

Abstract
Highly activated/expanded natural killer (NK) cells can be generated via stimulation with the HLA-deficient cell line K562 genetically modified to express 41BB-ligand and membrane-bound interleukin (IL)15. We tested the safety, persistence and activity of expanded NK cells generated from myeloma patients (auto-NK) or haplo-identical family donors (allo-NK) in heavily pretreated patients with high-risk relapsing myeloma. The preparative regimen comprised bortezomib only or bortezomib and immunosuppression with cyclophosphamide, dexamethasone and fludarabine. NK cells were shipped overnight either cryopreserved or fresh. In 8 patients, up to 1×108 NK cells/kg were infused on day 0 and followed by daily administrations of IL2. Significant in vivo expansion was observed only in the 5 patients receiving fresh products, peaking at or near day 7, with the highest NK cell counts in 2 subjects who received cells produced in a high concentration of IL2 (500 units/mL). Seven days after infusion, donor NK cells comprised > 90% of circulating leukocytes in fresh allo-NK cell recipients, and cytolytic activity against allogeneic myeloma targets was retained in vitro. Among the 7 evaluable patients, there were no serious adverse events that could be related to NK cell infusion. One patient had a partial response and in another the tempo of disease progression decreased; neither patient required further therapy for 6 months. In the 5 remaining patients, disease progression was not affected by NK cell infusion. In conclusion, infusion of large numbers of expanded NK cells was feasible and safe; infusing fresh cells was critical to their expansion in vivo.
Keywords: multiple myeloma, natural killer cells, immunotherapy
Introduction
Multiple myeloma (MM) is a malignancy of antibody producing plasma cells that proliferate in the bone marrow (BM) leading to anemia, lytic bone lesions, and high levels of circulating monoclonal immunoglobulin that can cause organ impairment.1 The integration of novel drugs into autologous-peripheral blood stem cell transplantation (auto-PBSCT) regimens has significantly improved outcomes. Unfortunately, despite this progress, the outcome for patients with gene expression profiling (GEP)-defined high-risk myeloma remains inferior, with the majority succumbing to chemotherapy-resistant disease.2,3 Thus novel treatment options are urgently needed for this patient population.
Natural killer (NK) cells are CD3−CD56+ lymphocytes that can directly kill abnormal or infected cells, in addition to secreting cytokines which enhance both innate and adaptive mechanisms of immunity.4 Target cell recognition by NK cells is regulated by a complex array of activating and inhibitory interactions. NKG2D, DNAX-accessory molecule-1 (DNAM-1), and natural cytotoxicity receptors all appear to play a central role in NK cell recognition of myeloma,5,6 while interactions between killer immunoglobulin-like receptors (KIR) and self-major histocompatibility complex class I antigens induce strong inhibitory signals that predominate.5
Several techniques have been developed to expand NK cells in vitro that allow for a higher therapeutic cell dose while boosting activity and in vivo proliferative potential.7–11 Among these methods, the stimulation of peripheral blood mononuclear cells (PBMC) with the human leukocyte antigen (HLA)-class I deficient cell line K562, genetically modified to express membrane-bound interleukin (IL)15 and 41BB-ligand (K562-mb15-41BBL), leads to vigorous proliferation of NK cells, requires a relatively short co-culture period of 7–9 days to produce clinically relevant NK cell numbers, and does not induce significant expansion of T cells.8,12,13 We recently reported that expanded NK cells can be prepared in this way from both healthy donors and myeloma patients.6 The expanded NK cells expressed high levels of cell surface molecules critical for activation and adhesion, including NKG2D, natural cytotoxicity receptors, DNAM-1, and intracellular adhesion molecule (ICAM)-1. Furthermore, they killed both allogeneic and autologous primary myeloma cell targets in vitro and inhibited the growth of the human myeloma cell line OPM2 in a NOD-SCID-hu IL2-Rγ chainnull murine model.6 Finally, others have demonstrated in a xenogeneic model that human NK cells expanded with K562-mb15-41BBL exhibit superior homing to marrow compared to IL2 activated NK cells.14
We and others have shown that NK cell activity can also be enhanced by bortezomib due to down regulation of cell surface HLA15 and increased expression of tumor necrosis factor-related apoptosis inducing ligand (TRAIL) receptors on myeloma.16 We therefore aimed to evaluate the safety, persistence, and anti-myeloma activity of expanded NK cell products given after bortezomib with or without lymphodepletion in patients with GEP defined high-risk relapsed MM.
Materials and methods
Patients and donors
Informed consent was obtained for patients and haplo-identical family donors in accordance with the Declaration of Helsinki. This study was approved by the UAMS Institutional Review Board and conducted under BB-IND 14560. Patients had GEP-defined high-risk MM relapsing after auto-PBSCT, proteasome inhibitors, immunomodulatory agents, and additional salvage maneuvers. Patients who lacked expression of any of the three major KIR-Ls (HLA-C group I, II, and/or –Bw4), and who had a haplo-identical family donor were considered for allo-NK cell therapy. Patients who expressed all three KIR-Ls or who did not have a suitable family donor received auto-NK cells. Standard European Group for Blood and Marrow Transplantation criteria were used to evaluate response.17 Common terminology criteria for adverse events (CTCAE) version 3.0 was used to define and grade adverse events.
Study design
PBMC were collected at UAMS by a steady state apheresis, cryopreserved at 5×107 total nucleated cells/mL, and shipped to the NHLBI-Production Assistance for Cell Therapy (PACT) site at the Center for Cell and Gene Therapy (CAGT) at Baylor College of Medicine for NK cell production under good manufacturing practice (GMP) conditions as previously described.13 NK cell products were shipped overnight to UAMS either cryopreserved in liquid nitrogen dry shippers (patients 1–3) or fresh (patients 4–8). Fresh NK cell products (patients 4–8) were formulated at 1×107/mL in 5% albumin (Baxter Healthcare Corporation). Quality control testing was performed as previously described13 with the following exceptions. For fresh products sterility culture results were not available until after infusion, and mycoplasma and cytotoxicity testing was performed on in-process cells.18 Products for the last two subjects were produced in a higher IL2 concentration of 500 units (U)/mL whereas all other products were generated in 10 U/mL IL2 (Novartis, Emeryville, CA). A portion of the apheresis product and the final expanded NK cell product were retained for laboratory studies.
The preparative regimen is depicted in Figure 1. Patients were premedicated with acetaminophen and diphenhydramine and a target dose of 2×107 – 1×108 CD3−56+ cells/kg ideal body weight was infused intravenously. The allowable T cell dose for allo-products was ≤ 5×105/kg CD3+56− cells. Following infusion and each day afterwards 3×106 U IL2 was given subcutaneously for a total of 13 doses. The timing of planned correlative studies for peripheral blood samples were as follows: pre-treatment (within 35 days of NK infusion), pre-infusion on day 0, and on days 1, 3, 7, 14, 30, 90 and 180 after NK cell infusion. Additional peripheral blood and bone marrow samples were collected from patients who consented to an additional research sample collection protocol.
Figure 1. Protocol schema.
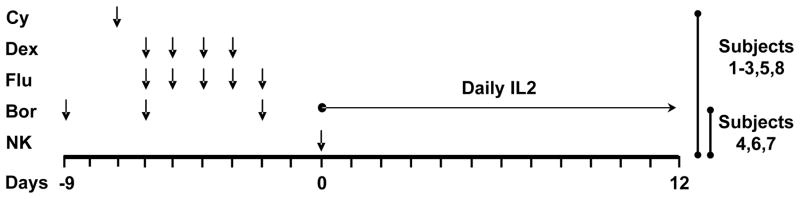
Bortezomib (Bor) was given (1.0mg/m2, day −9, −6, −2) to enhance NK cell therapy by down-regulating expression of HLA Class I,15 and up regulating TRAIL receptor on the myeloma cell surface. 16,46,47 Subjects 1, 2, 3, 5 and 8 also received immunosuppression with cyclophosphamide (Cy) 60 mg/kg IV on day −7, dexamethasone (Dex) 40 mg PO days −6 to −3, and fludarabine (Flu) 25 mg/m2 IV days −6 to −2 to reduce T regs and to prevent early rejection of infused NK cells. One administration of 2×107–1×108/kg of NK cells was given on day 0 and followed by 13 doses of daily IL2 (3×106 U, SC) to support the persistence and activity of NK cells in vivo.
