

Abstract
The hantaviral zoonotic diseases pose significant threat to human health due to the lack of potential antiviral therapeutics or a vaccine against hantaviruses. Sin Nombre hantavirus nucleocapsid protein (N) augments mRNA translation. N binds to both the mRNA 5′ cap and 40S ribosomal subunit via ribosomal protein S19 (RPS19). N with the assistance of viral mRNA 5′ untranslated region (UTR) preferentially favors the translation of downstream open reading frame. We identified and characterized the RPS19 binding domain at the N-terminus of N. Its deletion did not influence the secondary structure but impacted the conformation of trimeric N molecules. N variant lacking the RPS19 binding region was able to bind both the mRNA 5′ cap and panhandle like structure, formed by the termini of viral genomic RNA. In addition, N variant formed stable trimers similar to wild type N. Use of this variant in multiple experiments provided insights into the mechanism of ribosome loading during N-mediated translation strategy. Our studies suggest that N molecules individually associated with mRNA 5′ cap and RPS19 of the 40S ribosomal subunit undergo N-N interaction to facilitate the engagement of N associated ribosomes at the mRNA 5′ cap. These studies have revealed new targets for therapeutic intervention of hantavirus infection.
Keywords: Hantavirus cardiopulmonary syndrome, hemorrhagic fever with renal syndrome, hantavirus nucleocapsid protein, viral translation control, ribosomal protein S19
INTRODUCTION
Hantaviruses are negative strand emerging RNA viruses and members of the Bunyaviridae family. Humans are infected by hantaviruses through aerosolized excreta of infected rodent hosts causing a significant impact on human health [1, 2]. Hantavirus infection causes hemorrhagic fever with renal syndrome (HFRS) and hantavirus cardiopulmonary syndrome (HCPS) with mortalities of up to 15% and 50%, respectively [3]. Hantavirus particles are spherical in shape, harboring three genomic RNA segments S, M and L within a lipid bilayer [4]. The mRNAs derived from S, L and M segment encode viral nucleocapsid protein (N), viral RNA dependent RNA polymerase (RdRp) and glycoprotein precursor GPC, respectively. The GPC is post-translationally cleaved at a highly conserved WAASA motif, generating two glycoproteins Gn and Gc [5]. The characteristic feature of the hantaviral genome is the partially complementary nucleotides at the 5′ and 3′ termini of each of the three genome segments that undergo base pairing and form panhandle-like structures. N is a multifunctional protein primarily involved in encapsidation and packaging of the viral genome. However, recent studies have suggested that N plays diverse roles in the virus replication cycle [6–18]. The RNA binding domain of Hantaan virus N has been mapped to the central conserved region corresponding to the amino acids from 175 to 218 [19].
We have recently found that hantaviruses use a novel translation initiation strategy that likely lures the host translation apparatus for the preferential translation of viral mRNA [15, 20]. This translation mechanism is operated by N, which binds to mRNA 5′ cap and requires four nucleotides adjacent to the cap for high affinity binding [15]. In addition, N also binds to the 40S ribosomal subunit and loads it onto the 5′ terminus of capped mRNA. The efficient ribosome loading by N favored the translation of reporter capped transcripts in cells, although it is not clear if this translation mechanism also favors the translation of host cell transcripts [15]. Our published results suggest that N-mediated translation mechanism preferentially helps in the translation of viral mRNA by the selective binding of N to the heptanucleotide sequence GUAGUAG of the viral mRNA 5′ UTR [15, 20]. Hantaviral mRNAs contain the 5′ cap and lack the 3′ poly (A) tail. It has been reported that 3′ UTR of hantaviral small mRNA functionally replaces the poly(A) tail and stimulates the 5′ cap dependent translation [21]. The central component of N-mediated translation mechanism is the binding of N to the 40S ribosomal subunit via RPS19 [22]. Here in this manuscript, we identified and characterized the RPS19 binding domain in Sin Nombre hantavirus N. Our results shed light on the mechanism of ribosome loading during N-mediated translation strategy and reveal N-RPS19 interaction as novel target for therapeutic intervention of hantavirus diseases.
EXPERIMENTAL PROCEDURES
Reagents
PCR primers were from Integrated DNA Technologies. All restriction enzymes were from New England Biolabs. Anti-RPS19 and anti-His tag antibodies were from Santa Cruz. DNase I, T7 transcription reagents, rabbit reticulocyte lysates and RNAse inhibitor were from Promega. PCR reagents were from Invitrogen. The 5′ capping reagents were from Cell Script. DNA and RNA purification reagents were from QIAGEN. Radioactive 35S Methionine and 32P GTP were from Perkin Elmer. All other chemicals were purchased from Sigma.
Constructs
The plasmid pSNVN was used for the expression of wild type SNV N and was constructed as previously reported [13, 15, 23, 24]. All other deletion variants and pFLuc plasmid used in this study were prepared as discussed in Table S1. The plasmid pGL3 was from promega.
Expression and purification of hantavirus N protein
Wild type SNVN was expressed in E. coli as C-terminal His tagged protein and purified using NiNTA beads, as previously reported [25]. Same procedure was used for the purification of SNVNΔ151-175 variant. The purified proteins were quantified by Bradford assay and stored at −80 °C in 200 μl aliquots.
Semi-native gels
Semi-native SDS PAGE analysis was performed to analyze the migration pattern of SNVNΔ151-175 variant in comparison to wild type N. Purified SNVNΔ151-175 variant and wild type N proteins were resolved on 8% polyacrylamide gel containing 0.1% SDS. Denaturation of proteins was achieved by boiling the samples in loading buffer containing 2% SDS and 0.6% β-mercaptoethanol for 10 min, whereas this step was omitted for proteins that were separated under semi-native conditions. The proteins were separated at 60 V for 5 hours in SDS running buffer (25 mM Tris-HCl, 200 mM Glycine and 0.1 % SDS) before visualization using coomassie stain. Both wild type and variant N showed the formation of trimers and high molecular weight oligomers under native conditions, consistent with similar observations from other investigators [6].
Immunoprecipitation analysis
Immunoprecipitation analysis was carried out as previously described [22]. Briefly, cell lysates treated with RNase A were immunoprecipitated with the antibody of interest. The immunoprecipitated material was examined by western blot analysis using appropriate antibody.
T7 Transcription for RNA synthesis
The panhandle-like structure of viral S-segment RNA, capped and uncapped decameric RNA 5′GAUAUGUGAG3′ were synthesized by in vitro T7 transcription reaction, as previously reported [13, 24, 25].
5′ capping
The luciferase mRNA was 5′ capped using the ScriptCap m7G capping system (Cell Script Cat # C-SCCE0610), according to manufacturer’s instructions. Briefly, 50 μg of purified mRNA were added to the reaction mixture containing 50mM Tris HCl pH 8.0, 6mM KCl, 1.25mM MgCl2, 1mM GTP, 100μM S-adenosyl-methionine (SAM) and 40 units of the capping enzyme in a final volume of 100μl. The reaction mixture was incubated at 37 °C for 30 minutes, followed by purification of capped mRNA using RNeasy kit (Qiagen).
RNA Filter binding
Interaction of the wild type N or SNVNΔ151-175 variant with capped or uncapped decameric RNA was studied by filter binding assay as previously reported [24]. Briefly, a fixed concentration of RNA was incubated with increasing input concentrations of purified N and the mixture was filtered through nitrocellulose filter. The RNA retained on the filter was quantified by scintillation counter.
