

Abstract
Objective:
To expand the clinical phenotype of autosomal dominant congenital spinal muscular atrophy with lower extremity predominance (SMA-LED) due to mutations in the dynein, cytoplasmic 1, heavy chain 1 (DYNC1H1) gene.
Methods:
Patients with a phenotype suggestive of a motor, non–length-dependent neuronopathy predominantly affecting the lower limbs were identified at participating neuromuscular centers and referred for targeted sequencing of DYNC1H1.
Results:
We report a cohort of 30 cases of SMA-LED from 16 families, carrying mutations in the tail and motor domains of DYNC1H1, including 10 novel mutations. These patients are characterized by congenital or childhood-onset lower limb wasting and weakness frequently associated with cognitive impairment. The clinical severity is variable, ranging from generalized arthrogryposis and inability to ambulate to exclusive and mild lower limb weakness. In many individuals with cognitive impairment (9/30 had cognitive impairment) who underwent brain MRI, there was an underlying structural malformation resulting in polymicrogyric appearance. The lower limb muscle MRI shows a distinctive pattern suggestive of denervation characterized by sparing and relative hypertrophy of the adductor longus and semitendinosus muscles at the thigh level, and diffuse involvement with relative sparing of the anterior-medial muscles at the calf level. Proximal muscle histopathology did not always show classic neurogenic features.
Conclusion:
Our report expands the clinical spectrum of DYNC1H1-related SMA-LED to include generalized arthrogryposis. In addition, we report that the neurogenic peripheral pathology and the CNS neuronal migration defects are often associated, reinforcing the importance of DYNC1H1 in both central and peripheral neuronal functions.
Autosomal dominant congenital spinal muscular atrophy with lower extremity predominance (SMA-LED) has been described for decades1–4 and is characterized by congenital or early childhood onset, motor neuron degeneration, and lower limb–predominant weakness. Mutations in dynein, cytoplasmic 1, heavy chain 1 (DYNC1H1) (OMIM #158600) and bicaudal D homolog 2 (Drosophila) (BICD2) (OMIM #615290) underlie many cases of SMA-LED.5–9 Originally, 3 missense mutations in the tail domain of DYNC1H1 were reported as causative.8 Another tail domain mutation, p.His306Arg, was initially reported in a family with Charcot-Marie-Tooth type 2 in which some had proximal weakness10 and has since been found in a Japanese family with SMA-LED.11 DYNC1H1 mutations outside the tail domain have also been identified, primarily in patients with cognitive impairment and neuronal migration abnormalities.12,13 Malformations of cortical development (MCDs) were found in 10 individuals from 2 separate studies.14,15
Three tail domain mutations in the mouse homolog of DYNC1H1 have been described: “legs at odd angles” (Loa), “crawling” (Cra1), and “sprawling” (Swl),16,17 all sharing motor and sensory deficits and the Loa mice demonstrating cortical migration defects.18 Phenotypic differences among these models and the emerging diversity of reported patients, suggests that the human DYNC1H1 clinical spectrum extends beyond SMA-LED. This report describes 30 affected patients from 16 families with mutations in DYNC1H1. Although they are predominantly affected by SMA-LED, some have severe generalized arthrogryposis and one-third have cognitive impairment, which is frequently associated with MCDs.
METHODS
Subjects.
Patients with non–length-dependent motor neuronopathies predominantly affecting the lower limbs were identified at participating neuromuscular centers and referred for DYNC1H1 sequencing.
Standard protocol approvals, registrations, and patient consents.
Informed consent for clinical study and molecular genetic analysis was obtained from patients or their parents/legal guardians at all referring institutions. Local ethics committees approved this study.
Molecular genetic analysis.
DYNC1H1 sequencing.
Most subjects underwent Sanger sequencing of the tail domain of DYNC1H1 (exons 5–15) at either the Institute of Neurology, UCL, or at Washington University in St. Louis. Twelve primer pairs were used to amplify exons and flanking intronic sequences and were sequenced bidirectionally (primer details available on request). Five pedigrees (families UK4, 8, 9, US3, and NL1) had DYNC1H1 mutations identified from whole-exome analysis followed by Sanger sequencing validation.
Bioinformatics.
All identified variants were referenced to NM_001376.4 using HGVS (Human Genome Variation Society) nomenclature (http://www.hgvs.org/mutnomen) and compared against the National Heart, Lung, and Blood Institute Exome Sequencing Project, Exome Variant Server (http://evs.gs.washington.edu/EVS/). In silico analyses of mutations were performed using PolyPhen2 (http://genetics.bwh.harvard.edu/pph2/),19 SIFT (http://sift.jcvi.org/),20 PROVEAN (http://sift.jcvi.org/),21 MutationTaster (http://www.mutationtaster.org/),22 and MutationAssessor (http://mutationassessor.org/)23 (table e-1 on the Neurology® Web site at Neurology.org).
Haplotype analysis.
To determine whether the p.Val612Met mutation stems from a founder mutation or represents a mutational hotspot, 3 microsatellites flanking DYNC1H1 (D14S985, D14S1051, and D14S1007) and spanning 10.1 cM were genotyped in all carriers of this variant. Rare and novel polymorphisms segregating with the p.Val612Met variant in US4 were identified from exome sequencing data and genotyped in other pedigrees.
Clinical investigations.
Case-notes review.
