

Summary
R3 receptor tyrosine phosphatases (RPTPs) are characterized by extracellular domains composed solely of long chains of fibronectin type III repeats, and by the presence of a single phosphatase domain. There are five proteins in mammals with this structure, two in Drosophila, and one in Caenorhabditis elegans. R3 RPTPs are selective regulators of receptor tyrosine kinase (RTK) signaling, and a number of different RTKs have been shown to be direct targets for their phosphatase activities. Genetic studies in both invertebrate model systems and in mammals have shown that R3 RPTPs are essential for tubular organ development. They also have important functions during nervous system development. R3 RPTPs are likely to be tumor suppressors in a number of types of cancer.
Introduction
Mammalian receptor tyrosine phosphatases (RPTPs) have been subdivided into 8 ‘subtypes’ (R1–R8) based on their domain compositions (see (Tonks, 2006)). R3 RPTPs are characterized by extracellular (XC) domains composed solely of long chains of fibronectin type III (FNIII) repeats, and by the presence of a single phosphatase homology domain in their cytoplasmic regions. There are five proteins with this structure encoded in the human and mouse genomes, two in Drosophila, and one in the nematode Caenorhabditis elegans (Fig. 1).
Fig. 1. Structure and evolution of R3 RPTPs.
(A) The generic structure of an R3 RPTP. The XC domain consists of a long chain of FNIII repeats, followed by a single transmembrane domain and a single PTP homology domain. (B) Evolutionary tree of human, Drosophila, and C. elegans R3 RPTP PTP domain sequences.
R3 RPTPs appear to be selective regulators of receptor tyrosine kinase (RTK) signaling, and a number of different RTKs have been shown to be direct targets for their phosphatase activities. Because RTKs become autophosphorylated after ligand binding, and their phosphotyrosines are docking sites for downstream signaling proteins, dephosphorylation of the RTKs by R3 RPTPs would usually be expected to negatively regulate RTK signaling. However, if an R3 RPTP specifically targeted a phosphotyrosine residue that bound to a negative regulator of signaling, it could have a positive effect on RTK signaling.
Most R3 RPTPs have a C-terminal sequence that can be tyrosine-phosphorylated to form a binding site for the SH2 domains of Src-family TKs (SFKs). Binding of SFKs to this phosphotyrosine site disrupts interactions between their C-terminal phosphotyrosine residues (e.g., Y527 in chicken Src) and their SH2 domains, and this allows the phosphotyrosines to be accessible to dephosphorylation. Dephosphorylation of the C-terminal tyrosine is part of the process of SFK activation. Thus, R3 RPTPs can both negatively regulate RTKs and positively regulate SFKs (reviewed by (Matozaki et al., 2010)).
There are five R3 RPTP-like proteins in humans and mice: PTPRJ (DEP-1, CD148), PTPRB (VE-PTP), PTPRO (GLEPP1), PTPRH (SAP-1), and PTPRQ. The first four of these are tyrosine phosphatases. The PTPRQ protein, although its primary structure is very similar to that of the other R3 RPTPs, is a phosphatidylinositol (PI) phosphatase, and has little activity toward protein substrates. This has been shown to be due to mutations in PTPRQ that change its substrate binding properties. The replacement of the conserved WPD sequence with WPE, together with other changes, disorders the PTPRQ ‘M6 loop’ and flattens the catalytic pocket (Yu et al., 2013). PTPRQ is localized to the stereocilia of hair cells, and PTPRQ mutations cause deafness. (Pulido et al., 2013) have recently reviewed PTPRQ structure and function.
Drosophila has two R3 RPTPs, Ptp4E and Ptp10D. Ptp4E is very similar to Ptp10D, and was generated by a recent gene duplication (Jeon et al., 2008b). A third RPTP, Ptp52F, has an R3-like XC domain composed of FNIII repeats and a single PTP domain(Schindelholz et al., 2001), but it is not more closely related to R3 RPTPs than to other subtypes. Expansion of the vertebrate and fly R3 RPTP subfamilies occurred separately after the split between vertebrate and arthropod lineages, so there are no clear one-to-one orthologous relationships between Drosophila and mammalian R3 RPTPs (Fig. 1). However, the XC domains of the Drosophila R3 RPTPs are much more closely related to PTPRB than to other mammalian R3 RPTPs, suggesting that the three proteins might interact with similar ligands. C. elegans has a single R3 RPTP, DEP-1.
The (Matozaki et al., 2010) review provides detailed information and references for PTPRJ, PTPRB, PTPRO, and PTPRH. In this review, we describe the functions of R3 RPTPs in invertebrate models, which were not covered by (Matozaki et al., 2010), and examine some newer (post-2010) papers on vertebrate R3 RPTPs. Vertebrate and invertebrate R3 RPTPs have many properties in common. Both are selective regulators of RTK signaling, and both are required for development of tubular organs.
Regulation of receptor tyrosine kinase signaling by R3 RPTPs
Most of the known substrates of R3 RPTPs are RTKs, suggesting that a major function of this RPTP subtype is to regulate RTK signaling. Among the vertebrate R3 RPTPs, PTPRJ binds to and/or dephosphorylates the epidermal growth factor receptor (EGFR)(Tarcic et al., 2009); the hematopoietic Fms-like tyrosine kinase 3 (FLT3) (Arora et al., 2011); the platelet-derived growth factor receptor (PDGFR) (Jandt et al., 2003; Kappert et al., 2007); the vascular-endothelial growth factor receptor 2 (VEGFR2) (Grazia Lampugnani et al., 2003); the hepatocyte growth factor/scatter factor receptor, MET (Palka et al., 2003); and the glia-derived neurotrophic factor (GNDF) receptor, RET (Iervolino et al., 2006). Mammalian PTPRO dephosphorylates the TrkB and TrkC neurotrophin receptors, as well as RET (Gatto et al., 2013; Hower et al., 2009). Chick (but not mouse) PTPRO dephosphorylates the EphA and EphB RTKs(Gatto et al., 2013; Shintani et al., 2006), and zebrafish PTPRO dephosphorylates the fibroblast growth factor (FGF) receptor Fgfr1a(Liao et al., 2013). PTPRB binds to and dephosphorylates the angiopoietin (Ang) receptor RTK Tie-2, and dephosphorylates VEGFR2 (Hayashi et al., 2013; Mellberg et al., 2009; Winderlich et al., 2009).
Drosophila Ptp10D binds to EGFR (Jeon and Zinn, 2009) as does nematode DEP-1(Berset et al., 2005). Thus, interactions between R3 RPTPs and EGFR are conserved between vertebrates and invertebrates. The Drosophila R3 RPTPs also negatively regulate signaling by the FGFR and PDGFR/VEGFR orthologs, known as Breathless (Btl) and Pvr, but have not been shown to physically interact with them (Jeon et al., 2012; Jeon and Zinn, 2009b).
The in situ proximity ligation assay (in situ PLA) has recently been used to detect PTPRJ/FLT3 and PTPRB/VEGFR interactions (Bohmer et al., 2013; Hayashi et al., 2013; Mellberg et al., 2009). In situ PLA allows visualization of protein-protein interactions at endogenous levels using fluorescence microscopy. The assay uses antibodies against two candidate interacting proteins that are conjugated to DNA strands. These strands are brought into close proximity when the two antibodies bind to the same protein complex, and this enables synthesis of single stranded DNA by rolling circle amplification. The synthesized DNA is detected using fluorescently labeled oligonucleotides (reviewed by (Soderberg et al., 2008)). This assay is more sensitive than traditional biochemical methods in detecting protein complexes that are present at low levels, and it also provides information about their subcellular localizations. However, in situ PLA experiments can only show that two proteins are in close proximity, and cannot demonstrate that they bind directly to each other. Therefore, determining which protein-protein interactions are direct requires additional experiments.
