and Figure S4 of the Supporting Information).
Figure 7.
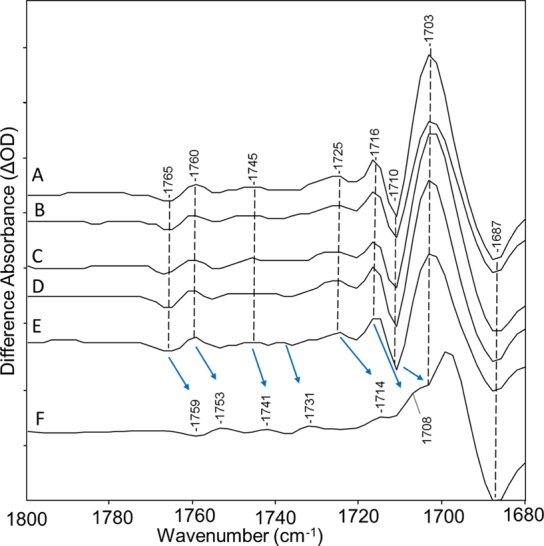
Spectra from Figure 2 over the 1680–1800 cm–1 region: (A) CaChR1 in H2O, (B) CaChR1 in H2O regenerated with A1 retinal, (C) CaChR1 in H2O regenerated with A2 retinal, (D) CaChR1 in H2O regenerated with [15-13C,15-2H]retinal, (E) CaChR1 in H2O regenerated with [14,15-2H2]retinal, and (F) CaChR1 in D2O. Arrows indicate shifts of difference bands due to H–D exchange. Y axis markers are approximately 0.5 mOD for all spectra. See the discussion in the Supporting Information regarding results shown in spectra B–F.
Glu169 and Asp299 are the CaChR1 residues homologous to the SB counterions Asp85 and Asp212 in BR, respectively. To identify possible contributions from Asp299 in this region, the conservative Asp → Glu substitution was made at position 299 (mutant D299E). This substitution, which adds an extra carbon in the side chain of Glu relative to Asp, often shifts the frequency of the C=O stretch vibration in the carboxylic acid (see, for example, ref (17)). As discussed below, this and other mutants studied still exhibit an all-trans to 13-cis isomerization and formation of a red-shifted P1 photoproduct as indicated from both the fingerprint and ethylenic stretching regions (Figure 8).
Figure 8.

Comparison of CaChR1 and the D299N, E169Q, and D299E mutants over the region from 800 to 1800 cm–1. The top four spectra were recorded in H2O and the bottom four in D2O. All spectra were acquired as described previously. Peaks labeled in bold are discussed in the text. Y axis markers are approximately 3 mOD for WT, 2 mOD for D299E, 2.5 mOD for D299N, and 1 mOD for E169Q. For detailed H–D exchange and site-directed mutagenesis procedures, see Materials and Methods.
As seen in Figure 9, the band at 1703 cm–1 downshifts to 1696 cm–1, indicating its assignment to Asp299 (and the 1696 cm–1 band to Glu299) [see also results from FTIR double-difference spectra (DDS) in the Supporting Information (Figure S6)]. Other changes in this region can also be attributed to this downshift and intensity cancellation of overlapping bands such as the increased intensity of the negative band at 1710 cm–1. Note that only the positive band at 1703 cm–1 appears to undergo a frequency shift due to this mutation, while other bands above 1700 cm–1 remain substantially unchanged.
Figure 9.

Comparison of CaChR1 and the D299N, E169Q, and D299E mutants over the region from 1680 to 1800 cm–1. The top four spectra were recorded in H2O and the bottom four in D2O. All spectra were acquired as described previously. Peaks labeled in bold are discussed in the text. Y axis markers are approximately 1.5, 1, 0.2, 0.4, 1.5, 0.3, 0.2, and 0.5 mOD for spectra from top to bottom, respectively. For detailed H–D exchange and site-directed mutagenesis procedures, see Materials and Methods.
These results and earlier studies that demonstrate that Glu299 exists in an ionized state at neutral pH16,21 are consistent with the assignment of the positive 1703 cm–1 band to protonation of Asp299 occurring during the CaChR1 → P1 transition. In particular, a hydrogen bonding change of Asp299 during the CaChR1 → P1 transition would be inconsistent with the finding that this group is unprotonated in the ground state.59 Furthermore, a negative band assigned to Asp299 is not found on the basis of the D299E mutation. However, we cannot exclude on the basis of these data the possibility that the positive band we assign in the difference spectrum reflects a partial protonation of Asp299.
Further support for this interpretation of the data is based on measurements of D299E in D2O, where the 1699 cm–1 band in WT (downshifted due to H–D exchange from 1703 cm–1) now appears due to the Asp → Glu substitution at an even lower frequency at 1692 cm–1. A strong negative band is also revealed in this mutant near 1699 cm–1, which most likely is due to the H–D exchange-induced downshift of the band in D299E in H2O near 1710 cm–1. In the WT sample in D2O, this band may be masked by the more intense positive band at 1699 cm–1 as indicated by the presence of a shoulder near this frequency. Again, other than the 1699 band cm–1 that can be assigned to the deuterated Asp299 (COOD group), other bands do not appear to be affected by the Asp → Glu substitution. For example, the positive 1716 cm–1 band for WT in H2O that downshifts to near 1708 cm–1 due to H–D exchange can also be seen in D299E in D2O at the same frequency.
