

ABSTRACT
Infections caused by highly successful clones of hospital-associated methicillin-resistant Staphylococcus aureus (HA-MRSA) are a major public health burden. The globally dominant sequence type 239 (ST239) HA-MRSA clone has persisted in the health care setting for decades, but the basis of its success has not been identified. Taking a collection of 123 ST239 isolates spanning 32 years, we have used population-based functional genomics to investigate the evolution of this highly persistent and successful clone. Phylogenetic reconstruction and population modeling uncovered a previously unrecognized distinct clade of ST239 that was introduced into Australia from Asia and has perpetuated the epidemic in this region. Functional analysis demonstrated attenuated virulence and enhanced resistance to last-line antimicrobials, the result of two different phenomena, adaptive evolution within the original Australian ST239 clade and the introduction of a new clade displaying shifts in both phenotypes. The genetic diversity between the clades allowed us to employ genome-wide association testing and identify mutations in other essential regulatory systems, including walKR, that significantly associate with and may explain these key phenotypes. The phenotypic convergence of two independently evolving ST239 clades highlights the very strong selective pressures acting on HA-MRSA, showing that hospital environments have favored the accumulation of mutations in essential MRSA genes that increase resistance to antimicrobials, attenuate virulence, and promote persistence in the health care environment. Combinations of comparative genomics and careful phenotypic measurements of longitudinal collections of clinical isolates are giving us the knowledge to intelligently address the impact of current and future antibiotic usage policies and practices on hospital pathogens globally.
IMPORTANCE
Methicillin-resistant Staphylococcus aureus (MRSA) is responsible for innumerable drug-resistant health care-associated infections globally. This study, the first to investigate the evolutionary response of hospital-associated MRSA (HA-MRSA) over many decades, demonstrates how MRSA can persist in a region through the reintroduction of a previously unrecognized distinct clade. This study also demonstrates the crucial adaptive responses of HA-MRSA to the highly selective environment of the health care system, the evolution of MRSA isolates to even higher levels of antibiotic resistance at the cost of attenuated virulence. However, in vivo persistence is maintained, resulting in a clone of HA-MRSA able to resist almost all antimicrobial agents and still cause invasive disease in the heavily compromised hosts found in modern health care settings.
INTRODUCTION
Staphylococcus aureus is one of the most common and problematic causes of infectious disease in humans. The emergence and spread of epidemic drug-resistant S. aureus clones have often been referred to as a wave-like phenomenon, with the first wave representing the development of penicillin-resistant S. aureus, the second wave representing that of methicillin-resistant S. aureus (MRSA), and the third and fourth waves representing the establishment of the highly successful hospital-associated MRSA (HA-MRSA) lineages that are encountered today and the appearance of genetically distinct MRSA clones in the community, respectively (1).
Genomic studies are providing new insights into the population structure of S. aureus, MRSA in particular, and have shown that the majority of HA-MRSA infections are the result of a small number of dominant, typically multidrug-resistant, clones that rarely cause disease outside the hospital setting (2, 3). One of the most successful and persistent clones is the globally dispersed, multidrug-resistant multilocus sequence type 239 (ST239), a naturally occurring hybrid having originated from a large recombination event involving descendants of the two major staphylococcal lineages, clonal complexes 8 and 30 (4). The earliest reports of MRSA ST239 date back to the late 1970s in Australia (5), the United Kingdom (6), and the United States (7). However, multidrug-resistant MRSA belonging to phage group III and possibly representing ST239 has been isolated in Australia since the 1960s (8). From the 1980s, ST239 had been reported as the causative agent of numerous epidemics in health care institutions worldwide; the number of regional names given to the ST239 clone attests to its success. These include, but are not limited to, the Brazilian, British epidemic (EMRSA-1, -4, -7, -9 and -11), Canadian epidemic (CMRSA-3), Hungarian, Portuguese, Nanjing/Taipei, Vienna, and Eastern Australian epidemic (EMRSA-Aus-2 and -Aus-3) clones.
Recent studies have explored the global population structure (9–11) and phylogeography (12) of ST239, as well as the role of recombination and the contribution of other mobile genetic elements to the spread and survival of this clone (11, 13, 14). However, the mechanisms underlying the successful adaptation and survival of ST239 MRSA within the health care environment, in particular, the phenotypic evolution of this clone, are poorly understood. There is a high prevalence of reduced glycopeptide susceptibility among HA-MRSA isolates in Australia (15), where ST239 has been a dominant MRSA clone for more than 30 years (16). Additionally, a recent study of S. aureus bacteremia isolates from Australia demonstrated higher vancomycin MICs for ST239 isolates than for other clones (17), suggesting that the ST239 clone may be adapted to enhanced antimicrobial resistance.
Using both genomic and integrated phenotypic approaches, we have explored the evolutionary dynamics of ST239 MRSA in Australia, revealing that two clades of this highly successful clone have evolved independently in our region, both contributing to enhanced antibiotic resistance and attenuated virulence driven by mutations in highly conserved essential regulatory genes. This work provides an in-depth insight into the persistence and evolution of HA-MRSA and highlights a bacterial population level response to the long-term and widespread use of antibiotics that is driving even higher levels of resistance.
RESULTS AND DISCUSSION
Global and regional phylogeny of ST239 MRSA.
To recover the population structure of Australian ST239 MRSA, as well as explore the evolution of this sequence type within a global phylogeny, we collected 87 ST239 isolates from health care institutions in Australia and New Zealand between 1980 and 2012. The majority of these isolates (n = 69) were recovered from cases of bacteremia or tissue infection. We supplemented this collection with the publicly available whole-genome sequence reads of a further 35 international ST239 isolates from the collection of Harris et al. (10). These additional isolates were recovered from Asia, Europe, South America, and North America between 1993 and 2007 and represented the phylogenetic diversity observed in their population model. Relevant isolate demographics and a summary of the sequencing results are available in Data Set S1 in the supplemental material.
Alignment of whole-genome sequence reads for all 123 isolates with reference ST239 isolate TW20 identified 2,716 core genome single nucleotide polymorphisms (SNPs). A maximum clade credibility (MCC) tree generated from these core genome SNPs is presented in Fig. 1A. The population structure proposed by the MCC tree is robust because of the strong branch support and the distinct grouping of isolates with shared geographic source creating regional clades, a finding consistent with previous ST239 phylogenetic models. These clades reflect the capacity of ST239 to undergo extensive local expansion but limited intercontinental transmission (9–11).
FIG 1 .