Medical records were reviewed for all affected individuals, and available subjects were re-examined. The patients followed at the Dubowitz Neuromuscular Center who presented with cognitive impairment and/or behavioral difficulties completed the Child Behavior Checklist (CBCL) and Conners 3 for parents questionnaires.24,25 Subjects with Conners t scores in the abnormal range (t < 65) on the hyperactivity and/or inattention indexes were categorized as meeting criteria for attention deficit hyperactivity disorder (ADHD). The CBCL is a parent-report measure, including scales indicating the presence of externalizing symptoms (including hyperactivity). Subjects with a cutoff score of 19 were considered as having externalizing symptoms.
Ancillary clinical testing.
Nerve conduction studies and EMG were conducted in 23 subjects. Muscle biopsies had been performed in 13 patients, with frozen sections stained according to standard procedures.26 MRI of the lower limbs was performed in 9 subjects using conventional T1-weighted spin-echo sequences according to reported methodology.27 Noncontrast images were obtained from the pelvis, thighs, and legs. Fifteen patients had undergone brain MRI.
RESULTS
Subjects and mutational spectrum.
Thirty subjects with DYNC1H1 mutations were included in this study (table 1), 25 of whom came from 11 different families and 5 of whom were apparently sporadic. Thirteen different DYNC1H1 mutations were found: 10 are novel, with 8 located in the tail domain and 2 in the motor domain (table 1, figure 1). Twenty-three cases demonstrated autosomal dominant inheritance with appropriate variant segregation. De novo heterozygous mutations were confirmed in 2 sporadic subjects. Four families carried the same p.Val612Met mutation, but haplotype analysis showed that the potentially shared region between US3 and the Spanish pedigree was <700 kb (0.9 cM) and argued against a common founder (supplemental material and table e-2. We found a codominant homozygous tail domain mutation (p.Arg399Gly) in UK1-II: both parents were heterozygous and the homozygous offspring was much more severely affected.
