

Abstract
Atherosclerosis is an inflammatory process that involves activation of matrix metalloproteinases (MMPs); MMPs degrade collagen and allow for smooth-muscle cell migration within a vessel. Moreover, this begets an accumulation of other cellular material, resulting in occlusion of the vessel and ischemic events to tissues in need of nutrients. Homocysteine has been shown to activate MMPs via an increase in oxidative stress and acting as a signaling molecule on receptors like the peroxisome proliferator activated receptor-γ and N-methyl-D-aspartate receptor. Nitric oxide has been shown to be beneficial in some cases of deactivating MMPs. However, in other cases, it has been shown to be harmful. Further studies are warranted on the scenarios that are beneficial versus destructive. Hydrogen sulfide (H2S) has been shown to decrease MMP activities in all cases in the literature by acting as an antioxidant and vasodilator. Various MMP-knockout and gene-silencing models have been used to determine the function of the many different MMPs. This has allowed us to discern the role that each MMP has in promoting or alleviating pathological conditions. Furthermore, there has been some study into the MMP polymorphisms that exist in the population. The purpose of this review is to examine the role of MMPs and their polymorphisms on the development of atherosclerosis, with emphasis placed on pathways that involve nitric oxide, hydrogen sulfide, and homocysteine.
Keywords: homocysteine, matrix metalloproteinases, oxidative stress, bone remodeling, collagen cross-linking, hydrogen sulfide, nitric oxide
Introduction
This paper focuses on the role of matrix metalloproteinases (MMPs) in promoting atherosclerosis. A brief discussion of the pathogenesis of atherosclerosis is included. There is also a discussion of homocysteine (Hcy) and its role in activating MMPs via oxidative stress and various receptor-mediated pathways, which include the peroxisome proliferator-activated receptor-γ and N-methyl-D-aspartate receptor. Moreover, there is some discussion about the role of nitric oxide in activating or deactivating MMPs, as it has been shown to do both in different circumstances. Further studies are warranted. Hydrogen sulfide (H2S) is also discussed in potential clinical applications to reduce oxidative stress and relax vessels, thereby increasing flow, and reducing susceptibility to ischemic events.
Several MMPs have been shown to be increased in cases of pathogenesis, including MMP-9, MMP-2, MMP-3, MMP-1, and MMP-8. However, MMP-10 has been sought as a potential alleviator of plaque, or a clot buster. Purified extract of MMP-10 may prove to be a modern-day aspirin, as it attacks the fibrinogen molecule and prevents its conversion to fibrin, which allows for the netting of platelets and resultant plaque occlusion. This is especially promising for patients. It is also very interesting to note the various ways that the vessel is remodeled when it is missing any particular MMP. Using delivery systems of small interfering ribonucleic acid (siRNA) may prove useful to decrease expression of a certain MMP under disease conditions.
Furthermore, there has been an investigation into the polymorphisms of MMPs that exist in the population. This can serve to determine the susceptibility that patients may have to atherosclerosis, as well as the prognosis of cardiac events. Further research is being conducted, and has found some conclusive results showing that there exist MMP polymorphisms that do pose this risk to atherosclerosis. These polymorphisms generally involve overactivation of a particular MMP. These exciting developments will all serve to improve the quality of life to the many patients in need of health care.
Atherosclerosis pathogenesis: a brief summary
Atherosclerosis was once thought to be caused in the same fashion as a pipe in one’s home that has been clogged by too much grease and oil; however, we now are aware that atherosclerosis is an inflammatory process that begins first with an insult on the endothelial lining within vessel walls.1 The process of atherosclerosis occurs in larger vessels that possess smooth-muscle layers that can become larger and encroach on the lumens of the vessels, thereby decreasing blood flow and inciting ischemic conditions. This process has a specific gene-expression profile; targeting these genes and manipulating their expressions is a priority in trying to reduce the magnitude of atherosclerosis that occurs.2 This is an extremely destructive process to all cells of the body that require nutrients and need to transport waste products out of the cell. With decreased transport from decreased flow, cells become damaged. Within coronary vessels, the heart may render some pain from insufficient blood supply and resultant death of the myocardium.
Atherosclerosis begins with a fatty streak within the intimal layer of the artery, due to an accumulation of lipid-engorged foam cells. These foam cells are macrophages that have engulfed oxidized low-density lipoprotein (LDL) cholesterol particles. As the cells become larger, they place pressure on the intimal layer, which encroaches on the lumen of the vessel and impedes flow.3 Moreover, there is a recruitment of even more inflammatory cells that can destroy vessel architecture, thereby promoting smooth muscle cell (SMC) migration. This SMC migration and increase of SMCs within the artery can further occlude the vessel. SMC proliferation via MMP activation is illustrated in Figure 1. There is also a connective tissue-cell layer of extracellular matrix that consists of collagen. VCAM-1 is known to be involved in this recruitment of inflammatory cells, as it is a protein that is involved in the adherence of these cells to the endothelial layer of vessel walls. Hence, VCAM-1 promotes attachment to vessel walls and entry within the inner architecture of the vessel of inflammatory cells and oxidized LDL cholesterol particles.4 This begets even further release of cytokine-signaling molecules that recruit more inflammatory cells. The inflammatory cells most involved are monocytes and T-cells that can release MCP-1.5 The more mature plaques contain dendritic cells, mast cells, B cells, and natural killer T-cells. Several of these cells are activated, and produce inflammatory cytokines like IFNγ and TNF.6 The anatomy of the plaque is made up of a thin fibrous cap, necrotic core, outward remodeling, and many inflammatory cells.7 The different mediators of atherosclerosis can be visualized in Figure 2.
Figure 1.
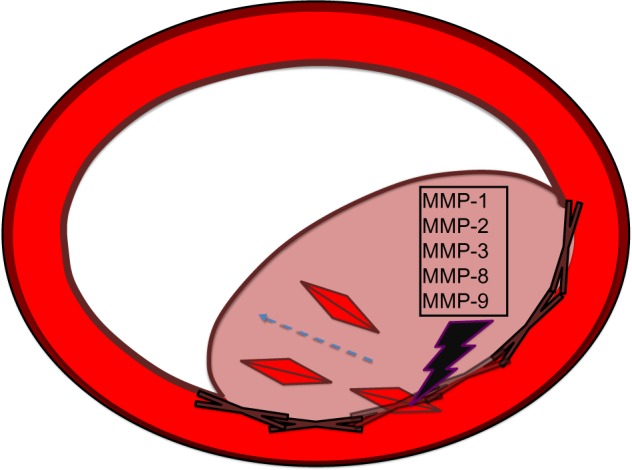
MMPs and smooth-muscle cell proliferation.
Notes: MMP-1, -2, -3, -8, and -9 have been shown to be involved in facilitating vascular remodeling by degrading the matrix separating the intima from the media and allowing smooth-muscle cell migration from the media to the intima. This smooth-muscle cell migration results in the occlusion of vessel walls and decreased blood flow to tissues in need of oxygen and nutrients carried by the blood.
Abbreviation: MMP, matrix metalloproteinase.
Figure 2.