Global phylogeny and molecular clock of ST239 MRSA. (A) MCC tree, generated in BEAST v1.7.5 from 2,716 SNPs in the core genome of 123 isolates, illustrating the global phylogeny of ST239 MRSA. Branches are colored to reflect statistical support; those possessing a posterior probability of <95% are red. Major branches with <95% support are annotated with their respective posterior probabilities. Branch annotations provide isolate names, countries, and years of recovery and are colored to reflect the source continents (yellow, Australasia; red, Europe; green, South America; blue, Asia). For Australian isolates, the state of origin is indicated by a colored box (see key). Australian clades are shaded and annotated as Clade 1 and Clade 2. (B) Root-to-tip divergence graph illustrating the strong correlation between year of recovery and branch length, estimated from an ML phylogenetic model. Data points representing Clade 1 and 2 isolates are blue and red, respectively. Trend lines are included for all of the data points (black), Clade 1 (blue), and Clade 2 (red) and have been extrapolated to estimate emergence dates.
The Australian ST239 isolates followed a similar pattern, grouping into two distinct clades on the MCC tree (annotated as Clades 1 and 2). The separation of these clades indicates the individual evolutionary paths along which they have developed, while the distance between them suggests that their core genomes are as different from each other as those of other continental clades. This reveals that ST239 MRSA has undergone concurrent but independent spread twice within Australia. Clade 1 isolates represented all of the regions sampled and 27 of the 32 years in the temporal span of the collection (1980 to 2007). Conversely, Clade 2 isolates were considerably less diverse; all were recovered in the Australian state of Victoria, the earliest of which was isolated in 2001. The difference in the demographic diversity of these two clades is consistent with Clade 1 being the original Australian ST239 clade and Clade 2 representing a more recent, previously unrecognized, reintroduction of ST239 into the Australian region. The more recent common ancestor shared by Clade 2 and the predominately Asian isolates, combined with the high posterior probability of major nodes connecting these groups, is highly supportive of an intercontinental transmission event between Asia and Australia, resulting in the local establishment of Clade 2. Furthermore, the only four Australian isolates that did not group with either large clade were located in a small, distinct clade close to Clade 2 and the Asian isolates. All four isolates were recovered in the Australian state of Victoria in 2000 and 2001, suggesting that more than one intercontinental transmission event may have occurred between the neighboring continents of Australia and Asia.
Genomic characterization of Australian ST239 MRSA.
To further investigate the molecular differences between the two Australian ST239 clades, as well as understand the impact of the introduction of Clade 2, we estimated the rate of nucleotide substitution, modeled the population size over time, and examined the conservation of protein coding sequences (CDS) among the clades.
Nucleotide substitution rate.
An initial global ST239 phylogeny, inferred by maximum-likelihood (ML) phylogenetic analysis, demonstrated a temporal gradient when rooted with a non-ST239 outgroup. This was further supported by the strong correlation between the branch length and the year of isolation (R2 = 0.8261, Fig. 1B). A comparison of the MCC and ML topologies is provided in Fig. S1 in the supplemental material. The Bayesian analysis estimated the mean rate of nucleotide substitutions for the ST239 clone at 1.6 × 10−6 substitutions per site per year (ss−1 year−1), with a 95% highest posterior density (HPD) interval of 1.2 × 10−6 to 2.0 × 10−6 ss−1 year−1. This rate is marginally slower than previous estimates of 3.3 × 10−6 (95% confidence interval, 2.5 × 10−6 to 4.0 × 10−6) ss−1 year−1 (10), and 3.4 × 10−6 (95% HPD interval, 2.7 × 10−6 to 4.1 × 10−6) ss−1 year−1 (12). Our estimate places the emergence of the ST239 clone in 1946 (95% HPD interval, 1885 to 1973). To ascertain the mutation rate and emergence date of each Australian clade, the Bayesian analysis was repeated by testing Clades 1 and 2 independently. Clade 1 was found to mutate at a mean rate of 9.2 × 10−7 (95% HPD interval, 5.8 × 10−7 to 1.3 × 10−6) ss−1 year−1, and Clade 2 was found to mutate at a mean rate of 1.3 × 10−6 (95% HPD interval, 6.5 × 10−7 to 2.0 × 10−6) ss−1 year−1. From this, we estimated that Clade 1 emerged in 1962 (95% HPD interval, 1919 to 1981) and Clade 2 emerged in 1986 (95% HPD interval, 1954 to 1998).
Population size.
To determine whether Clade 2 had contributed to the persistence of ST239 in the Australian health care environment, a Bayesian Skyline model was employed to estimate changes in the effective population size (EPS) of each clade over time (18). The EPS is estimated from the observed nucleotide variation (genetic diversity) in relation to the mutation rate and can be used to infer changes in the size of a population. These models, illustrated in Fig. 2A, suggested a steady increase in the EPS of Clade 1 from the early 1980s to the late 1990s, followed by a nearly 10-fold decrease over the next 10 years. This result was highly consistent with the reported dominance of the ST239 clone among HA-MRSA isolates in Australia over the last 30 years, as well as the subsequent decrease since 2005 (16). Intriguingly, the EPS of Clade 2 appeared stable throughout the modeled time frame of 2000 to 2012 and could be maintaining the ST239 population in Australia. This may partly explain why the ST239 clone still represents nearly half of HA-MRSA strains encountered in the health care environment nationally and 70% of those in the state of Victoria, where Clade 2 appears to be focused (19).
FIG 2 .
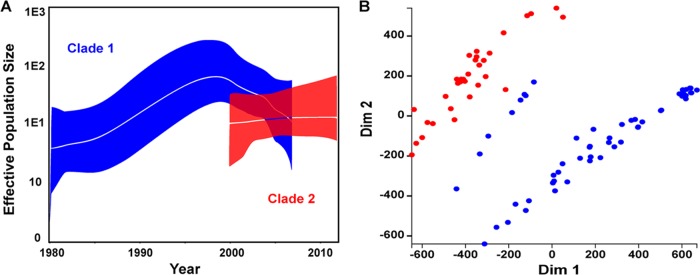
Genomic characterization of Australian ST239 clades. (A) Bayesian Skyline models illustrating the change in the EPS of Clades 1 (blue) and 2 (red). The clades were modeled separately, and their EPSs were overlaid onto a single graph. The median EPS is indicated by the central white line, and the 95% HPD interval is indicated by the colored zone. (B) MDS plot, based on protein ortholog clusters, illustrating the variation in gene content among the Australian isolates from Clades 1 (blue) and 2 (red). Dim, dimension.
Gene content variation.