Table 1.
Clinical and genetic features of patients with cytoplasmic dynein heavy chain 1 (DYNC1H1)-associated spinal muscular atrophy
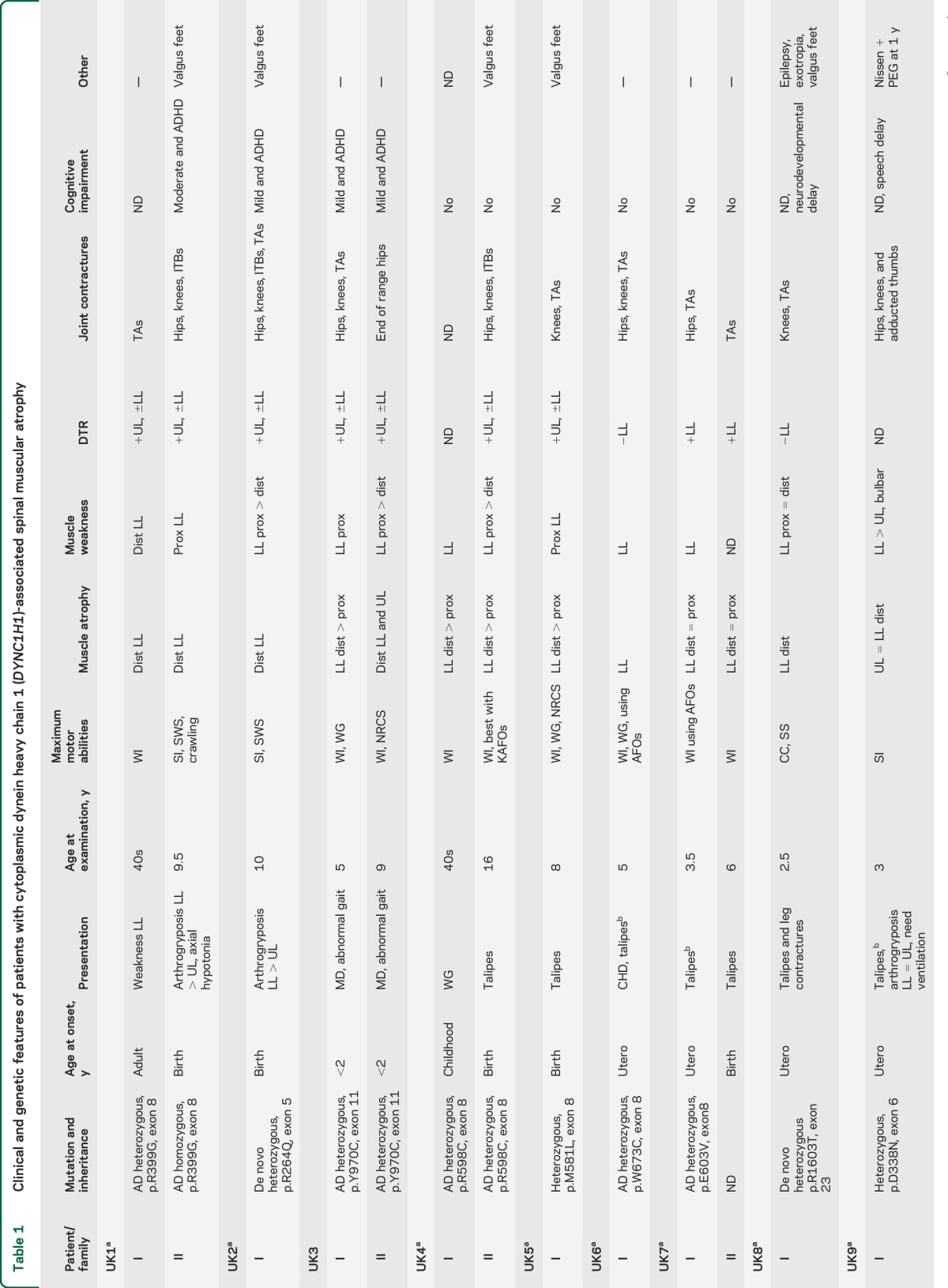
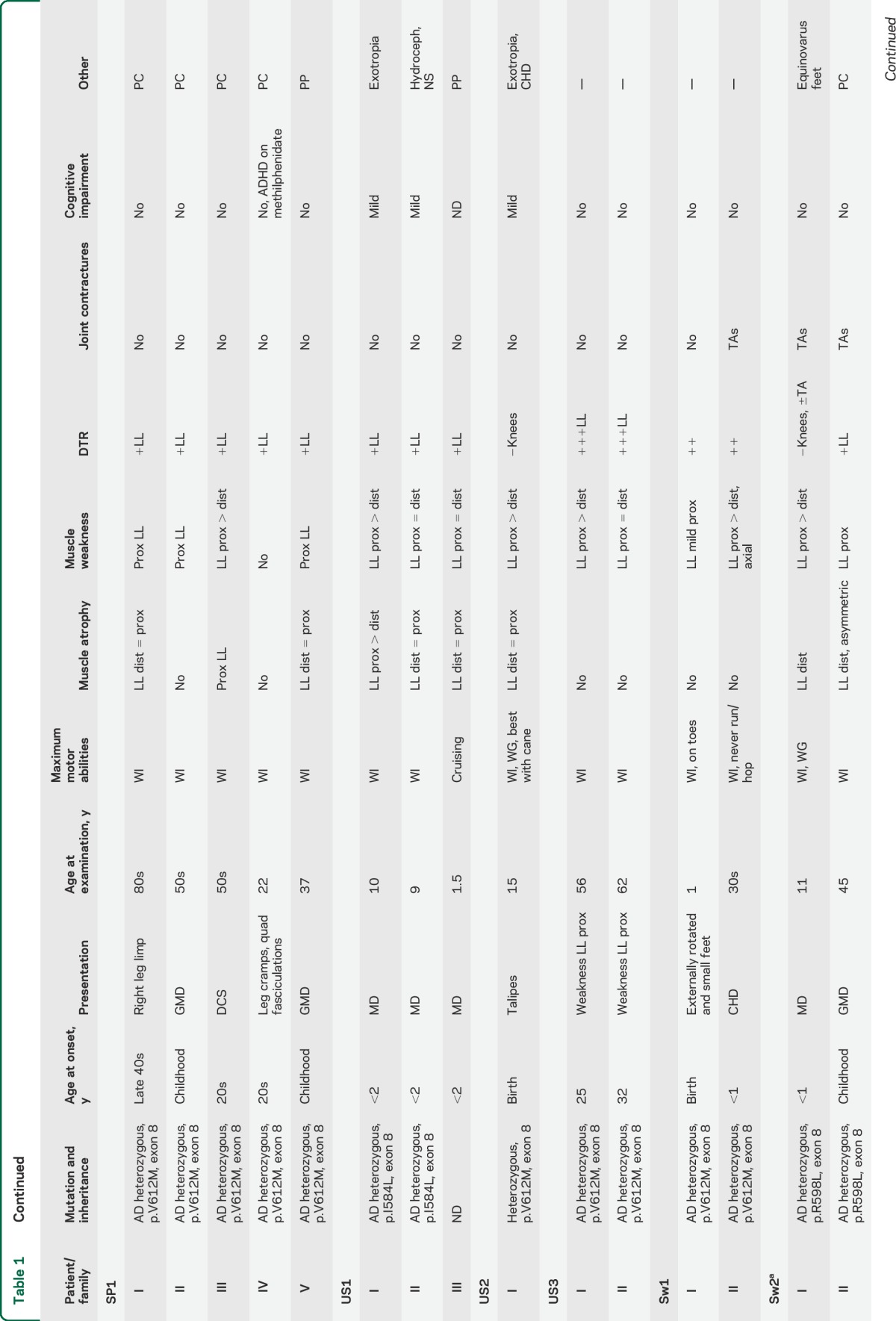

Figure 1. Position of mutations in DYNC1H1 relative to the structure of the dynein complex.

DYNC1H1 is a large gene encoding the heavy chain 1 of the cytoplasmic dynein protein complex, a ubiquitously expressed multisubunit molecular motor involved in retrograde axonal transport, cell migration, nucleokinesis, Golgi localization, and autophagy. The dynein complex consists of 2 heavy chains (dark blue), 2 intermediate chains (dark green), 4 light intermediate chains (light green), and a number of light chains (light blue). The tail domain, located in the N-terminus, is required for heavy chain dimerization. The dynein heavy chain motor domain (C-terminus) possesses adenosine triphosphate hydrolase activity and is required for movement along microtubules. This figure shows the position of all mutations described in this report and in the published literature. The mutations identified in this study to cause both SMA-LED and MCD can be seen to span the entire length of the protein. The cluster of mutations in the dimerization domain may be explained by the selective screening for mutations in this domain. *Novel mutation. CMT2 = Charcot-Marie-Tooth disease type 2; LD = learning disability with cognitive/behavioral impairment; MCD = malformation of cortical development; SMA-LED = spinal muscular atrophy with lower extremity predominance.
Thirteen of 14 families had mutations in the tail domain of DYNC1H1. It should be noted, however, that most cases only had this domain sequenced and additional subjects with mutations elsewhere in the gene may have been missed. Within the tail domain itself, however, a cluster of mutations occurs in exon 8, which encodes part of the tail dimerization domain. The distribution of DYNC1H1 mutations from this study and the literature is shown in figure 1.