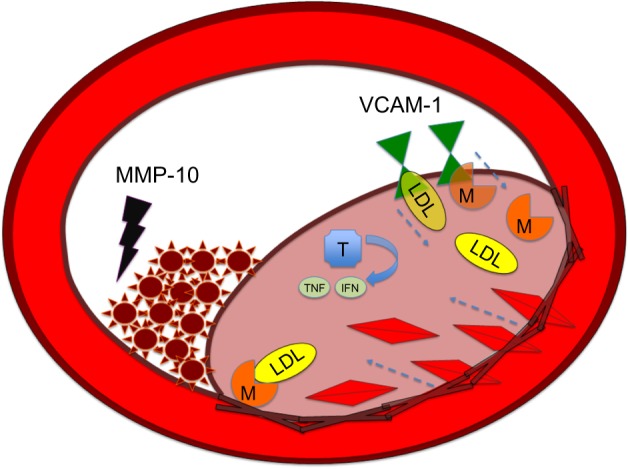
MMP-10 as clot destroyer in atherosclerosis.
Notes: LDL molecules and monocytes, including macrophages and T-cells, transcend the endothelial layer of the intima of blood vessels via transporters like VCAM-1. Macrophages within the intima will engulf oxidized LDL molecules and form foam cells. T-cells will release inflammatory cytokines, such as TNFα and IFNγ, that recruit molecules further into the intima. Smooth-muscle cells also proliferate into the intima layer from the media layer, which results in further occlusion of vessels. MMP-10 attacks the fibrinogen molecule and prevents its conversion to fibrin, which allows for the netting of platelets and resultant plaque occlusion.
Abbreviations: IFN, Interferon; LDL, low-density lipoprotein; M, macrophage; MMP, matrix metalloproteinase; T, T lymphocyte; TNF, tumor necrosis factor; VCAM-1, vascular cell adhesion molecule.
Human atherosclerosis is described via various types, including the milder forms I, II, III, and the more advanced forms IV, V, and VI via histological examination. The more advanced forms of plaque are vulnerable to rupture. Type I lesions have isolated macrophages and foam cells, while type II fatty-streak lesions have mainly intracellular lipid accumulation.8 Type III intermediate lesions have type II changes as well as small extracellular lipid pools, while type IV atheroma lesions have type II changes and core extracellular lipid.8 Type V fibroatheroma lesions have lipid cores and fibrotic layers; these layers may be either mainly calcific or fibrotic.8 Type VI complicated lesions have surface defects, hematoma/hemorrhage, and thrombus.8
MMPs and differential function of MMP-2 and MMP-9
MMPs are a very large family of calcium-dependent, zinc-containing endopeptidases. Their function is degrading extracellular matrix in various tissues. Their substrate includes collagen, elastin, gelatin, matrix glycoproteins, and proteoglycans. MMPs are known to be expressed in normal physiological conditions at some baseline level, but are differentially expressed in response to certain hormones, growth factors, and cytokines. Inhibitors of MMPs called tissue inhibitors of MMPs (TIMPs) control the enzyme. The MMPs are excreted by various tissues: connective tissue cells, proinflammatory cells of fibroblasts, osteoblasts, endothelial cells, macrophages, neutrophils, and lymphocytes. There are over 20 MMPs that have been identified, with differential expression of each found under varying pathological conditions and locations within vessels. By a series of knockout mouse and gene blockers, the function of each MMP continues to be discerned. The functions of MMP-2 and MMP-9 are of special concern, as these have been found in greater concentrations under pathological states, as detailed herein. One very recent study experimented with silencing MMP-2 expression as a means to decrease pathological remodeling. The delivery system used was amphipathic deoxycholic acid-modified polyethyleneimine. This was synthesized to be used as a carrier system for siRNA in order to prevent neointima formation secondary to migration of vascular SMCs (VSMCs).9 This work has profound implications for patient care. Developing these sorts of delivery systems to be localized in diseased vessels would serve to obliterate disease in a very specific way.
In a study of abdominal aortic aneurysms that were examined in humans after surgical operation, there were increased activities of MMP-9 and MMP-2; there were also increases in numbers of Mac-2-positive macrophages and cluster differentiation 3-positive T lymphocytes.10 From this, it may be inferred that MMP-9 and MMP-2 are involved in the dynamic process of aneurysm formation. It is known that MMP-2 plays a role in promoting atherosclerosis, since atherogenesis is reduced in MMP-2-deficient ApoE−/− mice, whereby LDL cholesterol is not properly processed and remains in the bloodstream.11 Using this type of knockout model is crucial to determining the role of MMP-2. However, the deletion of MMP-9 promotes plaque development, which suggests a beneficial role of MMP-9 in preventing plaque development.12
It was suggested from one study that MMP-2 and MMP-9 are involved in the neointima formation that is created by SMC migration from the media to the intima.13 This study used MMP-9-knockout mice in an experiment to determine MMP-9’s effect on venous grafts. Evidently, it appears as though MMP-2 is responsible for much of the remodeling that still occurs with the graft stresses even when MMP-9 is knocked out. The following remodeling of the vessel occurred: increase in collagen, sixfold increase in MMP-2 pro- and active forms, and earlier linear expansive remodeling. It would have been interesting to investigate how long these mice would live with this type of remodeled vessel versus the neointima formation that would otherwise occur. This is further elucidated in the following.
A similar result was obtained from a study using ApoE-deficient mice.14 It was discovered from this that inactivating MMP-8 resulted in a large reduction in atherosclerotic lesion development. For instance, there were significantly fewer macrophages, but the vessel had an increased collagen content. One great effect of knocking out MMP-8 as opposed to other MMPs is that although it is involved in proteolytic activity on matrix proteins and fibrillar collagens, it also has an effect on nonmatrix proteins, such as angiotensin I. The deactivation of MMP-8 prevents the cleavage of angiotensin I to angiotensin II, thereby decreasing blood pressure by decreasing vasoconstriction and afterload. Moreover, there was a decrease in VCAM-1, a vascular adhesion molecule involved in the leukocyte rolling and adhesion to the vascular endothelium, which is such a destructive process in the inflammatory atherosclerotic process. The decrease in this VCAM-1 molecule promotes a decrease in atherosclerosis.14
What if MMP-2 and MMP-9 were decreased in expression by some substance? Montelukast has been found to do such a thing in hyperlipidemic rabbits.15 This study discovered that the addition of montelukast reduced neointimal formation, decreased macrophage accumulation, and increased the number of SMCs without altering plasma lipid levels. Moreover, another substance that actually alters lipid levels, a statin, may be of even greater use, since it has also been shown to decrease MMP-2 and MMP-9 levels. It is unknown, however, whether this decrease in MMP-2 and MMP-9 levels is due to the decrease in overall atherosclerotic inflammatory process from which the increase in MMP-2 and MMP-9 naturally follows, or whether this statin has some direct role in decreasing expression.16 More experiments are necessary to determine this; however, these current results are promising for patients. In contrast to montelukast, which only decreases MMP-2 and MMP-9, it was shown that the rosuvastatin molecule decreases plasma total cholesterol, triglycerides, LDL cholesterol, and oxidized LDL. Therefore, the effect of rosuvastatin appears to be beneficial.