Fluorescence binding
Fluorescence binding studies were carried out as previously reported [29], except the percentage of wild type or variant N bound to panhandle-like RNA structure at each input concentration of the RNA was calculated from equation 1.
| (Eq.1) |
, Where ΔF is the change in fluorescence signal at 330 nm at each addition of RNA. ΔFmax is the same parameter when N is totally bound to the panhandle-like RNA structure. Double reciprocal plot (1/ΔF versus 1/Cp, [24]) was used to calculate the value of ΔFmax, using equation 2. Cp is the input RNA concentration.
| (Eq.2) |
. A plot of percent bound N versus RNA concentration was used for the calculation of apparent dissociation constant (Kd), which corresponds to the concentration of RNA required to obtain half saturation, assuming that complex formation obeys a simple bimolecular equilibrium. Stern volmer quenching analysis was carried out as previously reported [25]. The fluorescence studies of hydrophobic fluorophore bis-ANS were performed as previously reported [24].
Translation assays in rabbit reticulocyte lysates
Nuclease-treated rabbit reticulocyte lysates were used for the translation of mRNA in the presence and absence of either bacterially expressed and purified wild type N or N variant lacking the RPS19 binding domain as previously reported [15].
Luciferase assay
HeLa cells were co-transfected with pFLuc construct along with either pSNVN or pSNVNΔ151-175 and lysed 24 hours post-transfection. Cell lysis was carried out using luciferase cell lysis buffer (promega) and luciferase assays were performed according to the manufacturer’s protocol.
Flow cytometry
HeLa cells were co-transfected with pTGFP plasmid along with either pSNVN plasmid for the expression of wild type N or pSNVNΔ151-175 plasmid for the expression of SNVNΔ151-175 variant. Cells were trypsinized 24 hours post-transfection and examined for GFP expression using FACS analysis, as previously reported [26].
CD measurements
CD measurements for both wild type N and NΔ151-175 variant were carried out in 20 mM Na2HPO4 and 100 mM NaCl, PH 8.0, using a Jasco-720 spectropolarimeter (Japan Spectroscopic Co., Tokyo, Japan). A total of 100 scans from 190–260 nm with a 1 nm step were recorded at 25 °C for both the proteins, using a cuvette of 1 mm path length. Buffer base line was subtracted from both the spectra.
Biolayer Interferometry
Biolayer interferometry was used to monitor the binding affinities of both the wild type N and SNVNΔ151-175 variant with capped and uncapped decameric RNA using BLItz instrument (ForteBio). Briefly, RNA molecules were labeled with biotin (Roche) and loaded onto Streptavidin biosensors. After mounting, biosensors were equilibrated in RNA binding buffer and then dipped into either wild type N or SNVNΔ151-175 variant solutions of varying concentrations for the measurement of association and dissociation kinetics. The kinetic parameters Kass (association rate constant), Kdis (dissociation rate constant) and the binding affinities (Kd = Kdis/Kass) were calculated with the help of data analysis software (BLItZ Pro). All the experiments were performed at room temperature using different salt concentrations to confirm the specificity of the interaction.
RESULTS AND DISCUSSION
Mapping of RPS19 binding domain in SNV N
Since N binds to the 40S ribosomal subunit via RPS19, we asked whether N contains the RPS19 binding domain. We generated a panel of N variants lacking either 26 amino acids (NΔ403-428) or 82 amino acids (NΔ347-428) or 191 amino acids (NΔ238-428) or 254 amino acids (NΔ175-428) at the C-terminus of N (Fig 1A). All these variants contained a C-terminal His tag. HeLa cells were transfected with the plasmids expressing these variants and cell lysates were immunoprecipitated using anti-RPS19 antibody, as previously reported [22]. An examination by western blot analysis revealed that all these variants co-purified with RPS19 similar to wild type N (Fig 1B). An additional variant lacking 266 amino acids at the C-terminus (NΔ163-428) was not stably expressed in cells. This mutational analysis suggests that the domain mediating the N-RPS19 interaction is not located in the C-terminal 253 amino acids of N. This led to the search for the identification of RPS19 binding domain at the N-terminus of N. The trimerization and RNA binding domains spanning the regions from 373-421 and 175-217 amino acids, respectively (Fig 1A) have been reported to play crucial roles in the encapsidation and packaging of the viral genome [6, 7, 27]. It must be noted that N-terminus of N was also found to play a role in N trimerization [6]. Interestingly, the N variant (NΔ175-428) lacking both these domains strongly interacted with RPS19 (Fig 1B). This observation is consistent with our published data that N-RPS19 interaction is RNA independent [23]. In addition, the interaction of NΔ175-428 variant with RPS19 suggests that trimerization of N is not required for N-RPS19 interaction. This is consistent with our published data that N-RPS19 interaction occurs in 1:1 binding stoichiometry [23].
Figure 1.

Panels A and D Diagrammatic representation of wild type N and N variants used in this study. Thin line represents the amino acid sequence that was deleted from the wild type N protein. Cloning strategies for the construction of expression plasmids are shown in table S1. Panels B, C, E and F. HeLa cells in 60 mm dishes were transfected with either empty vector or plasmid expressing the N variant of interest. Cells were harvested thirty-six hours post transfection and lysed in 400 μl of RIPA buffer. 300 μl of the resulting cell lysates were incubated overnight at 4 °C with 1 μg of either anti-RPS19 antibody (panels B and E) or anti-His tag antibody (panels C and F) or IgG (negative control), followed by further incubation with 50 μl of washed sepharose G beads for six hours at 4 °C. Beads were washed three times with 1X PBS and incubated with 60 μl of SDS loading dye on boiling water bath. After brief centrifugation 30 μl of the supernatant were loaded on 12% SDS PAGE and examined by western blot (W.B) analysis using either anti-His tag antibody (panels B and E) or anti-RPS19 antibody (panels C and F). Input represents the 8–10% of the total cell lysate, loaded as positive control. It must be noted that same western blot for wild type N was shown in panels C and F. Since RPS19 is constitutively expressed in all cells, it was not possible to run a control in panels B and E showing that anti-RPS19 antibody does not bind to wild type N or N variant independent of RPS19. However, the reverse immunoprecipitation experiments performed in panels C and F clarify this issue. In addition, all variants were stably expressed in cells, except the variant (NΔ238-428) was slightly degraded. The degradation products also interacted with RPS19, which are not shown. Some of the variants shown in panels A and D were also used in our previous publication [36].
We generated three additional variants with intact C-terminal trimerization domain but the amino acid residues from either 175-364 (NΔ175-364) or 175-313 (NΔ175-313) or 175-262 (NΔ175-262) had been deleted (Fig 1A). These three middle deletion variants lacked the RNA binding domain. HeLa cells were transfected with plasmids expressing these variants and immunoprecipitation analysis was carried out as mentioned above. It is evident from Fig 1B that all three variants were still able to interact with RPS19 similar to wild type N, further confirming that N-RPS19 interaction is RNA independent. To further confirm the immunoprecipitation results from Fig 1B, we transfected HeLa cells with plasmids expressing either wild type N or N variants shown in Fig 1A. Cell lysates were immunoprecipitated with anti His-tag antibody and examined by western blot analysis using anti-RPS19 antibody. It is evident from this reverse immunoprecipitation experiment (Fig 1C) that both wild type N and N variants co-purified with RPS19, consistent with similar results from Fig 1B.
We next generated another panel of N variants lacking either 50 amino acids (NΔ1-50) or 100 amino acids (NΔ1-100) or 150 amino acids (NΔ1-150) or 175 amino acids (NΔ1-175) at the N terminus of N (Fig 1D). All these variants contained a C-terminal His tag and were stably expressed in HeLa cells. Cell lysates were immunoprecipitated as mentioned in Fig 1B. As shown in Fig 1E, the variants lacking either 50 or 100 or 150 amino acids at the N-terminus were able to interact with the RPS19. However, the deletion of N-terminal 175 amino acids abrogated the N-RPS19 interaction. A comparison of co-immunoprecipitation analysis between NΔ1-150 and NΔ1-175 variants suggests that RPS19 binding domain spans the region from 150-175 amino acids. To confirm this observation we generated another variant in which the region from 150-175 amino acids was deleted (SNVNΔ151-175). An examination by western blot analysis revealed that this variant does not interact with RPS19 (Fig 1E), suggesting that this region represents the RPS19 binding domain. To further confirm the immunoprecipitation results from Fig 1E, we performed reverse immunoprecipitation experiments as mentioned in Fig 1C. Consistent with the immunoprecipitation results from Fig 1E, we observed that except NΔ1-175 and NΔ151-175 all other variants showed interaction with RPS19 (Fig 1F).