Antibodies
See Table, Supplemental Digital Content 1, which lists all antibodies used in this study.
Flow cytometry
The absolute number of NK and T cells was determined in fresh PB and BM samples using antibodies to CD3, CD45, CD16 and CD56 and Trucount™ bead-containing tubes according to the manufacturer’s instructions (BD Biosciences, San Jose, CA). The percent of donor NK cells that were potentially alloreactive to recipient myeloma was determined using antibodies to CD3, CD56, NKG2A, KIR2DL1, KIR2DL2/3, and KIR3DL1 and were defined as CD3−56+ cells that were both positive for the KIR recognizing the ligand(s) missing in the recipient and also negative for NKG2A and KIRs recognizing ligands present in the recipient. Licensed NK were defined as alloreactive NK derived from a donor expressing the KIR-L missing in the recipient. CD3+/4+/25+/Foxp3+ T regulatory (T reg) cells were enumerated using the Human Regulatory T cell Staining Kit (eBiosciences, San Diego, CA). Samples were run on a FACSCalibur cytometer (BD Biosciences) and data was acquired using CellQuest software (BD Biosciences). Histogram overlays were created using FCS Express software (De Novo, Los Angeles, CA).
Chimerism studies
Chimerism was determined in DNA isolated from PBMC by quantifying short tandem repeats unique to the donor and recipient via polymerase chain reaction amplification and an automated nucleotide sequencer (UCSF, San Francisco, CA).
51Cr release assays
Standard 4 hour 51Cr release assays were performed and percent specific lysis calculated as previously described.6 Unmodified K562 cells were included as a positive control target and the myeloma cell line OPM2 was included when primary myeloma cells were unavailable (American Type Culture Collection, Manassas, VA). Effector cells were freshly cultured (not cryopreserved) expanded NK cells or NK cells enriched from PB samples or BM using microbeads coated with anti-CD56 antibody (Miltenyi Biotec). Research grade expanded NK cells were produced as previously described.6 CD138-positive plasma cells were purified from BM aspirates via positive selection with anti-CD138 coated microbeads (Miltenyi Biotec). After selection, purity of >90% was confirmed by flow cytometry. For blocking studies, isotype control antibody or anti-KIR antibody was added to effector cells. Control antibody, elotuzumab, or anti-HLA Class I antibody was added to 51Cr-labeled targets. Antibodies were left in the wells for the duration of the assay, at a final concentration of 10μg/mL.
Immunohistochemistry (IHC)
Immunostaining of acetic acid-zinc-formalin fixed and paraffin embedded BM biopsy specimens was performed with anti-CD57 antibody on 4μm thick sections to identify NK cells. All reactions were performed using an automated immunostainer (DakoCytomation, Carpenteria, CA) in conjunction with the Envision™-HRP detection system (Dako Cytomation) using diaminobenzidine as the chromogen. Both positive (normal tonsil) and negative (isotype-matched) controls were included. Nuclei were counterstained with hematoxylin. Sections were scanned using an Aperio ScanScope CS Digital image system and the images were processed by using Spectrum (Aperio Technologies, Vista, CA, USA).
Transforming growth factor β (TGFβ) assays
PBMCs and irradiated K562-mb15-41BBL cells were co-cultured in 24 well plates at a ratio of 2:1 in RPMI + 10% autologous serum + 30 U/mL IL2 and recombinant human TGFβ (5ng/mL, R&D Systems, Minneapolis, MN) or vehicle. Cultures were fed with half fresh media on day 3 or 4 and harvested on day 5. The fold change in NK cell number from baseline was determined by dividing the number of NK cells at harvest by the number of NK cells plated.
Western blot
NK cells were purified with an “untouched NK” isolation kit comprising a custom cocktail of antibodies to CD3, CD4, CD14, CD19, CD20, CD36, CD66b and glyA (Stem Cell Technologies, Vancouver, Canada). Purified NK cells (5 × 106) were frozen as dry pellets, thawed, lysed in RIPA buffer (Thermo Scientific) supplemented with Halt Protease and Phosphatase Inhibitor Cocktail (Thermo Scientific) and then cleared by centrifugation at 14,000g for 20 minutes at 4°C. Proteins were quantified using the BCA Protein Assay Kit (Thermo Scientific). Protein (20 μg) was heated at 95°C for 5 minutes, run on a 10% SDS-PAGE gel, and transferred to nitrocellulose membranes. Blots were blocked with 5% nonfat dry milk in tris-buffered saline-tween-20 (TBST, Thermo Scientific) and incubated with primary antibodies overnight at 4°C. Blots were then washed in TBST followed by incubation with species appropriate horse radish peroxidase (HRP)-conjugated secondary antibodies for 1 hour at room temperature. Dilutions of antibodies were 1:1000 for phospho-SMAD2/3 and 1:2000 for SMAD2/3, β-actin, and anti-rabbit IgG-HRP. To confirm equal loading of the protein on the gel, membranes were stripped and incubated with β-actin overnight at 4°C followed by incubation with HRP-conjugated secondary antibody for 1 hour at room temperature. Protein bands were visualized with SuperSignal West Femto chemiluminescent substrate (Thermo Scientific) and exposed to x-ray films.
ELISA
ELISAs for IL5, IL10, IL15, TGFβ, and IFNγ were performed for the indicated samples according to the manufacturer’s instructions (R&D Systems).
Statistical Analysis
A two-tailed paired Student’s t test was used to determine P values.
Results
Product characteristics: fresh products were superior to cryopreserved
Product Characteristics are listed in Table 1. In total, 13 clinical grade GMP products were generated for validation (N=2) and infusion (N=11). Eight subjects received their intended NK cell products. In one case, two production runs were required to generate the minimum dose, and after the second production run, the cell number infused was based on the allowable T cell dose (≤5×105/kg). One product was not infused because the subject had developed disease progression and infectious complications prior to the commencement of therapy, while another product derived from a patient with 21% CD138+ myeloma cells in the apheresis collection was not infused due to low viability (66.7%) and poor potency (26.0%). From 0.9–1.5×108 starting NK cells, 1.8–24 × 109 (median: 6.3×109) NK cells were produced. The fold NK-cell expansion was significantly lower for MM patients (N=6, median: 29 fold, range: 12–70 fold) than for healthy donors (HD) (N=7, median: 101 fold, range: 31–160 fold; p<0.03). At harvest, the median CD3−CD56+ NK cell purity was 78% (range: 52–90%). CD3 depletion increased NK cell purity of HD products to 93% (range: 86–99%) and resulted in a median CD3+CD56− T cell content of 0.1% (range: 0.04–1.02%). Overall, median viability was 93% (range: 67–100%), and potency (defined as lysis of K562 cells at a 20:1 E:T ratio) was 65% (range: 26–95%).
Table 1.
Product characteristics
| GMP Product | Subject | Allo/ Auto | NK Yield (×109) | Fold NK Expansion | Purity %CD3−56+ | Viability %7AAD- | Potency % Lysis | Recovery (%) |
|---|---|---|---|---|---|---|---|---|
| 1 | V | Allo | 10.0 | 111 | 93 | 91 | 92 | 61 |
| 2 | V | Allo | 9.1 | 101 | 93 | 97 | 79 | 100 |
| 3 | 1 | Auto | 6.3 | 70 | 88 | 83 | 65 | 65 |
| 4 | 2 | Allo | 3.9 | 31 | 86 | 96 | 74 | 100 |
| 5 | 2 | Allo | 4.6 | 31 | 89 | 89 | 74 | 100 |
| 6 | NI | Allo | 24.0 | 160 | 94 | 97 | 65 | 100 |
| 7 | 3 | Auto | 3.3 | 22 | 84 | 92 | 39 | 65 |
|
| ||||||||
| 8 | 4 | Auto | 1.9 | 13 | 68 | 98 | 58 | 87 |
| 9 | 5 | Allo | 13.3 | 89 | 95 | 93 | 77 | 151 |
| 10 | NI | Auto | 1.8 | 12 | 88 | 67 | 26 | 112 |
| 11 | 6 | Auto | 5.4 | 36 | 78 | 89 | 63 | 89 |
| 12 | 7 | Auto | 9.4 | 63 | 90 | 100 | 37 | 126 |
| 13 | 8 | Allo | 10.3 | 121 | 99 | 100 | 95 | 151 |
Cryopreserved products are above the dashed line, fresh products are below the dashed line. HD-derived NK were CD3 depleted. For products 1–7 NK yield, fold NK expansion, purity, viability and potency are from the time of harvest, prior to cryopreservation and recovery is 100 × (the number of NK cells after thaw ÷ the number of NK cells cryopreserved). For products 8–13 NK yield, fold NK expansion, purity, viability and potency are from the time of harvest, prior to shipping and recovery is 100 × (the number of NK cells received ÷ the number of NK cells shipped). Two productions runs were required to generate an adequate dose for treated subject #2. Potency was assessed as % lysis of K562 at 20:1 effector to target ratio. V, validation run; NI, not infused; Allo, allogeneic/healthy donor-derived NK cell product recipients; Auto, autologous/myeloma patient-derived product recipients;.
