

1. Introduction and Scope
The present advances in biology are related to progress in genomics, proteomics, and other “-omics”, including glycomics, working with a large amount of data regarding human and other genomes, protein expression, post-translational modifications of proteins, as well as a great diversity of glycan composition in glycoproteins, etc. Genomics, proteomics, and glycomics are intimately connected with each other at various levels. In their exponential growth, they require new integrative technologies for highly parallel analysis of genes and proteins of entire organisms.1 The current experimental techniques applied in these fields have been reviewed in a number of articles.1−13 Among them, finding a review on electrochemical (EC) techniques in proteomics is rather difficult.14 This is in contrast to a very large amount of reviews on EC analysis of nucleic acids and particularly on sensors and arrays applicable in genomics, which appeared in the recent decade.15−36 Also, reviews on EC analysis of glycoproteins are rather scarce, limited mostly to promising EC impedance spectroscopic detection of lectin-captured glycoproteins.37−42 Wider application of EC analysis in proteomics and biomedicine was hindered until recently by the absence of a sensitive EC reaction applicable to thousands of proteins existing in nature. However, interfacial electrochemistry of conjugated proteins containing nonprotein redox centers (such as some metalloproteins) allowing direct (i.e., unmediated) and reversible electron transfer between electrode and nonprotein component greatly advanced in recent decades.43−48 The number of metalloproteins in nature is very large; unfortunately, only a very small fraction among them was shown to yield such reversible electrochemistry (see section 3 for details). To make methods of EC analysis more convenient for application in biomedicine and in the above “-omics”, advances in both label-free and label-based EC methods of proteins and carbohydrate components of glycoproteins analysis are desirable.
In this Review, we wish to show that in recent years significant progress was done in the EC analysis of practically all proteins, based on the electroactivity of amino acid (aa) residues in proteins. Also, electrochemistry of polysaccharides, oligosaccharides, and glycoproteins greatly advanced in creating important steps for its larger application in the glycoprotein research. In recent decades, a great effort was devoted to the discovery and application of biomarkers for analysis of different diseases, including cancer.49−53 In the following paragraphs, special attention will be paid (i) to intrinsic electroactivity of peptides and proteins, including the sensitivity to changes in protein 3D structures (sections 4–6), as well as to recent advances in EC investigations of DNA–protein interactions (section 7), (ii) to intrinsic electroactivity of glycans and polysaccharides, advances in EC detection of lectin–glycoprotein interactions, and introduction of electroactive labels to polysaccharides and glycans (section 8), and finally (iii) to EC detection of protein biomarkers, based predominantly on application of antibodies in immunoassays, nucleic acid and peptide aptamers for construction of aptasensors, and lectin biosensors for detection of glycoprotein biomarkers (section 9).
1.1. Intrinsic Electroactivity of Proteins
Since the beginning of the 1970s, EC analysis of proteins focused on reversible processes of nonprotein components in conjugated proteins. This very interesting electrochemistry was reviewed in numerous articles43−48 and will be here only briefly mentioned in connection to proteins involved in the DNA repair (section 7). At the beginning of the 1980s, it was shown that tyrosine (Tyr) and tryptophan (Trp) residues in proteins produced voltammetric oxidation signals at carbon electrodes.54−56 In the first decade after this discovery, the oxidation signals of proteins exhibited only low sensitivity, but later by using different carbon electrodes and EC techniques, these signals became more useful tools in electrochemical protein analysis (section 4) and were applied in biomedical research. Recently, a simple label-free chronopotentiometric stripping (CPS) electrocatalytic method has been introduced (section 5), allowing the determination of practically any protein at low concentration, as well as recognition of changes in the protein structures (section 5.3), including those resulting from a single aa exchange (point mutations). The protein structure-sensitive analysis requires very fast potential changes (taking place at highly negative current densities), which can be hardly obtained using the usual voltammetric techniques. Special properties of CPS in relation to protein analysis are discussed in sections 5.1–5.3. For protein structure-sensitive analysis, thiol-modified liquid mercury or solid amalgam electrodes are convenient (section 5.4). CPS appeared particularly useful in the analysis of proteins important in biomedicine (section 6), including tumor suppressor p53 protein (section 6.2) and its sequence-specific interaction with DNA (section 7.5).
1.2. DNA–Protein Interactions
Until recently, EC methods were little used in DNA–protein interaction studies and were not included among the methods listed in handbooks on DNA–protein interaction analysis.57 In section 7, we review new EC methods dealing with DNA–protein interactions, which play significant roles in nature (e.g., sequence-specific transcription factors binding to DNA). These methods are based on different principles, and some of them show significant advantages over the methods commonly used in DNA–protein interaction studies. To our knowledge, section 7 represents the first comprehensive review on EC analysis of DNA–protein interactions. This section does not cover literature on DNA or RNA aptamers, which are mentioned only in relation to biomarkers in section 9.
1.3. Analysis of Glycoproteins
It is estimated that 70% of cytosolic proteins and 80% of membrane-bound proteins are glycosylated. It is thus very important to analyze glycans for better understanding of their role in the cell physiology and pathology and to develop novel and robust methods applicable in diagnostics. EC analysis of glycans is gaining increasing attention, providing an exceptionally low limit of detections (LODs) and in some cases also a label-free format of analysis. Moreover, EC analysis of glycans can be performed on various intact cell lines. Section 8 begins with a short historical overview on the development of EC methods of glycan analysis. It then continues with methods relying on a glycan release from glycoproteins. The most recent schemes, where EC detection platform can be applied for a direct glycoprofiling of glycoproteins and even intact cells (section 8.3.4), are discussed. A novel method of catalytic hydrogen evolution reaction in glucosamine-containing carbohydrates and selective modification of saccharides by osmium Os(VI) complexes has a potential for detection of glycoproteins in the future. Lectins, natural interacting partners of glycans, after being integrated into EC platforms of detection could analyze intact glycoproteins down to a single molecule level. Two EC detection platforms are discussed including label-free and label-based approaches, and some applications of such devices are provided.
1.4. Detection of Protein Biomarkers
Many diseases, including cancer, could be efficiently cured if early diagnosed. The diagnostics, among others, relies on detection of protein biomarkers, which circulate at elevated concentrations in body fluids sometimes even before any other symptoms appear. This makes early diagnostics extremely important not only because it saves and increases the quality of life, but also because it greatly decreases financial cost associated with the treatment of diseases in advanced stages.
Protein biomarkers can be detected using EC methods, offering such advantages as low cost, short time of analysis, and excellent sensitivity. In section 9, we describe a recent progress in development of strategies for ultrasensitive determination of protein biomarkers, including protein labeling. Many authors still use purified, commercially available samples, which can be important to demonstrate a proof of concept or to develop a novel strategy, but it is very important not to forget about application of real samples obtained from patients’ sera and other body fluids, where protein biomarkers can be often found at very low concentrations present in excess of a huge amount of interfering species.
The most current papers report on the development of immunoassays based on surface-immobilized antibodies (in most cases directly at the electrode) for binding of target biomarker. This binding is usually followed by introduction of labeled secondary antibody and by monitoring the resulting signal, which is greatly amplified through action of enzymes or nanoparticles (section 9.2). In the last years, synthetic aptamers (either nucleic acid- or peptide-based) with high affinity to various proteins are being more often used, having the same function as an immobilized antibody. In such a case, the aptamer-based platform is referred to as the aptasensor, instead of antibody-based immunoassays (or immunosensors) (sections 9.3 and 9.4). Last, if the protein biomarker is a glycoprotein (section 9.5), it can be advantageous to specifically capture its glycan part using lectins. Such lectin biosensors were constructed not only for detection of protein molecules, but also for identification of whole cancer cells.
Because we have touched on the topic of biosensors, we should make a short comment regarding correct terminology. Considering today’s literature on DNA or protein biosensors, as compared to the literature on sensors in general,58 one should, strictly speaking, refer to them more correctly as “sensing systems” or “bioelectrochemical assays”, because a true biosensor acquires information continuously, while a sensing system may do that in discrete steps.58 In this Review, we will use the more widespread, although not terminologically perfect, term “biosensor”, with its more relaxed definition stating that biosensors are devices that combine59 or integrate60 a biochemical recognition element with a signal conversion unit (transducer). This confusion in terminology was discussed in detail in our previous review.34
2. History of Protein Electrochemistry
Proteins were the first biomacromolecules that were analyzed by EC methods. In 1930, that is, only 8 years after J. Heyrovský’s invention of polarography, Heyrovský and Babička published their paper showing that albumins, in the presence of ammonium ions, produced the direct current (dc) polarographic “presodium wave” (Figure 1), for which catalytic evolution of hydrogen was responsible.61 Two years later, Herles and Vančura62 showed that the presodium wave was produced by various human body fluids, including blood serum and urine. Their work started several years earlier when J. Heyrovský gave a chance to young medical doctors to study polarographic activity of various human tissue liquids using the polarographic instrument in his laboratory. At that time, polarographs were not commercially available, and the young M.D.’s utilized their unique chance very efficiently. They described the body fluid-produced “presodium wave” as a cathodic wave occurring at potentials little more positive than the polarographic reduction wave of sodium ions and tentatively assigned this wave to proteins.
Figure 1.

Polarographic catalytic waves of human serum. (1) Pure supporting electrolyte, 0.1 M ammonia/ammonium chloride buffer; (2) the “presodium” catalytic wave, 400-times diluted human serum (A) in 0.1 M ammonia/ammonium chloride; (3) two-step reduction of Co(III), 1 mM Co(NH3)6Cl3 in 0.1 M ammonia/ammonium chloride; (4) the catalytic double-wave in Brdička solution, 1 mM Co(NH3)6Cl3 + 400-times diluted human serum (A′) in 0.1 M ammonia/ammonium chloride; recorded from 0 V vs mercury pool, 200 mV/abscissa. Adapted with permission from ref (64). Copyright 1933 Collection of Czechoslovak Chemical Communications.
Later, the “presodium wave” was characterized by Brdička63 in greater detail. He showed that the presence of ammonium ion was not essential for the reaction and concluded that the “presodium wave” was due to −SH groups in proteins similarly to his previously discovered electrocatalytic “double wave”, produced by thiols in the presence of cobalt ions.64 Comparison of the two polarographic responses of proteins showed that the “presodium wave” is much higher than the Brdička’s “double wave” and the former wave does not require the presence of cobalt ions in the electrolyte.65 It was concluded from a number of later studies (e.g., refs (66−69)) that the “presodium wave” did not depend specifically on the −SH or another group in the catalyst molecule, but rather on its structure, adsorbability, and other factors including solution composition, the electrode potential, and the rate of its change. In its 80 year history, the polarographic presodium wave was only occasionally utilized in the analysis of proteins. The dc polarographic version of this wave was too close to the background discharge and difficult to measure (Figure 1). The indentations of proteins obtained with oscillographic polarography (alternating current (ac) cyclic chronopotentiometry or cyclic reciprocal derivative chronopotentiometry70 according to the present nomenclature) were too close to the shining end point at negative potentials,71 and their evaluation was even more difficult than that of the dc polarographic presodium waves.
In contrast, Brdička’s double wave (Figure 1) was intensively applied in biochemistry, for several decades around the middle of the 20th century,72−75 particularly because of expected application of Brdička’s catalytic response in cancer diagnostics. After yielding some interesting data, the specificity of Brdička’s catalytic response turned out to be insufficient for cancer diagnostics, and the interest in Brdička’s catalytic response declined. In the 1970s, the attention of electrochemists turned to the electrochemistry of proteins containing a redox-active center (reviewed in refs (76−78)), and the outlooks for application of EC methods as tools for analysis of the majority of proteins important in molecular biology and biomedicine appeared grim.
3. Reversible Electrochemistry of Nonprotein Components in Conjugated Proteins
Among conjugated proteins (such as lipoproteins, glycoproteins, etc.), some contain one or more nonprotein redox centers, essential for direct electron transfer (DET) between the protein and its natural acceptor or donor (e.g., metalloproteins). Under conditions when the redox center is sufficiently close to the electrode, DET between this center and the electrode may take place (reviewed in refs (79−83)). The first papers on DET were published by the end of the 1970s.84−86 Pioneering studies described interaction of cytochrome c with either tin-doped indium oxide,86 4,4′-bipyridine-modified gold electrode,84 or mercury electrode.85 At present, this specialized branch of protein electrochemistry is well established as documented by numerous book chapters46,76−78,87−91 and reviews79,80,92−98 and a large number of original papers.
The DET rate from donor to acceptor is dependent on a number of factors, including orientation of the protein molecule at the surface, temperature, distance from the surface, reaction Gibbs energy, reorganization energy, etc. More efficient DET can be obtained by introducing a redox center into more hydrophobic regions far from aqueous environment, thus lowering the reorganization energy. For example, the reorganization energy for the solvent-accessible copper atom in redox pair Cu(1,10-phenantroline)22+/+ is 1.7 eV higher than that for copper embedded in the small protein azurin.99 Experiments with iron yielded similar results.100
Strict requirements for DET in proteins greatly limit application of DET phenomenon for analysis of proteins important in biomedicine. For example, numerous oncoproteins, such as tumor suppressor proteins p53,101,102 p63,103 and p73,104 contain metal ions playing important biological roles in their large molecules,101−104 but they do not yield any DET. However, such proteins are able to produce CPS responses based on catalytic hydrogen evolution reaction,105 sensitively reflecting changes in the protein structures as well as the presence and absence of metal ions in the molecules (section 5.3).
In this section, we shall only briefly summarize the electrochemistry of some conjugated (DNA repair) proteins containing [4Fe–4S] clusters, which have been utilized in EC studies of DNA–protein interactions (section 7). Such clusters were found in glycosylases involved in base excision repair (e.g., Endonuclease III, Endo III) and in a nucleotide excision repair (e.g., Xeroderma pigmentosum factor D, XPD). These enzymes are responsible for searching the genome for damaged bases/nucleotides and for enzymatic catalysis of their excision.106 After the damaged site is located (in a vast amount of intact bases), base excision repair enzymes flip their substrate into the protein active site, and catalyze rupture of the N-glycosidic bond between the damaged base and the DNA sugar–phosphate backbone. The searching process was studied in detail, but some aspects of this process in vivo have not been yet fully understood.107−112
Among glycosylases of this type, Endo III113 and a structurally similar MutY protein114 were identified. Endo III removes oxidized pyrimidines from DNA,107,115−122 while MutY removes adenine from 8-oxo-guanine:adenine mispaired bases.123−133 Crystal structures are available for free and DNA-bound EndoIII and MutY.114,119,120,134,135 In these structures, a [4Fe–4S] cluster is ligated by a cysteine (Cys) motif (C–X6–C–X2–C–X5–C). The [4Fe–4S] cluster is required for an enzyme acitivity and DNA binding, but not for protein folding and thermal stability of the protein.136−138
Earlier studies had shown that the [4Fe–4S]2+ cluster is not readily electro-oxidized nor reduced within the physiological potential range.113,139−142 Gorodetski et al.143 investigated Endo III at a highly oriented pyrolytic graphite electrode (HOPGE) using cyclic voltammetry (CV) and square wave voltammetry (SWV). On a bare HOPGE, they observed an irreversible anodic peak at 250 ± 30 mV against a normal hydrogen electrode, while at the DNA-modified HOPGE, a quasi-reversible redox couple was observed at 20 ± 10 mV (Figure 2). This peak reflected a DNA-bound protein redox process, and it did not result from Endo III interaction with DNA containing an abasic site, which was unable to take part in the DNA-mediated charge transport.34 Interaction of the above base excision repair and the nucleotide excision repair of DNA by proteins144 will be discussed in section 7.2.
Figure 2.
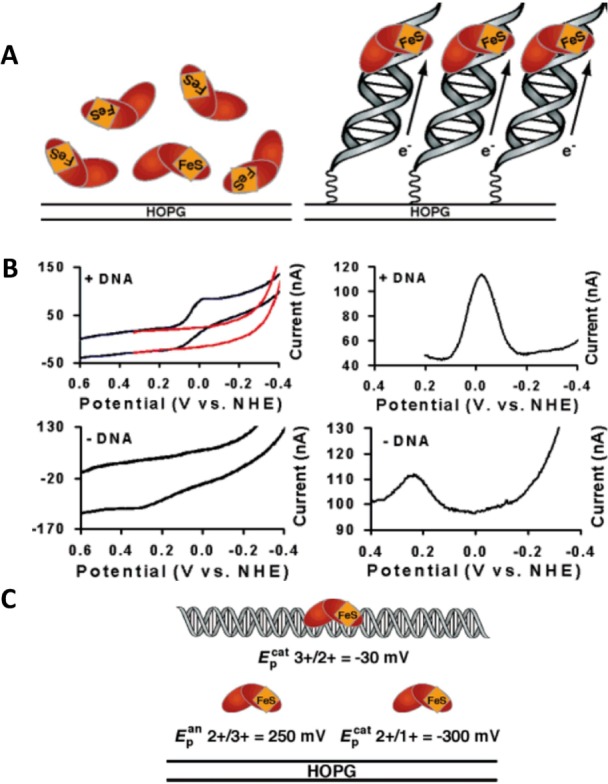
(A) Schematic representation of electrochemistry for endonuclease III (Endo III) on highly oriented pyrolytic graphite electrode (HOPGE) with and without modification with DNA. (B) Cyclic (left, 50 mV/s scan rate) and square wave voltammograms (right, 15 Hz) of 50 μM Endo III in 20 mM Na phosphate, 100 mM NaCl, 1 mM EDTA, 20% glycerol, pH 7.5. The top two panels show electrochemical responses of Endo III at a HOPGE modified with the sequence pyrene-(CH2)4-Pi-5′-AGT ACA GTC ATC GCG-3′ plus complement. Cyclic voltammograms of a HOPGE modified with DNA featuring an abasic site are in red (top left), where the abasic position corresponds to the complement of the italicized base. The bottom two panels show electrochemical responses of Endo III on a bare HOPGE. All runs were taken using the inverted drop cell electrode configuration vs Ag/AgCl reference and Pt auxiliary electrode. (C) Illustration of the potentials vs normal hydrogen electrode (NHE) of the couples of Endo III in the presence and absence of DNA. These values are obtained from SWV on a HOPGE and are averages of at least four trials each. Adapted with permission from ref (143). Copyright 2006 American Chemical Society.
4. Protein Oxidation at Carbon and Other Solid Electrodes
4.1. Free Amino Acids
More than 30 years ago, it was shown that free Cys, histidine (His), methionine (Met), Tyr, and Trp are oxidized at carbon electrodes.56,145 Other aa’s did not produce any oxidation signal at carbon electrodes in a pH range 4–10.54,55,146 Oxidation of the Tyr and Trp occurred at positive potentials far from zero. Application of new carbon-based nanomaterials for biosensing has attracted great attention (reviewed in refs (147−149)). Recently, it has been shown that a glassy carbon electrode (GCE) modified with multiwalled carbon nanotubes and gold nanorods allowed oxidation of free l-Cys at a very low anodic potential (0 V vs Ag/AgCl).150 A modified GCE and boron doped diamond electrode were used also in the analysis of Cys, Tyr, and Met.151,152 Shortly after the discovery of the electro-oxidation of some free aa’s,56,145 it was found that Tyr and Trp residues yield oxidation signals also in proteins.54−56
4.2. Peptides and Proteins
Protein electro-oxidation attracted greater attention in recent decades.146,153−158 SWV or CPS154,159 with efficient baseline correction yielded Tyr and Trp peaks that were better developed and allowed the determination of much lower peptide and protein concentrations than linear sweep voltammetry.146 Moreover, it was found that proteins are strongly adsorbed at carbon electrodes, which made it possible to prepare protein-modified electrodes without covalent binding of the protein to the surface.154 Using adsorptive transfer stripping voltammetry, microliter volumes of proteins were sufficient for the analysis at carbon electrodes.160 In contrast to metal electrodes, such as gold and mercury, at which thiol self-assembled monolayers (SAMs) can be easily formed,161 such SAMs do not form at carbon electrodes. However, tightly packed structures of DNA functionalized with pyrene at HOPGE were shown,162 and reduction of disulfide bonds incorporated in the DNA backbone was demonstrated.163 This system has not been so far widely applied in protein electrochemistry.
Oxidation schemes for Tyr and Trp proposed about three decades ago (Figure 3) are still used in the literature.164 The combination of EC oxidation of peptides and proteins with mass spectrometry (MS) recently revealed a specific cleavage of the peptide bond at the C-terminal side of Trp and Tyr residues.165,166 A set of Tyr and Trp-containing tripeptides (e.g., LYL, EYE, LWL) was studied, including the effect of adjacent aa residues. It was found that the ratio of oxidation and cleavage products is sequence-dependent and that the secondary chemical reactions occurring after the initial oxidation step are influenced by the adjacent aa residues.167 Control of the oxidation potential appears critical for avoiding dimer formation of Tyr and increasing hydroxylation of Trp. Working at pH values 1.9–3.1 resulted in optimal cleavage yields, but at basic pH’s no or a little cleavage took place. In proteomic experiments, usually enzymatic or chemical protein cleavage is used. Electrochemistry may offer a fast and simple instrumental alternative to these cleavage methods. The above-mentioned studies revealed complicated reaction schemes, but the primary step in Trp oxidation was in agreement with that proposed a long time ago (Figure 3). Using GCE, Enache and Brett168 investigated the pathways of EC oxidation of Trp and other indole-containing compounds with a substituent at C3 position. They found that oxidation of Trp occurs at the C2 position of the pyrrole ring followed by the hydroxylation at the C7 position of the indole benzene moiety in an irreversible pH-dependent process.
Figure 3.

Schemes of EC oxidation of tyrosine (Tyr) and tryptophan (Trp). Adapted with permission from ref (164). Copyright 2013 Wiley-VCH Verlag GmbH&Co.
By the end of the 20th century, it was believed that electroactivity of aa residues in proteins was limited to oxidation of Tyr and Trp. Recently, oxidation of His residues in a protein was reported at highly positive potentials on GCE.158,169 His oxidation peak was not observed in His-containing angiotensin peptides at a basal plane pyrolytic graphite,170 but it was not excluded that His oxidation may occur in proteins and peptides at GCE and other electrodes. Investigations of electroactivity of His residues in proteins are particularly interesting, because His-tags (usually short chains of six His residues) are frequently used to facilitate recombinant protein isolation.171 Moreover, attachment of His-tagged proteins on electrodes received recently special attention in relation to forming well-organized protein layers at electrified interfaces (reviewed in ref (172)). Very recently, it has been shown that using oxidation peaks of Tyr and Trp, subpicomole amounts of a potential cancer biomarker, protein AGR2,173 can be detected at carbon electrodes.174 The N-terminal His-tagged and non-His-tag forms of this protein were studied, and it was found that only the His-tagged form yielded a peak of histidine. Similar results were obtained with other His-tag containing and not-containing proteins, such as α-synuclein or cytochrom b5. It was concluded that His-tags in proteins influence the protein adsorption and orientation at the electrode surface and that the appearance of a His oxidation peak at carbon electrodes depends on many factors, including the number of His residues and their accessibility in the surface-attached protein molecule.
Oxidation processes of free Cys were reviewed,153 and oxidation of Cys in short peptides was reported.153,175,176 To our knowledge, oxidation of Cys residues in large proteins at carbon electrodes was however not shown.153 Nevertheless, Suprun et al.164 recently considered oxidation of Cys residues in several proteins, including bovine serum albumin (BSA) and human serum albumin (HSA), without bringing experimental evidence about oxidation of Cys residues in the complex protein molecules. Oxidation of Met in dipeptides was observed at boron doped diamond electrode at highly positive potentials, which depended on aa sequences.177 Also, oxidation of Met residues in proteins at carbon electrodes was reported,169,178 but unambiguous experimental evidence for this oxidation is still needed. With peptides, well-separated peaks of Tyr and Trp were obtained (Figure 4A). Also, a relatively small protein, lysozyme (containing 3 Tyr and 6 Trp residues), produced two separated peaks.146,154 In contrast, larger proteins containing both residue types produced mostly only a single peak (Figure 4B). Nitration of Tyr resulted in shifting of the Tyr oxidation peak to more positive potentials and formation of a reduction peak at ∼0.65 V. Also, nitrated BSA produced a peak at ∼0.75 V, which made it possible to discriminate it from unmodified native BSA.164 Ricin (RCA-60, ∼60 kDa), a deadly toxic glycoprotein,179,180 was recently analyzed at carbon, Au, and Pt electrodes.181 The metal electrodes were found inconvenient for this analysis because formation of Pt and Au oxides occurred at potentials where oxidation of RCA-60 took place. Using CV, SWV, or differential pulse voltammetry (DPV) and GCE, 200, or 100 μM RCA-60 produced in a wide pH range two peaks, probably due to oxidation of Tyr and Trp residues. Practical RCA-60 determination would require a combination of EC analysis with some separation technique, involving, for example, specific antibodies. Considering that RCA-60 contains Cys, Lys, His, and a large number of Arg residues (almost 5%), CPS electrocatalytic peak H (see section 5.2) would probably offer better sensitivity. Using this peak, it might be also possible to recognize reduction of the disulfide bond in RCA-60 (section 5.3), which results in separation of chains A and B and loss of the protein toxicity.182 EC analysis might be also useful for simple detection of glycan in the RCA-60 glycoprotein, using lectins and/or chemical modification (see sections 8.5 and 8.6).
Figure 4.
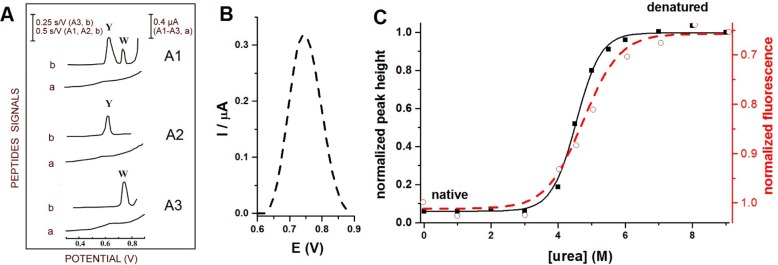
(A) Oxidation of Trp- and Tyr-containing peptides on carbon paste electrodes. (a) Differential pulse voltammograms and (b) chronopotentiograms for (A1) Tyr- and Trp-containing luteinizing hormone releasing hormone, (A2) Tyr-containing neurotensin, and (A3) Trp-containing bombesin. 10 nM peptide was adsorbed for 5 min at accumulation potential of 0.1 V followed by chronopotentiogram or DP voltammogram recording. CPS: Istr 5 μA; DPV scan rate, 5 mV/s. Y refers to Tyr and W to Trp residues. Adapted with permission from ref (159). Copyright 1996 Elsevier. (B) Oxidation peak of 2 μM human serum albumin (HSA) denatured in 8 M urea at glassy carbon electrode. (C) Dependences of square wave voltametric peak heights (−■−) and changes in fluorescence emission at 334 nm (− –○– −) on urea concentration. 1 μM HSA was incubated overnight with different urea concentrations (indicated in the figure) at 4 °C. Oxidation peak height of HSA denatured in 8 M urea was taken as 1. In the fluorescence measurements, intensity at 334 nm produced by 1 μM HSA incubated in the absence of urea was taken as 1. Adapted with permission from ref (218). Copyright 2012 Elsevier.
Recently, a signal enhancing system was developed to increase the aa and protein irreversible oxidation signals.183−186 This system relied on the electrocatalytic oxidation of Tyr mediated by phenoxazine or osmium bipyridine complexes (Figure 5). Using indium tin oxide (ITO) as the working electrode, detection of protein oxidative damage187 and phosphorylation of the Tyr residues in proteins,186 as well as ligand-protein binding184 and protein-conformation changes, were demonstrated.183
Figure 5.
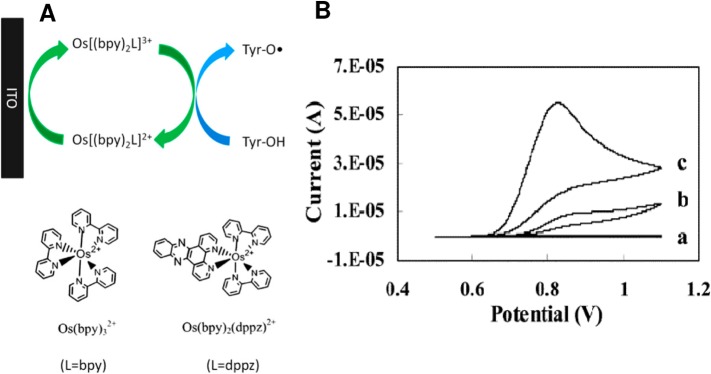
(A) Oxidation of tyrosine (Tyr) with signal enhancement. ITO, indium tin oxide electrode; Bipy, 2,2′-bipyridine; dppz, dipyrido [3,2-a:20,30-c] phenazine. (B) Cyclic voltammograms of (a) 5 μM Os(bpy)2dppz, (b) 2 mM Tyr, and (c) 5 μM Os(bpy)2dppz and 2 mM Tyr. Working electrode: ITO. Reference: Ag/AgCl/3 M KCl. Supporting electrolyte: 100 mM sodium phosphate, pH 7.3. Scan rate: 30 mV/s. Adapted with permission from ref (185) Copyright 2012 Elsevier.
Post-translation modification of proteins and particularly phosphorylation by kinases play important roles in many biological processes, such as cell cycle, differentiation, growth, and in apoptosis.188 During the phosphorylation, the γ-phosphoryl group of adenosine triphosphate (ATP) can be transferred to residues of serine, threonine, or Tyr.189 Abnormal protein phosphorylation is involved in many diseases, including cancer and neurodegenerative diseases.190 Different methods have been developed for studies of kinase-catalyzed protein phosphorylation (e.g., refs (191−193)), including relatively simple and inexpensive EC methods.164,185,186,194−200 First papers showed that phosphorylation of peptides and proteins resulted in a decrease of Tyr oxidation peak using screen-printed electrode.164,201 The difference between oxidation peaks of phosphorylated and nonphosphorylated Tyr was recently used by Li et al.202 to investigate activity and inhibition of protein kinase at a graphene-modified GCE. Graphene with its unique one atom thick structure, large specific surface area, excellent electric and thermal conductivity, and high mobility of charge carriers attracted great attention in recent years.203−205 However, skeptical opinion appeared regarding graphene application in various biosensors.206
An enhancement of Tyr peak Y through electrocatalytic oxidation reaction was observed at the graphene electrode.202 At this electrode, phosphorylated Tyr was inactive. Tyr oxidation peak Y was thus used to assay the Src kinase activity using peptide YIYGSFK as a substrate (phosphorylated predominantly at its N-terminus). Peak Y decreased with the logarithm of the kinase concentration from 0.26 to 33.79 nM, with a LOD of 0.087 nM, which was better than that reported previously.202 Using the same method, the kinase inhibition by low molecular weight PP2 inhibitor was followed, showing an increase of peak Y with increasing PP2 inhibitor concentration. The reported results are very interesting, but a biochemist might be not fully satisfied with the absence of control experiments in both the dependence of EC signal on the kinase and the PP2 inhibitor concentration. The enzymatic reaction was performed in a complex mixture containing the protein Src kinase and, in addition, different ions as well as 1 mM dithiothreitol and 50% glycerol. Using the same mixture with the inactivated kinase might be a proper control. More information on EC analysis of kinase activity can be found in section 9.1.1, describing the label-based approach.
4.2.1. Folded and Unfolded/Denatured Proteins
For several decades, it was unclear whether the protein oxidation peaks can be used to distinguish native from denatured proteins. Already in 1985 oxidation peaks of tobacco mosaic virus and its isolated protein in native and denatured forms were reported.207 Urea-denatured viral protein (at a concentration of 100 μg/mL) produced higher oxidation DPV peaks than the native protein. Urea was, however, not removed from the sample of the denatured protein prior to the DPV measurement. Later, differences in oxidation peak heights between native and denatured proteins were reported at various carbon electrodes in some studies, while other studies showed that oxidation peaks at carbon electrodes reflected poorly the changes in protein structures resulting from protein denaturation208 or a single aa exchange.105 Generally, oxidation responses of proteins at carbon electrodes were much less sensitive to changes in protein structures105,208 and redox states209 than reduction signals at bare and thiol-modified mercury electrodes (see section 5.3 and 5.4). Nevertheless, these protein oxidation signals were used in different sensors (reviewed in ref (210)) and for the development of various biomarker sensors (reviewed in refs (211) and (212)).
Over the recent years, various types of carbon electrodes have emerged,210,213−217 opening the door for a wider application of electroanalysis to diverse targets,76 including the biomacromolecules. Pyrolytic graphite,218 graphene,202 glassy carbon,218 as well as conductive diamond electrodes219 can be considered as convenient electrode materials for protein analysis.76 Polishing with abrasives can modify properties of carbon electrodes as it results in the formation of oxide groups and hydrophilic surfaces at these electrodes. In edge plane pyrolytic graphite electrodes (EPGE), edge planes are at the electrode surface and can be easily oxidized. It appears that EPGE may now represent an ideal material for electroanalytical purposes.214 Recently, it has been shown that by using these electrodes, the course of protein denaturation can be traced,218 yielding the results in good agreement with fluorescence emission at 334 nm (Figure 4C). Testing other carbon electrodes for their ability to distinguish native from denatured forms of HSA218 or α-2-macroglobulin220 showed GCE as a suitable material for this purpose (albeit worse than EPGE). Similarly, using boron doped diamond electrode, native and denatured BSA could be distinguished.219,220 Topal et al.220 showed oxidation signals of native and denatured macroglobulin at a gold electrode close to 0.7 V. However, a basal plane carbon electrode displayed poor ability to distinguish between these forms of HSA.221 Using EPGE, a number of proteins were tested, showing much higher oxidation peaks in denatured than in their native forms. In good agreement with the above results, treatment of natively unfolded α-synuclein protein222 with denaturing agents resulted only in a very small change in the oxidation peak.218 Treatment of this almost structureless protein with a denaturing agent can result only in small changes in accessibility of their aa residues (including accessibility of the Tyr residues responsible for the protein electro-oxidation). Oxidation peaks of proteins were shown to be able to follow aggregation (resulting in burying of aa residues) of amyloid proteins such as amyloid β-peptides223−225 and α-synuclein156,157,218,226 in relation to their roles in Alzheimer’s or Parkinson’s diseases, respectively (see section 6.1). Analysis of another type of post-translational modification, glycosylation by glycan assays, is described in section 8.
5. Label-Free Electrocatalysis in Peptides and Proteins
According to the review by Herzog and Arrigan153 in 2007, the electroactivity of nonconjugated proteins appeared rather poor. The authors deliberately omitted studies at mercury electrodes due to their claim that these electrodes were disappearing from laboratory benches and were generally unsuited for use in miniaturized and/or out-of-laboratory applications. Almost at the same time, it was shown that proteins yield chronopotentiometric signals at mercury and solid amalgam electrodes155,227−229 in a wide pH range.208,209,230−233 These signals appeared at highly negative potentials but were still well-separated from the background. They were not observed with mercury-free electrodes, suggesting that mercury electrodes can be especially useful in protein analysis.
5.1. Mercury-Containing Electrodes and Chronopotentiometry in Protein Analysis
5.1.1. Mercury Electrodes
The objection that mercury electrodes are unsuited for use in miniaturized and/or out-of-laboratory applications153 does not seem to have a solid ground because (a) miniaturization of Hg electrode was reported234−236 and (b) solid amalgam electrodes (SAEs) were miniaturized and SAE chips for protein analysis were developed.227,236 Moreover, regarding the electrode choice for protein analysis, other criteria should be considered as well. For example, the accessible potential window of a liquid mercury electrode and SAEs (roughly between 0 and –2 V against a saturated calomel electrode, SCE) greatly differs from that of most of the solid electrodes, such as carbon, gold, platinum, and silver (shifted by about 1 V in a positive direction as compared to Hg electrodes), making thus solid electrodes better suitable for studies of (irreversible) oxidation processes. However, Hg electrodes are more suited for studying reduction processes and particularly for processes involving catalytic hydrogen evolution in proteins, which has been observed solely with Hg electrodes (see below). An atomically smooth surface of liquid mercury makes it possible to prepare pinhole-free monolayers of thiolated DNAs237 and of other thiols161 to obtain chemically modified electrodes for protein analysis.105,228,238−240 Smooth surfaces can be prepared also by forming a miniature liquid Hg meniscus at SAE. Moreover, strong hydrophobicity and other properties of Hg electrodes differ from most of the solid electrodes predominantly used in EC analyses. With excellent reproducibility of their clean surfaces, liquid mercury electrodes still remain attractive for research purposes. It can be concluded from the recent development of electrochemistry of nonconjugated proteins that mercury electrodes will not soon disappear from laboratory benches as they open the door to new approaches in the EC analysis of biomacromolecules and particularly of nucleic acids,34,229,241,242 proteins,155,229,243 and carbohydrates.229,244−246 In the following paragraph, it will be shown that the combination of bare and chemically modified mercury electrodes with constant current chronopotentiometric stripping and electrocatalysis resulted in new methods of EC protein analysis applicable in biomedicine.
5.1.2. Chronopotentiometry
Contrary to voltammetry (in which current is recorded as a function of the potential applied to the working electrode), in chronopotentiometry (galvanostatic or controlled current methods) the electrode potential is measured as a function of time upon applying a current perturbation to the working electrode.247 Controlled-current methods were introduced a long time ago,248−250 and different current versus time programs were used for studying biomolecules.251 At present, the stripping mode of constant-current chronopotentiometric stripping (CPS) is gaining ground.155 The raw chronopotentiometric response (E–t curve) is of limited analytical interest, but the derivative of the E–t curve yielding a peak-shaped plot (dE/dt)−1 as a function of the potential E (Figure 6C,D) is calculated and usually used in protein analysis. In CPS, the applied current imposes a rate of charge flow across the electrode|solution interface, with total charge increasing linearly with time.
Figure 6.
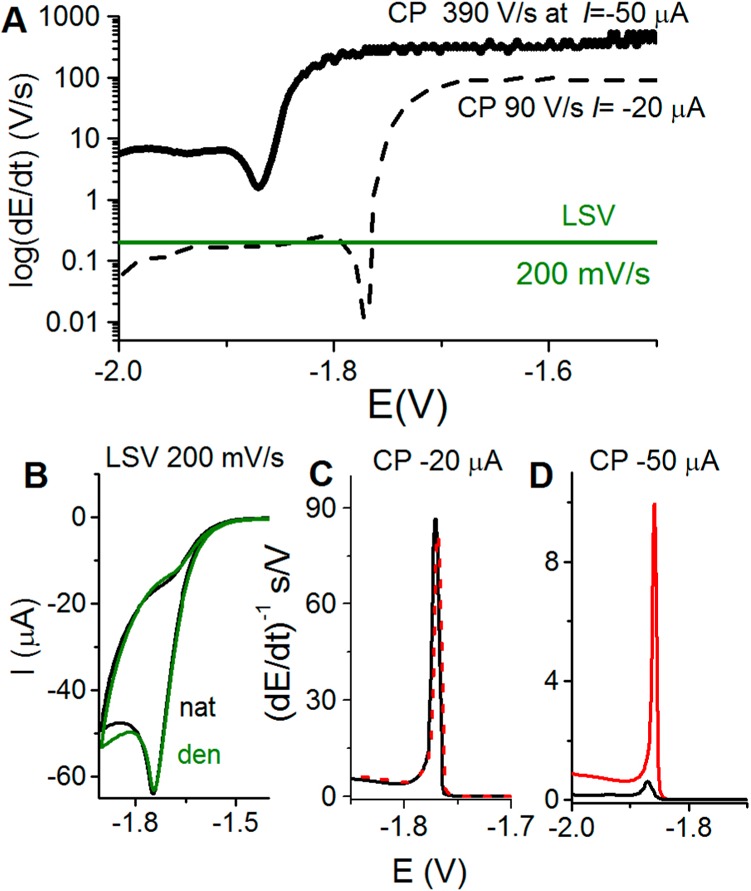
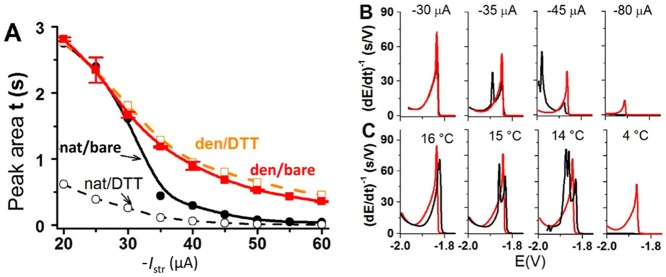
(A) Schematic representation of the rate of potential changes in chronopotentiometry (CP) at two different intensities as compared to voltammetry. In linear sweep voltammetry (LSV), the scan rate (chosen by an experimenter) is constant throughout the whole voltammogram recording. However, in CP the rate of potential changes is influenced by the current density. At constant electrode size, this density is determined by polarizing current intensity (I, chosen by an experimenter). In the absence of the electrode process, the potential changes very rapidly (e.g., 390 V/s at I = −50 μA), but it gets much slower in a narrow potential range where the electrode process (e.g., proton reduction and hydrogen evolution) is taking place. (B–D) In protein analysis, both native (nat, black) and denatured (den, green or red) proteins are firmly attached to the Hg electrode surface, and prolonged exposure of native folded protein to negative potentials (at low scan rates or I intensities) may result in its denaturation, indicated by almost the same (B) LSV or (C) CP responses. (D) At high current intensity in CP, for example, at I = −50 μA, fast potential changes (390 V/s) prevent protein from the denaturation at the negatively charged electrode surface, as indicated by a relatively small CP response of native (black) protein and a very large response of the denatured (red) protein.
As compared to voltammetric methods, CPS offers some advantages in protein analysis.252 For a given EC process, such as catalytic hydrogen evolution, chronopotentiometry yields better resolved peaks and lower background levels (baseline correction is usually not required) (Figure 6C,D), and polarization of the electrode may proceed in a very short time. Some part of these advantages may be due to the differential nature of the signal, but the main advantage lies in the way of the current perturbation itself. As mentioned above, the rate of the overall process in CPS is imposed by the value of the applied current and very fast potential changes can be reached (Figure 6), while in voltammetries the constant scan rate is used. Using CPS peak H in protein analysis (Figure 6D), it is important that the electrode potential shifts slower during an electrode process than in its absence, when potential changes can reach extremely high rates (e.g., about 390 V/s at stripping current, Istr −50 μA at 0.4 mm2 Hg electrode size, corresponding to a current density of −12.5 mA/cm2, Figure 6A). This feature is critical in protein structure analysis (sections 5.3 and 6), as well as in DNA–protein interaction studies (section 7.5). CPS peak H of proteins appears at highly negative potentials, such as −1.8 V (against Ag/AgCl electrode). To reach this potential, the surface-attached protein is exposed to the electric field effects at negative potentials causing unfolding/denaturation of the native protein238 or dissociation/disintegration of the DNA–protein complex.240 It has been shown that such damage to the surface-attached biomacromolecules depends strongly on the time of their exposure to negative potentials and can be avoided at highly negative Istr intensities (sections 5.3 and 7.5), inducing very high rates of potential changes and thus very short exposure times. Such negative Istr intensities (current densities) can be hardly used in low electron-yield protein electrode processes (e.g., in oxidation of protein Tyr or Trp residues, section 4), because the CPS signals decrease with increasing −Istr intensities, and at highly negative Istr intensities, they can disappear or become too low. In contrast, in high electron-yield electrocatalytic processes, such as those involving catalytic hydrogen evolution reaction, high −Istr intensities can be applied, and a well-developed peak H can be obtained using picomole amounts of proteins.230,231,238,239,253,254 In voltammetry, very short time scales can be also obtained using high scan rates, but a voltammetric peak of BSA analogous to CPS peak H shifts to negative potentials with the scan rate and merges with the background discharge at relatively low scan rates. Moreover, CPS analysis of proteins can mostly be performed under air.155,230,231,238,239,254
5.2. Peak H of Peptides and Proteins
By the end of the 1990s, CPS in combination with a hanging mercury drop electrode (HMDE) was applied in studies of peptides.255 Peak of vasopressin was observed at highly negative potentials well-separated from the background electrolyte. This peak was due to the catalytic hydrogen evolution reaction (CHER) and was denominated as peak H, as a tribute to Jaroslav Heyrovský, as well as due to its high sensitivity and hydrogen evolution. Peak H was much better developed and allowed the determination of lower concentrations of the peptides than a polarographic or a voltammetric presodium wave. Soon, CPS was shown to be the most sensitive EC label-free method allowing the determination of proteins at nanomolar and subnanomolar concentrations,155,253 suggesting a wide application of peak H in the analysis of peptides and proteins.105,208,209,230−233,239,254,256,257
5.2.1. Theory
The hydrogen evolution reaction and its reverse process of the hydrogen oxidation, as well as the CHER, have attracted great attention.258−263 The CHER can be described by the following equations:
| 1 |
| 2 |
where PH and P– stand for the protonated and unprotonated aa residues in the protein, respectively, BH is the acid component of the buffer solution, and B– is its conjugate base. The symbols in parentheses represent the state of the molecules ((aq) stands for aqueous, (surf) for surface confined, and (g) for gaseous). These reactions imply that the catalyst is the protein anchored at the electrode surface. Adsorptive transfer experiments155 suggest that the protein binding to the surface must be particularly strong, because the protein-modified electrode is washed, followed by immersion into the blank protein-free background electrolyte. CHER theories considered little the structure and properties of the protein catalyst and positioning and accessibility of catalytically active aa residues in the protein folded structure. Recently, homopolymers of different aa’s and peptides were studied to shed some light on this problem.170,264
Arginine (Arg), lysine (Lys), and Cys were found as catalytically active residues in proteins close to neutral pH.155,265,266 Under these conditions, His residues behaved as a much weaker catalyst than Cys, Arg, or Lys. Each of these aa types alone (bound in peptide chains of polyamino acids or peptides) was sufficient to catalyze hydrogen evolution and produce peak H at mercury electrodes.265,266 CHER in four angiotensin (AT) peptides containing Arg and His residues was studied in detail.170 At neutral pH, only one of them (ATIII) produced a well-developed CPS peak H (Figure 7A). This peptide contained one Arg and one His residue. ATII had the same sequence, but acidic aspartic acid (Asp) residue was added at the N-terminus (Figure 7B). ATI differed from ATII only by additional His and leucine (Leu) residues at the carboxyl end. ATIV was similar to ATIII but did not contain the Arg residue. These results suggested that Arg residue played a critical role in CHER, but the presence of the acidic Asp in its close neighborhood canceled the Arg catalytic activity. CPS behavior of hexaArg and hexaHis supported the importance of Arg in CHER as compared to the much smaller contribution of His (Figure 7A, inset).170 Studies of peptides and proteins showed that a particular contribution of the given aa residue to CHER depended on the electrolyte ionic conditions and accessibility of the aa in the surface-immobilized protein. Further work will be, however, necessary to better understand relations between the protein composition and structure, on one hand, and its CHER responses, on the other hand.
Figure 7.
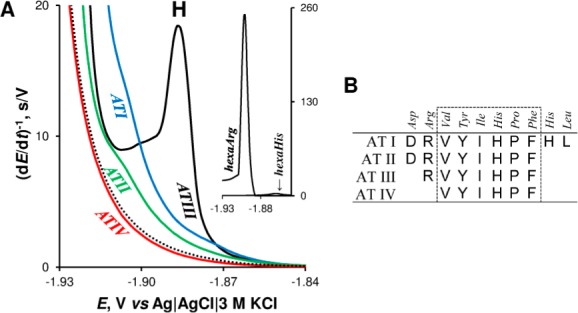
(A) CPS peaks H of 1 μM angiotensin peptides (AT) in McIlvaine buffer, pH 7 at HMDE; dotted line represents blank background electrolyte. Inset: CPS peak H of hexaArg and hexaHis. (B) Amino acids sequences of AT peptides. Adapted with permission from ref (170). Copyright 2013 Elsevier.
5.3. Protein Structure-Sensitive Electrocatalysis at Bare Mercury Electrodes
5.3.1. Determination of Peptide and Protein Redox States
Reduced state of intracellular proteins is frequently associated with their biological activities. The EC analyses of redox states were limited to low molecular weight thiols, such as glutathione and its fragments.267 These methods were based either on direct oxidation of thiols at solid electrodes (usually at large overpotentials close to 1 V) or on the formation of stable mercury thiolate complexes at mercury electrodes.268 The reduction of disulfide bonds of proteins at Hg electrodes was intensively studied (reviewed in refs (269−271)). However, little attention was paid to proteins, which require the reduced state for their biological function. Only recently have methods for the determination of the redox states of peptides and proteins based on CPS peak H been developed.
It was shown that reduced peptides adsorbed at positively charged HMDE produced substantially higher peak H than their oxidized forms.257 Similar behavior was observed with thioredoxin (a general protein disulfide reductase with a large number of biological functions).209 Large differences in the CPS responses of reduced and oxidized forms of peptides and proteins were explained by differences in chemisorption and orientation of the reduced compounds, combined with very fast potential changes in CPS not allowing significant changes in orientation of the species adsorbed at positively charged Hg surface. Peak H was used not only for the analysis of thioredoxin at nanomolar concentrations and for the determination of the thioredoxin redox states, but also to follow interactions of this protein with the product of lipid peroxidation such as 4-hydroxy-2-nonenal. CPS of thioredoxin at carbon electrodes (based on oxidation of Tyr and Trp residues) was less sensitive and did not allow discrimination between reduced and oxidized forms of thioredoxin and peptides.257,272
5.3.2. Are Proteins Denatured at Mercury Electrodes?
Early polarographic studies (with a dropping mercury electrode) of proteins indicated different Brdička’s catalytic responses of native and denatured (Cys-containing) proteins.72 These results were, however, not confirmed when metal solid electrodes76,273 or HMDE274 were used. Thus, any attempt to use a bare mercury electrode for protein structure analysis appeared ridiculous.
It was believed for a long time that routinely used metal electrodes such as gold, platinum, mercury, and silver led to denaturation and irreversible adsorption of the proteins at the electrode surfaces.76 Some papers claimed (e.g., refs (269) and (274)) that proteins were denatured upon adsorption at mercury electrodes producing adsorbed layers of uniform thickness. Few papers disagreed, however, with the above-mentioned conclusions.74,275 Recently, this problem was studied in greater detail using voltammetric and CPS methods in connection with HMDE and solid amalgam electrodes.208,230−233
It was found that, as compared to oxidation responses of proteins at carbon electrodes, peak H is more sensitive to local and global changes in the protein structures.105,208,209,230−233,239,254 For example, at weakly alkaline and neutral pH’s, large differences between peak H heights of native and denatured forms of BSA and some other proteins were observed.155,208,230−232 Native proteins produced very small signals, while their denatured forms yielded 10–50 times higher peaks208,230−233 (Figure 6D). These results were in qualitative agreement with the solution structure of native (ordered, folded) and denatured (disordered, unfolded) proteins, greatly differing in accessibility of aa residues (particularly hydrophobic aa’s are usually buried in the interior of the native folded protein molecule) and consequently in the protein orientation and adsorption at the electrode surface. However, very large differences between CPS signals of native and denatured proteins208,230−233 (Figure 6D) were at variance with the claimed denaturation of proteins attached to the mercury electrodes. Clearly, if a native protein was denatured at the electrode surface, its EC responses should not greatly differ from that of the protein, which was denatured in solution and adsorbed in its denatured form at the electrode surface.
5.3.3. Proteins Are Not Surface Denatured at Potentials Close to Potential of Zero Charge on Hg
Very small peaks of native proteins (Figures 8 and 9) suggested that no significant denaturation of the protein took place at the mercury electrode surface. Recently, it has been found208,231−233 that proteins are not denatured when adsorbed at the mercury electrode surface at the potential of zero charge (pzc) and at potentials positive of pzc.233 However, denaturation of proteins took place due to a prolonged exposure of protein at a negatively charged mercury surface.233
Figure 8.
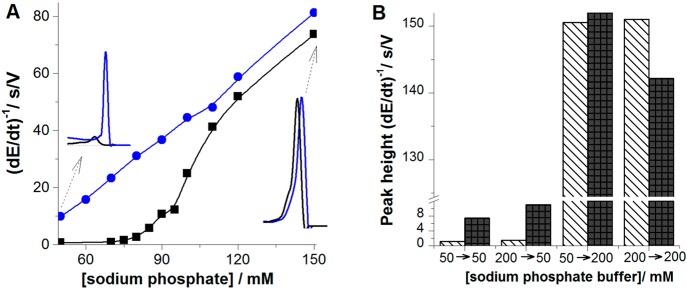
(A) Dependence of peak height of 100 nM native (black) and denatured (blue) BSA on concentration of sodium phosphate, pH 7 in the presence of 56 mM urea (black). Accumulation time tA of 60 s, accumulation potential EA of −0.1 V, stirring 1500 rpm, stripping current, Istr, of −30 μA. (B) Column graph showing peak H heights of native (stripped column) and denatured (black column) BSA obtained in AdT (ex situ) stripping experiment. 100 nM BSA was adsorbed at HMDE for tA of 60 s at EA of −0.1 V either from 50 mM or from 200 mM sodium phosphate, pH 7, and the BSA-modified electrode was transferred to the electrolytic cell with blank 50 or 200 mM sodium phosphate, pH 7, to record the chronopotentiogram. 50 → 200 indicates BSA adsorption from 50 mM phosphate, followed by a transfer of BSA-modified electrode to 200 mM phosphate in the electrolytic cell. Denaturation of 14.4 μM BSA in 0.1 M Tris-HCl, pH 7.3, with 8 M urea was performed overnight at 4 °C. The protein solution was then diluted by the background electrolyte to the final protein concentration (usually about 100 nM and immediately measured). Reprinted with permission from ref (232). Copyright 2009 Royal Society of Chemistry.
Figure 9.
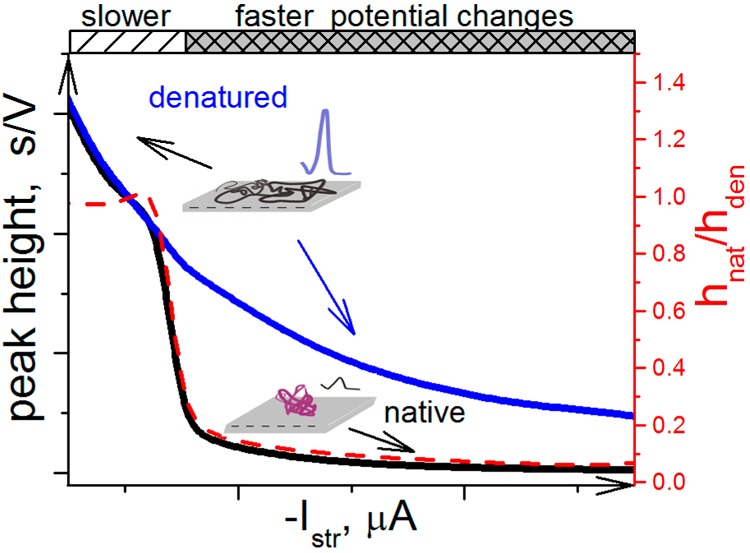
Schematic representation of the effect of the stripping current intensity (Istr) on peaks H of native and denatured proteins. The scheme demonstrates that at low Istr intensities, the surface-attached protein is denatured, producing almost the same peak H as the protein, which was denatured in solution by a chemical denaturation agent. The protein denaturation at the electrode surface is due to the prolonged effect of the electric field at negative potentials. At higher Istr intensities, the time of exposure of the protein to negative potentials is much shorter, causing a little harm to the surface-attached protein as manifested by a relatively small peak H.
5.3.4. Ionic Strength-Dependent Protein Denaturation at Negatively Charged Hg Electrode
Using peak H, an ionic strength-dependent structural transition in BSA at the HMDE surface was detected.232 In 50 mM sodium phosphate at pH 7.0, peak H of 100 nM denatured BSA was much higher than that of the native protein (Figure 8A). Increasing the phosphate concentration resulted at first only in small increase of peak H of native BSA, followed by a steep increase of this peak between 90 and 110 mM phosphate (not observed in the control urea-denatured BSA). At 200 mM phosphate concentration, peaks of native and urea-denatured BSA were almost the same (Figure 8A). Similar responses were observed with other proteins.
In principle, this transition could take place already during the adsorption of the BSA at the electrode charged to the accumulation potential −0.1 V or later in the course of electrode polarization to more negative potentials, subjecting the adsorbed protein to higher electric field strengths. To clarify this point, an adsorptive transfer analysis271,276 was performed, in which the BSA accumulation on the surface was separated from the electrode process (Figure 8B). After adsorbing BSA either from 50 mM or from 200 mM phosphate, and the transfer of the BSA-modified electrode to (blank) 50 mM phosphate, the great difference between peak heights of native and denatured BSA was retained,232 suggesting that no significant irreversible surface denaturation of BSA took place during the protein 60 s accumulation at EA −0.1 V. However, transfer of surface-immobilized BSA to 200 mM phosphate removed the large difference in the peak heights of native and denatured BSA, suggesting that surface denaturation occurred at potentials more negative than EA in a time interval ≪1 s. The absence of denaturation in proteins adsorbed at the Hg electrode at the pzc, and at potentials positive of pzc, was observed in a wide pH range.232,233
The abrupt increase of peak H (Figure 8A) was tentatively explained by the effect of strong electric field on the BSA immobilized at the negatively charged Hg surface,232 resembling thus the surface denaturation of double-stranded DNA at a negatively charged Hg and other surfaces.34 In the case of the BSA surface denaturation, formation of Hg–S bonds and hydrophobic interactions between the protein and hydrophobic Hg surface could be involved. Most probably, the electric field repulsed negatively charged protein segments from the surface and caused alteration of the charge distribution in the protein introduced by shifts in the acid–base equilibrium toward the ionized forms, the hydrogen bonds polarization, alignment of the molecular dipoles, and displacement of the charged residues.277 It cannot be excluded that protein structure changes controlled by ionic conditions and the electric field might take place also at cell surfaces and eventually play some biological role.
5.3.5. Interplay of Current Density, Ionic Conditions, and Temperature Affects the Structure of Surface-Attached Proteins
The above results suggested that the investigated proteins attached at Hg surfaces around the pzc did not denature under the usual experimental conditions, that is, close to neutral pH, moderate ionic strengths, and room temperature. However, in proteins attached to negatively charged Hg electrodes,238 denaturation took place depending on the time of exposure to the negative potentials. In CPS experiments, this exposure time depends on the current density (i.e., on polarizing current intensity at constant electrode size, see section 5.1). Thus, at high negative Istr intensities, the exposure to negative potentials is very short and the damage to protein structure is negligible, as documented by very small peak H of native protein as compared to a very large peak H of the protein denatured, for example, by urea or guanidinium (Figure 9). In contrast, at low negative Istr intensities, peaks H of native and denatured protein are either the same or differ only a little. The particular shape of the curve shown in Figure 9 may differ depending on temperature and ionic strength.238 At lower temperatures, longer exposure time periods (i.e., lower Istr intensities) are necessary to cause denaturation of the surface-attached protein. However, increasing the ionic strength makes the surface-attached protein more vulnerable to the effect of the electric field, and denaturation of the surface-attached protein may occur even at shorter time periods.
It can be concluded from the above-mentioned data that by using CPS peak H, bare Hg electrodes can be used to study changes in the protein structures under certain ionic conditions (e.g., at neutral pH and relatively low ionic strengths, Figure 8), while at diferrent ionic conditions protein surface denaturation at a negatively charged bare Hg electrode may take place even under very fast potential changes in CPS. For example, in 0.2 M sodium phosphate, pH 7 at Istr of −30 μA, it was not possible to discriminate between native and denatured BSA (Figure 8), because of the electric field-induced denaturation of the surface-immobilized protein.232 Recently, attempts have been made to decrease or eliminate protein-denaturing effects of the electric field, by using chemically modified Hg working electrodes.239
5.4. CPS Peak H at Thiol-Modified Mercury Electrodes
It has been shown that alkanethiols161,278 and thiolated DNA237 form in short time intervals impermeable, pinhole-free SAMs on Hg surfaces. Similar modification of gold electrodes has been widely used in the studies of conjugated proteins, yielding EC responses of nonprotein redox centers,77,78 but pinhole-free SAMs similar to those found at Hg electrodes have not been reported. Protein-catalyzed hydrogen evolution263 (responsible for peak H) has not been, however, observed at gold electrodes and other solid electrodes not containing Hg but solely with mercury-containing electrodes.155,263,271,279 To prevent direct contact of proteins with the metal surface, a dithiothreitol (DTT) SAM was formed at the HMDE surface (DTT-HMDE) and silver solid amalgam electrodes (DTT-AgSAE).239 DTT was chosen because this reducing agent in millimolar concentrations is frequently used for storage of reduced proteins.
5.4.1. Electric Field-Induced Denaturation of Surface-Immobilized BSA Is Strongly Decreased at DTT-Modified Hg Electrodes
It was shown that BSA and other proteins could be easily immobilized at DTT-HMDE and that denatured proteins produced high peak H. In contrast, native BSA (which underwent surface denaturation in 0.2 M sodium phosphate or McIlvaine buffer, pH 7, at the bare HMDE) displayed almost no peak at DTT-HMDE, suggesting the absence of BSA surface denaturation under the same conditions (Figure 10). Very small peak H in native BSA could be explained either by negligible adsorption of native BSA at the electrode and/or by inaccessibility of the catalytic aa residues in the surface-attached protein molecules. To solve this question, combined CPS and CV experiments were performed, which showed that at DTT-HMDE, native BSA was adsorbed to an extent similar to that of denatured BSA, but in CV, surface-immobilized native BSA was denatured during slow potential scanning to negative potentials, suggesting that much smaller CPS peak H height in native (than in denatured) BSA was predominantly due to inaccessibility of catalytic aa groups in the native surface-attached protein.
Figure 10.
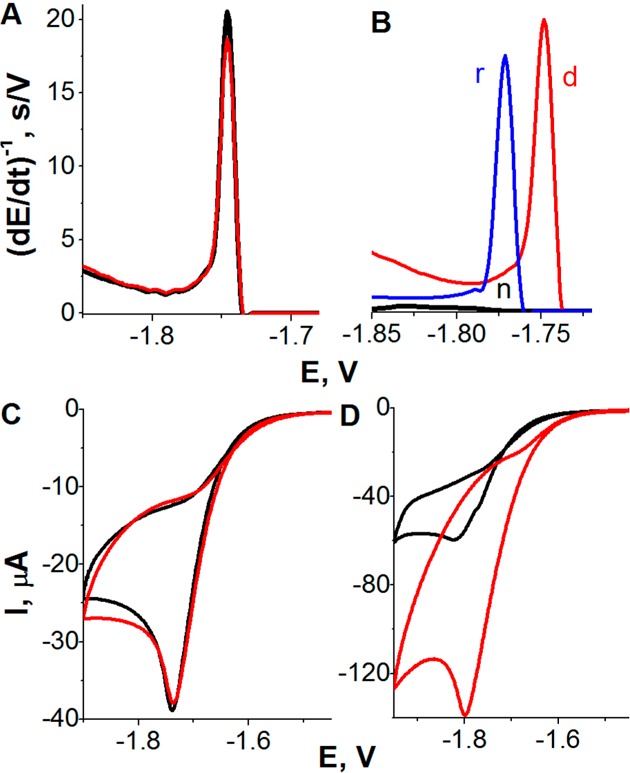
Constant current chronopotentiometric stripping (CPS) peak H of 100 nM native BSA (n, black), DTT-reduced BSA (r, blue), and guanidinium chloride (GdmCl)-denatured BSA (d, red) (A) at bare and (B) at DTT-modified HMDE (DTT-HMDE) in McIlvaine buffer, pH 7. GdmCl was present in all samples at nondenaturing (70 mM) concentration; conventional CPS measurements using stripping current, Istr, −70 μA. (C, D) Adsorptive transfer stripping cyclic voltammograms of native (black) and denatured BSA (red) at DTT-modified HMDE at scan rates of (C) 50 mV/s and (D) 1 V/s. In this experiment, 1 μM BSA was adsorbed at DTT-HMDE from McIlvaine buffer, pH 7, for tA 60 s. The adsorptive transfer procedure was applied to prevent additional BSA adsorption during the potential scanning. BSA was denatured with 6 M GdmCl. Adapted with permission from ref (239). Copyright 2010 American Chemical Society.
5.4.2. Prolonged Exposition of the DTT-SAM to Negative Potentials Disturbs DTT-SAM
At potential values, more positive than the reduction potential of the DTT Hg–S bond (∼ –0.65 V against Ag/AgCl), the densely packed DTT-SAM was impermeable to [Ru(NH3)6]3+.228 Prolonged exposure of the DTT-SAM to more negative potentials resulted in disturbance of the SAM, but under conditions of CPS (with very fast potential changes), the (reductively desorbed) SAM still protected the immobilized protein from surface-induced denaturation. In contrast, the usual slow scan voltammetry (scan rates between 50 mV/s and 1 V/s) displayed large disturbance of the densely packed DTT-SAM, and the adsorbed protein (in McIlvaine buffer, pH 7) was fully or partially denatured (Figure 10C,D). Exposure of BSA-modified DTT-HMDE to different potentials, EB for 60 s, followed by CPS measurement revealed three EB regions, in which BSA remained either native (region A, −0.1 to −0.3 V), was denatured (B, −0.35 to −1.4 V), or underwent desorption (C, at potentials more negative than −1.4 V).239
5.4.3. DTT-Hg Electrodes Can Be Used in the Analysis of Reduced and Oxidized Proteins
Chemical reduction of disulfide bonds in native BSA resulted in a large increase of peak H at both bare and DTT-modified HMDE, suggesting easier denaturation of reduced BSA at the electrode surface.231,239 Most probably, DTT chemisorbed at HMDE at millimolar concentrations adopted the “stand up” position with one of two thiol groups exposed to the solution.228 Nevertheless, adsorption of native BSA on DTT-HMDE did not result in a detectable increase of peak H, showing that no significant BSA reduction took place at the DTT-HMDE surface during a short contact of the protein with the DTT-SAM. Thus, the DTT-HMDE can be used for studies of both reduced and disulfide-containing proteins, but it can be expected that it will be particularly useful for the analysis of intracellular proteins in their reduced state, as documented by the analysis of glutathione-S-transferase (GST)239 and tumor suppressor protein p53105 (see section 6.2). Using DTT-AgSAE, results similar to those observed on DTT-HMDE228 were obtained, and, recently, AgSAE arrays were constructed.227,236
As compared to the established label-free optical methods for tracing the protein denaturation, such as fluorescence and circular dichroism spectroscopy, peak H is either more or equally sensitive to changes in the proteins structure.239 In EC adsorptive transfer experiments, 3–5 μL volumes of protein solution can easily be used. Moreover, miniaturization of SAEs can further decrease protein volumes necessary for the analysis. It can be expected that thiol-modified Hg electrodes in combination with CPS peak H will soon become an important tool in protein analysis.
5.4.4. Thiol SAMs at Hg Electrodes
Originally, the DTT-modified electrodes for protein analysis were prepared by forming first the DTT-SAM followed by immobilization of the respective protein on the SAM.239 Later, an easier way of attaching BSA and other proteins at HMDE or AgSAE based on adsorbing BSA and DTT together in a single step was proposed.228 Properties of the DTT layers, dependent on the DTT bulk concentration, were tested. Changes in the cyclic voltamograms of a redox couple of [Ru(NH3)6]3+/[Ru(NH3)6]2+ and in reduction of the Hg–S bonds (peak S, Figure 11A) suggested that DTT at lower concentrations was adsorbed as a dithiol with both −SH groups attached to the surface.228 When the electrode was incubated in a DTT solution with concentration between 200 and 900 μM of DTT, a change in the DTT-SAM occurred, and increasing the DTT concentration resulted in a densely packed pinhole-free layer237,280 in which the DTT molecules were probably bound to the electrode surface by a single −SH group, oriented perpendicularly to the surface. The amount of BSA molecules adsorbed at DTT-HMDE or coadsorbed with DTT at a bare HMDE appeared roughly the same, and thus a great majority of BSA molecules were attached to the DTT layer and not to the bare HMDE. This was explained by a faster diffusion and adsorption of DTT at HMDE as compared to slower diffusion of much larger BSA molecules, which were in solution at concentrations by 4 orders of magnitude lower than that of DTT.281,282 BSA was measured at relative high ionic strength, that is, under the conditions inducing BSA denaturation at bare HMDE.232
Figure 11.