PTPRJ (DEP-1) was identified as a specific negative regulator of FLT3 signaling through an siRNA screen of 20 RPTPs and PTPs (Arora et al., 2011). FLT3 is a class III RTK involved in hematopoietic differentiation, and mutations that constitutively activate FLT3 are common in acute myeloid leukemia. D→A (“substrate-trapping”) (Flint et al., 1997) and C→S (catalytically inactive) PTPRJ mutants formed stable complexes with FLT3, while wild-type PTPRJ did not (Arora et al., 2011). In situ PLA showed that PTPRJ and FLT3 interact in intact cells that express both proteins at endogenous levels. Complex formation was stimulated by FLT3 ligand, which would induce autophosphorylation of FLT3. Inhibition of FLT3’s kinase activity or inactivation of PTPRJ by oxidation disrupted complex formation, and knockdown of PTPRJ enhanced FLT3 signaling. These findings support a model in which PTPRJ is recruited to autophosphorylated FLT3 and turns off FLT3 signaling by dephosphorylating it (Bohmer et al., 2013).
Interactions of PTPRB (VE-PTP) with VEGFR2 were detected by (Mellberg et al., 2009) using in situ PLA, and a later paper from the same group further investigated these interactions and showed that they are partially dependent on Tie-2(Hayashi et al., 2013). They found that PTPRB and Tie-2 bind to each other, as reported by (Winderlich et al., 2009). However, anti-PTPRB could only precipitate VEGFR2 from lysates that had been enriched for Tie2-containing complexes by immunoprecipitation with anti-Tie-2. Anti-Tie-2 was able to precipitate VEGFR2 in the absence of PTPRB. Examination of these interactions by in situ PLA showed that PTPRB-VEGFR2 complexes at cell junctions are increased in number by VEGF and further increased by adding the Tie-2 ligand Ang. Knockdown of Tie-2 with siRNA reduces the number of complexes per cell by ~30%. These results are consistent with a model in which Tie-2 forms separate complexes with PTPRB and VEGFR2, and also brings together PTPRB and VEGFR2 to form a trimeric complex. Whether PTPRB also forms a separate dimeric complex with VEGFR2 is unclear(Hayashi et al., 2013).
Regulation of RPTP signaling by size exclusion
The data discussed above suggest that the XC domains of R3 RPTPs could be involved in interactions with RTK XC domains in the same cell (cis-interactions), although this has not been demonstrated in most cases. R3 RPTP XC domains also interact in cis and in trans with other coreceptors and ligands (Lee et al., 2013; Nawroth et al., 2002), but only a few of these have been identified. Recent studies on PTPRJ (CD148) in T cells show that the XC domain regulates access to substrates at the immunological synapse. The data suggest that RPTP XC domains can regulate signaling simply by virtue of their large sizes, without the necessity to interact with specific ligands (Cordoba et al., 2013). T-cell activation occurs when a major histocompatibility complex molecule presenting the appropriate peptide (pMHC) binds to a T-cell receptor (TCR). This causes relocalization and activation of membrane-bound signaling molecules, including the SFK Lck, which can bind to PTPRJ and is a substrate for its catalytic activity. The large XC domains of PTPRJ (and of another RPTP, CD45) appear to exclude these RPTPs from TCR ‘microclusters’. These represent sites of TCR-pMHC contact, where the membranes of the T-cell and of the pMHC-presenting cell are in close apposition. Expression of a truncated version of PTPRJ lacking most of the XC domain inhibited T-cell activation, and truncated PTPRJ was able to associate with TCR microclusters, presumably allowing it to access Lck and other substrates. However, a chimera in which the PTPRJ XC domain was replaced with the unrelated XC domain of CD43, which is of a similar size, did not cause inhibition and was excluded from TCR microclusters (Cordoba et al., 2013). These data suggest that the XC domain can regulate PTPRJ’s access to substrates simply by passive size exclusion.
Roles of R3 RPTPs in tubular organ development
In this section, we highlight recent data on PTPRB function during blood vessel development, and describe the functions of the Drosophila R3 RPTPs in regulating development of the tracheal (respiratory) system, which has many similarities to the mammalian vascular system. We also briefly review the functions of nematode DEP-1 in vulval development and of PTPRO in the kidney.
a. PTPRB and blood vessel development and function
PTPRB (VE-PTP) is selectively expressed in endothelial cells (Baumer et al., 2006). It has been shown to have two distinct roles in the vascular system, during angiogenesis and in regulating endothelial barrier function. Early in embryogenesis, blood vessel formation initiates by a process called vasculogenesis, wherein precursor cells called angioblasts aggregate and differentiate to form a rudimentary network of blood vessels, the primary vascular plexus. Subsequently, new vessels sprout from the primary vascular plexus by a process called sprouting angiogenesis, and the vascular system transforms from a primitive network of undifferentiated vessels into a hierarchal network of arteries, veins and capillaries. Angiogenesis is the predominant form of vascular growth from late embryogenesis onwards. It involves remodeling of existing branches, as well as cell proliferation and differentiation to accommodate addition of new blood vessels (reviewed by (Geudens and Gerhardt, 2011)).
Two endothelial-specific RTK families, the VEGF receptor and Tie receptor families, play key roles during vascular system development (for review see (Jeltsch et al., 2013)). VEGFR2 is required for both vasculogenesis and angiogenesis. It is expressed in the mesodermal cells that differentiate to become angioblasts, and mice that are mutant for VEGFR2 or its ligand VEGF-A die around E9.5 due to loss of endothelial cells. Later in development, VEGFR2 is expressed in cells undergoing angiogenesis, and is required for endothelial cell proliferation, sprouting of new branches, and maturation of sprouts into blood vessels (reviewed by (Olsson et al., 2006; Siekmann et al., 2013)). Tie receptors, on the other hand, are required primarily during angiogenesis but not during vasculogenesis. Mouse embryos bearing mutations eliminating the Tie-2 receptor or its ligand Ang1 display defective cardiac development and abnormal remodeling of the primary vascular plexus.
In PTPRB−/− null mutant embryos, formation of the primary vascular plexus is observed, suggesting that vasculogenesis is normal, but the subsequent remodeling phase of angiogenesis is aberrant (Dominguez et al., 2007). PTPRB mutant mice that express only the XC domain have similar defects. Both sets of mice die around E10 due to defects in formation of higher-order branched vascular networks (Baumer et al., 2006).
PTPRB is likely to regulate angiogenesis through its interactions with Tie-2, VEGFR2, and VE-cadherin at endothelial cell junctions. PTPRB binds to and dephosphorylates Tie-2 (Fachinger et al., 1999), and this dephosphorylation negatively regulates Tie-2 signaling (Winderlich et al., 2009). When cells were treated with antibodies against PTPRB, the PTPRB/Tie-2 complex was disrupted, phosphorylation of Tie-2 was increased, and Tie-2 signaling was upregulated. This caused increased endothelial cell proliferation and enlargement of vessels. A similar phenotype was observed in newborn mice injected with anti-PTPRB antibodies or with the Tie-2 activating ligand Ang1 (Thurston et al., 2005; Winderlich et al., 2009). Interestingly, an activating mutation in the Tie-2 kinase domain causes vascular defects in humans(Vikkula et al., 1996).
A direct physical interaction of PTPRB with VEGFR2 was not observed by co-immunoprecipitation (Fachinger et al., 1999). Tie2, however, interacts with both PTPRB and VEGFR2, and may act as a ‘bridge’ to facilitate formation of a trimeric VEGFR2/Tie2/PTPRB complex. In this complex, PTPRB can dephosphorylate and inactivate VEGFR2, and thus limit the duration and magnitude of the VEGFR2 signal. Dephosphorylation of activated VEGFR2 by PTPRB is important in limiting angiogenic sprouting, because PTPRB-deficient embryoid bodies displayed excess sprouting activity in response to VEGF, and the sprouts arising from mutant embryoid bodies had more phosphorylated VEGFR2 than wild-type sprouts(Hayashi et al., 2013).