To confirm that loss of interaction between RPS19 and SNVNΔ151-175 variant was due to the deletion of RPS19 binding domain and not due to misfolding of the deletion variant, we compared the CD spectra of both the purified wild type N and SNVNΔ151-175 variant (Fig 2A) in the wavelength region from 190–260 nm. As shown in Fig 2B, both the wild type and the variant exhibit the signatures of the alpha helical structure with two minima at 223 and 209 nm. Deletion of RPS19 domain induced a slight variation in the ellipticity with the retention of parental alpha helical characteristics of the CD spectrum. These observations confirm that loss of RPS19 binding activity of the SNVNΔ151-175 variant was due to the lack of RPS19 binding domain and not due to misfolding or dramatic alteration in its secondary structure. To determine whether RPS19 binding domain was sufficient for binding to RPS19, we fused the RPS19 binding domain at the C-terminus of GFP. The resulting fusion protein was expressed in HeLa cells. An examination of the cell lysate by immunoprecipitation analysis revealed that RPS19 binding domain facilitated the interaction between GFP and RPS19 (Fig 2C), suggesting that RPS19 binding domain is self sufficient to interact with RPS19. Sequence alignment of N from multiple hantavirus species revealed the high conservation of RPS19 binding domain in hantaviruses (Fig 2D).
Figure 2.

Panel A SDS-PAGE showing the purified wild type N and NΔ151-175 variant. Panel B. Far-UV CD spectra of the wild type N (19 μM) and NΔ151-175 variant (13 μM) in phosphate buffer, PH 8.0 at 25 °C. Panel C. HeLa cells were trasfected with plasmids expressing either GFP or GFP fused with the N-terminus of RSP19 binding domain of N protein (GFPN151-175). Cells were lysed 48 hours post-transfection and resulting lysates were immunoprecipitated using anti-RPS19 antibody. The immunoprecipitated material (IP) was examined by western blot analysis using anti-GFP antibody. The RPS19 antibody was run as control to show that secondary antibody used in western blot also detected the anti-RPS19 antibody used in immunoprecipitation. Panel D. Sequence alignment of RPS19 binding domain from several hantavirus species shows the domain is highly conserved.
Deletion of RPS19 binding domain abrogates N-mediated translation
To determine whether N-RPS19 interaction is required for N-mediated translation, we co-transfected cells with a reporter plasmid expressing either GFP or luciferase along with another plasmid expressing either wild type N or SNVNΔ151-175 variant. An examination of the reporter expression revealed that co-expression of wild type N enhanced both GFP and luciferase expression in cells, consistent with the previously reported translation activity of N [15]. However, SNVNΔ151-175 variant had no impact upon the reporter expression (Fig 3A and B), suggesting that N-RPS19 interaction is required for N-mediated translation mechanism. To further confirm that SNVNΔ151-175 variant was translationally inactive, a synthesized capped luciferase mRNA was translated in rabbit reticulocyte lysates in the presence of either wild type N or SNVNΔ151-175 variant. Again, wild type N facilitated the translation of luciferase mRNA (Fig 3C and D), consistent with our published data [15]. Interestingly, SNVNΔ151-175 variant inhibited the translation of luciferase mRNA in rabbit reticulocyte lysates (Fig 3C and D). To rule out the possibility that translation inhibition was due to the degradation of luciferase mRNA by exogenously added N variant in rabbit reticulocyte lysates, a capped luciferase mRNA was synthesized in vitro and radioactively labeled during synthesis. The radiolabelled mRNA was translated in rabbit reticulocyte lysates similar to Fig 3C (top). An examination of rabbit reticulocyte lysates on 4% acrylamide urea gel revealed that addition of purified wild type N or SNVNΔ151-175 variant did not compromise the integrity of luciferase mRNA in the translation mix (Fig 3C, bottom panel). N variant deficient in binding to RPS19 has active other functional domains. Due to its deficiency to interact with the ribosome, it is possible that most of the N variant at higher concentration was abundantly available to interact with the luciferase mRNA, which interfered in the ribosome engagement and resulted in translation inhibition. Alternatively, interference of N variant with other translation initiation factors at high input concentrations can’t be ruled out. Due to the higher abundance of capped transcripts and limited N protein expression, such type of interference is likely not possible in cells expressing SNVNΔ151-175 variant. Thus, unlike in vitro translation in rabbit reticulocyte lysates, SNVNΔ151-175 variant expression in cells did not noticeably abrogate the translation of reporter mRNAs (Fig 3A and B). Alternatively, it is possible that N variant bound to mRNA caps is displaced by cellular eIF4E, which compromises its dominant negative effect upon cellular translation.
Figure 3.

Panel A HeLa cells in twelve well plates were co-transfected with 0.1 μg of pFluc plasmid expressing firefly luciferase along with 0.5 μg of either empty vector (1) or pSNVN expressing wild type N (2) or pSNVNΔ151-175 expressing SNVNΔ151-175 variant of N (3). In (4) 0.1 μg of empty vector along with 0.5 μg of pSNV N were used for co-transfection as negative control. Cells were lysed 24 hours post-transfection and cell lysates were examined for luciferase expression. Luciferase signal in all samples was normalized to the signal in column 1. Error bars represent the standard deviation, calculated from three independent experiments. Panel B. HeLa cells in twelve well plates were co-transfected as mentioned in panel A, except pFLuc plasmid was replaced with pTGFP. Cells were trypsinized 24 hours post transfection and GFP expression was monitored by flow cytometry. Error bars represent the standard deviation, calculated from three independent experiments. Panel C. One μg of purified luciferase mRNA was translated in rabbit reticulocyte lysates in the absence (lane 5) or the presence of increasing concentrations of either purified wild type N (lanes 2 and 4) or SNVNΔ151-175 variant (lanes 1 and 3). Translation products were radiolabelled with S35 Methionine during synthesis. Twenty-five μl of translation mixture was separated on 10% SDS PAGE and examined by phosphorimage analysis (panel C top). To determine the integrity of luciferase mRNA in the translation reaction containing either wild type N or N variant, one μg of radiolabeleld luciferase mRNA was translated in rabbit reticulocyte lysates as mentioned above, except that S35 Methionine was not added to the translation mixture. Five μl of the translation mixture were separated on 5% acrylamide 8M urea denaturing gel and examined by phosphorimage analysis (panel C, bottom). Panel D. The intensity of bands in the Panel C was quantified, normalized to the intensity of the band in the lane 5 and plotted. Data from two independent experiments was used to generate error bars. Panel E: HeLa cells were cotransfecetd with GFP expression construct along with either empty vector (left three panels) or pSNVN (second panel from right) or pSNVNΔ151-175 (right panel) for the expression of wild type N and SNVNΔ151-175 variant, respectively. Eighteen hours post transfection, cells were incubated with either DMSO control or 4E1RCat for twelve more hours and visualized under fluorescence microscope.
To further examine the translation competence of SNVNΔ151-175 variant in vivo, cells expressing the GFP reporter from a transfected plasmid were treated with 4E1RCat, a chemical inhibitor that interferes in the formation of eIF4F cap binding complex and inhibits canonical translation of the host cell [28]. This treatment inhibited GFP reporter expression due to global shutdown of cap dependent host translation machinery. It is evident that HeLa cells expressing wild type N resisted the 4E1RCat induced shutdown of GFP expression due to independence of N-mediated translation strategy on eIF4F complex (Fig 3E). However, such resistance was not observed in cells expressing SNVNΔ151-175 variant. This observation clearly demonstrates that N-mediated translation strategy requires N-RPS19 interaction. Next series of experiments were aimed to demonstrate that translation deficiency of the N variant was due to the lack of RPS19 binding domain and not due to the deficiency in other functions of N, such as ability to bind mRNA cap and viral RNA panhandle-like structure.