Clinical spectrum of DYNC1H1 mutations.
To better characterize the phenotypic spectrum caused by DYNC1H1 mutations, clinical data for all affected individuals were obtained (table 1). Symptom onset ranged from in utero to late adulthood. Thirty-seven percent presented at birth with lower limb malformations, including 4 severe patients (UK1-II, UK2-I, UK8-I, and UK9-I) who presented at birth with congenital arthrogryposis affecting both the upper and lower limbs, with predominant lower limb involvement associated with respiratory and feeding difficulties in one case; 23% presented in infancy (motor delays and abnormal gait), and 17% in childhood (gross motor difficulties or frequent falls). One-fifth of subjects presented in adulthood with a variable degree of lower limb weakness. In family UK1-II with a codominant homozygous tail domain mutation (p.Arg399Gly), the heterozygous father showed lower limb denervation and positive family history for cognitive impairment, while the heterozygous mother only had positive family history for neurodevelopmental delay. Of note, the homozygous child of this couple was one of the most severely affected subjects: born with arthrogryposis, she never achieved independent ambulation and had mental retardation in association with ADHD. Four severely affected patients never walked. The remaining subjects achieved independent but abnormal ambulation, ranging from waddling to frequent falls and difficulties with stairs, and required bracing in some cases. None of the subjects lost ambulation with aging.
As in the original description of SMA-LED, most subjects showed more pronounced involvement of the lower limbs in comparison to the upper limbs. Although distal muscles were atrophic, weakness in proximal leg muscles predominated (figure 2). Muscle function was largely preserved in the arms and hands. Minimal distal hand weakness could be detected in a minority, but only the most affected subjects showed compromised hand function (UK2-I had tremor, grasp, and fine motor difficulties with wasting of the thenar eminence; UK9-I had tremor and adducted thumbs). Deep tendon reflexes were present in the upper limbs and diminished or absent in the lower limbs. Foot deformities (equinovarus or valgus feet and pes cavus or planus) and joint contractures (hips, knees, and ankles) were each detected in approximately half of the subjects.
Figure 2. Phenotypic characteristic of spinal muscular atrophy with lower extremity predominance caused by mutations in DYNC1H1.

Distal wasting in lower limbs, Achilles tendon tightness, and foot deformity (A, B) in case UK1-I at the age of approximately 50 years; hypotonic at birth and proximal contractures at birth (C), distal muscle wasting, proximal weakness, stands with support at 9 years, disproportion between trunk and legs (D, E) in patient UK1-II; arthrogryposis affecting upper and lower limbs and talipes at birth (F), good neck control at 5 months (G), standing with support at 3 years of age, disproportion between trunk and legs, feet deformity (H) in case UK2-I; standing unaided, disproportion between trunk and legs, hips and knees contractures, foot deformity (I) in case UK4-II at the age of 15 years; hyperlordosis, disproportion between trunk and legs (J) and proximal weakness (K), in case NL1-I at the age of 8 years; distal wasting (L) and pes cavus (M) at the age of approximately 40 years in case NL1-II. (N, O) Case SW1-I at age 12 years, feet deformity (operated), short legs, proximal > distal weakness.
Electrophysiology.
Nerve conduction studies and EMG were performed in 23 subjects and showed a motor neuropathy/neuronopathy without sensory involvement in all cases. All 4 subjects who had upper limb EMG showed neurogenic changes there as well, and 2 were also found to have neurogenic changes in the bulbar muscles (table e-3).
Muscle histology.
Thirteen subjects had undergone muscle biopsy, usually of a quadriceps muscle. Most biopsies showed chronic denervation but other heterogeneous features were also noted in some muscles (table e-3). Two previously reported cases8 showed type 2 muscle fiber predominance. Two biopsies from 2 members of family UK1 (carrying respectively a heterozygous and a homozygous p.Arg399Gly mutation) were initially considered compatible with a myopathic process because of the presence of core-like areas. Finally, in family US3, the presence of increased internal nuclei and rimmed vacuoles was also reported.
Muscle MRI.
MRI of the lower limb muscles in 9 subjects (6 children and 3 adults) demonstrated a common pattern of involvement. In the thigh, the quadriceps muscles showed diffuse involvement with selective sparing and relative hypertrophy of the adductor magnus and/or longus and of the semitendinosus muscles. In the lower leg, there was diffuse involvement with relative sparing of the anterior-medial muscles. In a few subjects with more mild lower leg involvement, the anterior-medial compartment appeared preserved compared with the posterior compartment (figure 3). One of our cases (UK4-II) was described before mutations in DYNC1H1 were found in SMA-LED.4
Figure 3. Muscle imaging of lower limbs.

All cases demonstrate a distinctive feature of diffuse involvement of the quadriceps muscles and relative sparing of the adductor compartments with relative hypertrophy of the adductor longus (single white arrow) and of the semitendinosus muscle (black arrow) at the thigh level, while at the calf level there was diffuse involvement (double white arrow) with sparing of the anterior-medial muscles. Cases SW2.I and NL1.I show a milder pattern of involvement at the calf level but with relative sparing of the medial-anterior compartment.
CNS involvement and brain imaging.
Ten subjects had some degree of cognitive impairment. Among the 5 subjects with mild to moderate cognitive impairment followed at the Dubowitz Neuromuscular Center, an association with ADHD traits, according to the scores in the CBCL and Conners 3 questionnaires, was documented in 4 subjects, one of whom was already treated with methylphenidate. Another 5 subjects had mild cognitive impairment and one subject had a diagnosis of ADHD, treated with methylphenidate, with normal cognitive ability (table 1).
