

Abstract
Staphylococcal enterotoxin B (SEB) is a major virulence factor for staphylococcal toxic shock syndrome (TSS). SEB activates a large subset of the T lymphocytic population, releasing proinflammatory cytokines. Blocking SEB-initiated toxicity may be an effective strategy for treating TSS. Using a process known as systematic evolution of ligands by exponential enrichment (SELEX), we identified an aptamer that can antagonize SEB with nanomolar binding affinity (Kd = 64 nM). The aptamer antagonist effectively inhibits SEB-mediated proliferation and cytokine secretion in human peripheral blood mononuclear cells. Moreover, a PEGylated aptamer antagonist significantly reduced mortality in a “double-hit” mouse model of SEB-induced TSS, established via sensitization with d-galactosamine followed by SEB challenge. Therefore, our novel aptamer antagonist may offer potential therapeutic efficacy against SEB-mediated TSS.
INTRODUCTION
Staphylococcal enterotoxins (SEs) are prototype superantigens excreted primarily by Staphylococcus aureus and Streptococcus pyogenes. SEs are potent activators of the immune system that can stimulate up to 40% of the naive T cell population and cause diverse human diseases, including food poisoning, autoimmune diseases, and toxic shock syndrome (TSS) (1). SEs can activate both monocytes/macrophages and T lymphocytes by directly binding to major histocompatibility complex class II (MHC II) molecules and to the variable region of the β-chain of T-cell receptors (TCR), without being taken up and presented by antigen-presenting cells (2).
These toxins lead to the proliferation of monocytes/macrophages and T lymphocytes and stimulate secretion of excess proinflammatory cytokines, including interferon gamma (IFN-γ), interleukin 2 (IL-2), and tumor necrosis factor alpha (TNF-α). Among these SEs, staphylococcal enterotoxin B (SEB) is considered an important cause of staphylococcal TSS, which is characterized by high fever, erythematous rash, hypotension, multiorgan system failure, and even death (3). Remarkably, SEB is highly heat resistant, easily transformed into an aerosol, and readily produced on a large scale using genetic engineering techniques. Humans are extremely susceptible to this superantigen, so SEB is often referred to as an “incapacitating agent” by the military, which may use this toxin as an antipersonnel aerosol or as a food supply contaminant (4).
At present, SEB is thought to be an effective treatment strategy for TSS by blocking early processes prior to T cell activation and initiation of the toxic cascade. Previous studies have suggested multiple approaches for interfering with SEB binding to MHC II and/or TCR. These include vaccine immunization to induce anti-SEB antibodies (5, 6), passive immunoprophylaxis with natural or artificial antibodies (7, 8), and the use of synthetic peptides (9) or receptor mimics (10, 11) as antagonists to prevent the formation of the MHC II–SEB–TCR complex. Research to identify new compounds with greater half-lives, improved SEB binding affinity, and less immunogenicity is ongoing.
Aptamers are artificial short, single-stranded oligonucleotides (ssDNA or ssRNA) that can form elaborate three-dimensional structures and specifically bind to desired targets with high affinity (12, 13). Because of their intrinsic properties, aptamers are nontoxic and nonimmunogenic. In addition, aptamers composed of modified nucleotides have a long in vivo half-life of hours to days.
During the past 2 decades, numerous aptamers have been developed to inhibit pathogenic antigen activity and many have substantial clinical potential for treating various diseases (14). Several researchers have reported on the selection of specific DNA aptamers against SEB (15–17). Methods using these aptamers as capture molecules have also been established to detect SEB in various samples (15, 18). However, as far as we know, no aptamer has been selected to inhibit the biological activity of SEB.
Therefore, we selected DNA aptamers directed against native SEB in vitro and identified aptamer antagonists capable of neutralizing SEBs by studying their therapeutic effects on SEB-mediated toxic shock (TSS) in vitro using human peripheral blood mononuclear cell (PBMC) cultures as well as a lethal murine model.
MATERIALS AND METHODS
SELEX (systematic evolution of ligands by exponential enrichment) library and primers.
The ssDNA library, which contained random 60-mer and fixed primer regions and all primers used, was described previously (see Table S1 in the supplemental material) (19). All oligonucleotides were synthesized and purified by high-performance liquid chromatography (HPLC) (Invitrogen, Guangzhou, China). For animal experiments, the aptamer candidate was conjugated at the 5′ end with 40-kDa polyethylene glycol (PEG). The PEGylated aptamer was prepared by treating 40-kDa PEG N-hydroxysuccinimide ester with 5′-amino and 3′-inverted dT modified aptamer candidates. All PEG-aptamer conjugates were purified by anion exchange and then by reverse-phase HPLC (Seebio Biotech, Shanghai, China).
Aptamer screening for specific SEB binding.
ssDNA aptamers specific to SEB were screened by a SELEX process as described previously, with minor modifications (20). Briefly, highly purified SEB (Toxin Technology, Sarasota, FL, USA) was coupled to M-270 carboxylic acid Dynabeads (DYNAL, Oslo, Norway) according to the manufacturer's instructions. Prior to selection, the ssDNA library was dissolved in 500 μl selection buffer (50 mM Tris HCl [pH 7.4], 5 mM KCl, 100 mM NaCl, 1 mM MgCl2), denatured by heating at 95°C for 5 min, and immediately placed on ice for 10 min. The denatured ssDNA library and a 5-fold molar excess of yeast tRNA (Invitrogen) were incubated with SEB (100 ng) immobilized on magnetic beads in a 500-μl reaction mixture and incubated at 37°C for 60 min with shaking.