(A) Chronopotentiograms of 100 nM native BSA coadsorbed with 60 μM DTT at HMDE. (B) Dependence of peak H and peak S heights obtained with BSA·DTT-HMDE on concentration of DTT. BSA·DTT-HMDE was prepared by coadsorption of 100 nM BSA and 1 mM DTT at HMDE followed by CPS peak H recording at stripping current, Istr, −70 μA. Adapted with permission from ref (228). Copyright 2013 Elsevier.
Figure 11 shows that peak H of native BSA decreased while peak S increased with increasing DTT concentration up to about 1 mM DTT. In contrast, peak H of urea-denatured BSA responded only little to changes in DTT concentration (not shown). It can be read from Figure 11, that when coadsorbing native BSA and DTT on HMDE, the DTT concentration should be well above 1 mM. Similar results were obtained with some other thiols.
5.5. Enzyme Activity at Hg Electrodes
Various enzymes showed a good activity at different surfaces and enzyme electrodes,88,92−95 enabling construction of various enzyme sensors,46,274 such as the well-known glucose biosensor.283,284 Already in 1977, it was shown by Bards’s group275 that urease and alcohol dehydrogenase retained their enzymatic activity, when adsorbed at bare mercury and solid amalgam electrodes. Later, the opinion prevailed that, when adsorbed at bare mercury and other metal electrodes, proteins were irreversibly denatured.76 Bard’s group also showed that prolonged exposure of the surface-attached urease to negative potentials resulted in the enzyme inactivation. This inactivation was explained by electroreduction of the disulfide groups in the protein.275 Recently, it has been shown238 that the original Bard’s finding regarding the urease enzymatic activity at Hg electrodes was correct275 and that exposure of the enzyme to negative potentials at bare HMDE resulted in disturbances of the structure of the surface-attached enzyme, as detected by CPS (Figure 12). The extent of the urease unfolding was time- and temperature-dependent, when the proteins were exposed to negative potentials (Figure 12B,C). Dependence of peak H on the negative polarizing current (−Istr, Figure 12A) showed secondary denaturation of the surface-attached urease during the CPS recording at Istr less negative than ∼ –30 μA. Urease enzymatic activity was observed also at a thiol-modified amalgam surface,238 and at this surface the protein was less vulnerable to the effects of the electric field. It was concluded that earlier observed loss of enzymatic activity, resulting from a 10 min exposure of the protein to −0.58 V vs Ag/AgCl/3 M KCl,275 was not due to reduction of the disulfide bonds as suggested by Santhanam et al.,275 because the enzyme showed the best activity in its reduced form, while its oxidation caused a decrease of its activity.238 Moreover, no disulfide bonds in native urease molecule were found.285 The loss of the enzyme activity at negative potentials probably resulted from the protein reorientation, after reduction of the Hg–S bonds (formed by accessible Cys residues), followed by prolonged electric field effect on the surface-attached protein.
Figure 12.
CPS responses of the native and denatured proteins at bare and DTT-modified mercury electrodes. (A) Dependence of CPS peak H area of 20 nM native (−●–, − –○– −) and denatured (−■–, − –□– −) urease on stripping current, Istr, at bare (solid line) and DTT-modified (dashed) HMDEs. Urease was adsorbed at accumulation potential of −0.1 V for accumulation time, tA, of 60 s from McIlvaine buffer, pH 7, with 26 mM GdmCl in a thermostated electrolytic cell at 25 °C, and CPS analysis proceeded at the given Istr. (B,C) Chronopotentiograms of 20 nM native (black) and denatured urease (red) on a bare HMDE at (B) different striping currents at 25 °C and (C) different temperatures using Istr −25 μA. Adapted with permission from ref (238). Copyright 2013 Elsevier.
5.6. Concluding Remarks
Section 5 represents the first comprehensive review of EC analysis of practically all proteins based on combination of CHER with CPS at mercury-containing electrodes. To obtain CPS protein CHER signal (peak H) under conditions close to physiological, the protein has to contain at least one type of catalytically active aa residue, that is, Arg, Lys, Cys, or His. Most probably, proteins not containing any of these residues, if any, are extremely rare. In other words, this CPS method can be applied for analysis of practically any protein in proteomics, biomedicine, and elsewhere. Another condition for obtaining protein peak H is the accessibility of the catalytically active aa residues for the electrode process. In a native folded protein, aa residues buried in the interior of the molecule and/or located far from the electrode surface may remain catalytically silent. However, they can become accessible after the protein denaturation. Using chemically modified electrodes (e.g., with different thiols) can help to change the orientation of protein molecules at the surface and the accessibility of some aa residues. Mechanistic aspects of the catalytic hydrogen evolution were elucidated >50 years ago,286 and even recent papers265,287 are behind the progress in understanding of redox processes in nonprotein components of conjugated proteins, including electron transfer80 and hydrogen tunneling in enzymes.83
Application of CPS to proteins <10 years ago opened the door not only for sensitive EC determination of nonconjugated proteins but also for a new type of protein structure-sensitive analysis based on the ability of the electric field forces to denature proteins attached to the negatively charged electrode surface. By adjusting the time intervals of the protein exposure to the electric field effects to milliseconds (by choosing the appropriate current density in CPS), as well as other experimental conditions, such as temperature and ionic strength, the surface-attached protein denaturation can be minimized and the stability of proteins as well as of DNA–protein complexes (section 7.5) at the surfaces can be investigated. We believe that CPS studies of nonconjugated proteins at Hg electrodes are a challenging field that deserves further attention of electrochemists and biochemists from both theoretical and experimental points of view.
6. Label-Free Protein Analysis in Biomedicine
Progress in understanding of changes in the protein structure at the mercury electrode surface made it possible to apply electrochemistry in studies of proteins important in biomedicine. For example, studies of α-synuclein (AS) protein, which is involved in Parkinson’s disease,288,289 showed that peak H at HMDE as well as oxidation signals at carbon electrodes could be used to study aggregation of this protein.156 HMDE was particularly sensitive to preaggregation changes,254 detected at short incubation time periods preceding AS oligomerization observable by a dynamic light scattering. EC studies of amyloid peptides involved in Alzheimer’s disease suggested that EC techniques have a good chance to become of great value also in better understanding of an aggregation process in Alzheimer’s disease.156,221 Application of electrochemistry in cancer research and especially studies of CPS responses of the tumor suppressor protein p53 and its mutants appear very interesting.105,240 The CPS responses of wild type and mutant proteins agreed well with changes of the X-ray crystal structures resulting from a single aa exchange in these proteins.105 As compared to X-ray crystal analysis, nuclear magnetic resonance, and other methods, the CPS analysis worked with picomole protein amounts and yielded instant results, suggesting that EC methods may complement classical methods of protein structure analysis in the future.
6.1. Neurodegenerative Diseases
Among a number of human diseases that are associated with protein misfolding,290,291 particular attention has been paid to a group of diseases in which protein conversions into insoluble fibrils play a critical role.292−295 The final forms of aggregated proteins have frequently a well-defined fibrillar nature denominated as amyloid. About 20 neurodegenerative diseases include Alzheimer’s, Parkinson’s, and Creutzfeldt–Jakob’s diseases. Alzheimer’s and Parkinson’s disease are the most common neurodegenerative disorders among the elderly. Aggregation processes of amyloid peptides and proteins can be induced in vitro and in vivo by a variety of agents and conditions. Numerous point mutations responsible for the disease were identified in the AS gene.296−298 Aggregation of amyloid peptides and proteins in vitro is commonly studied by several methods such as circular dichroism spectroscopy, light scattering methods, thioflavin T or Congo red fluorescence, electron microscopy, and atomic force microscopy.299−304 Aggregates of amyloid peptides and proteins involved in Parkinson’s and Alzheimer’s disease are highly polymorphic, with mature amyloid fibrils constituting the predominant structure in fully aggregated amyloids.299,300,302,305−310 Increasing evidence from studies in different organisms and in vitro systems indicated that intermediate aggregation products, such as soluble oligomers of amyloidogenic peptides and proteins, are responsible for amyloidosis311,312 and are the toxic agents.313,314
Parkinson’s disease is associated with the formation of amyloid fibrils of the AS. This 14 kDa protein (first described in 1988)315 is natively unfolded and comprises 140 aa residues contained in three regions: (i) aa’s 1–60 constitute the N-terminal region including the hexamer motif KTKEGV; (ii) the central region with highly amyloidogenic NAC sequence (aa’s 61–95), containing two additional KTKEGV sequences; and (iii) the C-terminal region (aa residues 96–140) rich in acidic residues and prolines, including three highly conserved Tyr residues;316 this region is presumably disordered under most conditions.
At first sight, EC might seem of little use in AS analysis, as this protein does not contain any redox center for reversible EC. Tryptophan is absent, but there are four Tyr residues that can undergo oxidation at carbon electrodes.154,271,317 To our knowledge, EC was applied in studies of AS for the first time in 2004.226 Among the EC methods tested, the best results were obtained with SWV oxidation peaks of Tyr at carbon paste electrodes and CPS electrocatalytic (reduction) peak H using HMDE. Both methods reflected fibrilization of AS in vitro by the decrease of their signals, but the changes in peak H were much larger than the decrease observed in the AS oxidation signals. EC studies were combined with atomic force microscopy and circular dichroism spectroscopy. Recently, it has been proposed that oligomeric intermediates rather than the fibrils themselves can be the pathogenic agents of Parkinson’s disease.301,318−321 Minor changes in the adsorption state of AS at solid spectroscopic graphite were followed through the shift of the Tyr oxidation potential, consistent with the compact and less-compact/unfolded AS.157 HMDE was particularly sensitive to preaggregation changes in AS. In an early stage of a standard aggregation assay, peak H increased and shifted to less negative potentials. In the following interval (in which a dynamic light scattering indicated AS oligomerization), peak H diminished, its potential shifted in the opposite direction, and AS adsorbability decreased. These early changes in the interfacial behavior of the protein were attributed to disruption of long-range interactions and to destabilization of AS, while the subsequent changes were related to the onset of oligomerization. EC methods (CPS analysis and alternating current voltammetry) together with SDS-PAGE, optical spectroscopy (UV/vis absorption, steady-state and dynamic fluorescence, and dynamic light scattering), MS, and atomic force microscopy were used for monitoring and characterization of stable covalent oligomeric species (dimers, trimers, and higher oligomers) produced in vitro by the photoinduced oxidative formation of side-chain tyrosyl radicals of AS. It was concluded that EC methods represent new and simple tools for the investigation of amyloid formation.322
It has been shown that various factors affect the progression of the AS and amyloid β (Aβ)-peptide fibrillation.294,300,311−314,323−326 Metals,327−330 organic solvents and acidic pH’s,305 pesticides and herbicides,331 and oxidative stress stimulate AS aggregation in vivo and in vitro.319,332 Inhibition of the AS aggregation can be observed in the presence of other factors,333,334 including certain flavonoids with therapeutic potential in Parkinson’s disease treatment.335−341 Among them, baicalein has shown a significant inhibitory effect on the AS aggregation.334−341 This flavonoid possesses antioxidant properties, and upon oxidation, it can form quinones interacting with AS and inhibiting its fibrillation.224 Chan et al. followed the AS aggregation in the presence of Cu(II) ions and baicalein (inhibiting the AS aggregation) by measuring changes in SWV Tyr oxidation peak Y at screen printed carbon electrodes.342 In an agreement with previous studies,226,254 this peak decreased in the course of AS aggregation. In the presence of baicalein, the AS oxidation peak greatly increased, probably due to the presence of overlapping oxidation peak of baicalein itself. Surprisingly, after 24 h of AS aggregation, peak Y heights observed in the absence or presence of 0.1 mM baicalein or 5 mM Cu(II) ions were about the same. A rather high concentration of AS (50 μM) was used for the analysis as compared to those used for EC aggregation studies of AS at Hg electrodes (2 μM or lower to measure CPS peak H).
In Alzheimer’s disease, the Aβ-peptides involved represent a paradigm for studies of amyloid formation and conformation.300 These peptides result from cleavage of the transmembrane amyloid precursor protein.300,343,344 Diagnosis of Alzheimer’s disease in patients is still difficult, and suitable biomarkers are sought. Protein tau and its phosphorylation have recently appeared as attractive candidates. This 50–65 kDa heat stable protein has many functions, including maintaince of structural integrity of microtubules.223,345−347 Tau binding and stabilization of microtubules is disturbed in cells affected by Alzheimer’s disease where tau hyper-phosphorylation and aggregation are evident. The role of Cu(II) ions in tau protein dysfunction has recently attracted attention. This protein was studied by some techniques such as surface plasmon resonance-based immunochip223 and also CV and X-ray photoelectron spectroscopy.348 Tau protein was immobilized at a gold electrode, and EC methods allowed the detection of Cu(II) ion binding and differentiation between normal tau and phosphorylated tau films and revealed ion displacement by Zn(II) ion for phosphorylated tau but not for normal tau films. The majority of information about the peptide assemblies was obtained from full-length Aβ-peptides Aβ1–42 and Aβ1–40.319,349−360 The aa sequence of the human Aβ1–42 is 1-DAEFRHDSGYEVHHQKLVFFAEDVGSNKGAIIGLMVGGVVIA-42.
In addition to the full-length Aβ-peptides, a number of Aβ-fragments generated in vitro were found to form amyloid fibrils. Among them, the Aβ16–22, included in the hydrophobic C-terminal region and containing the KLVFF core, represented one of the most studied fragments.361−369 It was suggested that an Aβ1–42 conformation with the C-terminus forming inside the wall of a hollow core and the N-terminus plays an important role in the peptide fibrilization.370
EC methods have been applied also in studies of Aβ-peptide aggregation,156 in addition to a variety of methods mentioned above. Interaction of amyloid peptides and proteins with lipid bilayers at Au and Hg371,372 electrodes was also studied. In contrast to the EC analysis of AS employing both Hg and carbon electrodes, EC analysis of Aβ-peptides relied predominantly on voltammetric oxidation signals of Tyr residues (peak Y) at carbon electrodes. Vestergaard et al.224 observed a decrease in Tyr voltammetric oxidation peak Y at GCE in the course of Aβ1–42 and Aβ1–40 aggregation and found a difference in the rate of aggregation of these two peptides. EC analysis was complemented with fluorescence of thioflavin T and atomic force microscopy analysis. Later, Aβ1–40 aggregation in the presence and absence of Zn(II), Cu(II), and Mg(II) ions was studied using SWV peak Y at GCE and electron microscopy.373 While Mg(II) ions were almost without effect, Zn(II) and Cu(II) stimulated the peptide aggregation. In the absence of metal ions, the time dependence of peak Y was sigmoidal, but in the presence of metal ions, the lag period almost disappeared as a result of rapid metal-induced aggregation. However, after prolonged aggregation (incubation of 50 μM Aβ1–40 for almost 150 h at 37 °C), peak Y heights of metal-treated peptides were significantly higher than this peak produced by Aβ1–40 peptide aggregated in the absence of metals. Electron microscopy displayed different morphologies of the peptide aggregates obtained in the presence and absence of Zn(II) or Cu(II). No precautions preventing bacterial growth (resulting from very long incubation time) were mentioned in this paper.373
Interactions of benzothiazole dyes (such as thioflavin T and [2-(4′-methylamino)phenyl) benzothiazole], BTA-1) with Aβ-peptides were well documented by fluorescence methods.300,374−379 Recently, interactions of these dyes with Aβ1–42 and Aβ1–40 peptides not containing oxidizable Tyr residues (aa sequences of the peptides in rats) were studied.380 Using DPV and CV, it was shown that thioflavin T and BTA-1 produce oxidation peaks at screen-printed carbon electrodes. Dramatically different behavior of oxidation peaks of positively charged thioflavin T and its neutral analog BTA-1 in the course of the Aβ-peptide aggregation was observed. Aggregation of these peptides in the presence of thioflavin T resulted in an unexpected increase of the oxidation thioflavin T peak after 24 h of incubation. The authors speculated that changes in the peak heights after longer incubation times resulted from changes in thioflavin T binding sites along the peptide β-sheets, while very early changes of the dye oxidation signals were due to oligomerization of the peptides.
Carbon nanotube (CNT)-modified electrodes are widely recognized for their electrical conductivity and catalytic effects.381,382 These electrodes were used to study Aβ1–42 peptide aggregation and disaggregation processes induced by the β-sheet breaker LPFFD.383 Tyr oxidation peaks were measured by DPV or CV at relatively low Aβ1–42 peptide concentrations (down to 10 μM) during the aggregation process in vitro. Antiaggregation effects of the peptide inhibitor LPFFD were observed in combination with conventional methods. In fibrils, Tyr residues were unexposed, while in earlier peptide forms exposition of Tyr was observed. The authors believe that this method opens a new avenue in EC studies of Aβ-peptides aggregation and better understanding of the mechanism that causes Alzheimer’s disease. In addition to typical Aβ-peptides, Tyr homopolymer (poly(Tyr)) was used as a model for studies of protein aggregation, which was accompanied by changes in Tyr oxidation signals at carbon electrodes.384 These changes were explained in terms of varying accessibility of Tyr residues for the EC oxidation.
In principle, two classes of in vitro measurements can be distinguished: (i) usual measurements in a bulk solution, for example, monitoring the fluorescence emission by amyloidophilic dyes,385−387 and (ii) recently applied techniques such as quartz crystal microbalance,388−393 surface plasmon resonance,394,395 or microcantilever measurements,388,391 allowing measurements of the growth of amyloid fibrils bound to a surface. In these techniques, reliable attachment of the peptide or protein to the surface plays a critical role.
The attachment of amyloid fibrils or other protein structures to matrixes can be achieved either via an amide coupling reaction394,395 or through antibody–antigen interactions.393 Alternatively, Cys-containing proteins can be attached to some metal surfaces such as gold or mercury via the Au–S or Hg–S bonds. This type of binding can be applied neither to Alzheimer’s Aβ-peptides nor to Parkinson’s AS protein, which do not contain Cys residues. Recently, a novel strategy was described, based on the attachment of small molecule linkers to protein fibrils, to immobilize the protein nanostructures to Au surfaces.396 This strategy involved the reaction of 2-iminothiolane and cystamine with the amyloid fibrils, which enabled their covalent linkage to gold surfaces via Au–S bond. Such attachment was irreversible, allowing quantitative analysis by biosensors that can revolutionize the way we look at a process of an amyloid growth. Moreover, this study contributed to a better understanding of the nature and relative importance of covalent versus noncovalent forces acting on protein superstructures at metal surfaces. This strategy appears attractive showing that the chemical reactions do not disturb the amyloid structures. In recent years, de novo designed peptides were synthesized, and their fibrilization and other properties were systematically studied.299
Cys residue can be easily added to Aβ-peptides in the course of their synthesis. Peptide-based octamer YYKLVFFC containing the KLVFF (Aβ16–20) core domain with additional terminal Tyr and Cys residues was designed in Hamley’s laboratory,397 to enable the peptide functionalization. Cys residue allowed covalent attachment to mercury and other metal surfaces as well as its chemical modification using its −SH group. The two Tyr residues served as fluorescence tags, potentially enabling bioconjugation, or responsiveness to enzyme (e.g., by phosphorylation/dephosphorylation reactions). The self-assembly of YYKLVFFC in solution has been investigated by various methods such as a variety of spectroscopic, scattering, and microscopic ones.398
The tendency of fibrils to spontaneous orientation was investigated in a detailed study with a focus on the orientation of the peptide alignment (in bulk solution and dried films) by X-ray diffraction and near-edge X-ray absorption fine structure polarized Raman spectroscopy, linear UV dichroism, and Fourier transform infrared spectroscopy.399 These approaches provided detailed information about the orientation of fibrils and of the constituent Tyr fluorophores. A similar approach was used to modify the Aβ12–28 peptide with a Cys residue at the C-terminal end with its subsequent immobilization onto gold electrodes.397 Using this platform, it was possible to electrochemically monitor the interaction of the peptide with Congo Red as well as with a β-sheet breaker peptide.
EC and interfacial properties of the above YYKLVFFC peptide and two other Aβ peptides YEVHHQKLVFF and KKLVFFA were investigated at carbon and predominantly at mercury electrodes. The interfacial behavior of these model amyloid peptides at the metal|aqueous solution interface was studied by voltammetric and CPS analysis. All three peptides adsorbed in a wide potential range exhibited different interfacial organizations depending on the electrode potential. At the least negative potentials, chemisorption of YYKLVFFC peptide occurred through the formation of a metal–sulfur bond. This bond was broken at about −0.6 V. The peptide then underwent a self-association at more negative potentials, leading to the formation of a “pit” characteristic for a 2D condensed film (Figure 13).221 Under the same conditions, the other peptides did not produce such a pit. Formation of the 2D condensed layer in YYKLVFFC peptide was supported by the time, potential, and temperature dependences of the interfacial capacity, and the presence of the 2D layer was reflected also by the peptide CPS signals due to the CHER. The ability of YYKLVFFC peptide to form the potential-dependent 2D condensed layer has not been reported for any other peptide or protein molecules. At this stage, it is not clear to which extent is the YYKLVFFC peptide 2D condensation related to the known oligomerization and aggregation of Alzheimer amyloid peptides.
Figure 13.
(A) Capacity–potential curves of 1 μM peptide YYKLVFFC (black), YEVHHQKLVFF (red), and KKLVFFA (blue) in 35 mM Na-phosphate, pH 7 (dashed line). Peptides were adsorbed at −0.1 V, for tA of 120 s at HMDE, followed by recording of ac voltammogram with a frequency of 150 Hz, amplitude of 5.0 mV, and a scan rate of 8.0 mV/s. (B) Capacity–potential curves of 1 μM peptide YYKLVFFC recorded in 35 mM phosphate buffer, pH 7.0 at different temperatures, as indicated on the graph. Accumulation potential EA of −0.5 V; accumulation time tA of 120 s. Adapted with permission from ref (221). Copyright 2013 Elsevier.
6.2. Tumor Suppressor Protein p53
Shortly after the invention of CPS protein analysis with DTT-modified Hg electrodes,239 the new method was applied for investigation of the tumor suppressor protein p53.105 This protein plays a critical role in the cellular responses to DNA damage by regulating the gene expression of factors involved in DNA repair, cell cycle, and apoptosis.400,401 p53 protein is inactivated by mutation in about one-half of human cancers. Most oncogenic mutations are located in the DNA-binding core domain of the protein.102,402 Better understanding of the molecular basis of p53 inactivation in cancer is essential for the development of novel anticancer strategies.403 The structural effects of many oncogenic p53 mutants were intensively studied by X-ray crystallography and complementary techniques (reviewed in ref (404)). The most frequent highly destabilized cancer-associated mutant, R175H, has, however, eluded a detailed structural characterization, indicating the need for complementary techniques to study this and other unstable mutants. R175H is located in the L2 loop of the DNA-binding domain in a vicinity of the zinc coordination sphere (Figure 14A). The mutation induced substantial structural perturbation in the zinc-binding region (Figure 14B), causing increased conformational flexibility and loss of the zinc ion and sequence-specific DNA-binding activity.404,405 Similar effects were found in the wild-type (wt) protein upon removal of the zinc ion.406
Figure 14.