To investigate larger genomic differences between the two clades, protein ortholog clustering was used to examine the variation in gene presence or absence. A multidimensional scaling (MDS) representation of these data illustrated discrete, clade-specific clustering (Fig. 2B), further highlighting the genomic divergence in Australian ST239 MRSA. A total of 67 orthologs were identified that differentiated the two clades, each being detected in a minimum of 50% of one clade and absent from all of the isolates of the other clade. It was noted that eight clade 1 isolates, all recovered in the early 1980s, appeared to share more protein orthologs with Clade 2, as indicated by their location in the MDS plot, than any other Clade 1 isolates. When these eight isolates are excluded, the number of orthologs that differentiate the two clades increased to 209. Of these, 123 (59%) were associated with the three prophages identified in TW20 (φSa1, φSa3, and φSaβ-like). Orthologs associated with these prophages were only identified in Clade 2 isolates and the eight Clade 1 isolates. The φSaβ-like prophage is of interest in the evolution of the ST239 clone because it carries the recently identified virulence determinant sasX. Located at the 3′ end of the φSPβ-like prophage, sasX encodes a small, surface-anchored protein that enhances nasal colonization, abscess formation, and tissue damage in the lungs during respiratory infection and has been implicated in the rapid spread of ST239 throughout Asia (13). Li et al. reported that the prevalence of sasX among ST239 MRSA in Asia increased substantially between 2003 and 2011, suggesting the preferential uptake of this mobile element. Local alignment identified sasX in all of the Clade 2 isolates, as well as the eight clade 1 isolates described above. While this may suggest a role for sasX in the evolution of Clade 2, its absence from most Clade 1 isolates indicates that it has not been transferred between the two clades. Furthermore, its presence among only a small number of early Clade 1 isolates suggests that it has been selectively lost, raising questions about the role of sasX in the evolution of ST239 MRSA in Australia. Nonetheless, the conservation of sasX among Clade 2 isolates is further evidence that this clade originated in Asia.
Collectively, these genomic analyses reveal that while both clades exist and have evolved within the same environment, at a molecular level, they are distinct. These differences are likely to have contributed to the successful expansion of Clade 2 after its reintroduction and the continued persistence of the ST239 clone in this region. These differences also explain some of the phenotypic variation detected in Australian ST239 MRSA.
Changing phenotype of Australian ST239 MRSA.
We took advantage of the 32-year time span of our Australian ST239 MRSA culture collection to identify changes in clinically relevant phenotypes (see Fig. S2 in the supplemental material).
Enhanced antimicrobial resistance.
Reduced glycopeptide susceptibility has been commonly reported in Australian MRSA isolates (15, 20). Intermediate-level resistance to vancomycin (vancomycin-intermediate S. aureus [VISA]) and teicoplanin was detected at low levels in our collection (15 and 3%, respectively). However, a further 19 isolates (21%) were identified as heterogeneous VISA (hVISA), indicating that more than a third (36%) of the collection had reduced glycopeptide susceptibility. Furthermore, daptomycin nonsusceptibility was detected in 15 isolates (17%). These proportions, although consistent with previous estimations of hVISA and VISA in our region (15), may be an overrepresentation due to the inclusion of a subset of isolates (n = 24) with known phenotypes of reduced susceptibility or nonsusceptibility to these agents. They have therefore been excluded from the following analyses of antimicrobial susceptibility and bacterial fitness.
A comparison of antimicrobial susceptibility over time and between phylogenetic clades is presented in Table 1. These analyses demonstrated that the average MIC of vancomycin, teicoplanin, and daptomycin for isolates recovered after 1990 was significantly higher than that for those isolates recovered during the 1980s (P = 0.003 to <0.001). A comparison of the average MICs for Clades 1 and 2 also showed a statistically significant difference, with Clade 2 consistently having a higher average MIC of all three antibiotics (P = 0.016 to <0.001). As Clade 2 contained only isolates recovered after 2000 and could therefore be creating a bias in the comparison of older and newer isolates, the comparison of temporal groups was repeated by testing Clade 1 independently of Clade 2. While there appeared to be a trend of increasing MICs of all three antibiotics (see Fig. S3 in the supplemental material), consistent with the shift observed in the wider data set, a statistically significant increase was only detected in the average MIC of daptomycin for Clade 1 isolates recovered after 1990 (P = 0.013), the P values for vancomycin and teicoplanin MICs being low but nonsignificant (P = 0.121 and 0.057, respectively).
TABLE 1 .
Comparison of the antimicrobial susceptibility and bacterial fitness of temporal and phylogenetic groupsa
| Group (no. of isolates tested) | MIC in µg/mL |
Bacterial Fitness |
||
|---|---|---|---|---|
| Vancomycin | Teicoplanin | Daptomycin | Doubling Time (mins) | |
| Isolates recovered during 1980s (19) | 1.09 | 0.81 | 0.24 | 39.35 |
| Isolates recovered after 1990 (44) | 1.45 | 1.49 | 0.51 | 45.62 |
| P = 0.003 | P < 0.001 | P < 0.001 | P < 0.001 | |
| Phylogenetic Clade 1 (38) | 1.18 | 0.97 | 0.35 | 43.90 |
| Phylogenetic Clade 2 (25) | 1.59 | 1.79 | 0.52 | 43.46 |
| P < 0.001 | P < 0.001 | P = 0.021 | P = 0.790 | |
| Clade 1 isolates recovered during 1980s (19) | 1.09 | 0.81 | 0.24 | 39.35 |
| Clade 1 isolates recovered after 1990 (19) | 1.28 | 1.17 | 0.50 | 48.46 |
| P = 0.121 | P = 0.057 | P = 0.013 | P < 0.001 | |
Each value is the mean result of the respective group. P values were determined with a Welsh two-sample t test with log transformations applied to MIC data prior to comparison.
There has been significant debate in the literature regarding “MIC creep” in S. aureus (21). Previous studies, while demonstrating increasing MICs for MRSA isolates over time, have not performed detailed genotyping to determine if the isolates being tested are related or not. Here, we provide an example of both phenomena contributing to “MIC creep” in one MRSA clone in a single region, adaptive evolution within a clade and the introduction of a new clade, resulting in high rates of VISA infections associated with enhanced morbidity and poor treatment responses (20, 22, 23).
GWAS.
To identify the genetic mediators of enhanced antimicrobial resistance, in particular to vancomycin, a genome-wide association study (GWAS) approach was implemented. These types of studies often require large numbers of isolates to detect associations, as the significance threshold is adjusted to account for multiple testing (critical P value, ≤0.05/n). Therefore, it was unsurprising that an initial comparison of the frequency of all of the core genome SNPs resulting in nonsynonymous amino acid changes between isolates with vancomycin MICs of ≤2 and ≥3 µg/ml (Fig. 3A) failed to detect any significant associations. Interestingly, the smallest P value achieved in this analysis was that of an SNP resulting in an RpoBH481Y amino acid substitution (P = 0.00029). This exact mutation within the beta subunit of the RNA polymerase has previously been demonstrated to lead to reduced vancomycin susceptibility by using genetic manipulation experiments (24) and was also identified in a recent GWAS of reduced glycopeptide susceptibility (25). In the Australian ST239 collection, this SNP was identified in three phylogenetically dispersed isolates with high vancomycin MICs. Furthermore, this analysis revealed a high number of SNPs within the essential two-component regulator walKR, all with very low P values (P = 0.00069).
FIG 3 .