Glu169 and Asp299 were also replaced with the neutral residues Gln and Asn, respectively (i.e., E169Q and D299N mutants, respectively). Previously, these mutants were studied in HEK293 cells using photoinduced charge displacement measurements59 as well as RRS and UV–visible absorption in detergent or reconstituted membranes16,21 (see Discussion and Conclusions). Strikingly, both neutral mutations have almost identical effects on both chromophore- and protein-assigned bands in both H2O and D2O (Figures 8 and 9). In the carboxylic acid C=O stretch region (Figure 9), the positive band at 1703 cm–1 disappears. In addition, positive/negative bands at 1716 and 1710 cm–1 are replaced by much broader positive/negative bands at 1718 and 1710 cm–1. The fact that the 1710 cm–1 band does not undergo a significant change in intensity as occurs in D299E despite the disappearance of the nearby 1703 cm–1 band indicates that a more intense negative band near this frequency also disappears. The broadening and slight shift to a higher frequency of the 1718 cm–1 band compared to the 1716 cm–1 band in WT indicate that this negative hidden band is most likely at a frequency higher than 1710 cm–1. FTIR DDS between these two mutants and WT (Figure S6 of the Supporting Information) also demonstrate the identical effects of both mutants in this region of the spectrum (see the Supporting Information). In addition, a negative band in the DDS appears at 1720 cm–1, which supports the existence of a hidden negative band in the 1710–1720 cm–1 region.
Similar effects are observed for these mutants in D2O compared to WT in D2O. The positive band at 1699 cm–1 disappears, and broad positive/negative bands appear near 1710 and 1699 cm–1, similar to the effects of these mutants in H2O but downshifted approximately 10 cm–1 due to H–D exchange. Importantly, no other bands in either H2O or D2O at a higher frequency are affected by these mutations, again supporting their assignment to other Asp or Glu residues in CaChR1. As described in Discussion and Conclusions, a simple model that would be consistent with all the observed changes induced by the three mutants studied is that in which a proton transfer occurs from Glu169 to Asp299 during the CaChR1 → P1 phototransition (Figure 10).
Figure 10.
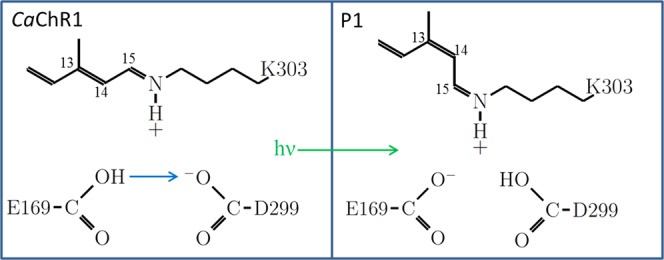
Schematic model showing Glu169 and Asp299 residues that interact with each other through a hydrogen bond formed by the carboxylic acid group of Glu169 and carboxylate group of Asp299. These residues together function as a counterion for the positively charged SB present in the unphotolyzed state of CaChR1. During the initial step in the CaChR1 photocycle (CaChR1 → P1) triggered by light absorption, an all-trans to 13-cis isomerization of the retinylidene chromophore occurs and a proton is transferred from Glu169 to Asp299.
Effects of Mutations at Glu169 and Asp299 in Other Spectral Regions
The appearance of a positive ethylenic band in the region from 1520 to 1530 cm–1 for all the mutants examined in both H2O and D2O provides strong evidence that these mutants produce a P1-like intermediate (Figure 8). In addition, the fingerprint region is highly characteristic of an all-trans to 13-cis isomerization for the primary transition, especially the appearance of the positive band near 1196 cm–1.
Interestingly, both neutral substitutions did not produce a downshift in the unphotolyzed state ethylenic frequency corresponding to an expected red-shift in the visible absorption wavelength, which would be expected if the putative counterion to the positively charged SB were neutralized as is observed in the case of BR.60 Instead, a small blue-shift can be deduced from the increase in the frequency of the ethylenic mode (Figure 8). A similar effect was also observed in the RRS of these mutants16 as well as directly from visible absorption measurements.21 As discussed recently, this absence of a red-shift is most likely a consequence of a neutral residue (Phe139) on helix B; in the case of most high-efficiency ChRs that do exhibit red-shifts, they have a positively charged Lys as the homologue.21 The largest upshift in frequency of the negative ethylenic band occurs for D299E (Figure 8) with a νC=C near 1547 cm–1, which agrees with a blue-shifted λmax near 505 nm and also with RRS measurements (J. I. Ogren et al., unpublished observations). A small negative band at 1182 cm–1 (Figure 8) is also observed, indicative of increased 13-cis isomer content in this mutant.
The negative band at 1553 cm–1 assigned to the amide II mode (see above) is also reduced or eliminated with the Asn and Gln substitutions. In the case of the D299E mutant, the band may still be present but is hidden by the upshift of the ethylenic band. In the case of the 1600–1700 cm–1 region, the small negative band at 1660 cm–1 in WT, most likely assigned to the amide I mode, is dramatically intensified along with a positive band near 1648 cm–1.
Interestingly, the bands assigned to weakly hydrogen bonded water molecules appear not to be significantly altered by these mutations as most clearly seen in the region of the OH stretch (Figure S7 of the Supporting Information). There is, however, a small upshift of 1–2 cm–1 in the frequency of these bands in the case of D299E. This indicates that the weakly hydrogen bonded waters do not strongly interact with Glu169 or Asp299 (see Discussion and Conclusions).
Discussion and Conclusions
To further investigate the molecular mechanism of ChR light activation, we have focused in this work on the structural changes that occur during the primary phototransition from the dark state to the red-shifted P1 intermediate of CaChR1, which is analogous to the well-known BR to K transition in the BR photocycle. Part of the motivation for this study is the recent finding based on near-IR confocal RRS measurements that the unphotolyzed state chromophore composition of CaChR1 is significantly different from that of CrChR2, which consists of a mixture of all-trans and 13-cis isomers.15 In contrast, CaChR1 resembles the pure all-trans retinal composition of many microbial rhodopsins such as BR and NpSRII.16 In addition, measurements of intramolecular proton transfers indicate that CaChR1 exhibits a fast outwardly directed photoinduced current with a rise time similar to that of M formation in BR, while other ChRs such as CrChR2 do not exhibit this current.59 Furthermore, recent time-resolved measurements of the CaChR1 photocycle (J. I. Ogren et al., unreported data) indicate that the decay time for the P1 intermediate is significantly longer than in CrChR2.61 Hence, it is important to further elucidate the differences in the light-activated ion gating mechanisms between CaChR1 and CrChR2 and a variety of other ChRs derived from green flagellate algae.