Nonbinding oligonucleotides were removed by washing the DNA-bead complex three times with selection buffer (0.2% bovine serum albumin [BSA]). SEB binding species were eluted by heating at 100°C for 5 min in 200 μl of sterile double-distilled water (ddH2O). ssDNA was amplified by PCR with fluorescein isothiocyanate (FITC)-labeled sense primers and biotin-labeled antisense primers (20 to 30 cycles of 0.5 min at 94°C, 0.5 min at 52°C, and 0.5 min at 72°C, followed by 5 min at 72°C). All PCR products were purified using a Biospin gel extraction kit (Bioer Technology, Hangzhou, China). From all purified PCR products from each SELEX round, large aliquots were alkaline denatured and used to prepare the aptamer strand using M-280 streptavidin Dynabeads (Dynal AS, Oslo, Norway) according to the manufacturer's instructions, and small aliquots were stored at −80°C prior to use.
To create aptamers with high affinity and specificity, we progressively increased the selective pressure by increasing the number of washes and KCl and by decreasing the ratio of ssDNA to SEB and the incubation time (see Table S2 in the supplemental material). The counterselection (depletion of aptamers that bind to nontarget molecules) was introduced against BSA-coupled or staphylococcal protein A (SPA)-coupled M-270 beads in the 7th, 9th, and 11th rounds. BSA was the blocking protein for SEB coupling, and SPA is a major protein of Staphylococcus aureus (21). To reduce the matrix binders, reaction tubes were blocked with 1% BSA–phosphate-buffered saline (PBS), and preselection steps with uncoupled streptavidin beads were performed.
Cloning, sequencing, and bioinformatic analysis.
After rounds of SELEX selection, the selected ssDNA pool was PCR amplified using unlabeled primers under the conditions described for screening. PCR products were purified and then cloned into pGEM-T vector (Promega, Madison, WI, USA) according to the manufacturer's instructions. The resulting products were transformed into Escherichia coli DH5α. Individual cultured colonies were chosen randomly, and their inserts were sequenced by Invitrogen Company. The aptamer sequences were analyzed by ClustalX software, and the secondary structure was predicted by a free-energy minimization algorithm according to Zuker (22) using the internet tool Mfold (http://mfold.rna.albany.edu/?q=mfold).
Aptamer binding assay for SEB.
A binding assay with fluorescently labeled ssDNA was performed to monitor the enrichment of each SELEX round and to evaluate aptamer binding affinity. In brief, fluorescently labeled ssDNA was thermally denatured in 200 μl of selection buffer and then incubated in the dark with SEB immobilized on magnetic beads at 37°C for 1 h with shaking. After incubation, nonbound ssDNA was collected, and SEB-bound ssDNA was eluted by heating at 100°C for 5 min in 200 μl of selection buffer. The fluorescent intensity of applied, nonbound, and eluted ssDNA was measured respectively using a TBS-380 minifluorometer (Turner Biosystems, Sunnyvale, CA). All binding assays were repeated three times. To monitor the enrichment of each SELEX round, the percent of ssDNA (200 nM) binding with SEB (20 μM) was calculated.
To determine the binding affinity of a selected aptamer, the binding assay was performed as described above but with increasing amounts of ssDNA aptamer (0 to 400 nM) and a constant amount of SEB (20 μM) for each assay. To calculate the dissociation constants (Kd) of the aptamers, the aptamer quantity bound to targets was plotted and data points were fitted to the equation y = Bmaxx/(Kd + x) via nonlinear regression analysis, using GraphPad Prism v5.01 (GraphPad, San Diego, CA).
Human PBMC proliferation assays.
To study the effect of aptamers on SEB-mediated T cell proliferation, an MTT [3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide] cell proliferation assay was performed with human PBMCs as described previously (23). Human PBMCs were isolated from heparinized venous blood by Ficoll-Hypaque density gradient centrifugation from healthy human donors. After erythrocytes had been lysed, PBMCs were cultured in RPMI 1640 medium (Gibco, Grand Island, NY, USA) supplemented with 2 mM l-glutamine, 100 U/ml penicillin, 100 mg/ml streptomycin, 10 mM HEPES, and 10% (vol/vol) fetal bovine serum (Gibco) at 37°C. PBMCs (105 cells/well) were plated with SEB (200 ng/ml) alone or combined with various concentrations of aptamer candidates in triplicate in a 96-well plate.
PBMCs cultivated in RPMI 1640 medium alone served as the negative control. Plates were incubated in a 5% CO2 humidified incubator at 37°C for 48 h. After stimulation, cell proliferation was measured by adding 20 μl of MTT solution (Sigma, St. Louis, MO, USA), prepared in PBS at 5 mg/ml (pH 7.2), to each well. After 2 h of incubation, 100 μl of sodium dodecyl sulfate-dimethylformamide (SDS-DMF) solution was added to each well, and the assay product was allowed to develop overnight. The plates were read at 570 nm. Data are expressed as stimulation indices (SI). The SI was calculated as the ratio of the absorbance at 570 nm of pools incubated with SEB to that of pools incubated with medium only.