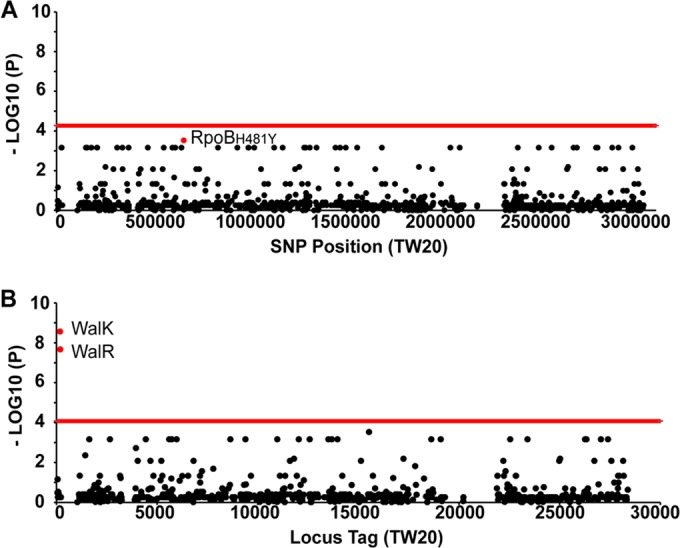
GWAS of reduced vancomycin susceptibility in ST239 MRSA. Manhattan plots illustrate the results of two GWASs comparing isolates with vancomycin MICs of ≤2 or ≥3 µg/ml. The red line indicates the threshold of significance (P value of ≤0.05/number of hypotheses tested), as determined by chi-square analysis. (A) Manhattan plot illustrating the associations between SNPs and vancomycin MICs. The single red data point represents the SNP resulting in an RpoBH481Y amino acid substitution. (B) Second Manhattan plot illustrating the associations of CDSs containing a SNP(s), independent of the SNP location within the locus, with vancomycin MICs. Highlighted red data points represent the walK and walR genes, the only significant associations identified in this analysis.
To identify whether walKR, or any other gene, is a hot spot for mutations associated with increased vancomycin MICs, a modified GWAS was performed. In this analysis, each CDS was classified as either “unaffected” or “affected”, the latter being assigned if a SNP was identified within the locus, regardless of its location. The frequencies of “affected” CDS in the same MIC groups described above were then compared (Fig. 3B). This showed that variant sequences of both walK and walR were highly associated with increased vancomycin MICs (P = 2.721 × 10−9 and 2.149 × 10−8, respectively). However, the exact mutations were highly variable, with three different walR mutations identified in four isolates and seven different walK mutations identified in seven isolates (see Table S1 in the supplemental material). The DNA sequences of rpoB and walKR are highly conserved in S. aureus; however, mutations in both loci have previously been linked to not only enhanced glycopeptide resistance (26) but also alterations in virulence and, in the case of rpoB, resistance to the host innate immune responses (27). The selective pressures driving resistance to last-line antimicrobials in Australian ST239 MRSA are therefore also likely to be having an impact on the fitness and virulence of this clone.
Relative bacterial fitness.
To determine whether there has also been a change in the relative fitness of Australian ST239 MRSA, the average replication rate of each isolate was measured (see Fig. S2 in the supplemental material). The mean doubling time of the Australian collection (n = 87) was 46.51 ± 8.48 min or 43.73 ± 6.61 min when the subset of isolates (n = 24) with known resistance phenotypes was excluded. This finding is similar to the previously published replication rate of five Australian ST239 isolates of 42.2 ± 6.5 min (28). A comparison of older and newer isolates identified a statistically significant increase in the mean doubling time of isolates recovered after 1990 (P < 0.001) (Table 1). This increase was also observed among newer Clade 1 isolates when they were tested independently (P < 0.001). No significant difference between the mean doubling times of Clades 1 and 2 was observed. However, Clade 2 isolates did demonstrate a significantly slower replication rate than Clade 1 isolates recovered in the 1980s (P = 0.017) but a significantly faster rate than those recovered after 1990 (P = 0.008). This suggested that reduced fitness among Australian ST239 MRSA was largely due to an increase in the replication rate of Clade 1. Furthermore, Clade 2, while demonstrating enhanced antimicrobial resistance, has not suffered the same loss of fitness, another possible contributor to its successful establishment within the Australian health care environment.
Evolution to attenuated virulence but preserved in vivo persistence in ST239 MRSA.
Reduced virulence has been demonstrated in HA-MRSA clones, particularly those that are multidrug resistant (29–31); however, the evolution of virulence over time has not been investigated. To determine the impact of hospital adaptation on the virulence of Australian ST239 MRSA, the activity of the accessory gene regulator (agr), the primary global virulence regulatory system in S. aureus, was measured, and the results were confirmed in a murine infection model.
δ-Hemolysin production.
The activity of the agr complex was assessed by determining the phenotypic production of δ-hemolysin (32). Of the 87 isolates in the collection, 40 (46%) demonstrated δ-hemolysis and were thus considered to have an active agr locus. The remaining 47 (54%) did not demonstrate δ-hemolysis and were therefore considered agr defective. A comparison of agr activity and the year of isolation showed a dramatic shift in the proportion of isolates with an active agr system over time. Nearly 84% (16/19) of the isolates recovered during the 1980s appeared to have an active agr locus. However, this dropped to 52% (12/23) of the isolates recovered between 1998 and 2002 and 31% (11/36) of those recovered between 2003 and 2007. All of the isolates recovered after 2007 appeared to be agr defective, although this time frame has been sparsely sampled. This suggests strong selection pressure against agr activity in the Australian health care environment and is consistent with the recent work of Paulander et al. (33) in which exposure to selected antimicrobials resulted in increased agr expression and reduced bacterial fitness. They proposed that certain antibiotics may be indirectly driving the selection of agr-defective isolates by imposing an RNAIII-associated fitness cost on isolates with functional loci. When the agr activities of the phylogenetic clades were compared, it became apparent that all Clade 2 isolates were potentially agr defective. When Clade 2 is excluded, a decrease in agr activity over time is still observed, the percentage of Clade 1 isolates with an active agr locus dropping from 84% to 75% (12/16) and then to 52% (11/21) for the pre-1990, 1998-to-2002, and 2003-to-2007 time frames, respectively.
Reduction of agr activity is most commonly associated with mutations in the agrABCD locus (32, 34). In the ST239 Australian collection, only eight SNPs and two single base insertions were identified within agrABCD but only seven of these resulted in nonsynonymous amino acid changes (see Table S2 in the supplemental material). Intriguingly, a total of 37 isolates were identified with a defective agr system but there was no mutation in agrABCD to explain the loss of function. When the presence of agrABCD mutation was compared to our regional phylogeny, it became apparent that the most common SNPs, resulting in predicted AgrCR6S and AgrCR393C changes, were found only within Clade 1 isolates (see Fig. S2 in the supplemental material). The predicted AgrCR6S change was identified in all Clade 1 isolates, regardless of agr activity. Alternatively, the AgrCR393C change was identified in a subset of five isolates, all agr defective and all belonging to a single subclade of Clade 1. The majority of the isolates that we observed with a defective agr system and no agr mutations identified belonged to Clade 2.