Cilostazol is a substance that was used with THP-1 cells to inhibit phosphodiesterase (PDE) type 3. Since invasion of monocytes into subendothelial space is an important initiator of early atherosclerosis, inhibiting this inflammatory process was of particular interest. Expression of MMP-9 and RNA was markedly inhibited by cilostazol. This is another indication that MMP-9 may play a proatherogenic role under normal conditions, since the antiatherogenic substance cilostazol inhibits the expression of this metalloproteinase.17
Another study served to show that MMP-2 and MMP-9 were both correlated with plaque instability.18 There was an increased correlation between an increase of MMP-2 expression with cap rupture, intraplaque hemorrhage, and thin fibrous cap. Moreover, it was discovered there was a correlation between increased MMP-9 expression and cap rupture, as well as a large lipid core.18
It was discovered that serum MMP-9 and TIMP-1 levels correlated also with individual MMP-1 and TIMP-1 ratios in the peripheral circulating monocytes. This study was performed in live patients who had either unstable angina pectoris or non-ST elevation myocardial infarction (NSTEMI). Compared to control patients in addition to those with stable angina, the unstable angina and NSTEMI group had higher MMP-9 levels in serum. Moreover, TIMP-1 levels were several times lower in those with STEMI than those with stable angina, unstable angina, or NSTEMI. Another study actually concluded that MMP-1 serum levels were associated with the greatest total plaque burden and significant association with noncalcified plaques.19 The noncalcified plaques are the most unstable, since the calcium serves to stabilize the plaque complex, which consists of platelets, cholesterol, and SMCs.
It was shown that MMP-10 participates in a fibrinolytic role to combat infarcted thrombus in blood vessels.20 Studies have suggested that MMPs are involved by attacking the fibrinogen molecule so that fibrinogen cannot go on to form fibrin.21,22 It has been shown that the thrombin molecule increases expression of MMP-10, most likely secondary to protease-activated receptor-1 activation.23,24 It was shown that activation of MMP-10 can influence the thickness of the arterial wall.25 Fibrinolysis is traditionally performed by the infusion of plasminogen activators that convert plasminogen to plasmin, thereby activating the thrombolytic pathway, which degrades the fibrin molecule. One experiment showed that MMP-10 enhanced the effect of fibrinolysis by preventing thrombin-activatable fibrinolysis-inhibitor activation.20 Perhaps the administration of concentrated MMP-10 may serve as a modern aspirin in preventing plaque formation and platelet aggregation.
When MMP-13 is inhibited, there is an increase in collagen accumulation in plaques.26 This feature in humans results in a resistance to rupture of the plaque, since the collagen fibers act to stabilize the plaque that has developed. From this, we can infer that MMP-13 acts to degrade collagen fibers of plaques, which may destabilize the plaque.26 This destabilization makes it susceptible to rupture.
One study used an interesting mouse model of LDL-R−/− ApoB (100/100), which is a knockout for the LDL receptor, allowing LDL cholesterol to remain longer in the bloodstream and to be oxidized and taken up by vessels in the inflammatory process of atherosclerosis. This study also found an increase of expression of MMP-2 and MMP-9 in advanced atherosclerotic lesions with macrophage infiltration. Moreover, there was a downregulation of MMP-8 and MMP-13, as well as a large amount of collagen observed in the vessels of this mouse model.27 The large amount of collagen was attributed to the downregulation of MMP-8 and MMP-13, which is thought to otherwise degrade this collagen.
Molecular effects of cigarette smoke on vascular cells were the focus of investigation for one study. Cigarette smoke is known to be a source of reactive oxygen species. This study showed that MMP-1, a collagenase expressed in atherosclerosis and aneurysms, was induced in the aortic endothelium of rabbits that were exposed to cigarette smoke.28 Cigarette-smoke extract as well as acrolein inhibited the mammalian target of rapamycin/p70S6K pathway in human endothelial cells; this chemical inhibition resulted in an elevation of MMP-1.28
One very interesting study examined the effects of mainstream cigarette smoke and angiotensin II-induced hypertension using the well-known knockout of ApoE−/− mice. Angiotensin II was used for treatment for 4 weeks, and then mice were also exposed to mainstream cigarette smoke or fresh air for 4 weeks. The angiotensin II treatment resulted in higher expression of MMP-2, -3, -8, -9, and -12. The smoke exposure resulted in a minor increase in those MMPs of the aorta in the hypertensive mice. This lends further credence toward raising awareness of the dangers of cigarette smoke.
Vein-graft failure is thought to be largely attributed to the activation of MMPs combined with the plasminogen activator and plasmin-mediated proteolysis. This plays a role in matrix degeneration and SMC migration. This process is thought to occlude the graft and make it no longer clinically useful. A nonviral expression vector that encoded the hybrid protein of TIMP-1 that inhibits MMP was created. Moreover, the viral vector also contained the protein ATF receptor-binding amino-terminal fragment of urokinase-type plasminogen activator. The final protein in this hybrid was BPTI, a potent protease inhibitor. This hybrid transfection resulted in a statistically and clinically significant reduction in neointima formation after 4 weeks. Therefore, these results provide for an excellent means of preventing vein-graft disease. This has excellent promise for patients who have diabetes and are constantly undergoing arteriovenous fistula revision or patients with peripheral arterial disease or coronary artery disease who need venous bypass. The grafts will last longer, and the patients will have a better quality of life.29 The electroporation of TIMP-1/ATF has been shown to inhibit vein-graft thickening of vein grafts placed in the carotid artery of hypercholesterolemic mice.30
Results from our lab examined blood flow and how that interplays with the elastin:collagen ratio as well as the MMP:TIMP ratio to maintain healthy vascular structure and function.31 Arterial and venous pressures were measured in Sprague Dawley rats. The aortic arch, femoral and carotid arteries, and abdominal vena cava were isolated to determine the expression of MMP-2, MMP-9, MMP-12, MMP-13, TIMP-1, TIMP-3, and TIMP-4. The results showed that MMP-9 and MMP-13 were highest in the carotid artery when compared to the other blood vessels.
It was also shown that MMP-3 acts in part to promote SMC migration and neointima formation, since MMP-3-knockout mice demonstrated reduced neointima formation.32 In this same experiment, it was shown that the addition of exogenous MMP-3 restored the migration of VSMCs. Moreover, MMP-9 knockout or silencing via siRNA also significantly decreased the migratory pattern of VSMCs.
The antibiotic and zinc chelator tetracycline is known to inhibit MMPs. A study has shown that various doses of tetracycline can inhibit MMP activity; however, it is unknown what the clinical implications of this would be in patients taking tetracycline in the long-term.33 In fact, this may be something to follow-up with patients on long-term antibiotic therapy for treatment against Propionibacterium acnes, the Gram-negative bacteria that is often killed with an oral dose of tetracycline that patients take daily. It would be interesting to do a prospective study on these patients for future coronary events. A clinical test that has been devised to check for progression of acute coronary syndrome in diabetics is to determine active MMP-9 levels within the bloodstream.