For cryopreserved products, median viability immediately after thawing was 94% (range: 75–99%), however thawed expanded NK failed to lyse K562 cells unless incubated overnight in IL2.13 Further, recovery of cryopreserved NK was extremely poor after overnight incubation (median 16%, range: 10–21%). We therefore validated shipment of fresh NK cell products. The median recovery for fresh clinical products after shipping was excellent (119%, range: 87–151%) as 4/6 products expanded further during shipping. We validated that expression of the key activating molecules NKG2D, NKp30, and NKp44 and activity against K562 were retained for up to 48 hours after formulation (see Figure, Supplemental Digital Content 2). Initially, NK cells were produced in low concentrations of IL2 to reduce potential T cell proliferation; however, we validated that increasing the IL2 concentration to 500 U/mL enhanced expression of NKp30 and cytolytic activity without significantly affecting the T cell content (see Figure, Supplemental Digital Content 3 A–C), and therefore the final two products were generated in high dose IL2. We also confirmed that the GMP grade NK cells produced in the higher concentration of IL2 and shipped in 5% albumin (no IL2) had similar lytic activity against K562 and OPM2 myeloma as small scale research grade NK cells (Research NK) not subjected to the stress of shipping (see Figure, Supplemental Digital Content 3D).
Clinical outcome and toxicity
Eight subjects with high-risk relapsed MM were treated on protocol (Table 2). All had abnormal metaphase cytogenetics and were high-risk in 70 (N=7) and/or 80 GEP models (N=8).2,19 Patients had received multiple lines of prior therapy including one (N=2), two (N=3) or three (N=3) prior auto-PBSCTs. The median platelet count at the time of enrollment was 59 × 103/μL blood (range: 22–220 × 103/μL) consistent with pre-existing marrow compromise due to extensive prior therapy. A comprehensive list of all toxicities are provided in Supplemental Digital Content 4, while Supplemental Digital Content 5 depicts the subject profile in terms of: chemotherapy/preparative regimen, platelet count at enrollment, duration of pancytopenia experienced, and whether stem cell rescue was required. The first and third treated patients (both autologous) experienced pancytopenia, though only the first subject required stem cell rescue. To enhance safety, subsequent autologous product recipients were prepared with Bor without Cy/Flu (patients 4, 6 and 7). One patient (subject 1) who had 3 prior auto-PBSCTs had a partial response after preparation with Bor/Flu/Cy and prior to NK infusion. The time to progression was 100 days but it was not feasible to determine whether the NK cells contributed to maintaining the partial response. In a second patient (subject 7) the tempo of the disease slowed and time to next treatment (TTNT) was 6 months. All others had progressive disease as indicated in Table 2. Subject 2 succumbed to MM despite further off protocol therapy. Two patients died on protocol; subject 5 died of pneumonia and septic shock 11 days after infusion and subject 8 died of persistent viral infection and PD (day 56). Graft versus host disease was not observed in any patients receiving allo-products. Six patients received all intended IL2 doses, but flu-like symptoms were observed. IL2 was stopped in patients 5 (on day 5) and 8 (on day 9) who developed infectious complications.
Table 2.
Patient characteristics and outcome
| Subject | Allo/ Auto | Age | Chemo | NK Manufacture | Dose ×107 NK/kg | Day 7 NK/μL (% donor) | Fold NK Day 7/Pre | Response | TTNT |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Auto | 73 | Bor/Cy/Flu | Frozen | 3.2 | NE | NE | PR (0) | 185 |
| 2 | Allo | 48 | Bor/Cy/Flu | Frozen | 3.4 | 9 (0%) | 0 | PD (20) Death (98) |
76 |
| 3 | Auto | 50 | Bor/Cy/Flu | Frozen | 2.2 | 82 | 1 | PD (20) | NE, LF |
|
| |||||||||
| 4 | Auto | 61 | Bor | Fresh Low IL2 |
6 | 512 | 37 | PD (23) | 79 |
| 5 | Allo | 64 | Bor/Cy/Flu | Fresh Low IL2 |
10 | 547 (92%) | 9 | NE Death (11) |
NA |
| 6 | Auto | 64 | Bor | Fresh Low IL2 |
8 | 438 | 5 | PD (12) | 48 |
| 7 | Auto | 68 | Bor | Fresh High IL2 |
10 | 5199 | 40 | PD (34) | 187 |
| 8 | Allo | 62 | Bor/Cy/Flu | Fresh High IL2 |
10 | 1855 (94%) | 21 | PD (32) Death (56) |
NA |
Subjects who received cryopreserved products are above the dashed line, those who received fresh products are below the dashed line. Autologous product recipients (Auto) below the dashed line were prepared with Bor without Cy/Flu. Absolute NK cell counts were obtained from PB samples. The absolute NK count was not evaluable for patient 1 due to low cell count, however the % NK cells and WBC count on day 7 was not higher than the baseline value. Fold NK change was calculated as the absolute number of NK cells in the blood on day 7 ÷ the number of NK cells pre-chemo. Allo, Allogeneic product recipients; Auto, Autologous product recipients; Bor, bortezomib; Cy, cyclophosphamide; Flu, fludarabine; PR, partial response (days since NK infusion); PD, progressing disease; TTNT, time to next treatment in days; NE, not evaluable; NA, not applicable; LF, lost to follow-up.
NK cells from fresh products expanded further in vivo and trafficked into the BM
PB NK cell counts remained low in the 3 patients who received cryopreserved NK cell products whereas robust in vivo expansion was observed in all 5 patients who received fresh products (Table 2). The median fold increase in NK from pre-treatment values for fresh products was 21 fold (range: 5–40 fold). NK cell counts peaked at day 7 in allo-NK recipients (Figures 2A, 2D), and chimerism studies confirmed that the circulating cells were of donor origin (Figure 2B, E). In auto-NK recipients NK cell counts peaked between days 7 and 9 (3A, 3C, 3E). In all 5 subjects who received fresh NK cells, the majority of circulating lymphocytes on day 7 were NK cells (57%, 92%, 64%, 79% and 98%). The highest number of circulating NK cells was observed in subjects 7 and 8 who received NK cells manufactured in high dose IL2 (500 U/mL). In particular, subject 7 had the most dramatic increase in NK cell counts, and circulating NK cell levels remained elevated through day 180 (539 NK/μL). Importantly, NK cells were also detected in the BM of 4/4 evaluable patients on day 5 (absolute number of NK cells/μL BM: 92, 453, 5198, 983) and IHC stains for CD57 confirmed the presence of NK cells in the BM of subject 7 (Figure 3G).
Figure 2. Allo-NK cells proliferated further after infusion and circulating NK retained lytic activity against myeloma cells.

A. The absolute number of NK cells per μL PB increased significantly post-infusion in subject 5. B. Chimerism studies confirmed that circulating PBMC in subject 5 were of donor origin. C. NK cells isolated from day 5 and day 7 post-infusion PB samples avidly killed subject 5 myeloma cells and K562 cells (51Cr release assay). D. Significantly higher NK cell levels were observed in the PB and BM of subject 8 who received NK produced in high IL2 (500 U/mL). E. The vast majority of post-infusion cells were of donor origin. F. Circulating NK cells obtained 14 days after infusion from the PB of subject 8 killed OPM2 myeloma and K562 cells (subject 8 myeloma was not available).