RNAIII expression.
To confirm these findings, reverse transcription-quantitative PCR was performed with a subset of isolates randomly selected from three groups, Clade 1 isolates recovered during the 1980s, Clade 1 isolates recovered after 1990, and Clade 2 isolates. RNAIII expression among the representative Clade 1 isolates revealed a dramatic reduction in the expression of the agr complex over time (Fig. 4A), consistent with the findings of the δ-hemolysin assay. Isolates recovered in the 1980s showed very high RNAIII expression, often above that of the strong positive control JKD6159, a highly virulent ST93 community MRSA isolate from Australia that produces large amounts of α-hemolysin (35), a product positively regulated by RNAIII. The discovery of isolates with expression levels higher than that of JKD6159 suggested that early ST239 isolates were potentially highly virulent but extended evolution within the hospital environment has led to less virulent phenotypes. Of these 12 isolates, only one produced a result inconsistent with the δ-hemolysin assay. BPH2070 showed no δ-hemolysis but expressed RNAIII at low levels. This suggests that a negative result in the initial δ-hemolysis screen may be indicative of both negative and very-low-level RNAIII expression. Of the four Clade 1 isolates that did not express RNAIII, only BPH2095 had an agr mutation that could account for the loss of activity, carrying a single base insertion within agrC resulting in a reading frameshift. No Clade 2 isolates demonstrated δ-hemolysis; however, four of the five representative isolates expressed RNAIII at low levels, much like BPH2070. The single Clade 2 isolate (BPH2016) that did not express RNAIII carried a single point mutation with a predicted AgrCP24S amino acid change. These results suggest that most Clade 2 isolates that do not possess an agrABCD mutation may express RNAIII at low levels. Furthermore, it highlights that at least three different mechanisms are contributing to attenuated virulence in the ST239 clone, mutation within the locus disrupting its activity, as well as external factors leading to both complete inactivity and reduced RNAIII expression.
FIG 4 .

Evolution of ST239 MRSA toward attenuated virulence. (A) Fold changes in RNAIII expression levels compared to control JKD6159, as determined by reverse transcription-quantitative PCR. Bar color indicates activity of the AGR complex as determined by the delta-hemolysin assay (black, active; grey, inactive). Colored boxes below isolate names indicate those isolates that were tested in the in vivo murine sepsis model; colors represent the temporal groups to which they belong (see panel B). (B) Results obtained with the in vivo sepsis model. (Left) Seven-day survival curves grouped by year of isolation as indicated in the key. Each group represents four bacterial isolates, each tested in 16 animals. The P value is for a comparison of Kaplan-Meier curves with a log-rank test. (Right) Bacterial burdens in the kidneys at 7 days postinjection. Each data point represents one animal infected with one isolate. The detection threshold is shown by the grey line; only values above this level are displayed.
In vivo sepsis model.
To determine whether Australian ST239 MRSA was indeed evolving toward attenuated virulence, eight of the 12 Clade 1 isolates, four from each temporal group, tested for RNAIII expression were also assessed in an in vivo murine sepsis model. As the reduction in RNAIII expression over time was observed only in Clade 1, Clade 2 isolates were not tested in this model. The results of the in vivo murine sepsis model demonstrated that Clade 1 isolates recovered in the 1980s were significantly more virulent (Fig. 4B), in terms of both survival (P = 0.0054) and weight loss at day 2 (P < 0.001), day 3 (P = 0.046), and day 4 (P = 0.0056), compared to newer clade 1 isolates (see Fig. S4 in the supplemental material), confirmation of the evolution of this clade toward attenuated virulence. Additionally, it has been well demonstrated that RNAIII expression contributes to virulence (35). Consistent with this, when isolates were regrouped on the basis of RNAIII expression, those with high-level expression were more virulent in the murine model than isolates with moderate or no RNAIII expression, as shown by both survival (P = 0.0027) and weight loss at day 2 (P < 0.0001) and day 4 (P = 0.0434) (see Fig. S4 in the supplemental material). Interestingly, there appeared to be no difference in virulence between isolates with moderate RNAIII expression and those with no RNAIII expression. The regulation of virulence in S. aureus is multifactorial, and these results could indicate that lower-level RNAIII expression may be insufficient in these isolates for the transcriptional regulation of downstream virulence determinants. Alternatively, isolates with defective agr loci may have compensatory mechanisms of pathogenesis that are independent of agr and have not been explored in this study.
Previous studies have shown that isolates with attenuated virulence are capable of persistent infection (27), with a viable bacterial cell count detectable in the kidneys at 7 days postinjection in a murine sepsis model. We also noted that our isolates were capable of persistence; however, it was not associated with a specific phenotypic group (Fig. 4B). Our results suggest that ST239 MRSA is capable of persistent infection regardless of its relative virulence level or evolutionary age.
Conclusions.
Using detailed genomic and phenotypic analyses, this study has revealed a very clear picture of the localized evolution of one of the most important global clones of HA-MRSA. The high resolving power of bacterial genomics has uncovered a previously unrecognized reimportation of ST239 MRSA into Australia, perpetuating the ST239 epidemic in our region, and demonstrated the genomic diversity between the cocirculating clades. Changes common to these two independent clades have revealed an adaptation toward increased antimicrobial resistance and a HA-MRSA clone that is primed for higher-level glycopeptide and daptomycin resistance. The apparent cost of these changes has been a significant reduction in the replication rate and virulence measured by analysis of doubling times, agr activity, and a murine sepsis model. However, these changes may actually represent coselected adaptive advantages, as the capacity of the ST239 clone to cause persistent invasive infection remains significant. These results shed new light on the complex evolution of S. aureus clones that predominate within the health care environment and illustrate how projects utilizing both comparative genomic and functional approaches are essential to understanding the impact of current antibiotic usage polices on hospital pathogens.
MATERIALS AND METHODS
Bacterial isolates.
This study utilized a temporal and geographically dispersed collection of 123 ST239 S. aureus isolates. The culture collection consisted of 87 isolates recovered from the Australia-New Zealand region between 1980 and 2012. For genomic analyses, this collection was supplemented with the genome sequences of an additional 35 isolates from the international collection of Harris et al. (10). Relevant isolate demographics are available in Data Set S1 in the supplemental material.
Genome sequencing and analyses.
Of the Australian isolates, 71 were sequenced in this study with MiSeq (Illumina) and a read length of 2 × 150 bp. The genome sequence reads of previously published strains are available under the following accession numbers: TW20, GenBank accession no. FN433596; the 35 international ST239 strains, Sequence Read Archive accession no. ERA000102; JKD6008, GenBank accession no. CP002120. Genomic and phylogenetic analyses were performed as described in Text S1 in the supplemental material.