Cytokine quantitation.
To measure cytokines, a standard curve was first prepared using known concentrations of recombination cytokines. After stimulation with SEB with or without the addition of the candidate aptamer, human TNF-α, IL-1β, IL-6, IL-2, and IFN-γ in culture supernatants from PBMCs were measured by enzyme-linked immunosorbent assay (ELISA) according to the kit instructions (R&D Systems, Minneapolis, MN, USA). Cytokines in each sample were measured from the standard curve, and the detection limit of all assays was 20 pg/ml.
Aptamer binding assay for the MHC II molecules.
The ability of aptamer A11 to bind to the MHC II molecules was measured with a competition assay as described previously (16). Human Raji cells (2 × 105 cells) were incubated in 100 μl of selection buffer (1% BSA) with various final concentration of SEB (0, 25, 50, 75, or 100 μg/ml) and with or without a 5-fold molar excess of A11 for 40 min on ice. Cells were washed in selection buffer and incubated with FITC-conjugated SEB (FITC-SEB; 50 μg/ml), prepared using a FluoReporter FITC protein labeling kit (AMRESCO, Solon, OH, USA). After extensive washing, Raji cells were resuspended and analyzed by flow cytometry. The surface fluorescence of Raji cells was measured as the mean fluorescence from three independent experiments.
Murine experiments.
To investigate the therapeutic efficacy of aptamers in vivo on SEB-induced TSS, a “double-hit” murine model was established (24). Pathogen-free female BALB/c mice 10 to 12 weeks of age were purchased from the Experimental Animal Centre of Fuzhou General Hospital of Nanjing Military Command (Fuzhou, China). The mice were housed under controlled conditions and fed commercial mouse chow and water. Groups of mice (n = 19) were sensitized by intraperitoneal injection of d-galactosamine (GalN; 20 mg/animal; Sigma, St. Louis, MO, USA) and then challenged by intraperitoneal injection of SEB (20 μg/animal) at 4 h. After treatment, mice fasted but were allowed ad libitum access to water.
For therapeutic investigations, PEGylated aptamer (14 nmol) was injected intravenously 1 h before or 2 h after SEB injection. All agents were dissolved in sterile nonpyrogenic saline solution (pH 7.2) and were injected intravenously via the tail vein at 10 μl/g. Mouse survival was calculated by Kaplan-Meier survival analyses. The IACUC of Fuzhou General Hospital of Nanjing Military Command approved all experimental procedures.
Statistical analysis.
Results are presented as means ± standard deviations (SD) and were analyzed for significant differences by the Student t test. Kaplan-Meier methods were used to compute survival analyses. A P value of <0.05 was considered statistically significant. All data were processed by SPSS 12.0 for Windows.
RESULTS
In vitro selection and sequence analysis of SEB binding aptamers. (i) Relative binding rate of ssDNA and SEB.
Eleven rounds of in vitro selection were used to isolate aptamers that could favorably bind to SEB. Figure 1 depicts the binding ratio of the ssDNA pool to SEB, which increased gradually with increasing number of selection rounds and then remained nearly constant in the last three rounds. This suggested that SEB-binding aptamers with good binding affinity were enriched, and the binding ability of selection pools plateaued after about 10 rounds of selection.
FIG 1.
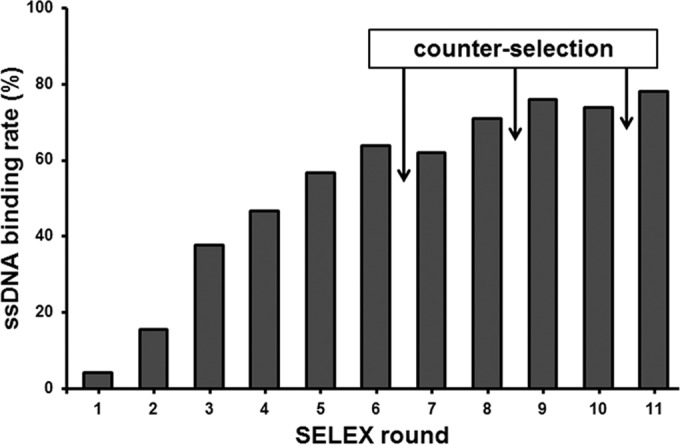
The abilities of ssDNA pools (200 ng) to bind to SEB (20 μM) were monitored by binding assay. The arrows indicate the counterselections performed using BSA and staphylococcal protein A (SPA). An increasing binding ability of the pool was achieved as the selection progressed.
(ii) Cloning, sequencing, and sequence analysis of oligonucleotide aptamers.
The enriched ssDNA pool of the 11th round was cloned into E. coli DH5α using the pGEM-T vector system. Forty individual clones were chosen randomly and sequenced. Multiple sequence alignments revealed that the primary clone structures were modestly enriched. In total, 6 nonredundant sequences were identified, and 13 sequences were identical to aptamer A1 and 8 variants of aptamer A1 which were highly homologous to aptamer A1, with only one or two base differences. Among the sequences, 5 A2, 3 A3, 2 A4, 1 A5, and 1 A6 aptamers were identified. Although each aptamer had the characteristic stem-loop or hairpin, their secondary structures predicted by software were significantly different. Although all aptamers can bind SEB (see Fig. S1 in the supplemental material), and some had dissociation constants in the low nanomolar range, none could inhibit SEB function except aptamer A2, which had a weak inhibitory effect (Fig. 2).