Structure of DNA-binding domain of p53. (A) Overall structure of T-p53C (PDB entry 1UOL).101 Sites of cancer mutations investigated in this study (V143A, R175H, F270L, and R273H) are highlighted as green stick models. (B) Close-up view of the zinc coordination sphere, with the four zinc ligands shown in magenta. (C–F) CPS peak H of wild type T-p53C (black) and mutant R175H (red) at DTT-HMDE in 50 mM phosphate, pH 7 at (C) 11.1 °C, (D) 13.9 °C, (E) 15.9 °C, and (F) 20.3 °C. (G–J) CPS peaks H of (G) wt, (H) R175H, (I) R273H, and (J) V143A treated by 0 mM (red), 5 mM (blue), 10 mM (green), and 20 mM (cyan) EDTA at 0 °C for 10 min. CPS measurements were performed at 18 °C. Adapted with permission from ref (105). Copyright 2011 American Chemical Society.
Superstable variants of wt p53 and mutant core domains (T-p53C), for which high-resolution structures are available, and which are better suited for physicochemical studies than the native p53 proteins,101,407 were used for the CPS analysis. Large differences between the CPS responses of wt and R175H proteins were found in a wide temperature range (Figure 14C–F). Below 13 °C, wt p53 did not produce any peak as compared to the mutant, which yielded a well-developed peak H (Figure 14C), suggesting better accessibility of aa residues involved in the electrocatalysis of the mutant protein. Between ∼16 and 20 °C, wt T-p53C yielded a single peak H, while the mutant displayed two peaks (Figure 14E, F). Wt and mutant R175H were treated with EDTA to remove zinc ion from their molecules. Treatment of wt T-p53C with 5 mM EDTA for 10 min resulted only in shifts of peak H1wt to positive potentials, while 20 mM EDTA produced in addition to peak H1wt a broad peak H2wt at −1.94 V, resembling thus the CPS profile of untreated R175H (Figure 14G). The same EDTA treatment of R175H was almost without effect on its CPS peaks (Figure 14H), in agreement with the expected absence of Zn2+ in this mutant protein. EDTA treatment of other mutants produced similar effects in their CPS profiles as in that of wt T-p53C (Figure 14I,J), albeit at lower EDTA concentrations. CPS profiles of all untreated mutants differed from that of the wt protein.
It was suggested that the sharp peak H1 is related to accessible electrocatalytically active groups in an ordered T-p53C layer. The broad peak H2 might involve distorted or partially unfolded protein regions, strongly influencing the structure of the layer at the electrode surface. The electrode process responsible for this peak required higher temperatures than peak H1, which was produced already at 8.6 °C in R175H and at 13.9 °C in wt T-p53C (Figure 14).105 Considering the relations between the appearance of peak H2 and the effect of the zinc removal from T-p53C by EDTA (Figure 14G,H), it was tentatively suggested that in R175H the Cys residues in the perturbed zinc-binding site (Cys176, Cys238, and Cys242) significantly change the structure of the adsorbed protein layer and contribute to CHER. Most of the results were obtained with DTT-modified HMDE, but similar differences between wt and mutant protein CPS profiles were achieved using DTT-AgSAE.105 However, the square wave voltammetric oxidation signals of wt and R175H mutant at carbon electrodes and cyclic voltammetric catalytic peaks close to −1.9 V at DTT-HMDE differed only a little.105
6.3. Analysis of Poorly Soluble Membrane Proteins
Among proteins occurring in mammals, about 30% are bound to membranes.408,409 Contrary to the rest of the proteins, which are mostly water-soluble and can easily be analyzed using EC techniques, membrane proteins are poorly soluble or insoluble in aqueous solutions and/or unstable outside the lipid membrane. Because of these problems, information about the EC behavior of membrane proteins was rather limited. Only recently has it been shown that by using the adsorptive transfer voltammetry and CPS, membrane proteins solubilized by a detergent can be attached to mercury or carbon electrodes and their reduction and oxidation responses studied with a protein-modified electrode immersed in a blank background electrolyte.410,411 In this way, a transmembrane protein Na+/K+-ATPase (NKA, a sodium–potassium pump) was attached to the electrodes and investigated in detail.410 Nonionic surfactant octaethylene glycol monododecyl ether (C12E8) was used for solubilization of NKA. Using C12E8, well-developed peak H, but not peak S, was obtained at HMDE (Figure 15A). In contrast, water-soluble cytoplasmic loop C45, which was analyzed in the absence of C12E8, yielded both peaks H and S (Figure 15B). It was concluded that C12E8 interfered with reduction of the Hg–S bond but not with CHER, responsible for peak H, under the given experimental conditions. NKA and its C45 loop were detectable at a femtomole level. At pyrolytic graphite electrodes, oxidation peaks of Tyr and Trp residues were obtained (see section 4).
Figure 15.
Chronopotentiograms of (A) Na+/K+-ATPase (NKA) and (B) its C45 loop. CPS experiment at HMDE, concentration of proteins: (A) 10 μM and (B) 500 nM, tA of 30 s; supporting electrolyte, Britton–Robinson buffer, pH 6.5; stripping current Istr, −10 μA. (C) CPS records of 5 μM C45 loop before (black) and after (red) incubation with cisplatin. (D) Dependence of peak heights (C45 peak S, C45 peak H2, and cis-Pt) on the concentration of cisplatin. Concentration of cisplatin (for C) was 40 μM, activated complex was used for all experiments. EC parameters: supporting electrolyte 0.2 M phosphate buffer, pH 7.4; tA of 30 s, Istr of −20 μA. “*” in inset of panel A: NKA transmembrane part. Adapted with permission from refs (410) and (412). Copyright 2012 Wiley-VCH Verlag GmbH&Co and 2012 Elsevier.
Changes in NKA structure in dependence on ATP binding were studied using adsorptive transfer and usual CPS at bare HMDE411 in the arrangement mentioned above. Peak H shifted to less negative potentials, and peak area increased with increasing ATP concentration, indicating better accessibility of proton-donating aa residues to the electrode surface, most probably resulting from opening of the cytoplasmic part of the NKA molecule. This behavior agreed well with changes in peak H of aqueous-soluble riboflavin-binding protein after riboflavin binding.256
The above techniques were utilized in the investigation of the effects of cisplatin binding to NKA.412 Earlier studies indicated that cisplatin could bind to proteins in blood or in cytosol.413−417 It was hypothesized that acute kidney failure accompanying cisplatin administration in cancer therapy might be related to inhibition of NKA.418−422 Testing of platinum drugs effects on NKA revealed almost no influence of carboplatin and oxaliplatin (known to be much less nephrotoxic than cisplatin) on NKA activity. In contrast, the cisplatin binding reduced NKA activity by 50%, and this inhibitory effect was substantially reduced in the presence of 0.5 mM DTT. EC studies of C45 loop showed that peak S decreased with increasing cisplatin concentration and its complete disappearance at 2-fold excess of cisplatin over C45, indicating cisplatin binding to Cys residues localized on the surface of C45. Disappearance of peak S was accompanied by a formation of peak H2 (not produced by intact C45), which grew with increasing cisplatin concentration. However, a decrease of peak H1 with increasing cisplatin concentration was observed (Figure 15C,D). EC studies were complemented by matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) MS experiments and determination of free −SH groups by Ellman’s reagent. It was concluded that (i) inhibition of NKA activity could be involved in nephrotoxicity in cisplatin-treated patients, and (ii) cisplatin binds to Cys residues in C45 loop and particularly to Cys367 close to the phosphorylation site. The results of the NKA analysis411 show that CPS analysis can be used not only for studies of structural changes and intermolecular interactions in water-soluble proteins at bare155,230,232,233,238,254,256,322 and thiol-modified electrodes,105,228,238,239 but also for the investigation of poorly soluble proteins.410,411,423
7. DNA–Protein Interactions
Interactions of proteins with DNA in cells control many aspects of DNA metabolism ranging from gene transcription, DNA replication, DNA-damage repair, or chromosome segregation to higher order structure organization of chromatin. Particularly, transcription factors constitute typically about 5% of the total number of genes in most species, highlighting the importance of proper gene expression regulation.11 Basal transcription factors (e.g., TATA-box binding protein, TBP) constitute general transcription complexes, which are regulated by intricate networks of transcription activators, repressors, and cofactors (e.g., immune responses converge on NF-κB and AP-1 transcription factors). Furthermore, the transcription regulation can be regulated through master transcription factors (e.g., factors initiating cell differentiation programs or “genome guardian” p53).102
A great number of protein complexes (e.g., a single-strand binding (SSB) complex) are involved in DNA replication, DNA repair, and homologous recombination.424 Given the high frequency of endogenous DNA damage, it is of great importance for every cell to have adequate repair mechanisms to keep them healthy. To deal with different kinds of base damage, cells are equipped with proteins recognizing chemically modified DNA and/or structurally altered DNA strands (e.g., MutY, MutS, HMG proteins). DNA-binding enzymes like helicases (e.g., XPD) and endonucleases (e.g., EndoIII) then assist in damage removal (e.g., base excision repair (BER) processes). Eventually, DNA polymerases and ligases finish the job by filling gaps and recovering DNA strands.425 On the basis of the nature of the damage, timing (through the cell cycle), and chromatin localization of the damage, the correct mechanisms must be employed again highlighting the importance of regulation at multiple levels. Deregulation of the above processes is deleterious for cells and organisms. Not surprisingly, many of the above protein–DNA complexes are involved in cell protection against tumorigenesis, underlining their importance for human health. It is therefore extremely important to investigate the mechanisms of DNA–protein binding and the nature of complexes formed between DNA and proteins. In the past two decades, we witnessed a great expansion from 100 to about 2500 of high-quality structures of DNA–protein complexes (including DNA-based aptamers; January 2014 in PDB, http://www.rcsb.org/pdb/; and in NPIDB, http://npidb.belozersky.msu.ru/).426 These structures have provided valuable insights into the principles of DNA–protein binding.
The phosphate backbone of DNA provides a negatively charged surface for the binding of basic side chains (Lys, Arg) of proteins.11,427 Such DNA–protein contacts are utilized by structure-specific (i.e., sequence nonspecific) proteins for DNA structure recognition. For the sequence-specific binding, these contacts stabilize the protein–DNA interaction mostly mediated by hydrogen bonds between bases and proteins. Base pairs inside the double-stranded (ds) DNA are accessible through either major or minor groove. Most sequence-specific binding proteins (e.g., transcription factors) approach the major groove (Figure 16A), which can easily accommodate an α-helix element of particular protein domain (e.g., helix-turn-helix). However, crystal structures provided insights also into special DNA–protein binding modes. For example, some proteins use β-ribbon or loop elements for major groove binding, and a few proteins bind into the minor groove. Particularly, TBP binding to minor groove partially unwinds and kinks DNA (Figure 16B), facilitating the initial step of transcription.428
Figure 16.
Different DNA–protein binding modes. (A) Most proteins insert helix element into the major groove of DNA molecule. For example, helical bZIP motif of the AP-1 transcription factor binds within the major groove of the specific DNA sequence (PDB: 1FOS).881 (B) Some proteins employ, however, β-sheets for their binding into the minor groove of DNA. For example, TBP protein binds to minor groove and partially unwinds and kinks DNA (PDB: 1YTB).428
In addition to the high-resolution X-ray crystal analysis, a number of methods have been used in studies of DNA–protein interactions.57 However, applications of EC analysis to studies of DNA–protein interactions were until recent years rather scarce. Considering that both DNA and proteins are electroactive and can be analyzed with high sensitivity, application of electrochemistry in DNA–protein analysis appears natural and very promising.
In difference to a large number of reviews on EC analysis of nucleic acids15−36 and proteins,44,155,210,229,263,429−432 comprehensive reviews on electrochemistry of nucleic acid–protein interactions are missing. Numerous reviews on DNA or ribonucleic acid (RNA) aptamers432−442 include interactions of such aptamers with proteins, but usually changes in EC signals of nucleic acids are measured. In this Review, nucleic acid and peptide aptamers will be mentioned only in connection with biomarkers (section 9). This section will focus on new ways of label-free EC analysis of DNA–protein interactions, which play significant roles in nature (such as DNA sequence-specific binding), and in which changes in the protein EC signals are monitored.
7.1. Early Work
To our knowledge, the first EC papers on DNA– or RNA–protein interactions were published more than 25 years ago. In 1986, the course of RNase cleavage of (a) RNA immobilized at the electrode and (b) in solution was investigated by voltammetry.276 More than 10 years later, DNA was immobilized at a gold electrode, and its enzymatic cleavage by deoxyribonuclease I (DNaseI) as well as formation of RNA duplexes at the electrode surface were monitored by quartz crystal microbalance.443 This work showed that surface-confined DNAs and RNAs could be manipulated at the gold electrode surface. Almost at the same time, cleavage of supercoiled DNA by DNaseI in solution and at the mercury electrode surface was investigated by ac voltammetry.444 Nicking of circular DNA resulted in a specific peak, which enabled investigation of the DNA enzymatic cleavage. Kinetics of the DNA cleavage at the electrode surface showed restricted accessibility of DNA at the surface as compared to solution cleavage. Cleavage of DNA adsorbed at positively charged electrode was inhibited, while at negatively charged electrode stimulation of the cleavage was observed. In all of these experiments, changes in DNA signals were measured, but in principle, EC analysis of DNA–protein interactions could rely also on changes in protein signals. Surprisingly, no attempt was made to use changes in Brdička’s catalytic response of Cys-containing proteins for this purpose (section 5.5). This might be due to alkaline pH used for Brdička’s catalytic response, which is not favorable for studies of DNA–protein interactions. In 2005, it was shown that thiol end-labeled DNAs produced catalytic peaks at potentials less negative than those of Cys-containing peptide 9-mer [Lys8]-vasopressin in cobalt-containing solutions.445 Using oligodeoxynucleotide HS-(TCC)7 and adsorptive transfer stripping DPV, this system was used at pH 7.5 to study H2A histone interaction with DNA,271 showing a decrease in the DNA signal after the histone binding. No decrease was observed when DNA and histone were incubated in 1 M NaCl, preventing DNA histone binding.
Among the first papers on EC detection of DNA–protein interactions, there was the method developed by Barton et al.446 This method was based on dsDNA-mediated charge transport.18,34,446 Binding of a base flipping enzyme, MHhaI, to dsDNA greatly decreased the EC signal of DNA labeled with daunomycin, indicating that this binding disturbed the DNA base stacking.447 Using a DNA repair enzyme MutY, which binds to 8-oxoG:A and G:A mismatches, single-base mismatches in DNA were detected.446 Shortly afterward, it was shown that the [4Fe–4S] cluster, contained in MutY, can be utilized in the detection of MutY–DNA interactions.448 As compared to free MutY, which shows no EC signal, the DNA-bound MutY displayed a reversible couple on cyclic voltammograms at gold electrodes. This method required relatively high concentrations of MutY (0.8 mM).448 These experiments marked the beginning of interesting research on DNA–protein interactions in Barton’s laboratory, which still continues (see section 7.2).
Almost at the same time, another technique was proposed by Palecek’s group,449 showing that binding of practically any protein to DNA can be detected using the so-called double-surface technique (DST) (reviewed in ref (34)) combined with EC detection of the protein electrocatalytic peak H (section 5.2). In DST, the DNA–protein interaction or DNA hybridization was performed at one surface (usually magnetic beads, reviewed in ref (450)) and the EC detection of these events at another surface (usually metal or carbon electrodes). This technique was proposed to overcome difficulties in DNA hybridization experiments using complex biological matrixes. Similarly to the work of Barton et al.,448 DNA repair protein was used to detect mismatched bases in DNA449 but instead of MutY, the MutS protein was used. MutS plays an important role in the DNA repair system in prokaryotic and eukaryotic cells,451−454 recognizing unpaired and mispaired bases in duplex DNA, and this protein was shown as a useful tool for the detection of point mutations.449,455−459 In DST experiments, biotinylated DNA was first attached to the magnetic beads, followed by their incubation with the MutS protein solution and magnetic separation of the DNA–MutS complexes (including extensive washing). Next, the protein was dissociated from its DNA complex and detected electrochemically at mercury electrodes using peak H (section 5.2). In this way, tens of attomoles of this protein were detected. The sensitivity of the determination at carbon electrodes was by 3 orders of magnitude lower.449 This highly sensitive label-free detection of MutS protein opened the door to the development of DNA chips for a high-throughput EC determination of proteins, as well as to the detection of point mutations and insertions/deletions in DNAs. These methods represented a new approach in the analysis of nucleic acid–protein interactions, which could be applied to both DNA and RNA, and to a large number of nucleic acid-binding proteins. DST methods were also applied using proteins (e.g., p53) attached to the beads and interacting with DNA in solution, followed by EC detection of DNA.460
In 2005, Kerman et al.461 used single-walled carbon nanotube (SWCNT)-modified screen-printed carbon electrodes to investigate interaction of the SSB protein with DNA. Bacterial SSB protein is a homotetrameric protein playing an important role in DNA replication, recombination, and repair.462 It binds selectively to ssDNA and facilitates DNA unwinding by helicases.463 Each monomer of the protein contains 3 Trp and 4 Tyr residues (section 4). DNA was bound to the SWCNT-carbon electrode via its end amino link, and the DNA-modified electrode was immersed into the SSB protein solution (10–50 μg/mL). Binding of the protein to ssDNA resulted in an appearance of the protein oxidation peak and in a decrease of the DNA guanine signal. No protein peak resulted from interaction of double-stranded DNA with the SSB protein solution.
7.2. DNA Charge Transport for DNA–Protein Sensing
The work of Barton’s group, which started with MHhaI–DNA interaction studies,446 has continued up to the present and was reviewed in 2012.464 Here, only a brief summary will be given, and recent results will be discussed. From the beginning, this group was primarily oriented to the merits of the DNA charge transport (CT)34,465 and only secondarily to the development of a method for detection and investigation of the DNA–protein interactions. They devoted their attention to the electronic properties of the DNA double helix and showed that DNA is an effective conductor of charge and that stacked bases function as the path of this conductivity.34,464,465 Electronic coupling of the donor and acceptor to the π-stack is required, and DNA CT is highly sensitive to the structural integrity of the stacked base pairs and can report the integrity changes on a single base pair level. Bases in the DNA double helix are stacked next to each other with an interplanar spacing similar to that of graphite, forming thus a conductive path of overlapping π-orbitals that extends along the DNA helical axis. Although the merits of the DNA CT were recently reviewed,34,465 a debate about various aspects of this CT has continued,466−469 suggesting that DNA CT and experimental arrangement of its EC studies are far from simple. Intercalation of the redox label, its charge, and the way of the label tethering to DNA as well as the structure of the DNA monolayer at the gold electrode surface are among the factors affecting the DNA CT.
After thorough studies of the DNA CT,464 Barton asked a question whether repair proteins could utilize DNA CT in signaling of the DNA damage throughout the genome. This question became even more attractive considering that a subset of DNA repair proteins contains redox [4Fe–4S] clusters. Examining metalloproteins from the BER proteins showed that [4Fe–4S]2+ clusters were not necessary in the protein folding process.106 Crystal structures of DNA complexes with two BER proteins (MutY and EndoIII) revealed the proximity of these clusters to the DNA backbone.120,134,470 Studies of these proteins at DNA-modified gold electrodes showed their midpoint potentials in the region potentially required for a biological redox switch and suggested much higher affinity for DNA in their oxidized state.18,467 These results helped to answer the above Barton’s question positively and to propose a model on how the BER proteins could cooperate in searching the genome and binding DNA to accomplish its eventual repair (Figure 17).
Figure 17.
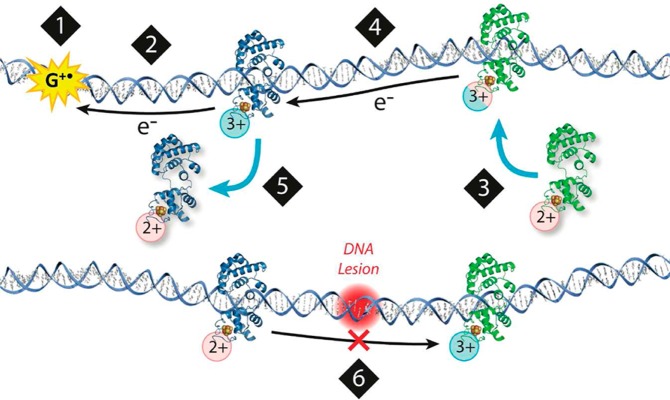
Model for a DNA-mediated search by repair proteins. (1) When the cell undergoes oxidative stress, guanine radicals are formed, triggering a repair protein to bind DNA. (2) DNA-binding protein is oxidized, releasing an electron that repairs the guanine radical. (3) Another repair protein binds to a distant site. As it binds to DNA, there is a shift in the redox potential of the protein, making it more easily oxidized. (4) The protein could then send an electron through the DNA base pair stack that travels to a distally bound protein, scanning the intervening region for damage. (5) If the base pair stack is intact, charge transport occurs between proteins. The repair protein that receives the electron is reduced and dissociates. (6) If a lesion is present (red), charge transport is attenuated, and the repair proteins will remain bound in the oxidized form and slowly proceed to the site of damage. Adapted with permission from ref (464). Copyright 2012 American Chemical Society.
We shall not explain here this model (which agrees well with the experimental data of Barton’s group) in greater detail, because its biological implications are out of the scope of this Review. Of course, it is an electrochemist’s wish to have a chance to make conclusions about biological processes in cells based on signals reflecting the DNA–protein interactions at electrode surfaces. Yet mostly real cell biology is much more complex than even the highly complicated electrode surfaces. Nevertheless, Barton’s model offers the interesting possibility of the DNA repair mechanism, which deserves further attention.
It is interesting that searching for damage in the genome is neither limited to BER glycosylases nor to the presence of [4Fe–4S] clusters in the proteins. Various [4Fe–4S] cluster-containing DNA binding proteins not related to DNA repair were identified.471 Testing of DNA-bound XPD protein (ATP-dependent helicase from Sulfolobus acidosaldarious) showed the same voltammetric peak potentials as those obtained with BER enzymes.472 Further work showed that the XPD redox signal responded to addition of ATP, suggesting that the helicase function can be investigated electrochemically. The [4Fe–4S] clusters in XPDs from archae appeared as relatively labile and not necessary to maintain the protein structure. A number of DNA-binding proteins involved in genome maintenance, such as FancJ, Dna2, primase, and DNA polymerases, were also shown to contain [4Fe–4S] clusters.473,474 The question of whether these proteins are involved in genome damage signaling has not been answered yet. The mechanism of bacterial ferritin Dps involvement in DNA protection was examined.475 It was shown that protective effects of Dps vary with iron content of the protein and that the DNA charge transport may be involved in the mechanism by which Dps protects the genome of pathogenic bacteria from a distance.
In the studies of DNA–protein interactions based on DNA CT, relatively high protein concentrations were used, making the methods poorly competitive, when compared to other methods not relying on electrochemistry,57 such as gel electrophoresis. Typically, a DNA-modified Au surface was prepared by self-assembly of thiolated DNAs to form a monolayer. Such DNA monolayer should offer good accessibility of relatively large protein molecules targeting specific DNA sequences. While certain control over the DNA surface density was possible, it was difficult to control the dispersion of DNA molecules within the film, where high density clusters of DNA molecules could be formed,476,477 in which the DNA-binding protein could hardly find its specific sequence or recognize the DNA structure. Only recently did Furst et al.478 propose an alternative approach to form a functionalized homogeneous DNA-free mixed SAM followed by conjugation of DNA to this SAM. They labeled DNA with a cyclooctyne moiety tethered to the DNA 5′-end, followed by modification of Au electrodes with an alcohol-terminated SAM doped with an azide-capped alkyl thiol. This process was followed by a copper-free click reaction in which cyclooctyne-labeled dsDNAs were coupled to the film via azide–alkyne cycloaddition. In this way, the tendency of DNA helices to cluster in the large high density domains was significantly decreased, allowing the formation of very low-density DNA monolayers containing as little as 5% of DNA in the surface layer. The low-density monolayers offered a better accessibility to interacting protein than conventional high-density films as demonstrated with detection of TBP binding at the nanomolar level.478
Using the two-electrode platform, this system was recently applied for the detection of mammalian DNA methyltransferase1 (DNMT1) in human tumors by Furst et al.479 Several methods of EC detection of DNA methylation are available,480 and some of them are based on differences of restriction cleavage of nonmethylated and methylated DNA.481 Because methylation does not affect CT, DNA methylation-sensitive restriction enzyme was used by Furst et al.479 to transform the DNA methylation state into an EC signal. The principle of this method is shown in Figure 18, showing clearly that methylation prevents DNA restriction cleavage (blue arrow) and does not thus interfere with the CT, while in the absence of methylation, restriction cleavage of DNA (red arrow) results in a significant decrease of DNA at the surface and in diminishing EC signal. It is important to note that in this method, biopsied tumor tissues and crude colorectal cancer cell lysates were used, thus making this new approach potentially applicable in cancer research and clinical testing.
Figure 18.
Electrochemical platform (right) and scheme (left) for the detection of human methyltransferase activity from crude cell lysates. (right) The electrochemical detection platform contains two electrode arrays, each with 15 electrodes (1 mm diameter each) in a 5 × 3 array. Multiple DNAs are patterned covalently to the substrate electrode by an electrochemically activated click reaction initiated with the patterning electrode array. Once a DNA array is established on the substrate electrode platform, electrocatalytic detection is then performed from the top patterning/detection electrode. (left) Overview of electrochemical detection scheme at each electrode of the 5 × 3 array. DNA, patterned onto the bottom electrode using the copper-activated click chemistry, is electrocatalytically detected from the top electrode using methylene blue (MB+) as the electrocatalyst and ferricyanide for amplification. Crude cell lysate is then added to the surface containing the patterned DNA. If methyltransferase (green) is present (blue arrows), the hemimethylated DNA on the electrode is methylated (green dot) by the methyltransferase to a fully methylated duplex; if methyltransferase is not present (red arrows), the hemimethylated DNA is not further methylated. A methylation-specific restriction enzyme, BssHII (purple), is then added. If the DNA is fully methylated (blue arrows), the electrochemical signal remains protected, and the DNA is not cleaved. However, if the DNA remains hemimethylated (red arrows), it is cut by the restriction enzyme, and the electrocatalytic signal associated with MB+ binding to DNA is diminished significantly. Adapted with permission from ref (479). Copyright 2014 Proceedings of the National Academy of Sciences of the United States of America.
7.3. Electrochemical Impedance Spectroscopy (EIS)
In the field of biosensing, EIS is particularly well suited for the detection of binding events on the electrode/transducer surface (reviewed in refs (482) and (483) and in section 8.5.1). This method was recently applied for a label-free detection of DNA–protein binding using thiolated DNA bound to gold electrodes, backfilled either with 6-mercapto-1-hexanol484 or with 4-mercapto-1-butanol.485 EIS was measured in the presence of K4Fe(CN)6/K3Fe(CN)6. Chang and Li484 investigated the TATA-box specific binding of TBP, known to use its β-sheet domain to bind the DNA minor groove (Figure 16B).486 They observed a significant impedance increase and obtained high sensitivity (down to 0.8 nM TBP) and a good specificity. They showed that formation of a DNA triplex (instead of duplex DNA) destabilized the DNA–protein complex, while binding of daunomycin to DNA duplex completely abolished the TBP binding to DNA. The presence of 5 μM BSA did not interfere with TBP binding.
Tersch and Lisdat485 studied the binding of a transcription factor NF-κB to the DNA recognition sequence GGGRNNYYCC (where R stands for purine and Y for a pyrimidine base). They found that incubation of the 25-mer DNA-modified electrode with 33 μg/mL NF-κB resulted in a decreased charge transfer resistance, while capacitance of the DNA–protein layer decreased only slightly. In the same experiment, in which DNA without the recognition sequence was used, no change in the impedance of the electrode was observed. In addition, cleavage of dsDNA by the restriction enzyme BamHI at the electrode was studied using EIS and CV with methylene blue end-labeled DNA. It was necessary to remove DTT (usually present in BamHI solutions) because it strongly affected the surface impedance. After treatment of dsDNA containing the restriction sequence (GGATCC), the charge transfer resistance strongly decreased in agreement with removal of the enzyme cleaved DNA fragments from the electrode. Similarly, CV reflected the DNA restriction cleavage by a decrease of the methylene blue peaks.
Tersch and Lisdat485 did not consider earlier results of Chang and Li,484 and we can only speculate about reasons of differences in their data. Figure 19A shows a relatively small decrease of the charge transfer resistance after binding of the NF-κB transcription factor to dsDNA, contrasting a large increase in the charge transfer resistance after TBP binding to DNA (Figure 19B), observed by Chang and Li.484 Experimental arrangements in Figure 19A and B somewhat differed: (a) screening the electrode with a different thiol and interference of DTT reported only in Tersch and Lisdat work;485 and (b) different potentials under which the EIS measurements were performed: Chang and Li measured at 0.2 V, while Tersch and Lisdat at open circuit potential. However, these differences need not represent the only reason for the very large difference between the EIS published by these authors (Figure 19). It is not excluded that the DNA binding modes and different properties of the TBP and NF-κB proteins contributed to the different results of their EIS measurements. The NF-κB p50 transcription factor recognizes specific DNA sequence through the major groove binding. In addition, a number of phosphate contacts stabilize the complex and embrace the DNA molecule inside the protein wrap. Tersch and Lisdat considered neutralization of this phosphate wrapping being behind the signal decrease.485 In contrast, the TBP binds DNA in an unusual way through the minor groove using a β-sheet motif (Figure 16B). The minor groove binding induces bending of the DNA by 80° and facilitates base pair melting. The increased impedance signal might be thus attributed to the exposition of bases. Such a conclusion would imply additional benefit of the EIS analysis, that is, the ability to reflect the protein–DNA binding mode. More work will be however necessary, including additional control experiments, to arrive to a more definite conclusion and to show what are the advantages and drawbacks of application of EIS to DNA–protein interaction studies.
Figure 19.

(A) Impedance spectra of a gold electrode with NF-κB-specific dsDNA before (a) and after (b) incubation with 33 μg/mL NF-κB p50, as measured by Tersch and Lisdat.485 (B) Nyquist plots of gold electrode modified with DNA duplexes before and after interaction with BSA and TBP, as reported by Chang and Li.484 Electrolyte: 10 mM PBS (pH 7.0) with 10 mM [Fe(CN)6]3–/4– and 10 mM NaCl. Frequency: from 0.1 Hz to 100 k Hz. Amplitude: 10 mV. Bias potential: 0.20 V. Adapted with permission from refs (485) and (484). Copyright 2011 and 2009 Elsevier.
7.4. E-DNA Sensors
E-DNA sensor was developed about 10 years ago in the laboratory of A. Heeger487 (reviewed in refs (25) and (34)) as a tool for the DNA nucleotide sequence analysis. Later, it was shown that this sensor could be used also as an aptamer-based, sensor detecting low molecular weight compounds and some proteins.488 Recently, it was shown that principles of these sensors could be used also in studies of naturally occurring DNA-binding proteins. The sensor is composed of a ds or single-stranded (ss) DNA probe, which is redox-labeled and covalently attached to an electrode. Upon protein binding, the signal produced by the redox label was attenuated, which was explained by reduction of the collision efficiency between the DNA probe label and the electrode by the bulky structure of the DNA-bound protein. Binding of TBP and MhaI methyltransferase to their specific recognition sequences in dsDNA, as well as bindings of SSB protein from E. coli and eukaryotic replication protein A to ssDNAs, were studied.496 The relative decrease of the label signal was rather small with short DNA probes (e.g., 20 bases) and increased significantly with the probe length (up to 70 bases). However, the absolute value of the label signal decreased with increasing probe length. It is generally accepted that DNA probe density at the electrode surface is important for the probe accessibility in different DNA interactions,34,489−494 and it may be particularly critical in DNA binding to bulky protein molecules.495,496 Accessibility of surface-attached DNA probes for protein interactions was tested using DNase I. As a result of the DNA enzymatic cleavage, the label was removed from the DNA probe and the signal decreased. It was shown that the accessibility of the DNA probes was (as expected) improved at lower probe densities.495 Yet even at the lowest densities investigated, a significant fraction of dsDNA probes remained inaccessible to the enzymatic cleavage. This might be due to formation of high-density clusters of dsDNA (see section 7.2). By linking an antigenic peptide epitope to the DNA probe, the E-DNA sensor supported detection of antibodies.497−499 In this way, proteins which do not bind DNA, can be detected by E-DNA sensor. This principle was recently applied for detection of protein IP-10, a biomarker (Interferon-γ inducible Protein-10 kDa or CXCL 10) for the diagnosis of inflammation.500 To detect this protein, methylene blue-labeled ssDNA was attached to Au electrode via a thiol group. This DNA was hybridized with peptide nucleic acid to which a 21-aa peptide binding element was grafted to recognize and bind IP-10 protein. The binding event was indicated by a decrease of a signal from the DNA label. The sensor worked in undiluted blood serum and did not require repetitive washing, showing properties that might be utilized in point-of-care diagnostics. The probability that, in E-sensor, redox impurities from the biological materials can give false positive signal is rather low. To produce such a signal, the impurity should remain firmly bound to the electrode surface resisting washing, and its response should appear at the same potentials as that of the DNA label.
7.5. CPS Peak H in Sensing of DNA–p53 Interaction
In the above methods, either changes in signals of the DNA label were measured or DNA itself served for the charge transfer through its duplex structure. Here, we will show that changes in CPS peak H can also indicate DNA–protein binding and provide information about the stability of the DNA–protein complex.
The tumor suppressor protein p53 binds DNA by several modes,501,502 and the sequence-specific binding of the p53 core domain (p53CD) to the dsDNA consensus (DNACON) site (involved in apoptosis and cell cycle arrest) was most intensively studied.503,504 This sequence contains two copies of the half-site 10-base pair motif RRRC(A/T)|(T/A)GYYY separated by up to 13 bp (vertical bar indicates the center of symmetry within the half site). The crystal structure showed binding of four p53 molecules to the DNACON sequence.503,504 Such binding caused bending and twisting of the DNA.505 It was found that wt and mutant p53CD produced CPS peak H at DTT-modified mercury electrodes, sensitively reflecting changes in the protein structure (section 6.2).105 The CPS peak H was utilized for investigation of the p53CD–DNA sequence specific binding,240 using the same arrangement as in free p53CD studies105 (section 6.2). The p53–DNA complex was prepared in the test tube using a usual DNA binding buffer459 and adsorbed at HMDE or SAE followed by the electrode washing and CPS recording with Istr −35 μA.240 The sequence-specific binding resulted in a disappearance or decrease of the p53CD peak H (Figure 20A). In contrast, the peak H was almost not influenced by p53CD interaction with a nonspecific DNA, not containing the consensus sequence (DNANON). Similarly, the complex of mutant protein R273H406 (not binding to DNA) displayed almost the same peak H as the free protein (Figure 20B). Removal of zinc from the wt p53CD (known to inhibit the DNA binding)105,406 resulted in the same effect.
Figure 20.
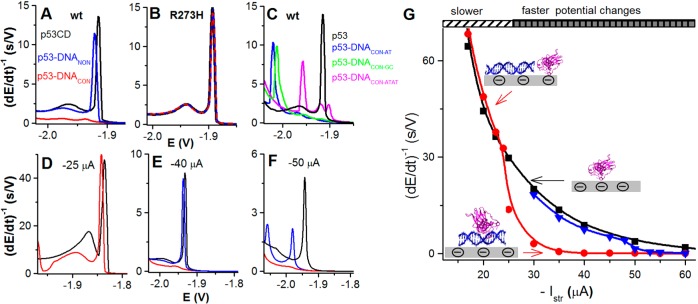
(A) Sequence-specific binding of wild-type p53 core domain (p53CD) to dsDNACON as detected by CPS at a DTT-modified hanging mercury drop electrode (DTT-HMDE) using Istr −35 μA at 21 °C. Free p53CD (black), sequence-specific p53CD–DNACON complex (red), and a mixture of p53CD with 40-mer dsDNA not containing the consensus sequence (blue, p53CD + dsDNANON). (B) Interaction of mutant p53CD R273H with dsDNACON (showing no DNA binding). Free p53CD R273H (black), p53CD R273H + dsDNACON (red), p53CD R273H + dsDNANON (blue). (C) Peak H of p53CD (black) and p53CD complexes with spacer-containing DNAs: DNACON–GC (green), DNACON–AT (blue), and DNACON–ATAT (magenta); Istr −35 μA at 21 °C. (D–F) Peak H of p53CD (black), p53CD–DNACON complex (red), and p53CD–DNANON (blue) at Istr of (D) −25 μA, (E) −40 μA, and (F) −50 μA. (G) Dependence of peak H1 height of free p53CD (black), p53CD–DNACON complex (red), and p53CD–DNANON (blue) on stripping current (−Istr). Adapted with permission from ref (240). Copyright 2014 Elsevier.
The decrease in the peak H height, resulting from the p53 binding to DNA, was related to changes in the accessibility of electrocatalytically active aa residues (particularly Lys, Arg, His, and Cys) at the electrode surface.155,170,264 Such changes may cause more difficult electrocatalysis, which can result in shifting of the peak H to more negative potentials and eventually can lead to almost complete disappearance of this peak.
High sensitivity of peak H for the p53CD sequence-specific DNA binding was observed only at a sufficiently negative Istr intensity (Figure 20A,E,F). For example, Istr–35 μA was sufficient for discrimination between a sequence-specific and a much weaker nonspecific p53CD binding (Figure 20A), while at Istr–25 μA, such discrimination was not possible (Figure 20D). Under these conditions, even the stable sequence-specific complex was disintegrated at the electrode surface, producing almost the same CPS response as free p53 protein. Nonspecific binding of p53 to DNANON resulted in a similar response even at a much higher Istr intensities (−40 μA; Figure 20E), but further increase of the –Istr intensities displayed a significant difference between the CPS responses of free p53 and p53–DNANON (Figure 20F), suggesting that even the weakly bound nonspecific complex was not completely disintegrated at the electrode surface, because of very short exposure to negative potentials. Earlier, it was shown that the DNA sequence and structure could modulate the affinity of p53 for its binding sites449,506 and that DNA sequences involved in a cell cycle arrest do not have spacers interspersed between the two half-sites and bind usually p53 with a higher affinity507,508 than DNA sequences involved in apoptosis and containing interspersed spacers between the two half-sites. Figure 20C displays CPS responses of free p53CD and compares them to the three different p53 sequence-specific complexes with spacer-containing DNAs. These data suggest that the DNA containing the longest ATAT spacer produced the CPS response closest to that of free p53 protein.
It appears that by controlling the temperature and/or Istr intensities in CPS, it will be possible to make a conclusion about the stabilities of different surface-attached p53CD–DNA complexes on the ground of their susceptibility to the electric field effects acting at the interface. Recently, DNACON bearing acrylamide or vinylsulfonamide groups covalently cross-linked with Cys residue of p53CD was prepared.509 A comparison of EC and other properties of p53CD–DNA complexes with covalently bound versus noncovalently bound DNACON appears of utmost interest.
7.6. p53–DNA Sequence-Specific Binding As Detected by Signals of Labeled DNA
The double-surface technique (DST) mentioned in section 7.1 was used for the detection of p53 protein sequence-specific binding to DNA. To increase the sensitivity of the detection, labeled DNAs were used.510−513 Tail-labeling of DNA with many nitrophenyl tags510 resulted in a significant increase in sensitivity as compared to previous label-free DNA analysis.460,514 However, this approach required an organic chemistry laboratory with highly qualified personnel. Recently, a much simpler DNA labeling technique, based on covalent binding of Os(VIII)bipy complex to thymine residues in a 20-mer tail, was used.460 Using the electrocatalytic adsorptive transfer stripping DPV, nanogram and subnanogram amounts of the DNA–Os(VIII)bipy adduct were determined. In competition experiments, relative affinities of different DNACON to the immobilized p53 protein were tested. DNA labeling consisted of mixing DNA with Os(VIII)bipy and dialysis after 2 h of reaction time at room temperature. DST with immobilized protein or DNA449,460 can be used practically to investigate any DNA (or RNA)–protein interactions. In its present form, this technique is laborious and not easy to use for highly parallel analysis. The combination of DST with microfluidic technique would help to overcome these problems.
7.7. Concluding Remarks
In the recent decade, electrochemistry significantly extended the field of DNA–protein interactions. Not only were new EC methods for DNA–protein binding detection developed,112,464,466−469,484,485 but also new approaches were offered including interesting views of DNA-mediated search by repair proteins464 and testing of DNA–protein complex stability at electrified interfaces.240 It can be expected that at least some of the EC methods and approaches will be further developed and new ones will appear. Great potentiality of EC analysis as a new tool for DNA–protein interaction studies will be utilized, and this analysis will become a useful technique among a number of well-established traditional methods. Investigation of multiprotein–DNA complexes playing important roles in transcription mechanisms515 and chromatin structure dynamics516 may represent a challenge for bioelectrochemists in the coming decades.
8. Electrochemical Analysis of Glycoproteins
8.1. Glycomics
Analysis of precisely controlled post-translational modifications of proteins,517 often with addition of only a small functionality (e.g., single phosphorylation), which can change activity of a kinase up to 108 times,518 is a big challenge for analytical chemistry at present. Enzymatic addition of a glycan (i.e., oligosaccharide chain covalently bound to proteins or lipids) in a process of glycosylation is a highly abundant form of co- and post-translational modification of proteins. Approximately 70% of cytosolic proteins and about 80% of membrane-bound human proteins are estimated to be glycosylated.519 Glycans take active part in many physiological processes,37,520−525 but they are also involved in pathological processes as well, triggered by adhesion of different pathogens to host tissues, in neurological disorders and in cancer.526−534 Better understanding of glycan involvement in pathological processes may be thus essential to cure diseases.535−542 Glycan changes can be used in early stage diagnostics of numerous diseases, including various types of cancer with already approved biomarkers.534,543−549 Moreover, many approaches previously applied for treatment of diseases are currently being revisited, taking into account glycan biorecognition to achieve high efficiency, low side effects, high serum half-life, or low cellular toxicity of drugs/therapeutics.526,550−553 The first antibody with controlled glycan composition was already launched to the pharmaceutical market, an achievement glorified by the authors as “a triumph for glyco-engineering”.554
Glycomics is aimed to reveal finely tuned biorecognition of glycans in the cells based on graded affinity, avidity, and multivalency of glycans.555,556 Glycans are preferential molecules for coding/storing biological information, because the number of possible unique sequences as compared to nucleic acids or proteins is staggering (Figure 21).557,558 The theoretical number of glycan hexamers is 8 orders larger in comparison to peptide hexamers and 11 orders of magnitude larger than nucleic acid sequences.559 The eukaryotic cellular glycome can contain up to 500 000 glycan containing biomolecules built from a pool of more than 5000 unique glycans.556,560 This glycan variation might be behind complexity of human phenotype, even though there is a relatively small human genome. Complexicity of glycans together with their similar physicochemical properties are the main reasons why progress in glycomics has lagged behind advances in analysis of DNA and proteins.561 Glycans can be formed from monosaccharide units with a different bond between them (branching). For example, sialic acids can be attached to galactose in three different ways (2–3, 2–6, or 2–8 bond).
Figure 21.

Graphical representation of complexity of glycans, showing variability of sugar building blocks, multiple branching, and attachment points. Adapted with permission from ref (37). Copyright 2013 Springer Science and Business Media.
The most important current analytical method for structural glycoprofiling of proteins is MS relying on different ionization techniques, mass analyzers, and detection platforms.12,562−565 The combination of MS with separation techniques including a diverse range of chromatographic approaches (reversed-phase, hydrophilic, graphitized carbon-based, and lectin affinity modes), gel electrophoresis and capillary electrophoresis, and integration of various glycan modifications/labeling steps has been successfully applied in the identification of glycan isomers, glycosylation sites, and in revealing glycan microheterogeneity.566,567 Moreover, such instrumental machinery can elucidate not only the structure of a glycan, but it can also characterize the corresponding protein. Instrumental techniques have been successfully applied in better understanding of progression of various forms of cancer or for the identification of new prospective glycan-containing biomarkers.566,568−571
Lectins are a heterogeneous group of proteins recognizing free or bound mono- and oligosaccharides or whole cells (Table 1).572,573 Lectins are not catalytically active nor involved in the immune system of higher organisms and have been found in viruses, bacteria, fungi, plants, and animals exhibiting similar sequence motifs. The lectin binding site is very often a shallow groove or a pocket displayed at the protein surface.519 Mostly four main aa residues are part of a biorecognition site in lectins such as asparagine, Asp, glycine (Arg in Con A), and an aromatic residue responsible for interaction with glycan via hydrogen and hydrophobic bonds.574 Ionic interactions are involved in the biorecognition of negatively charged glycans containing sialic acids. Lectins as natural glycocode readers559 are popularly applied in a wide range of traditional575−577 and advanced sensing protocols.37,38,40,41,527,529,578 Lectins are traditionally isolated from natural sources, but nowadays a recombinant DNA technology helps to produce lectins with high purity.527,578
Table 1. Specificity, Source, and Other Properties of the Most Commonly Used Lectinsa.
| lectin | specificity | source | source type | Mw (kDa) | pI | SUc | glycoprotein | metal ions |
|---|---|---|---|---|---|---|---|---|
| AAL | α-l-Fuc | Aleuria aurantia | F | 72 | 9 | 2 | ||
| Con A | α-d-Man, α-d-Glc | Canavalia ensiformis | P | 104 | 6.3–7 | 4 | Ca2+, Mn2+ | |
| MAA-I | Gal-β-(1–4)- GlcNAc | Maackia amurensis | P | 130 | 4.7 | 2 | yes | |
| MAA-II | Neu5Ac-α-(2–3)β-Gal | Maackia amurensis | P | 130 | 4.7 | 2 | yes | |
| PHA-L | branched β-(1–6)GlcNAc | Phaseolus vulgaris | P | 126 | 4.2–4.8 | 4 | yes | Ca2+, Mn2+ |
| PHA-E | oligo Man | Phaseolus vulgaris | P | 126 | 6–8 | 4 | yes | Ca2+, Mn2+ |
| SNA-Ib | Neu5Ac-α-(2–6)Gal | Sambucus nigra | P | 140 | 5.4–5.8 | 4 | yes | |
| SNA-II* | Gal, GalNAc | Sambucus nigra | P | |||||
| RCA-I/RCA120 | β-d-Gal | Ricinus communis | P | 120 | 7.8 | 2 | yes | |
| RCA-II/RCA60 | Gal-β-(1–4)- GalNAc | Ricinus communis | P | 60 | 7.1 | 1 | yes | |
| UEA-I | α-l-Fuc | Ulex europaeus | P | 63 | 4.5–5.1 | 2 | yes | Ca2+, Mn2+, Zn2+ |
| WGA | β-d-GlcNAc | Triticum vulgaris | P | 36 | >9 | 2 | no | Ca2+ |
Table based on ref (888).
SNA (EBL-elderberry bark lectin) is often available only as SNA without any specification, despite the different binding abilities of the two isolectins.
F, fungi; P, plant; SU, subunits; Gal, galactose; GalNAc, N-acetylgalactosamine; Glc, glucose; GlcNAc, N-acetylglucosamine; Fuc, fucose; Man, mannose; Neu5Ac, N-acetylneuraminic acid.
Lectins as natural glycan-recognizing proteins559,579,580 can be very helpful to fractionate or preconcentrate glycoproteins from other nonglycosylated proteins,566 which is essential for robust glycan measurements. Lectins of course can be applied in glycoprofiling in a direct way, that is, to be integrated into a microarray format of analysis with a fluorescent reading.527 An emerging lectin microarray-based technology offers highly parallel analysis with a minute sample consumption desperately needed for cost-effective monitoring of glycan changes.581,582 Therefore, direct analysis of intact glycoproteins/glycolipids and glycans on the surface of cells is possible.530,583−585 Moreover, this technology is routinely used in the analysis of glycan changes as a result of numerous diseases or for the discovery of new biomarkers. This technology can be thus complementary to the approach based on MS-based glycan analysis.529,584 Furthermore, the glycomic data set obtained from lectin microarrays together with microRNA data confirmed the involvement of microRNA in regulation of expression of glycan processing enzymes.586 Lectin microarrays are suitable for obtaining preliminary information about changed glycosylation rather than providing detailed information about the exact glycan structure/composition that can be acquired only by sophisticated instrumental analysis. Another drawback of lectin microarrays with fluorescent detection is a requirement to have lectins or a sample fluorescently labeled, which is an additional step resulting in analysis variability due to an uneven labeling efficiency of the molecules.527,529 Moreover, nonviable cells were found to exhibit stronger fluorescence as compared to viable ones, when assayed on lectin microarrays and fluorescent labeling of cells affected a biorecognition process on microarrays.527,529 Besides a requirement to use labels with lectin microarrays, this technique can not detect a low level of glycoproteins with a typical LOD in the sub nanomolar or low nanomolar level and offers only a narrow working concentration window.
Thus, alternative and more sensitive routes for glycoprofiling with application of lectins in combination with various transducing schemes are being sought.528,576,577 Application of nanotechnology, advanced surface modification protocols, and novel transducing schemes will help to overcome limitations of lectin microarrays. In particular, utilization of nanomaterials has a beneficial effect on sensitivity, detection limit, working concentration window of the device, and, in certain cases, also label-free mode of operation, and a real-time monitoring of a biorecognition process may be feasible.38−41,520,587−590 Advanced and highly sensitive detection schemes are therefore needed to detect very low concentrations of glycoproteins without a requirement to release glycans from the protein prior to analysis. Moreover, label-free methods for glycoprofiling are of particular interest, because the binding event is not compromised by the label. All of these requirements can be addressed by utilization of EC detection platforms offering low LOD, simple miniaturization, and integration into numerous multiplexed formats of analysis. In this section, a short historical overview of analysis of glycoproteins by various EC techniques, fulfilling “must-have attributes” of glycoprofiling, is provided together with application of glycoprofiling as a tool for diagnosis of various diseases.
8.2. EC Analysis of Glycoproteins
EC detection of glycoproteins either in the isolated state or present directly on the surface of various types of cells is the field developing at a tremendous pace with a focus to address limitations of the well-established technique of lectin microarrays for glycoprofiling.37,527,529 Section 8 provides a summary of glycan detection by EC lectin biosensors in various perspective label-free formats of analysis (EIS and capacitance-based detection) together with numerous reports relying on application of various labels. Moreover, approaches in which “artificial lectins” based on boronate derivatives were used are provided as well. A novel method of carbohydrate covalent modification by Os complexes and application of catalytic hydrogen evolution reactions in carbohydrate analysis are discussed. Additionally, analysis of a wide range of glycoprotein biomarkers by EC methods is extensively covered (section 9.5). This section provides the first comprehensive summary of the diverse range of possibilities that the EC detection platform can offer in glycan/carbohydrate analysis. The subject was previously partially covered with emphasis given on pulsed amperometric detection of carbohydrates released from glycoproteins.591 Short reviews were published with a focus on EIS-based glycan biosensing37−39 or pioneering work (until the end of the year 2010) in EC glycobiosensing.41 The most recent review only partly covered the subject by referring to only four relevant papers.42
8.3. Early Studies of Glycoproteins
In this section, only studies based on a (bio)recognition of a carbohydrate moiety on the glycoprotein surface by lectins and boronate derivatives integrated with EC detection will be discussed. A historical perspective gives an overview about the application of EC detection in the field of glycomics from the early 1980s. Analysis of mono-/oligosaccharides and detection of glycopeptides with some pretreatment steps are provided. Moreover, analysis of intact glycoproteins and determination of glycoproteins directly on the surface of various cells are described.
8.3.1. Analysis of Mono-/Oligosaccharides Released from Glycoproteins
Glycoproteins were detected in the past by a combination of various techniques. The first step was a cleavage of a glycan moiety from a protein or a peptide backbone either enzymatically (Figure 22) or chemically.592 The second step was separation of carbohydrates released from the glycan by chromatographic, electrophoretic, and other techniques.592 The final step was detection of separated carbohydrates by fluorescent or EC means.593 The main advantage of using EC detection of saccharides was a direct detection without any labeling needed, and thus a simpler, faster, and more efficient carbohydrate analysis was possible as compared to fluorescent/optical reading.49 In addition, EC analysis is among the most cost-effective and flexible analytical tools with instrumentation, which can be miniaturized and integrated with various separation techniques (i.e., capillary zone electrophoresis).49,593 There are intrinsic limitations of this approach including possible epimerization and degradation of carbohydrates at elevated pH needed for EC analysis and a need to know molar response for each carbohydrate to be assayed. Moreover, the method requires quite pure carbohydrates free from aa residues, peptides, and organic acids, to avoid interference with EC detection.594 Thus, the EC response to a particular carbohydrate had to be known in advance by quantification of carbohydrate standards, complicating acquisition of reliable data.594
Figure 22.
Enzymatic release of whole glycans or carbohydrates from glycoproteins. Abbreviation of enzymes: endo-β-GlcNAc-ase, endo-β-N-acetylglucosaminidase (endo H); PNGase F, peptide:N-glycosidase F (PNGase F); α-Man-ase, α-mannosidase; β-Man-ase, β-mannosidase; NANA-ase, N-acetylneuraminic acid hydrolase (sialidase); β-Gal-ase, β-galactosidase; and β-HexNac-ase, β-N-acetylhexosaaminidase.
EC analysis of carbohydrates in a reliable way was challenging for some time due to unwanted adsorption of products of carbohydrate oxidation on the electrode surface. More specifically, carbohydrates undergo electrocatalytic oxidation triggered by surface gold oxides generated electrochemically. In the O-transfer reaction, multiple electrons are exchanged between the electrode and carbohydrate with production of formate and smaller aldaric acids. Radical intermediates, formed through interfacial electrode reaction with carbohydrates, can rapidly foul the electrode.49 This problem was effectively solved in 1981, when Hughes and Johnson first introduced a pulsed amperometric detection (PAD) of carbohydrates at Pt electrodes using a triple-pulse potential waveform allowing detection of carbohydrates with frequency of at least 1 Hz.595
The technique further developed by Johnson’s group was soon applied in carbohydrate analysis using a commercially available instrumentation.596 The combination of a PAD detector with high-performance liquid chromatography offered a LOD for mono- and disaccharides at picomole levels, and its integration with capillary zone electrophoresis further reduced LOD down to femtomole range49,597 with a sample volume of 5–100 μL.591,595,598 Implementation of EC detection with capillary zone electrophoresis, however, required decoupling of the capillary zone electrophoresis and EC electronics to avoid electronic interference.599 Currently, EC analysis of carbohydrates is performed on electrodes made from transition metals (Cu, Ni, Co, and Ru) at a constant potential, lowering a risk for surface poisoning by carbohydrate oxidation byproducts.49
To better understand the structure of a glycan present on the surface of a glycoprotein, it was very important to identify glycosylation sites on the protein surface, to elucidate primary structure of the glycan, to determine the position of glycoside bonds, etc.49 This is why carbohydrate analysis by PAD had to be complemented with other instrumentation including nuclear magnetic resonance and an array of various techniques of MS and chromatography.12,565,592,600 A quadruple-potential waveform was introduced improving a long-term reproducibility of glycan analysis,601 when PAD was coupled with chromatographic separation.591 Despite continual effort in using PAD with separation techniques, the current instrumentation of choice is MS combined with liquid chromatography based on fluorescent detection or capillary electrophoresis-based laser-induced fluorescent detection (section 8.1).570
Biochemical characterization of glycans by enzymatic hydrolysis played a key role in revealing carbohydrate structure. For example, carbohydrates can be cleaved from the glycan structure one by one, that is, through the removal of terminal sialic acid by sialidase602 followed by removal of newly exposed galactose by β-galactosidase,603 and so on598 (Figure 22). The other way is to release an intact glycan from the protein en-block using glycopeptidases, that is, endo-β-N-acetylglucosaminidase (endo H) or peptide:N-glycosidase F (PNGase F).594,604,605 Both of these enzymes have the ability to provide information about the glycan structure, in which endo H hydrolyzes the linkage between the two core N-acetylglucosamine moieties in a high-mannose or hybrid glycan, while PNGase F cleaves the β-aspartylglucosaminyl bond of all known types of N-linked oligosaccharides (see Figure 22). Moreover, deglycosylation with PNGase F converts Asn residue to Asp, leading to a change in the mass detectable by MS.605 Usefulness of enzymes in structural characterization of released complex carbohydrates from the protein/peptide was proved by application of an enzyme array in digestion of a glycan with released monosaccharides detected by PAD.598,604 This approach has some limitations, such as a limited number of enzymes requiring high purity, problem distinguishing between similar mono-/oligosaccharides, and a need to work with pure carbohydrates rather than with complex mixtures.599,606 The O-linked carbohydrates are enzymatically released from the protein using Pronase digestion, that is, by a mixture of several proteases leaving glycan attached only to a single serine or threonine residue. It is worth mentioning that application of enzymes in glycan cleavage can be expensive, when extensive characterization of glycans has to be performed,570 and thus cost-effective methods are continuously evolving.12,566,570
8.3.2. Detection of Glycopeptides Cleaved from Glycoproteins
The first study that focused on the analysis of glycopeptides by PAD was published in 1988 by Hardy and Townsend, when a glycoprotein fetuin was cleaved into glycan-containing peptides.607 Quantitative analysis of glycopeptides by PAD was successfully validated by nuclear magnetic resonance.607 In the next study, PAD analysis of intact carbohydrates linked to asparagine was performed, and further information about a carbohydrate composition was revealed using enzymes.608 An alternative to PAD analysis of carbohydrates attached to biomolecules is a sinusoidal voltammetry,609,610 which can be compared to oscillographic polarography.611 EC analysis of carbohydrate still attached to a short peptide was successfully applied for glycoprofiling of pure glycoprotein (a coagulation factor VII) produced by a recombinant technology.612
8.3.3. Analysis of Intact Glycoproteins
Lectins (see section 8.4.1) are well suited to get preliminary information about spatial organization of a glycan moiety on the glycoproteins without glycan liberation.573,613,614 Thus, it is logical and practical to rely on their function to recognize glycans on glycoproteins in their natural, intact form. For example, 26 different lectins recognizing N-acetylgalactosamine were used in glycoprofiling of samples from patients suffering from breast cancer and showed subtle differences in the structure of complex glycans.600 Although antibodies raised against glycan moieties can detect glycans on glycoproteins with higher affinity and specificity as compared to lectins,529,535,537 their application in direct glycoprofiling of proteins is questionable due to their limited commercial availability.600 However, when available, they can be effective in early cancer detection.599,615,616 Most importantly, carbohydrate arrays can assist in finding novel glycan specificities of already utilized antibodies and lectins.599,617−621 Because lectins are almost exclusively applied in direct glycoprofiling of glycoproteins, details about their integration into EC formats of detection will be discussed in the forthcoming sections.
8.3.4. Glycoprofiling of Intact Cells
The first application of glycoprofiling of intact cells was published in 2001 with two completely different approaches. In the first one, Luong’s group applied EIS to study the attachment and growth of insect Spodoptera frugiperda Sf9 cells on eight modified gold electrodes present at the bottom of tissue culture wells.622 Because the cells were cultivated in the wells for up to 20 h, a change in impedance was not attributed only to the cell density on the electrode, but to the products of a metabolic activity as well. The experimental setup allowed one to monitor the growth inhibitory effect of key explosives with inhibitory constants calculated.622
In the second approach, a very interesting strategy was introduced,623 which involved a selective binding of six types of microbial cells to surfaces modified by 10 different lectins via glycans with subsequent EC monitoring of their respiratory activity by chronocoulometry. In the first step, the lectins determine the amount and cell types attached to the membrane. In the second step, the amount of bound viable cells was chronocoulometrically detected in the presence of two redox probes, menadione (reaching intracellular redox enzymes) and ferricyanide (a shuttle between menadione and the electrode). During the measurement, formate and succinate, substrates of enzymes involved in a respiratory chain, were present (Figure 23). This EC investigation is universal and can be applied to a diverse range of cells. Principal component analysis of the results showed that it is possible to distinguish between all microbial species tested in the study on an array of electrodes.623
Figure 23.
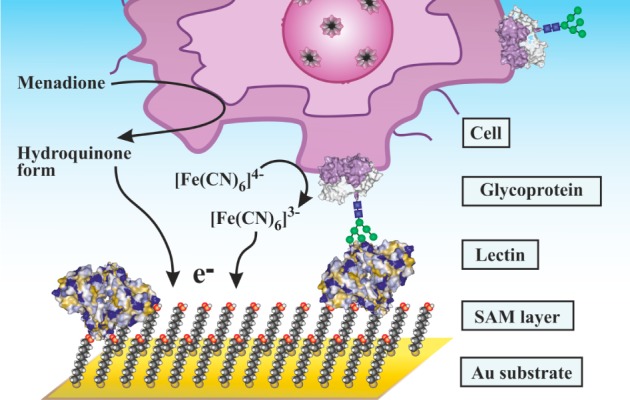
Schematic representation of EC detection of cells with lectins by monitoring of respiratory activity of the cells with a two-mediator system.
Later, the authors extended this approach to distinguish between four different E. coli subspecies on an array of 32 screen-printed carbon electrodes.624 Again, a principal component analysis helped to distinguish between all four E. coli subspecies based on differences in a respiratory activity after being bound to lectins.624 Moreover, mainly label-based approaches on how to detect intact cells of a different origin and status are discussed in section 9.5.5.
8.4. Biorecognition Molecules
8.4.1. Lectins – Useful Components for Glycoprofiling of Proteins
Lectins are proteins binding to free or bound mono- and oligosaccharides or whole cells. A binding site of lectin is a shallow groove or a pocket displayed at the protein surface with involvement of four main aa residues such as asparagine, Asp, glycine (Arg in Con A), and an aromatic residue responsible for interaction with glycan via hydrogen and hydrophobic bond, while ionic interactions are involved in biorecognition of negatively charged glycans containing sialic acids (see section 8.1). Thus, a binding preference of commonly used lectins is known quite well.37 However, unknown lectin binding specificity was revealed, when some glycan immobilization was controlled at a nanoscale.625−629 EC characterization of glycan surfaces can provide information about the quality of one- or two-component SAM deposited on gold surfaces, when probed by CV or EIS.630,631 An interesting approach on how to probe the specificity of lectins on carbohydrate modified interfaces was described recently. The glycan receptive interface was developed from a compound containing a thiol group on one end, glucose or galactose on the other end, and a middle part of the molecule containing a quinone redox moiety. Such a molecule after incubation on a gold surface formed a gold–thiol bond with glucose or galactose exposed to the solution. Quinone moiety of the molecule served for signal generation via CV or DPV. Upon incubation of the electrode with lectins, a decrease of EC signal was observed, enabling detection of two lectins, Con A and peanut agglutinin, down to micromolar concentration.632 A nanoscale-controlled interface formed on gold nanoparticle-modified electrodes by sequential deposition of thiolated saccharides and various blocking thiols (cysteamine, 11-mercaptoundecanoic acid, 6-mercaptohexanol, and 3-mercapto-1-propanesulfonate) was applied for investigation of Con A binding. The study showed that a blocking/diluting thiol had a detrimental effect on Con A binding to the surface-immobilized saccharide with the best blocking/diluting thiol (3-mercapto-1-propanesulfonate) allowing detection of Con A down to nanomolar level.633 Binding specificity of six different lectins on glucose- and galactose-modified graphene interfaces revealed that lectins can be detected down to low nanomolar level on “electrified” anthraquinonyl glycoside surfaces (Figure 24).634 Moreover, such a system was applied for analysis of a hepatoma cell line down to 1 × 104 cells/mL.634 When discussing graphene as a prospective transducing surface in glycan measurements, it is important to take into account that such material was proved to induce a strong interaction with Con A lectin, leading to lectin denaturation.635 Thus, a linker layer was suggested to be inserted between graphene and lectin ensuring lectin activity after binding.635 Applications of lectins in EC assays will be discussed in the forthcoming sections.
Figure 24.

Probing the specific sugar–lectin interactions on surface modified by (a,c) glucose and (b,d) galactose. The interaction between lectin and immobilized saccharide was detected by either (a,b) DPV or (c,d) EIS. Bare screen printed electrode was modified by graphene oxide (GO), then by antraquinone containing glucose (GO-GA1) or galactose (GO-GA2), and interaction between saccharide-containing surfaces was probed by two lectins: Con A, recognizing glucose; and peanut agglutinin, recognizing galactose. All experiments were performed in Tris-HCl (pH 7.0). Adapted with permission from ref (634). Copyright 2013 Macmillan Publishers Ltd.: Scientific Reports.
8.4.2. Lectin Engineering
A novel trend is to apply recombinant DNA technology for producing lectins with improved characteristics for construction of various lectin-based biodevices. Production of lectins by recombinant technology has distinct advantages as compared to lectin isolation and purification from natural sources such as time- and cost-effectiveness, high yields, low batch-to-batch variation, and high purity.527,636 Moreover, recombinant expression of lectins in prokaryotic hosts produces lectins without glycosylation. The presence of glycans on lectins can complicate glycoprofiling, when a sandwich configuration (primary lectin/glycoprotein/secondary lectin) is applied in sensing. Recombinant lectins can be produced with various tags applied for simple purification process, but applicable for an oriented lectin attachment on different surfaces, as well.580 Other attractive concepts of how to prepare lectins with advanced binding properties include multimerization of lectins. By incubation of biotinylated lectins with streptavidin, a 4–40-fold increase in sensitivity of analysis637 was observed (Figure 25A). Further, a redox switchable preparation of a lectin dimer from its monomers with an agglutination activity increased 16-fold as compared to a lectin monomer638 (Figure 25B). Introduction of a boronate moiety into lectin with affinity increasing 2–60-fold for its glycan analyte639 (Figure 25C) is another good example of improved lectin forms. Because such recombinant or modified lectins have been applied in bioanalysis relying on a fluorescent reading,640−642 they are not discussed here in detail, but can be combined with EC detection in the future for robust and advanced glycoprofiling.
Figure 25.