PTPRB also physically interacts with the endothelial-specific adhesion molecule VE-cadherin. VEGFR2 phosphorylated VE-cadherin in transfected fibroblasts, and coexpression of PTPRB reversed this phosphorylation (Nawroth et al., 2002). Addition of VEGF to endothelial cells caused dissociation of PTPRB from VE-cadherin and increased tyrosine phosphorylation of VE-cadherin, plakoglobin, and β–catenin, three major components of the endothelial cadherin complex (Nottebaum et al., 2008). A more recent paper (Hayashi et al., 2013) links PTRPB’s roles in controlling the activity of the trimeric VEGFR2/Tie2/PTPRB complex and in regulating phosphorylation of VE-cadherin. This paper provides evidence that in unstimulated endothelial cells the trimeric complex is located away from cell junctions, and that PTPRB at the junctions is associated with VE-cadherin and stabilizes junctions by maintaining the cadherin complex in a dephosphorylated state. Addition of VEGF causes the trimeric complex to translocate to cell junctions, where it can phosphorylate VE-cadherin and its associated proteins. Its activity at these junctions is limited by dephosphorylation of activated VEGFR2 by PTPRB. The level of VEGFR2 activity present in normal animals may cause limited phosphorylation of VE-cadherin, which destabilizes junctions in a controlled manner, allowing sprouts to mature into polarized and lumenized vessels. In vessels from PTPRB-deficient mice, VEGFR2 activity is increased, causing excess phosphorylation of the cadherin complex by VEGFR2. This leads to destabilization of cadherin junctions, and prevents normal polarization and lumen formation.
The detailed molecular mechanisms by which loss of PTRB affects lumen formation are unknown. However, (Bentley et al., 2014) have used computational modeling to show that differential dynamics of local adhesion mediated by VE-cadherin drive cell rearrangements that occur during sprouting and vessel maturation. Excess signaling through VEGFR2 abolishes differential adhesion and prevents normal tubulogenesis. These results suggest that PTPRB fine-tunes cell adhesion and facilitates formation of appropriately polarized and lumenized vessels by controlling the activities of VEGFR2 and Tie2 and the phosphorylation level of the VE-cadherin complex.
An important function of the endothelial walls of blood vessels is to regulate passage of molecules from blood into surrounding tissue. There are a variety of ways in which molecules or cells can pass through the barrier formed by these endothelial cells. One is to move through the endothelial cells via transcytosis, and another is via a paracellular pathway in which interactions among adhesion molecules ‘loosen’ or ‘tighten’ to allow cell junctions to open and close reversibly. In inflamed tissues, leukocytes preferentially cross the barrier by the paracellular pathway.
The role of PTPRB in endothelial barrier function has been recently reviewed (Kuppers et al., 2014; Vestweber et al., 2014). Knockdown of PTPRB decreases adhesion at VE-cadherin junctions. This increases barrier permeability and permits increased leukocyte transendothelial migration. VEGF induces endothelial permeability, and also causes dissociation of PRPRB from VE-cadherin (Nottebaum et al., 2008). This dissociation is required for opening of junctions, because forced binding of PTPRB to VE-cadherin blocks induction of vascular permeability by VEGF or by lipopolysaccharide (LPS), an inflammatory signal (Broermann et al., 2011). These data suggest that PTPRB maintains barrier integrity via its interactions with VE-cadherin, and that induction of vascular permeability by immune cells or by soluble factors requires dissociation of the PTPRB-VE-cadherin complex.
b. R3 RPTPs and development of the respiratory system in Drosophila
Like the mammalian vascular system, the Drosophila larval respiratory (tracheal) system is a highly branched tubular network that delivers oxygen to every cell in the animal. The logic of tracheal development has parallels to that of vascular development, in that tracheal cells migrate toward sources of the FGF ligand Branchless (Bnl), while vascular sprouts grow toward sources of VEGF. The pattern of Bnl expression in the embryo is genetically determined, and it directs primary and secondary tracheal branches to form a stereotyped network. Bnl, like VEGF, is also turned on when cells become hypoxic, and attracts terminal branches (tracheoles) to extend toward each hypoxic larval cell in order to supply it with oxygen.
Tube formation in the tracheal system involves complex morphogenetic events that differ between tube types. Multicellular tubes have lumens that are surrounded by the apical surfaces of several cells. Unicellular tubes are formed by rolling up of single cells to form ‘autocellular’ junctions with themselves. The branches formed by terminal cells have proximal segments that are unicellular tubes and distal segments that are ‘seamless’ tubes without junctions. The seamless tubes are intracellular structures that form within terminal cells. Many genes have been identified that affect the formation and morphology of tracheal tubes (reviewed by (Affolter and Caussinus, 2008; Caviglia and Luschnig, 2014; Schottenfeld et al., 2010; Zuo et al., 2013).
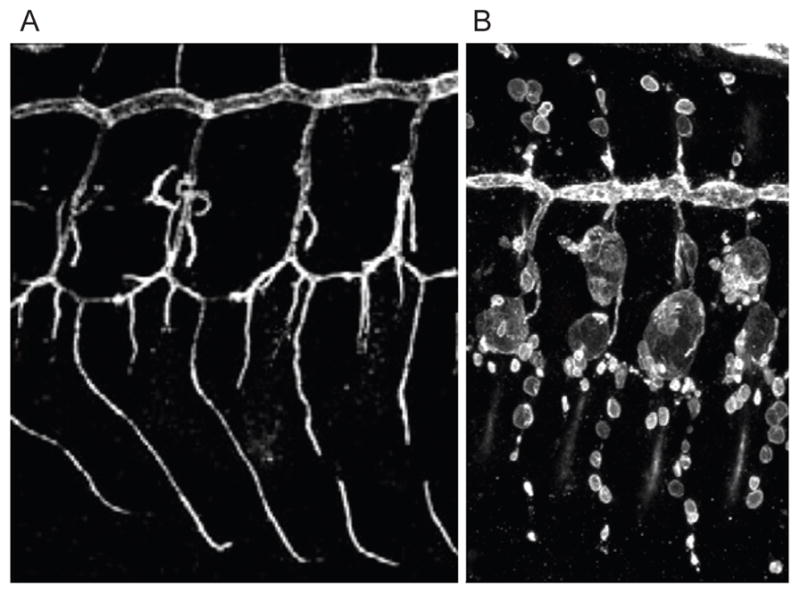
The two Drosophila R3 PTPs, Ptp4E and Ptp10D, have high sequence similarity (89% identity within the PTP domain). Ptp10D is the ancestral gene, being present in all insects, while Ptp4E is the result of a recent gene duplication(Jeon et al., 2008b). Ptp4E and Ptp10D single mutants are viable and fertile, but Ptp4E Ptp10D double mutants die from respiratory failure at the end of embryogenesis(Jeon and Zinn, 2009a). Ptp4E Ptp10D double mutants have defects in lumen formation that are specific to unicellular tubes and tracheoles; multicellular tubes are unaffected. Unicellular branches undergo a remodeling process during development in which cells that are originally positioned as pairs along the length of the branch slide past each other to take on an end-to-end configuration. During this process, junctions that were originally formed between cells transform into autocellular junctions. Terminal cells sprout branches containing seamless tubes late during development, after a continuous tracheal network has been built. In Ptp4E Ptp10D mutants, the cylindrical lumen within unicellular tubes is replaced by large bubble-like cysts. Even larger multicellular cysts develop at the junctions between unicellular tubes. Finally, seamless tubes are replaced by discontinuous bubbles (Fig. 2).
Fig. 2. Dramatic alterations of tracheal tube morphology produced by loss of R3 RPTP activity and hyperactivation of RTK signaling.

(A) Four segments of the normal tracheal network in a Drosophila embryo. (B) Four segments of the tracheal network in a Ptp4E Ptp10D double mutant embryo that also expresses an activated mutant EGFR (EgfrEllipse) in tracheal cells. Note that all tracheal branches are converted to large bubble-like cysts, except for the multicellular dorsal trunk (large horizontal tube at the top), which is relatively unaffected. Expression of EgfrEllipse in a wild-type embryo produces no cysts. For further information and diagrams and images of wild-type and mutant tracheae, see (Jeon et al., 2012; Jeon and Zinn, 2009).