Figure 3. Auto-NK cells exhibited robust in vivo expansion, trafficked into the marrow, and exerted enhanced killing of auto-targets when HLA/KIR interactions were blocked or when ADCC was induced.


A, C. Substantial in vivo expansion of NK cells was observed for subjects 4 and 6 who received fresh auto-NK cell products produced in 10 U/mL IL2. B, D. Circulating NK cells obtained from the PB 5 days after infusion did not induce significant in vitro killing of auto-myeloma cells although killing of K562 was high. E. The highest NK cell counts post infusion were observed in subject 7, who received NK produced in 500 U/mL IL2. F. Subject 7 myeloma cells were not available for in vitro assays until day 89; however OPM2 myeloma cells and K562 were avidly killed by BM-derived NK cells obtained 5 days after infusion. G. IHC staining further confirmed the presence of CD57+ lymphocytes in the BM of subject 7. Magnification is 400x. H. Enhanced NK cell mediated killing of auto-myeloma from subjects 4, 6, and 7 was achieved by inducing ADCC with elotuzumab (Elo) or blocking ligation of inhibitory receptors with antibodies to HLA Class I and KIR. E:T Ratio is 10:1.
Activity of NK cells prepared in vitro
Freshly prepared research grade donor NK cells were KIR phenotyped and tested for the ability to kill recipient myeloma (when available) and/or unrelated primary myeloma cells (Table 3). Allo-donor NK cells killed recipient myeloma for subjects 2 and 5 (primary myeloma from subject 8 was not available), and lysis of third party myeloma was observed in all 3 cases. Specific lysis of subject 5’s myeloma by the selected donor was particularly high. Interestingly, this donor had a high frequency of NK cells that expressed the inhibitory receptor for the KIR-L missing in subject 5, and lacked expression of other inhibitory KIRs and NKG2A, which would be expected to trigger lysis of the target according to the ‘missing self’ hypothesis.20 This donor also expressed all three KIR-Ls and thus these NK were functionally competent, or “licensed” to kill cells in which this ligand was absent.21 In contrast, auto-NK cell killing of recipient myeloma cells in vitro was low in 3/4 evaluable cases. The difference in killing of myeloma targets by allogeneic ENK was better than with recipient autologous ENK cells, although this did not reach statistical significance (p=0.099), which likely can be explained by small numbers. However, myeloma patient-derived NK cells also exhibited significant activity against allo-myeloma cells, demonstrating the capacity of these NK cells to kill myeloma. Further assays confirmed that killing of autologous myeloma from the three subjects treated with auto-NK could be enhanced by blocking inhibitory KIR receptor-HLA ligand interactions (Figure 3H). In the same assays we showed that antibody-dependent cellular cytotoxicity (ADCC) induced by the humanized antibody to CS1, elotuzumab, also augmented killing of primary myeloma cells by auto-NK. The increased killing of myeloma cells by allo donor cells and the data from our HLA/KIR blocking and ADCC studies leads us to postulate that the low level of auto-NK cell lysis in vitro may not be due to inherent myeloma resistance to NK cell-mediated killing but rather due to inhibitory receptor-self ligand interactions.
Table 3.
Expanded NK cell cytolytic activity
| Treated Subjects | Allo/ Auto | Recipient Missing KIR-L | Expanded NK
|
Specific Lysis (%)
|
||||
|---|---|---|---|---|---|---|---|---|
| CD3−56+ Potentially Alloreactive (%) | Potentially Alloreactive & licensed (%) | K562 | Donor NK vs. Recipient Myeloma | Donor NK vs. Unrelated Myeloma | Unrelated NK vs Recipient Myeloma | |||
|
|
|
|
|
|
|
|
|
|
| 1 | Auto | None | 0 | 0 | 81.1 | 8.3 | 30.9 | 19.0 |
| 2 | Allo | II, Bw4 | 17.4 | 13.2 | 84.4 | 45.7 | 19.0 | 50.1 |
| 3 | Auto | None | 0 | 0 | 78.2 | NA | 46.7 | NA |
| 4 | Auto | None | 0 | 0 | 71.1 | 8.2 | 58.7 | 18.0 |
| 5 | Allo | II | 33.4 | 33.4 | 71.2 | 74.3 | 49.5 | 63.1 |
| 6 | Auto | I | 27.1 | 0 | 77.0 | 5.4 | 51.0 | 49.9 |
| 7 | Auto | None | 0 | 0 | 81.7 | 45.0 | 28.5 | 58.7 |
| 8 | Allo | Bw4 | 12.5 | 0 | 80.4 | NA | 36.5 | NA |
Potentially alloreactive expanded NK cells were defined as CD3−56+ cells expressing KIR(s) recognizing the missing ligand(s) in the recipient and also negative for NKG2A and KIRs recognizing ligands present in the recipient. Licensed NK were defined as alloreactive NK derived from a donor expressing the KIR-L that was missing in the recipient. Allo, Allogeneic product recipients; Auto, Autologous product recipients; NA, Primary myeloma was not available from subjects 3 and 8.
Activity of NK cells isolated post infusion
We tested the cytolytic activity of post-infusion circulating NK cells against myeloma targets. Allo-donor NK cells obtained from subject 5 PB at two different post-infusion time points avidly killed recipient myeloma cells (Figure 2C). Subject 8 myeloma was not available; however we were able to confirm that day 14 PB-derived NK cells killed the surrogate allo-myeloma cell target OPM2 (Figure 2F). In contrast, NK cells isolated from the PB 5 days after infusion from auto-NK cell subjects 4 and 6 failed to induce significant lysis of auto-myeloma cells (Figures 3B, D). Subject 7 myeloma cells were not available until after progression but NK cells isolated from the BM of subject 7 five days after infusion killed OPM2 cells (Figure 3F).
Expanded NK cell immunophenotype
An example (subject 7) of the immunophenotype of NK cells pre- and post-expansion and after infusion is shown in Figure 4. Expanded NK cells exhibited very high CD56 expression. FcγRIIIa (CD16) was retained on the majority of cells, albeit at a reduced level. Dramatic and consistent up regulation of CD26 (dipeptidyl peptidase IV), a serine protease implicated in CD16 mediated killing,22 and CD70 (CD27 ligand) was observed after expansion. Cell surface expression of the adhesion molecule CD54, the early activation marker CD69, activating receptors NKp30, NKp44, and NKG2D and the apoptosis inducing ligand TRAIL (not shown) were also increased on expanded NK. CXCR3 expression increased and CD62L decreased on expanded NK while CXCR4 and CXCR6 were dim/negative on both resting and activated NK (data not shown). Inhibitory molecule expression was also evaluated. The number of cells expressing CD94 (not shown) did not increase and, although the number of NKG2A positive cells was higher after expansion, the increase was not significant (average increase 12%, N=5 pairs, p=0.07). GMP-expanded NK cells had lower expression of CD56, CD69, NKG2A, NKG2D, NKp30, and NKp44 compared to research grade NK cells, which may have been due to IL2 deprivation during overnight shipping.
Figure 4. Immunophenotype of NK cells pre-expansion, post-expansion, and post-infusion (subject 7).

CD56 histogram overlays are gated on lymphocytes, all others are gated on CD3−CD56+ lymphocytes. Thin lines are isotype controls and thick lines are the indicated marker. The samples shown are the apheresis product analyzed the day of collection (Aph), the clinical product post-shipping (GMP NK), small scale research grade NK generated at UAMS (Res Grade NK) and PB samples obtained at the post-infusion time points indicated.
After infusion, the changes in CD16 and CD56 expression induced by in vitro expansion reverted back to the levels seen in resting NK by day 30. Likewise, levels of CD26 and CD70 were high early post infusion, but returned to baseline by day 14 and day 30, respectively. The level of CD94 cell surface expression increased during the IL2 course in 4/5 evaluable patients.