Various ways to prepare modified lectins with improved binding properties. (A) Modification of a lectin by a biotin derivative, which upon incubation with a dye-labeled streptavidin forms a lectin multimer. Adapted with permission from ref (637). Copyright 2013 American Chemical Society. (B) A redox switchable formation of a lectin dimer involving a thiolated form of a lectin. Adapted with permission from ref (638). Copyright 2004 John Wiley & Sons. (C) A scheme of increased strength of interaction between BAD (boronic acid-decorated) lectin and a glycan with involvement of lectin binding site and boronate derivative in the biorecognition. Adapted with permission from ref (639). Copyright 2013 American Chemical Society.
8.5. Label-Free EC Detection
The usual EC techniques employed for a label-free analysis of intact glycoproteins include EIS (Figure 26) and capacitance sensing. A transducing mechanism of these techniques is described shortly below. At the end of this section, it will be shown that glucosamine-containing poly- and oligosaccharides catalyze hydrogen evolution at Hg electrodes.
Figure 26.
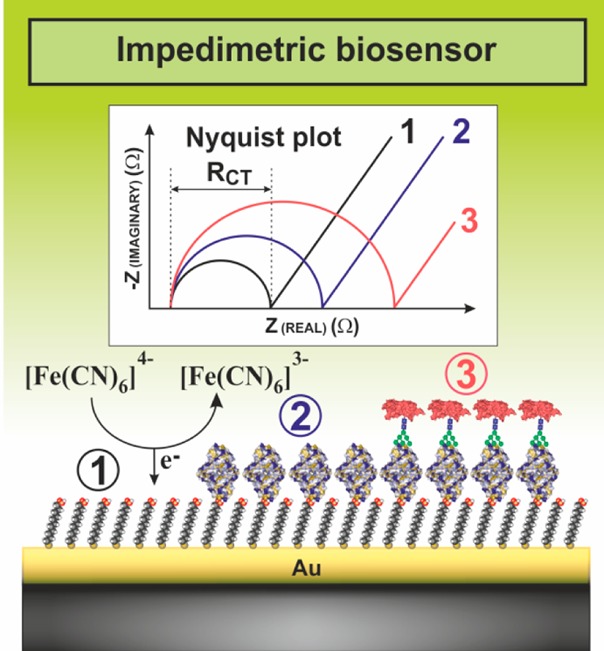
A typical interfacial layer of the EIS-based biosensor after SAM formation (1), immobilization of lectins (2), and biorecognition of a glycoprotein (3) with corresponding Nyquist plots showing shifts in the RCT value with an increased loading of the surface.
8.5.1. Electrochemical Impedance Spectrometry (EIS) of Glycoproteins
EIS as the most frequent label-free technique utilized in glycoprofiling is based on application of a small alternating potential amplitude to the electrode. This technique can provide interfacial characteristics utilizable in (bio)sensing by fitting Nyquist and Bode vector plots.37 The most often charge transfer resistance RCT extracted from a Nyquist plot (a diameter of a semicircle in Figure 26) is applied for biosensing purposes. EIS has been extensively used as an efficient tool for reliable analysis of surface conditions such as biorecognition binding events and desorption.434,483,643
In its simple form, EIS lacks selectivity. To apply EIS for analysis of biomacromolecules, sophisticated selectivity improvements based on biological recognition systems were elaborated. For example, in the case of detection of glycans in glycoproteins, the selectivity of the glycoprotein binding event relies on the specificity of the glycan binding to lectins, which can be immobilized on the electrode surface (see below). Nonspecific binding of other proteins and various compounds to the surface is prevented usually by screening the electrode with binary or ternary thiol layers including thiols with terminal betaine moiety.644 Electrode surfaces after immobilization of the biorecognition element can be blocked to prevent nonspecific interactions by filling the surface with some proteins such as bovine serum albumin, casein, or other molecules. In these arrangements, the specificity of the EIS analysis was greatly increased, allowing analysis of body fluids and cell lysates.
The first paper dealing with analysis of an intact glycoprotein by a label-free EC method based on EIS was published in 2006.645 Authors employed boronic acid and its derivatives, which have a high affinity toward carbohydrates and can be effectively applied for sensing purposes. Thin films of poly(aniline boronic acid) and poly(aminobenzoic acid boronate) electropolymerized on the surface of GCE were applied for biorecognition of glycoproteins. Horseradish peroxidase (HRP) and glucose oxidase were used as model glycoproteins, and the study showed that both proteins were effectively recognized by the boronic acid-containing films, but the sensor exhibited a nonspecific binding in the presence of BSA. The study showed that 5 μM glucose oxidase exhibited a shift in RCT from 5.1 to 6.3 kΩ,645 indicating that such a sensor is not very sensitive and cannot be applied in early detection of glycoprotein-based biomarkers. Application of lectins can significantly increase specificity of glycoprotein EIS-based analysis as compared to analysis based on utilization of boronic-acid functionalized film because lectins can specifically recognize different glycan structures (section 8.1).
Lectins started to be applied in combination with EIS measurement of glycoproteins in 2007.646 Con A lectin covalently attached to a one-component SAM layer of 11-mercaptoundecanoid acid (MUA) was tested for a specific detection of HRP. Interestingly, after covalent binding of Con A, RCT decreased from 15.6 kΩ (MUA SAM on gold) to 10.1 kΩ, and RCT further decreased to a value of 8.0 kΩ upon incubation with a micromolar concentration of HRP.646 This unexpected decrease of RCT after formation of protein layers is most likely a result of screening of a negative charge of pure MUA SAM by both proteins Con A and HRP.
Analysis of sialic acids is important because it is involved in numerous physiological and pathological processes.647 Thus, there is no doubt that lectin biosensors able to detect these carbohydrates can be useful for diagnostic purposes. The first paper dealing with detection of physiologically relevant glycoproteins (fetuin and asialofetuin) by EIS was published by the Joshi’s group.648 The lectin biosensors with either peanut agglutinin (galactose-specific) or Sambucus nigra agglutinin (SNA, sialic acid-specific) immobilized on printed circuit board electrodes were able to detect corresponding asialofetuin (predominantly without terminal sialic acid) or fetuin (with terminal sialic acid) in a short time (∼80 s). Both glycoproteins could be detected down to 150 fM level with a possibility to see microheterogeneity of glycan composition on fetuin by the biosensor relying on determination of changes in the RCT. Moreover, the authors noticed a difference in glycoprofiling of proteins depending on the provider of SNA lectin, suggesting that the lectin from different sources can contain different levels of SNA isoforms and the source of lectins has to be chosen with special care.648 This label-free concept of analysis was recommended for point-of-care ultrasensitive detection of early cancer stages because cancer biomarkers including carcinoembryonic antigen (CEA), carbohydrate antigen 125 (CA125), prostate specific antigen (PSA), and mucin 1 (MUC1) or carbohydrate antigen 15-3 (CA15-3) are glycosylated.648
Oliveira’s group was intensively involved in the application of EIS method for the detection of glycoproteins. In their first study, Con A and a lectin from Cratylia mollis CramoLL were immobilized on gold nanoparticles with polyvinyl butyral, and these complexes were subsequently adsorbed on the surface of gold electrodes with BSA (preventing nonspecific protein adsorption).649 EIS allowed detection of sub micromolar concentration of a glycoprotein ovalbumin with both lectins by monitoring changes in the RCT. The Con A biosensor offered a linear response toward ovalbumin up to 200 μg/mL, while the CramoLL biosensor only offered up to 100 μg/mL. A comparative investigation showed that CV can be applied for analysis of glycoprotein ovalbumin as well.649 CramoLL lectin in combination with EIS was successfully applied in glycoprofiling of various types of microbes, including Escherichia coli, Serratia marcescens, Salmonella enteric, and Klebsiella pneumonia, and CV investigation provided similar results.650 Two lectins, Con A and Ricinus communis agglutinin, integrated with EIS were employed for selective discrimination of prokaryotic strains (Escherichia coli DH5a, Enterobacter cloacae, and Bacillus subtilis) and eukaryotic cells (yeast Saccharomyces cerevisiae and human HeLa cell line) down to 1 × 103 cfu/mL (cfu = colony forming units).651
From EIS not only RCT but also capacitance of the surface can be read, which can be applied for evaluation of a specific binding.652 The surface of the silicon chip with an array of electrodes present in nanowells was applied for biosensing. Two lectins were covalently immobilized on such an interface, and the biosensor performance was tested with standard glycoproteins and a protein isolated from a cultured human pancreatic cancer cell line BXPC-3. The results obtained by EC investigation with both glycoproteins were in good agreement with glycan composition and affinity of lectins for a particular glycan. The biosensor exhibited high reliability of assays and a good agreement with enzyme-linked lectin assays (ELLA, enzyme-linked immuno sorbent assay, an (ELISA)-like method using lectins instead of antibodies). LOD of the biosensor for its analyte was 5 orders of magnitude lower, and the assay time of the biosensor was much shorter as compared to ELLA.652 In a series of recent publications from Tkáč’s group, a systematic effort was devoted to prepare highly sensitive lectin biosensors based on EIS for analysis of glycoproteins containing sialic acid.644,653−655 In the first report, density of a lectin covalently immobilized on a modified gold surface was controlled by adjusting the ratio of a functional thiol in a mixture with a diluting thiol.653 Surface patterning process was monitored by atomic force microscopy, showing differences in surface roughness caused by different molecules being attached to the surface (Figure 27). The lectin biosensor for analysis of sialic acid was able to detect two glycoproteins, fetuin (8.7% sialic acid) and asialofetuin (≤0.5% sialic acid), down to femtomolar concentration. The biosensor response to these two glycoprotein analytes was proportional to the content of sialic acid.653
Figure 27.

AFM images of the gold surfaces during a patterning procedure starting with the bare gold (upper left), the gold surface modified by a mixed SAM (upper right), the surface with covalently attached SNA I lectin (lower left), and the surface after being treated with a blocking agent (lower right). Scale of z-axis was adjusted in a way to clearly see topological features on the surface after each modification step. Adapted with permission from ref (653). Copyright 2013 Springer Science and Business Media.
Recently, a 3D lectin biosensor for analysis of glycoproteins containing sialic acid based on a gold nanoparticle-modified interface was developed.654 After a careful optimization of the 3D interface, it was possible to detect two glycoproteins containing sialic acid down to attomolar concentration, which is still the lowest LOD for analysis of glycoproteins. Again, the lectin biosensor was able to detect the level of sialic acid residues on fetuin and asialofetuin quantitatively, but the level of nonspecific interaction was quite high, reaching 23% of a specific signal.654 Finally, a direct comparison between sensitivity of the 2D biosensor versus 3D biosensor, taking into account the absolute amount of immobilized lectin, showed that a 3D configuration was by 61% more sensitive as compared to the 2D biosensor (Figure 28).655 Glycoproteins can be detected down to the attomolar level even on a 2D interface, when after binding of a glycoprotein to a lectin-modified gold surface, an additional lectin layer in a sandwich configuration is formed.644
Figure 28.

A graphical representation drawn to scale of interfaces applied to build (A) the 3D biosensor based on integrated 20 nm gold nanoparticles or (B) the 2D biosensor (upper image). The 3D biosensor was built on a planar gold surface by chemisorptions of 11-aminoundecanethiol for attachment of 20 nm gold nanoparticles (spheres). (A) On every gold nanoparticle, a mixed SAM composed of 11-mercaptoundecanoic acid (MUA) and 6-mercaptohexanol was formed for covalent immobilization of lectin. (B) The 2D biosensor was formed by incubation of a planar gold with MUA and mercaptohexanol for covalent attachment of a lectin. In the lower part of the figure, comparison of the response of the 2D and the 3D biosensor to its analyte fetuin (Fet) with concentration close to the LOD, represented in a Nyquist plot (left), and calibration graphs for detection of fetuin (Fet) by both biosensors (right) are shown. Adapted with permission from ref (655). Copyright 2014 Enterprise Strategy Group.
Besides all of the above-mentioned examples, when a Faradaic EIS was measured using a redox probe (i.e., a mixture of ferri- and ferrocyanide was applied for a biorecognition), it was also possible to utilize EIS in a non-Faradaic mode of operation without any redox probe present during measurements.656 In such a case, however, it is important to optimize ionic strength of the electrolyte employed for measurements. For example, an increase of a double layer thickness and thus sensing distance from the electrode surface, from ∼0.1 to ∼10 nm, can be achieved by changing the buffer concentration from 100 to 1 mM. This is why diluted phosphate buffer was used for analysis of ovarian cancer SKOV3 cells with a LOD of only 4 captured cells.656
EIS-based analysis of glycoproteins is the only exception, where LOD observed for analysis of glycoproteins is on the same level or better (fM to aM level) as compared to the LOD of protein analysis based on immunoassays.657−659 Even though there is so far only one study, when EIS-based lectin biosensor was applied in the analysis of complex samples such as human serum,644 recently developed DNA- and immuno-sensors working in saliva,660 bovine serum,661 and human serum659,662−669 indicate that sensitive and selective EIS-based lectin glycoprofiling in complex samples will be widespread in future diagnostics.
8.5.2. Capacitance Measurements
A novel capacitive EC measurement of biorecognition was introduced by Berggren in 1997.670 This method samples the current response triggered by a potentiostatic step (typically 50 mV or so in an amplitude) at a frequency of 50 kHz with a possibility to extract a value of capacitance of the layer.671 There is, however, a requirement to work with this step potential measurement in diluted electrolytes (i.e., 10 mM) to avoid fast current decay to get enough data points before current output decays to zero, and this is the main reason for high current sampling.671
This EC technique was successfully applied in the analysis of a glycoprotein, human immunoglobulin G (IgG), on a modified SAM layer with immobilized Con A.672 The lectin biosensor was able to detect its analyte down to 10 nM within 15 min in a flow injection mode, and the biorecognition could be followed in real time. Moreover, the native and aggregated form of the IgG could be distinguished by the biosensor, and aggregated IgG was detected with a concentration as low as 0.01% of the total IgG.672 Thus, the lectin biosensor can be effectively applied in a control of quality during heterologous proteins’ production.
A LOD obtained here for a step-potential capacitance method-based analysis of a glycoprotein is much higher as compared to detection of proteins with antibodies, when LOD in the sub femtomolar level670 or even sub attomolar level on a gold nanoparticle-modified interface673 was observed.
8.5.3. Catalytic Hydrogen Evolution in Polysaccharides
Very recently, a new possibility of EC analysis of polysaccharides (PS) and oligosaccharides (OLS) based on CHER appeared.246,263,674,675 This new method has been developed using free PS and OLS, but it has not been yet tested on glycans cleaved from glycoproteins. Until recently, label-free direct EC reduction or oxidation of PS and longer OLS under conditions close to physiological was not possible. In 2009, it was found that some sulfated PS are able to produce CHER at Hg electrodes.675 Using CPS, carrageen HPS peaks were obtained, similar to CPS peak H produced by proteins (see section 5.2). However, as compared to unfolded proteins, much larger PS concentrations were necessary to obtain peak HPS. Later, it was shown that using adsorptive transfer (ex situ) stripping, it was possible to adsorb sulfated PS directly from seawater and analyze them in buffered solutions.674
Very recent data suggest that the other types of OLS and PS, such as chitosan, have the ability to catalyze hydrogen evolution reaction as well.246,676−678 In the past decades, chitosan as a biodegradable material with many interesting properties (antimicrobial, anti-inflammatory, and anticholesterolemic activities) has attracted great attention, making it potentially useful in biomedicine, but also in other fields of everyday life.679 Chitosan occurs as a major structural component of the cell wall of some fungi, and it can be easily prepared by chemical deacetylation of more abundant chitin.677 Chitosan mostly exists as a random linear copolymer of d-glucosamine and N-acetyl-d-glucosamine units. Commonly, if the number of acetoamide groups is less than 30%, the PS is termed chitosan.676 Chitosan has been used for modification of various surfaces, including electrodes,680 but possibilities of its direct EC detection were rather limited.678−680 It has been shown that chitosan produces voltammetric and chronopotentiometric reduction peaks (in a wide pH range) at mercury and solid amalgam electrodes (Figure 29A–C) and can be determined down to concentrations of 1 μg/mL and below. Chitosan peaks occurring at highly negative potentials (Ep ≈ −1.8 to −1.9 V, Figure 29) are well separated from the background discharge. These peaks were assigned to the CHER and were much larger than those of carrageenans, suggesting that chitosan is a much better catalyst than carrageenans.
Figure 29.
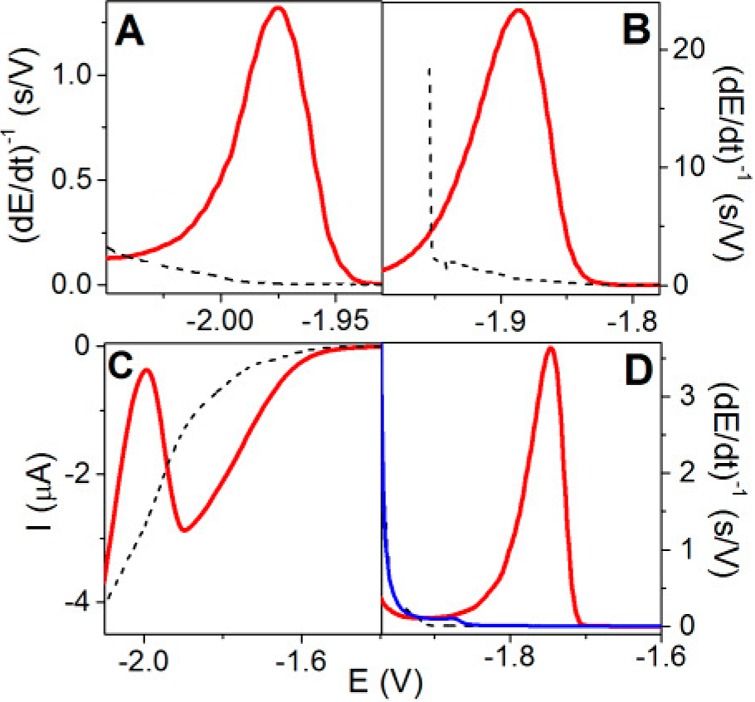
(A,B) CPS and (C) SWV curves of chitosan at mercury electrodes. (A,C) 10 μg/mL of chitosan at HMDE and (B) 15 μg/mL of chitosan at solid amalgam electrode. Accumulation time, tA, 60 s; stripping current intensity, (A) Istr, −70 μA; (B) Istr, −40 μA; (C) frequency 20 Hz; (D) CPS curves of 12 μM chitohexaose (red) and N,N′,N″,N‴,N‴′,N‴″-hexaacetylchitohexaose (blue); tA, 60 s; Istr, −40 μA. Background: 0.1 M sodium acetate, pH 5.2 (dashed). Adapted with permission from ref (246). Copyright 2014 Elsevier.
At a concentration of 10 μg/mL, chitosan produced a high SWV peak in 0.1 M sodium acetate, pH 5.2, while carrageenan yielded no signal under the same conditions. Chitosan oligomers yielded CHER peaks similar to those of chitosan, while acetylated (chitin) oligomers were inactive (Figure 29D). These results suggested that free −NH2 groups in glucosamine residues are responsible for the chitosan CHER. In agreement with this suggestion, chemical deacetylation of the above chitin oligosaccharides resulted in voltammetric and chronopotentiometric peaks similar to those of chitosan. In weakly acid media, chitosan hexamer was detectable at nanomolar concentrations, but it can be expected that more detailed studies will offer LODs at the picomolar level.
In contrast to chitosan, PS-containing acetylated glucosamine residues, such as hyaluronic acid, heparin, and chondroitin sulfate, did not produce any significant reduction signal (not shown) even at much higher concentrations than that of chitosan (Figure 29A). Finding the very strong ability of glucosamine-containing PS and OLS to catalyze hydrogen evolution opens the door for a simple label-free EC analysis of PS and OLS, including glycans of glycoproteins. Such glycans frequently contain N-acetylated glucosamine or galactosamine residues (not producing CHER signals), which may become electroactive as a result of chemical or enzymatic deacetylation (Figure 29D). More work will be, however, necessary to find out how useful will be the CHER signals in glycoprotein analysis.
8.6. Label-Based EC Detection
Label-free EC methods are simple, sensitive, and convenient for glycoprotein measurements. However, covalent and noncovalent labeling of the protein moieties in glycoproteins, as well as specific labeling of glycans either in their free state or directly in the glycoprotein, may have some advantages. Interestingly, DNA analysis started with label-free methods, and later label-based approaches prevailed.34 Different types of labels are available for EC detection of protein moieties in glycoproteins with enhanced sensitivity of detection. Besides traditional amplification agents such as HRP and alkaline phosphatase,681−685 nanomaterials such as metallic nanoparticles,686−688 carbon nanotubes,382,689 and quantum dots (QD)690,691 are applied for amplification of a binding event due to their attractive EC properties and electrocatalytic activities. Here, we introduce carbohydrate modification with Os(VI)L complexes (where L is a nitrogenous ligand), which can be used for labeling of only a glycan part of biomolecules such as glycoproteins and glycolipids, and which is less known in the glycomic community. An advantage of this labeling protocol is that a complex with a redox label can be formed after biorecognition, not disturbing the binding event.
8.6.1. Covalent Labeling with Os(VI) Complexes
Osmium tetroxide complexes, Os(VIII)L, binding covalently to pyrimidine residues in single-stranded DNA and RNA (Figure 30Aa) have shown their usefulness as probes of DNA structure in vitro (reviewed in ref (692)) and in cells (reviewed in ref (693)). In contrast, six-valent Os(VI)L complexes were shown to bind ribose (but not deoxyribose) in nucleosides692 (Figure 30Ab). It was found that products of this reaction were electroactive, displaying redox couples on CV of Os(VI)L-modified ribosides (Figure 30C) in a wide pH range (Figure 30D) at mercury and carbon electrodes, similar but not identical to those of the base-Os(VIII)-modified ribosides.694 In addition to redox couples seen on CV, reaction products of some Os(VI)L complexes yielded electrocatalytic peaks (see peak Vc in Figure 30C,D). Os(VI)L riboside reactions turned out to be useful for end-labeling of RNA and were utilized in sensing of microRNA.242,695,696
Figure 30.

(A) Reaction of Os(VIII)L and Os(VI)L complexes with different parts of a nucleoside showing Os(VI)L complex specifically modifying ribose moiety. (B) Fragment of Os(VI)L-modified dextran. (C) Adsorptive stripping cyclic voltammograms of 10 mM base-modified (blue) and sugar-modified thymine riboside (red), and sugar-modified adenine riboside (black). (D) Dependence on pH, 10 mM sugar-modified thymine riboside, HMDE, with stirring; Britton–Robinson buffer, pH 7.0; scan rate (C) 2 V/s, (D) 1 V/s; tA of 60 s; EA of 0 V, step potential 5 mV. Adapted with permission from ref (694). Copyright 2007 John Wiley & Sons, Inc.
It was also shown that the above reactions yield a redox active product, which could be useful in EC analysis of PS and OLS.229,244,697−699 Thus, PS lacking any redox moiety can be transformed into electroactive species by their reaction with six-valent Os(VI)L complexes (Figure 30B).694 Os(VI)L did not produce any electroactive adducts with DNA and proteins, suggesting high specificity in the glycoprotein measurements. The reaction is very simple and does not require any special equipment. The complex can be only mixed with the carbohydrate at room temperature, and the adduct is formed within hours. Using a ligand exchange process699 or elevated temperature245 (e.g., 75 °C), the reaction can proceed in about 15 min. Excess of the reagent may interfere with the EC determination, and mostly a purification step such as dialysis or separation on a chromatographic column or membrane is necessary. This step can be omitted when the adsorptive transfer stripping (ex situ) method276,700 is used with carbon electrodes.697 In adsorptive transfer stripping experiments, the electrode with strongly adsorbed PS adduct is washed to remove the weakly adsorbed Os(VI)L complex. The PS-modified electrode is then transferred to the electrolytic cell containing blank background electrolyte followed by voltammetric measurement. In this way, the purification step is avoided, and the PS adsorption can be performed from a small analyte drop (e.g., 3–10 μL, depending on the electrode size). Using adsorptive transfer stripping, an abundance of monomeric carbohydrates (e.g., glucose) does not interfere with PS determination as it can be easily washed away from the electrode.700
Properties and EC behavior of PS–Os(VI)L adducts can be significantly influenced by the nature of the chosen ligand. Table 2 shows some ligands that are useful in EC analysis of PS–Os(VI)L adducts. For example, by using Os(VI)bpds (bpds = bathophenanthrolinedisulfonic acid), negative charges can be introduced in the PS or OLS adducts. Using Os(VI)bipy, electrocatalytic peaks can be obtained allowing the determination of OLS at the picomolar level.244 The reaction of Os(VI)temed (Table 2) producing PS and OLS adducts appears particularly interesting. These adducts can be determined using adsorptive stripping method (in situ) at mercury electrodes directly in the reaction mixture, without any purification step because free Os(VI)temed adsorbs very weakly on Hg electrodes.700 Moreover, it has been shown that some glycan isomers can be distinguished on the ground of differences in the voltammetric responses of their Os(VI)L adducts.701
Table 2. Nitrogenous Ligands (L) Applied in Os(VI)L Complexes for Carbohydrate Modification.
Ligands shown in bold were mostly applied in PS modification.
8.6.1.1. Glycans Can Be Modified with Os(VI)L Directly in Glycoproteins and Recognized by Voltammetry from Nonglycosylated Proteins
Recently, it has been shown702 that by using Os(VI)L complexes, glycans can be modified directly in glycoproteins and detected voltammetrically at carbon electrodes (Figure 31A). Using square wave voltammetry and pyrolytic graphite electrode, linear dependence of peak heights on Os(VI)temed-modified avidin concentration was obtained between 100 nM and 1 μM (Figure 31B). In AdTS experiments (using 8 μL drops of Os(VI)temed-modified avidin), hundreds of femtomoles of avidin were sufficient for the analysis. The analysis could be performed directly in the reaction mixture when Os(VI)temed was used for the avidin modification. However, experiments with Os(VI)bipy required separation of the adduct from the reaction mixture. The combination of Os(VI)bipy with mercury electrodes resulted, however, in electrocatatalytic signals allowing sensitivity down to the picomolar level.
Figure 31.
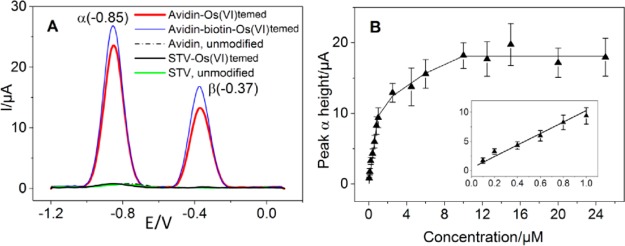
(A) AdTS SWV of unpurified 5 μM Os(VI)temed-treated avidin, complex avidin–biotin, and streptavidin (STV); (B) concentration dependence of peak α (EP ≈ −0.85 V) of avidin-Os(VI)temed, four replicative measurements. Inset: Detail of the concentration range 100–1000 nM. Adapted with permission from ref (702). Copyright 2014 Elsevier.
8.6.2. Covalent Labeling with Other Redox Labels
In 2006, Joseph Wang’s group introduced a concept of the lectin biosensor based on application of QD as a label in glycan measurements.703 Even though the biosensor was not exposed to glycoproteins as an analyte, the authors in the study selected important glycan determinants, which were conjugated to CdS QD. The lectin biosensor developed on a modified gold electrode was first incubated with a QD-conjugated glycan, and then this complex was displaced by an untagged sugar being a “sample”. Analysis of the amount of QD-conjugated glycan not displaced by the “sample” solution was performed via stripping voltammetric detection on a mercury coated GCE, with a LOD of 0.1 μM.703 Other forms of QDs composed of CdTe, ZnO, and CdSe were applied in glycoprofiling, as well.704−707
Ferrocene redox moiety has been successfully applied in labeling of Con A lectin708 or a gold nanoparticle coloaded with Con A709 for further glycoprofiling. Moreover, Con A was labeled with daunomycin for EC analysis of ovalbumin down to the sub nanomolar level710,711 or with Ru complex for detection of E. coli cells down to 127 cells/mL by electrochemiluminescence.712 Another way to use the redox labels is to attach them directly to the analyte, as in the case of electrostatic attraction between negatively charged ovomucoid and positively charged ZnO quantum dots. The labeled analyte was then detected by the biosensor with immobilized Con A down to the picomolar level.707 Label-based approaches have not been that often applied in the detection of isolated/purified glycoproteins, but rather for the analysis of glycoproteins attached to the surface of various types of cells (section 9.5.5).
It can be concluded that there is still plenty of room for improvement of label-based glycoprotein measurements with applications of lectins, when compared to the most sensitive protein assay schemes performed by antibodies, using various labels offering LODs in the femtomolar to attomolar levels.713−716
8.7. Concluding Remarks and Future Prospects
It is very important that a palette of currently used lectins mainly of a plant origin717 will be extended by lectins isolated from other higher organisms such as galectins and selectins.718,719 It will expand binding specificities for subtle changes in the glycan composition and will be essential for highly reliable diagnosis of pathological processes. Alternatively, artificial lectins such as lectin multimers, boronate-decorated lectins, or redox switchable lectins can be beneficial for the development of novel lectin-based EC biosensors. Recombinant technology will in the future undoubtedly allow us to expand the range of lectins produced with various tags for oriented and controlled immobilization of lectins as an important prerequisite for sensitive and selective glycan measurements. Additional options of how to increase the library of biorecognition elements are to produce highly stable DNA aptamers or lectin-like peptide aptamers with a high affinity for their glycan targets (section 9).
From a transducer point of view, it can be anticipated that many different ways of how nanomaterials can be integrated into the EC detection platform of detection will be developed. This can be done by direct modification of electroactive surfaces by nanomaterials or by advanced patterning protocol and by using nanomaterials as amplification tags, helping to produce lectin biosensors/biochips working in an ultrasensitive and selective way. Further, it can be anticipated that EC-based biosensors will compete in a future with instrumental techniques (MS, liquid chromatography, capillary electrophoresis) or lectin microarrays only if such devices are integrated into a biochip format offering multiplexed glycan measurements. At the same time, it will be desirable to develop redox switchable immobilization protocols for selective and controlled immobilization of lectins on electrodes within an array of electrodes.
Recent advances in synthetic chemistry applied for the synthesis of asymmetrically branched glycans,720 which resemble naturally occurring glycans, or better understanding of the enzymatic action of glycan processing enzymes,721 can assist in the development of bioreceptive surfaces applicable in finding novel glycan-binding biorecognition elements, including antibodies.
9. Detection of Biomarkers
Generally speaking, biomarkers for diseases may consist of any measurable or observable factors that indicate normal or disease-related processes in vivo or patient responses to therapy. Such biomarkers encompass, for example, physical symptoms, expressed proteins, mutated DNAs and RNAs, processes such as cell death or proliferation, and serum concentration of small molecules. Significant progress has been made in the identification of biomarkers by means of such technologies as DNA microarrays and proteomic approaches, including mass spectrometry, resulting in an increasing number of potential biomarker candidates. Proteomic and genomic analyses generate vast amounts of data, but these approaches do not appear to be the best suited for a routine cancer clinical testing due to their complexity. The laborious protocols have to be performed by highly skilled technicians to achieve acceptable reproducibility. If a complex pattern of gene expression is reduced to a few genes, biosensor-based detection may become advantageous for practical testing, because it is more user-friendly, faster, less expensive, and less technically demanding than microarray or proteomic analyses.722
The topic of EC detection of protein biomarkers has been the subject of several review articles,50−53 and here we wish to focus mostly on glycoprotein biomarkers and show that detection strategies were developed to recognize the protein part itself as well as a glycan part of the glycoproteins. We shall deal mostly with the detection of cancer-related protein biomarkers found at elevated levels in body fluids already during the early stages of cancer development, acting as indicators of the onset, progression, or recurrence of the cancer. Moreover, biomarkers for other diseases, such as diabetes, neurodegenerative diseases, AIDS, etc., will be also mentioned.
It is now well accepted that measurement of a single biomarker is often insufficient because of variations in the given protein expression among human population and also due to the fluctuation of biomarker levels within the single individual. Let us take prostate specific antigen (PSA) as an example. PSA is a glycoprotein secreted by a prostate gland, which is present in small amounts in serum of healthy men but becomes elevated in prostate cancer, the most common form of cancer in men in the U.S. and Europe.546 The majority of PSA in the blood is bound to serum proteins, but there are also tiny unbound amounts, referred to as free PSA; both of them together form total PSA. Testing of serum PSA was routinely applied for diagnosing prostate cancer and for monitoring the disease progress, with total PSA values below 4 ng/mL considered safe while levels above 10 ng/mL indicated the presence of a disease. However, besides prostate cancer, there are some benign conditions that may cause PSA levels to rise above the threshold level, including inflammation and enlargement of the prostate, or even prostate biopsies and surgeries (which give rise to false positives). Biopsy-detected prostate cancer was found among 15% of men that had otherwise normal PSA levels of 4 ng/mL or below (yielding false negatives).723 For these reasons, the U.S. Preventive Services Task Force and UK National Screening Committee do not recommend PSA screening anymore as it may lead to “overdiagnosis” or “overtreatment”. When the patient has borderline or moderately increased total PSA values, it may be useful to monitor the ratio of free PSA to total PSA and thus help to eliminate unnecessary biopsies in men with total PSA values above 4 ng/mL. This ratio decreases with increasing risk of prostate cancer. In addition, glycoprofiling of PSA with serum concentration in a gray zone (4–10 ng/mL) could be applied as a supplementary test in the diagnosis of this type of cancer.
It should be also noted that many protein biomarkers indicate more than one disease, and thus a single cancer biomarker is frequently not unique to a specific type of cancer. For these reasons, a combination of multiple biomarkers is preferred, which could result not only in improved accuracy, but also in the increase of a sample throughput and reduction of cost per test. Detection of panel of biomarkers is, however, complicated due to large variations in concentrations for different markers in serum, ranging from low picograms to hundreds of nanograms per mL. For example, the level of interleukin 6 (IL-6), a cytokine associated with different types of tumors, is about 1000-fold lower in healthy individuals than the level of PSA.724 This requires a development of a reliable detection method, spanning all of the concentration ranges for the chosen panel of biomarkers.
Contrary to nucleic acids, whose molecules can be recognized (and separated) on the ground of their complementarity, proteins do not possess such a unique system for specific recognition of individual protein types. Therefore, different strategies need to be employed for capturing specific proteins. The most widely used ones are based on application of (i) antibodies (in so-called immunoassays), (ii) nucleic acid aptamers, (iii) peptide aptamers, and (iv) glycan-binding lectins for detection of glycoproteins. All of these approaches will be described after a brief summary of protein labeling often used for biomarkers detection and for probing protein structures.
9.1. Labeling of Proteins
Usually, the label is introduced into the EC biomarker assay to greatly amplify the signal and thus to achieve substantially lower LODs. The vast majority of authors use enzymes or nanomaterials (especially nanoparticles) as labels in immunosensor or aptasensor format. Enzymes, such as horseradish peroxidase (HRP) and alkaline phosphatase (AP), provide high, steady, and reproducible signal amplification.725 If higher sensitivity is required, single enzyme approach may be combined with an additional amplification process (i.e., redox cycling), or using multienzyme labels per detection probe. As compared to enzymes, nanomaterials show better long-term stability and are more easily prepared. Nanomaterials used in EC assays mostly comprise various types of nanoparticles, carbon nanotubes (CNT), nanowires, or graphene sheets, which usually serve as immobilization substrates for accumulation of increased amounts of electroactive molecules, as well as catalysts or nanoelectrodes. It is common to combine both enzymes and nanomaterials, such as by loading multiple enzymes at the CNT surface, in a single assay to achieve even lower LODs. More details on the construction and application of biosensors using enzymes or nanomaterials as labels are given below in this section, where specific examples are described, and also in recent reviews.725−732
9.1.1. Chemical Modification of Proteins for Probing Their Structure and Activity
Chemical agent can serve not only as a signal amplifier, but also as a probe of protein structure (similarly to chemical probes of the DNA structure34). Although there are many reagents available for modification of aa residues in proteins, only few exhibit reasonable selectivity for a particular residue under conditions close to physiological. In most cases, the reagents are electrophilic molecules that target nucleophilic functional groups.733 These chemicals were utilized for probing accessibility of the reactive moieties upon changes in the protein structures due to unfolding, or intermolecular interactions. The probing of aa residues in proteins was used in connection with various detection techniques, including optical methods,734−739 chromatography combined with site-specific proteolysis,740−742 and mass spectrometric techniques.733,743−745
Literature devoted to EC analysis of chemically modified peptides or proteins for probing their structure is rather scarce. Several papers reported on the synthesis and applications of peptides using electrochemically active ferrocene derivatives.746−754 For instance, the approach chosen in Kraatz’s laboratory is based on the ability of kinases to transfer a redox-labeled phosphoryl group to surface-bound peptides that are highly specific substrates for the particular protein kinase. For this purpose, they mostly applied 5′-γ-ferrocenoyl-ATP (Fc-ATP) as a cosubstrate for peptide phosphorylation. After the Fc-phosphoryl group was transferred to the peptide, the presence of the redox active Fc group was detected electrochemically (Figure 32). The EC response enabled monitoring the kinase activity and its substrate, as well as the effect of small molecule inhibitors on protein phosphorylation. The authors developed peptide biosensors, for example, for papain,755 protein kinase C,754 serine/threonine kinase,756 sarcoma-related kinase,752,757 HIV enzymes,753,758 STAT3 dimerization,759 or even amyloid β peptides.760 More recently, this strategy was extended to an immunoarray format, in which authors utilized anti-Fc antibodies for visualization of the protein kinase-driven transfer of the phosphate group from Fc-ATP to the hydroxyl group of peptides or proteins.761
Figure 32.
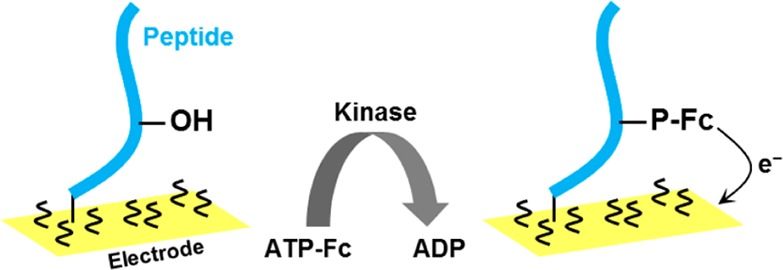
Protein kinase C-catalyzed phosphorylation of SIYRRGSRRWRKL peptide (with phosphorylated serine underlined) using ferrocene-labeled ATP (ATP-Fc) as a substrate. After a transfer of γ-phosphate-Fc group to the serine residue of the peptide, the surface-attached Fc groups are detected via EC techniques at thiol-modified gold electrodes. Adapted with permission from ref (754). Copyright 2008 Royal Society of Chemistry.
Another option is to probe the accessibility of specific Trp residues in proteins with a chemical agent combined with EC detection. Trp has a special status in several aspects (ref (762) and references therein) and is relatively rare in natural proteins. It often plays critical roles in the arrangement of the protein tertiary and quaternary structures,763−765 and is frequently found in active sites of enzymes and proteins containing specific binding sites for other molecules.735,740,741,766,767 The indole side group of Trp has a characteristic fluorescence spectrum, and can be utilized as intrinsic fluorophore in protein analysis.406,768,769 In addition, the Trp yields an electro-oxidation signal at carbon electrodes (see section 4).54,55,271,317,410 Several chemical agents are available, which react with the Trp indole group, including 2-hydroxy-5-nitrobenzyl bromide, sulfenyl halides, or 2-nitrobenzenesulfenyl chloride and N-bromosuccinimide.736,737,743,767 However, these agents exhibit reactivity also toward other aa residues and particularly cysteine. Increased specificity toward Trp can be achieved by optimization of reaction conditions. Chemical reactivity of individual Trp residues within a protein reflects their accessibility toward solvent, making it thus possible to distinguish between buried and exposed residues and/or monitor shielding of residues within the protein binding sites (e.g., in avidin–biotin).735,736,740,741,766,767
As compared to the above-mentioned chemical agents, a complex of eight-valent osmium tetroxide and 2,2′-bipyridine (Os(VIII)bipy) was shown to be a more specific electroactive label for Trp in peptides and proteins.770−772 Os(VIII)bipy forms a stable adduct with the Trp indole moiety,770,771,773 similar to adducts formed by pyrimidine residues in Os(VIII)bipy-treated nucleic acids.34,692,772 Analogically to Os(VIII)bipy-modified DNA, the Os(VIII)bipy-modified peptides (containing Trp) produce an electrocatalytic signal at the Hg electrode enabling their highly sensitive determination.770 Modification of several peptides with Os(VIII)bipy was recently studied using capillary electrophoresis and MALDI-TOF MS.771 Although several other aa residues in proteins also exhibit reactivity toward the osmium reagents,773 no other stable aa adducts (except Trp) bearing the Os moiety were identified. Oxidation products of cysteine or methionine (cysteic acid or methionine sulfone, respectively) were detected by MALDI-TOF MS,773 but they did not display EC signals characteristic for peptide Os(VIII)bipy adducts. Modification of protein Trp’s with Os(VIII)bipy can be analyzed by adsorptive transfer stripping voltammetry directly in the reaction mixture at carbon electrodes.774 Electrocatalysis of peptide/protein Os(VIII)bipy adducts at Hg electrodes is more sensitive but requires separation of the adduct from the reaction mixture. The technique was applied to monitor changes of accessibility of Trp residues during the formation of specific molecular complexes such as streptavidin–biotin under conditions close to physiological.
9.2. Immunoassays
Because principles, applications, as well as recent advances in immunoassays were thoroughly reviewed,728,732,775−777 we will limit ourselves only to a brief summary here. To our knowledge, first EC analysis of the antibody–antigen interaction was performed already in the 1950s by Breyer and Radcliff,778 who used polarography to detect interaction of azo-protein with rabbit antiserum. They observed a decrease of the azo-protein polarographic wave as a result of interaction of this protein with the specific antiserum. Today, most EC immunoassays are performed in an ELISA-like format, in which primary antibody (Ab1) immobilized at the immunosensor (mostly electrode) surface captures the protein analyte present in the sample, followed by the addition of an enzyme-labeled secondary antibody (Ab2) and EC detection of the enzymatic product. In this way, large amplification of the signal is obtained, because one molecule of the enzyme (per one molecule of the biomarker) leads to a generation of many electroactive molecules from an enzymatic reaction. It was Heineman and Halsall who in the middle of the 1980s developed sensitive enzyme-linked EC immunoassays for proteins and other molecules.779 They used AP enzyme yielding electroactive products, and obtained low LOD in the range of pg to ng/mL. Heineman’s team continued this research and was later among the first who used microfluidics in EC immunoassays of proteins. Limoges et al.780 elaborated theoretical analyses of amperometric and voltammetric AP-labeled immunosensor responses, and identified key factors behind a high sensitivity of detection. Besides AP, also HRP and glucose oxidase were shown to be suitable labels.776 The original Ab2/enzyme format has been increasingly replaced with more sophisticated amplification strategies in which the Ab2 is conjugated, for example, with nanoparticles (metal, iron oxide, semiconducting quantum dots, etc.), CNTs, multienzyme clusters, or even with their combinations, greatly amplifying the response (Figure 33).
Figure 33.