Phenotypic analyses.
The experimental methodology and analysis of antimicrobial susceptibility, growth rate, functionality of the agr locus and the in vivo murine sepsis model are described in Text S1 in the supplemental material. All animal studies were performed in accordance with the Animal Research Ethics Committee at the University of Melbourne.
Nucleotide sequence accession number.
The sequence reads determined in this study were submitted to the European Nucleotide Archive (ENA) under study number ERP009308, and can be accessed via http://www.ebi.ac.uk/ena/data/view/ERP009308.
SUPPLEMENTAL MATERIAL
All of the materials and methods used in this study. Download
Summary of study isolates and genome sequencing. The relevant demographic and whole-genome sequencing information of all of the isolates included in this study are shown. Download
Comparison of the global phylogeny of ST239 MRSA inferred by Bayesian and ML methods. This tanglegram, generated in Dendroscope v3.2.10, compares the topology of the ST239 MRSA global phylogeny inferred by Bayesian (A) and ML (B) analyses. Both trees are displayed as cladograms. Branch annotations provide isolate names, continents, and years recovered and are joined by a grey line to indicate their location within the respective trees. Download
Phenotypic characterization of Australian ST239 S. aureus strains. (A) Global ST239 phylogeny presented in Fig. 1A. Phenotypic data are presented for Australian isolates only, grouped into clades and ordered as they appear in the global phylogeny. Each isolate name is followed by the institution (three-letter code) at which it was isolated and then the year in which it was recovered. The Australian state is indicated by the colored box (see key). (B) Heat maps representing the isolate specific phenotypes for doubling time (minutes; gradient of red [32.3] to yellow [76.1]) and MICs (µg/ml; gradient of blue to red) of vancomycin (0.5 to 4.0), teicoplanin (0.25 to 12.0), and daptomycin (0.094 to 4.0). Reduced glycopeptide susceptibility is indicated by a grey (hVISA) or black (VISA) circle. On the right, the function of the agr, as determined by δ-hemolysin production, is shown. Mutations within the locus are displayed adjacent to the heat map (see key). Download
Increasing antimicrobial resistance of ST239 MRSA. The graphs illustrate changes in the MICs of vancomycin (A), teicoplanin (B), and daptomycin (C) for Australian ST239 MRSA over time. MICs are graphed on a log2 scale. The temporal and phylogenetic clade of each isolate is indicated by the inner and outline colors of the data point, respectively (see key). Log trend lines used to estimate the MIC changes over time across the whole data set (black), Clade 1 (blue), and Clade 2 (red) are included. Download
Evolution of ST239 S. aureus toward attenuated virulence. (A) Graphs showing the results of in vivo sepsis model analyses including 7-day survival curves and respective weight loss data for days 2, 3, and 4 postinjection, grouped by year of isolation. Each group represents four bacterial isolates, each tested in 16 animals. (B) Graphs illustrating the same data regrouped by RNAIII expression levels. The “high” and “medium” groups each represent three isolates, and the “low” group represents two isolates. In survival curves, P values are for comparisons of Kaplan-Meier curves with a log-rank test. In weight loss graphs, P values are for average weight loss comparisons of groups with an unpaired t test for year groups and one-way analysis of variance for RNAIII expression groups. Error bars show the standard errors of the means. (C) Bacterial burdens in the liver, kidneys, and spleen at 7 days postinjection. Each data point represents one animal infected with one isolate; however, each result is represented twice in the graph, once for the year group and once for each RNAIII expression group. The detection threshold is shown as a grey line; only values above this level are displayed. Download
WalKR mutations in Australian ST239 MRSA. All of the walKR SNPs identified in isolates of the Australian collection are listed alongside their respective phylogenetic clades, vancomycin MICs, and glycopeptide susceptibility phenotypes.
SNPs detected within the agr locus. All of the mutations detected within the agr locus of the Australian collection are listed, and the prevalence of each when isolates are divided by agr functionality, as determined by the production of delta-hemolysin, is shown.
ACKNOWLEDGMENTS
This work was funded by the National Health and Medical Research Council, Australia (fellowships APP1023526 to B.P.H., APP1008549 to T.P.S., and APP1061409 to K.E.H.).
We thank Elizabeth Grabsch of the Microbiology Department of Austin Health for assistance with the initial pulsed-field gel electrophoresis characterization of isolates.
Footnotes
Citation Baines SL, Holt KE, Schultz MB, Seemann T, Howden BO, Jensen SO, van Hal SJ, Coombs GW, Firth N, Powell DR, Stinear TP, Howden BP. 2015. Convergent adaptation in the dominant global hospital clone ST239 of methicillin-resistant Staphylococcus aureus. mBio 6(2):e00080-15. doi:10.1128/mBio.00080-15.
Contributor Information
Peter Gilligan, UNC Health Care System.
Vance G Fowler, Duke University Medical Center.