The FTIR-difference measurements reported here for the primary phototransition of CaChR1 and CrChR2 were taken at 80 K on protein reconstituted into bilayer lipid membranes as described in Materials and Methods. A number of methods were used to assign peaks in the spectra, including retinal isotope labeling, site-directed mutagenesis, and measurements of the effects of D2O and H218O substitutions on band frequency. Although no earlier FTIR-difference measurements have been reported for the primary phototransition of CaChR1, recent static FTIR-difference measurements were reported for CaChR1 at room temperature, which reflect mainly the CaChR1 → P2 transition.27,52 In addition, low-temperature FTIR-difference measurements have been reported for the primary phototransition of CrChR262,63 and C1C2, the chimera of CrChR1 and CrChR2 (see below).64
The picture that emerges from our study further supports the conclusion based on RRS16 that the unphotolyzed retinal compositions of CaChR1 and CrChR2 are significantly different, with CaChR1 containing an almost pure all-trans-retinal composition that photoisomerizes to a 13-cis configuration. In contrast, CrChR2 contains a mixed retinal composition with both species undergoing isomerization during the primary phototransition. It is important to note, however, that retinal extraction of the dark state of CaChR1 followed by high-pressure liquid chromatography indicates that CaChR1 also contains significant 13-cis-retinal (27%).41 The reason for this difference is unknown. However, possible reasons may be related to the use of detergent-solubilized protein for retinal extraction41 compared to our FTIR and earlier RRS measurements that were taken on CaChR1 reconstituted in a lipid bilayer membrane environment. In addition, thermal isomerization may occur during the extraction procedure that was performed awith the free aldehyde41 instead of with the more stable retinal-oxime.65,66
Overall, the FTIR differences reveal fundamental differences between the structural changes during the primary phototransition of these two ChRs from Chlamydomonas involving the retinylidene chromophore, peptide backbone, Asp/Glu residues, cysteine residues, and internal water molecules as summarized below.
1. Chromophore Structural Changes in the Primary Phototransition of CaChR1 Are More Similar to Those of BR Than Those of CrChR2
The FTIR-difference spectrum of CaChR1 is very similar to that measured in BR (Figure 1), particularly in the fingerprint region, where earlier RRS and FTIR-difference measurements show all-trans-retinal isomerizes to a 13-cis, C=N anti configuration. Furthermore, at 80 K the transition to the P1 state of CaChR1 is photoreversible, similar to those of BR and many other microbial rhodopsins.
In contrast, parallel photoreactions of both an all-trans- and 13-cis-retinal isomer appear to occur in the primary phototransition of CrChR2. In addition, this reaction is not fully photoreversible (see, for example, Figure S2 of the Supporting Information). This latter feature is understandable if the P1 photoproducts of the all-trans and 13-cis species of CrChR2 have different absorption properties and are thus not fully reversed to the same composition of retinal isomers as the original retinal species. In this regard, femtosecond time-resolved absorption measurements of CrChR2 reveal different P1-like intermediates in equilibrium after excitation for the first few picoseconds,67 which might reflect multiple species of the unphotolyzed state composition.
It is also noted that the C=N SB frequency assigned for the P1 intermediate is much higher than for the corresponding K intermediate of BR (1635 cm–1 vs 1609 cm–1) and the downshift from the unphotolyzed state much smaller (17 cm–1 vs 30 cm–1). One possible explanation is that different environments of the PSB exist in the photoactive site of the two primary photointermediates that, as discussed below, have different charges located on the respective PSB counterions (see point 4). For example, the C=N stretching vibration mixes strongly with the NH bending of the PSB,68 and thus, its frequency will be strongly affected by changes in hydrogen bonding to this group.
2. Structural Changes That Involve the Protein Backbone Are Different in CaChR1, CrChR2, and BR
The appearance during the primary phototransition of a negative band near 1553 cm–1 assigned to the amide II mode of CaChR1 along with the appearance of peaks assigned to amide I modes in the 1600–1700 cm–1 region provides strong evidence that the protein backbone of CaChR1 undergoes structural changes as early as the primary phototransition. Similarly, the primary phototransition of CrChR2 also involves significant but different protein backbone structural changes as indicated by the appearance of an intense negative band at 1664 cm–1 that is not present in the difference spectrum of CaChR1 (see Figure 1, Figure S2 of the Supporting Information, and ref (63)). Note, however, that a similar band appears near 1661 cm–1 in subsequent steps in the photocycle of CaChR1 (ref (41) and unreported data of J. I. Ogren et al.). There are also differences in other bands in the amide I region such as the appearance of a negative band at 1687 cm–1 in CaChR1 but not CrChR2. Overall, the appearance of amide I and amide II bands in the 80 K spectrum of CaChR1 compared to the absence of such contributions in BR (see Figure 1) indicates that retinal isomerization produces significantly more protein structural change in this protein as well as CrChR2.
