

Abstract
Finding robust biomarkers for Parkinson disease (PD) is currently hampered by inherent technical limitations associated with imaging or antibody-based protein assays. To circumvent the challenges, we adapted a staged pipeline, starting from our previous proteomic profiling followed by high-throughput targeted mass spectrometry (MS), to identify peptides in human cerebrospinal fluid (CSF) for PD diagnosis and disease severity correlation. In this multicenter study consisting of training and validation sets, a total of 178 subjects were randomly selected from a retrospective cohort, matching age and sex between PD patients, healthy controls, and neurological controls with Alzheimer disease (AD). From ∼14,000 unique peptides displaying differences between PD and healthy control in proteomic investigations, 126 peptides were selected based on relevance and observability in CSF using bioinformatic analysis and MS screening, and then quantified by highly accurate and sensitive selected reaction monitoring (SRM) in the CSF of 30 PD patients versus 30 healthy controls (training set), followed by diagnostic (receiver operating characteristics) and disease severity correlation analyses. The most promising candidates were further tested in an independent cohort of 40 PD patients, 38 AD patients, and 40 healthy controls (validation set). A panel of five peptides (derived from SPP1, LRP1, CSF1R, EPHA4, and TIMP1) was identified to provide an area under curve (AUC) of 0.873 (sensitivity = 76.7%, specificity = 80.0%) for PD versus healthy controls in the training set. The performance was essentially confirmed in the validation set (AUC = 0.853, sensitivity = 82.5%, specificity = 82.5%). Additionally, this panel could also differentiate the PD and AD groups (AUC = 0.990, sensitivity = 95.0%, specificity = 97.4%). Furthermore, a combination of two peptides belonging to proteins TIMP1 and APLP1 significantly correlated with disease severity as determined by the Unified Parkinson's Disease Rating Scale motor scores in both the training (r = 0.381, p = 0.038)j and the validation (r = 0.339, p = 0.032) sets. The novel panel of CSF peptides, if validated in independent cohorts, could be used to assist in clinical diagnosis of PD and has the potential to help monitoring or predicting disease progression.
Parkinson disease (PD)1, the second most common neurodegenerative disease after Alzheimer disease (AD), afflicts roughly 2% of persons over the age of 65 years (1, 2). Currently, PD diagnosis is mainly based on observation of the cardinal motor indicators of the disease, patient response to drug treatment, and medical history (3, 4). There is an appreciable misdiagnosis rate (4), particularly at early disease stages. Additionally, no objective measure of disease progression or treatment effects has been established. Thus, objective, reliable, and reproducible biomarkers are clearly needed to aid in the diagnosis of PD and tracking or predicting the disease progression.
The most sensitive tests developed to date are based on imaging modalities, which can detect functional and structural abnormalities even prior to the onset of motor dysfunction (5, 6). However, the usefulness of neuroimaging techniques is limited by high cost, limited accessibility, difficulty in reliable differentiation of PD from other atypical parkinsonian disorders and subjection to confounding factors such as medication and compensatory responses (4–7). Biochemical and molecular markers in cerebrospinal fluid (CSF) and other body fluids have also been actively investigated (5, 8–12). The most extensively studied candidate in CSF is probably α-synuclein, the major protein component of Lewy bodies and Lewy neurites, the pathological hallmarks of PD (2). The current consensus is that CSF α-synuclein concentrations are generally lower in patients with PD compared with controls (5, 8–10); the sensitivity and specificity, however, appear to be only moderate, and no correlation with PD severity or progression has been observed (8, 9). Notably, all these CSF protein markers are measured using antibody-based assays, which are often associated with relatively high variability, particularly when different detection techniques (different antibodies, sample preparation, calibrators, etc.) are used, leading to discrepant results across laboratories (5). It should also be stressed that this high variability in immunoassays is not unique to PD, because similar difficulty is encountered in AD and other related disorders (13, 14).
One strategy to avoid the inherent technical limitations associated with antibodies is to use alternative techniques in which unique peptides are selected and precisely quantified with mass spectrometry (MS) techniques, for example, accurate inclusion mass screening (AIMS) (15) and selected reaction monitoring (SRM) (16–18). To this end, in the last few years, we and others have utilized proteomic technologies to identify novel proteins and peptides associated with different disease states and stages (5, 6, 19–25). Using brain tissue or CSF, these unbiased proteomic profiling studies have revealed disease-related alterations in hundreds of peptides derived from many proteins (19–25). However, there are no quantitative assays for the majority of these candidate proteins/peptides, and development of such assays is limited by the lack of antibodies available for many of them. Thus, although a large library of potential peptide biomarkers has been developed, the vast majority never reach the stage of validation and clinical testing, hampered by the difficulty of de novo development of immunoassays, a process that is time consuming, prohibitively expensive to develop and very difficult to multiplex.
In this study, we aim to establish a PD biomarker identification and verification pipeline, with the goal of prioritizing candidates and swiftly developing reliable quantitative assays. We focused on identifying peptides by SRM and AIMS, because these targeted proteomic technologies have been proposed as the basis of a viable biomarker pipeline (16) and have become a powerful tool in biomarker discovery because of their high sensitivity, accuracy and specificity. SRM, in particular, has emerged as an alternative to immunoaffinity-based measurements of defined protein sets with excellent reproducibility across different laboratories and instrument platforms (17, 18). The staged pipeline in the current investigation (Fig. 1) includes: (1) data-dependent and bioinformatic prioritization of thousands of candidate biomarkers identified in our previous profiling studies, (2) de novo development of antibody-free multiplex SRM assays to reliably measure tens to hundreds of peptides simultaneously, and (3) multiplex biomarker verification studies allowing identification and validation of models or panels of candidates in independent sample sets, two of which were used in this study.
Fig. 1.
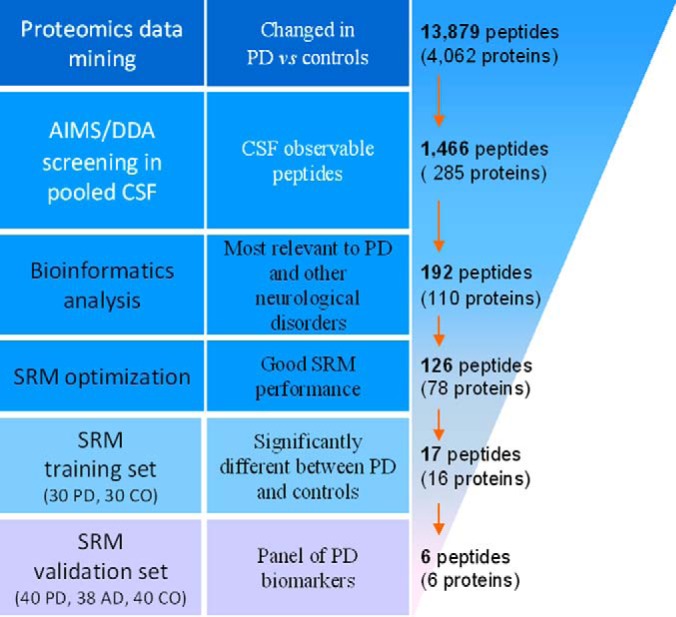
Overview of the workflow used for CSF peptide biomarker discovery and validation. AD, Alzheimer disease; AIMS, accurate inclusion mass screening; CO, healthy controls; DDA, data-dependent acquisition; PD, Parkinson disease; SRM, selected reaction monitoring.
EXPERIMENTAL PROCEDURES
Participants and CSF Sample Collection
A total of 178 subjects (70 PD, 38 AD, and 70 healthy controls) were randomly selected from a previously described well-characterized multicenter, retrospective cohort (8, 11), with age and sex matched between groups. Participating institutions include Baylor College of Medicine, Oregon Health and Science University, the University of California at San Diego, VA Puget Sound Health Care Systems at Seattle, and the University of Washington (UW). This study was approved by the Institutional Review Boards of all participating sites. All subjects provided informed consent and underwent evaluations consisting of medical history, physical and neurological examinations, laboratory tests, and neuropsychological assessments. The inclusion and exclusion criteria were previously described (8, 11) and a brief description is provided in the supplemental Methods. Thirty (30) patients with PD and 30 healthy controls were included as the training set in this study, and 40 patients with PD, 38 patients with AD, and 40 healthy controls were included as the validation set. Demographic information is listed in Table I for all subjects.