N variant lacking RPS19 binding domain binds mRNA 5′cap similar to wild type N
We have previously reported that N has distinct domains for binding to the mRNA cap and viral genomic RNA [24]. However, the cap-binding domain in N has not been mapped yet. The cap-binding domain is required to facilitate the translation of capped transcripts by the N-mediated translation mechanism. To ensure that lack of translation activity of N variant is due to the lack of RPS19 binding domain and not due to the defect in cap binding, we synthesized capped and uncapped decameric RNAs and studied their interaction with purified wild type N and SNVNΔ151-175 variant using RNA filter binding assay, as previously reported [24]. As shown in Fig 4 (panel A), the wild type N and SNVNΔ151-175 variant bound to the capped decameric RNA with a kd of 120 ± 0.6 nM and 131 ± 2 nM, respectively. In comparison, both wild type and variant N bound to the uncapped decamer with four fold lower affinity (Fig 4, panel B). These studies clearly demonstrate that the deletion of RPS19 binding domain does not interfere in the cap binding activity of N.
Figure 4.

Panels A and BBinding profiles for the interaction of wild type N (filled square) and SNVNΔ151-175 variant (filled circle) with a capped (panel A) and uncapped (panel B) decameric RNA. The binding profiles were generated using filter-binding assay and were used for the calculation of dissociation constants (Kd), as mentioned in experimental procedures section. Panels C, D, E & F: Representative BLI sensograms showing over time association and dissociation of capped RNA with wild type N (black line) or SNVNΔ151-175 variant (grey line) at 80 mM (4C) and 240 mM (4D), and uncapped RNA with wild type N (black line) or SNVNΔ151-175 protein (grey line) at 80 mM (4E) and 240 mM of NaCl concentration(4F). Each experiment was repeated at least twice.
To confirm the specificity for cap binding, we monitored the association and dissociation kinetics of both wild type N and SNVNΔ151-175 variant with the capped and uncapped decameric RNA at two different salt concentrations using Biolayer interferometry on BLItz (ForteBio) instrument. The nonspecific electrostatic interactions are salt sensitive and the dissociation constant (Kd) for such interactions increases at high salt concentrations. The representative kinetic profiles are shown in Fig 4 (Panels C–F) and the binding data is shown in table 1. It is evident from table 1B and Fig 4 (Panels C and D) that binding affinity of both wild type N and SNVNΔ151-175 variant with capped decameric RNA remains unaltered at two different salt concentration, confirming the specific binding. The Kd values calculated by filter binding assay Fig 4 (panels A and B) and interferometry (Table 1B) are in close agreement with each other. In comparison, the binding affinity of both the wild type and variant N with uncapped decameric RNA is decreased at high salt concentration (Table 1C, Fig 4E and F), confirming the nonspecific binding.
Table 1.
Binding parameters for the interaction of wild type N (wt-N) and SNVNΔ151-175 variant with RNA using Biolayer Interferometry.
| A: Binding of wt-N and SNVNΔ151-175 variant with viral S segment RNA panhandle-like structure-like structure | ||||
|---|---|---|---|---|
| Protein | NaCl (mM) | Kd ±SD (nM) | Kass (M−1s−1) | Kdis (s−1) |
| SNVN | 80 | 28.2 ± 1.2 | 2.15 × 104 | 0.59 × 10−3 |
| SNVN | 240 | 32.5 ± 1.2 | 2.88 × 104 | 0.91 × 10−3 |
| SNVNΔ151-175 | 80 | 31.8 ± 1.4 | 3.76 × 104 | 1.16 × 10−3 |
| SNVNΔ151-175 | 240 | 33.7 ± 1.0 | 4.73 × 104 | 1.56 × 10−3 |
| B: Binding of wt-N and SNVNΔ151-175 variant with capped RNA oligomer | ||||
|---|---|---|---|---|
| Protein | NaCl (mM) | Kd ±SD (nM) | Kass (M−1s−1) | Kdis (s−1) |
| SNVN | 80 | 105.9 ± 5.7 | 1.08 × 104 | 1.10 × 10−3 |
| SNVN | 240 | 107.8 ± 0.7 | 1.88 × 104 | 2.03 × 10−3 |
| SNVNΔ151-175 | 80 | 108.6 ± 4.5 | 1.65 × 104 | 1.87 × 10−3 |
| SNVNΔ151-175 | 240 | 110.4 ± 6.8 | 1.40 × 104 | 1.61 × 10−3 |
| C: Binding of wt-N and SNVNΔ151-175 variant with uncapped RNA oligomer | ||||
|---|---|---|---|---|
| Protein | NaCl (mM) | Kd ±SD (nM) | Kass (M−1s−1) | Kdis (s−1) |
| SNVN | 80 | 476.7 ± 6.8 | 5.25 × 103 | 2.57 × 10−3 |
| SNVN | 240 | 1309.0 ± 23.4 | 3.66 × 103 | 5.40 × 10−3 |
| SNVNΔ151-175 | 80 | 490.4 ± 29.2 | 6.55 × 103 | 3.35 × 10−3 |
| SNVNΔ151-175 | 240 | 1338.5 ± 84.1 | 2.96 × 103 | 4.13 × 10−3 |
N variant lacking RPS19 binding domain undergoes trimerization and specifically binds to the vRNA panhandle-like structure
Hantavirus N forms stable trimers both in vivo and in vitro. Although the role of trimers in N-mediated translation is not known, we wanted to determine whether both wild type N and SNVNΔ151-175 variant form trimers that are functionally similar. The main aim of this experiment was to demonstrate that translation inactivity of N variant was due to the lack of its RPS19 binding domain and not due to its defect to form functional trimers. An analysis by semi-native gels revealed that both wild type and SNVNΔ151-175 variant formed trimers (Fig 5A). To determine whether both the trimers were functionally similar, we purified wild type and SNVNΔ151-175 variant trimers on 10–60% sucrose gradient, as previously reported [25] and examined their binding activity with panhandle-like RNA structure using fluorescence spectroscopy, as previously reported [25]. The hallmark of functional N trimers is their specific binding to the viral RNA panhandle-like structure, as previously reported [25]. The panhandle-like RNA structure (Fig 5B) was synthesized by in vitro T7 transcription reaction, as previously reported [25]. Both wild type and variant N contain four tryptophan residues per polypeptide. The wild type trimeric N generated a tryptophan fluorescence spectrum with an emission peak at “338 nm” (Fig 5, panel C). In comparison, the emission peak of tryptophan fluorescence spectrum of SNVNΔ151-175 variant trimers is at “333 nm” (Fig 5, panel E). This suggests that wild type and variant N trimers are conformationally different and tryptophan residues in the later are more buried towards the hydrophobic microenvironment. It must be noted from Fig 2 that secondary structure of both wild type N and SNVNΔ151-175 variant is similar, yet they may form conformationally different trimeric molecules.
Figure 5.