Circulating cytokine levels
Prior to NK cell infusion, patients who received Flu had increased serum levels of the NK cell growth factor IL15 in comparison with patients who received bortezomib only (mean 57.6 vs. 3.4 pg/mL, respectively) which may have been due to chemotherapy-induced lymphopenia (Figure 5A). Peaks of IL5 and IL10 were observed on days 7–9 in 4/4 evaluated subjects, and seemed to coincide with in vivo NK cell expansion (Figure 5B, C). Circulating TGFβ levels (day 7 range: 1.9–20.8 ng/mL, Figure 5D) were similar to levels used in our TGFβ experiments (5 ng/mL), vide infra.
Figure 5. Serum levels of IL15, IL5, IL10, and TGFβ during the treatment course.

A. Circulating levels of IL15 were elevated in subjects who received Flu (N=5) compared to subjects who did not receive immunosuppression (N=3). B–D. IL5, IL10, and TGFβ levels during the treatment course for subjects 4, 6, 7 and 8 as indicated. Markers are open for auto-NK cell recipients and closed for allo-NK cell recipients. Cytokine levels for subjects who received immunosuppression with fludarabine (Flu) are depicted with dotted lines. Subject 5 was omitted from the IL5, IL10, TGFβ analysis as IL2 was stopped on day 5.
Mechanisms of suppression
IL2 administration has been shown to increase the frequency of suppressive T regulatory cells in cancer patients.23 While allo-NK subjects, who received immunosuppressive conditioning therapy, had few circulating T cells post NK cell infusion, an increase in absolute CD3+ T cell counts was observed in 2/3 recipients of fresh auto-NK cells during their IL2 course (Figure 6A) and the number of CD3+CD4+CD25+Foxp3+ T reg cells/μL blood also increased in auto-NK recipients, peaking 14 days after NK cell infusion, (Figure 6B) at which time the ratio of NK:T reg cells were lower than baseline values (Figure 6C). We therefore tested whether irradiated K562-mb15-41BBL feeder cell stimulation of PBMC, obtained from time points in which T reg cell levels were increased, led to further NK cell expansion, and whether depletion of CD25+ cells from these PBMC enhanced NK cell growth. CD25 depletion resulted in markedly enhanced NK cell fold expansion in subject 4, suggesting that T regs may have suppressed NK cell proliferation. In contrast, reduced expansion in CD25 depleted cultures was observed for subject 6, and no significant difference was demonstrated for subject 7 (Figure 6D). The levels of NK cell expansion were modest in both culture conditions for subject 7, perhaps in part due to the fact that thawed cells were utilized for this experiment.
Figure 6. Changes in T cell and T reg cell frequency.

Markers are open for auto-NK cell recipients and closed for allo-NK cell recipients. A. Shown is the fold T cell change from the pre-chemotherapy value for the indicated subjects. B. The frequency of T reg cells was determined for subjects with sufficient circulating T cells (auto-NK cell recipients who did not receive immunosuppression). CD3+CD4+CD25+Foxp3+ T reg cells increased during the IL2 course, peaking at day 14. C. The NK:T reg ratio was higher than day 0 at days 5, 9, and 30 but not on day 14 when T reg levels had peaked and NK cell levels had begun to decrease. D. PBMC obtained from time points in which T reg cell levels were increased from baseline (post NK infusion days 9 or 14) were thawed and then whole PBMC (CD25 replete) and PBMC depleted of CD25+ cells using anti-CD25 antibody-coated magnetic beads were stimulated with irradiated K562-mb15-41BBL cells in the presence of IL2 (300 U/mL). NK cells were enumerated 10 days later by multiplying the viable cell count in the cultures by the NK cell percentage as determined by flow cytometry. Five replicate expansion cultures were set up for each condition. The mean ± the standard deviation is shown in addition to significance values; n.s., not significant.
We also examined expanded NK cell sensitivity to TGFβ as it is produced by T regs24 and is a key mediator of immunosuppression in the myeloma bone marrow microenvironment.25 Our experiments confirmed that despite the strong activating stimulus of K562-mb15-41BB cells, TGFβ treatment reduced NK cell proliferation and suppressed IFNγ production (Figures 7A, B). Moreover, TGFβ treated, expanded NK cells exhibited significantly lower levels of NKG2D (p<0.005), NKp30 (p<0.02), NKp46 (p<0.02) and DNAM-1 (p<0.001), leading to inferior cytolytic activity (representative examples shown in Figures 7C, D). We observed SMAD3 phosphorylation in both resting NK and TGFβ-treated expanded NK cells, indicative of homeostatic cell cycle arrest and TGFβ suppression, respectively (Figure 7E).
Figure 7. K562-mb15-41BBL-stimulated NK cells are sensitive to TGFβ induced suppression.

A. Stimulation of fresh whole PBMC obtained on protocol days 7, 9 and 14 (subjects 7 and 8) with irradiated K562-mb15-41BBL cells resulted in a 3.0 – 4.9 fold increase in NK cells after 5 days of culture. Lower NK cell expansion was observed in replicate cultures in which 5ng/mL recombinant human TGFβ was added. B. TGFβ cultures had significantly lower IFNγ production compared to vehicle cultures. C. Cell surface expression of NKG2D, DNAM1, NKp30, and NKp46 were also reduced after TGFβ treatment. Solid lines are vehicle, dashed lines are TGFβ, thin lines are isotype control. One representative example of 6 experiments is shown. D. Cytolytic activity was diminished in NK from TGFβ cultures. E. Phosphorylation of SMAD3 was observed in highly purified NK cells from the pre-NK infusion apheresis product (resting, RNK) and expanded NK cells treated with TGFβ, but not in expanded NK cells treated with vehicle (cells were obtained from subject 7). ENK, expanded NK.
Discussion
In this study we demonstrated the feasibility and safety of administering high doses of ex vivo expanded NK cells to high-risk relapsed MM patients. NK cells were expanded from PBMCs by co-culture with K562-mb15-41BBL stimulator cells for a relatively short expansion period (8–9 days) and with minimal manipulation of the cultures.13 Importantly, large numbers of clinical grade NK cells from both MM patient- and donor-derived apheresis products were consistently generated, although less vigorous expansion was observed with patient-derived cells, likely due to extensive prior therapy. The presence of T cells at harvest was limited and CD3 depletion further increased the NK cell purity of allo-products.
Early NK cell products were cryopreserved to allow for completion of quality control testing prior to infusion. Unfortunately, upon thawing, cryopreserved products exhibited inferior recovery and potency, and survived poorly during further in vitro culture. In contrast, freshly formulated NK cells had excellent recovery and retained cytolytic ability after shipping. Additional advantages of using fresh cells include the elimination of thaw procedures and the convenience of the cells arriving ready to infuse, although the use of fresh cells make the coordination of culture initiation with the patient’s entry on the protocol crucial. Changes in release criteria relying on rapid and in process testing reduced the time from apheresis collection to NK infusion to 20 days, an important consideration when treating high-risk MM patients who can experience rapid disease progression. Most importantly, robust in vivo expansion was only seen after infusion of fresh NK cells. NK cells were observed at high levels not only in the blood but also in the marrow, despite low CXCR4 and CD62L expression. Chimerism studies for patients receiving allo-NK confirmed that virtually all of the circulating post-infusion cells were donor-derived. Our second critical process improvement, increasing the IL2 dose utilized for culture to 500 U/mL (originally limited to 10 U/mL in order to avoid outgrowth of T cells), further enhanced NK cell proliferation. We observed that fresh NK cells grown in high-dose IL2 exhibited massive expansion post-infusion, up to 40-fold, with NK cell counts remaining above baseline for up to 6 months. This change also led to significantly increased in vitro anti-myeloma activity and no excess T cell proliferation was observed in the cultures.
The activity of fresh NK was retained after infusion as both circulating PB- and BM-derived NK cells avidly killed allo-myeloma targets in vitro. However, the activity of auto-NK against recipient myeloma was suboptimal. This low in vitro activity was not due to inherent resistance to NK cell killing since the same targets were killed by allo-donor NK. Rather, blocking studies showed that interactions between self-HLA and inhibitory KIR were at least in part responsible for the reduced killing and significantly enhanced lysis of autologous targets was achieved by induction of ADCC. Unfortunately, the analysis of in vitro killing data after stratification of each non-autologous interaction was not possible due to the limited number of treated subjects.