Table I. Demographic data of participating subjects.
| Training set |
Validation set |
||||
|---|---|---|---|---|---|
| Con | PD | Con | PD | AD | |
| Subject number | 30 | 30 | 40 | 40 | 38 |
| Agea (range) | 65.9 ± 13.6 (42–89) | 60.3 ± 10.8 (41–80) | 66.8 ± 8.3 (55–87) | 64.6 ± 9.7 (45–83) | 69.0 ± 8.4 (52–82) |
| Sex (male:female) | 20:10 | 22:8 | 25:15 | 25:15 | 20:18 |
| UPDRS motora (range) | 19.7 ± 11.8 (1–54) | 22.7 ± 12.5 (6–50) | – | ||
| MMSEa (range) | 29.3 ± 1.2 (26–30) | 28.2 ± 3.5 (11–30) | 17.7 ± 5.8 (5–27) | ||
| Disease duration, yeara (range) | 6.9 ± 6.2 (1–22) | 8.8 ± 7.7 (1–42) | 6.4 ± 2.7 (1–14) | ||
a Mean±S.D.
AD, Alzheimer disease; Con, healthy control; MMSE, Mini Mental State Examination; PD, Parkinson disease; UPDRS, Unified Parkinson's Disease Rating Scale.
All CSF samples were obtained by lumbar puncture in the morning as described (8, 11) (details can also be found in the supplemental Methods). Similar CSF collection protocols and quality control procedures were followed at all participating centers, in particular, use of polypropylene collection and storage tubes, rapid separation into single use aliquots, and freezing of CSF samples, to minimize potential site variations. Indeed, no apparent site effects were observed on the concentrations of several potential PD CSF biomarkers (e.g. total α-synuclein and one phosphorylated form) in these CSF samples in our previous studies (8, 11, 26).
Proteomics Data Mining for PD-related Proteins/Peptides
Proteomic data were gathered from our previous quantitative PD-related proteomic studies, including a general human CSF profiling (19), two general human midbrain profiling studies (20, 21), general human frontal cortex profiling studies of PD progression (22–24), glycoprotein profiling studies of human CSF and frontal cortex (25), a phosphoprotein profiling of human frontal cortex (unpublished data), glyco- and phospho-protein profiling studies in a MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) monkey model (putamen; unpublished data). A total of 15 data sets were integrated: two on the human CSF proteome, four on the human midbrain (substantia nigra) proteome, six on the human frontal cortex proteome, and the remaining three on the monkey putamen proteome. Candidate peptides and their corresponding proteins were selected based on the following criteria: (1) identified more than once; (2) had a confidence level of ≥95%; (3) displayed ≥20% disease-associated change in at least one data set (PD versus control, ≥1.20 or ≤ 0.83). Peptides meeting these criteria were combined; human homologs were selected for peptides/proteins derived from monkey. A total of 13,879 unique peptides (derived from 4062 proteins) were identified (see a complete list in supplemental Table S1).
CSF Sample Processing
Proteins from CSF samples were precipitated using 20% trichloroacetic acid and solubilized in 8 m urea in 50 mm ammonium bicarbonate (pH 8.0). The protein concentration of individual samples was determined using the Pierce bicinchoninic acid (BCA) Protein Assay Kit (Thermo Scientific, Rockford, IL). The proteins were then reduced and alkylated using dithiothreitol and iodoacetamide, respectively, followed by trypsin digestion as described previously (24, 27).
For assay development, reference CSF samples were pooled from >5 healthy control samples collected at a UW clinic. For CSF observable peptide selection, tryptic digests of 990 μl reference CSF were desalted using 1cc Sep-Pak® Vac C18 cartridges (Waters, Milford, MA). Strong cation-exchange (SCX) fractionation was then carried out using a PolySulfo-ethyl A (200 × 2.1 mm × 5-μm, 300 Å) column (PolyLC, Columbia, MD) on a Biologic Duo-Flow LC system (Bio-Rad, Hercules, CA). Ten SCX pools were made for each CSF sample and desalted using C18 MicroSpin columns (The Nest Group, Southborough, MA) according to manufacturer's protocol. More details can be found in the supplemental Methods.
For SRM analysis of individual samples, 50 μl of CSF from each subject were precipitated, digested, desalted with a C18 MicroSpin column, and resuspended in 40 μl of 0.1% formic acid/2% acetonitrile plus 10 μl of pooled “heavy” peptides (see below).
Selection of CSF-observable Peptides
The +2 charge state for each of the 13,879 candidate peptides was used to generate two inclusion lists for targeted analysis on a Q-Exactive mass spectrometer (Thermo Scientific) based on the number of precursors that could be monitored in one run and the predicted retention time of the desired peptides. Liquid chromatography (LC) was performed using a Waters NanoAcquity UPLC; peptides were separated online with 75 μm i.d. × 20 cm home-packed fused silica columns (ReproSil-Pur C18-AQ, 3 μm, Dr Maisch GmBH, Ammerbuch-Entringen, Germany) with a 120 min 2–80% acetonitrile/water gradient containing 0.1% formic acid (see more details in the supplemental Methods). Each SCX pool was run four times: two data-dependent acquisition (DDA) runs, and two coupled with the inclusion lists.
MS/MS spectral data were converted to the mzML format and searched using the Trans-Proteomic Pipeline (TPP) (ver. 4.6.2; Institute for Systems Biology, Seattle, WA) against a human International Protein Index (IPI) sequence database (version 3.87) to identify proteins in each pool. Search results were then exported to the Skyline Targeted Proteomics Environment (v2.1) (McCoss Lab, University of Washington, Seattle, WA) (28) to create spectral libraries and for MS1 filtering to identify precursor ions. Peak detection and integration were also manually inspected and the precursors with a dot-product ratio of ≤0.94 were excluded. If no peptide in the targeted list of a protein of interest was identified but different peptide(s) derived from the same protein were observed, the targeted peptide(s) were replaced. Several observed peptides derived from proteins known to be critical in PD (e.g. DJ-1(2)), though not in the initial targeted list, were added. A total of 1466 unique peptides (285 proteins) that could be reliably observed in CSF were identified (see supplemental Table S2).
Bioinformatics Analysis
Ingenuity Pathway Analysis (IPA; Qiagen, Redwood City, CA) was performed on the integrated quantitative proteomic data to identify proteins involved in pathways affected in neurological disorders. IPA (http://www.ingenuity.com/) is a commercial tool that is based on a proprietary database to facilitate the identification of biological themes in proteomics or gene expression data. The identified proteins, together with the top ranked most PD-relevant proteins obtained from genecards.org (search using “Parkinson disease” as keyword), served as a reference list to select peptides for further SRM assay development and targeting.
SRM Analysis
All SRM analysis was performed on a TSQ Vantage triple quadrupole mass spectrometer (Thermo Scientific) coupled to a nanoAcquity UPLC (Waters). Reversed-phase chromatography was performed on capillary columns (75 μm × 20 cm; Polymicro Technologies, Phoenix, AZ) packed with 100 Å Magic C18 (Michrom/Bruker, Auburn, CA). Four (4) μl of tryptic digest were injected into a column and separated using a binary gradient (see supplemental Methods for more details). Scheduled SRM were performed with dwell times of 20 ms and retention time windows of 4 min and 5 min for the training set and the validation set, respectively.
Quantification of Target Peptides by SRM
To determine the levels of endogenous target peptides in CSF of PD and control subjects, 192 peptide standards (Thermo Scientific) corresponding to natural counterparts (“light” peptides) were synthesized with heavy isotopic lysine (13C615N2) or arginine (13C615N4) at the C termini (“heavy” peptides), pooled, and spiked into the digested, desalted CSF samples upon resolubilization. The “heavy” peptides were estimated to be 60–80% pure based on manufacturer's specifications and our independent quality checking using a 4800 Plus MALDI TOF/TOF (AB SCIEX, Framingham, MA). Collision energies (CE) were determined using the default formula from Thermo (0.034 × precursor mass m/z + 3.3140) and in the validation set they were further optimized with 10 additional CE steps (−5 to +5 V from the default, with 1V increments; see details in the supplemental Methods). The SRM transitions of the heavy and their corresponding light peptides were optimized and validated. For each peptide, the best three transitions, including three “Light” and three “Heavy” transitions were selected. One-hundred and twenty-six unique, reproducibly-detected peptides that showed good SRM transition signals and dot-product ratios >0.94 were identified (see supplemental Table S3) and targeted for SRM quantification in the training set consisting of 30 PD and 30 control subjects. Seventeen peptides (Table II) with significant changes in PD compared with controls in the training set were further validated in another set consisting of 40 PD, 38 AD, and 40 healthy subjects.