MRI of the brain showed similar abnormalities in 4 of 15 subjects. Each subject showed a common pattern of brain malformation resembling polymicrogyria, characterized by cortical nodularity or gyral overconvolutions (figure 4, table e-3), best seen in the frontal and perisylvian cortex. The sylvian fissures extended posteriorly as a parietal cleft in 3 subjects (unilateral in 1 and bilateral in 2). All subjects with abnormal brain MRI had cognitive impairment or neurodevelopmental delay and 3 had ADHD traits.
Figure 4. Brain imaging.

Brain MRI at the age of 3 years in case UK2.I shows a polymicrogyric pattern of frontal lobe cortex (A, B), sylvian fissure extending to the parietal lobe especially on the right side (C), and thin corpus callosum (D). Brain MRI at the age of 2 years in case UK3.I shows immature white matter, polymicrogyria-like pattern of sylvian and frontal cortex (A, B), posterior extension of sylvian fissure (C), and mild cerebellar hypoplasia (D). Brain MRI at the age of 4 years in case UK3.II shows a polymicrogyric pattern of the right frontal lobe (A, B), thin corpus callosum (C), and gyral overconvolution with posterior extension of the right sylvian fissure (D). All these subjects had underdeveloped white matter with thinning of the corpus callosum.
DISCUSSION
Autosomal dominant or sporadic congenital SMA-LED is characterized by nonprogressive congenital or early-onset lower limb–predominant weakness and wasting, and mutations in DYNC1H1 are a common cause of this disease. Mutations located in the tail domain of DYNC1H1 are mostly associated with SMA-LED8,13 or a Charcot-Marie-Tooth phenotype with proximal weakness,10 while mutations in the motor domain have previously only been identified in patients with cognitive impairment but no motor neuron disease.12,13 More recently, MCDs have been documented in 8 individuals carrying DYNC1H1 mutations, 2 of which were located in the tail domain. Of note, 3 subjects from that study had clinical features evocative of a neuropathy, although no detailed clinical or electrophysiologic information was provided.14 Two additional unrelated cases with novel DYNC1H1 mutations, located in the neck and motor domains, respectively, have been described in association with an SMA-LED phenotype and brain cortical malformation.15
In this study, we report a large cohort of children and adults affected by SMA-LED due to DYNC1H1 mutations, expanding the clinical spectrum to include severe cases with generalized arthrogryposis and milder cases with onset in adulthood. The detection of cognitive/behavioral impairment and cortical malformations in a proportion of affected individuals demonstrates the frequent co-occurrence of central and peripheral pathology. As in the earlier studies,8,28 a significant proportion of our subjects had clinically apparent symptom onset at birth or within the first years of life (63%); in about one-quarter of cases (7/30), onset was in adulthood. However, some patients in our case series were so severely affected that they never walked, and also had evidence of congenital involvement of the upper limbs and hands, expanding the spectrum of SMA-LED to encompass generalized neurogenic arthrogryposis. All the remaining subjects remained ambulant, confirming the stable course of the disease. Our cases showed pronounced involvement of the lower limbs in comparison to the upper limbs confirming previous reports7,8,11 but now adding that the upper extremities can be involved in severe cases.
Despite the clinical appearance of distal muscle wasting and foot deformities, the proximal muscles, especially the hip extensors, were the weakest. This is in contrast to the distal weakness reported in dominant SMA families with TRPV4 mutations, suggesting this is a key clinical aspect in the differential diagnosis.1,3,5,6 Joint contractures were not a predominant clinical feature of the original DYNC1H1 families, but were noted in almost half of our cohort (46.4%). In many cases, contractures were a presenting feature, were found almost exclusively in the lower limbs, and did not progress with time. In the most severe cases, the severity of the hips, knees, and ankles contractures contributed with the weakness to compromise the ability to walk.
Because the diagnosis was not obvious at presentation, several subjects underwent muscle biopsy. The histopathology showed variable features ranging from classic neurogenic features of fiber-type grouping and fascicular hypertrophy and atrophy, to type 1 or type 2 predominance with or without fiber atrophy as previously reported in molecularly unconfirmed SMA-LED kindreds.29 Excessive connective tissue was also present in several cases, a feature not seen in typical 5q-SMA. Of note, in a few cases, the predominance of slow fibers and the presence of “core-like” areas originally suggested a congenital myopathy. However, the presence of target fibers is a well-recognized feature in reinnervated muscle as well as a variety of other lesions, including moth-eaten fibers, mini-cores, larger cores, and even vacuolation. Such cases demonstrate the importance of considering clinical, electrophysiologic, and radiologic correlation with the biopsy findings.
The muscle MRI of the lower limbs showed a striking pattern in all patients who had this test, appearing to be highly suggestive of this condition. At the thigh level, there was selective sparing and relative hypertrophy of the adductor compartment and of the semitendinosus muscles. Before the identification of mutations in DYNC1H1 in SMA-LED, we described this imaging pattern in a cohort of 11 patients with genetically unclassified dominant congenital SMA with predominant involvement of the lower limbs.4 It is of interest that this imaging pattern is different from that described in another form of congenital SMA due to mutations in TRPV4,30 but is very similar to the pattern we recently described in association with mutations in BICD2 encoding a key adaptor protein that interacts with the dynein-dynactin motor complex leading to a combination of SMA-LED and upper motor neuron features.9 The consistent pattern of muscle imaging in our cohort suggests that the muscle MRI together with the clinical findings is a valuable tool to facilitate appropriate molecular analyses.