Scheme of amplification strategies for detection of protein cancer biomarkers in EC immunoassays. Surface-immobilized primary antibody (Ab1) captures an antigen, which is then detected using, for example, (a) a simple secondary antibody/enzyme (Ab2/enzyme) or (b) antibody/nanoparticle (Ab2/NP) bioconjugate, which are now often replaced with more sophisticated systems, in which the secondary antibody is coupled with, for example, (c) biotin/streptavidin/enzyme,882 (d) different quantum dots (Ab2/QD),883 (e) enzyme-modified carbon nanotubes (Ab2/CNT/multienzyme),689 (f) magnetic beads bearing cluster of enzymes (Ab2/MB/multienzyme),882 or (g) quantum dot-dendrimer nanocomposites (Ab2/dendrimer/QD).884
Typical materials for the preparation of the immunosensor surface are metals (e.g., gold, platinum), semiconductors (e.g., indium tin oxide), and carbon. One of the key factors determining the quality of the immunosensor is immobilization of the antibody. Depending on the surface, as well as on protein properties, different immobilization techniques were developed, including a simple physical adsorption, entrapment into a polymer matrix, covalent immobilization, or bioaffinity-based interactions (Figure 34). While physical adsorption is simple and quick, its weakness is nonspecificity and a random orientation of the immobilized antibody, greatly decreasing antigen binding. In the study by Salam and Tothill, covalent attachment led to a 250-fold improvement of a LOD as compared to a simple physical adsorption.781 However, covalent attachment may yield heterogeneous population of differently oriented proteins.782 For oriented antibody immobilization, site-specific noncovalent bioaffinity interactions are better suited, improving further efficiency of the antigen binding. An ideal orientation of the antibody is when the Fc region is in contact with the surface, and the Fab region (which binds the antigen) protrudes to the solution (Figure 34i). This can be achieved in several ways,732 such as via proteins A or G, His-tag system, DNA-directed antibody conjugation, avidin–biotin binding, etc. (Figure 34). Nanomaterials greatly enhance the sensitivity of the immunoassays, although the antibody orientation is usually random. Their main advantages lie in their large surface-to-volume ratio, faster reaction kinetics, and good compatibility with biomolecules.
Figure 34.
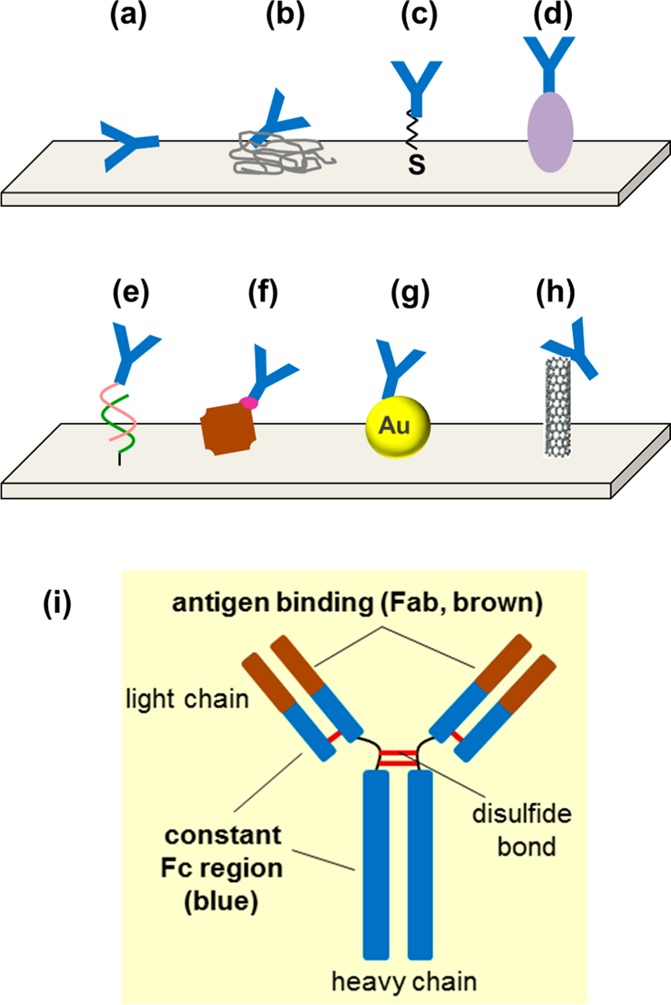
Examples of antibody immobilization to the surface. The binding can be achieved via, for example, (a) physical adsorption, (b) entrapment into a polymer matrix, (c) thiol groups, (d) protein A or G, (e) DNA-directed immobilization (by site-specific coupling of protein G to DNA oligonucleotide), (f) avidin–biotin system, (g) nanoparticle, or (h) carbon nanotubes (linked via carboxyl groups at CNT). (i) Structure of the antibody with Fab and Fc region.
Special care should be taken to minimize noise (usually caused by nonspecific adsorption of proteins or other molecules on the electrode or other surfaces involved in the assay), especially in label-free techniques such as EIS (section 8.5.1), because any adsorbed interfering molecule causes a change in the resulting signal (giving rise to false positives or negatives). Introduction of blocking reagents usually saturates remaining binding sites at the surface, leading to a formation of a dense gap-free layer. Such a layer not only improves binding capacity of antibodies to the antigens, but also enhances a long-term stability of the sensing layer. However, the blocking reagents, which are usually proteins (BSA, casein), detergents (Tween-20), or polymers (such as polyethylene glycol), require stringent washing following their addition, making the assay more time-consuming and complex. Recent progress in tailoring the gold electrodes with ternary thiol layers greatly improved the signal-to-noise ratio (S/N) in DNA hybridization sensors,783,784 and it can be expected that application of such layers will also improve the S/N in immunoassays.
9.2.1. Cancer Biomarkers
Several examples of EC immunoassays for cancer protein biomarkers detection are listed in Table 3. It should be noted that not all papers demonstrate detection of multiple biomarkers using samples from body fluids, but rather focus on a single protein, sometimes present in a plain buffer. Such studies may be useful from a methodological point of view, when demonstrating proof of concept, but they are not very relevant in clinical settings. New strategies should be therefore developed for measuring both high and low concentrations of different protein biomarkers in the same sample in the presence of thousands of other serum proteins, usually occurring at much higher concentrations.
Table 3. List of Immunoassay-Based EC Studies of Protein Biomarkers.
| biomarker | amplification strategy | LODa | glycoprotein | ref |
|---|---|---|---|---|
| PSA | Ab2/AP | 1.4 ng/mL | yes | (889) |
| PSA | Ab2/CNT/multi-HRP | 4 pg/mL | yes | (689) |
| PSA | MB/HRP | 0.5 pg/mL | yes | (794) |
| PSA | graphene/HRP/Ab2/Au NPs | 2 pg/mL | yes | (890) |
| PSA | MWCNT/AuNP/HRP | 0.4 pg/mL | yes | (891) |
| PSA | label-free (EIS) | 1 ng/mL | yes | (892) |
| PSA | Ab2/HRP/MWCNT | 5 pg/mL | yes | (797) |
| IL-8 | 8 pg/mL | no | ||
| PSA | multi-HRP/MB/Ab2 | 0.23 pg/mL | yes | (50) |
| IL-6 | 0.3 pg/mL | no | ||
| PSA | Ab2/HRP | 2 ng/mL | yes | (893) |
| CEA | 0.2 ng/mL | yes | ||
| CA 15-3 | 5.2 U/mL | yes | ||
| CEA | AuNP/Ag deposition | 0.5 pg/mL | yes | (894) |
| CEA | graphene/Au | 1 pg/mL | yes | (895) |
| AFP | microspheres/HRP | yes | ||
| CA 125 | Ab2/dendrimer/CdS QD | 0.005 U/mL | yes | (884) |
| CA 15-3 | Ab2/dendrimer/ZnS QD | 0.003 U/mL | yes | |
| CA 19-9 | Ab2/dendrimer/PbS QD | 0.002 U/mL | yes | |
| PSMA | virus matrix/PEDOT | 100 pM | yes | (896) |
| AFP | graphene/Au–Pd nanocrystals | 5 pg/mL | yes | (897) |
| IL-6 | multi-HRP | 10 fg/mL | no | (898) |
| IL-6 | Ab2/MB/multi-HRP | 10 fg/mL | no | (796) |
| IL-8 | 15 fg/mL | no | ||
| VEGF | 8 fg/mL | yes | ||
| VEGF-C | 60 fg/mL | yes | ||
| CA 125 | SPCE/sol–gel/Ab2/HRP | 0.5 U/mL | yes | (787) |
| CA 15-3 | 0.2 U/mL | yes | ||
| CA 19-9 | 0.3 U/mL | yes | ||
| CEA | 0.1 μg/L | yes |
PSA, prostate specific antigen; IL-6, interleukin 6; IL-8, interleukin 8; CEA, carcinoembryonic antigen; CA, cancer antigen; AFP, α-fetoprotein; CRP, C-reactive protein; LOD, limit of detection; PSMA, prostate specific membrane antigen; VEGF, vascular endothelial growth factor; Ab2, secondary antibody; AP, alkaline phosphatase; CNT, carbon nanotube; HRP, horseradish peroxidase; MB, magnetic bead; AuNP, gold nanoparticle; QD, quantum dot; PEDOT, poly(3,4-ethylenedioxythiophene).
Despite these difficulties, some encouraging results were already obtained. For instance, EC immunosensor arrays (ECIAs), based on miniaturization and low cost of the entire assay, were constructed for multiplexed EC immunoassay.785−790 In 2003, Kojima et al. designed ECIA for detection of two tumor markers: α-fetoprotein (AFP, a plasma protein elevated in several types of cancer) and β2 microglobulin.788 Shortly afterward, Wilson reported ECIAs for the simultaneous quantitative detection of seven tumor markers.790 Such ECIAs were prepared by immobilizing immunoreagents on photolithographic and sputter-deposited layers adhering to a glass substrate. In a different study, a multiplexed analysis of a tenascin C, glycoprotein present in the extracellular matrix expressed in adults in cancerous tissues (solid tumors like glioma or breast carcinoma), was performed on a chip having nine independent electrodes with immobilized primary antibodies.791 When probed with a HRP-labeled secondary antibody (against primary antibody), unoccupied binding sites produced an insoluble film, leading to high RCT observed with EIS. Conversely, the higher was the concentration of the analyte, the lower was the RCT of the electrode surface observed with EIS. The biosensor was able to detect 14 ng (48 fmol) of tenascin C, sufficient for clinical diagnostics.791
Immunosensing strategies are being increasingly coupled with various nanomaterials. For instance, in 2010, a graphene sheet sensor platform and functionalized carbon nanospheres labeled with HRP-secondary antibodies (Ab2) for detection of α-fetoprotein (AFP), a plasma protein elevated in several types of cancer, were proposed.792 Enhanced sensitivity was achieved by (i) using the multiconjugates of HRP-Ab2-carbon nanospheres onto the electrode surface through “sandwich” immunoreactions and (ii) functionalized graphene sheets-chitosan, which increased the surface area, capturing a large amount of primary antibodies and amplifying the detection response 7-fold, as compared to that without graphene modification and labeling. Rusling’s group has successfully designed several immunosensors for detection of various cancer biomarkers.793 By using conductive nanostructured electrode platform based on films of SWCNT forests,689 they achieved LOD for PSA of 4 pg/mL. About an 8-fold increase in sensitivity as compared to the above SWCNT forests was achieved by using 5 nm glutathione-decorated Au nanoparticles, containing carboxylate groups for attachment of large amounts of capture antibodies combined with multienzyme magnetic beads bioconjugate.794 Magnetic beads (1 μm in a diameter) contained 7500 HRP labels along with Ab2. Using this platform, LOD of 0.5 pg of PSA per mL in 10 μL of undiluted serum (corresponding to 5 fg of PSA in the analyzed sample) was achieved, what is near or below the normal serum levels of most cancer biomarkers. An excellent correlation with standard ELISA assays in cell lysates and sera of cancer patients was obtained. An ultrasensitive EC immunoassay protocol with signal amplification via formation of a sandwich configuration with Ab2 complexed to dendrimer-containing silver nanoparticles could detect PSA down to 10 fM.795 More recently, Rusling’s group has simultaneously detected four oral cancer biomarkers, IL-6, IL-8, vascular endothelial growth factor (VEGF), and VEGF-C, directly in diluted sera obtained from 78 oral cancer patients and 49 negative controls, with different serum levels for each biomarker (Figure 35).796 Good clinical sensitivity (aM levels) and specificity for early stage tumor detection was reached, and, again, results were confirmed by a good correlation with ELISA. Wan et al. have proposed an immunoarray using so-called universal nanoprobe consisting of multi-HRP labels and antirabbit antibodies loaded onto multiwalled carbon nanotubes (MWCNT) for simultaneous detection of PSA and IL-8.797 This strategy required three different antibodies, monoclonal antibody against the biomarker, which was immobilized at the sensor surface, polyclonal rabbit antibody against the biomarker recognizing a different epitope, and antirabbit antibodies attached to the universal nanoprobe, binding to polyclonal rabbit antibodies. In this way, multiple antigens could be detected with only a single labeled antibody, leading to a reasonable LOD of 5 pg/mL for PSA and 8 pg/mL for IL-8. Other nanomaterial-based strategies for EC detection of tumor biomarkers involved, for example, gold nanoparticle-modified screen-printed carbon electrodes,798,799 bilayer nano-Au and nickel hexacyanoferrates nanoparticles,800 nanosilver-doped DNA polyion complex membrane,784 bimetallic AuPt nanochains,637 or ferrocene liposomes combined with MWCNT.801
Figure 35.
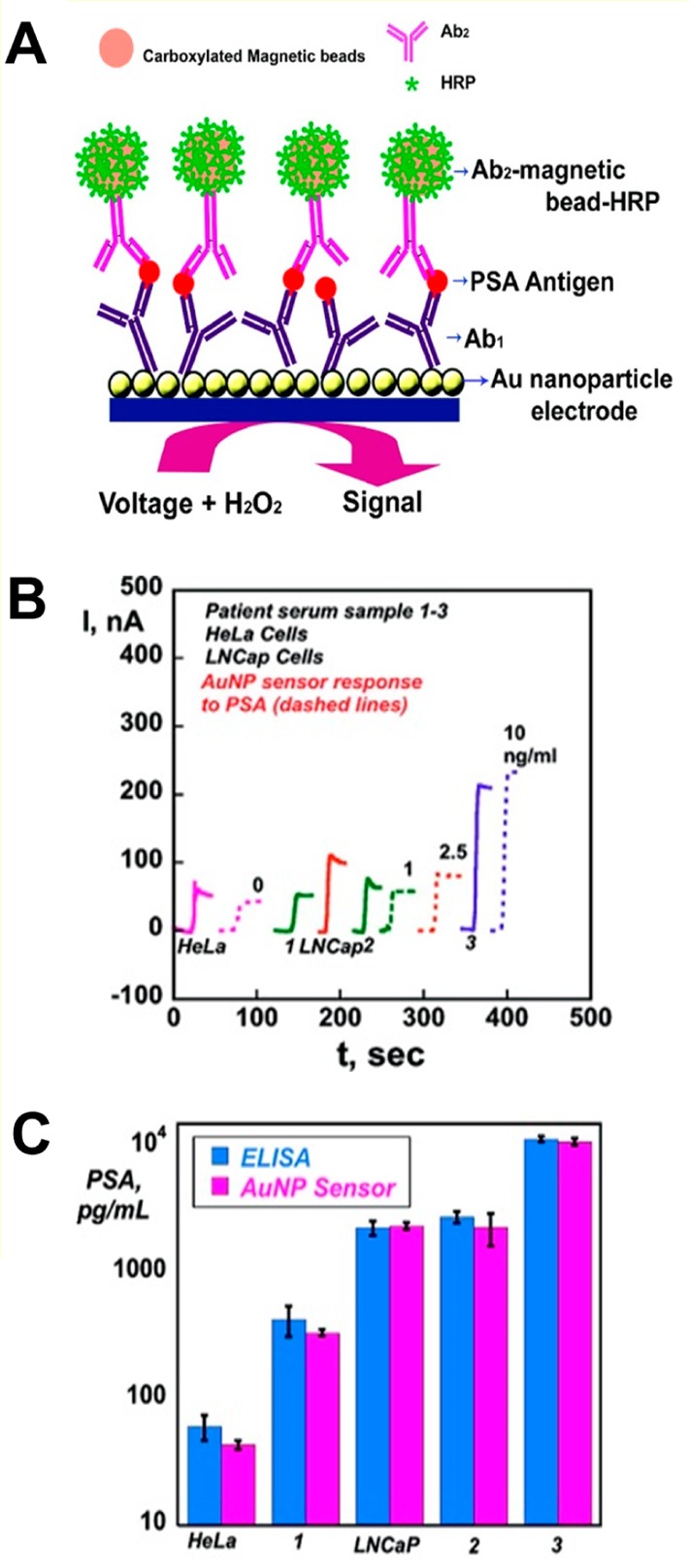
(A) Gold nanoparticle (AuNP)-based immunosensor. The immunosensor involves attached Ab1, which captures antigen from a sample, followed by incubation with Ab2-magnetic bead-HRP (Ab2-MB-HRP), providing multiple enzyme labels for each PSA bound. The detection step involves immersing the immunosensor into a buffer containing a mediator, applying voltage, and injecting H2O2. (B) Results for AuNP immunosensor incubated with PSA present in 10 μL of a calf serum (ng/mL labeled on curves, dashed lines), cell lysates (HeLa and LNCap cells), and human patient serum samples (1–3) (solid lines) for 1.25 h, followed by an injection of 10 μL of 4 pmol/mL of Ab2-MB-HRP. (C) Validation of AuNP sensor results for cell lysate and human serum samples by comparing against results from an ELISA determination (relative standard deviation ∼10%) for the same samples. Adapted with permission from ref (794). Copyright 2009 American Chemical Society.
9.2.1.1. Electric Field Effects
It has been shown that application of positive potentials to the electrode with immobilized DNA stimulates DNA hybridization, while a negative electric field destabilizes DNA duplex.34 Similarly, it has been observed that electric field can be utilized in EC immunosensors. Application of positive potentials greatly increased the rate of protein binding, while negative potentials were used to prevent a weak nonspecific binding, improving thus the S/N ratio.787 Originally, Wu et al.785 used the sandwich assay in combination with a screen-printed carbon electrode array and electron-transfer mediators to develop disposable ECIAs for a low-cost multiplexed immunoassay of proteins. The multiplexed immunoassay needed, however, a long incubation time due to the slow diffusion of an antigen in an unstirred layer necessary for the immunocomplex formation. Earlier, various technologies were explored to accelerate the immunoreaction on the surface, including low-power microwave radiation,802 magnetic stirring,803 and magnetic field combined with superparamagnetic labels.804 An electrophoresis-assisted optical immunoassay was developed to accelerate the protein transport.805−807 These technologies required additional equipment, resulting in the increased cost and inconvenience of assays. To speed the incubation, Wu et al.787 constructed integrated electric field-driven ECIAs for multiplexed immunoassay, in which antibodies against carbohydrate antigens CA 125 (used especially for monitoring of a therapy progress or recurrence of ovarian cancer), carcinoembryonic antigen (CEA, a surface glycoprotein elevated in colorectal carcinoma), CA 15-3 (a serum biomarker in breast cancer), and CA 19-9 were labeled with HRP and immobilized on biopolymer/sol–gel modified electrodes. In the presence of Au nanoparticles, the HRP showed enhanced EC responses. Formation of immunocomplexes resulted in a decrease in the EC signals, due to the impedance changes caused by the nonconductive immunocomplexes and blocking of the electron transfer between the electrode and HRP-labeled antibodies. The ECIA benefited from the electric field-driven incubation strategy, and the EC end-point detection of four protein biomarkers could be accomplished in less than 5 min. Literature on the electric field effects on proteins is rather scarce, as compared to a large number of papers on DNA, resulting from decades of studies of the electric field effects on DNA.34 Consequently, these effects on proteins are much less understood. The incubation time depended on the strength of an electric field/driving potential and on a solution pH.787 Shortening of the incubation time was explained by increased immunoreaction rates due to the faster transport of the antigens to the immunosensor interfaces. More work will be necessary to understand fully all aspects of the electric field effects on the surface protein–protein interactions.
9.2.2. Neurodegenerative Diseases
It was mentioned in section 6.1 that both oxidation signals at carbon electrodes and peak H at HMDE could be used to study aggregation of α-synuclein (AS), a neuronal protein involved in Parkinson’s disease. Its importance naturally led also to the development of other EC strategies for its analysis, such as a dual amplification strategy in which GCE was modified by gold nanoparticle-based dendrimer loaded with thionine.683 AS was nonspecifically bound to this surface, then incubated with a primary antibody, followed by an addition of secondary antibody labeled with HRP adsorbed on a gold nanoparticle surface. CV was used to monitor EC signal generated by the HRP in the presence of H2O2 with adsorbed thionine. LOD in a high femtomolar range for the analyte was 2–3 orders of magnitude lower as compared to the approach without amplification by a gold nanoparticle-loaded dendrimer or a secondary HRP-labeled antibody.
9.2.3. gp160 as a Marker of AIDS
An early detection of HIV infection is desirable, and thus highly effective bioanalytical methods have to be developed. One way to detect this virus is to look for antibodies against HIV in patients serum. However, these antibodies are usually not present in the serum in an initial highly infectious phase. Therefore, an alternative way is to look for glycoproteins on the surface of HIV virus such as gp160 and gp120.808 An immunoassay using antibodies raised against this antigen offered LOD down to the low picomolar level.809 The method relied on a nanostructured layer deposited on GCE working as a catalyst to reduce H2O2, followed by an amperometric monitoring of a current at −300 mV, which decreased with an increased concentration of gp160. Spiking of serum samples by this antigen revealed a recovery of 96–102% with results being in excellent agreement when compared to ELISA.809
9.3. Nucleic Acid Aptamers
The term aptamer was first used in 1990 by Ellington and Szostak to describe artificial RNA molecules binding to a small analyte.810 The name aptamer comes from the Latin expression aptus (to fit) and from the Greek word meros (part). Aptamers are single-stranded oligonucleotides with a size of 15–60 nucleotides selectively binding a wide range of biomolecules, including whole cells.811 DNA/RNA aptamers have several attractive properties such as a relative simplicity of chemical modification, simple regeneration/reusability, and thermal and chemical stability.812 The small size of DNA/RNA aptamers is a key in achieving high interfacial densities, resulting in the construction of selective and sensitive bioanalytical interfaces. As compared to DNA, RNA is more flexible from a structural point of view, and thus RNA aptamers can be theoretically raised against a wider range of analytes.813 However, a major limitation of using RNA is their susceptibility to chemical and/or enzymatic degradation and the time-consuming process of its preparation. Aptamers resistant to chemical/enzymatic degradation can be prepared by modifications of the DNA or RNA backbone or by introduction of modified bases.814
As compared to the immunoassays, literature on aptamer-based EC detection of protein biomarkers is rather scarce. Nevertheless, some recent papers appeared showing that aptamers might be also useful as biorecognition elements when constructing EC platforms suitable for clinical diagnostics (Figure 36).
Figure 36.
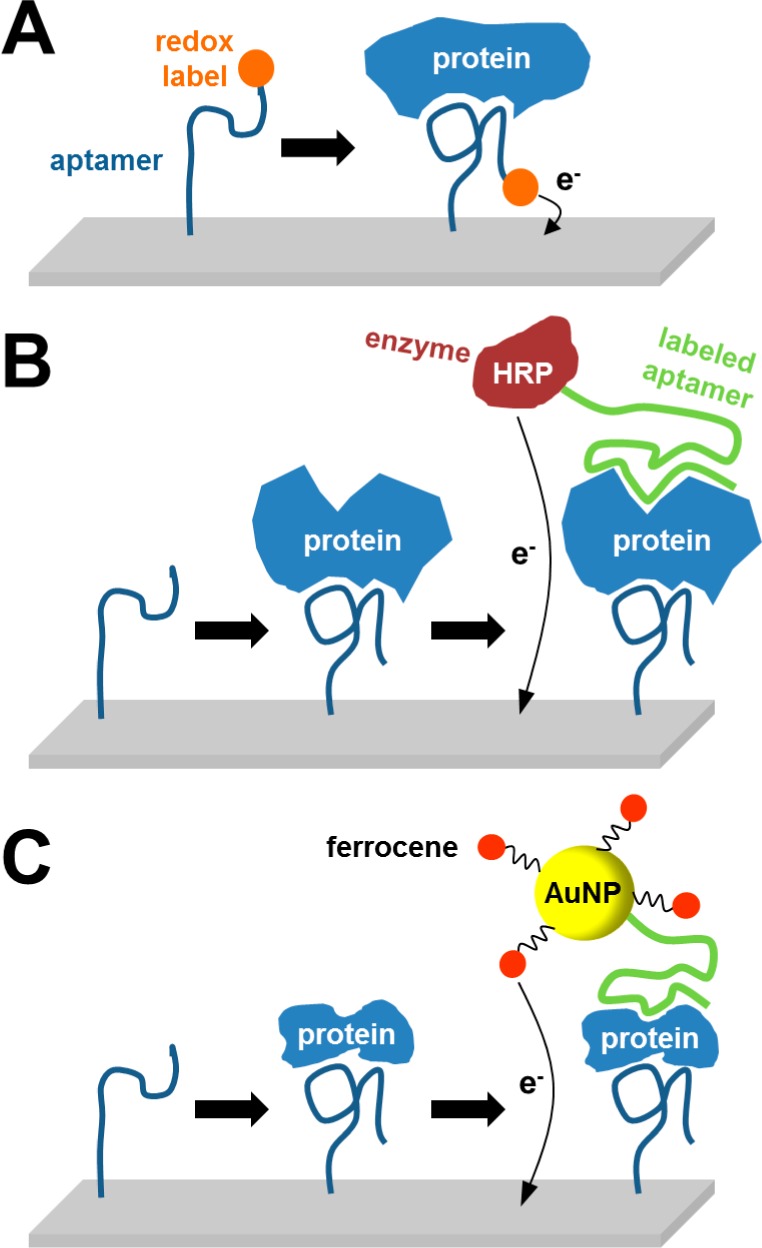
Different approaches for detection of proteins using nucleic acid aptamers. (A) Redox-labeled aptamer alters its conformation after aptamer–protein complex formation, positioning the label closer to the electrode.885 (B) Strategy employing two aptamers, electrode-immobilized aptamer for capturing the protein and a second aptamer labeled with enzyme for EC monitoring of enzymatic reaction.886 (C) Approach similar to that in (B), with the second aptamer being labeled with gold nanoparticle–ferrocene conjugate.887
Liu et al.815 developed an EC DNA aptamer-based biosensor for detection of interferon gamma (IFN-γ), the aberrant expression of which is associated with a number of autoinflammatory and autoimmune diseases. Thiolated DNA aptamer conjugated with methylene blue as a redox tag was immobilized on a gold electrode, followed by a binding of IFN-γ, which caused the aptamer hairpin to unfold, moving methylene blue away from the electrode and decreasing electron transfer rate. Using SWV, LOD was estimated as 0.06 nM.
Aptamers were employed also in a study aimed at the detection of human cellular prions, PrPC, playing a role in prion diseases.816 The biosensor comprised MWCNTs modified with polyamidoamine dendrimers, which in turn were coupled to DNA aptamers used as bioreceptors. The signal originated from a ferrocenyl redox marker incorporated between the dendrimer and the aptamer layer. Interaction of aptamers with prion proteins led to a change of the EC signal, with prion proteins detectable down to 1 pM concentration.
Besides protein molecules, aptasensors were utilized also in the detection of whole cancer cells with biomarkers exposed at the cell surface. Feng et al. reported an EC sensor for multiple cancer types using functionalized graphene and the aptamer AS1411, the first clinical trial II aptamer, with a high binding affinity and specificity to the overexpressed nucleolin on the cancer cell surface.817 The sensor having LOD as low as 1000 HeLa cells (using EIS) could be regenerated and reused. More recent work employing aptamer-aided recognition was described, in which a cancer cell-aptamer binding event mediated by an AP-catalyzed silver deposition reaction was followed by an EC detection.818 In this work, Ramos cells detected down to 10 cells were used as a model for possible analysis of blood cell cancer or Burkitt’s lymphoma. Zhang et al. studied the HL-60 leukemic cell line using a DNA aptamer against this cell line.819 DNA aptamer was first hybridized with capture DNA conjugated to gold nanoparticles. Upon cells binding to DNA aptamer, capture DNA attached to the gold nanoparticle was released from DNA aptamer and detected on GCE modified by CdS QDs and DNA complementary to capture DNA. After hybridization on a modified GCE, an energy transfer between gold nanoparticles and QDs was registered in the form of electrochemiluminescence at an applied potential of −1200 mV vs SCE. The signal increased with increasing cell number with LOD of 20 cells/mL.819
So far, DNA aptamers have not been used for EC analysis of glycoproteins via glycan recognition, but quite a few reports refer to effective production of DNA aptamers against glycan moieties of glycoproteins, for example, from Binghe Wang’s laboratory.820 An extended library consisting of modified nucleotides was used for the generation of DNA aptamers against glycoproteins such as a necrosis factor receptor superfamily member 9,821 vascular endothelial cell growth factor-165, or IFN-γ.822 Boronate affinity capillary proved to be an efficient tool for selection of novel DNA aptamers against glycoproteins with a quite high affinity (∼10 nM), and a process was completed within 2 days with a small consumption of glycoproteins or oligonucleotides (10–20 μL).823 The proof of the concept approach was applied for the selection of DNA aptamers against HRP, but can be applied to other glycoproteins with a diagnostic potential.823
Although the aptamer-based approach for sensing purposes is promising, further studies using real human samples are necessary. In such studies, modification of the aptasensor surface will be very important to avoid nonspecific adsorptions. In real biological samples, probably worse LODs will be obtained.
9.4. Peptide Aptamers
The term peptide aptamer was first applied by Colas in 1996824 to define a protein having a variable peptide sequence displayed on an inert scaffold protein. Peptide aptamers exhibit affinity constant toward protein analytes comparable to antibodies.811 Immobilization of peptide aptamers in a biochip format would generate an “army of terracotta soldiers” with unique “facial” features (binding sites). Such proteins exhibit a high solubility and chemical/thermal stability, a small uniform size of a scaffold protein, and peptide aptamers can be produced in a cost-effective way using heterologous techniques.813 Such proteins could be immobilized with high density on surfaces proving high sensitivity and selectivity of bioanalytical devices.811 Peptide aptamers are isolated from combinatorial libraries during a selection procedure, when the whole library is incubated with an analyte of interest, and thus there is no requirement to know the structure of the analyte and/or the mechanism of the binding in advance.825 The scaffold protein has to be pretreated to “silence” its original biological activities with a possibility to tolerate peptide sequences without changing its overall structure.826 Ferrigno’s group developed peptide aptamers based on a scaffold constructed by mutations of Stefin A protein (a cysteine protease inhibitor). This scaffold allows inserting more than one peptide sequence, and peptide aptamers are working well after being immobilized on various interfaces.827
The most systematic studies with the application of EC methods in combination with peptide aptamers were realized with peptide aptamers based on a Stefin A triple mutant scaffold. This scaffold is made from a Stefin A protein by introducing three point mutations to block its binding with its interaction partners. Various peptide inserts against CDK2 and CDK4 proteins (kinases playing an important role in the cell cycle),828−832 biomarkers of systemic sclerosis,833 or C-reactive protein834 were introduced into this scaffold protein. Moreover, a library of peptide aptamers based on Stefin A triple mutant scaffold can be applied for the identification of novel disease biomarkers and for the development of novel clinical assays.835
Estrela et al. developed an EC method of protein detection by utilizing peptide aptamers to sense a change in the interfacial charge as a result of a biorecognition.828−830 In the first study, such changes were monitored in a real-time by an open circuit potential measurement using an ultralow input bias current instrumentation amplifier.828 Three different peptide aptamers (pep2, pep6, and pep9) differing in peptide inserts and two analytes CDK2 and CDK4 (both present in complex cell lysates) were applied in the study. These label-free measurements were complemented by EIS assays with a soluble redox probe. Results indicated that a negative shift of an open circuit potential was consistent with an increase in Rct and a decrease in capacitance of the interfacial layer after analyte binding.828
The Davis group compared a bioanalytical performance of two different immobilized peptide aptamers with the performance of the immobilized antibody to detect C-reactive protein.834 Three detection techniques were applied in such a comparison, including fluorescent protein microarray, surface plasmon resonance, and EIS. In surface plasmon resonance and microarray configurations, antibody outperformed peptide aptamers from an analytical perspective. If EIS was applied for protein detection, increased assay sensitivity was observed for detection of C-reactive protein with immobilized peptide aptamers as compared to the device based on the immobilized antibody, but both biorecognition molecules were able to detect the analyte down to the 300 pM level.834
Three important challenges ahead in the field of multiplexed protein analysis have recently been postulated by Colas, including generation of highly stable, specific biorecognition molecules, production of high-density arrays, and a very sensitive detection platform.836 All of these challenges were addressed in a pioneering work from Walti’s group.832 The first challenge was addressed by utilization of peptide aptamers pep2 and pep9 based on a Stefin A triple mutant scaffold raised against proteins CDK2 and CDK4. Second, high density of immobilized peptide aptamers was achieved using an original masking/demasking procedure (Figure 37A).837 An array of 10 gold 20 μm wide microelectrodes 15 μm apart was first masked by a methyl-terminated polyethylene glycol-terminated thiol. On-demand immobilization of a peptide aptamer on electrode #1 within an array was performed in two steps. The first step involved application of a negative voltage of −1.4 V vs Ag/AgCl for 120 s where a reductive desorption of a thiol mask was applied on the electrode #1, while the rest of microelectrodes within an array were held at −0.2 V (Figure 37B). In the second step, a bare gold microelectrode #1 was covered by a peptide aptamer via a cysteine–gold bond (Figure 37C). The procedure could be repeated on the rest of gold microelectrodes to selectively pattern a whole array by different peptide aptamers (Figure 37D). The third challenge was addressed by an application of a label-free EIS, when a phase difference between an applied working potential and measured current, Φ(ω), was utilized for sensing purposes. A typical dependence of Φ(ω) as a function of applied frequency ω is shown in Figure 37E, indicating a binding of CDK2 to a pep9 modified surface, while a binding of CDK2 on pep2 and on a reference surface was negligible. The device, which was able to detect CDK2 in a clinically relevant concentration range from 25 pM to 100 nM, worked reliably well in a cell lysate. The authors concluded that biochip fabrication is scalable, enabling the generation of high-density, submicrometer electrode arrays, unlike the conventional printing pin-based technologies with resolutions of the order of 0.1 mm or more, for massively parallel, ultrasensitive, and label-free analysis of proteins present only in a single cell.832
Figure 37.
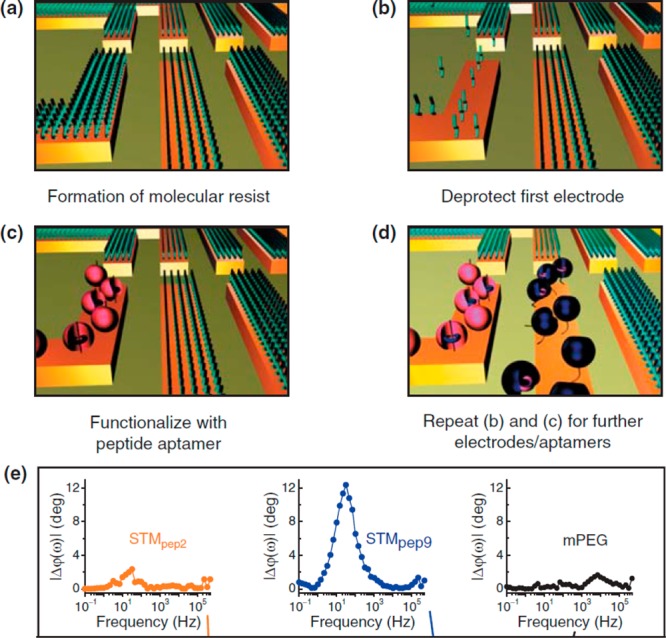
Electrochemically triggered immobilization of peptide aptamers within a biochip. (a) All microelectrodes are initially protected by a mask from mPEG, resisting protein adsorption. (b) Release of a mask from the electrode #1 by a highly negative voltage. (c) Funcionalization of the electrode #1 with a peptide aptamer. (d) Independent functionalization of the whole array by various peptide aptamers by repeating steps (a)–(c). (e) Analysis of CDK2 bound to pep2, pep9, or to a reference surface covered by mPEG as change in a phase shift, ϕ(ω), which is a phase difference between an applied working potential and measured current. A typical dependence of ϕ(ω) as a function of applied frequency ω used in the analysis is shown. mPEG is a methyl-terminated polyethylene glycol containing thiol. Reprinted with permission from ref (832). Copyright 2008 BioMed Central.
Even though peptide lectin aptamers have not been raised yet, it is possible that a restricted range of aa’s enriched in four aa types involved in glycan recognition by lectins will help to design such proteins with high selectivity and affinity toward glycoproteins in the future.
9.5. Analysis of Glycoprotein Biomarkers
In sections 9.2, 9.3, 9.4, we described strategies involving recognition of protein biomarkers by antibodies or aptamers, that is, strategies aimed at protein molecules themselves. In this section, we would like to pay attention to the application of EC methods in the analysis of glycoprotein biomarkers or in glycoprofiling of intact, mainly cancerous cells, utilizing recognition of glycan (nonprotein) part of the biomarker, as described more generally in section 8. Protein glycosylation, which is the most common post-translational modification in higher organisms including human, means that a wide range of different glycoproteins can be used as disease biomarkers for early detection of pathological processes. This is a relatively new, yet very interesting approach with a potential to develop assays for the detection of diseases such as diabetes, rheumatoid arthritis, infection by a Dengue virus, as well as various types of cancer.
9.5.1. Glycated Hemoglobin as a Diabetes Marker
Glycated hemoglobin is a marker of diabetes. In this disease, a continuously elevated concentration of glucose within the blood of patients with diabetes modifies the N-terminal valine of the β-chain of hemoglobin. Thus, monitoring of glycated hemoglobin can give information about a long-term progression of the disease, not influenced by a short-term variability in the glucose concentration. The clinical reference range is 4–20% of a glycated form of hemoglobin related to the total amount of hemoglobin with the amount above 7% indicating a pathological process. The 1% change in this percentage proportion corresponds to a fluctuation in mean blood glucose concentration of about 350 mg/L. Besides traditional methods of analysis of glycated hemoglobin,838 the EC way of determination can be a viable alternative. The first study for EC detection of glycated hemoglobin complexed with ferroceneboronic acid was published in 2008.839 The EC assay-based method could detect nanomolar level of glycated hemoglobin, and a 3-fold signal improvement was achieved using glucose oxidase immobilized on the electrode to enhance electron transfer rate between the electrode and a protein–ferrocene complex.839 Later, a poly(amidoamine) dendrimer layer formed on a modified gold electrode surface was applied to detect glycated hemoglobin with a percentage of 2.5–15% in a mixture with hemoglobin.840 Recently, glycated hemoglobin was successfully detected in whole blood. A blood sample was hemolyzed, and total hemoglobin was preconcentrated by Zn2+-induced precipitation and centrifugation to remove interfering glucose and glycoproteins including antibodies.841 The study applied the same transduction mechanism as discussed above.839
9.5.2. Rheumatoid Arthritis
Antibodies (particularly immunoglobulins G, IgG) circulating in the blood contain complex type N-glycans of a biantennary structure.842 IgG’s glycan is in healthy individuals often terminated with N-acetylneuraminic acid, and in patients with rheumatoid arthritis this glycan can be terminated in galactose or N-acetylglucosamine. The severity of the rheumatoid arthritis correlates with the extent of the IgG’s glycosylation change.520,644,843 Diluted serum samples from patients suffering from rheumatoid arthritis and healthy individual controls were analyzed by the EIS-based lectin biosensors.644 Three different lectins were covalently immobilized on SAM patterned gold surfaces to prepare three lectin biosensors for detection of sialic acid, galactose, and mannose/N-acetylglucosamine present on the surface of glycoproteins. The lectin biosensors could detect glycoproteins selectively down to the femtomolar level with the ability to distinguish between samples from healthy and sick individuals based on changes in the glycan composition on IgGs. Moreover, the lectin biosensors outperformed state-of-the-art lectin microarrays as a commonly used glycoprofiling tool in terms of wider linear range response and LOD. Thus, EIS-based lectin biosensors have a great potential for searching for new disease biomarkers, which are often present in complex samples at extremely low concentrations.644 A recent study suggests that other glycoproteins, such as ficolin 3, might be potential biomarkers for rheumatoid arthritis.844
9.5.3. Viral Glycoproteins
Dengue is a widely spread disease caused by RNA dengue virus. Four different virus serotypes can cause three clinically, pathologically, and epidemiologically distinct symptoms.845 EIS biosensor based on Con A lectin immobilized on a gold nanoparticles (AuNPs)-modified electrode exhibited different interactions with serum glycoproteins from patients suffering from the disease and control serum samples.845 Moreover, EIS-based Con A biosensor could distinguish the serum of healthy individuals from the serum of people suffering with different disease types by variations in RCT used for data evaluation of the biosensor.846 Moreover, when three parameters obtained from fitting EIS response by an equivalent circuit were plotted in a 3D graph, a distinct spatial localization of different samples within the graph was observed. In the next two studies, the authors managed to apply new lectins, CramoLL from Cratylia mollis(847) and BmoLL from Bauhinia monandra,848 in the detection of various dengue sera serotypes applicable in the analysis of an early stage of dengue virus infection.
Hong et al. prepared a nanostructured surface with immobilized Con A for analysis of Norovirus (causing gastroenteritis) viral particles in a sandwich format of analysis.849 The EC response (CV and EIS) of the biosensor was linear in the concentration range 102–106 copies/mL with LOD in real samples (lettuce extract) of 60 copies/mL. Analysis was completed within 1 h, and the biosensor was able to distinguish between the Norovirus particles and hepatitis A and E viral particles, respectively, with selectivity of about 98%.849
9.5.4. Cancer
An early diagnosis of any type of cancer requires identification of cancer biomarkers, an effort feasible to be achieved only by a multidisciplinary approach relying on an array of strategies, technologies, and methods.569,850,851 Such studies revealed that in cancer tissues, glycan structures of glycoproteins are altered. There are only few cancer glycoprotein biomarkers approved by the U.S. Food and Drug Administration for diagnostics of various cancer types (Table 3), but the reliability of many of these is questionable.
9.5.4.1. P-Glycoprotein
A multidrug resistance is the main reason for the limiting efficiency of chemotherapy to treat cancer. Resistance is the result of overexpression of few proteins, and among them the most important appears P-glycoprotein, a protein believed to function as an energy-dependent drug efflux pump lowering intracellular drug level.852 Thus, evaluation of the P-glycoprotein level on the cell membrane can predict efficiency of chemotherapy.684 Huangxian Ju’s group detected P-glycoprotein directly in an immunoassay format on the cell surface of K562/ADM leukemic cells with a primary monoclonal antibody followed by incubation with a secondary antibody (against primary antibody) conjugated with AP enzyme.684 The same experimental setup with a primary and a secondary antibody was later applied for analysis of human gastric BGC823 carcinoma cells via detection of P-glycoprotein.685 In this case, AP made an insoluble product on an electrode surface by hydrolysis of 5-bromo-4-chloro-3-indolyl phosphate, which made the DPV assay more sensitive. Interestingly, this approach was applied to calculate the number of P-glycoproteins as many as 4.7 × 107 molecules per one cell.685 Huangxian Ju’s group later applied EIS for a label-free detection of P-glycoprotein directly on the surface of K562/ADM leukemic cells by their incubation on the GCE modified by antibody against this protein.853 All of these assay protocols for detection of P-glycoprotein relied on antibodies, and comparison of cancer cell lines with other cells was not done.
The first paper using immobilized Con A lectin for attachment of cancerous HeLa cells with subsequent biorecognition of P-glycoprotein by HRP-labeled antibody was published in 2010.681 The DPV signal read at −250 mV vs SCE was generated by the action of HRP in the presence of H2O2 on the thionine-adsorbed surface, and the method allowed estimation of 4.0 × 1010 mannose moieties and 8.5 × 106 molecules of P-glycoprotein, respectively, on each HeLa cell. Interestingly, reversal of a multidrug resistant leukemia cell K562/ADM into a drug resistant one by addition of limonin agent was monitored by EC means. After addition of limonin into suspension of cancer cells, the efflux of an anticancer drug kaempferol from the cells decreased. Because this drug is redox active, its extracellular concentration was monitored by DPV at 150 mV vs SCE in a convenient way.854
9.5.4.2. PSA
Most EC studies on PSA detection were performed in an immunoassay format not involving recognition of glycan moieties (section 9.2). Recent studies, however, suggest that PSA from sera of patients with prostate cancer contain glycan with a higher amount of sialic acid as compared to PSA from healthy individuals or those having benign prostatic hyperplasia.543 Moreover, it was shown that a relative abundance of glycosylated isoforms of PSA may provide useful additional information for clinically relevant investigations.855 Despite these efforts, no EC-based biosensor so far has been described for the detection of PSA via recognition of a glycan moiety of PSA.
9.5.4.3. Carcinoembryonic Antigen
CEA is one of the most widely used tumor biomarkers worldwide and the most often utilized biomarker of colorectal cancer. Although it was shown that CEA (or a CEA-like protein) is present in healthy people, its concentration in people affected by cancer is approximately 60-fold higher as compared to healthy individuals.856 CEA is considered to be a biomarker also for other kinds of cancer including breast, lung, and pancreatic cancer.543
Again, we will discuss here methods of CEA detection relying on or involving (at least to some extent) its glycan part. For instance, EC immunosensor for analysis of CEA was built up on a layer of boronic acid containing SAM layer for selective attachment of CEA via its glycan moiety. In the next step, an anti-CEA (HRP-labeled) antibody was added. Finally, two different detection strategies were employed: either EIS assays or DPV technique detecting thionine via HRP at an applied potential of −210 mV. Both assay protocols allowed the detection of CEA down to 1 ng/mL (∼10 pM) concentration.857
An elegant strategy on how to detect a drug to cure a colorectal cancer, cetuximab, together with an analysis of a disease marker CEA in a single square wave voltammogram was based on the displacement of QD-labeled glycans by the target glycans using two different lectins, Con A and Euonymus europaeus lectin, as the recognition element.705 Upon incubation with the sample containing both the drug and the biomarker, QDs conjugates (ZnO/CEA and CdSe/cetuximab) were displaced from the surface, and released Zn2+ and Cd2+ ions were analyzed by stripping voltammetry at −1100 or −700 mV, respectively, vs SCE on a single GCE. Both glycoproteins were detected down to a concentration 1 order of magnitude lower than a required diagnostic cutoff value for both proteins.705 Even though this suspension array was not applied in real sample analysis, the authors suggested that this detection platform for simultaneous analysis of 5–6 proteins in a single run is possible.
CEA as a cancer biomarker was detected together with epithelial cell adhesion molecules directly on the surface of circulating cancer cells with antibodies attached to gold nanoparticles, while other circulating cells were not affected. EC detection of circulating cancer cells expressing both antigens was performed by the aid of an excellent electrocatalytic activity of gold nanoparticles toward hydrogen evolution detected chronoamperometrically at a negative voltage.858
9.5.4.4. α-Fetoprotein
The α-fetoprotein (AFP) is present in different forms of cancer and contains biantennary glycan chains of a complex type.575 AFP was detected with a label-free EIS-based biosensor with wheat-germ agglutinin immobilized covalently on oxidized SWCNTs deposited on a screen-printed carbon electrode down to the femtomolar level.859 Glycan composition of AFP was probed by four other lectins for detection of all carbohydrates present in the glycan, that is, mannose, sialic acid, galactose, N-acetylglucosamine, and N-acetylgalactosamine. Serum samples from healthy individuals and those suffering from cancer were diluted 1:100, and glycoprofiling showed changes in glycan composition as a result of liver cancer.859 It was concluded that samples from patients with cancer contain a higher amount of fucose and sialic acid on glycoproteins (such as AFP), as compared to samples from healthy individuals. This makes the strategy applicable for clinical diagnosis of an early stage of cancer.859
9.5.5. Analysis of Intact Cancerous Cells
9.5.5.1. Gastric Carcinoma Cells
An extremely sensitive lectin-based biosensor was prepared through attachment of gastric carcinoma cells on the surface of SWCNT-coated GCE modified by RGDS peptide, applied for nonspecific attachment of any type of cells via integrin receptors.860 Mannosyl groups present on the surface of cells were profiled by incubation with HRP-labeled Con A lectin, and the EC signal was generated by the oxidation of o-phenylendiamine by HRP in the presence of H2O2 using DPV (Figure 38). Both SWCNTs and HRP amplified the EC signal considerably with an ability to detect only 620 cells/mL or as few as 6 cells present in the 10 μL of the assayed liquid. The cells could be detected in a concentration up to 1 × 107 cells/mL, and an estimated amount of mannose molecules was 5.3 × 107 mannoses/cell.860
Figure 38.
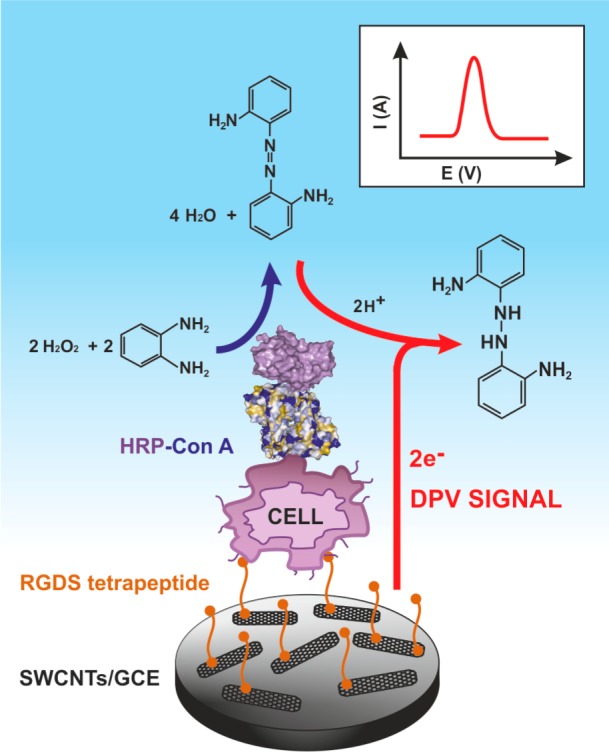
Analysis of a gastric carcinoma cell line after a nonspecific attachment of cells to a RGDS peptide. In the following step, a Con A–HRP conjugate was injected to probe glycans on the cell surface. A DPV signal was acquired by analysis of a product of oxidation of o-phenylendiamine by HRP in the presence of hydrogen peroxide. Adapted with permission from ref (860). Copyright 2008 American Chemical Society.
9.5.5.2. Hepatoma/Liver Carcinoma Cells
A human hepatoma carcinoma SMMC-7721 cell line was analyzed using photosensitive CdS-polyamidoamine nanocomposite film prepared by an electrodeposition method.861 In the presence of ascorbate, the photoexcitation of this modified electrode held at a potential of 0 V vs Ag/AgCl led to an anodic photocurrent. Con A lectin was immobilized on the surface of modified ITO electrode covalently via glutaraldehyde, and after the cells were bound to Con A, a change of the current could be applied for a signal reading with a LOD down to 5 × 103 cells/mL.861
A human liver Bel-7404 cancer cell line was detected by EIS sensing with Con A immobilized directly on a gold electrode, achieving LOD of 234 cells/mL.862 Much lower change of Rct was observed when the biosensor was incubated with normal liver cell line L02, because these cells contain a lower amount of a membrane-associated glycoprotein gp43. Finally, the biosensor exhibited a high reliability of sensing with a recovery index of 97–100%.862
9.5.5.3. Human Leukemic Cell Lines
Two different leukemic cell lines were analyzed in numerous studies with Con A lectin almost exclusively applied for biorecognition and quantitation of a number of mannose residues present on the surface of such cells. Human leukemic K562 cell line was detected by incubation with Con A covalently labeled by ferrocene carboxylic acid.708 These cells were detected down to 3 × 103 cells/mL, and EC assay protocol agreed with results obtained from flow cytometric detection.708 Later, a similar concept of analysis of K562 cells was based on Au nanoparticles loaded with Con A and ferrocene moieties.709 The Con A immobilized on a mixed SAM film recognized the cells, and the sandwich assay protocol was completed by incubation of a cell layer with modified gold nanoparticles. The biosensor could detect as few as 73 cells/mL.709 The same cell line was analyzed on a carbon nanohorn-modified GCE interfaced with RGDS peptide.682 A gold nanoparticle loaded with Con A and HRP formed a sandwich configuration for analysis of cells down to a concentration of 1.5 × 103 cells/mL.682 A sandwich configuration combined with electrochemiluminescent detection was applied for analysis of the K562 cell line.863 GCE modified by MWCNTs was applied for covalent immobilization of Con A, and after incubation with cells, a nanoprobe containing gold nanoparticles, Ru(bipy)32+-doped silica nanoparticles, and Con A was applied for signal generation. K562 cells could be detected down to 600 cells/mL. The biosensor was successfully applied to monitor dynamic changes in the expression of glycans on cell surface during a growth phase and to detect variation in the glycan composition as a result of an inhibitor action.863
Another sandwich configuration with electrochemiluminescent detection protocol allowed the detection of K562 cells down to 46 cells/mL.864 A screen-printed carbon electrode modified by a nanoporous gold film was applied for DNA aptamer immobilization.864 Carbon QDs deposited on ZnO nanospheres conjugated to Con A formed a sandwich configuration after cells were bound to the DNA aptamer. The biosensor reliability was proved by a recovery index between 92% and 106%, and the biosensor could be regenerated.864 EIS-based biosensor built on a GCE modified by MWCNTs with immobilized Con A was applied for the analysis of a K562 cell line down to a concentration of 1 × 104 cells/mL.865 The EC method was applied for monitoring of a dynamic glycan variation on K562 cells as a response to their exposure to drugs.865
In another approach, the EIS assay of K562 cells was integrated into a microfluidic array platform of three different immobilized lectins.866 ITO electrodes were used for EIS detection of a binding event, but also for cell counting using optical microscopy. Both methods of analysis showed that the best lectin for binding of K562 cells is wheat germ agglutinin, followed by Con A and PNA. The optical-EC microfluidic biochip was employed to evaluate the composition of cell surface glycans in response to the 3′-azido-3′-deoxythymidine drug.866
Human leukemic HL-60 cell lines were also detected in a sandwich configuration.704 Annexin V deposited on a 3D matrix formed on GCE through a modification with nitrogen-doped CNTs, and gold nanoparticles were utilized to capture human cells. In the next step, silica particles modified by few layers of CdTe QDs and Con A were injected over the cell surface for a signal generation. A stripping voltammetry applied for the detection of Cd2+ exhibited LOD of 1 × 103 cells/mL (48 apoptotic HL-60 cells) with a successful validation by a flow cytometry.704 Glycoprofiling of K562 cells can be done on lab-on-a-paper devices containing 3-D macroporous Au-paper electrode with EC or electrochemiluminescent detection.867 A sandwich format of analysis with application of lectins exhibited LOD of 4 cells with linear range spanning from 550 to 2.0 × 107 cells/mL.867 In the second case, aptamer-modified surface and Con A-conjugated porous AuPd alloy nanoparticles as nanolabels were applied for electrochemiluminescent signal readout system.868 This microfluidic paper-based origami cyto-device was applied in the analysis of K562 cells down to a concentration of 250 cells/mL.868
9.5.5.4. Intestinal/Colon Human Carcinoma Cells
A novel EC method for the detection of cancer cells was based on deposition of a quinone derivative on a gold electrode via gold–sulfur surface chemistry, making the electrode redox active.869 The DNA aptamer against lectin l-selectin then was adsorbed on the electrode, “shielding” a quinone redox activity, which resulted in a decreased current. In the subsequent step, incubation of the biosensor with l-selectin pushed a DNA aptamer away from the electrode, partly restoring redox activity of a quinone moiety. When intestinal human carcinoma LS180 cells were incubated with the biosensor, l-selectin was withdrawn by cells, leaving the surface of the biosensor blocked by the DNA aptamer. Thus, a decrease of the current response was again detected after cells were bound to l-selectin. The carcinoma cells were detected down to 1 × 103 cells/mL.869
9.5.5.5. Human Lung Cancer Cells
Quite a complex assay protocol was applied to achieve LOD down to 12 cell/mL for a lung cancer H1299 cell line.870 The assay procedure relied on CNT and gold nanoparticle-modified GCE interface with immobilized Con A. Such a surface was preincubated with CNTs loaded with gold nanoparticles, thionine (a redox probe), and mannose units. When the device was incubated with cancerous cells, the hybrid nanocomposite was displaced from the surface by the cells with a decrease in the current as a result of a lower amount of thionine present on the electrode surface (Figure 39). The 95-D cell line containing 2.8 × 108 mannose units per cell could be detected down to 580 cells/mL, while the H1299 cell line having 3.1 × 1010 mannose units per cell could be detected with unprecedented LOD of 12 cells/mL.870
Figure 39.
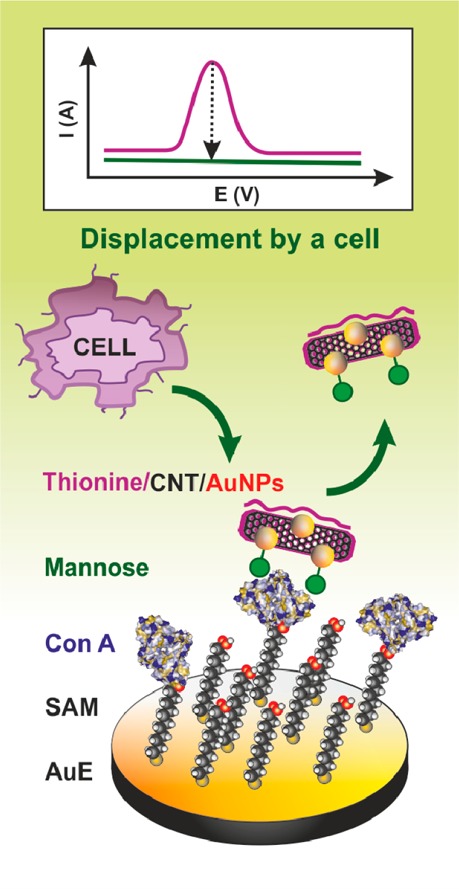
A displacement strategy for analysis of cells. Con A was immobilized on SAM layer by covalent coupling. A thionine/carbon nanotube (CNT)/gold nanoparticles (AuNPs) nanocomposite was then incubated with the biosensor, and displacement of the nanocomposite from the surface of the Con A biosensor after incubation with cells resulted in a decrease of EC response. Adapted with permission from ref (870). Copyright 2011 Royal Society of Chemistry.
9.5.5.6. Human Gastric Cancer Cells
Human gastric cancer cells were detected by an interesting competitive approach down to a concentration of 1.2 × 103 cells/mL.871 GCE was covered with CdS QDs and a mannan (mannose-containing polysaccharide) to selectively detect Con A lectin. First, cells were incubated with Con A in a solution, and then the solution was incubated with the GCE-modified surface to detect freely available residual Con A molecules not bound to the cells. After binding of Con A to the modified surface, electrochemiluminescent signal decreased proportionally with increasing amount of freely available Con A and thus increased with increasing concentration of cells. These measurements suggested that every cell contains 8.7 × 107 glycan molecules.871
Two lectins, Sambucus nigra agglutinin (SNA, for sialic acid) and Con A (for mannose), were used for the monitoring of changes in the expression of mannose and sialic acid in normal and cancer cells derived from human lung, liver, and prostate tissues.872 The lectins were covalently immobilized on the modified GCE, and after attachment of cells, a sandwich was completed by incubation of the surface with nanoparticles loaded by the lectin and thionine as a redox probe. Analysis of three different types of lung cancerous cells showed an increased expression of sialic acid on the cell surface, while a decrease in the amount of mannose units on the surface of the cell was observed at the same time, when compared to a normal cell line. The same glycosylation changes were detected on two different liver cancerous cell lines, as compared to normal cells. In the case of prostate cancer, a cancerous cell line showed an increase in the amount of both sialic acid and mannose, as compared to normal cells.872
Analysis of cancer CCRF-CEM cell line was possible with application of a supersandwich strategy for the amplification of signal.873 In this case, LOD of 50 cells/mL was achieved. GCE was patterned by CNTs, gold nanoparticles, and Con A to complete a receptive layer. The cells were incubated with this biosensor surface, and in a subsequent step a supersandwich was formed by binding of cells to DNA concatamer containing CdTe QDs. DNA concatamer with multiple target molecules and signal probes was formed by hybridization of three different oligonucleotide chains (Figure 40). Finally, cancer cells were detected by anodic stripping voltammetry of Cd2+ ions. A supersandwich biosensor configuration was 5.6-fold more sensitive as compared to a biosensor configuration using only a DNA aptamer–QD conjugate.873
Figure 40.
A supersandwich strategy for signal amplification in cancer cell analysis. Initially, GCE was modified by oxidized MWCNTs, dopamine, and AuNPs to which Con A was immobilized for detection of a CCRF-CEM cancerous cell line. DNA concatamer was first assembled from a capture DNA-aptamer (CDNA), a CdTe QD-labeled signal DNA (SDNA), and an auxiliary DNA (ADNA). When the cells were attached to the surface, they were probed by a preassembled concatamer, and finally a binding event was detected by an anodic stripping voltammetry of Cd2+. Adapted with permission from ref (873). Copyright 2013 American Chemical Society.
CCRF-CEM cells can be detected with a LOD of 10 cells/mL on GCE modified with a poly(amidoamine) dendrimer on a reduced graphene oxide (rGO) nanocomposite.874 After immobilization of Con A lectin and blocking with BSA, cells were successfully bound to a surface, and the DPV signal was amplified by application of AuNPs/aptamer/HRP nanoprobe in a sandwich format of analysis. In addition, at a concentration of 5.0 × 104 cells/mL, this biosensor clearly distinguished the CCRF-CEM from five other types of cell lines used in the study.874 The same cell line was detected in a sandwich configuration with LOD of 38 cells/mL using the biosensor having aptamer immobilized on a rGO-dendrimer-modified surface with electroluminescent detection.875
9.6. Active Glycoprofiling by Microengines
A remarkable way for active glycoprofiling of bacterial species in a wide range of environments was developed by Joseph Wang.876 Nanowire-based microengine with attached Con A lectin was self-propelled by the formation of oxygen bubbles from hydrogen peroxide as a fuel. The device was able to selectively pick up Escherichia coli cells, transport them, and finally release them by a change of pH.876 Alternatively, cargo could be on demand released by the interaction with saccharide.877 The device can be loaded with other cargos besides bacterial cells including polymeric drug carrying spheres having a therapeutic effect.876 The device can actively isolate microbial cells, when Con A lectin was substituted by boronic acid-containing film.878 Toxicity of hydrogen peroxide as a fuel was overcome, when magnetic field879 or ultrasound880 was applied to propel microengines. However, practical use and selectivity of such active glycoprofiling has yet to be proved.
9.7. Concluding Remarks
Although many EC strategies were developed for highly sensitive detection of various important biomarkers with good accuracy and selectivity, not many of them were actually validated against patient samples. Moreover, especially in cancer diagnostics it will be important to detect simultaneously a panel of 4–10 biomarkers to obtain more reliable results.53 EC methods are well-suited for point-of-care diagnostics, requiring inexpensive and simple instrumentation, which is easy to operate. EC sensors and assays for determination of protein biomarkers of cancer, AIDS, diabetes, or rheumatoid arthritis were reported mostly on the basis of application of antibodies, aptamers, or lectins for their capturing, often combined with nanomaterials to greatly enhance the sensitivity of the method. Despite all of these reports, much more work will be necessary to develop a reliable and commercially successful EC device for point-of-care diagnostics, applicable to clinical samples.
10. Conclusions
In this Review, we show that EC analysis of proteins is not limited to redox signals of nonprotein components of conjugated proteins and that some aa residues in proteins can contribute to protein reduction or oxidation signals. The development of a label-free EC method for analysis of practically all proteins represents a great challenge for electrochemistry to enter wide fields of proteomics and complement standard methods. High sensitivity of EC methods, simple miniaturization, and formation of chips for parallel analysis are among the advantages of EC analysis. These advantages are gradually finding ground in biomedicine. Very recently it has been shown899 that glycation (section 9.5.1) of BSA results in a decrease or disappearance of electrocatalytic peak H (section 5). Moreover, we show that EC analysis can be applied also in glycomics to analyze glycans directly in glycoproteins on the cell surface or after their isolation. In this direction, perhaps the greatest progress has been done in EIS detection of glycoprotein interactions with specific lectins. Moreover, it has been shown that some glucosamine-containing poly- and oligosaccharides are electroactive under conditions close to physiological and that most polysaccharides and glycans can be transformed into electrochemically active substances by a simple chemical modification. Detection of protein biomarkers is only briefly summarized, but special attention is paid to glycoprotein biomarkers and particularly to their glycan parts. It appears that finding differences in protein glycosylations may significantly increase the specificity of some biomarkers. Detection of a single biomarker usually provides only a very low specificity. Therefore, usually 4–10 biomarkers have to be detected to obtain good specificity and selectivity of detection. EC detection appears particularly advantageous for the preparation of low-density chips with this number of biomarkers.
An important take-home message is that, when considering protein EC analysis, not only voltammetry and EIS but also chronopotentiometry should be taken into account. Now it is clear that the old chronopotentiometry combined with adsorptive stripping, thiol-modified electrodes, and present instrumentation is particularly advantageous in protein structure-sensitive as well in DNA–protein complexes analyses. The combination of high current densities with high-electron yield electrocatalytic processes allows very fast potential changes, which can be utilized in studies of protein molecules and protein complex stability at electrode surfaces. Further development of this rapidly growing field may show the great usefulness of EC methods for biochemistry and molecular biology.
Acknowledgments
We are indebted to Dr. H. Cernocka, Dr. V. Dorcak, and Dr. Z. Pechan for critical reading of the manuscript. This work was supported by the Czech Science Foundation projects P301/11/2055 and 15-15479S (to E.P.), 14-24931P (to M.B.), 13-00956S (to V.O.), 13-00774S (to J.P.), by the European Regional Development Fund and the State Budget of the Czech Republic (RECAMO, CZ.1.05/2.1.00/03.0101), MH CZ - DRO (MMCI, 00209805), by Institutional support of Central European Institute of Technology (CZ.1.05/1.1.00/02.0068) to J.P., Slovak research and development agency APVV 0282-11 (to J.T. and T.B.), the European Research Council under the European Union’s Seventh Framework Program (FP/2007-2013)/ERC grant agreement no. 311532 (to J.T. and T.B.), the European Union’s Seventh Framework Programme for research, technological development and demonstration under grant agreement no. 317420 (to J.T.), and the National Priorities Research Program (Qatar National Research Fund), NPRP 6-381-1-078 (to J.T. and T.B.).
Glossary
Abbreviations
- aa
amino acid
- Aβ
amyloid β
- Ab
antibody
- ac
alternating current
- AFP
α-fetoprotein
- AgSAE
silver solid amalgam electrode
- AP
alkaline phosphatase
- Arg
arginine
- AS
α-synuclein
- Asp
aspartic acid
- AT
angiotensin
- ATP
adenosine triphosphate
- AuNP
gold nanoparticles
- BER
base excision repair
- BSA
bovine serum albumin
- CEA
carcinoembryonic antigen
- CHER
catalytic hydrogen evolution reaction
- CNT
carbon nanotube
- CON
consensus sequence
- Con A
lectin from Canavalia ensiformis
- CPNTs
carboxylated polypyrrole nanotubes
- CP
constant current chronopotentiometry
- CPS
constant current chronopotentiometric stripping
- CramoLL
lectin from Cratylia mollis
- Cys
cysteine
- CV
cyclic voltammetry
- CT
charge transport
- d.c
direct current
- DET
direct electron transfer
- DNA
DNA
- DNase I
deoxyribonuclease I
- DPV
differential pulse voltammetry
- ds
double-stranded
- DST
double-surface technique
- DTT
dithiothreitol
- EA
accumulation potential
- EC
electrochemical
- ECIA
electrochemical immunosensor arrays
- EIS
electrochemical impedance spectroscopy
- ELISA
enzyme-linked immuno sorbent assay
- ELLA
enzyme-linked lectin assays
- EndoIII
endonuclease III
- EP
peak potential
- GCE
glassy carbon electrode
- His
histidine
- HMDE
hanging mercury drop electrode
- HOPGE
highly oriented pyrolytic graphite electrode
- HRP
horseradish peroxidase
- HSA
human serum albumin
- IgG
immunoglobulin G
- Istr
stripping current
- ITO
indium tin oxide
- Leu
leucine
- LOD
limit of detection
- Lys
lysine
- MALDI–TOF
matrix-assisted laser desorption-ionization–time-of-flight
- Met
methionine
- MS
mass spectrometry
- MUA
mercaptoundecanoid acid
- MWCNT
multiwalled carbon nanotube
- NKA
sodium–potassium pump
- OLS
oligosaccharides
- Os(VIII)bipy
osmium tetroxide, 2,2′-bipyridine
- p53CD
core domain of p53 protein
- PAD
pulsed amperometric detection
- pcz
potential of zero charge
- PS
polysaccharides
- PSA
prostate specific antigen
- QD
quantum dot
- RCA-60
ricin
- RNA
ribonucleic acid
- SAE
solid amalgam electrode
- SAM
self-assembled monolayer
- SCE
saturated calomel electrode
- ss
single-stranded
- SNA
Sambucus nigra agglutinin
- SSB
single-strand binding complex
- SWV
square wave voltammetry
- SWCNT
single-walled carbon nanotube
- TBP
TATA-box binding protein
- T-p53C
superstable variants of p53 core domain
- Tyr
tyrosine
- Trp
tryptophan
- wt
wild type
- XPD
Xeroderma pigmentosum factor D
Biographies

Emil Paleček received his Ph.D. in biochemistry from Masaryk University in Brno, Czechoslovakia, in 1959. After working 5 years at the Institute of Biophysics of the Czechoslovak Academy of Sciences in Brno, he was a postdoctoral fellow with Professor Julius Marmur at the Graduate Department of Biochemistry, Brandeis University, Waltham, MA (1962–63). In 1967, he founded the Department of Biophysics of Macromolecules at the Institute of Biophysics in Brno, and in 1969 he was promoted to Associate Professor. In 1989, he became a Corresponding Member of the Czechoslovak Academy of Sciences and in 1994 a Founding Member of the Learned Society of the Czech Republic. From 1993–1997, he was a Member of the Academy Council, and in 2001–2005, and from 2013, a Member of the Scientific Board of the Academy of Sciences of the Czech Republic. He is Full Professor of Molecular Biology and Honorary Member of the Bioelectrochemical Society. Recently he was awarded the Medal of the Senate (Upper Chamber) of the Parliament of the Czech Republic and the State Prize “Czech Head” for his achievements in science. His research interests are in the structure and chemical reactivity of nucleic acids and in the electrochemistry of biomacromolecules and electrochemical biosensors.