Panel A Semi-native PAGE analysis to determine the trimerization of wild type N and SNVNΔ151-175 variant. Denatured proteins were loaded in lanes 2–3 and semi-native proteins were loaded in lanes 4–5. Wild type N is shown in lanes 2 and 4 and SNVNΔ151-175 variant is shown in lanes 3 and 5. Monomeric and trimeric forms are shown by arrow and their molecular weight matches well with the protein ladder shown. We also observed high molecular eight oligomers, which are not shown. Panel B. The sequence of SNV S-segment vRNA panhandle-like structure used in fluorescence binding. The RNA sequence was folded using mfold. The thin line represents the six nucleotide long uracil loop. Panels C and E. Purified trimeric wild type N (panel C) and SNVNΔ151-175 variant (panel E) at a concentration of 147 nM each in RNA binding buffer [25] were excited at 295 nm, and tryptophan fluorescence emission spectra were recorded from 310–500 nm (black line). The excitation and emission slit width for wild type N (panel C) was 5 nm. However, the excitation and emission slit width for SNVNΔ151-175 variant (Panel E) was 5 nm and 10 nm, respectively. All fluorescence experiments were carried out at room temperature. Fluorescence spectra at increasing concentrations of panhandle-like RNA structure are shown by arrows. Fluorescence signal from free buffer was subtracted from each fluorescence spectra. Panels D and F. Binding profile for the interaction of wild type trimeric N (panel D) and SNVNΔ151-175 variant (panel F) with the panhandle-like RNA structure. Using the fluorescence data from panels C and E, the fluorescence intensity at 330 nm was recorded at each input panhandle concentration and was used to plot binding profiles for the calculation of dissociation constant (Kd), as mentioned in Experimental procedures. The Kd values calculated from three independent experiments were averaged and reported in the panels D and F. Panels G & H: Representative BLI sensograms showing over time association and dissociation of panhandle-like RNA structure with wild type N (black line) or SNVNΔ151-175 variant (grey line) at 80mM (5G) and 240mM (5H). Each experiment was repeated at least twice.
In addition, the emission peak of free L-tryptophan is at 350 nm. The 12–17 nm blue shift in the emission peak of wild type and variant trimeric N compared to free L-tryptophan is twice the most monomeric proteins [29, 30], and is consistent with the shielding of tryptophan residues by a trimeric protein [25]. We monitored a change in the tryptophan fluorescence spectrum of both wild type and variant trimeric N with the addition of purified panhandle-like RNA structure. We observed consistent quenching of tryptophan fluorescence signal without any noticeable peak shift at increasing input concentrations of the RNA in both wild type and variant N (Fig 5, panels C and E). The fluorescence quenching data was used to generate the binding profile for the calculation of dissociation constant (Kd) (Fig 5, panels D and F), as described in experimental procedures. This analysis revealed that wild type trimeric N bound to vRNA panhandle-like structure with a Kd of 25.5 ± 0.1 nM (Fig 5D) and variant N bound to vRNA panhandle-like structure with a Kd of 29 ± 1.0 nM (Fig 5F). Similar results were previously reported for the wild type N protein [25]. Taken together the results from Fig 2 and Fig 5, it is clear that deletion of RPS19 binding domain does not noticeably alter the secondary structure of N protein, yet wild type N and SNVNΔ151-175 variant form trimers having different 3D structures without impacting their function, as evident from similar binding affinity for panhandle-like RNA structure (compare Fig 5D and F). To examine the specificity of N-panhandle interaction, we examined the binding of both wild type and N variant with the panhandle-like RNA structure at different salt concentrations, using Biolayer interferometry. As shown in Fig 5 (panels G and H) and table 1A that binding affinity of both wild type and variant N to panhandle-like RNA structure remained unaltered at varying salt concentrations, confirming the specific binding.
N variant trimers are conformationally distinct from the trimers of wild type N
To further demonstrate that deletion of RPS19 binding domain impacts the conformation of N protein trimer, we carried out Stern Volmer quenching analysis to monitor the accessibility of tryptophan residues of purified trimeric N variant to a neutral quencher (acrylamide) before and after binding to the panhandle-like RNA structure, and compared the results with wild type N. It is clear from Fig 6A and B, that data points fit to a single straight line, suggesting a single population of N molecules in the reaction. The Stern Volmer quenching analysis (Fig 6A and B) showed that tryptophan residues of wild type trimeric N before binding to the panhandle-like RNA structure are accessible to the acrylamide with a Ksv value of 6.4 ± 0.05. However, after binding to the panhandle-like RNA structure the Ksv value was reduced to 2.5 ± 0.03 (Fig 6A), consistent with 60% reduction in the accessibility of tryptophan residues to the acrylamide, as previously reported [12]. In comparison, the Ksv values for SNVNΔ151-175 variant before and after binding to the panhandle-like RNA structure were 3.25 ± 0.25 and 2.05 ± 0.13, respectively (Fig 6B), consistent with 40% reduction in the accessibility of tryptophan residues to the acrylamide after binding to panhandle-like structure. This observation suggests that both wild type and variant trimeric N likely undergo a panhandle induced conformational change during which tryptophan residues are buried inside the protein structure and are less accessible to the quencher. It is equally probable that tryptophan residues in the vicinity of the panhandle binding site of N are shielded by N-panhandle interaction. Interestingly, a comparison of Ksv values between wild type and variant N before binding to the panhandle-like RNA structure suggests that conformation of N variant trimers is distinct from the trimers of wild type N and tryptophane residues in the former are more buried inside the protein structure and are less accessible to the quencher. This is consistent with similar observation made in Fig 5 (panels C and E).
Figure 6.
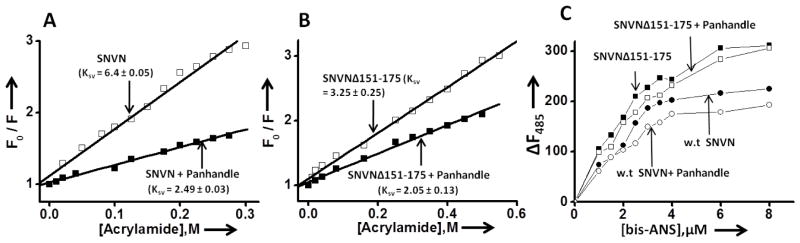
Stern Volmer quenching analysis of wild type (panel A) and SNVNΔ151-175 variant trimers (panel B) before (open square) and after (filled square) binding to the panhandle-like RNA structure. Panel C. Fluorescence titration of trimeric wild type N and SNVNΔ151-175 variant with hydrophobic fluorophore (bis-ANS) in RNA-binding buffer at room temperature under different conditions. The fluorophore was excited at 399 nm, and emission was recorded at 485 nm. Shown are the titration curves of bis-ANS binding with either free trimeric wild type N at a concentration of 100 nM (filled circle) or after incubation with 250 nM panhandle-like RNA structure at room temperature for half an hour (open circle) or free SNVNΔ151-175 variant at a concentration of 100 nM (filled square) or after incubation with 250 nM of panhandle-like RNA structure at room temperature for half an hour (open square).
We next employed bis-ANS as a probe to monitor the difference in the conformational changes, if any, between the trimers of wild type and variant N before and after binding to the panhandle-like RNA structure. Results are presented in Fig 6C. The increase in fluorescence signal of bis-ANS upon association with N is due to the binding of the probe at the hydrophobic pockets of the protein. The fluorescence signal of bis-ANS increases with higher magnitude after binding to the purified trimers of SNVNΔ151-175 variant in comparison to the trimers of wild type N, suggesting that N variant trimers have comparatively more number of binding sites for the hydrophobic probe bis-ANS. This again indicates that N variant trimers are conformationally distinct from the trimers of wild type N and is consistent with similar observations made in Fig 6 (A and B) and Fig 5 (C and E). In comparison with free SNVNΔ151-175 trimers, the fluorescence signal of bis-ANS decreases after association with SNVNΔ151-175 trimer-panhandle complex, suggesting that binding of panhandle-like RNA structure induces a conformational change in N variant trimers that decreases the number of binding sites for bis-ANS. A similar trend was observed for the binding of panhandle-like RNA structure to the trimers of wild type N, suggesting that panhandle binding likely induces similar structural alterations in both wild type and variant N trimers. Taken together the data from Fig 2, Fig 5 and Fig 6, it is evident that deletion of RPS19 binding domain does not have a noticeable impact upon the secondary structure of N protein. However, the trimers formed by wild type N and SNVNΔ151-175 variant are conformationaly different but functionally similar.