One study investigated the association of serum MMPs with carotid artery characteristics.34 From this study, it was found that plasma MMP-1, -3, and -7 were significantly higher among the high intima media-thickness group compared with the ones in the low intima media-thickness group. Moreover, fibrous cap thickness and measures of lipid core were highest in those with elevated TIMP-1 levels. In spite of this, another prospective study investigated the serum MMP-9 levels of a large group of patients (almost 10,000); this study determined that MMP-9 is associated with a risk of myocardial infarction (MI) and stroke. However, it was found not to be a strong independent risk factor for either MI or stroke.35
MMP polymorphisms and susceptibility to disease
It is very interesting to note the polymorphisms in the MMP9 gene in relation to susceptibility to MI. The results of a study indicated that the −1562C>T polymorphism may be associated with increased susceptibility to MI. Another single-nucleotide polymorphism, R279Q, was not shown to be significantly associated with MI.36 This study is a reminder to us how individual genetic profiles when placed under certain physiological stresses, such as obesity or smoking, may result in more profound pathology than would otherwise be noted. This same vulnerability was determined with MMP-7, which may play an important role in the development of vulnerable carotid plaques.37 Another study confirmed that MMP-7 was positively associated with carotid calcification.34 Furthermore, a study investigated haplotypes of MMP-1 and MMP-12 toward prediction of future clinical events in patients with coronary artery disease.38 This study found a trend for predicting events and prognosis, but it did not reach statistical significance due to low sample size. More studies are necessary to establish this. However, we cannot discount the importance of this study.
Another study investigated the MMP-8 polymorphism −381A/G as well as −799C/T. Interestingly, it was found in female patients with carotid atherosclerosis that there was a higher degree of the −381G allele; moreover, there was increased expression of MMP-8 protein.39 One other study has examined the role of MMP-8, and discovered that there was an increased expression of MMP-8 enzyme in carotid plaques.40 It was however correlated with C-reactive protein, urea, and aspartate transaminase and creatinine levels.
Studies examining polymorphisms of MMP-3 showed that there were significant differences in those possessing the MMP-3 5A allele in patients with acute MI compared to controls. There were statistically significant differences in plasma levels in those carrying the 5A allele of MMP-3 compared to those with the 6A allele.41 Other investigators found that MMP-3 polymorphism affected the severity and risk of coronary artery stenosis, and also that −16125A/6A polymorphism and the 6A/6A MMP-3 genotype were genetic susceptibility factors for coronary artery stenosis. However, it was not determined whether this affected disease severity. It is known that genetic variation in MMP-promoter regions can alter the transcription of MMP genes, and has been associated with coronary heart disease. These sorts of studies are in their infancy, and an expansion into exploring the genetic implications of susceptibility and prognosis for those with certain gene profiles is necessary.
Homocysteine increases reactive oxygen species, thereby increasing MMPs
Previous experiments have conclusively shown that Hcy, a metabolite of cysteine and methionine amino acids, activates MMPs.42,43 This molecule has been shown to be an independent risk factor for types of heart disease, such as atherosclerosis and coronary artery disease. Hcy has been shown to utilize the ERK pathway. This is a protein found in great abundance within many types of cells that is activated via phosphorylation.42 Moreover, it was found that calcium, protein, and protein kinase C were necessary for this activation pathway.42 Another study in rats has shown that MMP activity as measured by zymography is increased with chronic Hcy administration; moreover, administering the coenzyme to the methyltetrahydrofolate reductase (MTHFR) enzyme ameliorates this activity. This is because MTHFR is involved in the conversion of Hcy back to methionine, which does not have the same signaling properties as the Hcy molecule.44
A similar conclusion was reached in a study with rabbits. This study examined neointima formation in hyperhomocysteinemic subjects. There has been a long association with restenosis after angioplasty in those subjects who are under high Hcy conditions. Therefore, using folate to alleviate this condition was a point of interest in order to measure the parameter of neointima thickness. Indeed, it was shown that neointimal thickness, neointimal area, and media area were all higher in hyperhomocysteinemic rabbits. In addition to this, MMP-9 messenger RNA (mRNA) levels were significantly higher, an enzyme known to be involved in the atherosclerotic remodeling process. Moreover, treatment with folate alleviated the thickness of the neointima.45
Aorta MMP-2, MMP-9, and connexin 43 expression were shown to be increased in high Hcy-level conditions of mice. Moreover, the following physiological parameters were measured and shown to be elevated: aortic blood pressure and resistance, pulse rate, wall thickness, and extracellular collagen accumulation in the aortic wall. Moreover, the S-adenosylhomocysteine hydrolase inhibitor was administered to cystathionine β-synthase (CBS)-knockout mice, resulting in a reduction of all these destructive physiological parameters. Since the enzyme S-adenosylmethionine is involved in converting methionine to Hcy, inhibiting this process results in less conversion to Hcy. Therefore, by isolating different metabolic steps in Hcy metabolism, we may either reduce or elevate Hcy and examine physiological effects from this.46
Methionine is known to increase Hcy concentration within living organisms, since this is an amino acid and that is a precursor to Hcy. Moreover, experimenters have discovered that ICAM-1 and VCAM-1 were upregulated under high-Hcy conditions.47 These are proteins that are attached to the endothelial cell of vessels that facilitate the entrance of cells into the inner architecture of vessels and into the body tissues. This is especially important and beneficial in the process of infectious disease, such that leukocytes may find their disease-causing targets. However, in the inflammatory process of atherosclerosis, these proteins that facilitate the entrance of leukocytes also facilitate the occlusion of vessels; more inflammatory cells and substrate accumulate.
Nitric oxide in MMP activation and vasorelaxation
The role of nitric oxide is still being investigated. The substrates oxygen and L-arginine are known to be used by endothelial nitric oxide synthetase (eNOS), neuronal NOS, and inducible NOS (iNOS) enzymes to produce nitric oxide, which potentiates guanylyl cyclase to convert guanosine triphosphate to cyclic guanosine monophosphate (cGMP). cGMP is a molecule that allows for vascular relaxation. PDE-5 inhibits vasorelaxation, as its activation converts cGMP to GMP and potentiates vasoconstriction.48 This is the mechanism that it is thought to be involved in the balance of vasorelaxation/constriction involving nitric oxide utilizing the aerobic pathway. This is illustrated in Figure 3.
Figure 3.

Mechanism of vasodilation.
Notes: H2S can be acquired via various conversion steps of Hcy in an anaerobic pathway. Hcy is converted to cystathionine by CBS and then to L-cysteine by the CSE enzyme. L-Cysteine can be converted to H2S, which is used by adenylyl cyclase to convert ATP to cAMP involved in vasorelaxation. The substrates oxygen and L-arginine are known to be used by eNOS, nNOS, and iNOS enzymes to produce nitric oxide, which potentiates guanylyl cyclase to convert GTP to cGMP. cGMP is a molecule that allows for vascular relaxation. PDE5 inhibits vasorelaxation, as its activation converts cGMP to GMP and potentiates vasoconstriction.