After infusion NK cell levels decreased and activating receptors were down regulated. We considered the role of T reg cells as they can suppress T and NK cell proliferation by modulating available IL2,26 by secreting inhibitory cytokines,24 and via inhibitory cell-cell interactions.24,27,28 The frequency of T reg cells increased during the IL2 course in patients who did not receive the immunosuppressive conditioning regimen and depletion of cells expressing CD25 from post infusion PBMC enhanced NK cell proliferation in vitro for at least one subject. We further considered the inhibitory cytokine TGFβ, which has been shown to inhibit NK cell activating receptor expression,29,30 IFNγ production, and cytolytic function via SMAD3.31 We showed that expanded NK cells are sensitive to TGFβ suppression at concentrations comparable to that observed in patient serum. The fact that these activated NK cells are highly sensitive to TGFβ-induced suppression may be an important topic for further investigation in myeloma microenvironment models as the bone marrow niche is a rich source of TGFβ.32 Inadequately sustained activation signaling could have also contributed to the leveling off of proliferation in vivo at protocol days 9–14 (2.5–3 weeks after primary stimulation with K562-15-41BBL feeders). Fujisaki et al. have previously published that NK cells stimulated with IL2 and irradiated K562-mb15-41BBL feeder cells may undergo 10–30 population doublings before becoming senescent, typically 5–8 weeks from the initial stimulation.33 However, restimulation with K562-mb15-41BBL every 2–3 weeks is required to sustain high proliferation throughout this period.33 It was difficult to judge the clinical efficacy of the NK cells in this trial. Clearly, cryopreserved NK cells (patients 1–3) had little activity, while suboptimal doses of IL2 during manufacture may have hampered anti-myeloma efficacy in patients 4–6. Only 2 patients were treated with NK cells generated with the optimized procedure. Of these, the patient who received allo-NK cells died early due to chemotherapy toxicity, whilst the patient treated with auto-NK cells progressed slowly with time to next treatment being 6 months.
The duration of low counts for patients receiving Flu was 0 – 58, median 9 days. No prolonged cytopenias were observed in patients who received only bortezomib and ENK cells. All patients had been heavily treated and hematopoietic compromise was common as is reflected by the platelet counts at the time of enrollment (Supplemental Digital Content 5). Cy and Flu are well known to cause myelosuppression. Further, in vitro experiments showed only low level killing of purified CD34+ cells by ENK cells (data not shown). Taken together, these considerations suggest that any cytopenias were due to Cy/Flu rather than the infused ENK cells. Lymphodepleting therapy with Cy and Flu in these heavily pretreated patients was therefore clearly too toxic and has been adjusted so that future allo-NK patients will receive only Cy.
Few clinical trials testing ex vivo expanded NK cells have been completed. Barkholt et al. described one transient response in 5 advanced cancer patients treated in a dose escalation trial testing expanded allo-NK and NKT cell administrations after allogeneic transplant. However, since these products also contained significant numbers of T cells and post infusion the frequency of circulating T cells were much higher than NK cells, it may be difficult to ascertain the NK cell mediated effects in vivo.34 Another trial for patients with advanced malignancies utilized pentostatin to deplete T regs followed by bortezomib and escalating doses of auto-NK cells. This study established the safety of infusing up to 1×108 ex vivo expanded auto-NK cells/kg and reported stable disease in 7/14 treated patients with a 30% reduction in serum tumor markers for 2 patients and a minor response in a patient with metastatic kidney cancer.35,36 As with our study, significant increases of circulating NK cells occurred, peaking 7 days after infusion.
Efforts centered on IL2 stimulation to boost the activity of haplo-identical NK cells in AML patients have resulted in some impressive responses which closely correlated with in vivo expansion of the adoptively transferred cells. Investigators at the University of Minnesota treated patients with relapsed AML with IL2 activated CD3 depleted haplo-identical donor lymphocyte infusions observed significant in vivo expansion after Cy/Flu immunosuppression and complete hematologic remissions in 5/19 patients in whom expansion was demonstrated.37 A second study by this group in which total body irradiation was added to the immunosuppressive conditioning regimen and IL2 activated CD3 and CD19 depleted haplo-identical products were utilized further corroborated the correlation between in vivo expansion of allo-NK cells and outcome.38 Remarkably, 89% of the 19 patients who achieved NK cell expansion in vivo achieved leukemia clearance and 84% achieved remission versus 42% and 10%, respectively, for non-expanders.38
Our unique expansion method achieves marked expansion in vitro followed by very significant further increase in numbers post infusion, indicating extensive further expansion of NK in vivo. A number of different methods of expansion have been described including the use of genetically modified stimulator cells, provision of feeder cells and feeder free conditions. However, head to head comparisons have thus far not been performed. It is therefore not certain which method of expansion is optimal in terms of activating NK cells, further in vivo expansion, cytolytic ability, and long term persistence of NK cells.
In summary, production assistance by the NHLBI PACT program allowed for the rapid implementation of a novel therapy utilizing fresh NK cells for poor prognosis MM patients. We demonstrated superior expansion and activity of fresh NK cell products and the importance of using high concentrations of IL2 in NK cell generation. It is exciting that dramatic in vivo expansion was observed and that circulating NK retained the ability to kill allo-myeloma cells. Our preliminary results suggest that expanded NK cell immunotherapy, especially in the allo-setting, is deserving of further exploration and point toward a number of strategies for optimization. Substituting IL2 cytokine support with IL15 may enhance in vivo NK cell activity and persistence14 without inducing unwanted T reg proliferation.23 TGFβ blockade may further protect NK from T reg suppression and from suppression in the myeloma microenvironment.39 Enhanced targeting and anti-myeloma activity may be accomplished via incorporation of elotuzumab,40,41 inhibitory receptor blocking,42 or a combination of these two approaches,43,44 while genetic modification of NK with chimeric antigen receptors12,45 is another attractive option to ensure prolonged activation in vivo. Once proven effective in the relapsed setting, optimized NK cell therapy could be incorporated into the upfront treatment of high-risk myeloma.
Supplementary Material
Table of antibodies used in this study.
Cells formulated in 5% HSA were shipped overnight and the product bag was subsequently held at 2–11 degrees until testing was performed. No media or IL2 was added prior to analysis.
NK cell cultures were initiated with culture media supplemented with the indicated IL2 concentrations. A. Enhanced NKp30 cell surface expression was observed in NK cells generated in 500–1000 U/mL of IL2. B. Increased cytolytic activity against K562 target cells was observed at higher IL2 concentrations. C. Percent T cell and NK cell content were not significantly different in cultures initiated in 500 U/mL versus 10 U/mL IL2 (N=4 cultures, p=0.4 and 0.5, respectively). D. GMP grade NK cells produced in 500 U/mL IL2 and shipped overnight had similar lytic activity against K562 and OPM2 cells as small scale research grade NK (subject 7 data is shown).
Depicted are toxicities experienced by all treated subjects (N=8), according to category and grade. Toxicities were graded based upon CTCAE v4 guidelines. Values indicated are: number of subjects who experienced the toxicity (median duration in days, range). *One individual, no range. **All subjects experienced toxicity for same duration, no range.
Pancytopenia defined as both absolute neutrophil count (ANC) <500/μL and platelets (Plt) <20,000/μL. Subjects above the dotted line received ENK products which were shipped frozen and infused after thaw. Subjects below the dotted line received ENK products which were shipped and infused fresh. Bor, bortezomib; Cy, cyclophosphamide; Flu, fludarabine. Auto, autologous; allo, allogeneic.
Depicted are the average NK cell count (#/μL) for each of three manufacture conditions: 1) products which were expanded in low IL2 (10 U/mL) and infused post freeze-thaw, 2) products which were expanded in low IL2 (10 U/mL) and infused fresh, and 3) products which were expanded in high IL2 (500 U/mL) and infused fresh. This graph clearly demonstrates the dramatic difference in in vivo expansion after the adoption of a higher IL2 concentration in the ENK manufacture method
Acknowledgments
This work was supported by NIH grants CA55819 and CA134522. The clinical grade production of NK was supported by PACT grant NIH-NHLBI-N01 HB37163. The authors wish to acknowledge the MIRT and Cell Therapy Program staff for their contributions toward the clinical aspects of this study. We also acknowledge the UAMS Translational Research Institute, funded by the National Institutes of Health Clinical and Translational Science Award program, grants UL1TR000039 and KL2TR000063 for regulatory support, and the UAMS Experimental Pathology Core for technical assistance with histology. We thank our collaborators at CAGT, and St. Jude Children’s Research Hospital for technical advice.