Probing the mechanism of ribosome loading during N-mediated translation initiation
Consistent with our published data [15], we showed in Fig 3E that N is a surrogate of eIF4F cap binding. However, it is still unclear how N associated 40S ribosomal subunits are loaded on capped mRNAs during N-mediated translation initiation without the assistance of eIF4F cap binding complex. We propose that N molecules individually associated with RPS19 of the 40S ribosomal subunit and 5′ mRNA cap undergo N-N interaction and facilitate ribosome loading on capped transcripts (Fig 7F). It is also possible that additional N molecules serve as bridge between N molecules individually associated with mRNA cap and RPS19. In addition, it is equally probable that a single N molecule simultaneously binds to both the RPS19 using RPS19 binding domain and mRNA 5′ cap using cap-binding domain and facilitates ribosome loading without N-N interaction (Fig 7G). To indirectly test these models we co-transfected HeLa cells in six well plates with a fixed concentration of luciferase reporter plasmid along with a fixed concentration of pSNVN plasmid expressing wild type N and increasing concentrations of pSNVNΔ151-175 plasmid expressing N variant deficient in RPS19 binding (Fig 7A). Cells were lysed 24 hours post transfection and lysates were examined for the expression of both wild type and variant N by western blot analysis (Fig 7B). As expected, a gradual increase in pSNVNΔ151-175 plasmid during transfection resulted in the corresponding increase of N variant expression (Fig 7B and D). An examination of cell lysates for the luciferase expression revealed that wild type N facilitated the translation of luciferase, as expected (Fig 7A).
Figure 7.

Competition experiments to determine whether increased expression of SNVNΔ151-175 variant inhibits N-mediated translation mechanism in cells. Panel A. HeLa cells in twelve well plates were co-transfected with a fixed concentrations of pGL3 plasmid (0.1 μg) and pSNVN plasmid (0.5 μg) along with increasing concentrations of pSNVNΔ151-175 plasmid (0 to 1 μg). Total amount of plasmid DNA in each well was balanced to 2.0 μg using empty vector. Cells were lysed 24 hours post transfection and luciferase activity was monitored. Luciferase signal in all the samples was normalized to the signal in the first sample at left. Error bars represent the standard deviation from three independent experiments. Panel B. Cell lysates from panel (A) were examined for the expression of wild type N and SNVNΔ151-175 variant using anti-His tag antibody. Panel C. The experiment performed in panel C was similar to panel A, except pGL3 plasmid was replaced by pTGFP for the expression of GFP. Cells were trypsinized 24 hours post transfection and GFP expression was monitored using FACS analysis. Panel D. Cell lysates from panel C were examined for the expression of wild type N and SNVNΔ151-175 variant using anti-His tag antibody. Panel E. Translation of capped luciferase mRNA in rabbit reticulocyte lysates in the absence (lane 1) and the presence of fixed concentration of wild type N (0.1 μM) (lanes 2–7). The input concentration of SNVNΔ151-175 variant was gradually increased from 0.0 μM (lane 2) to 0.6 μM (lane 6). Luciferase was radiolabelled with S35 Methionine during synthesis and examined by phosphorimage analysis. Panel F. A hypothetical model in which N molecules individually associated with RPS19 and mRNA 5′ cap undergo N-N interaction and facilitate ribosome loading on capped transcript in the vicinity of 5′ cap. Panel G. A hypothetical model showing the simultaneous binding of single N molecule to both RPS19 and mRNA 5′ cap to facilitate the ribosome loading on a capped transcript. Panel H. HeLa cells in six well plates were transfected with plasmids expressing either wild type N (lanes 1 and 2) or SNVNΔ151-175 variant (lanes 3 and 4) or co-transfected with plasmids expressing both wild type N and SNVNΔ151-175 variant (lanes 5–8). Cell lysates were immunoprecipitated with either anti-RPS19 antibody or IgG as negative control. Immunoprecipitated material was examined by western blot analysis using anti-N antibody which detects both wild type and variant N. Cell lysates expressing wild type N, SNVNΔ151-175 variant or co-expressing both wild type and variant N were directly loaded in lanes 2, 4 and 6, respectively, as positive control. In lane 8, cell lysate containing both wild type and variant N was incubated with beads without anti-RPS19 antibody to determine the nonspecific binding of proteins with beads. Panel I. HeLa cells were transfected with a plasmid expressing Myc tagged SNV N (left two lanes) or co-transfected with plasmids expressing both Myc tagged SNVN and His tagged SNVN151-175 variant (right two lanes). Cell lysates were incubated with Ni-NTA beads and the pull down material (P.D) from washed beads was examined by western blot analysis using either anti-Myc antibody (top panel) or anti-His antibody (bottom panel).
If a single N molecule binds to both the mRNA cap and RPS19 to facilitate ribosome loading (Fig 7G), one would expect that increasing concentrations of SNVNΔ151-175 variant in cells will efficiently occupy the mRNA 5′ caps and competitively inhibit the engagement of wild type N associated ribosomes at the mRNA 5′ cap. Such competitive inhibition would selectively inhibit the N-mediated translation of luciferase transcripts in HeLa cells. It must be noted that expression of N variant alone did not effect the luciferase expression in cells, suggesting that capped luciferase mRNAs having N variant bound to their 5′ caps were not discriminated by the host cell translation machinery (Fig 3A). Interestingly, increasing SNVNΔ151-175 variant expression along with wild type N had no noticeable affect upon N-mediated translation of luciferase mRNAs in cells (Fig 7A). Similar results were obtained when experiments were performed with a GFP reporter (Fig 7C). These results suggest that reporter mRNAs with their caps occupied by N variant were still used by wild type N for translation. Since SNVNΔ151-175 variant undergoes intermolecular interaction and also binds mRNA cap with same affinity as wild type N, it is likely that SNVNΔ151-175 variant bound to mRNA cap undergoes intra-molecular interaction with wild type N molecule pre-associated with RPS19 in the 40S ribosomal subunit and thereby participates in translation. Under such circumstances, SNVNΔ151-175 variant is not expected to cause inhibition of N-mediated translation, consistent with the observations made in Figs 7A and 7C. This explanation supports the model shown in Fig 7F. To indirectly test this hypothesis, we transfected HeLa cells as mentioned above, except wild type N was expressed as C-terminally myc tagged fusion protein and SNVNΔ151-175 variant was expressed as C-terminally His tagged fusion protein. A pull-down analysis using NiNTA beads revealed that wild type and variant N undergo intra-molecular interaction, supporting the model shown in Fig 7F (Fig 7I).
To further determine whether N molecules individually associated with mRNA cap and RPS19 undergo N-N interaction and facilitate ribosome loading (Fig 7F), we translated a capped luciferase mRNA in rabbit reticulocyte lysates and examined the effect of purified wild type N and SNVNΔ151-175 variant upon luciferase expression (Fig 7E). A fixed concentration of luciferase test mRNA was incubated with increasing concentrations of SNVNΔ151-175 variant such that most mRNA caps were occupied by the N variant. On the other hand a fixed concentration of wild type N was incubated with rabbit reticulocyte lysates to allow the N-RPS19 interaction to take place. The two solutions were mixed together to allow translation to take place and luciferase expression was monitored by phosphorimage analysis (Fig 7E). It is evident from Fig 7 (panels E) that wild type N enhanced the translation of capped luciferase mRNA (lane 2). Interestingly, the increasing input concentrations of SNVNΔ151-175 variant did not affect the luciferase expression (lanes 3–7), suggesting that wild type N was able to engage the 40S ribosomal subunits on mRNA caps pre-occupied by SNVNΔ151-175 variant. Although this experiment supports the model shown in Fig 7F, the weak possibility that wild type N bound to RPS19 has strong affinity for mRNA caps and displaces pre-bound SNVNΔ151-175 variant from mRNA caps cannot be ruled out.