Abbreviations: Hcy, homocysteine; CBS, cystathionine β-synthase; CSE, cystathionine γ-lyase; ATP, adenosine triphosphate; cAMP, cyclic adenosine monophosphate; eNOS, endothelial nitric oxide synthetase; nNOS, neuronal NOS; iNOS, inducible NOS; GTP, guanosine triphosphate; cGMP, cyclic guanosine monophosphate; PDE, phosphodiesterase; ADMA, asymmetric dimethylarginine; DDAH, dimethylarginine dimethylaminohydrolase; MST, 3-mercaptopyruvate sulfurtransferase; GC, guanylyl cyclase; AC, adenylyl cyclase.
In one study investigating the effect of eNOS, resveratrol was administered to mice with carotid or aortic injury.49 Resveratrol was shown to decrease neointimal formation after injury of these vessels, but its effects were not realized after administration of the NO inhibitor N-nitro-L-arginine methyl ester (L-NAME). Moreover, its effects were not realized in eNOS knockout mice. Therefore, this shows a role that nitric oxide has in promoting pathological repair of damaged vessels.
In a study conducted on kidney nephron units, the human mesangial cell line was treated with different concentrations of cytokines and NO inhibitors. MMP-9 was examined by gelatin zymography along with TIMP-1 via Western blot. The stimulation of cells via TNFα or IL-1β led to a large increase in NO production that was attributed to increased expression of the iNOS enzyme.50 Treating cells with the iNOS inhibitor N6-(1-iminoethyl)-lysine hydrochloride resulted in an increase of MMP-9 production that was induced by TNFα. Moreover, NO donor sodium nitroprusside stimulated an increase in NO production in HMCL cells, which had an association with decreases in both basal levels of MMP-9 and in TNFα-stimulated levels of MMP-9.50
In another recent study from our lab, we found that in vitro, sodium nitroprusside, an NO donor, actually activated MMPs, as it likely combines with superoxide to form peroxynitrite, which cleaves latent MMP into active MMP. Therefore, without nitric oxide being used by various other enzymes in the harmony of a living organism, it can potentiate harmful effects. This was reversed using H2S, which is known to scavenge reactive oxygen species, thereby deactivating MMPs.51
One study examined the use of angiotensin-converting enzyme inhibitors in relation to alteration of expression of MMP-2 and TIMP-2 in human cardiac fibroblasts in the presence of IL-1β.52 This study showed that IL-1β increased MMP-2 activity and transcription as well as nitric oxide production from the supernatant of the culture medium. Moreover, these effects were inhibited by imidaprilat and the NO synthase inhibitor NG-methyl-L-arginine acetate salt. Therefore, a decrease in NO followed, with a decrease in MMP-2 activity. From this, one might conclude that nitric oxide in this case is harmful, since an increase in NO may result in an increase in MMP-2 activity.
NO is known to modify the activity of enzymes either directly on the physical enzyme itself or via some NO-dependent metabolic pathway. One study investigated the role of NO in the expression of MMPs within rat primary astrocytes.53 Inhibition of NO production using the nitric oxide synthase inhibitor L-NAME increased MMP-9 even further. The authors concluded from this that NO inhibits MMP-9 expression in vivo.53 Moreover, the authors even added exogenous NO donors sodium nitroprusside or S-nitroso-N-acetylpenicillamine, which showed an inhibition of MMP-9 expression in astrocytes. The authors also showed that the inhibitory effect of NO was mediated by downregulation of mRNA and protein levels of MMP-9, not by direct modification of enzymatic activity of MMP-9. In fact, these in vivo effects were thought to be mediated via the ERK½ pathway.53 This is consistent with other studies that have found utilization of the ERK pathway.42
Another study analyzed the effect of continuous treatment of an iNOS inhibitor as well as superoxide dismutase on MMP-9 activity.54 This study showed that the NO inhibitor reduced the MMP-9 enzyme and activity. This is in contrast to the beneficial effects NO is thought to exude. Moreover, this decrease in MMP-9 activity and protein level was associated with improvement of endothelium-dependent and independent coronary vasomotor function in the cardiac allografts.
Another study investigated the role of the NOS inhibitors aminoguanidine and baicalein, which resulted in a decrease in MMP production. Specifically, MMP-1 and MMP-2 production was reduced to approximately 40% of basal levels when treated with small micromolar amounts of baicalein in the culture media of ultraviolet-irradiated human dermal fibroblasts.55 Again, this makes us question what role NO has in activating or deactivating MMPs.
Hydrogen sulfide MMP-activation atherosclerosis
Hydrogen sulfide is both a vasodilator and antioxidant that leads to a decrease in MMP activity.56 H2S can be acquired via various conversion steps of Hcy in an anaerobic pathway. Hcy is converted to cystathionine by CBS, and then to L-cysteine by the cystathionine γ-lyase (CSE) enzyme. L-Cysteine can be converted to H2S, which is used by adenylyl cyclase to convert adenosine triphosphate to the cyclic adenosine monophosphate involved in vasorelaxation. This is illustrated in the schematic in Figure 3.
One pivotal study conducted in our lab investigated the effects of vascular damage on MMP expression and activity.57 Carotid artery injury was created by inserting a polyethylene-10 catheter and rotating it several times before removing and sealing the entry site. The following groups were used: control, damaged artery, control + H2S, and damage + H2S. Our results showed a large increase in p47, nitrotyrosine, MMP-9, MMP-12, and TIMP-3. Moreover, there was a decrease in superoxide dismutase 1 and MMP-2 levels in injured arteries. Treatment with H2S mitigated the vascular remodeling by normalizing the levels of redox stress and normalizing MMPs and TIMPs.
Moreover, H2S treatment decreased the activity of MMPs in cerebrovascular cells. Hydrogen sulfide is a metabolite of Hcy that is produced via the following enzymes, some of which have already been discussed: CBS, CSE, and 3-mercaptopyruvate sulfurtransferase. Using triple-gene therapy and increasing these enzymes, our lab showed a greater generation of H2S in the presence of Hcy; this was conducted using a plasmid that contained all three genes.56 The upregulation of MMP-13 and downregulation of TIMP-1 were normalized by overexpression of the three genes in the triple-gene therapy, thereby inhibiting the destructive remodeling effects.
One study investigated the CSE/H2S pathway, whereby Hcy is converted by CSE to H2S, in order to determine its role on neointimal formation and atherosclerosis.58 The SMCs and aortic explants from CSE-knockout mice showed more migration and outgrowth compared to that from wild-type mice. Moreover, exogenous sodium hydrosulfide H2S donor at concentrations of 100 μM resulted in significant attenuation of SMC migration, resulting in less vessel narrowing. Moreover, there was an increase in neointima formation in the carotid artery 4 weeks after ligation that was prevented by H2S donor.58 H2S establishes itself as a potential molecule that can be used in the clinical setting to help patients with atherosclerosis.