Table II. Test results of 17 CSF peptides.
| Peptide | Proteina | Training set |
Validation set |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Con | PD | PD/Con | p (PD vs Con)b | Con | PD | PD/Con | AD | p (PD vs Con; PD vs AD)c | ||
| AALEGFLAALQADPPQAERd | APLP1, Amyloid-like protein 1 (P51693) | 4.751 ± 1.804 | 4.199 ± 1.366 | 0.88 | 0.028 | 3.762 ± 1.663 | 2.446 ± 1.450 | 0.65 | 4.697 ± 1.720 | 0.001; <0.001 |
| GAYPLSIEPIGVRd | CP, Ceruloplasmin (P00450) | 2.314 ± 1.026 | 2.031 ± 0.539 | 0.88 | 0.041 | 2.374 ± 0.877 | 1.444 ± 0.996 | 0.61 | 2.780 ± 1.374 | 0.001; <0.001 |
| VIPGPPALTLVPAELVRd | CSF1R, Macrophage colony-stimulating factor 1 receptor (P07333) | 2.737 ± 1.165 | 2.357 ± 0.730 | 0.86 | 0.014 | 1.558 ± 0.660 | 1.002 ± 0.484 | 0.64 | 1.738 ± 0.745 | <0.001; <0.001 |
| DCNSLPGVMGTCKd | EPHA4, Ephrin type-A receptor 4 (P54764) | −2.058 ± 0.666 | −2.165 ± 0.489 | 1.05 | 0.031 | −2.322 ± 0.556 | −2.998 ± 0.549 | 1.29 | −2.783 ± 0.479 | <0.001; 0.179 |
| SERPPIFEIRd | LRP1, Prolow-density lipoprotein receptor-related protein 1 (Q07954) | −2.104 ± 0.442 | −2.303 ± 0.149 | 1.10 | 0.006 | −1.045 ± 0.245 | −1.205 ± 0.218 | 1.15 | −1.113 ± 0.165 | 0.003; 0.143 |
| AIPVAQDLNAPSDWDSRd | SPP1, Osteopontin (P10451) | 4.622 ± 2.090 | 2.657 ± 1.704 | 0.58 | <0.001 | 4.066 ± 1.189 | 2.697 ± 1.100 | 0.66 | 5.266 ± 1.110 | <0.001; <0.001 |
| EPGLCTWQSLRd | TIMP1, Metalloproteinase inhibitor 1 (P01033) | 1.093 ± 0.819 | 0.940 ± 0.570 | 0.86 | 0.049 | 0.391 ± 0.441 | 0.001 ± 0.456 | 0.002 | 0.157 ± 0.381 | <0.001; 0.244 |
| GIISALLVPPETEEAKd | APOB, Apolipoprotein B-100 (P04114) | −0.713 ± 0.420 | −0.848 ± 0.147 | 1.19 | 0.003 | −1.317 ± 0.244 | −1.275 ± 0.214 | 0.97 | −0.980 ± 0.306 | 0.745; <0.001 |
| TVPGASDLFYWPRd | GPR37, Prosaposin receptor GPR37 (O15354) | −1.155 ± 0.203 | −1.205 ± 0.148 | 1.04 | 0.040 | −1.250 ± 0.181 | −1.235 ± 0.151 | 0.99 | −1.310 ± 0.110 | 0.899; 0.078 |
| FATTFYQHLADSKd | SERPINC1, ntithrombin-III (P01008) | −2.087 ± 0.543 | −2.287 ± 0.328 | 1.10 | 0.010 | −2.426 ± 0.430 | −2.405 ± 0.579 | 0.99 | −2.303 ± 0.374 | 0.977; 0.608 |
| TSDQIHFFFAKd | SERPINC1, Antithrombin-III (P01008) | 0.570 ± 0.574 | 0.930 ± 0.295 | 1.63 | 0.003 | 0.668 ± 0.631 | 0.161 ± 0.694 | 0.24 | 1.036 ± 0.773 | 0.004; <0.001 |
| WQEEMELYRe | APOA1, Apolipoprotein A-I (P02647) | 0.021 ± 0.035 | 0.012 ± 0.003 | 0.58 | 0.004 | 52.490 ± 31.117 | 40.211 ± 37.188 | 0.77 | 34.615 ± 35.336 | 0.256; 0.756 |
| QPSSAFAAFVKe | C3, Complement C3 (P01024) | 0.122 ± 0.393 | 0.035 ± 0.028 | 0.29 | 0.005 | 2.636 ± 2.326 | 1.619 ± 2.878 | 0.61 | 2.695 ± 1.838 | 0.143; 0.121 |
| DPDITSSPIRe | CDH8, Cadherin-8 (P55286) | 0.000513 ± 0.000227 | 0.000440 ± 0.000219 | 0.86 | 0.028 | 0.000405 ± 0.000225 | 0.000400 ± 0.000556 | 0.99 | 0.000206 ± 0.000125 | 0.998; 0.047 |
| GLVSWGNIPCGSKe | KLK6, Kallikrein-6 (Q92876) | 0.191 ± 0.673 | 0.083 ± 0.025 | 0.43 | 0.017 | 64.762 ± 27.508 | 46.068 ± 22.327 | 0.71 | 51.273 ± 22.475 | 0.002; 0.611 |
| ATAVVDGAFKe | PRDX2, Peroxiredoxin-2 (P32119) | 0.479 ± 0.355 | 1.095 ± 1.844 | 2.29 | 0.012 | 0.324 ± 0.192 | 0.728 ± 0.599 | 2.25 | 0.499 ± 0.289 | <0.001; 0.035 |
| TITLEVEPSDTIENVKe | RPS27A, Ubiquitin-40S ribosomal protein S27a (P62979) | 0.030 ± 0.056 | 0.068 ± 0.042 | 2.27 | <0.001 | 9.620 ± 5.812 | 5.067 ± 2.405 | 0.53 | 10.884 ± 6.830 | 0.001; <0.001 |
a Gene symbol, protein name (UniProt Accession Number).
b Mann–Whitney U test.
c One-way ANOVA with Tukey's post-hoc test.
d Peptide data shown are normalized (transformed) light/heavy ratios, except for
e those from six (6) peptides that failed to meet our criteria (see Experimental Procedures) for robust and reliable detection and quantification in the training or validation set; the raw light/heavy ratios are shown for these six peptides. AD, Alzheimer disease; Con, healthy control; PD, Parkinson disease.
All SRM mass spectral data were processed using the Skyline software (v2.1). Typical settings applied were 0.055 Th match tolerance m/z and default peak boundary assignment informed by Savitzky-Golay smoothing. All peak boundaries were manually inspected and reassigned as needed to ensure correct peak detection and accurate integration. Information including peak area and area ratio of light/heavy peptide pair were exported for further analysis. Following peak detection and integration, peptides were considered “detectable” for each subject if 1) the peptide transitions had a signal-to-noise ratio of ≥3, and 2) at least two light SRM transitions and two heavy SRM transitions were observed. Peptides detected in less than 50% of the subjects were excluded.
Luminex Assay
CSF α-synuclein protein concentrations were assessed using our previously established Luminex immunoassay (8, 11).
Statistical Analysis
All analyses were performed in SPSS 18.0 (IBM, Chicago, IL) or Prism 4.0 (GraphPad Software, La Jolla, CA). Peptide data were normalized by the Box-Cox transformation (29) (SERPINC1-FAT, and EPHA4: λ = 0.1; SPP1, and LRP1, λ = 0.3; SERPINC1-TSD, APOB, TIMP1, and CSF1R: λ = 0.4; GPR37, and APLP1, λ = 0.5; CP, λ = 0.6), which represents a family of power transformations that incorporates and extends the traditional options to help researchers easily find the optimal normalizing transformation for each variable. CSF α-synuclein protein data were log10 transformed. The Mann-Whitney U test was used to examine median differences between PD and healthy control groups in the training set. One-way analysis of variance (ANOVA) followed by the Tukey test was used to compare differences between groups in the validation set, without or with (through general linear model analysis) controlling for potential confounding factors such as age, sex, and CSF total protein concentrations. Receiver Operating Characteristic (ROC) curves for individual peptide analytes were generated to evaluate their sensitivities and specificities in distinguishing PD from healthy or diseased (AD) control subjects. Logistic regression was used to determine the best linear combination of peptide analytes for predicting disease status (versus healthy or diseased controls), followed by ROC analysis on the linear combination. The “optimum” cutoff value for a ROC curve was defined as the value associated with the maximal sum of sensitivity and specificity. Additionally, relationships between the analytes and age, sex, and the Unified Parkinson's Disease Rating Scale (UPDRS) motor scores were analyzed with bivariate correlation using Pearson's correlation coefficients. Partial correlations between CSF peptide levels and UPDRS scores were also conducted while controlling for potential confounding factors such as sex and age of subjects. Stepwise multiple linear regression analysis was used to screen for the best predictors (linear combination of peptide analytes) that correlates the disease severity (UPDRS). Values with p < 0.05 were regarded as significant. Increased type I errors because of multiple comparisons were minimized by using the training and validation approach, in which a limited set of outcomes determined in the training set are repeated in the validation set, and considered significant only if the a priori determined outcomes are confirmed in the validation set.