In support of the ribosome loading model proposed in Fig 7F, we asked whether wild type N bound to RPS19 in the 40S ribosomal subunit interacts with SNVNΔ151-175 variant in cells co-expressing both wild type and variant N. HeLa cell lysates co-expressing both wild type and SNVNΔ151-175 variant were immunoprecipitated with anti-RPS19 antibody and immunoprecipitated material was examined for the presence of wild type and variant N by western blot analysis using anti-N antibody. As shown in Fig 7H, SNVNΔ151-175 variant co-purified using anti-RPS19 antibody only when both wild type and variant N were co-expressed in cells, suggesting that wild type N bound to RPS19 interacted with SNVNΔ151-175 variant. Since SNVNΔ151-175 variant binds to mRNA caps with same affinity as wild type N, it is highly likely that ribosome loading during N-mediated translation initiation follows a model proposed in Fig 7F.
The notion that viral nucleocapsid proteins are mainly required for packaging the viral genome has started to change due to the emergence of diverse roles of nucleocapsid proteins at different stages of virus replication cycle. For example, N has been suggested to play crucial roles in the cap-snatching mechanism of viral transcription initiation and preferential translation of viral mRNAs in infected cells [13, 15]. In addition, N has also been reported to directly interact with viral RdRp, actin filaments, apoptosis enhancers and components of SUMO-1 (small ubiquitin-like modifiers) pathway (reviewed in [31]). N must contain multifunctional domains to perform a series of diverse functions in the host cell during virus replication. Some of these important domains have recently been identified. For example, the domain mediating the RNA helix unwinding activity of N has been mapped to the N-terminus of N protein [32]. The RNA helix unwinding activity of N has been proposed to play a role during viral mRNA synthesis in conjunction with viral RdRp [14]. The RNA binding domain has been mapped to the center of N corresponding to the amino acids from 175-218 [19]. In addition, N is known to undergo trimerization and the domain mediating the intra-molecular interaction has been mapped to the C-terminus of N, corresponding to the amino acids from 373-421 [33]. The identification of RPS19 binding domain in N further demonstrates that nucleocapsid proteins contain multifunctional domains to perform multiple functions.
The capped viral transcripts have to effectively compete with the host cell mRNAs for the same translation machinery. For the efficient manufacture of viral proteins, viruses have evolved selfish strategies to favor the translation of their mRNAs in host cells during the course of infection (reviewed in [34]). For example, suspension of the cap-dependent translation initiation by picornaviruses or degradation of cellular mRNAs by herpes simplex virus are the well characterized viral strategies that make host translation machinery abundantly available for the translation of viral transcripts [34]. However, there are number of negative strand RNA viruses, including hantaviruses, which actively replicate in host cells without causing the host translation shutoff or cytopathic effects to the host cell [35]. Their active replication implies that viral mRNAs actively compete with host cell transcripts for the same translation machinery. We recently showed that hantaviruses may use N-mediated translation strategy to facilitate the translation of viral transcripts in the host cell [26]. We demonstrated that a monomeric N molecule binds to the mRNA 5′ cap and a trimeric N molecule binds to the heptanucleotide sequence GUAGUAG of the viral mRNA 5′ UTR [26]. We proposed that monomeric and trimeric N molecules individually associated with the 5′ cap and the heptanucleotide sequence undergo intra-molecular interaction and generate a tetrameric ribosome loading pad at which N-associated ribosomes are efficiently loaded by an unknown mechanism, leading to the efficient translation of viral mRNAs in comparison to the cellular transcripts that lack the viral heptanucleotide sequence in their 5′UTR [22]. We also proposed that N associated ribosomes remain dedicated for N-mediated translation mechanism [22].
Since N-mediated translation mechanism favors the translation of capped transcripts in general [15], it is yet unclear whether hantaviruses use this translation strategy to promote the translation of important cellular factors required for virus replication. The exact mechanism for ribosome loading on cellular or viral mRNAs by N-mediated translation mechanism is still unclear. However, the mechanism for ribosome loading in canonical host translation is well understood. During canonical translation initiation, the eIF4F complex composed of eIF4E, eIF4G and eIF4A is recruited at the mRNA cap. The eukaryotic initiation factor eIF3 serves as a bridge between eIF4F complex and the 40S ribosomal subunit during the engagement of ribosomes with mRNA caps. Although the role of eIF3 in N-mediated translation mechanism is still a mystery, the results from Fig 3E demonstrate that eIF4F complex is not required for N-mediated translation mechanism, consistent with previously reported similar findings [15]. The use of SNVNΔ151-175 variant in translation experiments performed in both in vivo and in vitro systems suggest that N molecules individually associated with the mRNA cap and the 40S ribosomal subunit likely undergo N-N interaction to engage the N-associated ribosomes at the mRNA 5′ cap (Fig 7F). These studies have led to the identification of multiple targets for the design of anti-hantaviral therapeutics. It is clear from Fig 7F that N-cap, N-N and N-RPS19 interactions play critical roles in N-mediated translation mechanism and hence represent novel targets for the development of antiviral therapeutics in future.
Supplementary Material
Acknowledgments
This work was supported by NIH grants RO1 AI095236-01 and 1R21 AI097355-01.
Abbreviations used
- RPS19
ribosomal protein S19
- N
Sin Nombre hantavirus nucleocapsid protein
- RdRp
hantavirus RNA dependent RNA polymerase
- GPC
glycoprotein precursor
- HFRS
hemorrhagic fever with renal syndrome
- HCPS
hantavirus cardiopulmonary syndrome
Footnotes
AUTHORS CONTRIBUTION
Mohammad A Mir supervised the research and wrote the manuscript. Absarul Haque helped in cloning. Erdong Cheng, Tania S. Bonny and Nilshad N. Salim did the experiments and provided the final figures (3A, 7A, 1B), (3C, 7E) and (5A), respectively for this publication. Rest all experiments were carried out by Safder S. Ganaie and he also provided rest all figures for the manuscript. All authors read the manuscript before publication.