The Ptp4E Ptp10D cyst phenotype is enhanced by expression of activated (CA) EGFR and suppressed by expression of dominant-negative (DN) EGFR. This suggests that the phenotype is partially due to an increase in EGFR activity, and that these RPTPs are negative regulators of EFGR signaling. Ptp10D binds to EGFR when the two proteins are coexpressed in transfected Drosophila cells. Expression of activated EGFR in wild-type embryos, however, did not generate any cysts, indicating that there are likely to be other RTK pathways that are affected in Ptp4E Ptp10D mutants(Jeon and Zinn, 2009a). To identify these, CA and DN mutants of two other RTKs expressed in tracheal cells, Btl (FGFR ortholog) and Pvr (PDGFR/VEGFR2 ortholog) were expressed in the Ptp4E Ptp10D background. All three CA mutants enhanced the phenotype, and all three DN mutants suppressed it. Experiments in which CA mutants of one RTK were competed against DN mutants of another by simultaneously expressing them in the Ptp4E Ptp10D background showed that the three RTKs have partially interchangeable activities. Increasing the activity of one RTK can compensate for the effects of reducing the activity of another. These results imply that SH2-domain downstream effectors that are required for the phenotype are likely to be able to interact with phosphotyrosines on all three RTKs.
The results described above suggest that the unique Ptp4E Ptp10D cyst phenotype is the result of simultaneous elevation of the activities of EGFR, FGFR, and Pvr, and possibly of other RTKs as well. The fact that expression of two or three RTK CA mutants together did not produce cysts (Jeon et al., 2012) would seem to be inconsistent with this model. However, because tyrosine phosphatase activity normally is greatly in excess of tyrosine kinase activity, removal of R3 RPTP control of RTK activity probably has a much greater effect on tyrosine phosphorylation than does expression of CA RTK mutants. This can be seen by comparing the dramatic effects on phosphotyrosine levels of treating cells with pervanadate, a tyrosine phosphatase inhibitor, vs. the relatively subtle effects of expressing an activated TK (see (Bugga et al., 2009) for an example of this).
RTKs can signal through a wide variety of pathways. Two pathways that are relevant to the Ptp4E Ptp10D cystic phenotype are the MAP kinase pathway and the Rho GTPase pathway. Expression of CA mutants of MAP kinase kinase kinase (MAPKKK; Phl in Drosophila) or of Rho1 in the Ptp4E Ptp10D background enhanced the phenotype, while expression of DN mutants of Rho or Rac suppressed it. Like CA RTKs, however, neither of these CA mutants could induce cyst formation in a wild-type background. These data suggest that the phenotype occurs through simultaneous activation of multiple downstream RTK signaling pathways(Jeon et al., 2012).
In Ptp4E Ptp10D mutants, the lumen in unicellular branches expands to 6 or more times its normal diameter, and has a spherical rather than a cylindrical shape. The normal lumen and the abnormal cysts are both apical compartments. We suggest that the phenotype arises from a failure to coordinate apical membrane expansion with the remodeling of the rest of the cell during its transformation into a tube. Perhaps there are common elements involved in the failure to form normal lumen in Ptp4E Ptp10D mutant tracheal branches and in PTPRB mutant blood vessels(Hayashi et al., 2013).
Two recent papers have identified new genes whose activities may be relevant to the control of tracheal lumen formation by R3 RPTPs. The first is a novel cytoplasmic Smad-like protein, Expansion (Exp). Remarkably, exp mutant embryos have cystic phenotypes restricted to unicellular and terminal branches that are identical to those of Ptp4E Ptp10D mutants, although the exp phenotype is weaker. Although extensive genetic screening has been performed for tracheal phenotypes, the Ptp4E Ptp10D cyst phenotype was unique until now. We had suggested that perhaps there was no single gene for which mutation could produce this phenotype, because it requires an increase in signaling by at least three RTKs. However, the discovery of exp shows that this is not the case. Exp is homologous to Smad proteins, which are downstream of TGF-β receptors, but Exp does not appear to participate in TGF-β signaling. Rather, genetic interaction studies showed that, like Ptp4E and Ptp10D, Exp is likely to be a negative regulator of RTK signaling (Iordanou et al., 2014).
Exp clearly regulates the same aspects of RTK signaling as the R3 RPTPs, and is probably downstream of RTKs and RPTPs, because overexpression of Exp partially suppressed the Ptp4E Ptp10D phenotype. Also, like the Ptp4E Ptp10D phenotype, the exp phenotype is enhanced by expression of CA MAPKKK and CA Rho1, and suppressed by DN Rho and Rac(Iordanou et al., 2014). These data show that Exp is a critical component of the pathway(s) regulated by RTKs and R3 RPTPs that are relevant to the normal formation of tracheal lumen.
The Ptp4E Ptp10D double mutation causes cystic lumen dilation phenotypes both in unicellular tubes, which have junctions, and in seamless tubes in terminal cells, which do not. Because these tubes are very different types of structures, it is possible that cyst formation occurs by different mechanisms in the two tube types. Mutations in wheezy, which encodes Germinal center kinase III (GCKIII) cause lumen dilation in terminal cells within the ‘transition zone’ where a unicellular tube with an autocellular junction transitions to a seamless tube. Only terminal cells were affected in wheezy mutants. GCKIII proteins are a subfamily of Ste20-related serine/threonine kinases. GCKIII binds to Cerebral cavernous malformation 3 (CCM3), a gene mutated in a human vascular disease characterized by dilation of cerebral capillaries. The authors identified a CCM3 ortholog in Drosophila, and showed that Ccm3 mutants have a similar tracheal lumen dilation phenotype. This provides a further parallel between the Drosophila tracheal system and the mammalian vascular system. Finally, the wheezy terminal cell lumen dilation phenotype is associated with extension of septate junctions into the transition zone, and loss-of-function mutations of septate junction components were able to suppress the phenotype(Song et al., 2013).
c. DEP-1 and development of the C. elegans vulva
The C. elegans hermaphrodite vulva is a tubular organ that connects the gonad to the exterior environment. It develops from a group of epithelial cells called vulval precursor cells (VPCs), initially located on the body wall, that migrate internally and connect to the somatic gonad to create an opening to the outside world. VPCs are equivalent until an EGF ligand, LIN-3, secreted from the anchor cell (AC) instructs them to take on different cell fates. The P6.p VPC located closest to AC receives the strongest EGF signal and takes on the primary cell fate, becoming the leading cell during the invagination process. The adjacent cells P5.p and P7.p receive intermediate levels of EGF signal, take on secondary cell fates, and become follower cells during vulval tube morphogenesis. These cell fate decisions are further reinforced by lateral inhibition through NOTCH/LIN-12 signaling, where P6.p inhibits P5.p and P7.p from taking on primary cell fates by upregulating expression of negative regulators of the EGFR/LET-23 pathway in P5.p and P7.p.
lip-1, which encodes a dual-specificity phosphatase that is the ortholog of vertebrate MKP-3, is one such negative regulator. The dep-1 mutation was isolated as an enhancer of lip-1. lip-1 and dep-1 mutant animals develop normal vulvae, while lip-1;dep-1 double mutants have defects in vulva development that resemble those produced by prolonged or hyperactivated EGFR signaling (Berset et al., 2005). Thus, DEP-1 and LIP-1 negatively regulate EGFR signaling in parallel pathways to regulate primary and secondary cell fate decisions. Interestingly, like Ptp4E and Ptp10D, DEP-1 and LIP-1 appear to have redundant activities with regard to tubular organ development. However, Ptp4E and Ptp10D are very similar to each other, whereas DEP-1 and LIP-1 are members of different phosphatase families.