RESULTS
Identification of CSF Peptide Biomarker Candidates
For biomarker candidate selection, we first integrated an extensive compilation of 15 PD-related quantitative proteomic data sets generated from our previous studies (references (19–25) and unpublished data). All analyses included samples from patients with PD as compared with age- and sex- matched healthy or diseased controls or monkeys treated with MPTP. A total of 13,879 unique PD-related peptides (corresponding to 4062 proteins) were identified (see supplemental Table S1). There are no traditional quantitative assays (e.g. ELISAs) for the majority of the candidates, severely limiting our ability to perform follow-up verification studies.
To triage and verify a large number of peptide or protein candidates in a manner that would enable us to test as many candidates as possible while containing costs, we adapted a strategically staged pipeline (16) using targeted proteomic technologies (Fig. 1). Specifically, the technology with the lesser expense and higher capacity to triage large numbers of candidates (data-dependent LC-ESI-MS/MS analysis, AIMS) was used first to determine whether the peptides can be reliably monitored in CSF. Digested pooled reference CSF samples were fractionated using SCX chromatography to further facilitate our sensitivity for detecting low abundance proteins during this initial analysis. A total of 1466 unique peptides (derived from 285 proteins) that could be reliably observed in CSF were empirically selected (MS1 filtering; supplemental Table S2).
Quantitative SRM Assay Development
The next step involved high-throughput quantification technology (SRM) to establish sensitive, accurate assays for candidate marker measurements. However, reagent costs (e.g. synthetic “heavy” standard peptides) limited the practicality of developing assays for all confirmed CSF candidates, necessitating a further prioritization step. For this, we used a bioinformatics approach (Ingenuity Pathway Analysis, ingenuity.com; and the GeneCards database, genecards.org) to identify the proteins that are most relevant to PD and other neurological disorders from the target list. A list of 192 peptides, representing 110 proteins, was generated (supplemental Table S3) to advance SRM assay development.
We configured these peptide candidates into a multiplex SRM assay with “heavy” peptides used as internal standards. Based on the performance of the assays, such as quality of light and heavy SRM transition signals, and reproducibility of the target peptides (CVs<20%), the target list was further narrowed down to 126 unique peptides (see supplemental Table S3).
Peptide Marker Discovery in the Training Set
A total of 60 subjects (30 PD, 30 controls) were used in the training set to identify altered peptides/proteins in PD as compared with healthy controls. The relative peptide levels (light/heavy ratios) were measured by using the optimized 126-plex SRM assay. Seventeen (17) peptides corresponding to 16 proteins showed significant differences between PD and controls (p < 0.05, Mann-Whitney) in the training set (Table II; see the complete dataset in supplemental Table S4A and box plots in supplemental Fig. S1). Six of them were later determined to fail to meet our criteria (see Experimental Procedures) for robust and reliable detection and quantification in the training or validation set, and thus were excluded from ROC and disease severity correlation analyses. Except for CP (r = 0.256, p = 0.048, Pearson), none of these 17 peptides significantly correlated with age of subjects. Similarly, only EPHA4 significantly correlated with sex of subjects in this training set (p = 0.041).
To further evaluate the sensitivity and specificity in PD diagnosis for these 17 peptides, alone or in combination, ROC analysis was performed. The best-performing individual peptides were SPP1 (AUC: 0.791; sensitivity: 90.0%; and specificity: 56.7%) and LRP1 (AUC: 0.706; sensitivity: 70.0%; and specificity: 70.0%). A logistic regression analysis identified a combination of five peptides (derived from proteins SPP1, LRP1, CSF1R, EPHA4, and TIMP1) that improved the AUC to 0.873 (sensitivity 76.7%, specificity 80.0%; see Table III and Fig. 2A). For comparison, we also measured α-synuclein protein concentrations using our established Luminex immunoassay (8, 11). In this training set, CSF α-synuclein produced a moderate AUC of 0.714 (sensitivity 86.7%, specificity 56.7%; see supplemental Fig. S2A), in line with previous reports (8, 9, 11).
Table III. AUC values, sensitivities, specificities, and cutoff points of the best discriminating parameters for PD versus healthy or diseased controls.
| Parameter | Training set |
Validation set |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| AUC | p | cutoff | Sens% | Spec% | AUC | p | Sens% | Spec% | |
| SPP1 | |||||||||
| PD vs Con | 0.791 | 1E-4 | 4.362 | 90.0 | 56.7 | 0.821 | 8E-7 | 77.5 | 75.0 |
| PD vs AD | 0.948 | 1E-11 | 87.5 | 97.4 | |||||
| LRP1 | |||||||||
| PD vs Con | 0.706 | 0.006 | −2.218 | 70.0 | 70.0 | 0.698 | 0.002 | 80.0 | 57.5 |
| PD vs AD | 0.662 | 0.014 | 70.0 | 63.2 | |||||
| SPP1, LRP1, CSF1R, EPHA4, and TIMP1 | |||||||||
| PD vs Con | 0.873 | 7E-7 | 0.5394 | 76.7 | 80.0 | 0.853 | 5E-8 | 82.5 | 82.5 |
| PD vs AD | 0.990 | 1E-13 | 95.0 | 97.4 | |||||
| 11-peptide panela | |||||||||
| PD vs Con | 0.982 | 1E-10 | 0.6761 | 90.0 | 96.7 | 0.932 | 3E-11 | 85.0 | 92.5 |
| PD vs AD | 1.000 | 3E-14 | 100.0 | 100.0 | |||||
a APLP1, APOB, CP, CSF1R, EPHA4, GPR37, LRP1, SERPINC1-FAT, SERPINC1-TSD, SPP1, and TIMP1. Note that the 11-peptide data needs to be interpreted with caution due to potential overfitting. AD, Alzheimer disease; AUC: area under curve; Con, healthy control; PD, Parkinson disease; Sens: sensitivity; Spec: specificity.
Fig. 2.

Panel of six CSF peptides for PD diagnosis and disease severity correlation. A panel of five CSF peptides (derived from SPP1, LRP1, CSF1R, EPHA4, and TIMP1) provided an area under curve (AUC) of 0.873 (sensitivity = 76.7%, specificity = 80.0%) in the training set (30 PD and 30 healthy controls) A, and 0.853 (sensitivity = 82.5%, specificity = 82.5%) in the validation set (40 PD and 40 healthy controls) B. See also Table III. A linear model of two CSF peptides (TIMP1 and APLP1) significantly correlated with disease severity as determined by the Unified Parkinson's Disease Rating Scale (UPDRS) motor scores in patients with PD in both the training (r = 0.381, p = 0.038) C, and the validation (r = 0.339, p = 0.032) D, sets. UPDRS_predict = −0.35*TIMP1–0.1*APLP1.
The correlation of levels of a single peptide or a combination of peptides with PD severity (as determined by UPDRS motor scores) was also examined in the training set, with or without controlling for potential confounding factors (e.g. sex of subjects). Although no peptides could be considered individually correlated with the UPDRS scores, a 2-peptide model (derived from TIMP1 and APLP1) was identified using linear regression analysis to significantly correlate with disease severity (r = 0.381, p = 0.038, Pearson; r = 0.381, p = 0.041, controlling for sex; Fig. 2C).
Promising Marker Confirmation in the Validation Set
To further evaluate the potential of the peptides identified in the training set as PD biomarkers and determine their specificity for PD, the 17 peptides were re-screened in the validation set consisting of 40 PD, 38 AD, and 40 healthy controls. The group differences between PD and healthy controls for eight of the 11 (six of the 17 peptides were excluded because of inconsistent assay performance) promising peptides were confirmed in this set (ANOVA, Table II; see also supplemental Table S4B and supplemental Fig. S1). Five of these eight peptides (APLP1, CP, CSF1R, SERPINC1-TSD, and SPP1) and APOB also displayed significant differences between PD and AD groups. The group differences remained largely unchanged after controlling for potential confounding factors including age, sex, and CSF total protein concentrations (general linear model analysis). None of these 17 peptides significantly correlated with age of subjects in the validation, or combined (training + validation) sets. The sex effects on EPHA4 were also not confirmed (p = 0.422); however, SERPINC1-FAT (p = 0.025), APOB (p = 0.040), LRP1 (p = 0.005) significantly correlated with sex in the validation set but not in the training set. Though these data indicate that overall the concentrations of the candidate peptide markers may not be substantially affected by sex of subjects, when the two sets were considered a one combined set, SERPINC1-FAT (p = 0.014), APOB (p = 0.045), EPHA4 (p = 0.037), and TIMP1 (p = 0.049) showed a significant correlation with sex.