References
- 1.Klempa B, Avsic-Zupanc T, Clement J, Dzagurova TK, Henttonen H, Heyman P, Jakab F, Kruger DH, Maes P, Papa A, Tkachenko EA, Ulrich RG, Vapalahti O, Vaheri A. Complex evolution and epidemiology of Dobrava-Belgrade hantavirus: definition of genotypes and their characteristics. Arch Virol. 2013;158:521–529. doi: 10.1007/s00705-012-1514-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vaheri A, Henttonen H, Voutilainen L, Mustonen J, Sironen T, Vapalahti O. Hantavirus infections in Europe and their impact on public health. Rev Med Virol. 2013;23:35–49. doi: 10.1002/rmv.1722. [DOI] [PubMed] [Google Scholar]
- 3.Schmaljohn CS, Hooper JW. Bunyaviridae: the viruses and their replication. In: Knipe DM, Howley PM, editors. Fields Virology. 4. Williams & Wilkins; Philadelphia, Lippincott: 2001. pp. 1581–1602. [Google Scholar]
- 4.Hepojoki J, Strandin T, Lankinen H, Vaheri A. Hantavirus structure--molecular interactions behind the scene. J Gen Virol. 2012;93:1631–1644. doi: 10.1099/vir.0.042218-0. [DOI] [PubMed] [Google Scholar]
- 5.Battisti AJ, Chu YK, Chipman PR, Kaufmann B, Jonsson CB, Rossmann MG. Structural studies of Hantaan virus. J Virol. 2011;85:835–841. doi: 10.1128/JVI.01847-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alfadhli A, Love Z, Arvidson B, Seeds J, Willey J, Barklis E. Hantavirus nucleocapsid protein oligomerization. J Virol. 2001;75:2019–2023. doi: 10.1128/JVI.75.4.2019-2023.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alfadhli A, Steel E, Finlay L, Bachinger HP, Barklis E. Hantavirus nucleocapsid protein coiled-coil domains. J Biol Chem. 2002;277:27103–27108. doi: 10.1074/jbc.M203395200. [DOI] [PubMed] [Google Scholar]
- 8.Blakqori G, Kochs G, Haller O, Weber F. Functional L polymerase of La Crosse virus allows in vivo reconstitution of recombinant nucleocapsids. J Gen Virol. 2003;84:1207–1214. doi: 10.1099/vir.0.18876-0. [DOI] [PubMed] [Google Scholar]
- 9.Bridgen A, Elliott RM. Rescue of a segmented negative-strand RNA virus entirely from cloned complementary DNAs. Proc Natl Acad Sci U S A. 1996;93:15400–15404. doi: 10.1073/pnas.93.26.15400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ikegami T, Peters CJ, Makino S. Rift valley fever virus nonstructural protein NSs promotes viral RNA replication and transcription in a minigenome system. J Virol. 2005;79:5606–5615. doi: 10.1128/JVI.79.9.5606-5615.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kohl A, Hart TJ, Noonan C, Royall E, Roberts LO, Elliott RM. A bunyamwera virus minireplicon system in mosquito cells. J Virol. 2004;78:5679–5685. doi: 10.1128/JVI.78.11.5679-5685.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mir MA, Brown B, Hjelle B, Duran WA, Panganiban AT. Hantavirus N protein exhibits genus-specific recognition of the viral RNA panhandle. J Virol. 2006;80:11283–11292. doi: 10.1128/JVI.00820-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mir MA, Duran WA, Hjelle BL, Ye C, Panganiban AT. Storage of cellular 5′ mRNA caps in P bodies for viral cap-snatching. Proc Natl Acad Sci U S A. 2008;105:19294–19299. doi: 10.1073/pnas.0807211105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mir MA, Panganiban AT. The bunyavirus nucleocapsid protein is an RNA chaperone: possible roles in viral RNA panhandle formation and genome replication. RNA. 2006;12:272–282. doi: 10.1261/rna.2101906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mir MA, Panganiban AT. A protein that replaces the entire cellular eIF4F complex. EMBO J. 2008;27:3129–3139. doi: 10.1038/emboj.2008.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pinschewer DD, Perez M, de la Torre JC. Role of the virus nucleoprotein in the regulation of lymphocytic choriomeningitis virus transcription and RNA replication. J Virol. 2003;77:3882–3887. doi: 10.1128/JVI.77.6.3882-3887.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Taylor SL, Frias-Staheli N, Garcia-Sastre A, Schmaljohn CS. Hantaan virus nucleocapsid protein binds to importin alpha proteins and inhibits tumor necrosis factor alpha-induced activation of nuclear factor kappa B. J Virol. 2009;83:1271–1279. doi: 10.1128/JVI.00986-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Taylor SL, Krempel RL, Schmaljohn CS. Inhibition of TNF-alpha-induced activation of NF-kappaB by hantavirus nucleocapsid proteins. Ann N Y Acad Sci. 2009;1171(Suppl 1):E86–93. doi: 10.1111/j.1749-6632.2009.05049.x. [DOI] [PubMed] [Google Scholar]
- 19.Severson W, Xu X, Kuhn M, Senutovitch N, Thokala M, Ferron F, Longhi S, Canard B, Jonsson CB. Essential amino acids of the hantaan virus N protein in its interaction with RNA. J Virol. 2005;79:10032–10039. doi: 10.1128/JVI.79.15.10032-10039.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mir MA, Panganiban AT. The triplet repeats of the Sin Nombre hantavirus 5′ untranslated region are sufficient in cis for nucleocapsid-mediated translation initiation. J Virol. 2010;84:8937–8944. doi: 10.1128/JVI.02720-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vera-Otarola J, Soto-Rifo R, Ricci EP, Ohlmann T, Darlix JL, Lopez-Lastra M. The 3′ untranslated region of the Andes hantavirus small mRNA functionally replaces the poly(A) tail and stimulates cap-dependent translation initiation from the viral mRNA. J Virol. 2010;84:10420–10424. doi: 10.1128/JVI.01270-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Haque A, Mir MA. Interaction of hantavirus nucleocapsid protein with ribosomal protein S19. J Virol. 2010;84:12450–12453. doi: 10.1128/JVI.01388-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cheng E, Haque A, Rimmer MA, Hussein IT, Sheema S, Little A, Mir MA. Characterization of the Interaction between hantavirus nucleocapsid protein (N) and ribosomal protein S19 (RPS19) J Biol Chem. 2011;286:11814–11824. doi: 10.1074/jbc.M110.210179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mir MA, Sheema S, Haseeb A, Haque A. Hantavirus nucleocapsid protein has distinct m7G cap- and RNA-binding sites. J Biol Chem. 2010;285:11357–11368. doi: 10.1074/jbc.M110.102459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mir MA, Panganiban AT. The hantavirus nucleocapsid protein recognizes specific features of the viral RNA panhandle and is altered in conformation upon RNA binding. J Virol. 2005;79:1824–1835. doi: 10.1128/JVI.79.3.1824-1835.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hussein IT, Cheng E, Ganaie SS, Werle MJ, Sheema S, Haque A, Mir MA. Autophagic clearance of Sin Nombre hantavirus glycoprotein Gn promotes virus replication in cells. J Virol. 2012;86:7520–7529. doi: 10.1128/JVI.07204-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xu X, Severson W, Villegas N, Schmaljohn CS, Jonsson CB. The RNA binding domain of the hantaan virus N protein maps to a central, conserved region. J Virol. 2002;76:3301–3308. doi: 10.1128/JVI.76.7.3301-3308.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cencic R, Hall DR, Robert F, Du Y, Min J, Li L, Qui M, Lewis I, Kurtkaya S, Dingledine R, Fu H, Kozakov D, Vajda S, Pelletier J. Reversing chemoresistance by small molecule inhibition of the translation initiation complex eIF4F. Proc Natl Acad Sci U S A. 2011;108:1046–1051. doi: 10.1073/pnas.1011477108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bougie I, Bisaillon M. Initial binding of the broad spectrum antiviral nucleoside ribavirin to the hepatitis C virus RNA polymerase. J Biol Chem. 2003;278:52471–52478. doi: 10.1074/jbc.M308917200. [DOI] [PubMed] [Google Scholar]
- 30.Weisshart K, Kuo AA, Painter GR, Wright LL, Furman PA, Coen DM. Conformational changes induced in herpes simplex virus DNA polymerase upon DNA binding. Proc Natl Acad Sci U S A. 1993;90:1028–1032. doi: 10.1073/pnas.90.3.1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hussein IT, Mir MA. How hantaviruses modulate cellular pathways for efficient replication? Front Biosci (Elite Ed) 2013;5:154–166. doi: 10.2741/e604. [DOI] [PubMed] [Google Scholar]
- 32.Brown BA, Panganiban AT. Identification of a region of hantavirus nucleocapsid protein required for RNA chaperone activity. RNA Biol. 2010;7:830–837. doi: 10.4161/rna.7.6.13862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Alminaite A, Backstrom V, Vaheri A, Plyusnin A. Oligomerization of hantaviral nucleocapsid protein: charged residues in the N-terminal coiled-coil domain contribute to intermolecular interactions. J Gen Virol. 2008;89:2167–2174. doi: 10.1099/vir.0.2008/004044-0. [DOI] [PubMed] [Google Scholar]
- 34.Gale M, Jr, Tan SL, Katze MG. Translational control of viral gene expression in eukaryotes. Microbiol Mol Biol Rev. 2000;64:239–280. doi: 10.1128/mmbr.64.2.239-280.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Muranyi W, Bahr U, Zeier M, van der Woude FJ. Hantavirus infection. J Am Soc Nephrol. 2005;16:3669–3679. doi: 10.1681/ASN.2005050561. [DOI] [PubMed] [Google Scholar]
- 36.Cheng E, Wang Z, Mir MA. Interaction between Hantavirus Nucleocapsid Protein (N) and RNA Dependent RNA Polymerase (RdRp) mutant reveals the requirement of N-RdRp interaction for Viral RNA Synthesis. J Virol. 2014 doi: 10.1128/JVI.00405-14. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.