In one-third of patients, we detected mild to moderate cognitive impairment and/or behavioral comorbidities consistent with ADHD traits. The prevalence of ADHD in our cohort is higher than that described in the general population31,32 and did not appear to be related to the severity of the motor impairment. Of note, cognitive involvement and ADHD are not features we have observed in similarly impaired patients affected by BICD2 mutations (personal observation of the authors). It has previously been reported that mutations in the motor domain of DYNC1H1 can cause severe intellectual disability associated with neuronal migration defects12,13 while only 2 cases have been described with learning difficulties associated with Charcot-Marie-Tooth disease type 2 due to a mutation in the tail domain of DYNC1H1.10 In our cohort, 9 of the 10 patients with cognitive impairment and/or behavioral comorbidities underwent brain MRI, which showed in 4 cases a pattern of cortical malformation. This is in keeping with recent reports that mutations in DYNC1H1 are a common cause of malformations of cortical development (apparently in isolation).13,14 Our cases not only indicate that there is often coexistence of central and peripheral pathology, but furthermore showed novel features such as an extended parietal cleft with posterior extension of the sylvian fissure and mild cerebellar hypoplasia.
Our findings confirm that heterozygous missense DYNC1H1 mutations can lead to a wide range of neuronal migration defects in association with a variable degree of cognitive/behavioral impairment. The Loa mice with a p.Phe580Tyr point mutation in the tail domain of DYNC1H1 show abnormalities of cortical and hippocampal development attributed to impaired radial migration and a reduction in axonal outgrowth.18 It is likely that the missense mutations identified in this study act in a similar way to the Loa mutation and impair cortical radial migration and lumbar motor neuron axonal outgrowth giving rise to the combined phenotype of SMA-LED and malformation of cortical development.
No clear genotype/phenotype correlations were found in relation to age at onset and severity, but in all cases the conditions remained essentially stable over time.
Our study provides additional information on the inheritance pattern of SMA-LED due to mutations in the tail and motor domains of DYNC1H1. We have reported both de novo mutations and for the first time a codominant homozygous mutation associated with a severe phenotype. We have also expanded the clinical spectrum, both regarding the range of severity to include generalized arthrogryposis and the age at onset, and provided further information on the long-term functional outcome. Furthermore, we confirm a characteristic pattern of muscle involvement both clinically and by MRI, which is useful diagnostically.
Supplementary Material
ACKNOWLEDGMENT
Data generated by the UK10K Consortium for one family were included in this study. A full list of the investigators who contributed data is available online at Neurology.org. Thalia Antoniadi and Sarah Burton-Jones from the Bristol Genetics Laboratory, North Bristol NHS Trust, Southmead Hospital Bristol, UK, are gratefully acknowledged for the diagnostic work on the molecular genetic confirmation in 2 families. The authors are grateful to the patients who took part in this study and their families.
GLOSSARY
- ADHD
attention deficit hyperactivity disorder
- BICD2
bicaudal D homolog 2 (Drosophila)
- CBCL
Child Behavior Checklist
- DYNC1H1
dynein, cytoplasmic 1, heavy chain 1
- LED
lower extremity predominance
- Loa
legs at odd angles
- MCD
malformation of cortical development
- SMA
spinal muscular atrophy
Footnotes
Supplemental data at Neurology.org
AUTHOR CONTRIBUTIONS
M. Scoto: oversaw the design of the study, wrote the first draft of the manuscript, contributed to data collection and to the analysis and interpretation of data. A.M. Rossor: oversaw the design of the study, contributed to the writing of the manuscript, contributed to data collection and to the analysis and interpretation of data, and gave final approval of the version to be published. M.B. Harms: oversaw the design of the study, contributed to the writing of the manuscript, contributed to data collection and to the analysis and interpretation of data, and gave final approval of the version to be published. S. Cirak: contributed to data collection and to the analysis and interpretation of data, contributed to revising the manuscript, and gave final approval of the version to be published. M. Calissano: contributed to data collection, contributed to revising the manuscript, and gave final approval of the version to be published. S. Robb: contributed to revising the manuscript and gave final approval of the version to be published. A.Y. Manzur: contributed to data collection, contributed to revising the manuscript, and gave final approval of the version to be published. A. Martínez Arroyo: contributed to data collection, contributed to revising the manuscript, and gave final approval of the version to be published. A. Rodriguez Sanz: contributed to data collection, contributed to revising the manuscript, and gave final approval of the version to be published. S. Mansour: contributed to data collection, contributed to revising the manuscript, and gave final approval of the version to be published. P. Fallon: contributed to data collection, contributed to revising the manuscript, and gave final approval of the version to be published. I. Hadjikoumi: contributed to data collection, contributed to revising the manuscript, and gave final approval of the version to be published. A. Klein: contributed to data collection, contributed to revising the manuscript, and gave final approval of the version to be published. M. Yang: contributed to data collection, contributed to revising the manuscript, and gave final approval of the version to be published. M. De Visser: contributed to data collection, contributed to revising the manuscript, and gave final approval of the version to be published. W.C.G. Overweg-Plandsoen: contributed to data collection, contributed to revising the manuscript, and gave final approval of the version to be published. F. Baas: contributed to revising the manuscript and gave final approval of the version to be published. J.P. Taylor: contributed to revising the manuscript and gave final approval of the version to be published. M. Benatar: contributed to data collection, contributed to revising the manuscript, and gave final approval of the version to be published. A.M. Connolly: contributed to revising the manuscript and gave final approval of the version to be published. M.T. Al-Lozi: contributed to revising the manuscript and gave final approval of the version to be published. J. Nixon: contributed to data collection, contributed to revising the manuscript, and gave final approval of the version to be published. C.G.E.L. de Goede: contributed to data collection, contributed to revising the manuscript, and gave final approval of the version to be published. A.R. Foley: contributed to data collection, contributed to revising the manuscript, and gave final approval of the version to be published. C. Mcwilliam: contributed to data collection, contributed to revising the manuscript, and gave final approval of the version to be published. M. Pitt: contributed to the analysis and interpretation of data, contributed to revising the manuscript, and gave final approval of the version to be published. C. Sewry: contributed to the analysis and interpretation of data, contributed to revising the manuscript, and gave final approval of the version to be published. R. Phadke: contributed to the analysis and interpretation of data, contributed to revising the manuscript, and gave final approval of the version to be published. M. Hafezparast: contributed to revising the manuscript and gave final approval of the version to be published. W.K. Chong: contributed to the analysis and interpretation of data, contributed to revising the manuscript, and gave final approval of the version to be published. E. Mercuri: contributed to the analysis and interpretation of data, contributed to revising the manuscript, and gave final approval of the version to be published. R.H. Baloh: contributed to revising the manuscript and gave final approval of the version to be published. M.M. Reilly: oversaw the design of the study, contributed to the analysis and interpretation of data, contributed to revising the manuscript, and gave final approval of the version to be published. F. Muntoni: oversaw the design of the study, contributed to data collection and to the analysis and interpretation of data, contributed to revising the manuscript, and gave final approval of the version to be published.