3. Structural Changes of Weakly Hydrogen Bonded Internal Water Molecules Are Different in CaChR1 and CrChR2
FTIR-difference bands due to structural changes of internal water molecules during the primary phototransition have not previously been identified for either CaChR1 or CrChR2, although they have been observed in the FTIR-difference spectrum during the primary phototransition of a chimera of CrChR1 and CrChR264 as well as many other microbial rhodopsins, including BR and archaerhodopsin-3 (AR3).36
As discussed in Results, a comparison of the bands appearing in the OH stretching region for weakly hydrogen bonded water molecules (sometimes termed water molecules with dangling OH groups) reveals distinct differences among CaChR1, CrChR2, and BR (see Figure 5 and ref (36)). This provides clear evidence that distinct differences occur in the environment and/or structural changes that these waters undergo during the primary phototransition. Most notably, much broader bands appear in CrChR2 than in CaChR1. The broader bands in CrChR2 indicate multiple subconformational states of the protein at least at 80 K, consistent with conclusions discussed above regarding the retinal configuration of CrChR2.
Bands in the region of weakly hydrogen bonded D2O from 2650 to 2700 cm–1 (OD stretch mode) have also been identified in the C1C2 chimera64 (whose three-dimensional structure has been determined at 2.3 Å resolution69). Particularly striking is the agreement of these bands for C1C2 (see Figure 5 of ref (69)) and the spectra shown in Figure S8 of the Supporting Information for CrChR2 (but not CaChR1). This indicates that the waters giving rise to these bands must exist in very similar environments and undergo similar changes during the primary phototransition. Because the first five TM helices in C1C2 are derived from CrChR1 and the last two from CrChR2, the water or waters in both proteins giving rise to these bands are likely to be interacting with residues in the last two helices. We also note that as in the case of C1C2, neutralization of D299 and E169 in CaChR1 does not alter significantly the frequency of the bands shown in Figure S7 of the Supporting Information assigned to a weakly hydrogen bonded water.
4. Asp299 Is Protonated during the Primary Phototransition
An important goal of this study is to understand the role of residues Glu169 and Asp299 in CaChR1, which are homologous to Asp85 and Asp212, respectively, in BR. In the case of BR, these two residues exist in an ionized form in both the BR light- and dark-adapted states and together serve along with a water molecule (W402) as a complex counterion to the Schiff base.70 Both residues also exist in an ionized form in the K intermediate because no bands can be assigned to these residues in the carboxylic acid region of the low-temperature BR → K FTIR-difference spectrum.16 Instead, a protonated form of Asp115 (BR numbering) located near the β-ionone ring of BR70 is perturbed, giving rise to negative/positive bands at (−)1741 and (+)1733 cm–1 (see Figure 1).16
A key question is whether the protonation states of Glu169 and Asp299 change during the primary phototransition in CaChR1. This would not be unreasonable because other protein structural changes that appear to be much larger than what occurs in BR are observed (see point 2 above). In this regard, we concluded on the basis of earlier visible absorption and RRS pH titrations16,21 that, unlike Asp85 in BR, at neutral pH the homologous residue Glu169 is protonated (i.e., neutral). In contrast, Asp299 exists in an ionized form similar to its homologue Asp212 in BR. Thus, unlike BR, in which both homologous residues are ionized in the unphotolyzed state and first photointermediate and thus cannot contribute to the BR → K difference spectrum, it is possible that both residues could contribute to the CaChR1 → P1 difference spectra. In particular, protonation of Asp299 would be expected to produce a positive band in the carboxylic acid region, whereas deprotonation of Glu169 would produce a negative band in this region. Alternatively, a perturbation of the hydrogen bonding of the protonated Glu169 would result in negative/positive bands in this region.
As described in Results, a positive band at 1703 cm–1 was identified and assigned to the protonation of Asp299 on the basis of a downshift of this band in the mutant D299E and the disappearance of this band in the mutant D299N. Thus, we conclude that during the CaChR1 → P1 transition, this group accepts a proton from an unidentified donor group.
One possible candidate for this donor group is Glu169, which as discussed above is protonated in the unphotolyzed state and located close to an Asp299 as revealed in the crystal structure of the C1C2 chimera.69 Such a deprotonation would be expected to result in a negative band in the CaChR1 → P1 difference spectrum. In fact, as discussed in Results, a negative band in WT is found to be located in the region from 1710 to 1720 cm–1. Such a band is also identified in the WT − mutants FTIR DDS (see Figure S6 of the Supporting Information).
In support of this assignment, the disappearance of both bands (positive band at 1703 cm–1 and negative band between 1710 and 1720 cm–1) in the E169Q and D299N mutants is expected on the basis of this model because the postulated proton transfer between Glu169 and Asp299 would be prevented, thus abolishing all bands associated with this transfer. In contrast, proton transfer could still occur in D299E, where Glu299 can still function as a proton acceptor and Glu169 as a donor, thus preserving the bands, albeit with a downshifted band assigned to Glu299, as observed. It is also noted that Glu169 is predicted to exist in an ionized form in the D299N mutant;16,21 thus, bands due to protonated Glu169 are not expected to contribute to the difference spectrum in this region.
The model also explains the nearly identical effects of the E169Q and D299N mutants on the FTIR-difference spectrum. In the carboxylic acid region, both mutants abolish the same bands in the difference spectrum associated with the postulated Glu169 to Asp299 proton transfer because they both prevent proton transfer. Furthermore, the more delocalized effects that occur due to this blocked proton transfer on chromophore and protein structural changes are expected to be similar.
Additional support for this model comes from the X-ray crystallographic structure of C1C2, which shows Glu169 and Asp299 are in a position to form an ionic interaction.69 Such a strong interaction of a carboxylate group (ionized E169) with a carboxylic acid (protonated D299) would account for the relatively low frequency of the 1703 cm–1 band assigned to Asp299 in the P1 state and Glu169 (1710 cm–1) in the unphotolyzed state as previously shown for carboxylate groups that act as H-bond donors.71 Importantly, the overall proton transfer from Glu169 to Asp299 would also preserve the charge neutrality of the active site that includes the PSB. It is possible that a small displacement of the positive charge due to all-trans to 13-cis isomerization of the chromophore could trigger such a redistribution of charge.