REFERENCES
- 1.Wyllie D, Paul J, Crook D. 2011. Waves of trouble: MRSA strain dynamics and assessment of the impact of infection control. J Antimicrob Chemother 66:2685–2688. doi: 10.1093/jac/dkr392. [DOI] [PubMed] [Google Scholar]
- 2.Enright MC, Robinson DA, Randle G, Feil EJ, Grundmann H, Spratt BG. 2002. The evolutionary history of methicillin-resistant Staphylococcus aureus (MRSA). Proc Natl Acad Sci U S A 99:7687–7692. doi: 10.1073/pnas.122108599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McAdam PR, Templeton KE, Edwards GF, Holden MT, Feil EJ, Aanensen DM, Bargawi HJ, Spratt BG, Bentley SD, Parkhill J, Enright MC, Holmes A, Girvan EK, Godfrey PA, Feldgarden M, Kearns AM, Rambaut A, Robinson DA, Fitzgerald JR. 2012. Molecular tracing of the emergence, adaptation, and transmission of hospital-associated methicillin-resistant Staphylococcus aureus. Proc Natl Acad Sci U S A 109:9107–9112. doi: 10.1073/pnas.1202869109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Robinson DA, Enright MC. 2004. Evolution of Staphylococcus aureus by large chromosomal replacements. J Bacteriol 186:1060–1064. doi: 10.1128/JB.186.4.1060-1064.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pavillard R, Harvey K, Douglas D, Hewstone A, Andrew J, Collopy B, Asche V, Carson P, Davidson A, Gilbert G, Spicer J, Tosolini F. 1982. Epidemic of hospital-acquired infection due to methicillin-resistant Staphylococcus aureus in major Victorian hospitals. Med J Aust 29:451–454. [PubMed] [Google Scholar]
- 6.Cookson BD, Phillips I. 1988. Epidemic methicillin-resistant Staphylococcus aureus. J Antimicrob Chemother 21:57–65. doi: 10.1093/jac/21.suppl_C.57. [DOI] [PubMed] [Google Scholar]
- 7.Dubin DT, Chikramane SG, Inglis B, Matthews PR, Stewart PR. 1992. Physical mapping of the mec region of an Australian methicillin-resistant Staphylococcus aureus lineage and a closely related American strain. J Gen Microbiol 138:169–180. doi: 10.1099/00221287-138-1-169. [DOI] [PubMed] [Google Scholar]
- 8.Rountree PM, Beard MA. 1968. Hospital strain of Staphylococcus aureus with particular reference to methicillin-resistant strains. Med J Aust 2:1163–1168. [DOI] [PubMed] [Google Scholar]
- 9.Smyth DS, McDougal LK, Gran FW, Manoharan A, Enright MC, Song J.-H, de Lencastre H, Robinson DA. 2010. Population structure of a hybrid clonal group of methicillin-resistant Staphylococcus aureus, ST239-MRSA-III. PLoS One 5:e8582. doi: 10.1371/journal.pone.0008582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Harris SR, Feil EJ, Holden MT, Quail MA, Nickerson EK, Chantratita N, Gardete S, Tavares A, Day N, Lindsay JA, Edgeworth JD, de Lencastre H, Parkhill J, Peacock SJ, Bentley SD. 2010. Evolution of MRSA during hospital transmission and intercontinental spread. Science 327:469–474. doi: 10.1126/science.1182395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Castillo-Ramírez S, Corander J, Marttinen P, Aldeljawi M, Hanage WP, Westh H, Boye K, Bentley SD, Parkhill J, Holden MT, Feil EJ, Feil EJ. 2012. Phylogeographic variation in recombination rates within a global clone of methicillin-resistant Staphylococcus aureus. Genome Biol 13:R126. doi: 10.1186/gb-2012-13-12-r126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gray RR, Tatem AJ, Johnson JA, Alekseyenko AV, Pybus OG, Suchard MA, Salemi M. 2011. Testing spatiotemporal hypothesis of bacterial evolution using methicillin-resistant Staphylococcus aureus ST239 genome-wide data within a Bayesian framework. Mol Biol Evol 28:1593–1603. doi: 10.1093/molbev/msq319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li M, Du X, Villaruz AE, Diep BA, Wang D, Song Y, Tian Y, Hu J, Yu F, Lu Y, Otto M. 2012. MRSA epidemic linked to a quickly spreading colonization and virulence determinant. Nat Med 18:816–819. doi: 10.1038/nm.2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Espedido BA, Steen JA, Barbagiannakos T, Mercer J, Paterson DL, Grimmond SM, Cooper MA, Gosbell IB, van Hal SJ, Jensen SO. 2012. Carriage of an ACME II variant may have contributed to methicillin-resistant Staphylococcus aureus sequence type 239-like strain replacement in Liverpool Hospital, Sydney, Australia. Antimicrob Agents Chemother 56:3380–3383. doi: 10.1128/AAC.00013-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Horne KC, Howden BP, Grabsch EA, Graham M, Ward PB, Xie S, Mayall BC, Johnson PD, Grayson ML. 2009. Prospective comparison of the clinical impacts of heterogeneous vancomycin-intermediate methicillin-resistant Staphylococcus aureus (MRSA) and vancomycin-susceptible MRSA. Antimicrob Agents Chemother 53:3447–3452. doi: 10.1128/AAC.01365-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Australian Group on Antibiotic Resistance 2004, posting date Staphylococcus aureus programme (SAP) hospital/community surveys, MRSA epidemiology and typing reports: 2003–2011. Australian Group on Antimicrobial Resistance, Perth, WA, Australia: http://www.agargroup.org/surveys. [Google Scholar]
- 17.Holmes NE, Turnidge JD, Munckhof WJ, Robinson JO, Korman TM, O’Sullivan MVN, Anderson TL, Roberts SA, Warren SJC, Coombs GW, Tan H.L, Gao W, Johnson PDR, Howden BP. 2014. Genetic and molecular predictors of high vancomycin minimum inhibitory concentration in Staphylococcus aureus bacteremia isolates. J Clin Microbiol 52:3384–3393. doi: 10.1128/JCM.01320-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Minin VN, Bloomquist EW, Suchard MA. 2008. Smooth Skyride through a rough Skyline: Bayesian coalescent-based inference of population dynamics. Mol Biol Evol 25:1459–1471. doi: 10.1093/molbev/msn090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Coombs GW, Nimmo GR, Pearson JC, Collignon PJ, Bell JM, McLaws ML, Christiansen KJ, Turnidge JD, Australian Group on Antimicrobial Resistance . 2013. Australian Group on Antimicrobial Resistance hospital-onset Staphylococcus aureus surveillance programme annual report, 2011.. Commun Dis Intell Q Rep 37:E210–E218 [DOI] [PubMed] [Google Scholar]
- 20.Howden BP, Davies JK, Johnson PD, Stinear TP, Grayson ML. 2010. Reduced vancomycin susceptibility in Staphylococcus aureus, including vancomycin-intermediate and heterogeneous vancomycin-intermediate strains: resistance mechanisms, laboratory detection, and clinical implications. Clin Microbiol Rev 23:99–139. doi: 10.1128/CMR.00042-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.van Hal SJ, Fowler VG Jr.. 2013. Is it time to replace vancomycin in the treatment of MRSA infections? Clin Infect Dis 56:1779–1788. doi: 10.1093/cid/cit178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Howden BP, Ward PB, Charles PG, Korman TM, Fuller A, du Cros P, Grabsch EA, Roberts SA, Robson J, Read K, Bak N, Hurley J, Johnson PD, Morris AJ, Mayall BC, Grayson ML. 2004. Treatment outcomes for serious infections caused by methicillin-resistant Staphylococcus aureus with reduced vancomycin susceptibility. Clin Infect Dis 38:521–528. doi: 10.1086/381202. [DOI] [PubMed] [Google Scholar]
- 23.Charles PG, Ward PB, Johnson PD, Howden BP, Grayson ML. 2004. Clinical features associated with bacteremia due to heterogeneous vancomycin-intermediate Staphylococcus aureus. Clin Infect Dis 38:448–451. doi: 10.1086/381093. [DOI] [PubMed] [Google Scholar]