In this validation set, the 5-peptide model (SPP1, LRP1, CSF1R, EPHA4, and TIMP1) produced an AUC of 0.854, and both sensitivity and specificity equal to 82.5% in differentiating patients with PD and healthy controls in the ROC analysis (Table III and Fig. 2B), confirming its diagnostic performance observed in the training set. Furthermore, this peptide panel could also differentiate the PD and AD groups well (AUC = 0.990, sensitivity = 95.0%, specificity = 97.4%; see Table III and supplemental Fig. S3). In contrast, the CSF α-synuclein protein measured with the immunoassay did not perform well in differentiating PD and healthy controls in this validation set (AUC = 0.570, sensitivity = 70.0%, specificity = 37.5%; see supplemental Fig. S2B).
The 2-peptide model (TIMP1 and APLP1) identified in the training set was also validated in its significant correlation with UPDRS motor scores (r = 0.339, p = 0.032, Pearson; r = 0.336, p = 0.036, controlling for sex; Fig. 2D). Interestingly, TIMP1 alone correlated with the disease severity in this validation set (r = −0.368, p = 0.019, Pearson; r = −0.376, p = 0.018, controlling for sex).
DISCUSSION
Biomarker discovery in PD and other neurodegenerative disorders has been quite challenging and ideal biomarkers are still an unmet clinical need. In this study, we employed a targeted approach, and established a staged pipeline to facilitate biomarker discovery and validation. We report a panel consisting of five peptides/proteins (SPP1, LRP1, CSF1R, EPHA4, and TIMP1) with fair robustness in regard to specificity and sensitivity in differentiating PD from healthy and diseased (AD) controls. Additionally, we report a 2-peptide/protein model (TIMP1 and APLP1) that significantly correlates with disease severity as measured by UPDRS motor scores.
The first major achievement of this study is to establish a pipeline that distills proteomic profiling results to a reasonable number of peptides that can be followed practically. This process is quite important, because the “-omics” based discovery experiments are fraught with false discoveries resulting from biological variability and the large number of hypotheses being tested in small numbers of samples (16), and thus validation studies using independent methods must be performed to verify the clinical utility of a candidate. The validation process often requires development and optimization of protein-specific assays, frequently depending on immunologic reagents that may be unavailable, poorly characterized, or insufficiently specific. Even if good antibody pairs are available, high multiplexing (>tens of simultaneous assays) of traditional immunoassays is still challenging, largely because of antibody cross-reactions and matrix effects (30). Therefore, many candidates are not carried through the validation stage because the traditional processes are simply too labor-intensive and/or require impractical sample volumes to be a reasonable tool for clinical investigation. In the current investigation, with a staged pipeline we were able to not only follow up and test a far larger number of candidates than would have been possible using conventional methods, but also successfully identify a novel panel of candidate peptide markers for PD diagnosis and severity correlation, thus marking a substantial improvement over the current state of PD biomarker evaluation.
The proteins in both panels/models we identified in this study are differentially regulated in diseased states versus healthy and/or diseased (AD) controls, and have been mechanistically implicated in various neurodegenerative processes. Of particular interest, osteopontin (SPP1; included in the 5-peptide model for diagnosis) is a glycosylated phosphoprotein expressed in neuronal cell bodies that seems to act like a double-edged sword in neurodegenerative disorders–it can be toxic to neurons and cause cell death in some instances, but is neuroprotective in others (31). Iczkiewicz and colleagues demonstrated that osteopontin expression was decreased in substantia nigra of MPTP-treated primates and in PD (32). In contrast, Maetzler et al. found it was enriched in the neuromelanin containing zone of post mortem human PD brain tissue and present in Lewy bodies, but was absent from control brain tissue (33). The same authors also showed CSF and serum osteopontin concentrations were higher in PD patients than controls (33). A more recent study reported increased CSF and plasma concentrations in mild cognitive impairment and AD patients (34). Interestingly, the peptide (AIPVAQDLNAPSDWDSR, unmodified) used in our study displayed lower levels in PD; this could be related to the changes in post-translational modifications (e.g. the increase of the extent of modifications exceeds the increase of the total protein levels, resulting in a decrease of the unmodified peptide) and should be further investigated.
Three of the other candidates (LRP1, CSFR1, and EPHA4) are receptors implicated in signaling pathways associated with neurodegeneration, in particular with inflammatory response. The low density lipoprotein receptor-related protein 1 (LRP1) is a cell surface receptor expressed in different brain cell types including endothelial cells and neurons; it can regulate amyloid-β and other ligands trafficking into the cell and clearance from the brain (35, 36), and maintain brain lipid homeostasis and associated synaptic and neuronal integrity (37). Increased expression of LRP1 was observed in PD brain (38) and functional soluble LRP1 was also detected in human brain tissue and CSF (39). Colony-stimulating factor receptor 1 (CSFR1) is the receptor for colony stimulating factor 1 and interleukin-34 (IL-34), which are key regulators of the monocyte/macrophage lineage (40). In a mouse model lacking CSFR1, neurons are more susceptible to cell death and neurodegeneration after excitotoxic injury, suggesting involvement of CSFR1 signaling in their survival (40). In addition, CSFR1 is required for the development of microglia, brain development, and maintenance of normal brain structure (41). Ephrin type-A receptor 4 (EPHA4) is a member of the A subclass of Ephrin receptor tyrosine kinases and interacts with both A-type and B-type ephrins (42). EPHA4 signaling through its ephrin ligands has been implicated in guiding axons during neural development, synapse formation, and regulation of long-term synaptic plasticity and memory (43). Recently, it has been found to be decreased in Huntington disease CSF (44) and also to be able to modulate the vulnerability of motor neurons to axonal degeneration in amyotrophic lateral sclerosis in animal models and in humans (42). However, no direct evidence yet has been discovered on the implications of EPHA4 signaling pathway in PD pathogenesis.
Tissue inhibitor of metalloproteinases-1 (TIMP1) is included in both the panel for PD diagnosis and the model for disease severity correlation. This protein is abundantly expressed and functions primarily to inhibit a large class of matrix metalloproteinases (MMPs) (45). TIMP1 has been shown to be neuroprotective in various systems, which indirectly supports the damaging role of MMP-3 (46, 47). MMP-3 could cleave α-synuclein to remove the negative charges in the C-terminal portion making it more hydrophobic and prone to aggregation (46). Likewise, DJ-1, another protein implicated with PD (2), could also be cleaved by MMP-3 (46). TIMP1 has been detected in human CSF (48, 49) and our finding of lower TIMP1 concentrations in CSF of PD patients is in line with the association of the aberrant and excessive activity of MMPs with the neurodegenerative processes in the PD brain (50).
Amyloid-like protein 1 (APLP1), another protein/peptide in the model for PD severity correlation, is also implicated in neurodegeneration. APLP1 is localized in the cerebral cortex postsynaptic density of rats and humans and its expression was reported to be increased in synaptic development, suggesting a role in synaptogenesis, or synaptic maturation (51). Increased APLP1 expression and neurodegeneration were found in the frontal cortex of manganese-exposed nonhuman primates (52), which could be a compensatory event. Soluble forms of APLP1 (53) and more interestingly, three APLP1-derived amyloid-beta-like peptides (54), were previously observed in human CSF.
Together, these proteins represent an important step forward in PD biomarker discovery. To date, the best-performing PD biomarkers (e.g. α-synuclein (8, 9, 11)) are those identified based on their known roles in disease processes, but none have provided sufficient and validated sensitivity/specificity to be of clinical use for diagnosis or progression. Although further testing (e.g. against different control groups) and larger-scale, independent validations are needed, the panel established here performed better than CSF α-synuclein and likely performs at least similarly to other best-established markers, suggesting that the strategy developed here, that is, selection of targets from proteomic studies based on biological plausibility performance in MS-based assays, could be an efficient way to expand the search for feasible biomarkers. Additionally, the candidate CSF peptides/proteins were selected from those more relatively specific to PD (as compared with AD) in our proteomic studies, and thus the performance of the CSF peptide/protein panel on differential diagnosis between PD and AD further confirmed the efficiency of our strategy. Furthermore, two unique peptides related to neurodegeneration appeared to be related to PD severity, which can be potentially used in PD progression or drug treatment assessment, if the observations can be validated in longitudinal cohosts, especially those prospective ones.