Interestingly, this model is supported by photoinduced channel currents measured for CaChR1 and its mutants expressed in HEK293 cells.59 For example, current generation measured during formation of the P2 intermediate is accelerated in the D299N mutant and slowed significantly in the E169Q mutant. The interpretation of these data is that Glu169 serves as the primary acceptor group for the SB proton and Asp299 can act as an alternate acceptor,59 albeit much less efficiently. This model would then necessitate that Glu169 be deprotonated prior to the formation of P2. In particular, because Glu169 is predicted to be protonated in the dark state of CaChR1, its deprotonation at P1 would allow it to act as a proton acceptor during P2 formation.
We also note that a critical feature that has been proposed to explain the difference between CaChR1 and CrChR2 is the existence of the neutral residue Phe132 in CaChR1 (CaChR1 numbering), which is replaced by the positively charged residue Lys132 in CrChR2 (CaChR1 numbering).21 Importantly, this residue is in a position to modulate the pKa of Glu169 that as discussed above is predicted to be protonated at neutral pH in CaChR1. The presence of a positively charged lysine at this position in CrChR2 could lower the pKa so that this group would exist in an ionized state in the unphotolyzed state of this protein, thereby altering the active site and subsequent protein conformational changes observed even during the primary phototransition as deduced in this work.
Acknowledgments
We thank Erica Saint-Claire, Dan Russano, and Jihong Wang who participated in the early stages of this project.
Glossary
Abbreviations
- ChRs
channelrhodopsins
- RRS
resonance Raman spectroscopy
- FTIR
Fourier transform infrared
- DDS
double-difference spectra
- CaChR1
channelrhodopsin-1 from C. augustae
- CrChR2
channelrhodopsin-2 from C. reinhardtii
- BR
bacteriorhodopsin
- H–D exchange
hydrogen–deuterium exchange
- SB
Schiff base
- C1C2
chimera of CrChR1 and CrChR2
- PSB
protonated Schiff base
- ECPL
E. coli polar lipids.
Supporting Information Available
(1) Comparison of the retinal structures of CaChR1 and CrChR2 from the fingerprint region along with photoreversibility of the two primary photointermediates; (2) assignment of bands in the 1700–1800 cm–1 region to carboxylic acid C=O stretching vibrations; and (3) figures showing a comparison of CrChR2 reconstituted in both DMPC and ECPL in both H2O and D2O (over the 800–1800 cm–1 region) (Figure S1); a comparison of CaChR1 and CrChR2 “first push” differences and averaged difference data (Figure S2); a comparison of BR and BR regenerated with the isotope-labeled retinals used for CaChR1 (Figure S3); a comparison of the carboxyl stretch region (1680–1800 cm–1) for CaChR1 and CrChR2 (Figure S4); a comparison of the isotope-labeled retinal-regenerated CaChR1, A2-regenerated CaChR1, WT CaChR1 with H–D exchange, and WT CaChR1 with H218O exchange shown over the weak hydrogen bonding region (3600–3700 cm–1) (Figure S5); FTIR double-difference spectra for the WT and mutants measured in H2O in the 1680–1800 cm–1 region (Figure S6); a comparison of WT, E169Q, D299N, and D299E CaChR1 in H2O and WT, E169Q, and D299N CaChR1 in D2O in the OH stretch region (3600–3700 cm–1) (Figure S7); and a comparison of CrChR2 in DMPC and ECPL with CaChR1 and C1C2 (from ref (64)) in D2O over the 2500–2700 cm–1 region (Figure S8). This material is available free of charge via the Internet at http://pubs.acs.org.
This work was supported by National Science Foundation Grant CBET-1264434 and National Institutes of Health Grant 5R01EY21022 to K.J.R. and National Institutes of Health Grant R01GM027750, funding from the Hermann Eye Fund, and Endowed Chair AU-0009 from the Robert A. Welch Foundation to J.L.S.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Zhang F.; Vierock J.; Yizhar O.; Fenno L. E.; Tsunoda S.; Kianianmomeni A.; Prigge M.; Berndt A.; Cushman J.; Polle J.; Magnuson J.; Hegemann P.; Deisseroth K. (2011) The microbial opsin family of optogenetic tools. Cell 147, 1446–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spudich J. L.; Sineshchekov O. A.; Govorunova E. G. (2014) Mechanism divergence in microbial rhodopsins. Biochim. Biophys. Acta 1837, 546–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deisseroth K. (2010) Controlling the brain with light. Sci. Am. 303, 48–55. [DOI] [PubMed] [Google Scholar]
- Deisseroth K. (2011) Optogenetics. Nat. Methods 8, 26–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diester I.; Kaufman M. T.; Mogri M.; Pashaie R.; Goo W.; Yizhar O.; Ramakrishnan C.; Deisseroth K.; Shenoy K. V. (2011) An optogenetic toolbox designed for primates. Nat. Neurosci. 14, 387–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alivisatos A. P.; Andrews A. M.; Boyden E. S.; Chun M.; Church G. M.; Deisseroth K.; Donoghue J. P.; Fraser S. E.; Lippincott-Schwartz J.; Looger L. L.; Masmanidis S.; McEuen P. L.; Nurmikko A. V.; Park H.; Peterka D. S.; Reid C.; Roukes M. L.; Scherer A.; Schnitzer M.; Sejnowski T. J.; Shepard K. L.; Tsao D.; Turrigiano G.; Weiss P. S.; Xu C.; Yuste R.; Zhuang X. (2013) Nanotools for neuroscience and brain activity mapping. ACS Nano 7, 1850–1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyden E. S.; Zhang F.; Bamberg E.; Nagel G.; Deisseroth K. (2005) Millisecond-timescale, genetically targeted optical control of neural activity. Nat. Neurosci. 8, 1263–1268. [DOI] [PubMed] [Google Scholar]
