

Abstract
The ability of the innate immune system to trigger an adaptive T cell response is critical to resolution of infection with the fungal pathogen Histoplasma capsulatum. However, the signaling pathways and cell types involved in the recognition of and response to this respiratory pathogen remain poorly defined. Here, we show that MyD88, an adaptor protein vital to multiple innate immune pathways, is critically required for the host response to Histoplasma. MyD88-deficient (MyD88−/−) mice are unable to control the fungal burden and are more sensitive to Histoplasma infection than wild-type, Dectin-1−/−, or interleukin 1 receptor-deficient (IL-1R−/−) mice. We found that MyD88 is necessary for the production of key early inflammatory cytokines and the subsequent recruitment of inflammatory monocytes to the lung. In both our in vitro and ex vivo analyses, MyD88 was intrinsically required in dendritic cells and alveolar macrophages for initial cytokine production. Additionally, MyD88-deficient bone marrow-derived dendritic cells fail to efficiently control fungal growth when cocultured with primed splenic T cells. Surprisingly, mice that lack MyD88 only in dendritic cells and alveolar macrophages are competent for early cytokine production and normal survival, indicating the presence of compensatory and redundant MyD88 signaling in other cell types during infection. Ultimately, global MyD88 deficiency prevents proper T cell activation and gamma interferon (IFN-γ) production, which are critical for infection resolution. Collectively, this work reveals a central role for MyD88 in coordinating the innate and adaptive immune responses to infection with this ubiquitous fungal pathogen of humans.
INTRODUCTION
Histoplasma capsulatum is the most common cause of fungal respiratory infections in immunocompetent hosts in the United States (1–3). The organism exists in the environment in a sporulating filamentous form that is easily aerosolized and inhaled by the mammalian host. Inside the host, fungal cells convert into a pathogenic yeast form that is able to evade immune defenses by replicating within macrophages. In a healthy host, the adaptive immune response is critical for bringing the disease under control, and individuals with defects in adaptive immunity frequently fail to contain Histoplasma infections and succumb to disseminated disease (1, 4, 5).
Successful activation of an adaptive immune response depends on the early innate events that occur during microbial infection (6). In general, these events are initiated by resident immune cells in the lung, including alveolar macrophages and dendritic cells, which recognize and respond to invading pathogens by directly controlling pathogen growth, secreting antimicrobial products, and producing proinflammatory cytokines, ultimately leading to an adaptive T cell response (7). During Histoplasma infection, both Th1 and Th17 responses contribute to the activation of macrophages to restrict and control fungal growth (8–12). If early innate immune events fail to occur and the appropriate immune response is disrupted, infection can continue unchecked and lead to disseminated disease and mortality. In the case of infection by Histoplasma, the precise events required to initiate an appropriate innate immune response and curtail disease progression remain poorly defined.
In a healthy host, resolution of Histoplasma infection requires a Th1 CD4+ T cell response (13). Depleting either CD4+ T cells or gamma interferon (IFN-γ) leads to rapid dissemination of the pathogen and host mortality (9–13). Recruitment and activation of CD4+ T cells is dependent on the complex cascade of events underlying the innate immune response. Multiple cytokines, including tumor necrosis factor alpha (TNF-α), interleukin 12 (IL-12), IL-1β, CCL2, and granulocyte-macrophage colony-stimulating factor (GM-CSF), are all produced early during infection and promote the recruitment, activation, and/or maturation of a diverse group of immune cells, including monocytes, neutrophils, and T cells (8, 10). While it is known that neutralizing these cytokines exacerbates disease (14–17), the specific cell types and signaling pathways involved in pathogen recognition and subsequent initiation of the innate immune response to Histoplasma are still being explored. Recent work implicates the C-type lectin receptors Dectin-1 and Dectin-2 in the recognition of and response to Histoplasma (18). Nonetheless, much remains to be understood about signaling pathways that are activated by Histoplasma during infection. Since MyD88 is a central adaptor protein in multiple immune recognition and signaling pathways, we chose to explore its role in the host immune response to Histoplasma.
MyD88 mediates both pathogen recognition through Toll-like receptors (TLRs) and cytokine signaling through the IL-1 and IL-18 receptors (IL-1R and IL-18R) (19, 20). In many infectious-disease models, perturbing MyD88 signaling causes defects in early immune events that lead to decreased induction of the adaptive immune response and increased host sensitivity to infection. For example, in infection models of fungal pathogens, such as Candida albicans and Aspergillus fumigatus, MyD88 deficiency causes a block in the production of proinflammatory cytokines early during infection, decreased neutrophil recruitment, and decreased T cell activation (21, 22). Here, we demonstrate that MyD88 signaling is critical for mounting the appropriate immune response to Histoplasma. In particular, we provide direct evidence that MyD88 is necessary for both the early production of proinflammatory cytokines by alveolar macrophages and dendritic cells and the subsequent recruitment of inflammatory monocytes. Ultimately, deficiency in MyD88 severely hinders the development of an appropriate T cell response to Histoplasma infection, culminating in an increased fungal burden and host mortality.