FIG 2.
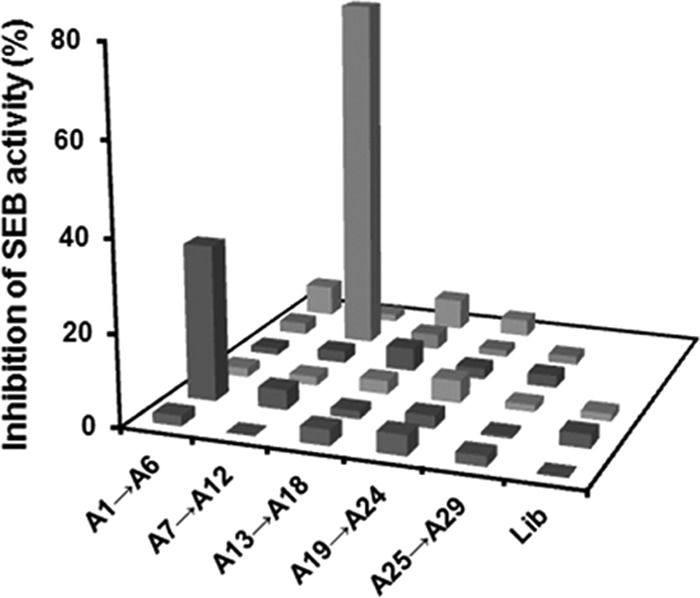
Inhibition of human PBMC proliferation stimulated with SEB by candidate aptamers. Each value is the mean from three experiments. Lib, the original synthetic ssDNA library.
Screening functional aptamers by PBMC proliferation assay.
To identify functional aptamers that can inhibit SEB superantigen activity, 40 individual clones from the 10th round were sequenced further as mentioned above. In total, 29 nonredundant sequences were identified; A1 to A6 were from the 11th round, and A7 to A29 were from the 10th round. Because superantigens can polyclonally activate T cells, these 29 oligonucleotides were all chemically synthesized and were tested for inhibition of SEB-mediated proliferation of human PBMCs. Data show that SEB-mediated proliferation of PBMCs decreased most significantly by A11 in a dose-response manner, with maximal inhibition (93%) achieved at 20 times the molarity of SEB compared to controls incubated with toxin alone (P < 0.05). At the same concentration, A2 achieved 34% inhibition (P < 0.05). However, the other 27 did not significantly inhibit binding (Fig. 2). A2 and A11 both bound with high affinity to SEB, with dissociation constants of 26 nM and 64 nM, respectively (Fig. 3A). Although no significant between-aptamer sequence similarity was found, secondary structures of A11 and A2 were predicted to be similar, and both had the characteristic stem-loop (Fig. 3B). Thus, the candidate aptamer A11 could potentially inhibit superantigen activity of SEB in vitro.
FIG 3.
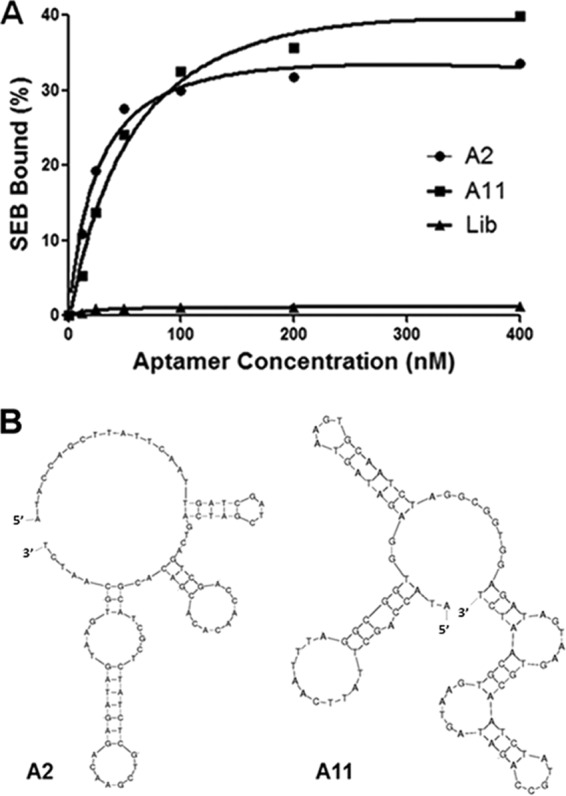
Dissociation constants and predicted structure of A2 and A11. (A) Binding curve of the aptamers A2 and A11 with SEB. The dissociation constants (Kd) were calculated by nonlinear regression analysis. (B) Comparison of a secondary structure prediction for the aptamers A2 and A22. Structures were predicted by Mfold.
Inhibition of SEB-mediated cytokine release from human PBMCs.