- Chow B. Y.; Boyden E. S. (2013) Optogenetics and translational medicine. Sci. Transl. Med. 5, 177ps175. [DOI] [PubMed] [Google Scholar]
- Zhang F.; Wang L. P.; Boyden E. S.; Deisseroth K. (2006) Channelrhodopsin-2 and optical control of excitable cells. Nat. Methods 3, 785–792. [DOI] [PubMed] [Google Scholar]
- Klapoetke N. C. (2014) Independent Optical Excitation of Distinct Neural Populations. Nat. Methods 11, 338–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arenkiel B. R.; Peca J.; Davison I. G.; Feliciano C.; Deisseroth K.; Augustine G. J.; Ehlers M. D.; Feng G. (2007) In vivo light-induced activation of neural circuitry in transgenic mice expressing channelrhodopsin-2. Neuron 54, 205–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gradinaru V.; Mogri M.; Thompson K. R.; Henderson J. M.; Deisseroth K. (2009) Optical deconstruction of parkinsonian neural circuitry. Science 324, 354–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokaia M.; Sorensen A. T. (2011) The treatment of neurological diseases under a new light: The importance of optogenetics. Drugs Today (Barc) 47, 53–62. [DOI] [PubMed] [Google Scholar]
- Hou S. Y.; Govorunova E. G.; Ntefidou M.; Lane C. E.; Spudich E. N.; Sineshchekov O. A.; Spudich J. L. (2012) Diversity of Chlamydomonas channelrhodopsins. Photochem. Photobiol. 88, 119–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nack M.; Radu I.; Bamann C.; Bamberg E.; Heberle J. (2009) The retinal structure of channelrhodopsin-2 assessed by resonance Raman spectroscopy. FEBS Lett. 583, 3676–3680. [DOI] [PubMed] [Google Scholar]
- Ogren J. I.; Mamaev S.; Russano D.; Li H.; Spudich J. L.; Rothschild K. J. (2014) Retinal chromophore structure and Schiff base interactions in red-shifted channelrhodopsin-1 from Chlamydomonas augustae. Biochemistry 53, 3961–3970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braiman M. S.; Mogi T.; Marti T.; Stern L. J.; Khorana H. G.; Rothschild K. J. (1988) Vibrational spectroscopy of bacteriorhodopsin mutants: Light-driven proton transport involves protonation changes of aspartic acid residues 85, 96, and 212. Biochemistry 27, 8516–8520. [DOI] [PubMed] [Google Scholar]
- Braiman M. S.; Mogi T.; Stern L. J.; Hackett N. R.; Chao B. H.; Khorana H. G.; Rothschild K. J. (1988) Vibrational spectroscopy of bacteriorhodopsin mutants: I. Tyrosine-185 protonates and deprotonates during the photocycle. Proteins 3, 219–229. [DOI] [PubMed] [Google Scholar]
- Fahmy K.; Weidlich O.; Engelhard M.; Sigrist H.; Siebert F. (1993) Aspartic acid-212 of bacteriorhodopsin is ionized in the M and N photocycle intermediates: An FTIR study on specifically 13C-labeled reconstituted purple membranes. Biochemistry 32, 5862–5869. [DOI] [PubMed] [Google Scholar]
- Bamann C.; Gueta R.; Kleinlogel S.; Nagel G.; Bamberg E. (2010) Structural guidance of the photocycle of channelrhodopsin-2 by an interhelical hydrogen bond. Biochemistry 49, 267–278. [DOI] [PubMed] [Google Scholar]
- Li H.; Govorunova E. G.; Sineshchekov O. A.; Spudich J. L. (2014) Role of a helix B lysine residue in the photoactive site in channelrhodopsins. Biophys. J. 106, 1607–1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becher B. M.; Cassim J. Y. (1975) Improved Isolaton Procedures for the Purple Membrane of Halobacterium halobium. Prep. Biochem. 5, 161–178. [DOI] [PubMed] [Google Scholar]
- Pardoen J. A.; van den Berg E. M. M.; Winkel C.; Lugtenburg J. (1986) Recl. Trav. Chim. Pays-Bas 105, 92–98. [Google Scholar]
- Bergo V.; Amsden J. J.; Spudich E. N.; Spudich J. L.; Rothschild K. J. (2004) Structural changes in the photoactive site of proteorhodopsin during the primary photoreaction. Biochemistry 43, 9075–9083. [DOI] [PubMed] [Google Scholar]
- Furutani Y.; Sudo Y.; Wada A.; Ito M.; Shimono K.; Kamo N.; Kandori H. (2006) Assignment of the hydrogen-out-of-plane and -in-plane vibrations of the retinal chromophore in the K intermediate of pharaonis phoborhodopsin. Biochemistry 45, 11836–11843. [DOI] [PubMed] [Google Scholar]
- Ikeda D.; Furutani Y.; Kandori H. (2007) FTIR study of the retinal Schiff base and internal water molecules of proteorhodopsin. Biochemistry 46, 5365–5373. [DOI] [PubMed] [Google Scholar]