Like mammalian DEP-1 and Drosophila Ptp10D, C. elegans DEP-1 binds directly to EGFR and is likely to function by regulating EGFR autophosphorylation in response to EGF stimulation(Berset et al., 2005). More recent data also implicate Rho signaling in development of lumen in the vulva. This may represent a further parallel to the Drosophila tracheal system. VPCs undergo three rounds of division, and the progenitors of primary and secondary cell fated VPCs form donut-shaped (toroidal) cells that build the walls of the vulva. EGFR signaling in primary toroids inhibits activation of the Rho kinase (ROK) LET-502, in order to allow expansion of the dorsal lumen that is closest to the junction with the somatic gonad. NOTCH/LIN-12 signaling in the secondary toroids activates ROK to induce actomyosin-mediated contractions that direct the vulva to grow dorsally towards the somatic gonad(Farooqui et al., 2012).
d. PTPRO and kidney filtration
PTPRO, also known as glomerular epithelial protein 1 (GLEPP1) is expressed in podocytes, which are specialized epithelial cells that surround the capillaries in the renal glomerulus. Podocytes play a critical role in filtration. The foot processes of podocytes interdigitate with those of neighboring podocytes to cover the surface of capillaries. In the spaces between the interdigitating foot processes are found a meshwork of cell adhesion molecules such as nephrins and cadherins that act as a sieve to allow passage of small solutes and retention of proteins and other large molecules. This molecular sieve is called the slit diaphragm. PTPRO’s functions in podocytes were reviewed by (Matozaki et al., 2010). Briefly, PTPRO is important for proper function of the slit diaphragm. In PTPRO mutant mice, podocyte foot processes have abnormal morphologies and the glomerular filtration rate is decreased (Wharram et al., 2000). PTPRO is likely to regulate slit diaphragm function by phosphorylation of tyrosine residues on the cytoplasmic domains of slit diaphragm components like nephrin and P-cadherin.
R3 RPTPs and neural development
Of the four vertebrate R3 RPTPs, only PTPRO has been shown to have clear functions during neural development. Here we discuss the neural functions of the Drosophila R3 RPTPs, which were not reviewed by (Matozaki et al., 2010), as well as three newer papers on neuronal PTPRO (Gatto et al., 2013; Liao et al., 2013; Tchetchelnitski et al., 2014).
a. Ptp10D and development of the embryonic Drosophila CNS
In Drosophila, five of the six RPTPs are selectively expressed in neurons, and their functions have primarily been studied in the context of nervous system development. Ptp10D is expressed only on central nervous system (CNS) axons and tracheal (respiratory) cells during embryonic development (Tian et al., 1991; Yang et al., 1991). The other R3 RPTP, Ptp4E, appears to be expressed in most cells at roughly equal levels, although Ptp4E mRNA is enriched in the gut in late embryos (Jeon et al., 2008a; Oon et al., 1993).
Null mutations in three of the six Drosophila Rptp genes (Lar, Ptp52F, Ptp69D) confer lethality and produce embryonic axon guidance phenotypes. Mutations in the other three (Ptp4E, Ptp10D, Ptp99A) produce no known embryonic phenotypes, and mutant adults are viable and have no obvious defects. The viability of Ptp4E and Ptp10D mutants is due to redundancy between these closely related R3 RPTPs, because Ptp4E Ptp10D double mutants die at the end of embryogenesis(Jeon et al., 2008a, b; Jeon and Zinn, 2009b). The only single mutant phenotype that has been reported for an R3 RPTP is a defect in long-term memory formation in Ptp10D mutant adults (Qian et al., 2007).
Due to genetic redundancy, the embryonic functions of R3 RPTPs have primarily been studied by making double and triple mutants lacking expression of multiple RPTPs. These studies revealed that Ptp10D and the Type IIa RPTP Ptp69D have partially redundant roles in preventing longitudinal axons in the CNS from abnormally crossing the ventral midline of the embryo. In Ptp10D Ptp69D double mutants, many axons that would normally extend anteriorly or posteriorly in the longitudinal tracts instead grow across the midline within the anterior and posterior commissures (Sun et al., 2000; Sun et al., 2001). Interestingly, double mutants (Ptp4E Ptp10D) lacking both R3 RPTPs, as well as Ptp4E Ptp69D double mutants, do not have strong axon guidance phenotypes. Ptp4E Ptp10D Ptp69D triple mutants have phenotypes that are very similar to Ptp10D Ptp69D double mutants (Jeon et al., 2008a). These data suggest that Ptp10D and Ptp69D share some substrate(s) whose dephosphorylation is essential in preventing abnormal midline crossing. Despite its very similar catalytic domain sequence, Ptp4E cannot compensate for the absence of Ptp10D in Ptp10D Ptp69D double mutants. This may indicate that Ptp4E cannot dephosphorylate these putative neuronal Ptp10D/Ptp69D substrate(s), perhaps because it has a different substrate specificity or because it is not expressed at sufficiently high levels on axons to be able to compensate for the absence of Ptp10D. We favor the latter explanation, because Ptp4E and Ptp10D appear to target the same RTKs in tracheal cells (Jeon et al., 2012; Jeon and Zinn, 2009). The TKs whose signaling is regulated by Ptp10D and Ptp69D in neurons have not been identified.
A recent paper identified a cell-surface ligand/coreceptor required for Ptp10D function, Stranded at second (Sas) (Lee et al., 2013). The XC domain of Ptp10D binds to Sas in vitro and on the surfaces of cultured cells. Sas is a large single-pass transmembrane protein containing von Willebrand C (VWC) domains and FNIII repeats in its XC region. It has a short cytoplasmic domain containing putative SH2 and PTB domain binding sites. Sas has no clear vertebrate ortholog, although there are many vertebrate cell-surface proteins that contain VWC and FNIII domains.
Sas is expressed on both neurons and glia in the embryo, and has distinct roles in these two cell types. Neuronal Sas is required for Ptp10D’s functions in preventing abnormal midline crossing by longitudinal axons. The evidence for this is that sas Ptp69D double mutants, like Ptp10D Ptp69D double mutants, have ectopic midline crossing phenotypes, and these are rescued by restoring expression of Sas in neurons. Glial Sas interacts in trans with neuronal Ptp10D, and this interaction affects glial migration and morphogenesis. Ptp10D negatively regulates Sas signaling in glia. This is demonstrated by findings that: 1) glial organization is disrupted in embryos in which Sas is overexpressed in glia, and, 2) this glial Sas gain-of-function phenotype is enhanced by removal of Ptp10D from neurons (Lee et al., 2013).
b. PTPRO in the vertebrate nervous system
PTPRO is involved in motor and retinal axon guidance in the chick, and can dephosphorylate the Eph and TrkC RTKs (Hower et al., 2009; Shintani et al., 2006; Stepanek et al., 2005). Its functions in mouse neural development have been less well understood. However, a recent paper shows that target innervation by trigeminal ganglion (TG) neurons is altered in PTPRO−/− embryos (Gatto et al., 2013). In particular, one of the arbors of the ophthalmic branch of the TG is more complex and covers a bigger area in mutant animals. The TG contains TrkA+ (nociceptive), TrkB+ and Ret+ (mechanoreceptive), and TrkC+ neurons. PTPRO is primarily expressed in TrkB+ and Ret+ neurons, and cultured TG neurons from PTPRO−/− embryos extend longer axons than wild-type neurons in response to BDNF (the ligand for the TrkB RTK) and to GDNF (the ligand for the Ret RTK), consistent with this expression pattern. PTPRO colocalizes with TrkB and Ret when expressed in HeLa cells, and can dephosphorylate both of these RTKs. Interestingly, mouse PTPRO does not appear to affect Eph signaling in TG or retinal neurons, and has a reduced ability to dephosphorylate an Eph RTK in HEK293 cells relative to chick PTPRO. These data suggest that the mammalian and avian PTPROs may have evolved to have different substrate specificities and functions (Gatto et al., 2013).