To confirm the inhibitory role of A11, the effect of A11 on the production of proinflammatory cytokines was examined by ELISA in human PBMCs incubated with SEB. Figure 4 shows that A11 dose-dependently reduced cytokine production from PBMCs incubated with SEB, compared to controls incubated with toxin alone (P < 0.05). TNF-α, IL-1β, IL-6, IL-2, and IFN-γ in culture supernatants were reduced by 77, 97, 64, 96, and 99%, respectively, at the same concentration (16 nM) at which maximal inhibition of SEB-induced PBMC proliferation response was achieved. Furthermore, inhibitory effects of A11 on SEB-mediated PBMC proliferation or cytokine release were not due to cytotoxic effect of aptamers; PBMCs had similar viability with aptamer addition. In addition, inhibitory effects were not mediated by an SEB-independent mechanism; A11 did not influence activation of PHA-induced stimulation of PBMC proliferation (data not shown).
FIG 4.
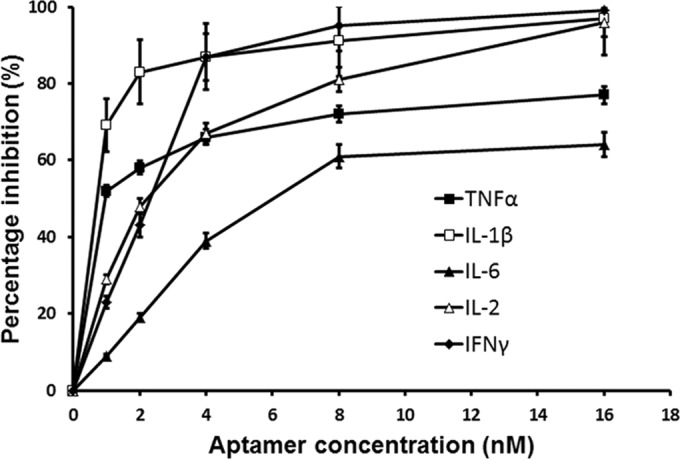
Inhibition of TNF-α, IL-1β, IL-6, IL-2, and IFN-γ production by human PBMCs stimulated with SEB by various concentrations of A11. Each value is the mean ± SD from three experiments.
A11 binding to MHC II molecules.
The inhibitory activity of A11 may arise from its ability to bind to SEB or to MHC II molecules. To clarify this, MHC II binding ability of A11 was estimated using a competitive binding assay against FITC-SEB. As shown in Fig. 5, SEB but not a mixture of SEB and A11 competed effectively with FITC-SEB for binding to MHC II-bearing Raji cells. Thus, the binding target of A11 was SEB, not MHC II molecules.
FIG 5.
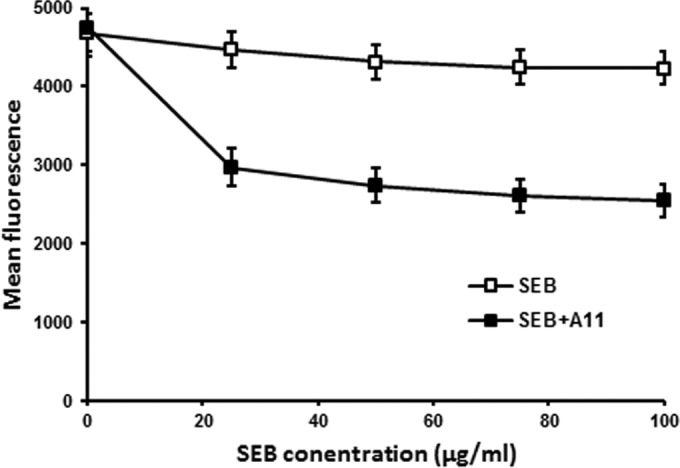
Binding ability of A11 to the MHC II molecules. The binding of FITC-SEB to MHC class II-bearing Raji cells was competed by SEB or A11. Each value is the mean ± SD from three experiments.
Modified optimization of A11.
To prolong in vivo A11 retention time, a PEGylated form of A11 (PEGA11) was prepared by modifying A11 at the 5′ end with a 40-kDa PEG. An aptamer binding assay indicated that PEGylation did not decrease the affinity for binding of A11 to SEB. PEGA11 had a dissociation constant of 83.5 nM in selection buffer, and in vitro, PEGA11 blocked the SEB-induced PBMC proliferation response more efficiently than did A11. At a relatively low molar concentration, PEGA11 had greater inhibitory activity for SEB (Fig. 6). Meanwhile, PEG alone did not bind to SEB or inhibit its activity.
FIG 6.
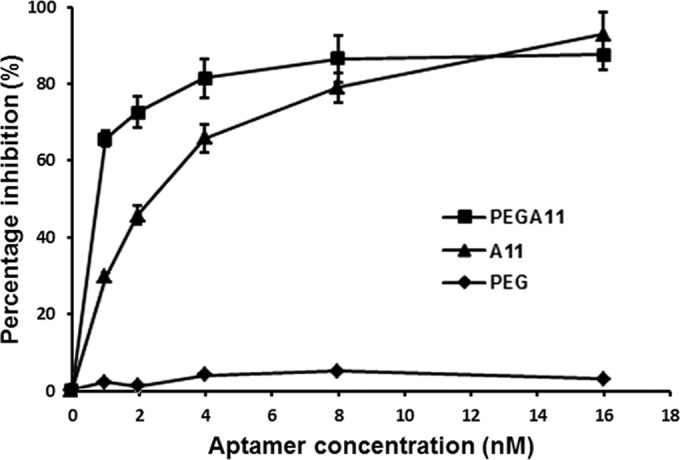
Inhibition of proliferation of human PBMCs stimulated with SEB by various concentrations of PEGA11. Each value is the mean ± SD from three experiments.