One concern when using combinations of multiple markers in a relatively small number of subjects is overfitting, which means that if one investigates enough classification rules then, by chance, one of them is likely to perform well (55). To avoid overfitting, one approach is to use a training sample set to formulate the classification rules and a test/validation sample set to create the definitive ROC curve (55), as we did in the current study. Additionally, we chose to reduce the peptide numbers in the model as much as possible and present the results from a 5-peptide model instead of using more peptides. In fact, if all 11 “good” peptides were included in the model, a nearly perfect diagnosis would be achieved in the training set (AUC: 0.982; sensitivity: 90.0%; and specificity: 96.7%), with an AUC of 0.932 (sensitivity: 85.0%; specificity: 92.5%) in the validation set (Table III). Although similar results have been considered acceptable, including those reported recently in Nature Medicine (56), we took a more conservative approach because, without pathology confirmation, the typical correct rate of clinical PD diagnosis is no more than 90% even at major medical centers (4). Therefore, a biomarker model perfectly matching the imperfect clinical diagnosis is unlikely to correctly reflect the underlying disease status and should be used with great caution.
The panel of candidate PD biomarkers identified in the current study does not contain peptides from several known potential PD CSF biomarkers (e.g. α-synuclein and DJ-1 (8, 9, 11)). This is not unexpected, because: (1) some proteins may not be readily identified in an unbiased proteomic profiling because of their low abundance or modifications (α-synuclein, which is usually highly modified (2, 57), was not even included in the initial candidate list compiled from our previous proteomic profiling studies); and (2) the goal of the current investigation was to reveal not only novel but also robust markers, and therefore, the experimental conditions were geared toward markers that are readily and reproducibly quantified in a multiplex assay in CSF. Consequently, some of the peptides (e.g. those from DJ-1), even though they were included in the list, failed to pass our rigorous pipeline selection criteria. That said, a peptide from complement factor C3 (C3), a potential marker identified in CSF in our previous study (58), did appear as one of the 17 peptides identified in the training set. In further investigations, we will try to optimize the conditions to include not only these robust biomarker candidates but also known potential markers such as α-synuclein and DJ-1 for an “ideal” panel of markers to be used clinically. Additionally, although the identified panel of CSF peptides/proteins could differentiate PD and AD well, to fully confirm disease specificity of these potential markers, a larger cohort including related disease controls (e.g. those with multiple system atrophy or progressive supranuclear palsy) will be needed to further test their usage in PD differential diagnosis.
In conclusion, through a staged pipeline and high-throughput SRM quantification, a panel of candidate peptide biomarkers has been identified in CSF to provide good diagnostic sensitivity/specificity for PD and correlation with disease severity. These results, if validated in independent studies, particularly those with samples collected prospectively, could be used to assist in clinical diagnosis of PD and have the potential to help monitoring or predicting disease progression.
Supplementary Material
Acknowledgments
We thank Drs. HyeJin Hwang and Jianpeng Zhang for their contributions to the early proteomic profiling and data analysis, Mr. Michael J Hipp for his assistance in sample preparation and data analysis, and Mr. Allion A Salvador for his kind help in peptide picking. We also deeply appreciate the patients and participants for their generous participation and donation of samples.
Footnotes
Author contributions: JZ conceived and supervised the project; MS performed some of the initial proteomic profiling, assisted in experimental design and execution as well as data interpretation during the pipeline establishment, performed statistical analyses with PA, and drafted the manuscript with JZ, RD, and PA; JM helped to establish the pipeline in the initial phases and performed most of the peptide picking, CSF observable peptide selection, and SRM assay optimization experiments; RD assisted in SRM assays and data interpretation; PA performed most of the SRM data analysis and assisted in statistical analyses; YZ performed most of the SRM data acquirements and assisted in data analysis; CP was involved in the initial proteomic profiling and assisted in SRM assays; XL performed some of the initial proteomic profiling and assisted in peptide picking; TKB performed the IPA analysis; TS assisted in data interpretation and copyediting; CPZ, ERP, SCH, JFQ, and DRG were responsible for patient characterization and sample collection; all authors critically reviewed the manuscript.
* This study was supported by generous grants from the National Institutes of Health (NIH) (U01 NS082137, P42 ES004696-5897, P30 ES007033-6364, R01 AG033398, R01 ES016873, R01 ES019277, R01 NS057567, and P50 NS062684-6221 to JZ, R01 NS065070 to CPZ, and P50 AG005131 to DRG), and partially by a pilot study award from the NIH-sponsored ADRC at the UW (P50 AG003156-30) and a National Institute of Neurological Disorders and Stroke/NIH award R21 NS085425 to MS. It was also supported in part by the University of Washington's Proteomics Resource (UWPR95794), the National Institute Of Environmental Health Sciences of the NIH under Award Number P30 ES007033, and the National Natural Science Foundation of China (NSFC) projects (31200105 and 31470238). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH and other sponsors.
 This article contains supplemental Methods, Figs. S1 to S3, and Tables S1 to S4.
This article contains supplemental Methods, Figs. S1 to S3, and Tables S1 to S4.
1 The abbreviations used are:
- PD
- Parkinson disease
- AD
- Alzheimer disease
- AIMS
- accurate inclusion mass screening
- APLP1
- amyloid-like protein 1
- AUC
- area under curve
- CSF
- cerebrospinal fluid
- CSF1R
- Macrophage colony-stimulating factor 1 receptor
- Con
- healthy control
- CP
- ceruloplasmin
- EPHA4
- ephrin type-A receptor 4
- LRP1
- prolow-density lipoprotein receptor-related protein 1
- MMSE
- Mini Mental State Examination
- MS
- mass spectrometry
- ROC
- receiver operating characteristic
- SCX
- strong cation-exchange
- SPP1
- osteopontin
- SRM
- selected reaction monitoring
- TIMP1
- metalloproteinase inhibitor 1
- UPDRS
- Unified Parkinson's Disease Rating Scale.
REFERENCES
- 1. Lang A. E., Lozano A. M. (1998) Parkinson's disease. First of two parts. N. Engl. J. Med. 339, 1044–1053 [DOI] [PubMed] [Google Scholar]
- 2. Thomas B., Beal M. F. (2007) Parkinson's disease. Hum. Mol. Genet. 16 Spec No 2, R183–R194 [DOI] [PubMed] [Google Scholar]
- 3. Jankovic J. (2008) Parkinson's disease: clinical features and diagnosis. J. Neurol. Neurosurg. Psychiatry 79, 368–376 [DOI] [PubMed] [Google Scholar]
- 4. Tolosa E., Wenning G., Poewe W. (2006) The diagnosis of Parkinson's disease. Lancet Neurol. 5, 75–86 [DOI] [PubMed] [Google Scholar]
- 5. Schapira A. H. (2013) Recent developments in biomarkers in Parkinson disease. Curr. Opin. Neurol. 26, 395–400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. O'Keeffe G. C., Michell A. W., Barker R. A. (2009) Biomarkers in Huntington's and Parkinson's disease. Ann. N.Y. Acad. Sci. 1180, 97–110 [DOI] [PubMed] [Google Scholar]
- 7. Booij J., Berendse H. W. (2011) Monitoring therapeutic effects in Parkinson's disease by serial imaging of the nigrostriatal dopaminergic pathway. J. Neurol. Sci. 310, 40–43 [DOI] [PubMed] [Google Scholar]