Ján Tkáč received his Ph.D. degree in biotechnology in 2000 from Slovak University of Technology in Bratislava, Slovakia, and D.Sc. degree in analytical chemistry in 2011 from Slovak Academy of Sciences in Bratislava, Slovakia. He has done a few postdoctoral stays at Linkoping University, Sweden (2001–2003), Lund University, Sweden (2003–2006), and Oxford University, UK (2006–2008). Currently, he is a Head of Department of Glycobiotechnology and Head of the scientific council at the Institute of Chemistry, Slovak Academy of Sciences in Bratislava, Slovakia. His research activities cover label-free and label-based platforms of detection, nanoscale surface patterning protocols, catalytic biosensors and biofuel cells, glycan biochips/biosensors, and lectin/antibody-based affinity biosensors/biochips. He was the recipient of an Individual Marie-Curie Fellowship (2003–2006), and currently he is the holder of an ERC Starting Grant (2013–2017).

Martin Bartošík received his Ph.D. degree in biophysics at Masaryk University in cooperation with the Institute of Biophysics, both in Brno, Czech Republic. He is currently a junior scientist at the Masaryk Memorial Cancer Institute (Brno), where his research interests include electrochemical bioassays for detection of cancer biomarkers and study of antitumor drugs properties. He spent 5 months at the Department of Nanoengineering at the University of California, San Diego, where he was engaged in the construction of DNA hybridization chips and arrays.

Tomáš Bertók obtained M.Sc. (2010) and Ph.D. degrees (2014) at the Faculty of Chemical and Food Technology, Slovak University of Technology in Bratislava, Slovakia. His work is focused on the preparation of nanostructured biointerfaces integrated into biosensors/biochips with subsequent application in clinical diagnostics and glycomics. He is actively publishing in the area of biosensors and bioanalytical methods and working at the Institute of Chemistry, Slovak Academy of Sciences in Bratislava, Slovakia, as a junior research scientist.

Veronika Ostatná defended her Ph.D. Thesis “Study of mechanism of DNA–proteins interaction at the surfaces” at Faculty of Mathematics, Physics and Informatics of Comenius University in Bratislava in 2006. She studied DNA–protein interactions (including aptamers) using electrochemical and other methods. She designed one of the first electrochemical aptamer-sensors. She has been working on Prof. Paleček’s team at the Institute of Biophysics AS CR since 2005. Her research interests include electrochemisty of proteins and DNA, protein/protein, and DNA/protein interactions, where some new interesting approaches were developed.