Protection and rescue of mice from SEB-mediated TSS.
Next, we confirmed an inhibitory role for A11 on SEB in vivo using a “double-hit” murine model of SEB-induced TSS as described in Materials and Methods. SEB killed 50% of the animals 36 h after injection. In contrast, PEGylated A11 injected (intraperitoneally) 30 min prior to SEB injection allowed more than 90% of the mice to survive the lethal challenge (P < 0.05) (Fig. 7). Surviving mice appeared to be indistinguishable from GalN-sensitized mice with respect to appetite and behavior. Moreover, PEGylated A11—if injected within 2 h after toxin exposure—could rescue mice from a lethal shock (P < 0.05) (Fig. 7). Thus, A11 can effectively inhibit SEB superantigen activity in vivo and may be a potential superantigen antagonist for protecting and treating TSS.
FIG 7.
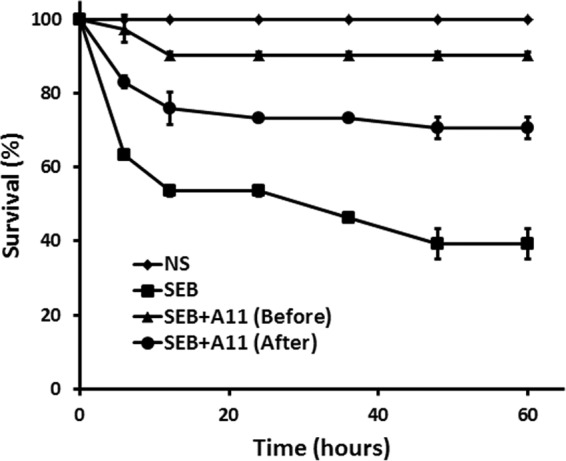
Survival analysis of GalN-sensitized mice treated with normal saline (NS), SEB, SEB plus A11 at 1 h before SEB, or SEB plus A11 at 2 h after SEB. Survival time up to 60 h after SEB challenge is shown. Each value is the mean ± SD from two experiments.
DISCUSSION
Aptamers, similar to antibodies, can be used to inhibit or antagonize superantigen activity and as such may be promising therapeutics for treating various diseases. To treat bacterial infection, functional aptamers have been selected to bind with antigens (25), toxins (26, 27), drug-resistant enzymes (28), and cells (29) of pathogenic bacteria. Here, we report the creation of a functional aptamer that can inhibit SEB superantigen activity.
In general, selecting an aptamer as a therapeutic agent is more difficult than selecting one as a recognition element. To inhibit superantigen activity, a functional aptamer should bind to or near the target's active site with high affinity. For example, Chang's group screened aptamer antagonists against human Toll-like receptor 2 by sequencing >400 clones from four IP-SELEX rounds (30). Although strategies such as competitive elution and counterselection may increase the chance of achieving functional aptamers (31), given the low molecular weight of SEB, we chose aptamers against SEB using the classic SELEX strategy and screened functional aptamers from more candidate aptamer clones. Among the 50 clones sequenced from the 11th SELEX round, 6 nonredundant aptamers were identified. Although several of these aptamers could bind SEB with high affinity, none could achieve the desired inhibition of SEB-mediated PBMC proliferation. Given that the functional aptamers with relatively low affinity might be lost in the elution and PCR steps of SELEX, we screened functional aptamers from the previous SELEX round. Finally, aptamer clone A11 was identified in the 10th SELEX round and this decreased SEB-mediated PBMC proliferation significantly, suggesting it may be a specific antagonist of SEB.
In in vitro functional analysis, A11 inhibited SEB-mediated expression of human genes for TNF-α, IL-1β, IL-6, IL-2, and IFN γ, cytokines that mediate shock, implying that A11 could significantly inhibit SEB activity at the beginning of the toxicity cascade, before the pathological activation of T cells. Furthermore, A11 did not inhibit T cell proliferation mediated by either TSS toxin 1 (TSST-1) or SEA (data not shown). The high specificity of aptamers may limit their application against bacterial infections, but it may also decrease side effects (32). To acquire aptamer antagonists with broad spectra against SEs, selecting aptamers that bind to highly conserved regions of SEs or combining aptamers specific to different SEs may be a viable approach.
Because SEB pathology in vivo is related to excessive production of inflammatory cytokines, A11 may be curative against SEB-induced TSS in vivo. However, therapeutic aptamers alone are not sufficiently stable for in vivo delivery, so they are usually modified or conjugated to vehicles that are resistant to nucleases to improve their residence time (33). Considering that PEGylation is one of the most commonly used modifications of aptamers (34, 35), we analyzed the therapeutic effects of PEGylated A11 in vivo. PEGylated A11 could significantly prevent and rescue a mouse model from SEB-mediated TSS, implying that the affinity of A11 for its targets remained high in vivo and did not decrease after PEGylation. A11 proved to inhibit SEB superantigen activity in both cultured human PBMCs and the murine model, implying that A11's protective effects are not MHC restricted and that the binding site of A11 might be the SEB active site. This finding is consistent with results from competitive binding assays against FITC-SEB, and these data offer a foundation for further clinical applications of A11 as an SEB antagonist.