Another study from our lab showed that H2S decreased the expression of MMP-9 activation while inducing the expression of MMP-2 in a pressure-overload aortic-banding experiment in mice. The aortic band increases afterload on the heart that creates hypertrophy, subsequent fibrosis, and eventual heart failure as the pump becomes less efficient at pumping blood. The results showed that H2S was able to induce MMP-2 and promote VEGF synthesis and angiogenesis. Moreover, it suppressed MMP-9 and TIMP-3 levels, thereby inhibiting antiangiogenic factors and also inhibiting the process of fibrosis and heart-pump destruction that results from increased fibrosis created by activated MMP-9 enzymes.59
One experiment used a hypoxic pulmonary artery treated with H2S to study effects on various parameters. It was found that H2S was a beneficial molecule that reduced the expression of collagen I and III, elastin, TGFβ3 protein, and procollagen I and III mRNA.60
Conclusion
Hcy is known to promote oxidative stress and to act as a signaling molecule to activate MMPs that are involved in pathological remodeling. Nitric oxide, in some cases, has been shown to be beneficial in alleviating atherosclerosis. Moreover, H2S is of special promise in deactivating MMPs and reducing the atherosclerotic burden. There are over 20 MMPs involved in remodeling of vessels, and their roles in doing that have been explored via several gene-knockout and -silencing models. Exogenous administration of MMP-10 may actually be beneficial to vessels occluded with plaque. Genetic predisposition to overactivated MMPs is being explored by determining various polymorphism variations in the population and examining susceptibility to disease.
Acknowledgment
Part of this study was supported by NIH grants HL-71010; HL-74185; HL-88012, and NS-51568.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Urso C, Caimi G. Oxidative stress and endothelial dysfunction. Minerva Med. 2011;102(1):59–77. Italian. [PubMed] [Google Scholar]
- 2.Van Assche T, Hendrickx J, Crauwels HM, et al. Transcription profiles of aortic smooth muscle cells from atherosclerosis-prone and - resistant regions in young apolipoprotein E-deficient mice before plaque development. J Vasc Res. 2011;48(1):31–42. doi: 10.1159/000317398. [DOI] [PubMed] [Google Scholar]
- 3.Filipovic N, Rosic M, Tanaskovic I, Parodi O, Fotiadis D. Computer simulation and experimental analysis of LDL transport in the arteries. Conf Proc IEEE Eng Med Biol Soc. 2011;2011:195–198. doi: 10.1109/IEMBS.2011.6090031. [DOI] [PubMed] [Google Scholar]
- 4.Yang H, Zhang L, Xiong X, Liu Y. Specific adhesion and accumulation of VCAM-1-targeted ultrasound microbubbles to inflammatory endothelial cells under hemodynamic shear flow simulation. J Control Release. 2011;152(Suppl 1):e227–e229. doi: 10.1016/j.jconrel.2011.09.027. [DOI] [PubMed] [Google Scholar]
- 5.Zhang X, Liu X, Shang H, Xu Y, Qian M. Monocyte chemoattractant protein-1 induces endothelial cell apoptosis in vitro through a p53-dependent mitochondrial pathway. Acta Biochim Biophys Sin (Shanghai) 2011;43(10):787–795. doi: 10.1093/abbs/gmr072. [DOI] [PubMed] [Google Scholar]
- 6.Xiao N, Yin M, Zhang L, et al. Tumor necrosis factor-alpha deficiency retards early fatty-streak lesion by influencing the expression of inflammatory factors in apoE-null mice. Mol Genet Metab. 2009;96(4):239–244. doi: 10.1016/j.ymgme.2008.11.166. [DOI] [PubMed] [Google Scholar]
- 7.Razavian M, Tavakoli S, Zhang JS, et al. Atherosclerosis plaque heterogeneity and response to therapy detected by in vivo molecular imaging of matrix metalloproteinase activation. J Nucl Med. 2011;52(11):1795–1802. doi: 10.2967/jnumed.111.092379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stary HC, Chandler AB, Dinsmore RE, et al. A definition of advanced types of atherosclerotic lesions and a histological classification of atherosclerosis. A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Arterioscler Thromb Vasc Biol. 1995;15(9):1512–1531. doi: 10.1161/01.atv.15.9.1512. [DOI] [PubMed] [Google Scholar]
- 9.Kim D, Lee D, Jang YL, et al. Facial amphipathic deoxycholic acid- modified polyethyleneimine for efficient MMP-2 siRNA delivery in vascular smooth muscle cells. Eur J Pharm Biopharm. 2012;81(1):14–23. doi: 10.1016/j.ejpb.2012.01.013. [DOI] [PubMed] [Google Scholar]
- 10.Kaneko H, Anzai T, Horiuchi K, et al. Tumor necrosis factor-alpha converting enzyme is a key mediator of abdominal aortic aneurysm development. Atherosclerosis. 2011;218(2):470–478. doi: 10.1016/j.atherosclerosis.2011.06.008. [DOI] [PubMed] [Google Scholar]
- 11.Kuzuya M, Nakamura K, Sasaki T, Cheng XW, Itohara S, Iguchi A. Effect of MMP-2 deficiency on atherosclerotic lesion formation in ApoE-deficient mice. Arterioscl Thromb Vasc Biol. 2006;26(5):1120–1125. doi: 10.1161/01.ATV.0000218496.60097.e0. [DOI] [PubMed] [Google Scholar]
- 12.Johnson JL, George SJ, Newby AC, Jackson CL. Divergent effects of matrix metalloproteinases 3, 7, 9, and 12 on atherosclerotic plaque stability in mouse brachiocephalic arteries. Proc Natl Acad Sci U S A. 2005;102(43):15575–15580. doi: 10.1073/pnas.0506201102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thomas AC, Newby AC. Effect of matrix metalloproteinase-9 knockout on vein graft remodelling in mice. J Vasc Res. 2010;47(4):299–308. doi: 10.1159/000265564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Laxton RC, Hu Y, Duchene J, et al. A role of matrix metalloproteinase-8 in atherosclerosis. Circ Res. 2009;105(9):921–929. doi: 10.1161/CIRCRESAHA.109.200279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu D, Ge S, Zhou G, et al. Montelukast inhibits matrix metalloproteinases expression in atherosclerotic rabbits. Cardiovasc Drugs Ther. 2009;23(6):431–437. doi: 10.1007/s10557-009-6211-6. [DOI] [PubMed] [Google Scholar]
- 16.Guo H, Shi Y, Liu L, Sun A, Xu F, Chi J. Rosuvastatin inhibits MMP-2 expression and limits the progression of atherosclerosis in LDLR-deficient mice. Arch Med Res. 2009;40(5):345–351. doi: 10.1016/j.arcmed.2009.07.006. [DOI] [PubMed] [Google Scholar]