- 8. Hong Z., Shi M., Chung K. A., Quinn J. F., Peskind E. R., Galasko D., Jankovic J., Zabetian C. P., Leverenz J. B., Baird G., Montine T. J., Hancock A. M., Hwang H., Pan C., Bradner J., Kang U. J., Jensen P. H., Zhang J. (2010) DJ-1 and alpha-synuclein in human cerebrospinal fluid as biomarkers of Parkinson's disease. Brain 133, 713–726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mollenhauer B., Locascio J. J., Schulz-Schaeffer W., Sixel-Döring F., Trenkwalder C., Schlossmacher M. G. (2011) alpha-Synuclein and tau concentrations in cerebrospinal fluid of patients presenting with parkinsonism: a cohort study. Lancet Neurol. 10, 230–240 [DOI] [PubMed] [Google Scholar]
- 10. Kang J. H., Irwin D. J., Chen-Plotkin A. S., Siderowf A., Caspell C., Coffey C. S., Waligorska T., Taylor P., Pan S., Frasier M., Marek K., Kieburtz K., Jennings D., Simuni T., Tanner C. M., Singleton A., Toga A. W., Chowdhury S., Mollenhauer B., Trojanowski J. Q., Shaw L. M. (2013) Association of cerebrospinal fluid beta-amyloid 1–42, t-tau, p-tau181, and alpha-synuclein levels with clinical features of drug-naive patients with early Parkinson disease. JAMA Neurol. 70, 1277–1287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shi M., Bradner J., Hancock A. M., Chung K. A., Quinn J. F., Peskind E. R., Galasko D., Jankovic J., Zabetian C. P., Kim H. M., Leverenz J. B., Montine T. J., Ginghina C., Kang U. J., Cain K. C., Wang Y., Aasly J., Goldstein D., Zhang J. (2011) Cerebrospinal fluid biomarkers for Parkinson disease diagnosis and progression. Ann. Neurol. 69, 570–580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hall S., Ohrfelt A., Constantinescu R., Andreasson U., Surova Y., Bostrom F., Nilsson C., Hakan W., Decraemer H., Nagga K., Minthon L., Londos E., Vanmechelen E., Holmberg B., Zetterberg H., Blennow K., Hansson O. (2012) Accuracy of a panel of five cerebrospinal fluid biomarkers in the differential diagnosis of patients with dementia and/or parkinsonian disorders. Arch. Neurol. 69, 1445–1452 [DOI] [PubMed] [Google Scholar]
- 13. Mattsson N., Blennow K., Zetterberg H. (2010) Inter-laboratory variation in cerebrospinal fluid biomarkers for Alzheimer's disease: united we stand, divided we fall. Clin. Chem. Lab. Med. 48, 603–607 [DOI] [PubMed] [Google Scholar]
- 14. Mattsson N., Andreasson U., Persson S., Carrillo M. C., Collins S., Chalbot S., Cutler N., Dufour-Rainfray D., Fagan A. M., Heegaard N. H., Robin Hsiung G. Y., Hyman B., Iqbal K., Lachno D. R., Lleo A., Lewczuk P., Molinuevo J. L., Parchi P., Regeniter A., Rissman R., Rosenmann H., Sancesario G., Schroder J., Shaw L. M., Teunissen C. E., Trojanowski J. Q., Vanderstichele H., Vandijck M., Verbeek M. M., Zetterberg H., Blennow K., Kaser S. A. (2013) CSF biomarker variability in the Alzheimer's Association quality control program. Alzheimers Dement. 9, 251–261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jaffe J. D., Keshishian H., Chang B., Addona T. A., Gillette M. A., Carr S. A. (2008) Accurate inclusion mass screening: a bridge from unbiased discovery to targeted assay development for biomarker verification. Mol. Cell. Proteomics 7, 1952–1962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Whiteaker J. R., Lin C., Kennedy J., Hou L., Trute M., Sokal I., Yan P., Schoenherr R. M., Zhao L., Voytovich U. J., Kelly-Spratt K. S., Krasnoselsky A., Gafken P. R., Hogan J. M., Jones L. A., Wang P., Amon L., Chodosh L. A., Nelson P. S., McIntosh M. W., Kemp C. J., Paulovich A. G. (2011) A targeted proteomics-based pipeline for verification of biomarkers in plasma. Nat. Biotechnol. 29, 625–634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Addona T. A., Abbatiello S. E., Schilling B., Skates S. J., Mani D. R., Bunk D. M., Spiegelman C. H., Zimmerman L. J., Ham A. J., Keshishian H., Hall S. C., Allen S., Blackman R. K., Borchers C. H., Buck C., Cardasis H. L., Cusack M. P., Dodder N. G., Gibson B. W., Held J. M., Hiltke T., Jackson A., Johansen E. B., Kinsinger C. R., Li J., Mesri M., Neubert T. A., Niles R. K., Pulsipher T. C., Ransohoff D., Rodriguez H., Rudnick P. A., Smith D., Tabb D. L., Tegeler T. J., Variyath A. M., Vega-Montoto L. J., Wahlander A., Waldemarson S., Wang M., Whiteaker J. R., Zhao L., Anderson N. L., Fisher S. J., Liebler D. C., Paulovich A. G., Regnier F. E., Tempst P., Carr S. A. (2009) Multi-site assessment of the precision and reproducibility of multiple reaction monitoring-based measurements of proteins in plasma. Nat. Biotechnol. 27, 633–641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Selevsek N., Matondo M., Sanchez Carbayo M., Aebersold R., Domon B. (2011) Systematic quantification of peptides/proteins in urine using selected reaction monitoring. Proteomics 11, 1135–1147 [DOI] [PubMed] [Google Scholar]
- 19. Abdi F., Quinn J. F., Jankovic J., McIntosh M., Leverenz J. B., Peskind E., Nixon R., Nutt J., Chung K., Zabetian C., Samii A., Lin M., Hattan S., Pan C., Wang Y., Jin J., Zhu D., Li G. J., Liu Y., Waichunas D., Montine T. J., Zhang J. (2006) Detection of biomarkers with a multiplex quantitative proteomic platform in cerebrospinal fluid of patients with neurodegenerative disorders. J. Alzheimers Dis. 9, 293–348 [DOI] [PubMed] [Google Scholar]
- 20. Jin J., Hulette C., Wang Y., Zhang T., Pan C., Wadhwa R., Zhang J. (2006) Proteomic identification of a stress protein, mortalin/mthsp70/GRP75: relevance to Parkinson disease. Mol. Cell. Proteomics 5, 1193–1204 [DOI] [PubMed] [Google Scholar]
- 21. Kitsou E., Pan S., Zhang J., Shi M., Zabeti A., Dickson D. W., Albin R., Gearing M., Kashima D. T., Wang Y., Beyer R. P., Zhou Y., Pan C., Caudle W. M., Zhang J. (2008) Identification of proteins in human substantia nigra. Proteomics Clin. Appl. 2, 776–782 [DOI] [PubMed] [Google Scholar]
- 22. Pan S., Shi M., Jin J., Albin R. L., Lieberman A., Gearing M., Lin B., Pan C., Yan X., Kashima D. T., Zhang J. (2007) Proteomics identification of proteins in human cortex using multidimensional separations and MALDI tandem mass spectrometer. Mol. Cell. Proteomics 6, 1818–1823 [DOI] [PubMed] [Google Scholar]
- 23. Shi M., Jin J., Wang Y., Beyer R. P., Kitsou E., Albin R. L., Gearing M., Pan C., Zhang J. (2008) Mortalin: a protein associated with progression of Parkinson disease? J. Neuropathol. Exp. Neurol. 67, 117–124 [DOI] [PubMed] [Google Scholar]
- 24. Shi M., Bradner J., Bammler T. K., Eaton D. L., Zhang J., Ye Z., Wilson A. M., Montine T. J., Pan C., Zhang J. (2009) Identification of glutathione S-transferase pi as a protein involved in Parkinson disease progression. Am. J. Pathol. 175, 54–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hwang H., Zhang J., Chung K. A., Leverenz J. B., Zabetian C. P., Peskind E. R., Jankovic J., Su Z., Hancock A. M., Pan C., Montine T. J., Pan S., Nutt J., Albin R., Gearing M., Beyer R. P., Shi M., Zhang J. (2010) Glycoproteomics in neurodegenerative diseases. Mass Spectrom. Rev. 29, 79–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wang Y., Shi M., Chung K. A., Zabetian C. P., Leverenz J. B., Berg D., Srulijes K., Trojanowski J. Q., Lee V. M., Siderowf A. D., Hurtig H., Litvan I., Schiess M. C., Peskind E. R., Masuda M., Hasegawa M., Lin X., Pan C., Galasko D., Goldstein D. S., Jensen P. H., Yang H., Cain K. C., Zhang J. (2012) Phosphorylated alpha-synuclein in Parkinson's disease. Sci. Transl. Med. 4, 121ra120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lin X., Cook T. J., Zabetian C. P., Leverenz J. B., Peskind E. R., Hu S. C., Cain K. C., Pan C., Edgar J. S., Goodlett D. R., Racette B. A., Checkoway H., Montine T. J., Shi M., Zhang J. (2012) DJ-1 isoforms in whole blood as potential biomarkers of Parkinson disease. Sci. Rep. 2, 954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. MacLean B., Tomazela D. M., Shulman N., Chambers M., Finney G. L., Frewen B., Kern R., Tabb D. L., Liebler D. C., MacCoss M. J. (2010) Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 26, 966–968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Box G. E. P., Cox D. R. (1964) An analysis of transformations. J. R. Stat. Soc. B. 26, 211–252 [Google Scholar]
- 30. Ellington A. A., Kullo I. J., Bailey K. R., Klee G. G. (2010) Antibody-based protein multiplex platforms: technical and operational challenges. Clin. Chem. 56, 186–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Carecchio M., Comi C. (2011) The role of osteopontin in neurodegenerative diseases. J. Alzheimers Dis. 25, 179–185 [DOI] [PubMed] [Google Scholar]