Supplementary Material
ACKNOWLEDGMENTS
This research was supported by the National Natural Science Foundation of China (grants 81101215 and 81271928), Fujian Provincial Natural Science Foundation (grants 2012Y0058 and 2013Y0075), and Science Foundation of Nanjing Military Command (grant 10Z029).
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.04414-14.
REFERENCES
- 1.Fraser JD, Proft T. 2008. The bacterial superantigen and superantigen-like proteins. Immunol Rev 225:226–243. doi: 10.1111/j.1600-065X.2008.00681.x. [DOI] [PubMed] [Google Scholar]
- 2.Sundberg EJ, Deng L, Mariuzza RA. 2007. TCR recognition of peptide/MHC class II complexes and superantigens. Semin Immunol 19:262–271. doi: 10.1016/j.smim.2007.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Libbey JE, Cusick MF, Fujinami RS. 2014. Role of pathogens in multiple sclerosis. Int Rev Immunol 33:266–283. doi: 10.3109/08830185.2013.823422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lindsay CD, Griffiths GD. 2013. Addressing bioterrorism concerns: options for investigating the mechanism of action of Staphylococcus aureus enterotoxin B Hum Exp Toxicol 32:606–619. doi: 10.1177/0960327112458941. [DOI] [PubMed] [Google Scholar]
- 5.Hudson LC, Seabolt BS, Odle J, Bost KL, Stahl CH, Piller KJ. 2013. Sublethal staphylococcal enterotoxin B challenge model in pigs to evaluate protection following immunization with a soybean-derived vaccine. Clin Vaccine Immunol 20:24–32. doi: 10.1128/CVI.00526-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Visvanathan K, Charles A, Bannan J, Pugach P, Kashfi K, Zabriskie JB. 2001. Inhibition of bacterial superantigens by peptides and antibodies. Infect Immun 69:875–884. doi: 10.1128/IAI.69.2.875-884.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Varshney AK, Wang X, Scharff MD, MacIntyre J, Zollner RS, Kovalenko OV, Martinez LR, Byrne FR, Fries BC. 2013. Staphylococcal enterotoxin B-specific monoclonal antibody 20B1 successfully treats diverse Staphylococcus aureus infections. J Infect Dis 208:2058–2066. doi: 10.1093/infdis/jit421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.LeClaire RD, Hunt RE, Bavari S. 2002. Protection against bacterial superantigen staphylococcal enterotoxin B by passive vaccination. Infect Immun 70:2278–2281. doi: 10.1128/IAI.70.5.2278-2281.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Arad G, Levy R, Hillman D, Kaempfer R. 2000. Superantigen antagonist protects against lethal shock and defines a new domain for T-cell activation. Nat Med 6:414–421. doi: 10.1038/74672. [DOI] [PubMed] [Google Scholar]
- 10.Buonpane RA, Churchill HR, Moza B, Sundberg EJ, Peterson ML, Schlievert PM, Kranz DM. 2007. Neutralization of staphylococcal enterotoxin B by soluble, high-affinity receptor antagonists. Nat Med 13:725–729. doi: 10.1038/nm1584. [DOI] [PubMed] [Google Scholar]
- 11.Hong-Geller E, Mollhoff M, Shiflett PR, Gupta G. 2004. Design of chimeric receptor mimics with different TcRVbeta isoforms. Type-specific inhibition of superantigen pathogenesis. J Biol Chem 279:5676–5684. doi: 10.1074/jbc.M309388200. [DOI] [PubMed] [Google Scholar]
- 12.Ellington AD, Szostak JW. 1992. Selection in vitro of single-stranded DNA molecules that fold into specific ligand-binding structures. Nature 355:850–852. doi: 10.1038/355850a0. [DOI] [PubMed] [Google Scholar]
- 13.Tuerk C, Gold L. 1990. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 249:505–510. doi: 10.1126/science.2200121. [DOI] [PubMed] [Google Scholar]
- 14.Santosh B, Yadava PK. 2014. Nucleic acid aptamers: research tools in disease diagnostics and therapeutics. Biomed Res Int 2014:540451. doi: 10.1155/2014/540451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bruno JG, Kiel JL. 2002. Use of magnetic beads in selection and detection of biotoxin aptamers by electrochemiluminescence and enzymatic methods. Biotechniques 32:178–180, 182–183. [DOI] [PubMed] [Google Scholar]
- 16.Purschke WG, Radtke F, Kleinjung F, Klussmann S. 2003. A DNA Spiegelmer to staphylococcal enterotoxin B. Nucleic Acids Res 31:3027–3032. doi: 10.1093/nar/gkg413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.DeGrasse JA. 2012. A single-stranded DNA aptamer that selectively binds to Staphylococcus aureus enterotoxin B. PLoS One 7:e33410. doi: 10.1371/journal.pone.0033410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Temur E, Zengin A, Boyaci IH, Dudak FC, Torul H, Tamer U. 2012. Attomole sensitivity of staphylococcal enterotoxin B detection using an aptamer-modified surface-enhanced Raman scattering probe. Anal Chem 84:10600–10606. doi: 10.1021/ac301924f. [DOI] [PubMed] [Google Scholar]