- Lorenz-Fonfria V. A.; Resler T.; Krause N.; Nack M.; Gossing M.; Fischer von Mollard G.; Bamann C.; Bamberg E.; Schlesinger R.; Heberle J. (2013) Transient protonation changes in channelrhodopsin-2 and their relevance to channel gating. Proc. Natl. Acad. Sci. U.S.A. 110, E1273–E1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagley K.; Dollinger G.; Eisenstein L.; Singh A. K.; Zimanyi L. (1982) Fourier transform infrared difference spectroscopy of bacteriorhodopsin and its photoproducts. Proc. Natl. Acad. Sci. U.S.A. 79, 4972–4976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothschild K.; Marrero H. (1982) Infrared evidence that the Schiff base of bacteriorhodopsin is protonated: bR570 and K intermediates. Proc. Natl. Acad. Sci. U.S.A. 79, 4045–4049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothschild K. J.; Marrero H.; Braiman M.; Mathies R. (1984) Primary photochemistry of bacteriorhodopsin: Comparison of Fourier transform infrared difference spectra with resonance Raman spectra. Photochem. Photobiol. 40, 675–679. [DOI] [PubMed] [Google Scholar]
- Siebert F.; Maentele W. (1983) Investigation of the primary photochemistry of bacteriorhodopsin by low-temperature Fourier-transform infrared spectroscopy. Eur. J. Biochem. 130, 565–573. [DOI] [PubMed] [Google Scholar]
- Bergo V.; Spudich E. N.; Spudich J. L.; Rothschild K. J. (2002) A Fourier transform infrared study of Neurospora rhodopsin: Similarities with archaeal rhodopsins. Photochem. Photobiol. 76, 341–349. [DOI] [PubMed] [Google Scholar]
- Bergo V.; Spudich E. N.; Spudich J. L.; Rothschild K. J. (2003) Conformational changes detected in a sensory rhodopsin II-transducer complex. J. Biol. Chem. 278, 36556–36562. [DOI] [PubMed] [Google Scholar]
- Smith S. O.; Pardoen J. A.; Lugtenburg J.; Mathies R. A. (1987) Vibrational analysis of the 13-cis-retinal chromophore in dark-adapted bacteriorhodopsin. J. Phys. Chem. 91, 804–819. [Google Scholar]
- Smith S. O.; Braiman M. S.; Myers A. B.; Pardoen J. A.; Courtin J. M. L.; Winkel C.; Lugtenburg J.; Mathies R. A. (1987) Vibrational analysis of the all-trans-retinal chromophore in light-adapted bacteriorhodopsin. J. Am. Chem. Soc. 109, 3108–3125. [Google Scholar]
- Clair E. C.; Ogren J. I.; Mamaev S.; Kralj J. M.; Rothschild K. J. (2012) Conformational changes in the archaerhodopsin-3 proton pump: Detection of conserved strongly hydrogen bonded water networks. J. Biol. Phys. 38, 153–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amsden J. J.; Kralj J. M.; Bergo V. B.; Spudich E. N.; Spudich J. L.; Rothschild K. J. (2008) Different structural changes occur in blue- and green-proteorhodopsins during the primary photoreaction. Biochemistry 47, 11490–11498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelhard M.; Scharf B.; Siebert F. (1996) Protonation changes during the photocycle of sensory rhodopsin II from Natronobacterium pharaonis. FEBS Lett. 395, 195–198. [DOI] [PubMed] [Google Scholar]
- Rothschild K. J.; Bousche O.; Braiman M. S.; Hasselbacher C. A.; Spudich J. L. (1988) Fourier transform infrared study of the halorhodopsin chloride pump. Biochemistry 27, 2420–2424. [DOI] [PubMed] [Google Scholar]
- Rothschild K. J.; Roepe P.; Lugtenburg J.; Pardoen J. A. (1984) Fourier transform infrared evidence for Schiff base alteration in the first step of the bacteriorhodopsin photocycle. Biochemistry 23, 6103–6109. [DOI] [PubMed] [Google Scholar]
- Muders V.; Kerruth S.; Lorenz-Fonfria V. A.; Bamann C.; Heberle J.; Schlesinger R. (2014) Resonance Raman and FTIR spectroscopic characterization of the closed and open states of channelrhodopsin-1. FEBS Lett. 588, 2301–2306. [DOI] [PubMed] [Google Scholar]
- Sineshchekov O. A.; Govorunova E. G.; Wang J.; Spudich J. L. (2012) Enhancement of the long-wavelength sensitivity of optogenetic microbial rhodopsins by 3,4-dehydroretinal. Biochemistry 51, 4499–4506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker F. S. (1983) Applications of infrared, Raman and resonance Raman spectroscopy in biochemistry, Plenum Press, New York. [Google Scholar]
- Smith S. O.; Myers A. B.; Mathies R. A.; Pardoen J. A.; Winkel C.; van den Berg E. M.; Lugtenburg J. (1985) Vibrational analysis of the all-trans retinal protonated Schiff base. Biophys. J. 47, 653–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith S. O.; Myers A. B.; Pardoen J. A.; Winkel C.; Mulder P. P.; Lugtenburg J.; Mathies R. (1984) Determination of retinal Schiff base configuration in bacteriorhodopsin. Proc. Natl. Acad. Sci. U.S.A. 81, 2055–2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kluge T.; Olejnik J.; Smilowitz L.; Rothschild K. J. (1998) Conformational changes in the core structure of bacteriorhodopsin. Biochemistry 37, 10279–10285. [DOI] [PubMed] [Google Scholar]
- Kandori H. (2000) Role of internal water molecules in bacteriorhodopsin. Biochim. Biophys. Acta 1460, 177–191. [DOI] [PubMed] [Google Scholar]
- Fischer W. B.; Sonar S.; Marti T.; Khorana H. G.; Rothschild K. J. (1994) Detection of a water molecule in the active-site of bacteriorhodopsin: Hydrogen bonding changes during the primary photoreaction. Biochemistry 33, 12757–12762. [DOI] [PubMed] [Google Scholar]
- Luecke H.; Schobert B.; Richter H. T.; Cartailler J. P.; Lanyi J. K. (1999) Structural changes in bacteriorhodopsin during ion transport at 2 Å resolution. Science 286, 255–261. [DOI] [PubMed] [Google Scholar]