- 17.Chuang SY, Yang SH, Chen TY, Pang JH. Cilostazol inhibits matrix invasion and modulates the gene expressions of MMP-9 and TIMP-1 in PMA-differentiated THP-1 cells. Eur J Pharmacol. 2011;670(2–3):419–426. doi: 10.1016/j.ejphar.2011.08.040. [DOI] [PubMed] [Google Scholar]
- 18.Heo SH, Cho CH, Kim HO, et al. Plaque rupture is a determinant of vascular events in carotid artery atherosclerotic disease: involvement of matrix metalloproteinases 2 and 9. J Clin Neurol. 2011;7(2):69–76. doi: 10.3988/jcn.2011.7.2.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lehrke M, Greif M, Broedl UC, et al. MMP-1 serum levels predict coronary atherosclerosis in humans. Cardiovasc Diabetol. 2009;8:50. doi: 10.1186/1475-2840-8-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Orbe J, Barrenetxe J, Rodriguez JA, et al. Matrix metalloproteinase-10 effectively reduces infarct size in experimental stroke by enhancing fibrinolysis via a thrombin-activatable fibrinolysis inhibitor-mediated mechanism. Circulation. 2011;124(25):2909–2919. doi: 10.1161/CIRCULATIONAHA.111.047100. [DOI] [PubMed] [Google Scholar]
- 21.Bini A, Wu D, Schnuer J, Kudryk BJ. Characterization of stromelysin 1 (MMP-3), matrilysin (MMP-7), and membrane type 1 matrix metalloproteinase (MT1-MMP) derived fibrin(ogen) fragments D-dimer and D-like monomer: NH2-terminal sequences of late-stage digest fragments. Biochemistry. 1999;38(42):13928–13936. doi: 10.1021/bi991096g. [DOI] [PubMed] [Google Scholar]
- 22.Hiller O, Lichte A, Oberpichler A, Kocourek A, Tschesche H. Matrix metalloproteinases collagenase-2, macrophage elastase, collagenase-3, and membrane type 1-matrix metalloproteinase impair clotting by degradation of fibrinogen and factor XII. J Biol Chem. 2000;275(42):33008–33013. doi: 10.1074/jbc.M001836200. [DOI] [PubMed] [Google Scholar]
- 23.Orbe J, Rodríguez JA, Calvayrac O, et al. Matrix metalloproteinase-10 is upregulated by thrombin in endothelial cells and increased in patients with enhanced thrombin generation. Arterioscler Thromb Vasc Biol. 2009;29(12):2109–2116. doi: 10.1161/ATVBAHA.109.194589. [DOI] [PubMed] [Google Scholar]
- 24.Lorente L, Martín MM, Labarta L, et al. Matrix metalloproteinase-9, -10, and tissue inhibitor of matrix metalloproteinases-1 blood levels as biomarkers of severity and mortality in sepsis. Crit Care. 2009;13(5):R158. doi: 10.1186/cc8115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Orbe J, Montero I, Rodríguez JA, Beloqui O, Roncal C, Páramo JA. Independent association of matrix metalloproteinase-10, cardiovascular risk factors and subclinical atherosclerosis. J Thromb Haemost. 2007;5(1):91–97. doi: 10.1111/j.1538-7836.2006.02276.x. [DOI] [PubMed] [Google Scholar]
- 26.Quillard T, Tesmenitsky Y, Croce K, et al. Selective inhibition of matrix metalloproteinase-13 increases collagen content of established mouse atherosclerosis. Arterioscler Thromb Vasc Biol. 2011;31(11):2464–2472. doi: 10.1161/ATVBAHA.111.231563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wågsäter D, Zhu CY, Björkegren J, Skogsberg J, Eriksson P. MMP-2 and MMP-9 are prominent matrix metalloproteinases during atherosclerosis development in the LdlrApob100/100 mouse. Int J Mol Med. 2011;28(2):247–253. doi: 10.3892/ijmm.2011.693. [DOI] [PubMed] [Google Scholar]
- 28.Lemaître V, Dabo AJ, D’Armiento J. Cigarette smoke components induce matrix metalloproteinase-1 in aortic endothelial cells through inhibition of mTOR signaling. Toxicol Sci. 2011;123(2):542–549. doi: 10.1093/toxsci/kfr181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Eefting D, Seghers L, Grimbergen JM, et al. A novel urokinase receptor-targeted inhibitor for plasmin and matrix metalloproteinases suppresses vein graft disease. Cardiovasc Res. 2010;88(2):367–375. doi: 10.1093/cvr/cvq203. [DOI] [PubMed] [Google Scholar]
- 30.Eefting D, de Vries MR, Grimbergen JM, Karper JC, van Bockel JH, Quax PH. In vivo suppression of vein graft disease by nonviral, electroporation-mediated, gene transfer of tissue inhibitor of metalloproteinase-1 linked to the amino terminal fragment of urokinase (TIMP-1. ATF), a cell-surface directed matrix metalloproteinase inhibitor. J Vasc Surg. 2010;51(2):429–437. doi: 10.1016/j.jvs.2009.09.026. [DOI] [PubMed] [Google Scholar]
- 31.Basu P, Sen U, Tyagi N, Tyagi SC. Blood flow interplays with elastin: collagen and MMP:TIMP ratios to maintain healthy vascular structure and function. Vasc Health Risk Manag. 2010;6:215–228. doi: 10.2147/vhrm.s9472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Johnson JL, Dwivedi A, Somerville M, George SJ, Newby AC. Matrix metalloproteinase (MMP)-3 activates MMP-9 mediated vascular smooth muscle cell migration and neointima formation in mice. Arterioscler Thromb Vasc Biol. 2011;31(9):e35–e44. doi: 10.1161/ATVBAHA.111.225623. [DOI] [PubMed] [Google Scholar]
- 33.Bench TJ, Jeremias A, Brown DL. Matrix metalloproteinase inhibition with tetracyclines for the treatment of coronary artery disease. Pharmacol Res. 2011;64(6):561–566. doi: 10.1016/j.phrs.2011.05.002. [DOI] [PubMed] [Google Scholar]
- 34.Gaubatz JW, Ballantyne CM, Wasserman BA, et al. Association of circulating matrix metalloproteinases with carotid artery characteristics: the Atherosclerosis Risk in Communities Carotid MRI Study. Arterioscler Thromb Vasc Biol. 2010;30(5):1034–1042. doi: 10.1161/ATVBAHA.109.195370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jefferis BJ, Whincup P, Welsh P, et al. Prospective study of matrix metalloproteinase-9 and risk of myocardial infarction and stroke in older men and women. Atherosclerosis. 2010;208(2):557–563. doi: 10.1016/j.atherosclerosis.2009.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang L, Ma YT, Xie X, et al. Interaction between MMP-9 gene polymorphisms and smoking in relation to myocardial infarction in a Uighur population. Clin Appl Thromb Hemost. 2012;18(1):72–78. doi: 10.1177/1076029611412365. [DOI] [PubMed] [Google Scholar]
- 37.Hu XF, Jin XP, Hu PY, et al. Association of a functional polymorphism in the MMP7 gene promoter with susceptibility to vulnerable carotid plaque in a Han Chinese population. Clin Chem Lab Med. 2011;49(10):1735–1741. doi: 10.1515/CCLM.2011.241. [DOI] [PubMed] [Google Scholar]