Mouse PTPRO is required for survival and axonal projection of a subset of dorsal root ganglion (DRG) neurons (Gonzalez-Brito and Bixby, 2009). A recent paper, however, analyzed several RPTPs expressed in DRG neurons, and found that shRNA knockdown of PTPRO or of two different Type IIa RPTPs did not measurably affect Trk signaling (Tchetchelnitski et al., 2014). These data suggest that, as in Drosophila (Sun et al., 2000), the R3 RPTP PTPRO might function redundantly with one or more Type IIa RPTPs in regulating Trk signaling and neural development. However, this has not yet been proven.
Finally, a recent study of zebrafish PTPRO found that morpholino knockdown caused cerebellar defects. Both granule and Purkinje neurons were reduced in number. FGF8 is required for cerebellar development, and dusp6, a gene whose expression is controlled by FGF signaling, is upregulated when PTPRO is knocked down. These results suggested that PTPRO might regulate the FGF receptor RTK. This was confirmed by studies in transfected 293T cells showing that coexpression of PTPRO reduces tyrosine phosphorylation of Fgfr1a (Liao et al., 2013). FGFRs had not been previously implicated as targets of vertebrate R3 RPTPs, so this provides a further parallel between the vertebrate and Drosophila systems, in which Ptp10D and Ptp4E regulate signaling by the FGFR ortholog in tracheal cells.
R3 RPTPs and cancer
PTPRJ is implicated in a variety of cancers in mice and humans (for a recent review see (Hendriks and Pulido, 2013)). PTPRJ was identified as a candidate for the Suppressor of colon cancer 1 (Scc1) locus in the mouse, but PTPRJ knockout mice do not spontaneously develop cancer. No mutations affecting the PTPRJ coding sequence have yet been found in human cancer, although loss or silencing of one copy of PTPRJ is very common in many tumor types. Another R3 RPTP, PTPRH, is upregulated in human colon and pancreatic tumors, suggesting that its activity might favor tumor formation rather than suppress it. However, loss of PTPRH inhibited tumorigenesis in a mouse model of colon cancer (reviewed by (Matozaki et al., 2010)).
A recent paper showed that PTPRB qualifies as a genuine tumor suppressor in a rare tumor type, angiosarcoma, which is associated with exposure to ionizing radiation. PTPRB was the most frequently mutated gene in angiosarcomas, with probable loss-of-function PTPRB alleles being found in 10 of 39 (26%) of tumors. In four of these tumors, mutations in both copies of PTPRB were found, and in two cases both mutations were truncating. These data suggest that PTPRB is a recessive tumor suppressor gene for this tumor type. PTPRB is a negative regulator of angiogenesis, and small-molecule inhibitors of the VEGFR2 RTK suppressed the increased angiogenesis caused by siRNA knockdown of PTPRB in human umbilical vein endothelial cell cultures. These results suggest that VEGFR2 inhibitors might be useful in treating the subset of angiosarcomas that harbor PTPRB mutations (Behjati et al., 2014).
Acknowledgments
This work was supported by an NIH R21 grant to K.Z., HD0773367. We thank Peter Lee for discussions and communication of unpublished data, and Matthew Scott for hosting M.J. in his laboratory and for helpful discussions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Affolter M, Caussinus E. Tracheal branching morphogenesis in Drosophila: new insights into cell behaviour and organ architecture. Development. 2008;135:2055–2064. doi: 10.1242/dev.014498. [DOI] [PubMed] [Google Scholar]
- Arora D, Stopp S, Bohmer SA, Schons J, Godfrey R, Masson K, Razumovskaya E, Ronnstrand L, Tanzer S, Bauer R, et al. Protein-tyrosine phosphatase DEP-1 controls receptor tyrosine kinase FLT3 signaling. J Biol Chem. 2011;286:10918–10929. doi: 10.1074/jbc.M110.205021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumer S, Keller L, Holtmann A, Funke R, August B, Gamp A, Wolburg H, Wolburg-Buchholz K, Deutsch U, Vestweber D. Vascular endothelial cell-specific phosphotyrosine phosphatase (VE-PTP) activity is required for blood vessel development. Blood. 2006;107:4754–4762. doi: 10.1182/blood-2006-01-0141. [DOI] [PubMed] [Google Scholar]
- Behjati S, Tarpey PS, Sheldon H, Martincorena I, Van Loo P, Gundem G, Wedge DC, Ramakrishna M, Cooke SL, Pillay N, et al. Recurrent PTPRB and PLCG1 mutations in angiosarcoma. Nat Genet. 2014;46:376–379. doi: 10.1038/ng.2921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentley K, Franco CA, Philippides A, Blanco R, Dierkes M, Gebala V, Stanchi F, Jones M, Aspalter IM, Cagna G, et al. The role of differential VE-cadherin dynamics in cell rearrangement during angiogenesis. Nat Cell Biol. 2014;16:309–321. doi: 10.1038/ncb2926. [DOI] [PubMed] [Google Scholar]
- Berset TA, Hoier EF, Hajnal A. The C. elegans homolog of the mammalian tumor suppressor Dep-1/Scc1 inhibits EGFR signaling to regulate binary cell fate decisions. Genes Dev. 2005;19:1328–1340. doi: 10.1101/gad.333505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohmer SA, Weibrecht I, Soderberg O, Bohmer FD. Association of the protein-tyrosine phosphatase DEP-1 with its substrate FLT3 visualized by in situ proximity ligation assay. PLoS One. 2013;8:e62871. doi: 10.1371/journal.pone.0062871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broermann A, Winderlich M, Block H, Frye M, Rossaint J, Zarbock A, Cagna G, Linnepe R, Schulte D, Nottebaum AF, et al. Dissociation of VE-PTP from VE-cadherin is required for leukocyte extravasation and for VEGF-induced vascular permeability in vivo. The Journal of experimental medicine. 2011;208:2393–2401. doi: 10.1084/jem.20110525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bugga L, Ratnaparkhi A, Zinn K. The cell surface receptor Tartan is a potential in vivo substrate for the receptor tyrosine phosphatase Ptp52F. Mol Cell Biol. 2009;29:3390–3400. doi: 10.1128/MCB.01764-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caviglia S, Luschnig S. Tube fusion: making connections in branched tubular networks. Seminars in cell & developmental biology. 2014;31:82–90. doi: 10.1016/j.semcdb.2014.03.018. [DOI] [PubMed] [Google Scholar]
- Cordoba SP, Choudhuri K, Zhang H, Bridge M, Basat AB, Dustin ML, van der Merwe PA. The large ectodomains of CD45 and CD148 regulate their segregation from and inhibition of ligated T-cell receptor. Blood. 2013;121:4295–4302. doi: 10.1182/blood-2012-07-442251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez MG, Hughes VC, Pan L, Simmons M, Daly C, Anderson K, Noguera-Troise I, Murphy AJ, Valenzuela DM, Davis S, et al. Vascular endothelial tyrosine phosphatase (VE-PTP)-null mice undergo vasculogenesis but die embryonically because of defects in angiogenesis. Proc Natl Acad Sci U S A. 2007;104:3243–3248. doi: 10.1073/pnas.0611510104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fachinger G, Deutsch U, Risau W. Functional interaction of vascular endothelial-protein-tyrosine phosphatase with the angiopoietin receptor Tie-2. Oncogene. 1999;18:5948–5953. doi: 10.1038/sj.onc.1202992. [DOI] [PubMed] [Google Scholar]