Footnotes
Authorship
FvR designed the clinical study; FvR, SS, designed the correlative studies; NL, CR, AG, optimized production and generated the clinical grade products; FvR, SS wrote the paper; FvR, SS, JL, AG, KS, EW, JK, JS, SP, JE, SY, WB, MR performed research, collected data, analyzed and interpreted data; FvR, BN, SW, MF and BB enrolled and treated patients on research protocols; DC provided the K562-mb15-41BB cells developed at St Jude Children’s Research Hospital.
Conflict-of-interest disclosure
There are no relevant conflicts of interest to disclose.
References
- 1.Palumbo A, Anderson K. Multiple myeloma. N Engl J Med. 2011;364:1046–1060. doi: 10.1056/NEJMra1011442. [DOI] [PubMed] [Google Scholar]
- 2.Shaughnessy JD, Jr, Zhan F, Burington BE, et al. A validated gene expression model of high-risk multiple myeloma is defined by deregulated expression of genes mapping to chromosome 1. Blood. 2007;109:2276–2284. doi: 10.1182/blood-2006-07-038430. [DOI] [PubMed] [Google Scholar]
- 3.Nair B, van Rhee F, Shaughnessy JD, Jr, et al. Superior results of Total Therapy 3 (2003–33) in gene expression profiling-defined low-risk multiple myeloma confirmed in subsequent trial 2006–66 with VRD maintenance. Blood. 2010;115:4168–4173. doi: 10.1182/blood-2009-11-255620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Caligiuri MA. Human natural killer cells. Blood. 2008;112:461–469. doi: 10.1182/blood-2007-09-077438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carbone E, Neri P, Mesuraca M, et al. HLA class I, NKG2D, and natural cytotoxicity receptors regulate multiple myeloma cell recognition by natural killer cells. Blood. 2005;105:251–258. doi: 10.1182/blood-2004-04-1422. [DOI] [PubMed] [Google Scholar]
- 6.Garg TK, Szmania SM, Khan JA, et al. Highly activated and expanded natural killer cells for multiple myeloma immunotherapy. Haematologica. 2012;97:1348–1356. doi: 10.3324/haematol.2011.056747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alici E, Sutlu T, Bjorkstrand B, et al. Autologous antitumor activity by NK cells expanded from myeloma patients using GMP-compliant components. Blood. 2008;111:3155–3162. doi: 10.1182/blood-2007-09-110312. [DOI] [PubMed] [Google Scholar]
- 8.Fujisaki H, Kakuda H, Shimasaki N, et al. Expansion of highly cytotoxic human natural killer cells for cancer cell therapy. Cancer Res. 2009;69:4010–4017. doi: 10.1158/0008-5472.CAN-08-3712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Berg M, Lundqvist A, McCoy P, Jr, et al. Clinical-grade ex vivo-expanded human natural killer cells up-regulate activating receptors and death receptor ligands and have enhanced cytolytic activity against tumor cells. Cytotherapy. 2009;11:341–355. doi: 10.1080/14653240902807034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carlens S, Gilljam M, Chambers BJ, et al. A new method for in vitro expansion of cytotoxic human CD3−CD56+ natural killer cells. Hum Immunol. 2001;62:1092–1098. doi: 10.1016/s0198-8859(01)00313-5. [DOI] [PubMed] [Google Scholar]
- 11.Shah N, Martin-Antonio B, Yang H, et al. Antigen presenting cell-mediated expansion of human umbilical cord blood yields log-scale expansion of natural killer cells with anti-myeloma activity. PLoS One. 2013;8:e76781. doi: 10.1371/journal.pone.0076781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Imai C, Iwamoto S, Campana D. Genetic modification of primary natural killer cells overcomes inhibitory signals and induces specific killing of leukemic cells. Blood. 2005;106:376–383. doi: 10.1182/blood-2004-12-4797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lapteva N, Durett AG, Sun J, et al. Large-scale ex vivo expansion and characterization of natural killer cells for clinical applications. Cytotherapy. 2012;14:1131–1143. doi: 10.3109/14653249.2012.700767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tolar J, Curtsinger J, McElmurry R, et al. Optimal xenogeneic adoptive transfer of human NK cells: Fresh NK cells and IL-15 administration are superior to frozen NK cells and IL2. Blood ASH Annual Meeting Abstracts. 2012;120:346. [Google Scholar]
- 15.Shi J, Tricot GJ, Garg TK, et al. Bortezomib down-regulates the cell-surface expression of HLA class I and enhances natural killer cell-mediated lysis of myeloma. Blood. 2008;111:1309–1317. doi: 10.1182/blood-2007-03-078535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lundqvist A, Abrams SI, Schrump DS, et al. Bortezomib and depsipeptide sensitize tumors to tumor necrosis factor-related apoptosis-inducing ligand: a novel method to potentiate natural killer cell tumor cytotoxicity. Cancer Res. 2006;66:7317–7325. doi: 10.1158/0008-5472.CAN-06-0680. [DOI] [PubMed] [Google Scholar]
- 17.Blade J, Samson D, Reece D, et al. Criteria for evaluating disease response and progression in patients with multiple myeloma treated by high-dose therapy and haemopoietic stem cell transplantation. Myeloma Subcommittee of the EBMT. European Group for Blood and Marrow Transplant. Br J Haematol. 1998;102:1115–1123. doi: 10.1046/j.1365-2141.1998.00930.x. [DOI] [PubMed] [Google Scholar]
- 18.Lapteva N, Szmania S, van Rhee F, et al. Clinical Grade Purification and Expansion of Natural Killer Cells. In: Talmadge J, Whiteside T, editors. Crit Rev Oncog. Vol. 19. Begell House, Inc; 2014. pp. 121–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shaughnessy JD, Jr, Qu P, Usmani S, et al. Pharmacogenomics of bortezomib test-dosing identifies hyperexpression of proteasome genes, especially PSMD4, as novel high-risk feature in myeloma treated with Total Therapy 3. Blood. 2011;118:3512–3524. doi: 10.1182/blood-2010-12-328252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ljunggren HG, Karre K. In search of the ‘missing self’: MHC molecules and NK cell recognition. Immunol Today. 1990;11:237–244. doi: 10.1016/0167-5699(90)90097-s. [DOI] [PubMed] [Google Scholar]
- 21.Kim S, Poursine-Laurent J, Truscott SM, et al. Licensing of natural killer cells by host major histocompatibility complex class I molecules. Nature. 2005;436:709–713. doi: 10.1038/nature03847. [DOI] [PubMed] [Google Scholar]
- 22.Madueno JA, Munoz E, Blazquez V, et al. The CD26 antigen is coupled to protein tyrosine phosphorylation and implicated in CD16-mediated lysis in natural killer cells. Scand J Immunol. 1993;37:425–429. doi: 10.1111/j.1365-3083.1993.tb03313.x. [DOI] [PubMed] [Google Scholar]
- 23.Ahmadzadeh M, Rosenberg SA. IL-2 administration increases CD4+CD25hi Foxp3+ regulatory T cells in cancer patients. Blood. 2006;107:2409–2414. doi: 10.1182/blood-2005-06-2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wahl SM, Wen J, Moutsolpoulos NM. The kiss of death: interrupted by NK-cell close encounters of another kind. Trends Immunol. 2006;27:161–164. doi: 10.1016/j.it.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 25.Cook G, Campbell JD, Carr CE, et al. Transforming growth factor beta from multiple myeloma cells inhibits proliferation and IL-2 responsiveness in T lymphocytes. J Leukoc Biol. 1999;66:981–988. doi: 10.1002/jlb.66.6.981. [DOI] [PubMed] [Google Scholar]
- 26.Gasteiger G, Hemmers S, Firth MA, et al. IL-2-dependent tuning of NK cell sensitivity for target cells is controlled by regulatory T cells. J Exp Med. 2013;210:1167–1178. doi: 10.1084/jem.20122462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ghiringhelli F, Ménard C, Terme M, et al. CD4+CD25+ regulatory T cells inhibit natural killer cell functions in a transforming growth factor–β–dependent manner. J Exp Med. 2005;202:1075–1085. doi: 10.1084/jem.20051511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Trzonkowski P, Szmit E, Mysliwska J, et al. CD4+CD25+ T regulatory cells inhibit cytotoxic activity of T CD8+ and NK lymphocytes in the direct cell-to-cell interaction. Clin Immunol. 2004;112:258–267. doi: 10.1016/j.clim.2004.04.003. [DOI] [PubMed] [Google Scholar]