- 38.Jguirim-Souissi I, Jelassi A, Slimani A, et al. Matrix metalloproteinase-1 and matrix metalloproteinase-12 gene polymorphisms and the outcome of coronary artery disease. Coron Artery Dis. 2011;22(6):388–393. doi: 10.1097/MCA.0b013e3283478d40. [DOI] [PubMed] [Google Scholar]
- 39.Djurić T1, Stanković A, Končar I, et al. Association of MMP-8 promoter gene polymorphisms with carotid atherosclerosis: preliminary study. Atherosclerosis. 2011;219(2):673–678. doi: 10.1016/j.atherosclerosis.2011.08.025. [DOI] [PubMed] [Google Scholar]
- 40.Djurić T, Zivković M, Stanković A, et al. Plasma levels of matrix metalloproteinase-8 in patients with carotid atherosclerosis. J Clin Lab Anal. 2010;24(4):246–251. doi: 10.1002/jcla.20393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ghaderian SM, Najar RA, Panah AS. Genetic polymorphisms and plasma levels of matrix metalloproteinases and their relationships with developing acute myocardial infarction. Coron Artery Dis. 2010;21(6):330–335. doi: 10.1097/MCA.0b013e32833ce065. [DOI] [PubMed] [Google Scholar]
- 42.Moshal KS, Sen U, Tyagi N, et al. Regulation of homocysteine-induced MMP-9 by ERK1/2 pathway. Am J Physiol Cell Physiol. 2006;290(3):C883–C891. doi: 10.1152/ajpcell.00359.2005. [DOI] [PubMed] [Google Scholar]
- 43.Bescond A, Augier T, Chareyre C, Garçon D, Hornebeck W, Charpiot P. Influence of homocysteine on matrix metalloproteinase-2: activation and activity. Biochem Biophys Res Commun. 1999;263(2):498–503. doi: 10.1006/bbrc.1999.1391. [DOI] [PubMed] [Google Scholar]
- 44.Miller A, Mujumdar V, Palmer L, Bower JD, Tyagi SC. Reversal of endocardial endothelial dysfunction by folic acid in homocysteinemic hypertensive rats. Am J Hypertens. 2002;15(2 Pt 1):157–163. doi: 10.1016/s0895-7061(01)02286-5. [DOI] [PubMed] [Google Scholar]
- 45.Liu F, Zhang J, Yu S, et al. Effect of folate on neointima formation and matrix metalloproteinase-9 expression after balloon injury in hyperhomocysteinemic rabbits. Int J Cardiol. 2008;131(1):59–65. doi: 10.1016/j.ijcard.2007.08.138. [DOI] [PubMed] [Google Scholar]
- 46.Ovechkin AV, Tyagi N, Sen U, et al. 3-Deazaadenosine mitigates arterial remodeling and hypertension in hyperhomocysteinemic mice. Am J Physiol Lung Cell Mol Physiol. 2006;291(5):L905–L911. doi: 10.1152/ajplung.00543.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sen U, Tyagi N, Kumar M, Moshal KS, Rodriguez WE, Tyagi SC. Cystathionine-beta-synthase gene transfer and 3-deazaadenosine ameliorate inflammatory response in endothelial cells. Am J Physiol Cell Physiol. 2007;293(6):C1779–C1787. doi: 10.1152/ajpcell.00207.2007. [DOI] [PubMed] [Google Scholar]
- 48.Rybalkin SD, Yan C, Bornfeldt KE, Beavo JA. Cyclic GMP phosphodiesterases and regulation of smooth muscle function. Circ Res. 2003;93(4):280–291. doi: 10.1161/01.RES.0000087541.15600.2B. [DOI] [PubMed] [Google Scholar]
- 49.Breen DM, Dolinsky VW, Zhang H, et al. Resveratrol inhibits neointimal formation after arterial injury through an endothelial nitric oxide synthase-dependent mechanism. Atherosclerosis. 2012;222(2):375–381. doi: 10.1016/j.atherosclerosis.2012.03.021. [DOI] [PubMed] [Google Scholar]
- 50.Nee L, O’Connell S, Nolan S, Ryan MP, McMorrow T. Nitric oxide involvement in TNF-alpha and IL-1 beta-mediated changes in human mesangial cell MMP-9 and TIMP-1. Nephron Exp Nephrol. 2008;110(2):e59–e66. doi: 10.1159/000158524. [DOI] [PubMed] [Google Scholar]
- 51.Vacek TP, Qipshidze N, Tyagi SC. Hydrogen sulfide and sodium nitroprusside compete to activate/deactivate MMPs in bone tissue homogenates. Vasc Health Risk Manag. 2013;9:117–123. doi: 10.2147/VHRM.S39508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Guo XG, Uzui H, Mizuguchi T, Ueda T, Chen JZ, Lee JD. Imidaprilat inhibits matrix metalloproteinase-2 activity in human cardiac fibroblasts induced by interleukin-1β via NO-dependent pathway. Int J Cardiol. 2008;126(3):414–420. doi: 10.1016/j.ijcard.2007.08.134. [DOI] [PubMed] [Google Scholar]
- 53.Shin CY, Lee WJ, Choi JW, et al. Down-regulation of matrix metalloproteinase-9 expression by nitric oxide in lipopolysaccharide-stimulated rat primary astrocytes. Nitric Oxide. 2007;16(4):425–432. doi: 10.1016/j.niox.2007.03.004. [DOI] [PubMed] [Google Scholar]
- 54.Egi K, Conrad NE, Kwan J, Schulze C, Schulz R, Wildhirt SM. Inhibition of inducible nitric oxide synthase and superoxide production reduces matrix metalloproteinase-9 activity and restores coronary vasomotor function in rat cardiac allografts. Eur J Cardiothorac Surg. 2004;26(2):262–269. doi: 10.1016/j.ejcts.2004.04.037. [DOI] [PubMed] [Google Scholar]
- 55.Choe TB, Lee BC, Park IC. Inhibition of matrix metalloproteinase-1 and -2 expression using nitric oxide synthase inhibitors in UV-irradiated human dermal fibroblasts. J Cosmet Sci. 2003;54(3):229–238. [PubMed] [Google Scholar]
- 56.Sen U, Sathnur PB, Kundu S, et al. Increased endogenous H2S generation by CBS, CSE, and 3MST gene therapy improves ex vivo renovascular relaxation in hyperhomocysteinemia. Am J Physiol Cell Physiol. 2012;303(1):C41–C51. doi: 10.1152/ajpcell.00398.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vacek TP, Gillespie W, Tyagi N, Vacek JC, Tyagi SC. Hydrogen sulfide protects against vascular remodeling from endothelial damage. Amino Acids. 2010;39(5):1161–1169. doi: 10.1007/s00726-010-0550-2. [DOI] [PubMed] [Google Scholar]
- 58.Yang G, Li H, Tang G, et al. Increased neointimal formation in cystathionine gamma-lyase deficient mice: role of hydrogen sulfide in α5β11-integrin and matrix metalloproteinase-2 expression in smooth muscle cells. J Mol Cell Cardiol. 2012;52(3):677–688. doi: 10.1016/j.yjmcc.2011.12.004. [DOI] [PubMed] [Google Scholar]
- 59.Givvimani S, Munjal C, Gargoum R, et al. Hydrogen sulfide mitigates transition from compensatory hypertrophy to heart failure. J Appl Physiol. 2011;110(4):1093–1100. doi: 10.1152/japplphysiol.01064.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hongfang J, Cong B, Zhao B, et al. Effects of hydrogen sulfide on hypoxic pulmonary vascular structural remodeling. Life Sci. 2006;78(12):1299–1309. doi: 10.1016/j.lfs.2005.07.009. [DOI] [PubMed] [Google Scholar]