- 32. Iczkiewicz J., Jackson M. J., Smith L. A., Rose S., Jenner P. (2006) Osteopontin expression in substantia nigra in MPTP-treated primates and in Parkinson's disease. Brain Res. 1118, 239–250 [DOI] [PubMed] [Google Scholar]
- 33. Maetzler W., Berg D., Schalamberidze N., Melms A., Schott K., Mueller J. C., Liaw L., Gasser T., Nitsch C. (2007) Osteopontin is elevated in Parkinson's disease and its absence leads to reduced neurodegeneration in the MPTP model. Neurobiol. Dis. 25, 473–482 [DOI] [PubMed] [Google Scholar]
- 34. Sun Y., Yin X. S., Guo H., Han R. K., He R. D., Chi L. J. (2013) Elevated osteopontin levels in mild cognitive impairment and Alzheimer's disease. Mediators Inflamm. 2013:615745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zlokovic B. V., Deane R., Sagare A. P., Bell R. D., Winkler E. A. (2010) Low-density lipoprotein receptor-related protein-1: a serial clearance homeostatic mechanism controlling Alzheimer's amyloid beta-peptide elimination from the brain. J. Neurochem. 115, 1077–1089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kanekiyo T., Cirrito J. R., Liu C. C., Shinohara M., Li J., Schuler D. R., Shinohara M., Holtzman D. M., Bu G. (2013) Neuronal clearance of amyloid-beta by endocytic receptor LRP1. J. Neurosci. 33, 19276–19283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Liu Q., Trotter J., Zhang J., Peters M. M., Cheng H., Bao J., Han X., Weeber E. J., Bu G. (2010) Neuronal LRP1 knockout in adult mice leads to impaired brain lipid metabolism and progressive, age-dependent synapse loss, and neurodegeneration. J. Neurosci. 30, 17068–17078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wilhelmus M. M., Bol J. G., Van Haastert E. S., Rozemuller A. J., Bu G., Drukarch B., Hoozemans J. J. (2011) Apolipoprotein E and LRP1 increase early in Parkinson's disease pathogenesis. Am. J. Pathol. 179, 2152–2156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Liu Q., Zhang J., Tran H., Verbeek M. M., Reiss K., Estus S., Bu G. (2009) LRP1 shedding in human brain: roles of ADAM10 and ADAM17. Mol. Neurodegener. 4, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Luo J., Elwood F., Britschgi M., Villeda S., Zhang H., Ding Z., Zhu L., Alabsi H., Getachew R., Narasimhan R., Wabl R., Fainberg N., James M. L., Wong G., Relton J., Gambhir S. S., Pollard J. W., Wyss-Coray T. (2013) Colony-stimulating factor 1 receptor (CSF1R) signaling in injured neurons facilitates protection and survival. J. Exp. Med. 210, 157–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Erblich B., Zhu L., Etgen A. M., Dobrenis K., Pollard J. W. (2011) Absence of colony stimulation factor-1 receptor results in loss of microglia, disrupted brain development, and olfactory deficits. PLoS One 6, e26317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Van Hoecke A., Schoonaert L., Lemmens R., Timmers M., Staats K. A., Laird A. S., Peeters E., Philips T., Goris A., Dubois B., Andersen P. M., Al-Chalabi A., Thijs V., Turnley A. M., van Vught P. W., Veldink J. H., Hardiman O., Van Den Bosch L., Gonzalez-Perez P., Van Damme P., Brown R. H., Jr., van den Berg L. H., Robberecht W. (2012) EPHA4 is a disease modifier of amyotrophic lateral sclerosis in animal models and in humans. Nat. Med. 18, 1418–1422 [DOI] [PubMed] [Google Scholar]
- 43. Klein R. (2009) Bidirectional modulation of synaptic functions by Eph/ephrin signaling. Nat. Neurosci. 12, 15–20 [DOI] [PubMed] [Google Scholar]
- 44. Fang Q., Strand A., Law W., Faca V. M., Fitzgibbon M. P., Hamel N., Houle B., Liu X., May D. H., Poschmann G., Roy L., Stuhler K., Ying W., Zhang J., Zheng Z., Bergeron J. J., Hanash S., He F., Leavitt B. R., Meyer H. E., Qian X., McIntosh M. W. (2009) Brain-specific proteins decline in the cerebrospinal fluid of humans with Huntington disease. Mol. Cell. Proteomics 8, 451–466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Porter J. F., Sharma S., Wilson D. L., Kappil M. A., Hart R. P., Denhardt D. T. (2005) Tissue inhibitor of metalloproteinases-1 stimulates gene expression in MDA-MB-435 human breast cancer cells by means of its ability to inhibit metalloproteinases. Breast Cancer Res. Treat. 94, 185–193 [DOI] [PubMed] [Google Scholar]
- 46. Kim E. M., Hwang O. (2011) Role of matrix metalloproteinase-3 in neurodegeneration. J. Neurochem. 116, 22–32 [DOI] [PubMed] [Google Scholar]
- 47. Ashutosh, Chao C., Borgmann K., Brew K., Ghorpade A. (2012) Tissue inhibitor of metalloproteinases-1 protects human neurons from staurosporine and HIV-1-induced apoptosis: mechanisms and relevance to HIV-1-associated dementia. Cell Death Dis. 3, e332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bjerke M., Zetterberg H., Edman A., Blennow K., Wallin A., Andreasson U. (2011) Cerebrospinal fluid matrix metalloproteinases and tissue inhibitor of metalloproteinases in combination with subcortical and cortical biomarkers in vascular dementia and Alzheimer's disease. J. Alzheimers Dis. 27, 665–676 [DOI] [PubMed] [Google Scholar]
- 49. Lorenzl S., Albers D. S., LeWitt P. A., Chirichigno J. W., Hilgenberg S. L., Cudkowicz M. E., Beal M. F. (2003) Tissue inhibitors of matrix metalloproteinases are elevated in cerebrospinal fluid of neurodegenerative diseases. J. Neurol. Sci. 207, 71–76 [DOI] [PubMed] [Google Scholar]
- 50. Kim Y. S., Joh T. H. (2012) Matrix Metalloproteinases, New Insights into the Understanding of Neurodegenerative Disorders. Biomol. Ther. 20, 133–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kim T. W., Wu K., Xu J. L., McAuliffe G., Tanzi R. E., Wasco W., Black I. B. (1995) Selective localization of amyloid precursor-like protein 1 in the cerebral cortex postsynaptic density. Brain Res. Mol. Brain Res. 32, 36–44 [DOI] [PubMed] [Google Scholar]
- 52. Guilarte T. R., Burton N. C., Verina T., Prabhu V. V., Becker K. G., Syversen T., Schneider J. S. (2008) Increased APLP1 expression and neurodegeneration in the frontal cortex of manganese-exposed non-human primates. J. Neurochem. 105, 1948–1959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Paliga K., Peraus G., Kreger S., Durrwang U., Hesse L., Multhaup G., Masters C. L., Beyreuther K., Weidemann A. (1997) Human amyloid precursor-like protein 1–cDNA cloning, ectopic expression in COS-7 cells, and identification of soluble forms in the cerebrospinal fluid. Eur. J. Biochem. 250, 354–363 [DOI] [PubMed] [Google Scholar]
- 54. Yanagida K., Okochi M., Tagami S., Nakayama T., Kodama T. S., Nishitomi K., Jiang J., Mori K., Tatsumi S., Arai T., Ikeuchi T., Kasuga K., Tokuda T., Kondo M., Ikeda M., Deguchi K., Kazui H., Tanaka T., Morihara T., Hashimoto R., Kudo T., Steiner H., Haass C., Tsuchiya K., Akiyama H., Kuwano R., Takeda M. (2009) The 28-amino acid form of an APLP1-derived Abeta-like peptide is a surrogate marker for Abeta42 production in the central nervous system. EMBO Mol. Med. 1, 223–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Baker S. G., Kramer B. S., Srivastava S. (2002) Markers for early detection of cancer: statistical guidelines for nested case-control studies. BMC Med. Res. Methodol. 2, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Mapstone M., Cheema A. K., Fiandaca M. S., Zhong X., Mhyre T. R., Macarthur L. H., Hall W. J., Fisher S. G., Peterson D. R., Haley J. M., Nazar M. D., Rich S. A., Berlau D. J., Peltz C. B., Tan M. T., Kawas C. H., Federoff H. J. (2014) Plasma phospholipids identify antecedent memory impairment in older adults. Nat. Med., in press (doi: 10.1038/nm.3466) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Beyer K. (2006) Alpha-synuclein structure, posttranslational modification and alternative splicing as aggregation enhancers. Acta Neuropathol. 112, 237–251 [DOI] [PubMed] [Google Scholar]
- 58. Wang Y., Hancock A. M., Bradner J., Chung K. A., Quinn J. F., Peskind E. R., Galasko D., Jankovic J., Zabetian C. P., Kim H. M., Leverenz J. B., Montine T. J., Ginghina C., Edwards K. L., Snapinn K. W., Goldstein D. S., Shi M., Zhang J. (2011) Complement 3 and factor H in human cerebrospinal fluid of Parkinson's disease, multiple system atrophy, and Alzheimer's disease. Am. J. Pathol. 178, 1509–1516 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.