- 29.Castriconi R, Cantoni C, Della Chiesa M, et al. Transforming growth factor beta 1 inhibits expression of NKp30 and NKG2D receptors: consequences for the NK-mediated killing of dendritic cells. Proc Natl Acad Sci U S A. 2003;100:4120–4125. doi: 10.1073/pnas.0730640100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee JC, Lee KM, Kim DW, et al. Elevated TGF-beta1 secretion and down-modulation of NKG2D underlies impaired NK cytotoxicity in cancer patients. J Immunol. 2004;172:7335–7340. doi: 10.4049/jimmunol.172.12.7335. [DOI] [PubMed] [Google Scholar]
- 31.Trotta R, Dal Col J, Yu J, et al. TGF-beta utilizes SMAD3 to inhibit CD16-mediated IFN-gamma production and antibody-dependent cellular cytotoxicity in human NK cells. J Immunol. 2008;181:3784–3792. doi: 10.4049/jimmunol.181.6.3784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Roodman GD. Mechanisms of bone metastasis. N Engl J Med. 2004;350:1655–1664. doi: 10.1056/NEJMra030831. [DOI] [PubMed] [Google Scholar]
- 33.Fujisaki H, Kakuda H, Imai C, et al. Replicative potential of human natural killer cells. Br J Haematol. 2009 doi: 10.1111/j.1365-2141.2009.07667.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Barkholt L, Alici E, Conrad R, et al. Safety analysis of ex vivo-expanded NK and NK-like T cells administered to cancer patients: a phase I clinical study. Immunotherapy. 2009;1:753–764. doi: 10.2217/imt.09.47. [DOI] [PubMed] [Google Scholar]
- 35.Smith A, Khuu H, Betters DM, et al. Adoptive transfer of escalating doses of ex vivo expanded autologous natural killer (NK) cells in patients with advanced malignancies following bortezomib treatment to sensitize to NK-TRAIL cytotoxicity. Blood ASH Annual Meeting Abstracts. 2010;116:4296. [Google Scholar]
- 36.Lundqvist A, Berg M, Smith A, et al. Bortezomib Treatment to Potentiate the Anti-tumor Immunity of Ex-vivo Expanded Adoptively Infused Autologous Natural Killer Cells. J Cancer. 2011;2:383–385. doi: 10.7150/jca.2.383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Miller JS, Soignier Y, Panoskaltsis-Mortari A, et al. Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood. 2005;105:3051–3057. doi: 10.1182/blood-2004-07-2974. [DOI] [PubMed] [Google Scholar]
- 38.Cooley S, Foley B, Verneris MR, et al. Haploidentical Natural Killer (NK) Cells Expanding In Vivo After Adoptive Transfer Exhibit Hyperfunction That Partially Overcomes Self Tolerance and Leads to Clearance of Refractory Leukemia. ASH Annual Meeting Abstracts. 2011;118:355. [Google Scholar]
- 39.Wilson EB, El-Jawhari JJ, Neilson AL, et al. Human Tumour Immune Evasion via TGF-β Blocks NK Cell Activation but Not Survival Allowing Therapeutic Restoration of Anti-Tumour Activity. PLoS One. 2011;6:e22842. doi: 10.1371/journal.pone.0022842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Szmania S, Greenway A, Lingo J, et al. Impact Of Elotuzumab Therapy On Circulating and Ex Vivo Activated/Expanded Autologous Natural Killer (Auto-ENK) Cell Activity. Blood ASH Annual Meeting Abstracts. 2013;122:5389. [Google Scholar]
- 41.Collins SM, Bakan CE, Swartzel GD, et al. Elotuzumab directly enhances NK cell cytotoxicity against myeloma via CS1 ligation: evidence for augmented NK cell function complementing ADCC. Cancer Immunol Immunother. 2013;62:1841–1849. doi: 10.1007/s00262-013-1493-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Benson DM, Jr, Bakan CE, Zhang S, et al. IPH2101, a novel anti-inhibitory KIR antibody, and lenalidomide combine to enhance the natural killer cell versus multiple myeloma effect. Blood. 2011;118:6387–6391. doi: 10.1182/blood-2011-06-360255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kohrt HE, Thielens A, Marabelle A, et al. Anti-KIR antibody enhancement of anti-lymphoma activity of natural killer cells as monotherapy and in combination with anti-CD20 antibodies. Blood. 2014;123:678–686. doi: 10.1182/blood-2013-08-519199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chan WK, Kung Sutherland M, Li Y, et al. Antibody-dependent cell-mediated cytotoxicity overcomes NK cell resistance in MLL-rearranged leukemia expressing inhibitory KIR ligands but not activating ligands. Clin Cancer Res. 2012;18:6296–6305. doi: 10.1158/1078-0432.CCR-12-0668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chu J, Deng Y, Benson DM, et al. CS1-specific chimeric antigen receptor (CAR)-engineered natural killer cells enhance in vitro and in vivo antitumor activity against human multiple myeloma. Leukemia. 2013 doi: 10.1038/leu.2013.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lundqvist A, Yokoyama H, Smith A, et al. Bortezomib treatment and regulatory T-cell depletion enhance the antitumor effects of adoptively infused NK cells. Blood. 2009;113:6120–6127. doi: 10.1182/blood-2008-11-190421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hallett WH, Ames E, Motarjemi M, et al. Sensitization of tumor cells to NK cell-mediated killing by proteasome inhibition. J Immunol. 2008;180:163–170. doi: 10.4049/jimmunol.180.1.163. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table of antibodies used in this study.
Cells formulated in 5% HSA were shipped overnight and the product bag was subsequently held at 2–11 degrees until testing was performed. No media or IL2 was added prior to analysis.
NK cell cultures were initiated with culture media supplemented with the indicated IL2 concentrations. A. Enhanced NKp30 cell surface expression was observed in NK cells generated in 500–1000 U/mL of IL2. B. Increased cytolytic activity against K562 target cells was observed at higher IL2 concentrations. C. Percent T cell and NK cell content were not significantly different in cultures initiated in 500 U/mL versus 10 U/mL IL2 (N=4 cultures, p=0.4 and 0.5, respectively). D. GMP grade NK cells produced in 500 U/mL IL2 and shipped overnight had similar lytic activity against K562 and OPM2 cells as small scale research grade NK (subject 7 data is shown).
Depicted are toxicities experienced by all treated subjects (N=8), according to category and grade. Toxicities were graded based upon CTCAE v4 guidelines. Values indicated are: number of subjects who experienced the toxicity (median duration in days, range). *One individual, no range. **All subjects experienced toxicity for same duration, no range.
Pancytopenia defined as both absolute neutrophil count (ANC) <500/μL and platelets (Plt) <20,000/μL. Subjects above the dotted line received ENK products which were shipped frozen and infused after thaw. Subjects below the dotted line received ENK products which were shipped and infused fresh. Bor, bortezomib; Cy, cyclophosphamide; Flu, fludarabine. Auto, autologous; allo, allogeneic.
Depicted are the average NK cell count (#/μL) for each of three manufacture conditions: 1) products which were expanded in low IL2 (10 U/mL) and infused post freeze-thaw, 2) products which were expanded in low IL2 (10 U/mL) and infused fresh, and 3) products which were expanded in high IL2 (500 U/mL) and infused fresh. This graph clearly demonstrates the dramatic difference in in vivo expansion after the adoption of a higher IL2 concentration in the ENK manufacture method