STUDY FUNDING
Matthew Harms is grateful for support from the NIH/National Institute of Neurological Disorders and Stroke (K08-NS075094) and UL1 RR024992 from the National Center for Research Resources (NCRR), a component of the NIH, and NIH Roadmap for Medical Research. Its contents are solely the responsibility of the authors and do not necessarily represent the official view of NCRR or NIH. Francesco Muntoni is funded by Great Ormond Street Hospital (GOSH) Children's Charity and the GOSH Biomedical Research Center, and gratefully acknowledges the MRC Center (G0601943) and the Muscular Dystrophy Campaign Centre grants. Mary M. Reilly is grateful to the Medical Research Council (MRC), MRC Center grant (G0601943), and the National Institutes of Neurological Diseases and Stroke and Office of Rare Diseases (U54NS065712) for their support. Part of this work was undertaken at University College London Hospitals/University College London, which received a proportion of funding from the Department of Health's National Institute for Health Research Biomedical Research Centers funding scheme. The Neuromuscular MRC Center Biobank is also gratefully acknowledged. Alexander M. Rossor is grateful for his fellowship funding from the National Institutes of Neurological Diseases and Stroke and Office of Rare Diseases (U54NS065712). He has also been in receipt of an IPSEN clinical research fellowship. Funding for UK10K was provided by the Wellcome Trust under award WT091310.
DISCLOSURE
M. Scoto and A. Rossor report no disclosures relevant to the manuscript. M. Harms has received honoraria from Genetech for speaking, grants from the National Institute of Neurological Disorders and Stroke, Ultragenyx Pharmaceuticals, and Biogen Idec, and nonfinancial research support from ISIS Pharmaceuticals and Oxford Nanopore, all for efforts unrelated to the present study. S. Cirak, M. Calissano, S. Robb, A. Manzur, A. Martínez Arroyo, A. Rodriguez Sanz, S. Mansour, P. Fallon, I. Hadjikoumi, A. Klein, M. Yang, M. De Visser, W. Overweg-Plandsoen, F. Baas, J. Taylor, M. Benatar, A. Connolly, M. Al-Lozi, J. Nixon, C. de Goede, A. Foley, C. Mcwilliam, M. Pitt, C. Sewry, R. Phadke, M. Hafezparast, W. Chong, E. Mercuri, and R. Baloh report no disclosures relevant to the manuscript. M. Reilly serves on the editorial boards of Neuromuscular Disorders, Journal of Neurology, Neurosurgery and Psychiatry, and the Journal of the Peripheral Nervous System and the editorial advisory board of Brain. F. Muntoni serves on scientific advisory boards for Pfizer and has served in the past for Roche, PTC, Summit, Italfarmaco, and Sarepta Therapeutics; he serves on the editorial board of Neuromuscular Disorders and Neuropediatrics; his institution (UCL) receives research support from the European Union, the Medical Research Council, the Wellcome Trust, the Association Française Contre les Myopathies (AFM), the Muscular Dystrophy Campaign, the Great Ormond Street Hospital (GOSH) Biomedical Research Centre, GSK, Genethon, and NIH. His institutions (UCL and GOSH) receive funding for clinical trials from GSK, Trophos, Prosensa, the British Heart Foundation, Summit, and has received funding for trials from AVI BioPharma and PTC Therapeutics. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Fleury P, Hageman G. A dominantly inherited lower motor neuron disorder presenting at birth with associated arthrogryposis. J Neurol Neurosurg Psychiatry 1985;48:1037–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Frijns CJ, Van Deutekom J, Frants RR, Jennekens FG. Dominant congenital benign spinal muscular atrophy. Muscle Nerve 1994;17:192–197. [DOI] [PubMed] [Google Scholar]
- 3.Van der Vleuten AJ, van Ravenswaaij-Arts CM, Frijns CJ, et al. Localisation of the gene for a dominant congenital spinal muscular atrophy predominantly affecting the lower limbs to chromosome 12q23-q24. Eur J Hum Genet 1998;6:376–382. [DOI] [PubMed] [Google Scholar]
- 4.Mercuri E, Messina S, Kinali M, et al. Congenital form of spinal muscular atrophy predominantly affecting the lower limbs: a clinical and muscle MRI study. Neuromuscul Disord 2004;14:125–129. [DOI] [PubMed] [Google Scholar]