- Garczarek F.; Gerwert K. (2006) Functional waters in intraprotein proton transfer monitored by FTIR difference spectroscopy. Nature 439, 109–112. [DOI] [PubMed] [Google Scholar]
- Shibata M.; Kandori H. (2005) FTIR studies of internal water molecules in the Schiff base region of bacteriorhodopsin. Biochemistry 44, 7406–7413. [DOI] [PubMed] [Google Scholar]
- Lorenz Fonfria V. A.; Muders V.; Schlesinger R.; Herbele J. (2014) Changes in the hydrogen-bonding strength of internal water molecules and cysteine residues in the conductive state of channelrhodopsin-1. J. Chem. Phys. 141, 22D507. [DOI] [PubMed] [Google Scholar]
- Alben J. O.; Bare G. H.; Bromberg P. A. (1974) Sulfhydryl groups as a new molecular probe at the α1β1 interface in hemoglobin using Fourier transformed infrared spectroscopy. Nature 252, 736–738. [DOI] [PubMed] [Google Scholar]
- Bare G. H.; Alben J. O.; Bromberg P. A. (1975) Sulfhydryl groups in hemoglobin. New molecular probe at the α1β1 interface studied by Fourier transform infrared spectroscopy. Biochemistry 14, 1578–1583. [DOI] [PubMed] [Google Scholar]
- Li H. C.; Wurrey C. J.; Thomas G. J. (1992) Cysteine conformation and sulfhydryl interactions in proteins and viruses. 2. Normal coordinate analysis of the cysteine side chain in model compounds. J. Am. Chem. Soc. 114, 7463–7469. [Google Scholar]
- Rath P.; Bovee-Geurts P. H.; DeGrip W. J.; Rothschild K. J. (1994) Photoactivation of rhodopsin involves alterations in cysteine side chains: Detection of an S-H band in the Meta I → Meta II FTIR difference spectrum. Biophys. J. 66, 2085–2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergo V. B.; Ntefidou M.; Trivedi V. D.; Amsden J. J.; Kralj J. M.; Rothschild K. J.; Spudich J. L. (2006) Conformational changes in the photocycle of Anabaena sensory rhodopsin: Absence of the Schiff base counterion protonation signal. J. Biol. Chem. 281, 15208–15214. [DOI] [PubMed] [Google Scholar]
- Barth A. (2000) The infrared absorption of amino acid side chains. Prog. Biophys. Mol. Biol. 74, 141–173. [DOI] [PubMed] [Google Scholar]
- Sineshchekov O. A.; Govorunova E. G.; Wang J.; Li H.; Spudich J. L. (2013) Intramolecular proton transfer in channelrhodopsins. Biophys. J. 104, 807–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rath P.; Marti T.; Sonar S.; Khorana H. G.; Rothschild K. J. (1993) Hydrogen bonding interactions with the Schiff base of bacteriorhodopsin. Resonance Raman spectroscopy of the mutants D85N and D85A. J. Biol. Chem. 268, 17742–17749. [PubMed] [Google Scholar]
- Verhoefen M. K.; Bamann C.; Blocher R.; Forster U.; Bamberg E.; Wachtveitl J. (2010) The photocycle of channelrhodopsin-2: Ultrafast reaction dynamics and subsequent reaction steps. ChemPhysChem 11, 3113–3122. [DOI] [PubMed] [Google Scholar]
- Ritter E.; Stehfest K.; Berndt A.; Hegemann P.; Bartl F. J. (2008) Monitoring light-induced structural changes of Channelrhodopsin-2 by UV-visible and Fourier transform infrared spectroscopy. J. Biol. Chem. 283, 35033–35041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radu I.; Bamann C.; Nack M.; Nagel G.; Bamberg E.; Heberle J. (2009) Conformational changes of channelrhodopsin-2. J. Am. Chem. Soc. 131, 7313–7319. [DOI] [PubMed] [Google Scholar]
- Ito S.; Kato H. E.; Taniguchi R.; Iwata T.; Nureki O.; Kandori H. (2014) Water-containing hydrogen-bonding network in the active center of channelrhodopsin. J. Am. Chem. Soc. 136, 3475–3482. [DOI] [PubMed] [Google Scholar]
- Groenendijk G. W.; De Grip W. J.; Daemen F. J. (1980) Quantitative determination of retinals with complete retention of their geometric configuration. Biochim. Biophys. Acta 617, 430–438. [DOI] [PubMed] [Google Scholar]
- Groenendijk G. W.; Jansen P. A.; Bonting S. L.; Daemen F. J. (1980) Analysis of geometrically isomeric vitamin A compounds. Methods Enzymol. 67, 203–220. [DOI] [PubMed] [Google Scholar]
- Scholz F.; Bamberg E.; Bamann C.; Wachtveitl J. (2012) Tuning the primary reaction of channelrhodopsin-2 by imidazole, pH, and site-specific mutations. Biophys. J. 102, 2649–2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aton B.; Doukas A. G.; Narva D.; Callender R. H.; Dinur U.; Honig B. (1980) Resonance Raman studies of the primary photochemical event in visual pigments. Biophys. J. 29, 79–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato H. E.; Zhang F.; Yizhar O.; Ramakrishnan C.; Nishizawa T.; Hirata K.; Ito J.; Aita Y.; Tsukazaki T.; Hayashi S.; Hegemann P.; Maturana A. D.; Ishitani R.; Deisseroth K.; Nureki O. (2012) Crystal structure of the channelrhodopsin light-gated cation channel. Nature 482, 369–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luecke H.; Schobert B.; Richter H. T.; Cartailler J. P.; Lanyi J. K. (1999) Structure of bacteriorhodopsin at 1.55 Å resolution. J. Mol. Biol. 291, 899–911. [DOI] [PubMed] [Google Scholar]
- Iliadis G.; Zundel G.; Brzezinski B. (1994) Aspartic proteinases: Fourier transform IR studies of the aspartic carboxylic groups in the active site of pepsin. FEBS Lett. 352, 315–317. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.