- Farooqui S, Pellegrino MW, Rimann I, Morf MK, Muller L, Frohli E, Hajnal A. Coordinated lumen contraction and expansion during vulval tube morphogenesis in Caenorhabditis elegans. Dev Cell. 2012;23:494–506. doi: 10.1016/j.devcel.2012.06.019. [DOI] [PubMed] [Google Scholar]
- Flint AJ, Tiganis T, Barford D, Tonks NK. Development of “substrate-trapping” mutants to identify physiological substrates of protein tyrosine phosphatases. Proc Natl Acad Sci U S A. 1997;94:1680–1685. doi: 10.1073/pnas.94.5.1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatto G, Dudanova I, Suetterlin P, Davies AM, Drescher U, Bixby JL, Klein R. Protein tyrosine phosphatase receptor type O inhibits trigeminal axon growth and branching by repressing TrkB and Ret signaling. J Neurosci. 2013;33:5399–5410. doi: 10.1523/JNEUROSCI.4707-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geudens I, Gerhardt H. Coordinating cell behaviour during blood vessel formation. Development. 2011;138:4569–4583. doi: 10.1242/dev.062323. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Brito MR, Bixby JL. Protein tyrosine phosphatase receptor type O regulates development and function of the sensory nervous system. Molecular and cellular neurosciences. 2009;42:458–465. doi: 10.1016/j.mcn.2009.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grazia Lampugnani M, Zanetti A, Corada M, Takahashi T, Balconi G, Breviario F, Orsenigo F, Cattelino A, Kemler R, Daniel TO, et al. Contact inhibition of VEGF-induced proliferation requires vascular endothelial cadherin, beta-catenin, and the phosphatase DEP-1/CD148. J Cell Biol. 2003;161:793–804. doi: 10.1083/jcb.200209019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi M, Majumdar A, Li X, Adler J, Sun Z, Vertuani S, Hellberg C, Mellberg S, Koch S, Dimberg A, et al. VE-PTP regulates VEGFR2 activity in stalk cells to establish endothelial cell polarity and lumen formation. Nature communications. 2013;4:1672. doi: 10.1038/ncomms2683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendriks WJ, Pulido R. Protein tyrosine phosphatase variants in human hereditary disorders and disease susceptibilities. Biochimica et biophysica acta. 2013;1832:1673–1696. doi: 10.1016/j.bbadis.2013.05.022. [DOI] [PubMed] [Google Scholar]
- Hower AE, Beltran PJ, Bixby JL. Dimerization of tyrosine phosphatase PTPRO decreases its activity and ability to inactivate TrkC. J Neurochem. 2009;110:1635–1647. doi: 10.1111/j.1471-4159.2009.06261.x. [DOI] [PubMed] [Google Scholar]
- Iervolino A, Iuliano R, Trapasso F, Viglietto G, Melillo RM, Carlomagno F, Santoro M, Fusco A. The receptor-type protein tyrosine phosphatase J antagonizes the biochemical and biological effects of RET-derived oncoproteins. Cancer research. 2006;66:6280–6287. doi: 10.1158/0008-5472.CAN-06-0228. [DOI] [PubMed] [Google Scholar]
- Iordanou E, Chandran RR, Yang Y, Essak M, Blackstone N, Jiang L. The novel Smad protein Expansion regulates the receptor tyrosine kinase pathway to control Drosophila tracheal tube size. Dev Biol. 2014;393:93–108. doi: 10.1016/j.ydbio.2014.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jandt E, Denner K, Kovalenko M, Ostman A, Bohmer FD. The protein-tyrosine phosphatase DEP-1 modulates growth factor-stimulated cell migration and cell-matrix adhesion. Oncogene. 2003;22:4175–4185. doi: 10.1038/sj.onc.1206652. [DOI] [PubMed] [Google Scholar]
- Jeltsch M, Leppanen VM, Saharinen P, Alitalo K. Receptor tyrosine kinase-mediated angiogenesis. Cold Spring Harbor perspectives in biology. 2013;5 doi: 10.1101/cshperspect.a009183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon M, Nguyen H, Bahri S, Zinn K. Redundancy and compensation in axon guidance: genetic analysis of the Drosophila Ptp10D/Ptp4E receptor tyrosine phosphatase subfamily. Neural development. 2008a;3:3. doi: 10.1186/1749-8104-3-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon M, Nguyen H, Bahri S, Zinn K. Redundancy and compensation in axon guidance: genetic analysis of the Drosophila Ptp10D/Ptp4E receptor tyrosine phosphatase subfamily. Neural development. 2008b;3:3. doi: 10.1186/1749-8104-3-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon M, Scott MP, Zinn K. Interactions between Type III receptor tyrosine phosphatases and growth factor receptor tyrosine kinases regulate tracheal tube formation in Drosophila. Biology Open. 2012;1:548–558. doi: 10.1242/bio.2012471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon M, Zinn K. Receptor tyrosine phosphatases control tracheal tube geometries through negative regulation of Egfr signaling. Development. 2009a;136:3121–3129. doi: 10.1242/dev.033597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon M, Zinn K. Receptor tyrosine phosphatases control tracheal tube geometries through negative regulation of Egfr signaling. Development. 2009b;136:3121–3129. doi: 10.1242/dev.033597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kappert K, Paulsson J, Sparwel J, Leppanen O, Hellberg C, Ostman A, Micke P. Dynamic changes in the expression of DEP-1 and other PDGF receptor-antagonizing PTPs during onset and termination of neointima formation. FASEB J. 2007;21:523–534. doi: 10.1096/fj.06-6219com. [DOI] [PubMed] [Google Scholar]
- Kuppers V, Vockel M, Nottebaum AF, Vestweber D. Phosphatases and kinases as regulators of the endothelial barrier function. Cell and tissue research. 2014;355:577–586. doi: 10.1007/s00441-014-1812-1. [DOI] [PubMed] [Google Scholar]
- Lee HK, Cording A, Vielmetter J, Zinn K. Interactions between a Receptor Tyrosine Phosphatase and a Cell Surface Ligand Regulate Axon Guidance and Glial-Neuronal Communication. Neuron. 2013;78:813–826. doi: 10.1016/j.neuron.2013.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao WH, Cheng CH, Hung KS, Chiu WT, Chen GD, Hwang PP, Hwang SP, Kuan YS, Huang CJ. Protein tyrosine phosphatase receptor type O (Ptpro) regulates cerebellar formation during zebrafish development through modulating Fgf signaling. Cell Mol Life Sci. 2013;70:2367–2381. doi: 10.1007/s00018-013-1259-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matozaki T, Murata Y, Mori M, Kotani T, Okazawa H, Ohnishi H. Expression, localization, and biological function of the R3 subtype of receptor-type protein tyrosine phosphatases in mammals. Cell Signal. 2010;22:1811–1817. doi: 10.1016/j.cellsig.2010.07.001. [DOI] [PubMed] [Google Scholar]
- Mellberg S, Dimberg A, Bahram F, Hayashi M, Rennel E, Ameur A, Westholm JO, Larsson E, Lindahl P, Cross MJ, et al. Transcriptional profiling reveals a critical role for tyrosine phosphatase VE-PTP in regulation of VEGFR2 activity and endothelial cell morphogenesis. FASEB J. 2009;23:1490–1502. doi: 10.1096/fj.08-123810. [DOI] [PubMed] [Google Scholar]
- Nawroth R, Poell G, Ranft A, Kloep S, Samulowitz U, Fachinger G, Golding M, Shima DT, Deutsch U, Vestweber D. VE-PTP and VE-cadherin ectodomains interact to facilitate regulation of phosphorylation and cell contacts. Embo J. 2002;21:4885–4895. doi: 10.1093/emboj/cdf497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nottebaum AF, Cagna G, Winderlich M, Gamp AC, Linnepe R, Polaschegg C, Filippova K, Lyck R, Engelhardt B, Kamenyeva O, et al. VE-PTP maintains the endothelial barrier via plakoglobin and becomes dissociated from VE-cadherin by leukocytes and by VEGF. The Journal of experimental medicine. 2008;205:2929–2945. doi: 10.1084/jem.20080406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsson AK, Dimberg A, Kreuger J, Claesson-Welsh L. VEGF receptor signalling - in control of vascular function. Nature reviews. 2006;7:359–371. doi: 10.1038/nrm1911. [DOI] [PubMed] [Google Scholar]