- 24.Matsuo M, Hishinuma T, Katayama Y, Cui L, Kapi M, Hiramatsu K. 2011. Mutation of RNA polymerase β subunit (rpoB) promotes hVISA-to-VISA phenotypic conversion of strain Mu3. Antimicrob Agents Chemother 55:4188–4195. doi: 10.1128/AAC.00398-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Alam MT, Petit RA III, Crispell EK, Thornton TA, Conneely KN, Jiang Y, Satola SW, Read TD. 2014. Dissecting vancomycin-intermediate resistance in Staphylococcus aureus using genome-wide association. Genome Biol Evol 6:1174–1185. doi: 10.1093/gbe/evu092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Howden BP, McEvoy CR, Allen DL, Chua K, Gao W, Harrison PF, Bell J, Coombs G, Bennett-Wood V, Porter JL, Robins-Browne R, Davies JK, Seemann T, Stinear TP. 2011. Evolution of multidrug resistance during Staphylococcus aureus infection involves mutation of the essential two component regulator WalKR. PLoS Pathog 7:e1002359. doi: 10.1371/journal.ppat.1002359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gao W, Cameron DR, Davies JK, Kostoulias X, Stepnell J, Tuck KL, Yeaman MR, Peleg AY, Stinear TP, Howden BP. 2013. The rpoB H481Y rifampicin resistance mutation and an active stringent response reduce virulence and increase resistance to innate immune responses in Staphylococcus aureus. J Infect Dis 207:929–939. doi: 10.1093/infdis/jis772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Okuma H, Iwakawa K, Turnidge JD, Grubb WB, Bell JM, O’Brien FG, Coombs GW, Pearman JW, Tenover FC, Kapi M, Tiensasitorn C, Ito T, Hiramatsu K. 2002. Dissemination of new methicillin-resistant Staphylococcus aureus clones in the community. J Clin Microbiol 40:4289–4294. doi: 10.1128/JCM.40.11.4289-4294.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rudkin JK, Edwards AM, Bowden MG, Brown EL, Pozzi C, Waters EM, Chan WC, Williams P, O’Gara JP, Massey RC. 2012. Methicillin resistance reduces the virulence of healthcare-associated methicillin-resistant Staphylococcus aureus by interfering with the agr quorum sensing system. J Infect Dis 205:798–806. doi: 10.1093/infdis/jir845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Howden BP, Johnson PD, Ward PB, Stinear TP, Davies JK. 2006. Isolates with low-level vancomycin resistance associated with persistent methicillin-resistant Staphylococcus aureus bacteremia. Antimicrob Agents Chemother 50:3039–3047. doi: 10.1128/AAC.00422-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sakoulas G. 2006. The accessory gene regulator (agr) in methicillin-resistant Staphylococcus aureus: role in virulence and reduced susceptibility to glycopeptide antibiotics. Drug Discov Today Dis Mech 3:287–294. doi: 10.1016/j.ddmec.2006.06.007. [DOI] [Google Scholar]
- 32.Herbert S, Ziebandt A.-K, Ohlsen K, Schäfer T, Hecker M, Albrecht D, Novick R, Götz F. 2010. Repair of global regulators in Staphylococcus aureus 8325 and comparative analysis with other clinical isolates. Infect Immun 78:2877–2889. doi: 10.1128/IAI.00088-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Paulander W, Nissen Varming A, Bæk KT, Haaber J, Frees D, Ingmer H. 2013. Antibiotic-mediated selection of quorum-sensing-negative Staphylococcus aureus. mBio 3:e00459–e00412. doi: 10.1128/mBio.00459-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Moore PC, Lindsay JA. 2001. Genetic variation among hospital isolates of methicillin-sensitive Staphylococcus aureus: evidence for horizontal transfer of virulence genes. J Clin Microbiol 39:2760–2767. doi: 10.1128/JCM.39.8.2760-2767.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chua KY, Monk IR, Lin Y.-H, Seemann T, Tuck KL, Porter JL, Stepnell J, Coombs GW, Davies JK, Stinear TP, Howden BP. 2014. Hyperexpression of α-hemolysin explains enhanced virulence of sequence type 93 community-associated methicillin-resistant Staphylococcus aureus. BMC Microbiol 14:31. doi: 10.1186/1471-2180-14-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
All of the materials and methods used in this study. Download
Summary of study isolates and genome sequencing. The relevant demographic and whole-genome sequencing information of all of the isolates included in this study are shown. Download
Comparison of the global phylogeny of ST239 MRSA inferred by Bayesian and ML methods. This tanglegram, generated in Dendroscope v3.2.10, compares the topology of the ST239 MRSA global phylogeny inferred by Bayesian (A) and ML (B) analyses. Both trees are displayed as cladograms. Branch annotations provide isolate names, continents, and years recovered and are joined by a grey line to indicate their location within the respective trees. Download
Phenotypic characterization of Australian ST239 S. aureus strains. (A) Global ST239 phylogeny presented in Fig. 1A. Phenotypic data are presented for Australian isolates only, grouped into clades and ordered as they appear in the global phylogeny. Each isolate name is followed by the institution (three-letter code) at which it was isolated and then the year in which it was recovered. The Australian state is indicated by the colored box (see key). (B) Heat maps representing the isolate specific phenotypes for doubling time (minutes; gradient of red [32.3] to yellow [76.1]) and MICs (µg/ml; gradient of blue to red) of vancomycin (0.5 to 4.0), teicoplanin (0.25 to 12.0), and daptomycin (0.094 to 4.0). Reduced glycopeptide susceptibility is indicated by a grey (hVISA) or black (VISA) circle. On the right, the function of the agr, as determined by δ-hemolysin production, is shown. Mutations within the locus are displayed adjacent to the heat map (see key). Download
Increasing antimicrobial resistance of ST239 MRSA. The graphs illustrate changes in the MICs of vancomycin (A), teicoplanin (B), and daptomycin (C) for Australian ST239 MRSA over time. MICs are graphed on a log2 scale. The temporal and phylogenetic clade of each isolate is indicated by the inner and outline colors of the data point, respectively (see key). Log trend lines used to estimate the MIC changes over time across the whole data set (black), Clade 1 (blue), and Clade 2 (red) are included. Download
Evolution of ST239 S. aureus toward attenuated virulence. (A) Graphs showing the results of in vivo sepsis model analyses including 7-day survival curves and respective weight loss data for days 2, 3, and 4 postinjection, grouped by year of isolation. Each group represents four bacterial isolates, each tested in 16 animals. (B) Graphs illustrating the same data regrouped by RNAIII expression levels. The “high” and “medium” groups each represent three isolates, and the “low” group represents two isolates. In survival curves, P values are for comparisons of Kaplan-Meier curves with a log-rank test. In weight loss graphs, P values are for average weight loss comparisons of groups with an unpaired t test for year groups and one-way analysis of variance for RNAIII expression groups. Error bars show the standard errors of the means. (C) Bacterial burdens in the liver, kidneys, and spleen at 7 days postinjection. Each data point represents one animal infected with one isolate; however, each result is represented twice in the graph, once for the year group and once for each RNAIII expression group. The detection threshold is shown as a grey line; only values above this level are displayed. Download
WalKR mutations in Australian ST239 MRSA. All of the walKR SNPs identified in isolates of the Australian collection are listed alongside their respective phylogenetic clades, vancomycin MICs, and glycopeptide susceptibility phenotypes.
SNPs detected within the agr locus. All of the mutations detected within the agr locus of the Australian collection are listed, and the prevalence of each when isolates are divided by agr functionality, as determined by the production of delta-hemolysin, is shown.