- 5.Deng HX, Klein CJ, Yan J, et al. Scapuloperoneal spinal muscular atrophy and CMT2C are allelic disorders caused by alterations in TRPV4. Nat Genet 2010;42:165–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Auer-Grumbach M, Olschewski A, Papić L, et al. Alterations in the ankyrin domain of TRPV4 cause congenital distal SMA, scapuloperoneal SMA and HMSN2C. Nat Genet 2010;42:160–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Harms MB, Allred P, Gardner R, Jr, et al. Dominant spinal muscular atrophy with lower extremity predominance: linkage to 14q32. Neurology 2010;75:539–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Harms MB, Ori-McKenney KM, Scoto M, et al. Mutations in the tail domain of DYNC1H1 cause dominant spinal muscular atrophy. Neurology 2012;78:1714–1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Oates EC, Rossor AM, Hafezparast M, et al. Mutations in BICD2 cause dominant congenital spinal muscular atrophy and hereditary spastic paraplegia. Am J Hum Genet 2013;92:965–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weedon MN, Hastings R, Caswell R, et al. Exome sequencing identifies a DYNC1H1 mutation in a large pedigree with dominant axonal Charcot-Marie-Tooth disease. Am J Hum Genet 2011;89:308–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tsurusaki Y, Saitoh S, Tomizawa K, et al. A DYNC1H1 mutation causes a dominant spinal muscular atrophy with lower extremity predominance. Neurogenetics 2012;13:327–332. [DOI] [PubMed] [Google Scholar]
- 12.Vissers LE, de Ligt J, Gilissen C, et al. A de novo paradigm for mental retardation. Nat Genet 2010;42:1109–1112. [DOI] [PubMed] [Google Scholar]
- 13.Willemsen MH, Vissers LE, Willemsen MA, et al. Mutations in DYNC1H1 cause severe intellectual disability with neuronal migration defects. J Med Genet 2012;49:179–183. [DOI] [PubMed] [Google Scholar]
- 14.Poirier K, Lebrun N, Broix L, et al. Mutations in TUBG1, DYNC1H1, KIF5C and KIF2A cause malformations of cortical development and microcephaly. Nat Genet 2013;45:639–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fiorillo C, Moro F, Yi J, et al. Novel dynein DYNC1H1 neck and motor domain mutations link distal SMA and abnormal cortical development. Hum Mutat 2014;35:298–302. 10.1002/humu.22491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hafezparast M, Klocke R, Ruhrberg C, et al. Mutations in dynein link motor neuron degeneration to defects in retrograde transport. Science 2003;300:808–812. [DOI] [PubMed] [Google Scholar]
- 17.Dupuis L, Fergani A, Braunstein KE, et al. Mice with a mutation in the dynein heavy chain 1 gene display sensory neuropathy but lack motor neuron disease. Exp Neurol 2009;215:146–152. [DOI] [PubMed] [Google Scholar]
- 18.Ori-McKenney KM, Vallee RB. Neuronal migration defects in the Loa dynein mutant mouse. Neural Dev 2011;6:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods 2010;7:248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc 2009;4:1073–1081. [DOI] [PubMed] [Google Scholar]
- 21.Choi Y, Sims GE, Murphy S, et al. Predicting the functional effect of amino acid substitutions and indels. PLoS One 2012;7:e46688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schwarz JM, Cooper DN, Schuelke M, et al. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods 2014;11:361–362. [DOI] [PubMed] [Google Scholar]
- 23.Reva BA, Antipin YA, Sander C. Determinants of protein function revealed by combinatorial entropy optimization. Genome Biol 2007;8:R232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Achenbach T, Rescorla L. Child Behavior Checklist. Burlington, VT: ASEBA; 2000. [Google Scholar]
- 25.Keith LC. CPRS-R: L, CTRS-R: Conners 2000. Conners Rating Scales–Revised: Technical Manual. Toronto: Multi-Health System Inc.; 2000. [Google Scholar]
- 26.Dubowitz V, Sewry C, Oldfors A. Muscle Biopsy: A Practical Approach, 4th ed Oxford: Elsevier; 2013. [Google Scholar]
- 27.Mercuri E, Pichiecchio A, Counsell S, et al. A short protocol for muscle MRI in children with muscular dystrophies. Eur J Paediatr Neurol 2002;6:305–307. [DOI] [PubMed] [Google Scholar]
- 28.Nishimura AL, Mitne-Neto M, Silva HC, et al. A mutation in the vesicle-trafficking protein VAPB causes late-onset spinal muscular atrophy and amyotrophic lateral sclerosis. Am J Hum Genet 2004;75:822–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reddel S, Ouvrier RA, Nicholson G, et al. Autosomal dominant congenital spinal muscular atrophy—a possible developmental deficiency of motor neurones? Neuromuscul Disord 2008;18:530–535. [DOI] [PubMed] [Google Scholar]
- 30.Astrea G, Brisca G, Fiorillo C, et al. Muscle MRI in TRPV4-related congenital distal SMA. Neurology 2012;78:364–365. [DOI] [PubMed] [Google Scholar]
- 31.Polanczyk G, de Lima MS, Horta BL, et al. The worldwide prevalence of ADHD: a systematic review and metaregression analysis. Am J Psychiatry 2007;164:942–948. [DOI] [PubMed] [Google Scholar]
- 32.Rappley MD. Clinical practice: attention deficit-hyperactivity disorder. N Engl J Med 2005;352:165–173. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.