- Oon SH, Hong A, Yang X, Chia W. Alternative splicing in a novel tyrosine phosphatase gene (DPTP4E) of Drosophila melanogaster generates two large receptor-like proteins which differ in their carboxyl termini. J Biol Chem. 1993;268:23964–23971. [PubMed] [Google Scholar]
- Palka HL, Park M, Tonks NK. Hepatocyte growth factor receptor tyrosine kinase met is a substrate of the receptor protein-tyrosine phosphatase DEP-1. J Biol Chem. 2003;278:5728–5735. doi: 10.1074/jbc.M210656200. [DOI] [PubMed] [Google Scholar]
- Pulido R, Stoker AW, Hendriks WJ. PTPs emerge as PIPs: protein tyrosine phosphatases with lipid-phosphatase activities in human disease. Hum Mol Genet. 2013;22:R66–76. doi: 10.1093/hmg/ddt347. [DOI] [PubMed] [Google Scholar]
- Qian M, Pan G, Sun L, Feng C, Xie Z, Tully T, Zhong Y. Receptor-like tyrosine phosphatase PTP10D is required for long-term memory in Drosophila. J Neurosci. 2007;27:4396–4402. doi: 10.1523/JNEUROSCI.4054-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelholz B, Knirr M, Warrior R, Zinn K. Regulation of CNS and motor axon guidance in Drosophila by the receptor tyrosine phosphatase DPTP52F. Development. 2001;128:4371–4382. doi: 10.1242/dev.128.21.4371. [DOI] [PubMed] [Google Scholar]
- Schottenfeld J, Song Y, Ghabrial AS. Tube continued: morphogenesis of the Drosophila tracheal system. Current opinion in cell biology. 2010;22:633–639. doi: 10.1016/j.ceb.2010.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shintani T, Ihara M, Sakuta H, Takahashi H, Watakabe I, Noda M. Eph receptors are negatively controlled by protein tyrosine phosphatase receptor type O. Nat Neurosci. 2006;9:761–769. doi: 10.1038/nn1697. [DOI] [PubMed] [Google Scholar]
- Siekmann AF, Affolter M, Belting HG. The tip cell concept 10 years after: new players tune in for a common theme. Exp Cell Res. 2013;319:1255–1263. doi: 10.1016/j.yexcr.2013.01.019. [DOI] [PubMed] [Google Scholar]
- Soderberg O, Leuchowius KJ, Gullberg M, Jarvius M, Weibrecht I, Larsson LG, Landegren U. Characterizing proteins and their interactions in cells and tissues using the in situ proximity ligation assay. Methods. 2008;45:227–232. doi: 10.1016/j.ymeth.2008.06.014. [DOI] [PubMed] [Google Scholar]
- Song Y, Eng M, Ghabrial AS. Focal defects in single-celled tubes mutant for Cerebral cavernous malformation 3, GCKIII, or NSF2. Dev Cell. 2013;25:507–519. doi: 10.1016/j.devcel.2013.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stepanek L, Stoker AW, Stoeckli E, Bixby JL. Receptor tyrosine phosphatases guide vertebrate motor axons during development. J Neurosci. 2005;25:3813–3823. doi: 10.1523/JNEUROSCI.4531-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Q, Bahri S, Schmid A, Chia W, Zinn K. Receptor tyrosine phosphatases regulate axon guidance across the midline of the Drosophila embryo. Development. 2000;127:801–812. doi: 10.1242/dev.127.4.801. [DOI] [PubMed] [Google Scholar]
- Sun Q, Schindelholz B, Knirr M, Schmid A, Zinn K. Complex genetic interactions among four receptor tyrosine phosphatases regulate axon guidance in Drosophila. Molecular and cellular neurosciences. 2001;17:274–291. doi: 10.1006/mcne.2000.0939. [DOI] [PubMed] [Google Scholar]
- Tarcic G, Boguslavsky SK, Wakim J, Kiuchi T, Liu A, Reinitz F, Nathanson D, Takahashi T, Mischel PS, Ng T, et al. An unbiased screen identifies DEP-1 tumor suppressor as a phosphatase controlling EGFR endocytosis. Curr Biol. 2009;19:1788–1798. doi: 10.1016/j.cub.2009.09.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tchetchelnitski V, van den Eijnden M, Schmidt F, Stoker AW. Developmental co-expression and functional redundancy of tyrosine phosphatases with neurotrophin receptors in developing sensory neurons. International journal of developmental neuroscience: the official journal of the International Society for Developmental Neuroscience. 2014;34:48–59. doi: 10.1016/j.ijdevneu.2014.01.005. [DOI] [PubMed] [Google Scholar]
- Thurston G, Wang Q, Baffert F, Rudge J, Papadopoulos N, Jean-Guillaume D, Wiegand S, Yancopoulos GD, McDonald DM. Angiopoietin 1 causes vessel enlargement, without angiogenic sprouting, during a critical developmental period. Development. 2005;132:3317–3326. doi: 10.1242/dev.01888. [DOI] [PubMed] [Google Scholar]
- Tian SS, Tsoulfas P, Zinn K. Three receptor-linked protein-tyrosine phosphatases are selectively expressed on central nervous system axons in the Drosophila embryo. Cell. 1991;67:675–685. doi: 10.1016/0092-8674(91)90063-5. [DOI] [PubMed] [Google Scholar]
- Tonks NK. Protein tyrosine phosphatases: from genes, to function, to disease. Nature reviews. 2006;7:833–846. doi: 10.1038/nrm2039. [DOI] [PubMed] [Google Scholar]
- Vestweber D, Wessel F, Nottebaum AF. Similarities and differences in the regulation of leukocyte extravasation and vascular permeability. Seminars in immunopathology. 2014;36:177–192. doi: 10.1007/s00281-014-0419-7. [DOI] [PubMed] [Google Scholar]
- Vikkula M, Boon LM, Carraway KL, 3rd, Calvert JT, Diamonti AJ, Goumnerov B, Pasyk KA, Marchuk DA, Warman ML, Cantley LC, et al. Vascular dysmorphogenesis caused by an activating mutation in the receptor tyrosine kinase TIE2. Cell. 1996;87:1181–1190. doi: 10.1016/s0092-8674(00)81814-0. [DOI] [PubMed] [Google Scholar]
- Wharram BL, Goyal M, Gillespie PJ, Wiggins JE, Kershaw DB, Holzman LB, Dysko RC, Saunders TL, Samuelson LC, Wiggins RC. Altered podocyte structure in GLEPP1 (Ptpro)-deficient mice associated with hypertension and low glomerular filtration rate. The Journal of clinical investigation. 2000;106:1281–1290. doi: 10.1172/JCI7236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winderlich M, Keller L, Cagna G, Broermann A, Kamenyeva O, Kiefer F, Deutsch U, Nottebaum AF, Vestweber D. VE-PTP controls blood vessel development by balancing Tie-2 activity. J Cell Biol. 2009;185:657–671. doi: 10.1083/jcb.200811159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang XH, Seow KT, Bahri SM, Oon SH, Chia W. Two Drosophila receptor-like tyrosine phosphatase genes are expressed in a subset of developing axons and pioneer neurons in the embryonic CNS. Cell. 1991;67:661–673. doi: 10.1016/0092-8674(91)90062-4. [DOI] [PubMed] [Google Scholar]
- Yu KR, Kim YJ, Jung SK, Ku B, Park H, Cho SY, Jung H, Chung SJ, Bae KH, Lee SC, et al. Structural basis for the dephosphorylating activity of PTPRQ towards phosphatidylinositide substrates. Acta crystallographica Section D, Biological crystallography. 2013;69:1522–1529. doi: 10.1107/S0907444913010457. [DOI] [PubMed] [Google Scholar]
- Zuo L, Iordanou E, Chandran RR, Jiang L. Novel mechanisms of tube-size regulation revealed by the Drosophila trachea. Cell and tissue research. 2013;354:343–354. doi: 10.1007/s00441-013-1673-z. [DOI] [PMC free article] [PubMed] [Google Scholar]