Jan Paleček received his Ph.D. in biochemistry at the University of Vienna, Austria, in 1997. He then spent 4 years of his postdoctoral training at the Institute of Biophysics in the lab of his father Professor Emil Paleček (Czech Republic), working on DNA-binding properties of the tumour suppressor p53 complex. In 2002, he started his research on another DNA-binding complex, SMC5/6, in the lab of Professor Alan R. Lehmann at University of Sussex (Genome Damage and Stability Centre, Brighton, UK). In 2006, he returned back to Czech Republic and founded his own group at the Masaryk University in Brno, where he was promoted to Associate Professor in 2012. His research interests are in architecture and function of protein complexes associated with chromatin.
The authors declare no competing financial interest.
References
- Yates J. R.; Osterman A. L. Chem. Rev. 2007, 107, 3363. [Google Scholar]
- Hawkins R. D.; Hon G. C.; Ren B. Nat. Rev. Genet. 2010, 11, 476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabilloud T.; Chevallet M.; Luche S.; Lelong C. J. Proteomics 2010, 73, 2064. [DOI] [PubMed] [Google Scholar]
- Fang X. P.; Balgley B. M.; Lee C. S. Electrophoresis 2009, 30, 3998. [DOI] [PubMed] [Google Scholar]
- Gstaiger M.; Aebersold R. Nat. Rev. Genet. 2009, 10, 617. [DOI] [PubMed] [Google Scholar]
- Aebersold R.; Goodlett D. R. Chem. Rev. 2001, 101, 269. [DOI] [PubMed] [Google Scholar]
- Aebersold R.; Mann M. Nature 2003, 422, 198. [DOI] [PubMed] [Google Scholar]
- Domon B.; Aebersold R. Science 2006, 312, 212. [DOI] [PubMed] [Google Scholar]
- Gorg A.; Weiss W.; Dunn M. J. Proteomics 2004, 4, 3665. [DOI] [PubMed] [Google Scholar]
- MacBeath G. Nat. Genet. 2002, 32, 526. [DOI] [PubMed] [Google Scholar]
- Stormo G. D.Introduction to Protein −DNA Interactions. Structure, Thermodynamics, and Bioinformatics; Cold Spring Harbor Laboratory Press: New York, 2013. [Google Scholar]
- Alley W. R.; Mann B. F.; Novotny M. V. Chem. Rev. 2013, 113, 2668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novotny M. V.; Alley W. R. Curr. Opin. Chem. Biol. 2013, 17, 832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billova S.; Palecek E. In Electrochemistry of Nucleic Acids and Proteins. Towards Electrochemical Sensors for Genomics and Proteomics; Palecek E., Scheller F., Wang J., Eds.; Elsevier: Amsterdam, 2005. [Google Scholar]
- Abu Salah K.; Alrokyan S. A.; Khan M. N.; Ansari A. A. Sensors 2010, 10, 963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonanni A.; del Valle M. Anal. Chim. Acta 2010, 678, 7. [DOI] [PubMed] [Google Scholar]
- Carrara S. Sensors 2010, 10, 526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genereux J. C.; Boal A. K.; Barton J. K. J. Am. Chem. Soc. 2010, 132, 891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hocek M.; Fojta M. Chem. Soc. Rev. 2011, 40, 5802. [DOI] [PubMed] [Google Scholar]
- Hvastkovs E. G.; Buttry D. A. Analyst 2010, 135, 1817. [DOI] [PubMed] [Google Scholar]
- Choi S.; Goryll M.; Sin L. Y. M.; Wong P. K.; Chae J. Microfluid. Nanofluid. 2011, 10, 231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs C. B.; Peairs M. J.; Venton B. J. Anal. Chim. Acta 2010, 662, 105. [DOI] [PubMed] [Google Scholar]
- Li D.; Song S. P.; Fan C. H. Acc. Chem. Res. 2010, 43, 631. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Kim E.; Ghodssi R.; Rubloff G. W.; Culver J. N.; Bentley W. E.; Payne G. F. Biofabrication 2010, 2, 022002. [DOI] [PubMed] [Google Scholar]
- Lubin A. A.; Plaxco K. W. Acc. Chem. Res. 2010, 43, 496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mir M.; Homs A.; Samitier J. Electrophoresis 2009, 30, 3386. [DOI] [PubMed] [Google Scholar]
- Mir M.; Martinez-Rodriguez S.; Castillo-Fernandez O.; Homs-Corbera A.; Samitier J. Electrophoresis 2011, 32, 811. [DOI] [PubMed] [Google Scholar]
- Pedrero M.; Campuzano S.; Pingarron J. M. Anal. Methods 2011, 3, 780. [Google Scholar]
- Pumera M.; Ambrosi A.; Bonanni A.; Chng E. L. K.; Poh H. L. TrAC, Trends Anal. Chem. 2010, 29, 954. [Google Scholar]
- Shao Y. Y.; Wang J.; Wu H.; Liu J.; Aksay I. A.; Lin Y. H. Electroanalysis 2010, 22, 1027. [Google Scholar]
- Tosar J. P.; Branas G.; Laiz J. Biosens. Bioelectron. 2010, 26, 1205. [DOI] [PubMed] [Google Scholar]
- Wang J. ChemPhysChem 2009, 10, 1748. [DOI] [PubMed] [Google Scholar]
- Wei F.; Lillehoj P. B.; Ho C. M. Pediatr. Res. 2010, 67, 458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palecek E.; Bartosik M. Chem. Rev. 2012, 112, 3427. [DOI] [PubMed] [Google Scholar]
- Campuzano S.; Kuralay F.; Wang J. Electroanalysis 2012, 24, 483. [Google Scholar]
- Merkoci A. Electroanalysis 2013, 25, 15. [Google Scholar]
- Bertok T.; Katrlik J.; Gemeiner P.; Tkac J. Microchim. Acta 2013, 180, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham S.; Gerlach J. Q.; Kane M.; Joshi L. Analyst 2010, 135, 2471. [DOI] [PubMed] [Google Scholar]
- Gerlach J. Q.; Cunningham S.; Kane M.; Joshi L. Biochem. Soc. Trans. 2010, 38, 1333. [DOI] [PubMed] [Google Scholar]
- Reuel N. F.; Mu B.; Zhang J.; Hinckley A.; Strano M. S. Chem. Soc. Rev. 2012, 41, 5744. [DOI] [PubMed] [Google Scholar]
- Sanchez-Pomales G.; Zangmeister R. A. Int. J. Electrochem. 2011, 2011, 1. [Google Scholar]
- Zeng X.; Andrade C. S.; Oliveira M. L.; Sun X.-L. Anal. Bioanal. Chem. 2012, 402, 3161. [DOI] [PubMed] [Google Scholar]
- Sim S.; Asakura N. Electrochem. Commun. 2013, 34, 161. [Google Scholar]
- Shleev S.; Tkac J.; Christenson A.; Ruzgas T.; Yaropolov A. I.; Whittaker J. W.; Gorton L. Biosens. Bioelectron. 2005, 20, 2517. [DOI] [PubMed] [Google Scholar]
- Schneider E.; Clark D. S. Biosens. Bioelectron. 2013, 39, 1. [DOI] [PubMed] [Google Scholar]
- Ferapontova E. E.; Shleev S.; Ruzgas T.; Stoica L.; Christenson A.; Tkac J.; Yaropolov A. I.; Gorton L. In Electrochemistry of Nucleic Acids and Proteins – Towards Electrochemical Sensors for Genomics and Proteomics; Palecek E., Scheller F., Wang J., Eds.; Elsevier: Amsterdam, 2005. [Google Scholar]
- Armstrong F. A.; Butt J. N.; Sucheta A. Methods Enzymol. 1993, 227, 479. [DOI] [PubMed] [Google Scholar]
- Armstrong F. A.; Heering H. A.; Hirst J. Chem. Soc. Rev. 1997, 26, 169. [Google Scholar]
- Baldwin R. P. J. Pharm. Biomed. Anal. 1999, 19, 69. [DOI] [PubMed] [Google Scholar]
- Chikkaveeraiah B. V.; Mani V.; Patel V.; Gutkind J. S.; Rusling J. F. Biosens. Bioelectron. 2011, 26, 4477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo X. L.; Davis J. J. Chem. Soc. Rev. 2013, 42, 5944. [DOI] [PubMed] [Google Scholar]
- Rusling J. F. Anal. Chem. 2013, 85, 5304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusling J. F.; Kumar C. V.; Gutkind J. S.; Patel V. Analyst 2010, 135, 2496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brabec V.; Mornstein V. Biochim. Biophys. Acta 1980, 625, 43. [DOI] [PubMed] [Google Scholar]
- Brabec V.; Mornstein V. Biophys. Chem. 1980, 12, 159. [DOI] [PubMed] [Google Scholar]
- Malfoy B.; Reynaud J. A. J. Electroanal. Chem. 1980, 114, 213. [Google Scholar]
- Moss T.; Leblanc B.. DNA–Protein Interactions: Principles and Protocols; Humana Press: New York, 2009. [Google Scholar]
- Janata J. Chem. Rev. 2008, 108, 327. [DOI] [PubMed] [Google Scholar]
- Rasooly A.; Herold K. E. J. AOAC Int. 2006, 89, 873. [PubMed] [Google Scholar]
- Labuda J.; Brett A. M. O.; Evtugyn G.; Fojta M.; Mascini M.; Ozsoz M.; Palchetti I.; Palecek E.; Wang J. Pure Appl. Chem. 2010, 82, 1161. [Google Scholar]
- Heyrovsky J.; Babicka J. Collect. Czech. Chem. Commun. 1930, 2, 370. [Google Scholar]
- Herles F.; Vancura A. Bull. Int. Acad. Sci. Boheme 1932, 33, 119. [Google Scholar]
- Brdicka R. Collect. Czech. Chem. Commun. 1936, 8, 366. [Google Scholar]
- Brdicka R. Collect. Czech. Chem. Commun. 1933, 5, 112. [Google Scholar]
- Jurka E. Collect. Czech. Chem. Commun. 1939, 11, 243. [Google Scholar]
- Bednarski T. M.; J J. J. Am. Chem. Soc. 1967, 89, 1552. [DOI] [PubMed] [Google Scholar]
- Kolthoff I. M.; Yamashita K.; Tan B. J. Electroanal. Chem. 1975, 63, 393. [Google Scholar]
- Millar G. J. Biochem. J. 1953, 53, 385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Stackelberg M.; Hans W.; Jensch W. Z. Electrochem. 1958, 62, 839. [Google Scholar]
- Britz D. Int. J. Electrochem. Sci. 2006, 1, 379. [Google Scholar]
- Kalvoda R.Techniques of Oscillographic Polarography; Elsevier Publ. Co.: Amsterdam, 1965. [Google Scholar]
- Brezina M.; Zuman P.. Polarography in Medicine, Biochemistry and Pharmacy; Interscience: New York, 1958. [Google Scholar]
- Zuman P.; Palecek E. In Electrochemistry of Nucleic Acids and Proteins. Towards Electrochemical Sensors for Genomics and Proteomics; Palecek E., Scheller F., Wang J., Eds.; Elsevier: Amsterdam, 2005. [Google Scholar]
- Ruttkay-Nedecky G.; Takacova M.; Vesela V. In Charge and Field Effects in Biosystems; Allen M. J., Usherwood P. N. R., Eds.; Abacus Press: Wellingborough, 1984. [Google Scholar]
- Minevich I.; Tur’Yan Y. I. J. Solid State Electrochem. 2013, 17, 1529. [Google Scholar]
- Armstrong F. A. In Bioelectrochemistry; Wilson G. S., Ed.; Wiley-VCH: Weinheim, 2002; Vol. 9. [Google Scholar]
- Hammerich O.; Ulstrup J.. Bioinorganic Electrochemistry; Springer: Dordrecht, Netherlands, 2008. [Google Scholar]
- Wackerbarth H.; Zhang J.; Grubb M.; Glargaard H.; Ooi B. L.; Christensen H. E. M.; Ulstrup J. In Electrochemistry of Nucleic Acids and Proteins. Towards Electrochemical Sensors for Genomics and Proteomics; Palecek E., Scheller F., Wang J., Eds.; Elsevier: Amsterdam, 2005. [Google Scholar]
- Yu F.; Cangelosi V. M.; Zastrow M. L.; Tegoni M.; Plegaria J. S.; Tebo A. G.; Mocny C. S.; Ruckthong L.; Qayyum H.; Pecoraro V. L. Chem. Rev. 2014, 114, 3495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler J. R.; Gray H. B. Chem. Rev. 2013, 114, 3369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migliore A.; Polizzi N. F.; Therien M. J.; Beratan D. N. Chem. Rev. 2014, 114, 3381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartosík M.; Doneux T.; Pechan Z.; Ostatna V.; Palecek E. In Encyclopedia of Analytical Chemistry; Meyers R. E., Ed.; John Wiley and Sons: Chichester, 2011; Supplementary Vols. S1–S3. [Google Scholar]
- Layfield J. P.; Hammes-Schiffer S. Chem. Rev. 2013, 114, 3466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddowes M. J.; Hill H. A. O. J. Chem. Soc., Chem. Commun. 1977, 21, 771. [Google Scholar]
- Niki K.; Yagi T.; Inokuchi H.; Kimura K. J. Am. Chem. Soc. 1979, 101, 3335. [Google Scholar]
- Yeh P.; Kuwana T. Chem. Lett. 1977, 10, 1145. [Google Scholar]
- Winkler J. R.; Dunn A. R.; Hess C. R.; Gray H. B. In Bioinorganic Electrochemistry; Hammerich O., Ulstrup J., Eds.; Springer: Dordrecht, 2008. [Google Scholar]
- Willner B.; Willner I. In Bioinorganic Electrochemistry; Hammerich O., Ulstrup J., Eds.; Springer: Dordrecht, 2008. [Google Scholar]
- Butt J. N.; Armstrong F. A. In Bioinorganic Electrochemistry; Hammerich O., Ulstrup J., Eds.; Springer: Dordrecht, 2008. [Google Scholar]
- Warsinke A.; Stocklein W.; Leupold E.; Micheel E.; Scheller F. W. In Electrochemistry of Nucleic Acids and Proteins. Towards Electrochemical Sensors for Genomics and Proteomics; Palecek E., Scheller F., Wang J., Eds.; Elsevier: Amsterdam, 2005. [Google Scholar]
- Wollenberger U. In Comprehensive Analytical Chemistry; Gorton L., Ed.; Elsevier: Amsterdam, 2005. [Google Scholar]
- Armstrong F. A.; Wilson G. S. Electrochim. Acta 2000, 45, 2623. [Google Scholar]
- Ghindilis A. L.; Atanasov P.; Wilkins E. Electroanalysis 1997, 9, 661. [Google Scholar]
- Gorton L. Electroanalysis 1995, 7, 23. [Google Scholar]
- Léger C.; Bertrand P. Chem. Rev. 2008, 108, 2379. [DOI] [PubMed] [Google Scholar]
- Willner I.; Katz E. Angew. Chem., Int. Ed. 2000, 39, 1180. [DOI] [PubMed] [Google Scholar]
- Zhang J. D.; Kuznetsov A. M.; Medvedev I. G.; Chi Q. J.; Albrecht T.; Jensen P. S.; Ulstrup J. Chem. Rev. 2008, 108, 2737. [DOI] [PubMed] [Google Scholar]
- Zhang J. D.; Chi Q. J.; Hansen A. G.; Jensen P. S.; Salvatore P.; Ulstrup J. FEBS Lett. 2012, 586, 526. [DOI] [PubMed] [Google Scholar]
- Chi Q. J.; Zhang J. D.; Nielsen J. U.; Friis E. P.; Chorkendorff I.; Canters G. W.; Andersen J. E. T.; Ulstrup J. J. Am. Chem. Soc. 2000, 122, 4047. [Google Scholar]
- Winkler J. R.; Wittung-Stafshede P.; Leckner J.; Malmstrom B. G.; Gray H. B. Proc. Natl. Acad. Sci. U.S.A. 1997, 94, 4246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joerger A. C.; Allen M. D.; Fersht A. R. J. Biol. Chem. 2004, 279, 1291. [DOI] [PubMed] [Google Scholar]
- Joerger A. C.; Fersht A. R. Annu. Rev. Biochem. 2008, 77, 557. [DOI] [PubMed] [Google Scholar]
- Davison T. S.; Vagner C.; Kaghad M.; Ayed A.; Caput D.; Arrowsmith C. H. J. Biol. Chem. 1999, 274, 18709. [DOI] [PubMed] [Google Scholar]
- Senoo M.; Seki N.; Ohira M.; Sugano S.; Watanabe M.; Inuzuka S.; Okamoto T.; Tachibana M.; Tanaka T.; Shinkai Y.; Kato H. Biochem. Biophys. Res. Commun. 1998, 248, 603. [DOI] [PubMed] [Google Scholar]
- Palecek E.; Ostatna V.; Cernocka H.; Joerger A. C.; Fersht A. R. J. Am. Chem. Soc. 2011, 133, 7190. [DOI] [PubMed] [Google Scholar]
- David S. S.; Wiliams S. D. Chem. Rev. 1998, 98, 1221. [DOI] [PubMed] [Google Scholar]
- Breimer L. H.; Lindahl T. J. Biol. Chem. 1984, 259, 5543. [PubMed] [Google Scholar]
- Francis A. W.; David S. S. Biochemistry 2003, 42, 801. [DOI] [PubMed] [Google Scholar]
- Gerland U.; Moroz J. D.; Hwa T. Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 12015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gowers D. M.; Halford S. E. EMBO J. 2003, 22, 1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halford S. E.; Szczelkun M. D. Eur. Biophys. J. Biophys. Lett. 2002, 31, 257. [DOI] [PubMed] [Google Scholar]
- Sontz P. A.; Mui T. P.; Fuss J. O.; Tainer J. A.; Barton J. K. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham R. P.; Asahara H.; Bank J. F.; Scholes C. P.; Salerno J. C.; Surerus K.; Munck E.; McCracken J.; Peisach J.; Emptage M. H. Biochemistry 1989, 28, 4450. [DOI] [PubMed] [Google Scholar]
- Guan Y.; Manuel R. C.; Arvai A. S.; Parikh S. S.; Mol C. D.; Miller J. H.; Lloyd S.; Tainer J. A. Nat. Struct. Biol. 1998, 5, 1058. [DOI] [PubMed] [Google Scholar]
- Boorstein R. J.; Hilbert T. P.; Cadet J.; Cunningham R. P.; Teebor G. W. Biochemistry 1989, 28, 6164. [DOI] [PubMed] [Google Scholar]
- Demple B.; Linn S. Nature 1980, 287, 203. [DOI] [PubMed] [Google Scholar]
- Dizdaroglu M.; Laval J.; Boiteux S. Biochemistry 1993, 32, 12105. [DOI] [PubMed] [Google Scholar]
- Katcher H. L.; Wallace S. S. Biochemistry 1983, 22, 4071. [DOI] [PubMed] [Google Scholar]
- Kuo C. F.; McRee D. E.; Fisher C. L.; Ohandley S. F.; Cunningham R. P.; Tainer J. A. Science 1992, 258, 434. [DOI] [PubMed] [Google Scholar]
- Thayer M. M.; Ahern H.; Xing D.; Cunningham R. P.; Tainer J. A. EMBO J. 1995, 14, 4108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner J. R.; Blount B. C.; Weinfeld M. Anal. Biochem. 1996, 233, 76. [DOI] [PubMed] [Google Scholar]
- Weiss R. B.; Duker N. J. Nucleic Acids Res. 1986, 14, 6621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulychev N. V.; Varaprasad C. V.; Dorman G.; Miller J. H.; Eisenberg M.; Grollman A. P.; Johnson F. Biochemistry 1996, 35, 13147. [DOI] [PubMed] [Google Scholar]
- Francis A. W.; Helquist S. A.; Kool E. T.; David S. S. J. Am. Chem. Soc. 2003, 125, 16235. [DOI] [PubMed] [Google Scholar]
- Gogos A.; Cillo J.; Clarke N. D.; Lu A. L. Biochemistry 1996, 35, 16665. [DOI] [PubMed] [Google Scholar]
- Chepanoske C. L.; Porello S. L.; Fujiwara T.; Sugiyama H.; David S. S. Nucleic Acids Res. 1999, 27, 3197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu A. L.; Tsaiwu J. J.; Cillo J. J. Biol. Chem. 1995, 270, 23582. [DOI] [PubMed] [Google Scholar]
- Manuel R. C.; Czerwinski E. W.; Lloyd R. S. J. Biol. Chem. 1996, 271, 16218. [DOI] [PubMed] [Google Scholar]
- Michaels M. L.; Cruz C.; Grollman A. P.; Miller J. H. Proc. Natl. Acad. Sci. U.S.A. 1992, 89, 7022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaels M. L.; Miller J. H. J. Bacteriol. 1992, 174, 6321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaels M. L.; Tchou J.; Grollman A. P.; Miller J. H. Biochemistry 1992, 31, 10964. [DOI] [PubMed] [Google Scholar]
- Nghiem Y.; Cabrera M.; Cupples C. G.; Miller J. H. Proc. Natl. Acad. Sci. U.S.A. 1988, 85, 2709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidmar J. J.; Cupples C. G. Can. J. Microbiol. 1993, 39, 892. [DOI] [PubMed] [Google Scholar]
- Fromme J. C.; Banerjee A.; Huang S. J.; Verdine G. L. Nature 2004, 427, 652. [DOI] [PubMed] [Google Scholar]
- Fromme J. C.; Verdine G. L. EMBO J. 2003, 22, 3461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golinelli M. P.; Chmiel N. H.; David S. S. Biochemistry 1999, 38, 6997. [DOI] [PubMed] [Google Scholar]
- Messick T. E.; Chmiel N. H.; Golinelli M. P.; Langer M. R.; Joshua-Tor L.; David S. S. Biochemistry 2002, 41, 3931. [DOI] [PubMed] [Google Scholar]
- Porello S. L.; Cannon M. J.; David S. S. Biochemistry 1998, 37, 6465. [DOI] [PubMed] [Google Scholar]
- Asahara H.; Wistort P. M.; Bank J. F.; Bakerian R. H.; Cunningham R. P. Biochemistry 1989, 28, 4444. [DOI] [PubMed] [Google Scholar]
- Fu W. G.; Ohandley S.; Cunningham R. P.; Johnson M. K. J. Biol. Chem. 1992, 267, 16135. [PubMed] [Google Scholar]
- Ohandley S.; Scholes C. P.; Cunningham R. P. Biochemistry 1995, 34, 2528. [DOI] [PubMed] [Google Scholar]
- Xing D. X.; Dorr R.; Cunningham R. P.; Scholes C. P. Biochemistry 1995, 34, 2537. [DOI] [PubMed] [Google Scholar]
- Gorodetsky A. A.; Boal A. K.; Barton J. K. J. Am. Chem. Soc. 2006, 128, 12082. [DOI] [PubMed] [Google Scholar]
- Grodick M. A.; Segal H. M.; Zwang T. J.; Barton J. K. J. Am. Chem. Soc. 2014, 136, 6470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynaud J. A.; Malfoy B.; Canesson P. J. Electroanal. Chem. 1980, 114, 195. [Google Scholar]
- Brabec V.; Vetterl V.; Vrana O. In Experimental Techniques in Bioelectrochemistry; Brabec V., Walz D., Milazzo G., Eds.; Birghauser Verlag: Basel, 1996. [Google Scholar]
- Yang W. R.; Ratinac K. R.; Ringer S. P.; Thordarson P.; Gooding J. J.; Braet F. Angew. Chem., Int. Ed. 2010, 49, 2114. [DOI] [PubMed] [Google Scholar]
- Chen Y.; Vedala H.; Kotchey G. P.; Audfray A.; Cecioni S.; Imberty A.; Vidal S.; Star A. ACS Nano 2012, 6, 760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgakilas V.; Otyepka M.; Bourlinos A. B.; Chandra V.; Kim N.; Kemp K. C.; Hobza P.; Zboril R.; Kim K. S. Chem. Rev. 2012, 112, 6156. [DOI] [PubMed] [Google Scholar]
- Silva F. D. D.; da Silva M. G. A.; Lima P. R.; Meneghetti M. R.; Kubota L. T.; Goulart M. O. F. Biosens. Bioelectron. 2013, 50, 202. [DOI] [PubMed] [Google Scholar]
- Liu X.; Luo L. Q.; Ding Y. P.; Kang Z. P.; Ye D. X. Bioelectrochemistry 2012, 86, 38. [DOI] [PubMed] [Google Scholar]
- Enache T. A.; Oliveira-Brett A. M. Bioelectrochemistry 2011, 81, 46. [DOI] [PubMed] [Google Scholar]
- Herzog G.; Arrigan D. W. M. Analyst 2007, 132, 615. [DOI] [PubMed] [Google Scholar]
- Palecek E.; Jelen F.; Teijeiro C.; Fucík V.; Jovin T. M. Anal. Chim. Acta 1993, 273, 175. [Google Scholar]
- Palecek E.; Ostatna V. Electroanalysis 2007, 19, 2383. [Google Scholar]
- Veloso A.; Kerman K. Anal. Bioanal. Chem. 2013, 405, 5725. [DOI] [PubMed] [Google Scholar]
- Lopes P.; Dyrnesli H.; Lorenzen N.; Otzen D.; Ferapontova E. E. Analyst 2014, 139, 749. [DOI] [PubMed] [Google Scholar]
- Enache T. A.; Oliveira-Brett A. M. Bioelectrochemistry 2013, 89, 11. [DOI] [PubMed] [Google Scholar]
- Cai X. H.; Rivas G.; Farias P. A. M.; Shiraishi H.; Wang J.; Palecek E. Anal. Chim. Acta 1996, 332, 49. [Google Scholar]
- Wang J.; Rivas G.; Cai X. H.; Chicharro M.; Farias P. A. M.; Palecek E. Electroanalysis 1996, 8, 902. [Google Scholar]
- Finklea H. O. In Encyclopedia of Analytical Chemistry; Meyers R. A., Ed.; Wiley: New York, 2000; Vol. 11. [Google Scholar]
- Gorodetsky A. A.; Barton J. K. Langmuir 2006, 22, 7917. [DOI] [PubMed] [Google Scholar]
- Gorodetsky A. A.; Barton J. K. J. Am. Chem. Soc. 2007, 129, 6074. [DOI] [PubMed] [Google Scholar]
- Suprun E. V.; Zharkova M. S.; Morozevich G. E.; Veselovsky A. V.; Shumyantseva V. V.; Archakov A. I. Electroanalysis 2013, 25, 2109. [Google Scholar]
- Permentier H. P.; Bruins A. P. J. Am. Soc. Mass Spectrom. 2004, 15, 1707. [DOI] [PubMed] [Google Scholar]
- Permentier H. P.; Jurva U.; Barroso B.; Bruins A. P. Rapid Commun. Mass Spectrom. 2003, 17, 1585. [DOI] [PubMed] [Google Scholar]
- Roeser J.; Permentier H. P.; Bruins A. P.; Bischoff R. Anal. Chem. 2010, 82, 7556. [DOI] [PubMed] [Google Scholar]
- Enache T. A.; Oliveira-Brett A. M. Electroanalysis 2011, 23, 1337. [Google Scholar]
- Oliveira S. C. B.; Santarino I. B.; Oliveira-Brett A. M. Electroanalysis 2013, 25, 1029. [Google Scholar]
- Dorcak V.; Ostatna V.; Palecek E. Electrochem. Commun. 2013, 31, 80. [Google Scholar]
- Bell M. R.; Engleka M. J.; Malik A.; Strickler J. E. Protein Sci. 2013, 22, 1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley C.; Holtmann D.; Mangold K.-M.; Schrader J. Colloids Surf., B 2011, 88, 539. [DOI] [PubMed] [Google Scholar]
- Brychtova V.; Vojtesek B.; Hrstka R. Cancer Lett. 2011, 304, 1. [DOI] [PubMed] [Google Scholar]
- Ostatná V.; Vargová V.; Hrstka R.; Ďurech M.; Vojtěšek B.; Paleček E. Electrochim. Acta 2014, 150, 218. [Google Scholar]
- Sestakova I.; Kopanica M.; Havran L.; Palecek E. Electroanalysis 2000, 12, 100. [Google Scholar]
- Sestakova I.; Mader P. Cell. Mol. Biol. 2000, 46, 257. [PubMed] [Google Scholar]
- Berges J.; de Oliveira P.; Fourre I.; Houee-Levin C. J. Phys. Chem. B 2012, 116, 9352. [DOI] [PubMed] [Google Scholar]
- Chiku M.; Nakamura J.; Fujishima A.; Einaga Y. Anal. Chem. 2008, 80, 5783. [DOI] [PubMed] [Google Scholar]
- Federation of American scientists Biological Agent Fact Sheet–Ricin; http://www.fas.org/resource/08262005181228.pdf, October 21, 2014.
- Schweizerische Eidgenossenschaft, FACT SHEET Ricin; http://www.labor-spiez.ch/en/dok/fa/pdf/fact_sheet_ricin_2010_en.pdf, October 21, 2014.
- Ribeiro W. F.; da Costa D. J. E.; Lourenco A. S.; Lopes I. C.; de Medeiros E. P.; Salazar-Banda G. R.; do Nascimento V. B.; de Araujo M. C. U. Analyst 2013, 138, 4565. [DOI] [PubMed] [Google Scholar]
- Severino L. S.; Auld D. L.; Baldanzi M.; Candido M. J. D.; Chen G.; Crosby W.; Tan D.; He X.; Lakshmamma P.; Lavanya C.; Machado O. L. T.; Mielke T.; Milani M.; Miller T. D.; Morris J. B.; Morse S. A.; Navas A. A.; Soares D. J.; Sofiatti V.; Wang M. L.; Zanotto M. D.; Zieler H. Agron. J. 2012, 104, 853. [Google Scholar]
- Guo L. H.; Qu N. Anal. Chem. 2006, 78, 6275. [DOI] [PubMed] [Google Scholar]
- Chen Y. M.; Guo L. H. J. Environ. Sci. (Beijing, China) 2009, 21, 373. [DOI] [PubMed] [Google Scholar]
- Wei M. Y.; Guo L. H.; Famouri P. TrAC, Trends Anal. Chem. 2012, 39, 130. [Google Scholar]
- Yang Y.; Guo L. H.; Qu N.; Wei M. Y.; Zhao L. X.; Wan B. Biosens. Bioelectron. 2011, 28, 284. [DOI] [PubMed] [Google Scholar]
- Qu N.; Guo L. H.; Zhu B. Z. Biosens. Bioelectron. 2011, 26, 2292. [DOI] [PubMed] [Google Scholar]
- Sebolt-Leopold J. S.; English J. M. Nature 2006, 441, 457. [DOI] [PubMed] [Google Scholar]
- Cohen P. Nat. Rev. Drug Discovery 2002, 1, 309. [DOI] [PubMed] [Google Scholar]
- Flajolet M.; He G.; Heiman M.; Lin A.; Nairn A. C.; Greengard P. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 4159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dziadosz M.; Lessig R.; Bartels H. J. Chromatogr. B: Anal. Technol. Biomed. Life Sci. 2012, 893, 77. [DOI] [PubMed] [Google Scholar]
- Mori T.; Inamori K.; Inoue Y.; Han X.; Yamanouchi G.; Niidome T.; Katayama Y. Anal. Biochem. 2008, 375, 223. [DOI] [PubMed] [Google Scholar]
- Tan Y. J.; Sui D. X.; Wang W. H.; Kuo M. H.; Reid G. E.; Bruening M. L. Anal. Chem. 2013, 85, 5699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji J.; Yang H.; Liu Y.; Chen H.; Kong J. L.; Liu B. H. Chem. Commun. 2009, 12, 1508. [DOI] [PubMed] [Google Scholar]
- Martic S.; Beheshti S.; Rains M. K.; Kraatz H. B. Analyst 2012, 137, 2042. [DOI] [PubMed] [Google Scholar]
- Wang C. L.; Wei L. Y.; Yuan C. J.; Hwang K. C. Anal. Chem. 2012, 84, 971. [DOI] [PubMed] [Google Scholar]
- Wang J.; Cao Y.; Li Y.; Liang Z. Q.; Li G. X. J. Electroanal. Chem. 2011, 656, 274. [Google Scholar]
- Xu S. J.; Liu Y.; Wang T. H.; Li J. H. Anal. Chem. 2010, 82, 9566. [DOI] [PubMed] [Google Scholar]
- Xu X. H.; Nie Z.; Chen J. H.; Fu Y. C.; Li W.; Shen Q. P.; Yao S. Z. Chem. Commun. 2009, 45, 6946. [DOI] [PubMed] [Google Scholar]
- Kerman K.; Kraatz H. B. Biosens. Bioelectron. 2009, 24, 1484. [DOI] [PubMed] [Google Scholar]
- Kerman K.; Vestergaard M.; Tamiya E. Anal. Chem. 2007, 79, 6881. [DOI] [PubMed] [Google Scholar]
- Li B.; Shi X.; Gu W.; Zhao K.; Chen N.; Xian Y. Analyst 2013, 138, 7212. [DOI] [PubMed] [Google Scholar]
- Geim A. K.; Novoselov K. S. Nat. Mater. 2007, 6, 183. [DOI] [PubMed] [Google Scholar]
- Jiao L. Y.; Wang X. R.; Diankov G.; Wang H. L.; Dai H. J. Nat. Nanotechnol. 2010, 5, 321. [DOI] [PubMed] [Google Scholar]
- Shen J. F.; Hu Y. H.; Li C.; Qin C.; Ye M. X. Small 2009, 5, 82. [DOI] [PubMed] [Google Scholar]
- Janata J. ECS Solid State Lett. 2012, 1, M29. [Google Scholar]
- Ruttkay-Nedecky G.; Brabec V. Gen. Physiol. Biophys. 1985, 4, 393. [PubMed] [Google Scholar]
- Ostatna V.; Kuralay F.; Trnkova L.; Palecek E. Electroanalysis 2008, 20, 1406. [Google Scholar]
- Dorcak V.; Palecek E. Anal. Chem. 2009, 81, 1543. [DOI] [PubMed] [Google Scholar]
- Vestergaard M.; Kerman K.; Tamiya E. Sensors 2007, 7, 3442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H.; Jiang C. M.; Yu C.; Zhang S.; Liu B. H.; Kong J. L. Biosens. Bioelectron. 2009, 24, 3399. [DOI] [PubMed] [Google Scholar]
- Li J. P.; Li S. H.; Yang C. F. Electroanalysis 2012, 24, 2213. [Google Scholar]
- Aragay G.; Pons J.; Merkoci A. Chem. Rev. 2011, 111, 3433. [DOI] [PubMed] [Google Scholar]
- Banks C. E.; Compton R. G. Analyst 2006, 131, 15. [DOI] [PubMed] [Google Scholar]
- Centi S.; Laschi S.; Mascini M. Bioanalysis 2009, 1, 1271. [DOI] [PubMed] [Google Scholar]
- McCreery R. L. Chem. Rev. 2008, 108, 2646. [DOI] [PubMed] [Google Scholar]
- Svancara I.; Walcarius A.; Kalcher K.; Vytras K. Cent. Eur. J. Chem. 2009, 7, 598. [Google Scholar]
- Ostatna V.; Cernocka H.; Kurzatkowska K.; Palecek E. Anal. Chim. Acta 2012, 735, 31. [DOI] [PubMed] [Google Scholar]
- Chiku M.; Ivandini T. A.; Kamiya A.; Fujishima A.; Einaga Y. J. Electroanal. Chem. 2008, 612, 201. [Google Scholar]
- Topal B. D.; Özkan S. A.; Uslu B. J. Electroanal. Chem. 2014, 719, 14. [Google Scholar]
- Kurzatkowska K.; Ostatna V.; Hamley I. W.; Doneux T.; Palecek E. Electrochim. Acta 2013, 106, 43. [Google Scholar]
- Uversky V. N.; Eliezer D. Curr. Protein Pept. Sci. 2009, 10, 483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vestergaard M.; Kerman K.; Kim D. K.; Hiep H. M.; Tamiya E. Talanta 2008, 74, 1038. [DOI] [PubMed] [Google Scholar]
- Vestergaard M.; Kerman K.; Saito M.; Nagatani N.; Takamura Y.; Tamiya E. J. Am. Chem. Soc. 2005, 127, 11892. [DOI] [PubMed] [Google Scholar]
- Hung V. W. S.; Masoom H.; Kerman K. J. Electroanal. Chem. 2012, 681, 89. [Google Scholar]
- Masarik M.; Stobiecka A.; Kizek R.; Jelen F.; Pechan Z.; Hoyer W.; Jovin T. M.; Subramaniam V.; Palecek E. Electroanalysis 2004, 16, 1172. [Google Scholar]
- Juskova P.; Ostatna V.; Palecek E.; Foret F. Anal. Chem. 2010, 82, 2690. [DOI] [PubMed] [Google Scholar]
- Ostatna V.; Cernocka H.; Palecek E. Bioelectrochemistry 2012, 87, 84. [DOI] [PubMed] [Google Scholar]
- Palecek E.; Bartosik M.; Ostatna V.; Trefulka M. Chem. Rec. 2012, 12, 27. [DOI] [PubMed] [Google Scholar]
- Ostatna V.; Dogan B.; Uslu B.; Ozkan S.; Palecek E. J. Electroanal. Chem. 2006, 593, 172. [Google Scholar]
- Ostatna V.; Palecek E. Electrochim. Acta 2008, 53, 4014. [Google Scholar]
- Palecek E.; Ostatna V. Chem. Commun. 2009, 13, 1685. [DOI] [PubMed] [Google Scholar]
- Palecek E.; Ostatna V. Analyst 2009, 134, 2076. [DOI] [PubMed] [Google Scholar]
- Trnkova L.; Novotny L.; Serrano N.; Klosova K.; Polaskova P. Electroanalysis 2010, 22, 1873. [Google Scholar]
- Laborda E.; Suwatchara D.; Batchelor-McAuley C.; Compton R. G. J. Electroanal. Chem. 2013, 704, 102. [Google Scholar]
- Coldrick Z.; Steenson P.; Millner P.; Davies M.; Nelson A. Electrochim. Acta 2009, 54, 4954. [Google Scholar]
- Ostatna V.; Palecek E. Langmuir 2006, 22, 6481. [DOI] [PubMed] [Google Scholar]
- Cernocka H.; Ostatna V.; Palecek E. Anal. Chim. Acta 2013, 789, 41. [DOI] [PubMed] [Google Scholar]
- Ostatna V.; Cernocka H.; Palecek E. J. Am. Chem. Soc. 2010, 132, 9408. [DOI] [PubMed] [Google Scholar]
- Palecek E.; Cernocka H.; Ostatna V.; Navratilova L.; Brazdova M. Anal. Chim. Acta 2014, 828, 1. [DOI] [PubMed] [Google Scholar]
- Palecek E. Electroanalysis 2009, 21, 239. [Google Scholar]
- Bartosik M.; Trefulka M.; Hrstka R.; Vojtesek B.; Palecek E. Electrochem. Commun. 2013, 33, 55. [Google Scholar]
- Palecek E.; Heyrovsky M.; Janik B.; Kalab D.; Pechan Z. Collect. Czech. Chem. Commun. 2009, 74, 1739. [Google Scholar]
- Palecek E.; Trefulka M. Analyst 2011, 136, 321. [DOI] [PubMed] [Google Scholar]
- Trefulka M.; Palecek E. Bioelectrochemistry 2012, 88, 8. [DOI] [PubMed] [Google Scholar]
- Palecek E.; Rimankova L. Electrochem. Commun. 2014, 44, 59. [Google Scholar]
- Herman H. B.; Bard A. J. Anal. Chem. 1964, 36, 510. [Google Scholar]
- Bard A. J.; Faulkner L. R.. Electrochemical Methods: Fundamentals and Applications; Wiley: New York, 2000. [Google Scholar]
- Heyrovsky J. Chem. Listy 1941, 35, 155. [Google Scholar]
- Heyrovsky J.; Forejt J. Z. Phys. Chem. 1943, 193, 77. [Google Scholar]
- Palecek E. Nature 1960, 188, 656. [DOI] [PubMed] [Google Scholar]
- Kalvoda R. Electroanalysis 2000, 12, 1207. [Google Scholar]
- Kizek R.; Trnkova L.; Palecek E. Anal. Chem. 2001, 73, 4801. [DOI] [PubMed] [Google Scholar]
- Palecek E.; Ostatna V.; Masarik M.; Bertoncini C. W.; Jovin T. M. Analyst 2008, 133, 76. [DOI] [PubMed] [Google Scholar]
- Tomschik M.; Havran L.; Fojta M.; Palecek E. Electroanalysis 1998, 10, 403. [Google Scholar]
- Bartosik M.; Ostatna V.; Palecek E. Bioelectrochemistry 2009, 76, 70. [DOI] [PubMed] [Google Scholar]
- Dorcak V.; Palecek E. Electroanalysis 2007, 19, 2405. [Google Scholar]
- Heyrovsky J.; Kuta J.. Principles of Polarography; Academic Press: New York, 1965. [Google Scholar]
- Conway B. E.; Tilak B. V. Electrochim. Acta 2002, 47, 3571. [Google Scholar]
- Kibler L. A. ChemPhysChem 2006, 7, 985. [DOI] [PubMed] [Google Scholar]
- Koper M. T. M.; Bouwman E. Angew. Chem., Int. Ed. 2010, 49, 3723. [DOI] [PubMed] [Google Scholar]
- Markovic N. M.; Ross P. N. Surf. Sci. Rep. 2002, 45, 121. [Google Scholar]
- Heyrovsky M. Electroanalysis 2004, 16, 1067. [Google Scholar]
- Vargova V.; Zivanovic M.; Dorcak V.; Palecek E.; Ostatna V. Electroanalysis 2013, 25, 2130. [Google Scholar]
- Doneux T.; Dorcak V.; Palecek E. Langmuir 2010, 26, 1347. [DOI] [PubMed] [Google Scholar]
- Zivanovic M.; Aleksic M.; Ostatna V.; Doneux T.; Palecek E. Electroanalysis 2010, 22, 2064. [Google Scholar]
- Dorcak V.; Mader P.; Vesela V.; Fedurco M.; Sestakova I. Chem. Anal. (Warsaw) 2007, 52, 979. [Google Scholar]
- Heyrovsky M.; Mader P.; Vavricka S.; Vesela V.; Fedurco M. J. Electroanal. Chem. 1997, 430, 103. [Google Scholar]
- Honeychurch M. J. Bioelectrochem. Bioenerg. 1997, 44, 13. [Google Scholar]
- Palecek E. In Topics in Bioelectrochemistry and Bioenergetics; Milazzo G., Ed.; J. Wiley: London, 1983; Vol. 5. [Google Scholar]
- Palecek E. In Electrochemistry of Nucleic Acids and Proteins. Towards Electrochemical Sensors for Genomics and Proteomics; Palecek E., Scheller F., Wang J., Eds.; Elsevier: Amsterdam, 2005. [Google Scholar]
- Dorcak V.; Bartosik M.; Ostatna V.; Palecek E.; Heyrovsky M. Electroanalysis 2009, 21, 662. [Google Scholar]
- Roscoe S. G. In Modern Aspects of Electrochemistry; Bockris J. O. M., Conway B. E., White R. E., Eds.; Plenum Press: New York, 1996; Vol. 29. [Google Scholar]
- Scheller F.; Janchen M.; Prumke H.-J. Biopolymers 1975, 14, 1553. [DOI] [PubMed] [Google Scholar]
- Santhanam K. S. V.; Jespersen N.; Bard A. J. J. Am. Chem. Soc. 1977, 99, 274. [DOI] [PubMed] [Google Scholar]
- Palecek E.; Postbieglová I. J. Electroanal. Chem. 1986, 214, 359. [Google Scholar]
- Wackerbarth H.; Hildebrandt P. ChemPhysChem 2003, 4, 714. [DOI] [PubMed] [Google Scholar]
- Muskal N.; Mandler D. Curr. Sep. 2000, 19, 49. [Google Scholar]
- Banica F. G.; Ion A. In Encyclopedia of Analytical Chemistry; Meyers R. A., Ed.; John Wiley and Sons: Chichester, 2000. [Google Scholar]
- Brucknerlea C.; Kimmel R. J.; Janata J.; Conroy J. F. T.; Caldwell K. Electrochim. Acta 1995, 40, 2897. [Google Scholar]
- Creczynski-Pasa T. B.; Millone M. A. D.; Munford M. L.; de Lima V. R.; Vieira T. O.; Benitez G. A.; Pasa A. A.; Salvarezza R. C.; Vela M. E. Phys. Chem. Chem. Phys. 2009, 11, 1077. [DOI] [PubMed] [Google Scholar]
- Young M. E.; Carroad P. A.; Bell R. L. Biotechnol. Bioeng. 1980, 22, 947. [Google Scholar]
- Heller A.; Feldman B. Chem. Rev. 2008, 108, 2482. [DOI] [PubMed] [Google Scholar]
- Wang J. Chem. Rev. 2008, 108, 814. [DOI] [PubMed] [Google Scholar]
- Balasubramanian A.; Ponnuraj K. J. Mol. Biol. 2010, 400, 274. [DOI] [PubMed] [Google Scholar]
- Heyrovsky J.; Kuta J.. Principles of Polarography; Academic Press: New York, 1965. [Google Scholar]
- Doneux T.; Ostatna V.; Palecek E. Electrochim. Acta 2012, 56, 9337. [Google Scholar]
- Lansbury P. T. Proc. Natl. Acad. Sci. U.S.A. 1999, 96, 3342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breydo L.; Wu J. W.; Uversky V. N. Biochim. Biophys. Acta, Mol. Basis Dis. 2012, 1822, 261. [DOI] [PubMed] [Google Scholar]
- Uversky V. N. FEBS J. 2010, 277, 2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uversky V. N. Biopolymers 2013, 99, 870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makin O. S.; Serpell L. C. FEBS J. 2005, 272, 5950. [DOI] [PubMed] [Google Scholar]
- Rambaran R. N.; Serpell L. C. Prion 2008, 2, 112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caughey B.; Lansbury P. T. Annu. Rev. Neurosci. 2003, 26, 267. [DOI] [PubMed] [Google Scholar]
- Lansbury P. T.; Lashuel H. A. Nature 2006, 443, 774. [DOI] [PubMed] [Google Scholar]
- Kruger R.; Kuhn W.; Muller T.; Woitalla D.; Graeber M.; Kosel S.; Przuntek H.; Epplen J. T.; Schols L.; Riess O. Nat. Genet. 1998, 18, 106. [DOI] [PubMed] [Google Scholar]
- Polymeropoulos M. H.; Lavedan C.; Leroy E.; Ide S. E.; Dehejia A.; Dutra A.; Pike B.; Root H.; Rubenstein J.; Boyer R.; Stenroos E. S.; Chandrasekharappa S.; Athanassiadou A.; Papapetropoulos T.; Johnson W. G.; Lazzarini A. M.; Duvoisin R. C.; DiIorio G.; Golbe L. I.; Nussbaum R. L. Science 1997, 276, 2045. [DOI] [PubMed] [Google Scholar]
- Zarranz J. J.; Alegre J.; Gomez-Esteban J. C.; Lezcano E.; Ros R.; Ampuero I.; Vidal L.; Hoenicka J.; Rodriguez O.; Atares B.; Llorens V.; Tortosa E. G.; del Ser T.; Munoz D. G.; de Yebenes J. G. Ann. Neurol. 2004, 55, 164. [DOI] [PubMed] [Google Scholar]
- Hamley I. W. Angew. Chem., Int. Ed. 2007, 46, 8128. [DOI] [PubMed] [Google Scholar]
- Miller Y.; Ma B.; Nussinov R. Chem. Rev. 2010, 110, 4820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertoncini C. W.; Jung Y. S.; Fernandez C. O.; Hoyer W.; Griesinger C.; Jovin T. M.; Zweckstetter M. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoyer W. G.; Cherny D.; Subramaniam V.; Jovin T. M. J. Mol. Biol. 2004, 340, 127. [DOI] [PubMed] [Google Scholar]
- Antony T.; Hoyer W.; Cherny D.; Heim G.; Jovin T. M.; Subramaniam V. J. Biol. Chem. 2003, 278, 3235. [DOI] [PubMed] [Google Scholar]
- Harper J.; Lansbury P. Annu. Rev. Biochem. 1997, 66, 385. [DOI] [PubMed] [Google Scholar]
- Hoyer W.; Antony T.; Cherny D.; Heim G.; Jovin T. M.; Subramaniam V. J. Mol. Biol. 2002, 322, 383. [DOI] [PubMed] [Google Scholar]
- Conway K. A.; Harper J. D.; Lansbury P. T. Biochemistry 2000, 39, 2552. [DOI] [PubMed] [Google Scholar]
- Ding T.; Lee S.; Rochet J.; Lansbury P. Biochemistry 2002, 41, 10209. [DOI] [PubMed] [Google Scholar]
- Dobson C. Nature 2003, 426, 884. [DOI] [PubMed] [Google Scholar]
- Lashuel H. A.; Hartley D.; Petre B. M.; Walz T.; Lansbury P. T. Nature 2002, 418, 291. [DOI] [PubMed] [Google Scholar]
- Uversky V. J. Biomol. Struct. Dyn. 2003, 21, 211. [DOI] [PubMed] [Google Scholar]
- Kirkitadze M. D.; Bitan G.; Teplow D. B. J. Neurosci. Res. 2002, 69, 567. [DOI] [PubMed] [Google Scholar]
- Walsh D. M.; Selkoe D. J. Protein Pept, Lett. 2004, 11, 213. [DOI] [PubMed] [Google Scholar]
- Hardy J.; Selkoe D. J. Science 2002, 297, 353. [DOI] [PubMed] [Google Scholar]
- Kayed R.; Head E.; Thompson J. L.; McIntire T. M.; Milton S. C.; Cotman C. W.; Glabe C. G. Science 2003, 300, 486. [DOI] [PubMed] [Google Scholar]
- Maroteaux L.; Campanelli J. T.; Scheller R. H. J. Neurosci. 1988, 8, 2804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uversky V. N.; Li J.; Souillac P.; Millett I. S.; Doniach S.; Jakes R.; Goedert M.; Fink A. L. J. Biol. Chem. 2002, 277, 11970. [DOI] [PubMed] [Google Scholar]
- Cai X.; Rivas G.; Farias P. A. M.; Shiraishi H.; Wang J.; Palecek E. Electroanalysis 1996, 8, 753. [Google Scholar]
- Bertoncini C. W.; Fernandez C. O.; Griesinger C.; Jovin T. M.; Zweckstetter M. J. Biol. Chem. 2005, 280, 30649. [DOI] [PubMed] [Google Scholar]
- Conway K. A.; Lee S. J.; Rochet J. C.; Ding T. T.; Williamson R. E.; Lansbury P. T. Proc. Natl. Acad. Sci. U.S.A. 2000, 97, 571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dedmon M. M.; Lindorff-Larsen K.; Christodoulou J.; Vendruscolo M.; Dobson C. M. J. Am. Chem. Soc. 2005, 127, 476. [DOI] [PubMed] [Google Scholar]
- Goldberg M. S.; Lansbury P. T. Nat. Cell Biol. 2000, 2, E115. [DOI] [PubMed] [Google Scholar]
- Borsarelli C. D.; Falomir-Lockhart L. J.; Ostatna V.; Fauerbach J. A.; Hsiao H. H.; Urlaub H.; Palecek E.; Jares-Erijman E. A.; Jovin T. M. Free Radical Biol. Med. 2012, 53, 1004. [DOI] [PubMed] [Google Scholar]
- Lalowski M.; Golabek A.; Lemere C. A.; Selkoe D. J.; Wisniewski H. M.; Beavis R. C.; Frangione B.; Wisniewski T. J. Biol. Chem. 1996, 271, 33623. [DOI] [PubMed] [Google Scholar]
- Maury C. P. J. J. Intern. Med. 2009, 265, 329. [DOI] [PubMed] [Google Scholar]
- Selkoe D. J. Nat. Cell Biol. 2004, 6, 1054. [DOI] [PubMed] [Google Scholar]
- Sunde M.; Blake C. C. F. Q. Rev. Biophys. 1998, 31, 1. [DOI] [PubMed] [Google Scholar]
- Binolfi A.; Lamberto G. R.; Duran R.; Quintanar L.; Bertoncini C. W.; Souza J. M.; Cervenansky C.; Zweckstetter M.; Griesinger C.; Fernandez C. O. J. Am. Chem. Soc. 2008, 130, 11801. [DOI] [PubMed] [Google Scholar]
- Rao B. J. Mol. Neurosci. 2008, 35, 273. [DOI] [PubMed] [Google Scholar]
- Barnham K. J.; Bush A. I. Curr. Opin. Chem. Biol. 2008, 12, 222. [DOI] [PubMed] [Google Scholar]
- Uversky V. N.; Li J.; Fink A. L. J. Biol. Chem. 2001, 276, 44284. [DOI] [PubMed] [Google Scholar]
- Le Couteur D. G.; McLean A. J.; Taylor M. C.; Woodham B. L.; Board P. G. Biomed. Pharmacother. 1999, 53, 122. [DOI] [PubMed] [Google Scholar]
- Ruf R. A. S.; Lutz E. A.; Zigoneanu I. G.; Pielak G. J. Biochemistry 2008, 47, 13604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leong S. L.; Pham C. L. L.; Galatis D.; Fodero-Tavoletti M. T.; Perez K.; Hill A. F.; Masters C. L.; Ali F. E.; Barnham K. J.; Cappai R. Free Radical Biol. Med. 2009, 46, 1328. [DOI] [PubMed] [Google Scholar]
- Beyer K.; Ariza A. Curr. Med. Chem. 2008, 15, 2748. [DOI] [PubMed] [Google Scholar]
- Meng X. Y.; Munishkina L. A.; Fink A. L.; Uversky V. N. Biochemistry 2009, 48, 8206. [DOI] [PubMed] [Google Scholar]
- Hong D. P.; Fink A. L.; Uversky V. N. J. Mol. Biol. 2008, 383, 214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi M.; Kim J.; Kobayashi N.; Han S.; Nakamura C.; Ikebukuro K.; Sode K. Biochem. Biophys. Res. Commun. 2006, 349, 1139. [DOI] [PubMed] [Google Scholar]
- Ono K.; Yamada M. J. Neurochem. 2006, 97, 105. [DOI] [PubMed] [Google Scholar]
- Porat Y.; Abramowitz A.; Gazit E. Chem. Biol. Drug Des. 2006, 67, 27. [DOI] [PubMed] [Google Scholar]
- Li J.; Zhu M.; Rajamani S.; Uversky V. N.; Fink A. L. Chem. Biol. 2004, 11, 1513. [DOI] [PubMed] [Google Scholar]
- Zhu M.; Rajamani S.; Kaylor J.; Han S.; Zhou F. M.; Fink A. L. J. Biol. Chem. 2004, 279, 26846. [DOI] [PubMed] [Google Scholar]
- Chan T.; Chow A. M.; Tang D. W. F.; Li Q.; Wang X. Y.; Brown I. R.; Kerman K. J. Electroanal. Chem. 2010, 648, 151. [Google Scholar]
- Inouye H.; Fraser P. E.; Kirschner D. A. Biophys. J. 1993, 64, 502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtenthaler S. F. J. Neurochem. 2011, 116, 10. [DOI] [PubMed] [Google Scholar]
- Reiniger L.; Lukic A.; Linehan J.; Rudge P.; Collinge J.; Mead S.; Brandner S. Acta Neuropathol. 2011, 121, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandelkow E. Nature 1999, 402, 588. [DOI] [PubMed] [Google Scholar]
- Weingarten M. D.; Lockwood A. H.; Hwo S. Y.; Kirschner M. W. Proc. Natl. Acad. Sci. U.S.A. 1975, 72, 1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martic S.; Rains M. K.; Kraatz H. B. Anal. Biochem. 2013, 442, 130. [DOI] [PubMed] [Google Scholar]
- Antzutkin O. N.; Balbach J. J.; Leapman R. D.; Rizzo N. W.; Reed J.; Tycko R. Proc. Natl. Acad. Sci. U.S.A. 2000, 97, 13045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antzutkin O. N.; Balbach J. J.; Tycko R. Biophys. J. 2003, 84, 3326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antzutkin O. N.; Leapman R. D.; Balbach J. J.; Tycko R. Biochemistry 2002, 41, 15436. [DOI] [PubMed] [Google Scholar]
- Balbach J. J.; Petkova A. T.; Oyler N. A.; Antzutkin O. N.; Gordon D. J.; Meredith S. C.; Tycko R. Biophys. J. 2002, 83, 1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bucciantini M.; Giannoni E.; Chiti F.; Baroni F.; Formigli L.; Zurdo J. S.; Taddei N.; Ramponi G.; Dobson C. M.; Stefani M. Nature 2002, 416, 507. [DOI] [PubMed] [Google Scholar]
- El-Agnaf O. M. A.; Nagala S.; Patel B. P.; Austen B. M. J. Mol. Biol. 2001, 310, 157. [DOI] [PubMed] [Google Scholar]
- Goldsbury C.; Frey P.; Olivieri V.; Aebi U.; Muller S. A. J. Mol. Biol. 2005, 352, 282. [DOI] [PubMed] [Google Scholar]
- Luhrs T.; Ritter C.; Adrian M.; Riek-Loher D.; Bohrmann B.; Doeli H.; Schubert D.; Riek R. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 17342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson M. P. Nature 2004, 431, 107. [Google Scholar]
- Petkova A. T.; Ishii Y.; Balbach J. J.; Antzutkin O. N.; Leapman R. D.; Delaglio F.; Tycko R. Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 16742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petkova A. T.; Yau W. M.; Tycko R. Biochemistry 2006, 45, 498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh D. M.; Selkoe D. J. Neuron 2004, 44, 181. [DOI] [PubMed] [Google Scholar]
- Lu Y.; Derreumaux P.; Guo Z.; Mousseau N.; Wei G. H. Proteins: Struct., Funct., Bioinf. 2009, 75, 954. [DOI] [PubMed] [Google Scholar]
- Liang Y.; Pingali S. V.; Jogalekar A. S.; Snyder J. P.; Thiyagarajan P.; Lynn D. G. Biochemistry 2008, 47, 10018. [DOI] [PubMed] [Google Scholar]
- Krone M. G.; Hua L.; Soto P.; Zhou R. H.; Berne B. J.; Shea J. E. J. Am. Chem. Soc. 2008, 130, 11066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohrig U. F.; Laio A.; Tantalo N.; Parrinello M.; Petronzio R. Biophys. J. 2006, 91, 3217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favrin G.; Irback A.; Mohanty S. Biophys. J. 2004, 87, 3657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santini S.; Mousseau N.; Derreumaux P. J. Am. Chem. Soc. 2004, 126, 11509. [DOI] [PubMed] [Google Scholar]
- Santini S.; Wei G. H.; Mousseau N.; Derreumaux P. Structure 2004, 12, 1245. [DOI] [PubMed] [Google Scholar]
- Klimov D. K.; Thirumalai D. Structure 2003, 11, 295. [DOI] [PubMed] [Google Scholar]
- Ma B. Y.; Nussinov R. Proc. Natl. Acad. Sci. U.S.A 2002, 99, 14126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R.; Hu X. Y.; Khant H.; Ludtke S. J.; Chiu W.; Schmid M. F.; Frieden C.; Lee J. M. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 4653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Protopapa E.; Maude S.; Aggeli A.; Nelson A. Langmuir 2009, 25, 3289. [DOI] [PubMed] [Google Scholar]
- Protopapa E.; Ringstad L.; Aggeli A.; Nelson A. Electrochim. Acta 2010, 55, 3368. [Google Scholar]
- Geng J.; Yu H. J.; Ren J. S.; Qu X. G. Electrochem. Commun. 2008, 10, 1797. [Google Scholar]
- Fodera V.; Groenning M.; Vetri V.; Librizzi F.; Spagnolo S.; Cornett C.; Olsen L.; van de Weert M.; Leone M. J. Phys. Chem. B 2008, 112, 15174. [DOI] [PubMed] [Google Scholar]
- Khurana R.; Coleman C.; Ionescu-Zanetti C.; Carter S. A.; Krishna V.; Grover R. K.; Roy R.; Singh S. J. Struct. Biol. 2005, 151, 229. [DOI] [PubMed] [Google Scholar]
- Krebs M. R. H.; Bromley E. H. C.; Donald A. M. J. Struct. Biol. 2005, 149, 30. [DOI] [PubMed] [Google Scholar]
- Levine H. Amyloid 2005, 12, 5. [DOI] [PubMed] [Google Scholar]
- Mathis C. A.; Bacskai B. J.; Kajdasz S. T.; McLellan M. E.; Frosch M. P.; Hyman B. T.; Holt D. P.; Wang Y. M.; Huang G. F.; Debnath M. L.; Klunk W. E. Bioorg. Med. Chem. Lett. 2002, 12, 295. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Rodriguez C.; de Groot N. S.; Rimola A.; Alvarez-Larena A.; Lloveras V.; Vidal-Gancedo J.; Ventura S.; Vendrell J.; Sodupe M.; Gonzalez-Duarte P. J. Am. Chem. Soc. 2009, 131, 1436. [DOI] [PubMed] [Google Scholar]
- Veloso A. J.; Hung V. W. S.; Sindhu G.; Constantinof A.; Kerman K. Anal. Chem. 2009, 81, 9410. [DOI] [PubMed] [Google Scholar]
- Bollo S.; Ferreyra N. F.; Rivas G. A. Electroanalysis 2007, 19, 833. [Google Scholar]
- Wang J. Electroanalysis 2005, 17, 7. [Google Scholar]
- de la Fuente E.; Adura C.; Kogan M. J.; Bollo S. Electroanalysis 2012, 24, 938. [Google Scholar]
- Loksztejn A.; Dzwolak W.; Krysinski P. Bioelectrochemistry 2008, 72, 34. [DOI] [PubMed] [Google Scholar]
- Hawe A.; Sutter M.; Jiskoot W. Pharm. Res. 2008, 25, 1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luhrs T. T.; Zahn R.; Wuthrich K. J. Mol. Biol. 2006, 357, 833. [DOI] [PubMed] [Google Scholar]
- Kusumoto Y.; Lomakin A.; Teplow D. B.; Benedek G. B. Proc. Natl. Acad. Sci. U.S.A. 1998, 95, 12277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White D. A.; Buell A. K.; Dobson C. M.; Welland M. E.; Knowles T. P. J. FEBS Lett. 2009, 583, 2587. [DOI] [PubMed] [Google Scholar]
- Buell A. K.; Tartaglia G. G.; Birkett N. R.; Waudby C. A.; Vendruscolo M.; Salvatella X.; Welland M. E.; Dobson C. M.; Knowles T. P. J. ChemBioChem 2009, 10, 1309. [DOI] [PubMed] [Google Scholar]
- Hovgaard M. B.; Dong M. D.; Otzen D. E.; Besenbacher F. Biophys. J. 2007, 93, 2162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowles T. P. J.; Shu W. M.; Devlin G. L.; Meehan S.; Auer S.; Dobson C. M.; Welland M. E. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 10016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuno H.; Mori K.; Okada T.; Yokoyama Y.; Suzuki H. Chem. Biol. Drug Des. 2007, 69, 356. [DOI] [PubMed] [Google Scholar]
- Kotarek J. A.; Johnson K. C.; Moss M. A. Anal. Biochem. 2008, 378, 15. [DOI] [PubMed] [Google Scholar]
- Hu W. P.; Chang G. L.; Chen S. J.; Kuo Y. M. J. Neurosci. Methods 2006, 154, 190. [DOI] [PubMed] [Google Scholar]
- Cannon M. J.; Papalia G. A.; Navratilova I.; Fisher R. J.; Roberts L. R.; Worthy K. M.; Stephen A. G.; Marchesini G. R.; Collins E. J.; Casper D.; Qiu H. W.; Satpaev D.; Liparoto S. F.; Rice D. A.; Gorshkova II; Darling R. J.; Bennett D. B.; Sekar M.; Hommema E.; Liang A. M.; Day E. S.; Inman J.; Karlicek S. M.; Ullrich S. J.; Hodges D.; Chu T.; Sullivan E.; Simpson J.; Rafique A.; Luginbuhl B.; Westin S. N.; Bynum M.; Cachia P.; Li Y. J.; Kao D.; Neurauter A.; Wong M.; Swanson M.; Myszka D. G. Anal. Biochem. 2004, 330, 98. [DOI] [PubMed] [Google Scholar]
- Buell A. K.; White D. A.; Meier C.; Welland M. E.; Knowles T. P. J.; Dobson C. M. J. Phys. Chem. B 2010, 114, 10925. [DOI] [PubMed] [Google Scholar]
- Partovi-Nia R.; Beheshti S.; Qin Z.; Mandal H. S.; Long Y.-T.; Girault H. H.; Kraatz H.-B. Langmuir 2012, 28, 6377. [DOI] [PubMed] [Google Scholar]
- Hamley I. W.; Castelletto V.; Moulton C.; Myatt D.; Siligardi G.; Oliveira C. L. P.; Pedersen J. S.; Abutbul I.; Danino D. Macromol. Biosci. 2010, 10, 40. [DOI] [PubMed] [Google Scholar]
- Hamley I. W.; Nutt D. R.; Brown G. D.; Miravet J. F.; Escuder B.; Rodriguez-Llansola F. J. Phys. Chem. B 2010, 114, 940. [DOI] [PubMed] [Google Scholar]
- Vogelstein B.; Lane D.; Levine A. J. Nature 2000, 408, 307. [DOI] [PubMed] [Google Scholar]
- Vousden K. H.; Prives C. Cell 2009, 137, 413. [DOI] [PubMed] [Google Scholar]
- Petitjean A.; Mathe E.; Kato S.; Ishioka C.; Tavtigian S. V.; Hainaut P.; Olivier M. Hum. Mutat. 2007, 28, 622. [DOI] [PubMed] [Google Scholar]
- Joerger A. C.; Fersht A. R. Cold Spring Harbor Perspect. Biol. 2010, 2, a000919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joerger A. C.; Fersht A. R. Oncogene 2007, 26, 2226. [DOI] [PubMed] [Google Scholar]
- Butler J. S.; Loh S. N. Biochemistry 2003, 42, 2396. [DOI] [PubMed] [Google Scholar]
- Bullock A. N.; Henckel J.; Fersht A. R. Oncogene 2000, 19, 1245. [DOI] [PubMed] [Google Scholar]
- Nikolova P. V.; Henckel J.; Lane D. P.; Fersht A. R. Proc. Natl. Acad. Sci. U.S.A. 1998, 95, 14675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mol. Cell. Proteomics 2010, 9, 427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fruh V.; Ijzerman A. P.; Siegal G. Chem. Rev. 2011, 111, 640. [DOI] [PubMed] [Google Scholar]
- Zatloukalova M.; Orolinova E.; Kubala M.; Hrbac J.; Vacek J. Electroanalysis 2012, 24, 1758. [Google Scholar]
- Vacek J.; Zatloukalova M.; Havlikova M.; Ulrichova J.; Kubala M. Electrochem. Commun. 2013, 27, 104. [Google Scholar]
- Huliciak M.; Vacek J.; Sebela M.; Orolinova E.; Znaleziona J.; Havlikova M.; Kubala M. Biochem. Pharmacol. 2012, 83, 1507. [DOI] [PubMed] [Google Scholar]
- Dabrowiak J. C.Metals in Medicine; John Wiley & Sons, Ltd.: Chichester, 2009. [Google Scholar]
- Hartinger C. G.; Ang W. H.; Casini A.; Messori L.; Keppler B. K.; Dyson P. J. J. Anal. At. Spectrom. 2007, 22, 960. [Google Scholar]
- Hartinger C. G.; Tsybin Y. O.; Fuchser J.; Dyson P. J. Inorg. Chem. 2008, 47, 17. [DOI] [PubMed] [Google Scholar]
- Ishikawa T.; Aliosman F. J. Biol. Chem. 1993, 268, 20116. [PubMed] [Google Scholar]
- Odenheimer B.; Wolf W. Inorg. Chim. Acta, Bioinorg. Chem. 1982, 66, L41. [Google Scholar]
- Barabas K.; Milner R.; Lurie D.; Adin C. Vet. Comp. Oncol. 2008, 6, 1. [DOI] [PubMed] [Google Scholar]
- Dzagnidze A.; Katsarava Z.; Makhalova J.; Liedert B.; Yoon M. S.; Kaube H.; Limmroth V.; Thomale J. J. Neurosci. 2007, 27, 9451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller R. P.; Tadagavadi R. K.; Ramesh G.; Reeves W. B. Toxins 2010, 2, 2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pabla N.; Dong Z. Kidney Int. 2008, 73, 994. [DOI] [PubMed] [Google Scholar]
- Ries F.; Klastersky J. Am. J. Kidney Dis. 1986, 8, 368. [DOI] [PubMed] [Google Scholar]
- Vecerkova R.; Hernychova L.; Dobes P.; Vrba J.; Josypcuk B.; Bartosik M.; Vacek J. Anal. Chim. Acta 2014, 830, 23. [DOI] [PubMed] [Google Scholar]
- Krejci L.; Altmannova V.; Spirek M.; Zhao X. L. Nucleic Acids Res. 2012, 40, 5795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedberg E. C.; Lehmann A. R.; Fuchs R. P. P. Mol. Cell 2005, 18, 499. [DOI] [PubMed] [Google Scholar]
- Kirsanov D. D.; Zanegina O. N.; Aksianov E. A.; Spirin S. A.; Karyagina A. S.; Alexeevski A. V. Nucleic Acids Res. 2013, 41, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohs R.; Jin X.; West S. M.; Joshi R.; Honig B.; Mann R. S. Annu. Rev. Biochem. 2010, 79, 233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y.; Geiger J. H.; Hahn S.; Sigler P. B. Nature 1993, 365, 512. [DOI] [PubMed] [Google Scholar]
- Du P.; Liu S. N.; Wu P.; Cai C. X. Electrochim. Acta 2007, 52, 6534. [Google Scholar]
- Lojou E.; Bianco P. Electroanalysis 2004, 16, 1113. [Google Scholar]
- Lu Y. D.; Xu J. J.; Liu B. H.; Kong J. L. Biosens. Bioelectron. 2007, 22, 1173. [DOI] [PubMed] [Google Scholar]
- Wang J. Electroanalysis 2007, 19, 769. [Google Scholar]
- Citartan M.; Gopinath S. C. B.; Tominaga J.; Tan S. C.; Tang T. H. Biosens. Bioelectron. 2012, 34, 1. [DOI] [PubMed] [Google Scholar]
- Daniels J. S.; Pourmand N. Electroanalysis 2007, 19, 1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han K.; Liang Z. Q.; Zhou N. D. Sensors 2010, 10, 4541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hianik T. In Electrochemical Sensor Analysis; Alegret S., Merkoci A., Eds.; Elsevier: Amsterdam, 2007; Vol. 49. [Google Scholar]
- Hianik T. In Electrochemical Sensor Analysis; Alegret S., Merkoci A., Eds.; Elsevier: Amsterdam, 2007; Vol. 49. [Google Scholar]
- Hianik T.; Wang J. Electroanalysis 2009, 21, 1223. [Google Scholar]
- Hong P.; Li W. L.; Li J. M. Sensors 2012, 12, 1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sassolas A.; Blum L. J.; Leca-Bouvier B. D. Electroanalysis 2009, 21, 1237. [Google Scholar]
- Strehlitz B.; Nikolaus N.; Stoltenburg R. Sensors 2008, 8, 4296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y.; Cheng G. F.; He P. G.; Fang Y. Z. Electroanalysis 2009, 21, 1251. [Google Scholar]
- Wang J.; Jiang M.; Palecek E. Bioelectrochem. Bioenerg. 1999, 48, 477. [DOI] [PubMed] [Google Scholar]
- Fojta M.; Kubicarova T.; Palecek E. Electroanalysis 1999, 11, 1005. [Google Scholar]
- Ostatna V.; Jelen F.; Hianik T.; Palecek E. Electroanalysis 2005, 17, 1413. [Google Scholar]
- Boon E. M.; Pope M. A.; Williams S. D.; David S. S.; Barton J. K. Biochemistry 2002, 41, 8464. [DOI] [PubMed] [Google Scholar]
- Rajski S. R.; Kumar S.; Roberts R. J.; Barton J. K. J. Am. Chem. Soc. 1999, 121, 5615. [Google Scholar]
- Boon E. M.; Barton J. K. Bioconjugate Chem. 2003, 14, 1140. [DOI] [PubMed] [Google Scholar]
- Palecek E.; Masarik M.; Kizek R.; Kuhlmeier D.; Hassmann J.; Schulein J. Anal. Chem. 2004, 76, 5930. [DOI] [PubMed] [Google Scholar]
- Palecek E.; Fojta M. Talanta 2007, 74, 276. [DOI] [PubMed] [Google Scholar]
- Jiricny J. Mutat. Res., DNA Repair 1998, 409, 107. [DOI] [PubMed] [Google Scholar]
- Jiricny J. Curr. Biol. 2000, 10, R788. [DOI] [PubMed] [Google Scholar]
- Lamers M. H.; Perrakis A.; Enzlin J. H.; Winterwerp H. H. K.; de Wind N.; Sixma T. K. Nature 2000, 407, 711. [DOI] [PubMed] [Google Scholar]
- Obmolova G.; Ban C.; Hsieh P.; Yang W. Nature 2000, 407, 703. [DOI] [PubMed] [Google Scholar]
- Bi L. J.; Zhou Y. F.; Zhang X. E.; Deng J. Y.; Zhang Z. P.; Xie B.; Zhang C. G. Anal. Chem. 2003, 75, 4113. [DOI] [PubMed] [Google Scholar]
- Behrensdorf H. A.; Pignot M.; Windhab N.; Kappel A. Nucleic Acids Res. 2002, 30, e64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachadyn P.; Stanislawska A.; Kur J. Nucleic Acids Res. 2000, 28, e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su X. D.; Robelek R.; Wu Y. J.; Wang G. Y.; Knoll W. Anal. Chem. 2004, 76, 489. [DOI] [PubMed] [Google Scholar]