- 19.Wang KY, Zeng YL, Yang XY, Li WB, Lan XP. 2011. Utility of aptamer-fluorescence in situ hybridization for rapid detection of Pseudomonas aeruginosa. Eur J Clin Microbiol Infect Dis 30:273–278. doi: 10.1007/s10096-010-1074-0. [DOI] [PubMed] [Google Scholar]
- 20.Niazi JH, Lee SJ, Kim YS, Gu MB. 2008. ssDNA aptamers that selectively bind oxytetracycline. Bioorg Med Chem 16:1254–1261. doi: 10.1016/j.bmc.2007.10.073. [DOI] [PubMed] [Google Scholar]
- 21.Pauli NT, Kim HK, Falugi F, Huang M, Dulac J, Henry Dunand C, Zheng NY, Kaur K, Andrews SF, Huang Y, DeDent A, Frank KM, Charnot-Katsikas A, Schneewind O, Wilson PC. 2014. Staphylococcus aureus infection induces protein A-mediated immune evasion in humans. J Exp Med 211:2331–2339. doi: 10.1084/jem.20141404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zuker M. 2003. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Krakauer T, Buckley M, Issaq HJ, Fox SD. 2010. Rapamycin protects mice from staphylococcal enterotoxin B-induced toxic shock and blocks cytokine release in vitro and in vivo. Antimicrob Agents Chemother 54:1125–1131. doi: 10.1128/AAC.01015-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nagaki M, Muto Y, Ohnishi H, Yasuda S, Sano K, Naito T, Maeda T, Yamada T, Moriwaki H. 1994. Hepatic injury and lethal shock in galactosamine-sensitized mice induced by the superantigen staphylococcal enterotoxin B. Gastroenterology 106:450–458. [DOI] [PubMed] [Google Scholar]
- 25.Pan Q, Zhang XL, Wu HY, He PW, Wang F, Zhang MS, Hu JM, Xia B, Wu J. 2005. Aptamers that preferentially bind type IVB pili and inhibit human monocytic-cell invasion by Salmonella enterica serovar typhi. Antimicrob Agents Chemother 49:4052–4060. doi: 10.1128/AAC.49.10.4052-4060.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Challa S, Tzipori S, Sheoran A. 2014. Selective evolution of ligands by exponential enrichment to identify RNA aptamers against Shiga toxins. J Nucleic Acids 2014:214929. doi: 10.1155/2014/214929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vivekananda J, Salgado C, Millenbaugh NJ. 2014. DNA aptamers as a novel approach to neutralize Staphylococcus aureus alpha-toxin. Biochem Biophys Res Commun 444:433–438. doi: 10.1016/j.bbrc.2014.01.076. [DOI] [PubMed] [Google Scholar]
- 28.Kim SK, Sims CL, Wozniak SE, Drude SH, Whitson D, Shaw RW. 2009. Antibiotic resistance in bacteria: novel metalloenzyme inhibitors. Chem Biol Drug Design 74:343–348. doi: 10.1111/j.1747-0285.2009.00879.x. [DOI] [PubMed] [Google Scholar]
- 29.Chen F, Zhang X, Zhou J, Liu S, Liu J. 2012. Aptamer inhibits Mycobacterium tuberculosis (H37Rv) invasion of macrophage. Mol Biol Rep 39:2157–2162. doi: 10.1007/s11033-011-0963-3. [DOI] [PubMed] [Google Scholar]
- 30.Chang YC, Kao WC, Wang WY, Wang WY, Yang RB, Peck K. 2009. Identification and characterization of oligonucleotides that inhibit Toll-like receptor 2-associated immune responses. FASEB J 23:3078–3088. doi: 10.1096/fj.09-129312. [DOI] [PubMed] [Google Scholar]
- 31.Kulbachinskiy AV. 2007. Methods for selection of aptamers to protein targets. Biochemistry (Mosc) 72:1505–1518. doi: 10.1134/S000629790713007X. [DOI] [PubMed] [Google Scholar]
- 32.Kanwar JR, Shankaranarayanan JS, Gurudevan S, Kanwar RK. 2014. Aptamer-based therapeutics of the past, present and future: from the perspective of eye-related diseases. Drug Discov Today 19:1309–1321. doi: 10.1016/j.drudis.2014.02.009. [DOI] [PubMed] [Google Scholar]
- 33.Kong HY, Byun J. 2013. Nucleic Acid aptamers: new methods for selection, stabilization, and application in biomedical science. Biomol Ther 21:423–434. doi: 10.4062/biomolther.2013.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Diener JL, Daniel Lagasse HA, Duerschmied D, Merhi Y, Tanguay JF, Hutabarat R, Gilbert J, Wagner DD, Schaub R. 2009. Inhibition of von Willebrand factor-mediated platelet activation and thrombosis by the anti-von Willebrand factor A1-domain aptamer ARC1779. J Thromb Haemost 7:1155–1162. doi: 10.1111/j.1538-7836.2009.03459.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Floege J, Ostendorf T, Janssen U, Burg M, Radeke HH, Vargeese C, Gill SC, Green LS, Janjic N. 1999. Novel approach to specific growth factor inhibition in vivo: antagonism of platelet-derived growth factor in glomerulonephritis by aptamers. Am J Pathol 154:169–179. doi: 10.1016/S0002-9440(10)65263-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.