- Palecek E.; Brazda V.; Jagelska E.; Pecinka P.; Karlovska L.; Brazdova M. Oncogene 2004, 23, 2119. [DOI] [PubMed] [Google Scholar]
- Nemcova K.; Havran L.; Sebest P.; Brazdova M.; Pivonkova H.; Fojta M. Anal. Chim. Acta 2010, 668, 166. [DOI] [PubMed] [Google Scholar]
- Kerman K.; Morita Y.; Takamura Y.; Tamiya E. Anal. Bioanal. Chem. 2005, 381, 1114. [DOI] [PubMed] [Google Scholar]
- Chase J. W.; Williams K. R. Annu. Rev. Biochem. 1986, 55, 103. [DOI] [PubMed] [Google Scholar]
- Meyer R. R.; Laine P. S. Microbiol. Rev. 1990, 54, 342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sontz P. A.; Muren N. B.; Barton J. K. Acc. Chem. Res. 2012, 45, 1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genereux J. C.; Barton J. K. Chem. Rev. 2010, 110, 1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abi A.; Ferapontova E. E. J. Am. Chem. Soc. 2012, 134, 14499. [DOI] [PubMed] [Google Scholar]
- Boal A. K.; Genereux J. C.; Sontz P. A.; Gralnick J. A.; Newman D. K.; Barton J. K. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 15237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boal A. K.; Yavin E.; Lukianova O. A.; O’Shea V. L.; David S. S.; Barton J. K. Biochemistry 2005, 44, 8397. [DOI] [PubMed] [Google Scholar]
- Pheeney C. G.; Arnold A. R.; Grodick M. A.; Barton J. K. J. Am. Chem. Soc. 2013, 135, 11869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porello S. L.; Williams S. D.; Kuhn H.; Michaels M. L.; David S. S. J. Am. Chem. Soc. 1996, 118, 10684. [Google Scholar]
- Jervis A. J.; Crack J. C.; White G.; Artymiuk P. J.; Cheesman M. R.; Thomson A. J.; Le Brun N. E.; Green J. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 4659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mui T. P.; Fuss J. O.; Ishida J. P.; Tainer J. A.; Barton J. K. J. Am. Chem. Soc. 2011, 133, 16378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White M. F.; Dillingham M. S. Curr. Opin. Struct. Biol. 2012, 22, 94. [DOI] [PubMed] [Google Scholar]
- Netz D. J.; Stith C. M.; Stumpfig M.; Kopf G.; Vogel D.; Genau H. M.; Stodola J. L.; Lill R.; Burgers P. M.; Pierik A. J. Nat. Chem. Biol. 2011, 8, 125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold A. R.; Barton J. K. J. Am. Chem. Soc. 2013, 135, 15726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy J. N.; Cheng A. K. H.; Yu H. Z.; Bizzotto D. J. Am. Chem. Soc. 2009, 131, 4042. [DOI] [PubMed] [Google Scholar]
- Sam M.; Boon E. M.; Barton J. K.; Hill M. G.; Spain E. M. Langmuir 2001, 17, 5727. [Google Scholar]
- Furst A. L.; Hill M. G.; Barton J. K. Langmuir 2013, 29, 16141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furst A. L.; Muren N. B.; Hill M. G.; Barton J. K. Proc. Natl. Acad. Sci. U.S.A. 2014, 111, 14985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartosik M.; Fojta M.; Palecek E. Electrochim. Acta 2012, 78, 75. [Google Scholar]
- Hou P.; Ji M. J.; Ge C. W.; Shen J. Y.; Li S.; He N. Y.; Lu Z. H. Nucleic Acids Res. 2003, 31, e92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisdat F.; Schafer D. Anal. Bioanal. Chem. 2008, 391, 1555. [DOI] [PubMed] [Google Scholar]
- Katz E.; Willner I. Electroanalysis 2003, 15, 913. [Google Scholar]
- Chang H. X.; Li J. H. Electrochem. Commun. 2009, 11, 2101. [Google Scholar]
- Tersch C.; Lisdat F. Electrochim. Acta 2011, 56, 7673. [Google Scholar]
- Kim J. L.; Burley S. K. Nat. Struct. Biol. 1994, 1, 638. [DOI] [PubMed] [Google Scholar]
- Fan C. H.; Plaxco K. W.; Heeger A. J. Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 9134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swensen J. S.; Xiao Y.; Ferguson B. S.; Lubin A. A.; Lai R. Y.; Heeger A. J.; Plaxco K. W.; Soh H. T. J. Am. Chem. Soc. 2009, 131, 4262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S.; Phillips M. F.; Cerrina F.; Smith L. M. Langmuir 2009, 25, 6570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kjallman T. H. M.; Peng H.; Soeller C.; Travas-Sejdic J. Anal. Chem. 2008, 80, 9460. [DOI] [PubMed] [Google Scholar]
- Lee C. Y.; Gong P.; Harbers G. M.; Grainger D. W.; Castner D. G.; Gamble L. J. Anal. Chem. 2006, 78, 3316. [DOI] [PubMed] [Google Scholar]
- Peterson A. W.; Heaton R. J.; Georgiadis R. M. Nucleic Acids Res. 2001, 29, 5163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricci F.; Lai R. Y.; Heeger A. J.; Plaxco K. W.; Sumner J. J. Langmuir 2007, 23, 6827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White R. J.; Phares N.; Lubin A. A.; Xiao Y.; Plaxco K. W. Langmuir 2008, 24, 10513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biagiotti V.; Porchetta A.; Desiderati S.; Plaxco K. W.; Palleschi G.; Ricci F. Anal. Bioanal. Chem. 2012, 402, 413. [DOI] [PubMed] [Google Scholar]
- Ricci F.; Bonham A. J.; Mason A. C.; Reich N. O.; Plaxco K. W. Anal. Chem. 2009, 81, 1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cash K. J.; Ricci F.; Plaxco K. W. Chem. Commun. 2009, 41, 6222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cash K. J.; Ricci F.; Plaxco K. W. J. Am. Chem. Soc. 2009, 131, 6955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White R. J.; Kallewaard H. M.; Hsieh W.; Patterson A. S.; Kasehagen J. B.; Cash K. J.; Uzawa T.; Soh H. T.; Plaxco K. W. Anal. Chem. 2012, 84, 1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonham A. J.; Paden N. G.; Ricci F.; Plaxco K. W. Analyst 2013, 138, 5580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E.; Deppert W. Cell Death Differ. 2006, 13, 885. [DOI] [PubMed] [Google Scholar]
- Ma B.; Levine A. J. Nucleic Acids Res. 2007, 35, 7733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavletich N. P.; Chambers K. A.; Pabo C. O. Genes Dev. 1993, 7, 2556. [DOI] [PubMed] [Google Scholar]
- Beno I.; Rosenthal K.; Levitine M.; Shaulov L.; Haran T. E. Nucleic Acids Res. 2011, 39, 1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagaich A. K.; Zhurkin V. B.; Durell S. R.; Jernigan R. L.; Appella E.; Harrington R. E. Proc. Natl. Acad. Sci. U.S.A. 1999, 96, 1875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitayner M.; Rozenberg H.; Rohs R.; Suad O.; Rabinovich D.; Honig B.; Shakked Z. Nat. Struct. Mol. Biol. 2010, 17, 423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian H.; Wang T.; Naumovski L.; Lopez C. D.; Brachmann R. K. Oncogene 2002, 21, 7901. [DOI] [PubMed] [Google Scholar]
- Weinberg R. L.; Veprintsev D. B.; Bycroft M.; Fersht A. R. J. Mol. Biol. 2005, 348, 589. [DOI] [PubMed] [Google Scholar]
- Dadova J.; Orsag P.; Pohl R.; Brazdova M.; Fojta M.; Hocek M. Angew. Chem., Int. Ed. 2013, 52, 10515. [DOI] [PubMed] [Google Scholar]
- Horakova P.; Macickova-Cahova H.; Pivonkova H.; Spacek J.; Havran L.; Hocek M.; Fojta M. Org. Biomol. Chem. 2011, 9, 1366. [DOI] [PubMed] [Google Scholar]
- Nemcova K.; Sebest P.; Havran L.; Orsag P.; Fojta M.; Pivonkova H. Anal. Bioanal. Chem. 2014, 406, 5843. [DOI] [PubMed] [Google Scholar]
- Riedl J.; Menova P.; Pohl R.; Orsag P.; Fojta M.; Hocek M. J. Org. Chem. 2012, 77, 8287. [DOI] [PubMed] [Google Scholar]
- Riedl J.; Pohl R.; Ernsting N. P.; Orsag P.; Fojta M.; Hocek M. Chem. Sci. 2012, 3, 2797. [Google Scholar]
- Masarik M.; Cahova K.; Kizek R.; Palecek E.; Fojta M. Anal. Bioanal. Chem. 2007, 388, 259. [DOI] [PubMed] [Google Scholar]
- He Y.; Fang J.; Taatjes D. J.; Nogales E. Nature 2013, 495, 481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov D.; Nasmyth K. Cell 2005, 122, 849. [DOI] [PubMed] [Google Scholar]
- Jenuwein T.; Allis C. D. Science 2001, 293, 1074. [DOI] [PubMed] [Google Scholar]
- Jeffrey P. D.; Russo A. A.; Polyak K.; Gibbs E.; Hurwitz J.; Massague J.; Pavletich N. P. Nature 1995, 376, 313. [DOI] [PubMed] [Google Scholar]
- Arnaud J.; Audfray A.; Imberty A. Chem. Soc. Rev. 2013, 42, 4798. [DOI] [PubMed] [Google Scholar]
- Varki A.Essentials of Glycobiology; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, 2009. [PubMed] [Google Scholar]
- Ritchie G. E.; Moffatt B. E.; Sim R. B.; Morgan B. P.; Dwek R. A.; Rudd P. M. Chem. Rev. 2002, 102, 305. [DOI] [PubMed] [Google Scholar]
- McReynolds K. D.; Gervay-Hague J. Chem. Rev. 2007, 107, 1533. [DOI] [PubMed] [Google Scholar]
- Dalziel M.; Crispin M.; Scanlan C. N.; Zitzmann N.; Dwek R. A. Science 2014, 343, 1235681. [DOI] [PubMed] [Google Scholar]
- Novokmet M.; Lukic E.; Vuckovic F.; Duric Z.; Keser T.; Rajsl K.; Remondini D.; Castellani G.; Gasparovic H.; Gornik O.; Lauc G. Sci. Rep. 2014, 4, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee S.; Zheng H.; Derebe M. G.; Callenberg K. M.; Partch C. L.; Rollins D.; Propheter D. C.; Rizo J.; Grabe M.; Jiang Q.-X. Nature 2014, 477, 103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alizadeh A. A.; Eisen M. B.; Davis R. E.; Ma C.; Lossos I. S.; Rosenwald A.; Boldrick J. C.; Sabet H.; Tran T.; Yu X.; Powell J. I.; Yang L.; Marti G. E.; Moore T.; Hudson J.; Lu L.; Lewis D. B.; Tibshirani R.; Sherlock G.; Chan W. C.; Greiner T. C.; Weisenburger D. D.; Armitage J. O.; Warnke R.; Levy R.; Wilson W.; Grever M. R.; Byrd J. C.; Botstein D.; Brown P. O.; Staudt L. M. Nature 2000, 403, 503. [DOI] [PubMed] [Google Scholar]
- Gemeiner P.; Mislovicova D.; Tkac J.; Svitel J.; Patoprsty V.; Hrabarova E.; Kogan G.; Kozar T. Biotechnol. Adv. 2009, 27, 1. [DOI] [PubMed] [Google Scholar]
- Hirabayashi J. Nat. Chem. Biol. 2009, 5, 198. [DOI] [PubMed] [Google Scholar]
- Katrlik J.; Svitel J.; Gemeiner P.; Kozar T.; Tkac J. Med. Res. Rev. 2010, 30, 394. [DOI] [PubMed] [Google Scholar]
- Krishnamoorthy L.; Bess J. W.; Preston A. B.; Nagashima K.; Mahal L. K. Nat. Chem. Biol. 2009, 5, 244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schauer R.; Kamerling J. P. ChemBioChem 2011, 12, 2246. [DOI] [PubMed] [Google Scholar]
- Song X.; Lasanajak Y.; Xia B.; Heimburg-Molinaro J.; Rhea J. M.; Ju H.; Zhao C.; Molinaro R. J.; Cummings R. D.; Smith D. F. Nat. Methods 2011, 8, 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaishnava S.; Yamamoto M.; Severson K. M.; Ruhn K. A.; Yu X.; Koren O.; Ley R.; Wakeland E. K.; Hooper L. V. Science 2011, 334, 255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svarovsky S. A.; Joshi L. Anal. Methods 2014, 6, 3918. [Google Scholar]
- Burton D. R.; Poignard P.; Stanfield R. L.; Wilson I. A. Science 2012, 337, 183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doores K. J.; Fulton Z.; Hong V.; Patel M. K.; Scanlan C. N.; Wormald M. R.; Finn M. G.; Burton D. R.; Wilson I. A.; Davis B. G. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 17107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pejchal R.; Doores K. J.; Walker L. M.; Khayat R.; Huang P.-S.; Wang S.-K.; Stanfield R. L.; Julien J.-P.; Ramos A.; Crispin M.; Depetris R.; Katpally U.; Marozsan A.; Cupo A.; Maloveste S.; Liu Y.; McBride R.; Ito Y.; Sanders R. W.; Ogohara C.; Paulson J. C.; Feizi T.; Scanlan C. N.; Wong C.-H.; Moore J. P.; Olson W. C.; Ward A. B.; Poignard P.; Schief W. R.; Burton D. R.; Wilson I. A. Science 2011, 334, 1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anthony R. M.; Kobayashi T.; Wermeling F.; Ravetch J. V. Nature 2011, 475, 110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anthony R. M.; Nimmerjahn F.; Ashline D. J.; Reinhold V. N.; Paulson J. C.; Ravetch J. V. Science 2008, 320, 373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J.-H.; Resende R.; Wennekes T.; Chen H.-M.; Bance N.; Buchini S.; Watts A. G.; Pilling P.; Streltsov V. A.; Petric M.; Liggins R.; Barrett S.; McKimm-Breschkin J. L.; Niikura M.; Withers S. G. Science 2013, 340, 71. [DOI] [PubMed] [Google Scholar]
- Klein F.; Halper-Stromberg A.; Horwitz J. A.; Gruell H.; Scheid J. F.; Bournazos S.; Mouquet H.; Spatz L. A.; Diskin R.; Abadir A.; Zang T.; Dorner M.; Billerbeck E.; Labitt R. N.; Gaebler C.; Marcovecchio P. M.; Incesu R.-B.; Eisenreich T. R.; Bieniasz P. D.; Seaman M. S.; Bjorkman P. J.; Ravetch J. V.; Ploss A.; Nussenzweig M. C. Nature 2012, 492, 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F.; Ravetch J. V. Science 2011, 333, 1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E. H.; Misek D. E. Int. J. Proteomics 2011, 2011, 601937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandler K. B.; Goldman R. Mol. Cell. Proteomics 2013, 12, 836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferens-Sieczkowska M.; Kowalska B.; Kratz E. M. Biomarkers 2013, 18, 10. [DOI] [PubMed] [Google Scholar]
- Gilgunn S.; Conroy P. J.; Saldova R.; Rudd P. M.; O’Kennedy R. J. Nat. Rev. Urol. 2013, 10, 99. [DOI] [PubMed] [Google Scholar]
- Badr H. A.; AlSadek D. M.; Darwish A. A.; ElSayed A. I.; Bekmanov B. O.; Khussainova E. M.; Zhang X.; Cho W. C.; Djansugurova L. B.; Li C.-Z. Expert Rev. Proteomics 2014, 11, 227. [DOI] [PubMed] [Google Scholar]
- Cho W. C. Expert Rev. Proteomics 2014, 11, 131. [DOI] [PubMed] [Google Scholar]
- National Research Council. Transforming Glycoscience: A Roadmap for the Future; The National Academies Press: Washington, DC, 2012. [PubMed] [Google Scholar]
- Beck A.; Sanglier-Cianferani S.; Van Dorsselaer A. Anal. Chem. 2012, 84, 4637. [DOI] [PubMed] [Google Scholar]
- Schmaltz R. M.; Hanson S. R.; Wong C.-H. Chem. Rev. 2011, 111, 4259. [DOI] [PubMed] [Google Scholar]
- van Bueren J. J. L.; Rispens T.; Verploegen S.; van der Palen-Merkus T.; Stapel S.; Workman L. J.; James H.; van Berkel P. H. C.; van de Winkel J. G. J.; Platts-Mills T. A. E.; Parren P. W. H. I. Nat. Biotechnol. 2011, 29, 574. [DOI] [PubMed] [Google Scholar]
- Song R.; Oren D. A.; Franco D.; Seaman M. S.; Ho D. D. Nat. Biotechnol. 2013, 31, 1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck A.; Reichert J. M. mAbs 2012, 4, 419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman R.; Raguram S.; Venkataraman G.; Paulson J. C.; Sasisekharan R. Nat. Methods 2005, 2, 817. [DOI] [PubMed] [Google Scholar]
- Cummings R. D.; Pierce J. M. Chem. Biol. 2014, 21, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis B. G. Chem. Rev. 2002, 102, 579. [DOI] [PubMed] [Google Scholar]
- Gamblin D. P.; Scanlan E. M.; Davis B. G. Chem. Rev. 2008, 109, 131. [DOI] [PubMed] [Google Scholar]
- Gabius H.-J.; Andre S.; Jimenez-Barbero J.; Romero A.; Solis D. Trends Biochem. Sci. 2011, 36, 298. [DOI] [PubMed] [Google Scholar]
- Cummings R. D. Mol. BioSyst. 2009, 5, 1087. [DOI] [PubMed] [Google Scholar]
- Bertozzi C. R.; Kiessling L. L. Science 2001, 291, 2357. [DOI] [PubMed] [Google Scholar]
- Furukawa J.-I.; Fujitani N.; Shinohara Y. Biomolecules 2013, 3, 198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakus J. F.; Mahal L. K. Annu. Rev. Anal. Chem. 2011, 4, 367. [DOI] [PubMed] [Google Scholar]
- Smith D. F.; Cummings R. D. Mol. Cell. Proteomics 2013, 12, 902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mechref Y.; Novotny M. V. Chem. Rev. 2002, 102, 321. [DOI] [PubMed] [Google Scholar]
- Novotny M. V.; Alley W. R. Jr. Curr. Opin. Chem. Biol. 2013, 17, 832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Defaus S.; Gupta P.; Andreu D.; Gutierrez-Gallego R. Analyst 2014, 139, 2944. [DOI] [PubMed] [Google Scholar]
- Alley W. R.; Madera M.; Mechref Y.; Novotny M. V. Anal. Chem. 2010, 82, 5095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mechref Y.; Hu Y.; Garcia A.; Hussein A. Electrophoresis 2012, 33, 1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novotny M. V.; Alley W. R. Jr.; Mann B. F. Glycoconjugate J. 2013, 30, 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra I.; Alley W. R.; Goetz J. A.; Vasseur J. A.; Novotny M. V.; Jacobson S. C. J. Proteome Res. 2013, 12, 4490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharon N.; Lis H. Glycobiology 2004, 14, 53R. [DOI] [PubMed] [Google Scholar]
- Lis H.; Sharon N. Chem. Rev. 1998, 98, 637. [DOI] [PubMed] [Google Scholar]
- Jin S.; Cheng Y.; Reid S.; Li M.; Wang B. Med. Res. Rev. 2010, 30, 171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner G. A. Clin. Chim. Acta 1992, 208, 149. [DOI] [PubMed] [Google Scholar]
- Mislovicova D.; Gemeiner P.; Kozarova A.; Kozar T. Biologia 2009, 64, 1. [Google Scholar]
- Mislovicova D.; Katrlik J.; Paulovicova E.; Gemeiner P.; Tkac J. Colloids Surf., B 2012, 94, 163. [DOI] [PubMed] [Google Scholar]
- Bucko M.; Mislovicova D.; Nahalka J.; Vikartovska A.; Sefcovicova J.; Katrlik J.; Tkac J.; Gemeiner P.; Lacik I.; Stefuca V.; Polakovic M.; Rosenberg M.; Rebros M.; Smogrovicova D.; Svitel J. Chem. Pap. 2012, 66, 983. [Google Scholar]
- Murphy P.; André S.; Gabius H.-J. Molecules 2013, 18, 4026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira C.; Teixeira J. A.; Domingues L. Crit. Rev. Biotechnol. 2013, 33, 66. [DOI] [PubMed] [Google Scholar]
- Ribeiro J. P.; Mahal L. K. Curr. Opin. Chem. Biol. 2013, 17, 827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilobello K. T.; Mahal L. K. Curr. Opin. Chem. Biol. 2007, 11, 300. [DOI] [PubMed] [Google Scholar]
- Hirabayashi J.; Kuno A.; Tateno H. Electrophoresis 2011, 32, 1118. [DOI] [PubMed] [Google Scholar]
- Hirabayashi J.; Yamada M.; Kuno A.; Tateno H. Chem. Soc. Rev. 2013, 42, 4443. [DOI] [PubMed] [Google Scholar]
- Krishnamoorthy L.; Mahal L. K. ACS Chem. Biol. 2009, 4, 715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agrawal P.; Kurcon T.; Pilobello K. T.; Rakus J. F.; Koppolu S.; Liu Z.; Batista B. S.; Eng W. S.; Hsu K.-L.; Liang Y. Proc. Natl. Acad. Sci. U.S.A. 2014, 111, 4338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuel N. F.; Ahn J.-H.; Kim J.-H.; Zhang J.; Boghossian A. A.; Mahal L. K.; Strano M. S. J. Am. Chem. Soc. 2011, 133, 17923. [DOI] [PubMed] [Google Scholar]
- Tkac J.; Davis J. J. In Engineering the Bioelectronic Interface: Applications to Analyte Biosensing and Protein Detection; Davis J. J., Ed.; Royal Society of Chemistry: Cambridge, 2009. [Google Scholar]
- Reuel N. F.; Grassbaugh B.; Kruss S.; Mundy J. Z.; Opel C.; Ogunniyi A. O.; Egodage K.; Wahl R.; Helk B.; Zhang J.; Kalcioglu Z. I.; Tvrdy K.; Bellisario D. O.; Mu B.; Blake S. S.; Van Vliet K. J.; Love J. C.; Wittrup K. D.; Strano M. S. ACS Nano 2013, 7, 7472. [DOI] [PubMed] [Google Scholar]
- Mu B.; Zhang J.; McNicholas T. P.; Reuel N. F.; Kruss S.; Strano M. S. Acc. Chem. Res. 2014, 47, 979. [DOI] [PubMed] [Google Scholar]
- Rohrer J. S.; Basumallick L.; Hurum D. Biochemistry (Moscow) 2013, 78, 697. [DOI] [PubMed] [Google Scholar]
- Dwek R. A.; Edge C. J.; Harvey D. J.; Wormald M. R.; Parekh R. B. Annu. Rev. Biochem. 1993, 62, 65. [DOI] [PubMed] [Google Scholar]
- Rudd P. M.; Gulle G. R.; Kuster B.; Harvey D. J.; Opdenakker G.; Dwek R. A. Nature 1997, 388, 205. [DOI] [PubMed] [Google Scholar]
- Lee Y. C. Anal. Biochem. 1990, 189, 151. [DOI] [PubMed] [Google Scholar]
- Hughes S.; Johnson D. C. Anal. Chim. Acta 1981, 132, 11. [Google Scholar]
- Johnson D. C.; LaCourse W. R. Anal. Chem. 1990, 62, 589A. [Google Scholar]
- Colon L. A.; Dadoo R.; Zare R. N. Anal. Chem. 1993, 65, 476. [Google Scholar]
- Sun M.; Lee C. S. Biotechnol. Bioeng. 1998, 57, 545. [PubMed] [Google Scholar]
- Jelinek R.; Kolusheva S. Chem. Rev. 2004, 104, 5987. [DOI] [PubMed] [Google Scholar]
- Brooks S. Mol. Biotechnol. 2009, 43, 76. [DOI] [PubMed] [Google Scholar]
- Rocklin R. D.; Clarke A. P.; Weitzhandler M. Anal. Chem. 1998, 70, 1496. [Google Scholar]
- Aubeck R.; Eppelsheim C.; Brauchle C.; Hampp N. Analyst 1993, 118, 1389. [DOI] [PubMed] [Google Scholar]
- Pasek M.; Duk M.; Podbielska M.; Sokolik R.; Szechiński J.; Lisowska E.; Krotkiewski H. Glycoconjugate J. 2006, 23, 463. [DOI] [PubMed] [Google Scholar]
- Rudd P. M.; Dwek R. A. Curr. Opin. Biotechnol. 1997, 8, 488. [DOI] [PubMed] [Google Scholar]
- Spellman M. W. Anal. Chem. 1990, 62, 1714. [DOI] [PubMed] [Google Scholar]
- Geyer H.; Geyer R. Biochim. Biophys. Acta, Proteins Proteomics 2006, 1764, 1853. [DOI] [PubMed] [Google Scholar]
- Hardy M. R.; Townsend R. R. Proc. Natl. Acad. Sci. U.S.A. 1988, 85, 3289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L. M.; Yet M. G.; Shao M. C. FASEB J. 1988, 2, 2819. [DOI] [PubMed] [Google Scholar]
- Singhal P.; Kuhr W. G. Anal. Chem. 1997, 69, 3552. [DOI] [PubMed] [Google Scholar]
- Singhal P.; Kuhr W. G. Anal. Chem. 1997, 69, 4828. [DOI] [PubMed] [Google Scholar]
- Heyrovsky J. Chem. Listy 1941, 35, 155. [Google Scholar]
- Weber P. L.; Kornfelt T.; Klausen N. K.; Lunte S. M. Anal. Biochem. 1995, 225, 135. [DOI] [PubMed] [Google Scholar]
- Simanek E. E.; McGarvey G. J.; Jablonowski J. A.; Wong C.-H. Chem. Rev. 1998, 98, 833. [DOI] [PubMed] [Google Scholar]
- Dam T. K.; Brewer C. F. Chem. Rev. 2002, 102, 387. [DOI] [PubMed] [Google Scholar]
- Dwek M. V.; Ross H. A.; Leathem A. J. C. Proteomics 2001, 1, 756. [DOI] [PubMed] [Google Scholar]
- Chen S.; LaRoche T.; Hamelinck D.; Bergsma D.; Brenner D.; Simeone D.; Brand R. E.; Haab B. B. Nat. Methods 2007, 4, 437. [DOI] [PubMed] [Google Scholar]
- Wang L.; Cummings R. D.; Smith D. F.; Huflejt M.; Campbell C. T.; Gildersleeve J. C.; Gerlach J. Q.; Kilcoyne M.; Joshi L.; Serna S.; Reichardt N.-C.; Parera Pera N.; Pieters R.; Eng W.; Mahal L. K. Glycobiology 2014, 24, 507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geissner A.; Anish C.; Seeberger P. H. Curr. Opin. Chem. Biol. 2014, 18, 38. [DOI] [PubMed] [Google Scholar]
- Arthur C. M.; Cummings R. D.; Stowell S. R. Curr. Opin. Chem. Biol. 2014, 18, 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galban-Horcajo F.; Halstead S. K.; McGonigal R.; Willison H. J. Curr. Opin. Chem. Biol. 2014, 18, 78. [DOI] [PubMed] [Google Scholar]
- Palma A. S.; Feizi T.; Childs R. A.; Chai W.; Liu Y. Curr. Opin. Chem. Biol. 2014, 18, 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luong J. H. T.; Habibi-Rezaei M.; Meghrous J.; Xiao C.; Male K. B.; Kamen A. Anal. Chem. 2001, 73, 1844. [DOI] [PubMed] [Google Scholar]
- Ertl P.; Mikkelsen S. R. Anal. Chem. 2001, 73, 4241. [DOI] [PubMed] [Google Scholar]
- Ertl P.; Wagner M.; Corton E.; Mikkelsen S. R. Biosens. Bioelectron. 2003, 18, 907. [DOI] [PubMed] [Google Scholar]
- Spinelli N.; Defrancq E.; Morvan F. Chem. Soc. Rev. 2013, 42, 4557. [DOI] [PubMed] [Google Scholar]
- Reichardt N. C.; Martin-Lomas M.; Penades S. Chem. Soc. Rev. 2013, 42, 4358. [DOI] [PubMed] [Google Scholar]
- Gingras M.; Chabre Y. M.; Roy M.; Roy R. Chem. Soc. Rev. 2013, 42, 4823. [DOI] [PubMed] [Google Scholar]
- Martos-Maldonado M. C.; Casas-Solvas J. M.; Quesada-Soriano I.; García-Fuentes L.; Vargas-Berenguel A. Langmuir 2013, 29, 1318. [DOI] [PubMed] [Google Scholar]
- Kikkeri R.; Kamena F.; Gupta T.; Hossain L. H.; Boonyarattanakalin S.; Gorodyska G.; Beurer E.; Coullerez G. r.; Textor M.; Seeberger P. H. Langmuir 2009, 26, 1520. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Luo S.; Tang Y.; Yu L.; Hou K.-Y.; Cheng J.-P.; Zeng X.; Wang P. G. Anal. Chem. 2006, 78, 2001. [DOI] [PubMed] [Google Scholar]
- Szunerits S.; Niedziolka-Jonsson J.; Boukherroub R.; Woisel P.; Baumann J.-S.; Siriwardena A. Anal. Chem. 2010, 82, 8203. [DOI] [PubMed] [Google Scholar]
- He X.-P.; Wang X.-W.; Jin X.-P.; Zhou H.; Shi X.-X.; Chen G.-R.; Long Y.-T. J. Am. Chem. Soc. 2011, 133, 3649. [DOI] [PubMed] [Google Scholar]
- Loaiza O. A.; Lamas-Ardisana P. J.; Jubete E.; Ochoteco E.; Loinaz I.; Cabañero G. n.; García I.; Penadés S. Anal. Chem. 2011, 83, 2987. [DOI] [PubMed] [Google Scholar]
- Li Z.; Deng S.-S.; Zang Y.; Gu Z.; He X.-P.; Chen G.-R.; Chen K.; James T. D.; Li J.; Long Y.-T. Sci. Rep. 2013, 3, 2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alava T.; Mann J. A.; Théodore C.; Benitez J. J.; Dichtel W. R.; Parpia J. M.; Craighead H. G. Anal. Chem. 2013, 85, 2754. [DOI] [PubMed] [Google Scholar]
- Geisler C.; Jarvis D. L. Glycobiology 2011, 21, 988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Z.; Partyka K.; McDonald M.; Brouhard E.; Hincapie M.; Brand R. E.; Hancock W. S.; Haab B. B. Anal. Chem. 2013, 85, 1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald R. E.; Hughes D. J.; Davis B. G. Angew. Chem., Int. Ed. 2004, 43, 3025. [DOI] [PubMed] [Google Scholar]
- Lu Y. W.; Chien C. W.; Lin P. C.; Huang L. D.; Chen C. Y.; Wu S. W.; Han C. L.; Khoo K. H.; Lin C. C.; Chen Y. J. Anal. Chem. 2013, 85, 8268. [DOI] [PubMed] [Google Scholar]
- Hsu K.-L.; Gildersleeve J. C.; Mahal L. K. Mol. BioSyst. 2008, 4, 654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Propheter D. C.; Hsu K.-L.; Mahal L. K. ChemBioChem 2010, 11, 1203. [DOI] [PubMed] [Google Scholar]
- Propheter D. C.; Mahal L. K. Mol. BioSyst. 2011, 7, 2114. [DOI] [PubMed] [Google Scholar]
- Guan J.-G.; Miao Y.-Q.; Zhang Q.-J. J. Biosci. Bioeng. 2004, 97, 219. [DOI] [PubMed] [Google Scholar]
- Bertok T.; Klukova L.; Sediva A.; Kasak P.; Semak V.; Micusik M.; Omastova M.; Chovanova L.; Vlcek M.; Imrich R.; Vikartovska A.; Tkac J. Anal. Chem. 2013, 85, 7324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S.; Bakovic L.; Chen A. J. Electroanal. Chem. 2006, 591, 210. [Google Scholar]
- Liu S.; Wang K.; Du D.; Sun Y.; He L. Biomacromolecules 2007, 8, 2142. [DOI] [PubMed] [Google Scholar]
- Angata T.; Varki A. Chem. Rev. 2002, 102, 439. [DOI] [PubMed] [Google Scholar]
- La Belle J. T.; Gerlach J. Q.; Svarovsky S.; Joshi L. Anal. Chem. 2007, 79, 6959. [DOI] [PubMed] [Google Scholar]
- Oliveira M. D. L.; Correia M. T. S.; Coelho L. C. B. B.; Diniz F. B. Colloids Surf., B 2008, 66, 13. [DOI] [PubMed] [Google Scholar]
- Oliveira M. D. L.; Andrade C. A. S.; Correia M. T. S.; Coelho L. C. B. B.; Singh P. R.; Zeng X. J. Colloid Interface Sci. 2011, 362, 194. [DOI] [PubMed] [Google Scholar]
- Xi F.; Gao J.; Wang J.; Wang Z. J. Electroanal. Chem. 2011, 656, 252. [Google Scholar]
- Nagaraj V. J.; Aithal S.; Eaton S.; Bothara M.; Wiktor P.; Prasad S. Nanomedicine 2010, 5, 369. [DOI] [PubMed] [Google Scholar]
- Bertok T.; Gemeiner P.; Mikula M.; Gemeiner P.; Tkac J. Microchim. Acta 2013, 180, 151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertok T.; Sediva A.; Katrlik J.; Gemeiner P.; Mikula M.; Nosko M.; Tkac J. Talanta 2013, 108, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertok T.; Sediva A.; Vikartovska A.; Tkac J. Int. J. Electrochem. Sci. 2014, 9, 890. [PMC free article] [PubMed] [Google Scholar]
- Venkatanarayanan A.; Keyes T. E.; Forster R. J. Anal. Chem. 2013, 85, 2216. [DOI] [PubMed] [Google Scholar]
- Dijksma M.; Kamp B.; Hoogvliet J. C.; van Bennekom W. P. Anal. Chem. 2001, 73, 901. [DOI] [PubMed] [Google Scholar]
- Bart M.; Stigter E. C. A.; Stapert H. R.; de Jong G. J.; van Bennekom W. P. Biosens. Bioelectron. 2005, 21, 49. [DOI] [PubMed] [Google Scholar]
- Luo X.; Xu M.; Freeman C.; James T.; Davis J. J. Anal. Chem. 2013, 85, 4129. [DOI] [PubMed] [Google Scholar]
- Kiilerich-Pedersen K.; Daprà J.; Cherré S.; Rozlosnik N. Biosens. Bioelectron. 2013, 49, 374. [DOI] [PubMed] [Google Scholar]
- Cui H.; Li S.; Yuan Q.; Wadhwa A.; Eda S.; Chambers M.; Ashford R.; Jiang H.; Wu J. Analyst 2013, 138, 7188. [DOI] [PubMed] [Google Scholar]
- Xu Q.; Davis J. J. Electroanalysis 2014, 26, 1249. [Google Scholar]
- Chen W.; Lu Z.; Li C. M. Anal. Chem. 2008, 80, 8485. [DOI] [PubMed] [Google Scholar]
- Bryan T.; Luo X.; Bueno P. R.; Davis J. J. Biosens. Bioelectron. 2013, 39, 94. [DOI] [PubMed] [Google Scholar]
- Chen X.; Wang Y.; Zhou J.; Yan W.; Li X.; Zhu J.-J. Anal. Chem. 2008, 80, 2133. [DOI] [PubMed] [Google Scholar]
- Xu M.; Luo X.; Davis J. J. Biosens. Bioelectron. 2013, 39, 21. [DOI] [PubMed] [Google Scholar]
- Lee S.; Song K. M.; Jeon W.; Jo H.; Shim Y. B.; Ban C. Biosens. Bioelectron. 2012, 35, 291. [DOI] [PubMed] [Google Scholar]
- Luo X.; Xu Q.; James T.; Davis J. J. Anal. Chem. 2014, 86, 5553. [DOI] [PubMed] [Google Scholar]
- Kongsuphol P.; Ng H. H.; Pursey J. P.; Arya S. K.; Wong C. C.; Stulz E.; Park M. K. Biosens. Bioelectron. 2014, 61, 274. [DOI] [PubMed] [Google Scholar]
- Berggren C.; Johansson G. Anal. Chem. 1997, 69, 3651. [DOI] [PubMed] [Google Scholar]
- Berggren C.; Bjarnason B.; Johansson G. Electroanalysis 2001, 13, 173. [Google Scholar]
- Labib M.; Hedstrom M.; Amin M.; Mattiasson B. Biotechnol. Bioeng. 2009, 104, 312. [DOI] [PubMed] [Google Scholar]
- Loyprasert S.; Hedstrom M.; Thavarungkul P.; Kanatharana P.; Mattiasson B. Biosens. Bioelectron. 2010, 25, 1977. [DOI] [PubMed] [Google Scholar]
- Strmecki S.; Plavsic M. Electrochem. Commun. 2012, 18, 100. [Google Scholar]
- Strmecki S.; Plavsic M.; Cosovic B.; Ostatna V.; Palecek E. Electrochem. Commun. 2009, 11, 2032. [Google Scholar]
- Roberts G. A. F.Chitin Chemistry; MacMillan: Indianapolis, IN, 1992. [Google Scholar]
- Saburo M.; Minoru M.; Yoshiharu O.; Hiroyuki S.; Yoshihiro S. In Material Science of Chitin and Chitosan; Uragami T., Tokura S., Eds.; Springer: Tokyo, 2006. [Google Scholar]
- Kumar M. N. V. R.; Muzzarelli R. A. A.; Muzzarelli C.; Sashiwa H.; Domb A. J. Chem. Rev. 2004, 104, 6017. [DOI] [PubMed] [Google Scholar]
- Polysaccharide Building Blocks; Habibi Y., Lucia L. A., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, 2012. [Google Scholar]
- Suginta W.; Khunkaewla P.; Schulte A. Chem. Rev. 2013, 113, 5458. [DOI] [PubMed] [Google Scholar]
- Zhang J. J.; Cheng F. F.; Zheng T. T.; Zhu J. J. Anal. Chem. 2010, 82, 3547. [DOI] [PubMed] [Google Scholar]
- Ding L.; Ji Q.; Qian R.; Cheng W.; Ju H. Anal. Chem. 2010, 82, 1292. [DOI] [PubMed] [Google Scholar]
- An Y.; Jiang X.; Bi W.; Chen H.; Jin L.; Zhang S.; Wang C.; Zhang W. Biosens. Bioelectron. 2012, 32, 224. [DOI] [PubMed] [Google Scholar]
- Du D.; Ju H.; Zhang X.; Chen J.; Cai J.; Chen H. Biochemistry 2005, 44, 11539. [DOI] [PubMed] [Google Scholar]
- Shao M.-L.; Bai H.-J.; Gou H.-L.; Xu J.-J.; Chen H.-Y. Langmuir 2009, 25, 3089. [DOI] [PubMed] [Google Scholar]
- Daniel M. C.; Astruc D. Chem. Rev. 2004, 104, 293. [DOI] [PubMed] [Google Scholar]
- Katz E.; Willner I.; Wang J. Electroanalysis 2004, 16, 19. [Google Scholar]
- Soleymani L.; Fang Z.; Sargent E. H.; Kelley S. O. Nat. Nanotechnol. 2009, 4, 844. [DOI] [PubMed] [Google Scholar]
- Yu X.; Munge B.; Patel V.; Jensen G.; Bhirde A.; Gong J. D.; Kim S. N.; Gillespie J.; Gutkind J. S.; Papadimitrakopoulos F.; Rusling J. F. J. Am. Chem. Soc. 2006, 128, 11199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill R.; Zayats M.; Willner I. Angew. Chem., Int. Ed. 2008, 47, 7602. [DOI] [PubMed] [Google Scholar]
- Penn S. G.; He L.; Natan M. J. Curr. Opin. Chem. Biol. 2003, 7, 609. [DOI] [PubMed] [Google Scholar]
- Palecek E. Methods Enzymol. 1992, 212, 139. [DOI] [PubMed] [Google Scholar]
- Palecek E. Methods Enzymol. 1992, 212, 305. [DOI] [PubMed] [Google Scholar]
- Trefulka M.; Ostatna V.; Havran L.; Fojta M.; Palecek E. Electroanalysis 2007, 19, 1281. [Google Scholar]
- Bartosik M.; Hrstka R.; Palecek E.; Vojtesek B. Anal. Chim. Acta 2014, 813, 35. [DOI] [PubMed] [Google Scholar]
- Bartosik M.; Hrstka R.; Palecek E.; Vojtesek B. Electroanalysis 2014, 26, 2558. [Google Scholar]
- Trefulka M.; Palecek E. Electroanalysis 2009, 21, 1763. [Google Scholar]
- Trefulka M.; Bartosik M.; Palecek E. Electrochem. Commun. 2010, 12, 1760. [Google Scholar]
- Trefulka M.; Palecek E. Electroanalysis 2013, 25, 1813. [Google Scholar]
- Trefulka M.; Palecek E. Electroanalysis 2010, 22, 1837. [Google Scholar]
- Trefulka M.; Palecek E. Chem. Pap. 2014, 10.1515/Chempap-2015-0010. [DOI] [Google Scholar]
- Trefulka M.; Paleček E. Electrochem. Commun. 2014, 48, 52. [Google Scholar]
- Dai Z.; Kawde A.-N.; Xiang Y.; La Belle J. T.; Gerlach J.; Bhavanandan V. P.; Joshi L.; Wang J. J. Am. Chem. Soc. 2006, 128, 10018. [DOI] [PubMed] [Google Scholar]
- Zhang J.-J.; Zheng T.-T.; Cheng F.-F.; Zhang J.-R.; Zhu J.-J. Anal. Chem. 2011, 83, 7902. [DOI] [PubMed] [Google Scholar]
- Yang C.; Xu C.; Wang X.; Hu X. Analyst 2012, 137, 1205. [DOI] [PubMed] [Google Scholar]
- Kuramitz H.; Mawatari Y.; Ikeuchi M.; Kutomi O.; Hata N.; Taguchi S.; Sugawara K. Anal. Sci. 2012, 28, 77. [DOI] [PubMed] [Google Scholar]
- Yang C.; Gu B.; Xu C.; Xu X. J. Electroanal. Chem. 2011, 660, 97. [Google Scholar]
- Xue Y.; Ding L.; Lei J.; Ju H. Biosens. Bioelectron. 2010, 26, 169. [DOI] [PubMed] [Google Scholar]
- Ding C.; Qian S.; Wang Z.; Qu B. Anal. Biochem. 2011, 414, 84. [DOI] [PubMed] [Google Scholar]
- Sugawara K.; Yugami A.; Kadoya T.; Hosaka K. Talanta 2011, 85, 425. [DOI] [PubMed] [Google Scholar]
- Sugawara K.; Yugami A.; Kadoya T.; Kuramitz H.; Hosaka K. Analyst 2012, 137, 3781. [DOI] [PubMed] [Google Scholar]
- Yang H.; Wang Y.; Qi H.; Gao Q.; Zhang C. Biosens. Bioelectron. 2012, 35, 376. [DOI] [PubMed] [Google Scholar]
- Wang J.; Liu G.; Jan M. R. J. Am. Chem. Soc. 2004, 126, 3010. [DOI] [PubMed] [Google Scholar]
- Das J.; Aziz M. A.; Yang H. J. Am. Chem. Soc. 2006, 128, 16022. [DOI] [PubMed] [Google Scholar]
- Nam J. M.; Thaxton C. S.; Mirkin C. A. Science 2003, 301, 1884. [DOI] [PubMed] [Google Scholar]
- Hou S. Y.; Chen H. K.; Cheng H. C.; Huang C. Y. Anal. Chem. 2006, 79, 980. [DOI] [PubMed] [Google Scholar]
- Gerlach J. Q.; Kilcoyne M.; Joshi L. Anal. Methods 2014, 6, 440. [Google Scholar]
- Kamiya Y.; Satoh T.; Kato K. Biochim. Biophys. Acta, Gen. Subj. 2012, 1820, 1327. [DOI] [PubMed] [Google Scholar]
- Kletter D.; Singh S.; Bern M.; Haab B. B. Mol. Cell. Proteomics 2013, 12, 1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z.; Chinoy Z. S.; Ambre S. G.; Peng W.; McBride R.; de Vries R. P.; Glushka J.; Paulson J. C.; Boons G.-J. Science 2013, 341, 379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rillahan C. D.; Antonopoulos A.; Lefort C. T.; Sonon R.; Azadi P.; Ley K.; Dell A.; Haslam S. M.; Paulson J. C. Nat. Chem. Biol. 2012, 8, 661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasooly A. Biosens. Bioelectron. 2006, 21, 1847. [DOI] [PubMed] [Google Scholar]
- Thompson I. M.; Pauler D. K.; Goodman P. J.; Tangen C. M.; Lucia M. S.; Parnes H. L.; Minasian L. M.; Ford L. G.; Lippman S. M.; Crawford E. D.; Crowley J. J.; Coltman C. A. N. Engl. J. Med. 2004, 350, 2239. [DOI] [PubMed] [Google Scholar]
- Nakashima J.; Tachibana M.; Horiguchi Y.; Oya M.; Ohigashi T.; Asakura H.; Murai M. Clin. Cancer Res. 2000, 6, 2702. [PubMed] [Google Scholar]
- Yang H. Curr. Opin. Chem. Biol. 2012, 16, 422. [DOI] [PubMed] [Google Scholar]
- Gan T.; Hu S. S. Microchim. Acta 2011, 175, 1. [Google Scholar]
- Kuila T.; Bose S.; Khanra P.; Mishra A. K.; Kim N. H.; Lee J. H. Biosens. Bioelectron. 2011, 26, 4637. [DOI] [PubMed] [Google Scholar]
- Pei X. M.; Zhang B.; Tang J.; Liu B. Q.; Lai W. Q.; Tang D. P. Anal. Chim. Acta 2013, 758, 1. [DOI] [PubMed] [Google Scholar]
- Ricci F.; Adornetto G.; Palleschi G. Electrochim. Acta 2012, 84, 74. [Google Scholar]
- Diaconu I.; Cristea C.; Harceaga V.; Marrazza G.; Berindan-Neagoe I.; Sandulescu R. Clin. Chim. Acta 2013, 425, 128. [DOI] [PubMed] [Google Scholar]
- Tang D. P.; Cui Y. L.; Chen G. A. Analyst 2013, 138, 981. [DOI] [PubMed] [Google Scholar]
- Wan Y.; Su Y.; Zhu X. H.; Liu G.; Fan C. H. Biosens. Bioelectron. 2013, 47, 1. [DOI] [PubMed] [Google Scholar]
- Mendoza V. L.; Vachet R. W. Mass Spectrom. Rev. 2009, 28, 785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klymchenko A. S.; Avilov S. V.; Demchenko A. P. Anal. Biochem. 2004, 329, 43. [DOI] [PubMed] [Google Scholar]
- Paus E. Biochim. Biophys. Acta 1978, 533, 446. [DOI] [PubMed] [Google Scholar]
- Spande T. F.; Witkop B. Methods Enzymol. 1967, 11, 498. [Google Scholar]
- Spande T. F.; Witkop B. Methods Enzymol. 1967, 11, 528. [Google Scholar]
- Takaoka Y.; Ojida A.; Hamachi I. Angew. Chem., Int. Ed. 2013, 52, 4088. [DOI] [PubMed] [Google Scholar]
- Salmain M.; Jaouen G. C. R. Chim. 2003, 6, 249. [Google Scholar]
- Gitlin G.; Bayer E. A.; Wilchek M. Biochem. J. 1988, 256, 279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gitlin G.; Bayer E. A.; Wilchek M. Biochem. J. 1988, 250, 291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gitlin G.; Bayer E. A.; Wilchek M. Biochem. J. 1990, 269, 527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuyama H.; Watanabe M.; Toda C.; Ando E.; Tanaka K.; Nishimura O. Rapid Commun. Mass Spectrom. 2003, 17, 1642. [DOI] [PubMed] [Google Scholar]
- Strohalm M.; Kodicek M.; Pechar M. Biochem. Biophys. Res. Commun. 2003, 312, 811. [DOI] [PubMed] [Google Scholar]
- Konermann L.; Stocks B. B.; Pan Y.; Tong X. Mass Spectrom. Rev. 2010, 29, 651. [DOI] [PubMed] [Google Scholar]
- Bediako-Amoa I.; Sutherland T. C.; Li C. Z.; Silerova R.; Kraatz H. B. J. Phys. Chem. B 2004, 108, 704. [Google Scholar]
- DiGleria K.; Hill H. A. O.; Wong L. L. FEBS Lett. 1996, 390, 142. [DOI] [PubMed] [Google Scholar]
- Kraatz H. B. J. Inorg. Organomet. Polym. Mater. 2005, 15, 83. [Google Scholar]
- Lo K. K. W.; Ng D. C. M.; Lau J. S. Y.; Wu R. S. S.; Lam P. K. S. New J. Chem. 2003, 27, 274. [Google Scholar]
- Plumb K.; Kraatz H. B. Bioconjugate Chem. 2003, 14, 601. [DOI] [PubMed] [Google Scholar]
- Xu Y. M.; Saweczko P.; Kraatz H. B. J. Organomet. Chem. 2001, 637, 335. [Google Scholar]
- Martic S.; Labib M.; Kraatz H.-B. Analyst 2011, 136, 107. [DOI] [PubMed] [Google Scholar]
- Kerman K.; Kraatz H.-B. Analyst 2009, 134, 2400. [DOI] [PubMed] [Google Scholar]
- Song H.; Kerman K.; Kraatz H.-B. Chem. Commun. 2008, 4, 502. [DOI] [PubMed] [Google Scholar]
- Mahmoud K. A.; Kraatz H.-B. Chem.—Eur. J. 2007, 13, 5885. [DOI] [PubMed] [Google Scholar]
- Kerman K.; Song H.; Duncan J. S.; Litchfield D. W.; Kraatz H.-B. Anal. Chem. 2008, 80, 9395. [DOI] [PubMed] [Google Scholar]
- Martic S.; Labib M.; Freeman D.; Kraatz H.-B. Chem.—Eur. J. 2011, 17, 6744. [DOI] [PubMed] [Google Scholar]
- Labib M.; Shipman P. O.; Martic S.; Kraatz H.-B. Electrochim. Acta 2011, 56, 5122. [Google Scholar]
- Martic S.; Rains M. K.; Haftchenary S.; Shahani V. M.; Kraskouskaya D.; Ball D. P.; Gunning P. T.; Kraatz H. B. Mol. BioSyst. 2014, 10, 576. [DOI] [PubMed] [Google Scholar]
- Beheshti S.; Martic S.; Kraatz H.-B. ChemPhysChem 2012, 13, 542. [DOI] [PubMed] [Google Scholar]
- Martic S.; Gabriel M.; Turowec J. P.; Litchfield D. W.; Kraatz H.-B. J. Am. Chem. Soc. 2012, 134, 17036. [DOI] [PubMed] [Google Scholar]
- Samanta U.; Pal D.; Chakrabarti P. Proteins: Struct., Funct., Genet. 2000, 38, 288. [PubMed] [Google Scholar]
- Blake C. C. F.; Koenig D. F.; Mair G. A.; North A. C. T.; Phillips D. C.; Sarma V. R. Nature 1965, 206, 757. [DOI] [PubMed] [Google Scholar]
- Chilkoti A.; Tan P. H.; Stayton P. S. Proc. Natl. Acad. Sci. U.S.A. 1995, 92, 1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sano T.; Cantor C. R. Proc. Natl. Acad. Sci. U.S.A. 1995, 92, 3180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maenaka K.; Kawai G.; Watanabe K.; Sunada F.; Kumagai I. J. Biol. Chem. 1994, 269, 7070. [PubMed] [Google Scholar]
- Spande T. F.; Witkop B. Methods Enzymol. 1967, 11, 522. [Google Scholar]
- Kurzban G. P.; Gitlin G.; Bayer E. A.; Wilchek M.; Horowitz P. M. Biochemistry 1989, 28, 8537. [DOI] [PubMed] [Google Scholar]
- Kurzban G. P.; Gitlin G.; Bayer E. A.; Wilchek M.; Horowitz P. M. J. Protein Chem. 1990, 9, 673. [DOI] [PubMed] [Google Scholar]
- Billova S.; Kizek R.; Palecek E. Bioelectrochemistry 2002, 56, 63. [DOI] [PubMed] [Google Scholar]
- Sedo O.; Billova S.; Pena-Mendez E. M.; Palecek E.; Havel J. Anal. Chim. Acta 2004, 515, 261. [Google Scholar]
- Fojta M.; Kostecka P.; Pivonkova H.; Horakova P.; Havran L. Curr. Anal. Chem. 2011, 7, 35. [Google Scholar]
- Deetz J. S.; Behrman E. J. Int. J. Pept. Protein Res. 1981, 17, 495. [DOI] [PubMed] [Google Scholar]
- Fojta M.; Billova S.; Havran L.; Pivonkova H.; Cernocka H.; Horakova P.; Palecek E. Anal. Chem. 2008, 80, 4598. [DOI] [PubMed] [Google Scholar]
- Dey R. S.; Bera R. K.; Raj C. R. Anal. Bioanal. Chem. 2013, 405, 3431. [DOI] [PubMed] [Google Scholar]
- Diaz-Gonzalez M.; Gonzalez-Garcia M. B.; Costa-Garcia A. Electroanalysis 2005, 17, 1901. [Google Scholar]
- Liu G. D.; Lin Y. H. Talanta 2007, 74, 308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breyer B.; Radcliff F. J. Nature 1951, 167, 79. [DOI] [PubMed] [Google Scholar]
- Heineman W. R.; Halsall H. B. Anal. Chem. 1985, 57, 1321. [DOI] [PubMed] [Google Scholar]
- Limoges B.; Marchal D.; Mavre F.; Saveant J. M. J. Am. Chem. Soc. 2008, 130, 7276. [DOI] [PubMed] [Google Scholar]
- Salam F.; Tothill I. E. Biosens. Bioelectron. 2009, 24, 2630. [DOI] [PubMed] [Google Scholar]
- MacBeath G.; Schreiber S. L. Science 2000, 289, 1760. [DOI] [PubMed] [Google Scholar]
- Campuzano S.; Kuralay F.; Lobo-Castanon M. J.; Bartosik M.; Vyavahare K.; Palecek E.; Haake D. A.; Wang J. Biosens. Bioelectron. 2011, 26, 3577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J.; Campuzano S.; Halford C.; Haake D. A.; Wang J. Anal. Chem. 2010, 82, 8830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J.; Tang J. H.; Dai Z.; Yan F.; Ju H. X.; El Murr N. Biosens. Bioelectron. 2006, 22, 102. [DOI] [PubMed] [Google Scholar]
- Wu J.; Yan F.; Tang J. H.; Zhai C.; Ju H. X. Clin. Chem. 2007, 53, 1495. [DOI] [PubMed] [Google Scholar]
- Wu J.; Yan F.; Zhang X. Q.; Yan Y. T.; Tang J. H.; Ju H. X. Clin. Chem. 2008, 54, 1481. [DOI] [PubMed] [Google Scholar]
- Kojima K.; Hiratsuka A.; Suzuki H.; Yano K.; Ikebukuro K.; Karube I. Anal. Chem. 2003, 75, 1116. [DOI] [PubMed] [Google Scholar]
- Wilson M. S. Anal. Chem. 2005, 77, 1496. [DOI] [PubMed] [Google Scholar]
- Wilson M. S.; Nie W. Y. Anal. Chem. 2006, 78, 2507. [DOI] [PubMed] [Google Scholar]
- Steude A.; Schmidt S.; Robitzki A. A.; Panke O. Lab Chip 2011, 11, 2884. [DOI] [PubMed] [Google Scholar]
- Du D.; Zou Z. X.; Shin Y. S.; Wang J.; Wu H.; Engelhard M. H.; Liu J.; Aksay I. A.; Lin Y. H. Anal. Chem. 2010, 82, 2989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusling J. F.; Bishop G. W.; Doan N. M.; Papadimitrakopoulos F. J. Mater. Chem. B 2014, 2, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mani V.; Chikkaveeraiah B. V.; Patel V.; Gutkind J. S.; Rusling J. F. ACS Nano 2009, 3, 585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei X.; Xu Z.; Zhang J.; Liu Z.; Tian J. Anal. Methods 2013, 5, 3235. [Google Scholar]
- Malhotra R.; Patel V.; Chikkaveeraiah B. V.; Munge B. S.; Cheong S. C.; Zain R. B.; Abraham M. T.; Dey D. K.; Gutkind J. S.; Rusling J. F. Anal. Chem. 2012, 84, 6249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan Y.; Deng W.; Su Y.; Zhu X.; Peng C.; Hu H.; Peng H.; Song S.; Fan C. Biosens. Bioelectron. 2011, 30, 93. [DOI] [PubMed] [Google Scholar]
- Ravalli A.; Pilon Dos Santos G.; Ferroni M.; Faglia G.; Yamanaka H.; Marrazza G. Sens. Actuators, B 2013, 179, 194. [Google Scholar]
- Florea A.; Taleat Z.; Cristea C.; Mazloum-Ardakani M.; Sandulescu R. Electrochem. Commun. 2013, 33, 127. [Google Scholar]
- Yuan Y. R.; Yuan R.; Chai Y. Q.; Zhuo Y.; Miao X. M. J. Electroanal. Chem. 2009, 626, 6. [Google Scholar]
- Viswanathan S.; Rani C.; Vijay Anand A.; Ho J. A. A. Biosens. Bioelectron. 2009, 24, 1984. [DOI] [PubMed] [Google Scholar]
- Aslan K.; Geddes C. D. Anal. Chem. 2005, 77, 8057. [DOI] [PubMed] [Google Scholar]
- Tanaka K.; Imagawa H. Talanta 2005, 68, 437. [DOI] [PubMed] [Google Scholar]
- Driskell J. D.; Uhlenkamp J. M.; Lipert R. J.; Porter M. D. Anal. Chem. 2007, 79, 4141. [DOI] [PubMed] [Google Scholar]
- Morozov V. N.; Groves S.; Turell M. J.; Bailey C. J. Am. Chem. Soc. 2007, 129, 12628. [DOI] [PubMed] [Google Scholar]
- Morozov V. N.; Morozova T. Y. Anal. Chem. 2003, 75, 6813. [DOI] [PubMed] [Google Scholar]
- Morozov V. N.; Morozova T. Y. Anal. Chim. Acta 2006, 564, 40. [DOI] [PubMed] [Google Scholar]
- Oh S. K.; Cruikshank W. W.; Raina J.; Blanchard G. C.; Adler W. H.; Walker J.; Kornfeld H. J. Acquired Immune Defic. Syndr. 1992, 5, 251. [PubMed] [Google Scholar]
- Gan N.; Wang L.-Y.; Li T.-H.; Zheng L.; Wang F. Chin. J. Anal. Chem. 2009, 37, 1125. [Google Scholar]
- Ellington A. D.; Szostak J. W. Nature 1990, 346, 818. [DOI] [PubMed] [Google Scholar]
- Mascini M.; Palchetti I.; Tombelli S. Angew. Chem., Int. Ed. 2012, 51, 1316. [DOI] [PubMed] [Google Scholar]
- Keefe A. D.; Pai S.; Ellington A. Nat. Rev. Drug Discovery 2010, 9, 537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruigrok V. J. B.; Levisson M.; Eppink M. H. M.; Smidt H.; van der Oost J. Biochem. J. 2011, 436, 1. [DOI] [PubMed] [Google Scholar]
- Tolle F.; Mayer G. Chem. Sci. 2013, 4, 60. [Google Scholar]
- Liu Y.; Tuleouva N.; Ramanculov E.; Revzin A. Anal. Chem. 2010, 82, 8131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miodek A.; Castillo G.; Hianik T.; Korri-Youssoufi H. Anal. Chem. 2013, 85, 7704. [DOI] [PubMed] [Google Scholar]
- Feng L.; Chen Y.; Ren J.; Qu X. Biomaterials 2011, 32, 2930. [DOI] [PubMed] [Google Scholar]
- Yi Z.; Li X.-Y.; Gao Q.; Tang L.-J.; Chu X. Analyst 2013, 138, 2032. [DOI] [PubMed] [Google Scholar]
- Zhang H.-R.; Xia X.-H.; Xu J.-J.; Chen H.-Y. Electrochem. Commun. 2012, 25, 112. [Google Scholar]
- Patel V.; Hood B. L.; Molinolo A.; Lee N. H.; Conrads T. P.; Braisted J. C.; Krizman D. B.; Veenstra T. D.; Gutkind J. S. Clin. Cancer Res. 2008, 14, 1002. [DOI] [PubMed] [Google Scholar]
- Vaught J. D.; Bock C.; Carter J.; Fitzwater T.; Otis M.; Schneider D.; Rolando J.; Waugh S.; Wilcox S. K.; Eaton B. E. J. Am. Chem. Soc. 2010, 132, 4141. [DOI] [PubMed] [Google Scholar]
- Kimoto M.; Yamashige R.; Matsunaga K.-I.; Yokoyama S.; Hirao I. Nat. Biotechnol. 2013, 31, 453. [DOI] [PubMed] [Google Scholar]
- Nie H.; Chen Y.; Lü C.; Liu Z. Anal. Chem. 2013, 85, 8277. [DOI] [PubMed] [Google Scholar]
- Colas P.; Cohen B.; Jessen T.; Grishina I.; McCoy J.; Brent R. Nature 1996, 380, 548. [DOI] [PubMed] [Google Scholar]
- Tkac J.; Bertok T.; Nahalka J.; Gemeiner P. In Lectins: Methods and Protocols; Hirabayashi J., Ed.; Humana Press: New York, 2014. [Google Scholar]
- Woodman R.; Yeh J. T. H.; Laurenson S.; Ferrigno P. K. J. Mol. Biol. 2005, 352, 1118. [DOI] [PubMed] [Google Scholar]
- Stadler L. K. J.; Hoffmann T.; Tomlinson D. C.; Song Q. F.; Lee T.; Busby M.; Nyathi Y.; Gendra E.; Tiede C.; Flanagan K.; Cockell S. J.; Wipat A.; Harwood C.; Wagner S. D.; Knowles M. A.; Davis J. J.; Keegan N.; Ferrigno P. K. Protein Eng., Des. Sel. 2011, 24, 751. [DOI] [PubMed] [Google Scholar]
- Estrela P.; Paul D.; Li P.; Keighley S. D.; Migliorato P.; Laurenson S.; Ferrigno P. K. Electrochim. Acta 2008, 53, 6489. [Google Scholar]
- Wang L.; Estrela P.; Huq E.; Li P.; Thomas S.; Ferrigno P. K.; Paul D.; Adkin P.; Migliorato P. Microelectron. Eng. 2010, 87, 753. [Google Scholar]
- Estrela P.; Paul D.; Song Q.; Stadler L. K. J.; Wang L.; Huq E.; Davis J. J.; Ferrigno P. K.; Migliorato P. Anal. Chem. 2010, 82, 3531. [DOI] [PubMed] [Google Scholar]
- Johnson S.; Evans D.; Laurenson S.; Paul D.; Davies A. G.; Ko Ferrigno P.; Walti C. Anal. Chem. 2008, 80, 978. [DOI] [PubMed] [Google Scholar]
- Evans D.; Johnson S.; Laurenson S.; Davies A. G.; Ko Ferrigno P.; Walti C. J. Biol. 2008, 7, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straw S.; Ferrigno P. K.; Song Q.; Tomlinson D.; Del Galdo F. J. Biomed. Sci. Eng. 2013, 6, 32. [Google Scholar]
- Johnson A.; Song Q.; Ferrigno P. K.; Bueno P. R.; Davis J. J. Anal. Chem. 2012, 84, 6553. [DOI] [PubMed] [Google Scholar]
- Song Q.; Stadler J.; Kurt L.; Peng J.; Ferrigno P. K. Faraday Discuss. 2011, 149, 79. [DOI] [PubMed] [Google Scholar]
- Colas P. J. Biol. 2008, 7, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walti C.; Wirtz R.; Germishuizen W. A.; Bailey D. M.; Pepper M.; Middelberg A. P.; Davies A. G. Langmuir 2003, 19, 981. [Google Scholar]
- Bennett C. M.; Guo M.; Dharmage S. C. Diabetic Med. 2007, 24, 333. [DOI] [PubMed] [Google Scholar]
- Son S. Y.; Yoon H. C. Biochip J. 2008, 2, 116. [Google Scholar]
- Song S. Y.; Yoon H. C. Sens. Actuators, B 2009, 140, 233. [Google Scholar]
- Song S. Y.; Han Y. D.; Park Y. M.; Jeong C. Y.; Yang Y. J.; Kim M. S.; Ku Y.; Yoon H. C. Biosens. Bioelectron. 2012, 35, 355. [DOI] [PubMed] [Google Scholar]
- Parekh R.; Dwek R.; Sutton B.; Fernandes D.; Leung A.; Stanworth D.; Rademacher T.; Mizuochi T.; Taniguchi T.; Matsuta K. Nature 1985, 316, 452. [DOI] [PubMed] [Google Scholar]
- Bondt A.; Selman M. H. J.; Deelder A. M.; Hazes J. M. W.; Willemsen S. P.; Wuhrer M.; Dolhain R. J. E. M. J. Proteome Res. 2013, 12, 4522. [DOI] [PubMed] [Google Scholar]
- Roy S.; Biswas S.; Saroha A.; Sahu D.; Das H. R. Clin. Biochem. 2013, 46, 160. [DOI] [PubMed] [Google Scholar]
- Oliveira M. D. L.; Correia M. T. S.; Diniz F. B. Synth. Met. 2009, 159, 2162. [Google Scholar]
- Oliveira M. D. L.; Correia M. T. S.; Diniz F. B. Biosens. Bioelectron. 2009, 25, 728. [DOI] [PubMed] [Google Scholar]
- Oliveira M. D. L.; Nogueira M. L.; Correia M. T. S.; Coelho L. C. B. B.; Andrade C. A. S. Sens. Actuators, B 2011, 155, 789. [Google Scholar]
- Andrade C. A. S.; Oliveira M. D. L.; de Melo C. P.; Coelho L. C. B. B.; Correia M. T. S.; Nogueira M. L.; Singh P. R.; Zeng X. J. Colloid Interface Sci. 2011, 362, 517. [DOI] [PubMed] [Google Scholar]
- Hong S. A.; Kwon J.; Kim D.; Yang S. Biosens. Bioelectron. 2015, 64, 338. [DOI] [PubMed] [Google Scholar]
- Tousi F.; Hancock W. S.; Hincapie M. Anal. Methods 2011, 3, 20. [DOI] [PubMed] [Google Scholar]
- Lazar I. M.; Lee W.; Lazar A. C. Electrophoresis 2013, 34, 113. [DOI] [PubMed] [Google Scholar]
- Sulova Z.; Mislovicova D.; Gibalova L.; Vajcnerova Z.; Polakova E.; Uhriik B.; Tylkova L.; Kovarova A.; Sedlak J.; Breier A. J. Proteome Res. 2008, 8, 513. [DOI] [PubMed] [Google Scholar]
- Hao C.; Yan F.; Ding L.; Xue Y.; Ju H. Electrochem. Commun. 2007, 9, 1359. [Google Scholar]
- Zhao J.; Zhu L.; Li X.; Bo B.; Shu Y.; Li G. Electrochem. Commun. 2012, 23, 56. [Google Scholar]
- Li Y.; Tian Y.; Rezai T.; Prakash A.; Lopez M. F.; Chan D. W.; Zhang H. Anal. Chem. 2010, 83, 240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffy M. J. Clin. Chem. 2001, 47, 624. [PubMed] [Google Scholar]
- Zhang X.; Wu Y.; Tu Y.; Liu S. Analyst 2008, 133, 485. [DOI] [PubMed] [Google Scholar]
- Maltez-da Costa M.; de la Escosura-Muniz A.; Nogues C.; Barrios L.; Ibanez E.; Merkoci A. Nano Lett. 2012, 12, 4164. [DOI] [PubMed] [Google Scholar]
- Yang H.; Li Z.; Wei X.; Huang R.; Qi H.; Gao Q.; Li C.; Zhang C. Talanta 2013, 111, 62. [DOI] [PubMed] [Google Scholar]
- Cheng W.; Ding L.; Lei J.; Ding S.; Ju H. Anal. Chem. 2008, 80, 3867. [DOI] [PubMed] [Google Scholar]
- Qian Z.; Bai H.-J.; Wang G.-L.; Xu J.-J.; Chen H.-Y. Biosens. Bioelectron. 2010, 25, 2045. [DOI] [PubMed] [Google Scholar]
- Hu Y.; Zuo P.; Ye B.-C. Biosens. Bioelectron. 2013, 43, 79. [DOI] [PubMed] [Google Scholar]
- Chen Z.; Liu Y.; Wang Y.; Zhao X.; Li J. Anal. Chem. 2013, 85, 4431. [DOI] [PubMed] [Google Scholar]
- Zhang M.; Liu H.; Chen L.; Yan M.; Ge L.; Ge S.; Yu J. Biosens. Bioelectron. 2013, 49, 79. [DOI] [PubMed] [Google Scholar]
- Xue Y.; Bao L.; Xiao X.; Ding L.; Lei J.; Ju H. Anal. Biochem. 2011, 410, 92. [DOI] [PubMed] [Google Scholar]
- Cao J.-T.; Hao X.-Y.; Zhu Y.-D.; Sun K.; Zhu J.-J. Anal. Chem. 2012, 84, 6775. [DOI] [PubMed] [Google Scholar]
- Su M.; Ge L.; Kong Q.; Zheng X.; Ge S.; Li N.; Yu J.; Yan M. Biosens. Bioelectron. 2015, 63, 232. [DOI] [PubMed] [Google Scholar]
- Wu L.; Ma C.; Ge L.; Kong Q.; Yan M.; Ge S.; Yu J. Biosens. Bioelectron. 2015, 63, 450. [DOI] [PubMed] [Google Scholar]
- Shao Z.; Li Y.; Yang Q.; Wang J.; Li G. Anal. Bioanal. Chem. 2010, 398, 2963. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Teng Y.; Fu Y.; Zhang S.; Wang T.; Wang C.; Jin L.; Zhang W. Chem. Sci. 2011, 2, 2353. [Google Scholar]
- Han E.; Ding L.; Jin S.; Ju H. Biosens. Bioelectron. 2011, 26, 2500. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Teng Y.; Fu Y.; Xu L.; Zhang S.; He B.; Wang C.; Zhang W. Anal. Chem. 2010, 82, 9455. [DOI] [PubMed] [Google Scholar]
- Liu H.; Xu S.; He Z.; Deng A.; Zhu J.-J. Anal. Chem. 2013, 85, 3385. [DOI] [PubMed] [Google Scholar]
- Chen X.; Wang Y.; Zhang Y.; Chen Z.; Liu Y.; Li Z.-L.; Li J. Anal. Chem. 2014, 86, 4278. [DOI] [PubMed] [Google Scholar]
- Chen X.; He Y.; Zhang Y.; Liu M.; Liu Y.; Li J. Nanoscale 2014, 6, 11196. [DOI] [PubMed] [Google Scholar]
- Campuzano S.; Orozco J.; Kagan D.; Guix M.; Gao W.; Sattayasamitsathit S.; Claussen J. C.; Merkoci A.; Wang J. Nano Lett. 2012, 12, 396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H.; Li Y.; Sun K.; Fan J.; Zhang P.; Meng J.; Wang S.; Jiang L. J. Am. Chem. Soc. 2013, 135, 7603. [DOI] [PubMed] [Google Scholar]
- Kuralay F.; Sattayasamitsathit S.; Gao W.; Uygun A.; Katzenberg A.; Wang J. J. Am. Chem. Soc. 2012, 134, 15217. [DOI] [PubMed] [Google Scholar]
- Wang J.; Gao W. ACS Nano 2012, 6, 5745. [DOI] [PubMed] [Google Scholar]
- Garcia-Gradilla V.; Orozco J.; Sattayasamitsathit S.; Soto F.; Kuralay F.; Pourazary A.; Katzenberg A.; Gao W.; Shen Y.; Wang J. ACS Nano 2013, 7, 9232. [DOI] [PubMed] [Google Scholar]
- Glover J. N.; Harrison S. C. Nature 1995, 373, 257. [DOI] [PubMed] [Google Scholar]
- Munge B. S.; Krause C. E.; Malhotra R.; Patel V.; Gutkind J. S.; Rusling J. F. Electrochem. Commun. 2009, 11, 1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G. D.; Wang J.; Kim J.; Jan M. R.; Collins G. E. Anal. Chem. 2004, 76, 7126. [DOI] [PubMed] [Google Scholar]
- Tang D. P.; Hou L.; Niessner R.; Xu M. D.; Gao Z. Q.; Knopp D. Biosens. Bioelectron. 2013, 46, 37. [DOI] [PubMed] [Google Scholar]
- Radi A. E.; Sanchez J. L. A.; Baldrich E.; O’Sullivan C. K. J. Am. Chem. Soc. 2006, 128, 117. [DOI] [PubMed] [Google Scholar]
- Mir M.; Vreeke M.; Katakis L. Electrochem. Commun. 2006, 8, 505. [Google Scholar]
- Shu H. W.; Wen W.; Xiong H. Y.; Zhang X. H.; Wang S. F. Electrochem. Commun. 2013, 37, 15. [Google Scholar]
- Vector Laboratories, Table of Lectin Properties; https://www.vectorlabs.com/data/brochures/K4-K7.pdf, December 2, 2014.
- Zani A.; Laschi S.; Mascini M.; Marrazza G. Electroanalysis 2011, 23, 91. [Google Scholar]
- Yan M.; Zang D.; Ge S.; Ge L.; Yu J. Biosens. Bioelectron. 2012, 38, 355. [DOI] [PubMed] [Google Scholar]
- Akter R.; Rahman M. A.; Rhee C. K. Anal. Chem. 2012, 84, 6407. [DOI] [PubMed] [Google Scholar]
- Chiriaco M. S.; Primiceri E.; Montanaro A.; de Feo F.; Leone L.; Rinaldi R.; Maruccio G. Analyst 2013, 138, 5404. [DOI] [PubMed] [Google Scholar]
- Fragoso A.; Latta D.; Laboria N.; von Germar F.; Hansen-Hagge T. E.; Kemmner W.; Gartner C.; Klemm R.; Drese K. S.; O’Sullivan C. K. Lab Chip 2011, 11, 625. [DOI] [PubMed] [Google Scholar]
- Lin D.; Wu J.; Wang M.; Yan F.; Ju H. Anal. Chem. 2012, 84, 3662. [DOI] [PubMed] [Google Scholar]
- Tang J.; Tang D. P.; Niessner R.; Chen G. N.; Knopp D. Anal. Chem. 2011, 83, 5407. [DOI] [PubMed] [Google Scholar]
- Mohan K.; Donavan K. C.; Arter J. A.; Penner R. M.; Weiss G. A. J. Am. Chem. Soc. 2013, 135, 7761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L. F.; Li S. J.; He J.; Tian G. H.; Wei Q.; Li H. Biosens. Bioelectron. 2013, 49, 222. [DOI] [PubMed] [Google Scholar]
- Tang C. K.; Vaze A.; Rusling J. F. Lab Chip 2012, 12, 281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Havlikova M.; Zatloukalova M.; Ulrichova J.; Dobes P.; Vacek J. Anal. Chem. 2015, 87, 1757. [DOI] [PubMed] [Google Scholar]