MATERIALS AND METHODS
Strains and culture conditions.
Histoplasma yeast cells were grown in Histoplasma macrophage medium (HMM) (23). Liquid cultures were grown in an orbital shaker at 37°C with 5% CO2. HMM agarose plates were incubated in a humidified chamber at 37°C with 5% CO2. At the start of these experiments, a large stock of Histoplasma strain G217B, designated G217B-AC, was stored at −80°C in 50% glycerol; cells from this stock were used for all experiments. Cells were inoculated from frozen stock onto HMM plates 3 weeks before each experiment. One week before infection, the strain was inoculated from solid medium into liquid HMM and passaged at 1:25 every 3 days. In preparation for infection of both mice and in vitro cell cultures, mid-logarithmic-phase cultures were washed once with phosphate-buffered saline (PBS), sonicated for 3 s on setting 2 using a Fisher Scientific Sonic Dismembrator Model 100, and counted using a hemacytometer to determine the cell number.
Mice.
Female C57BL/6J, IL-1R-deficient (IL-1R−/−) (strain 003245; B6.129S7-Il1r1tm1Imx/J) and MyD88−/− [strain 009088; B6.129P2(SJL)-Myd88tm1.1Defr/J] mice were originally purchased from Jackson Laboratory. Dectin-1−/− mice were obtained from Chad Steele at the University of Alabama, Birmingham, AL. C57BL/6Tac mice, the wild-type (WT) control for Dectin-1−/− animals, were purchased from Taconic Farms. Mice carrying floxed alleles of MyD88 (MyD88fl/fl) (Jackson strain 008888; B6.129P2-Myd88tm1Defr/J), MyD88fl/fl × CD11cCre [Jackson strain 008068; B6.Cg-Tg(Itgax-cre)1-1Reiz/J], MyD88fl/fl × LysMCre (Jackson strain 004781; B6.129P2-Lyz2tm1(cre)Ifo/J), and MyD88fl/fl × VavCre [Jackson strain 008610; B6.Cg-Tg(Vav1-cre)A2Kio/J] mice were gifts from Anthony DeFranco (24, 25). All animals were bred and maintained in a specific-pathogen-free facility at the University of California, San Francisco (UCSF). All mouse experiments were performed in compliance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee at the University of California, San Francisco.
Mouse infections.
Eight- to 12-week-old, age-matched female mice were anesthetized with isoflurane and infected intranasally with 1.8 × 104 yeast cells of wild-type (G217B-AC) Histoplasma strain. The mice were monitored daily for symptoms of disease (i.e., weight loss, lack of activity/response to stimulus, panting, or lack of grooming). For survival experiments, mice were sacrificed after they exhibited 3 days of sustained weight loss of >15% of their maximum weight in conjunction with one other symptom of disease. For fungal-burden experiments, lungs and spleens were harvested from mice, and the homogenate was plated on brain heart infusion (BHI) agar plates and grown for 10 days at 30°C to enumerate CFU.
Cytokine analysis.
Lung and spleen homogenates from infected mice were homogenized in 1 ml PBS containing 1× cOmplete Ultra protease inhibitor (Roche). The homogenate was centrifuged at 4°C, and the resulting supernatant was sterilized using a 0.2-μm CoStar Spin-X centrifuge tube (Corning). For cytokine analysis of cells in culture, the supernatant was sterile filtered using AcroPrep 96-well filter plates (Pall). Mouse Cytometric Bead Array Flex Sets (BD) were used according to the manufacturer's instructions to determine the concentrations (pg/ml) of IL-6, TNF-α, keratinocyte chemoattractant (KC), IL-1β, IFN-γ, IL-17, and MCP-1/CCL2. IL-12p70 levels were quantified using a Ready-Set-Go! ELISA kit (eBioscience).
Isolation of lung cells.
Mouse lungs were perfused with PBS and digested in Hanks buffered salt solution (HBSS) containing 80 U/ml DNase (Sigma; D4527) and 2 mg/ml collagenase D (Roche) for 30 min at 37°C. After digestion, the lungs were dissociated using a GentleMacs Tissue Dissociator (Miltenyi). Red blood cells were hypotonically lysed, and the remaining cells were filtered through a 70-μm cell strainer (BD).
Flow cytometry.
Dissociated lung cells (2 × 106 to 4 × 106) were resuspended in PBS, stained with Fixable Viability Dye eFluor 450 (eBiosciences) for 20 min, and then washed and resuspended in PBS containing 1% heat-inactivated fetal bovine serum (FBS), 1 mM EDTA, 10 μg/ml anti-CD16/32 (Fc block), and 0.1% sodium azide. The cells were stained for 30 min with appropriate antibodies, fixed in 1× BD Stabilizing Fix, and stored at 4°C until analysis on an LSR II (BD). The antibodies used to identify macrophages, neutrophils, monocytes, and dendritic cells were as follows: peridinin chlorophyll protein (PerCp)-Cy5.5-CD11c, phycoerythrin (PE)-Cy7-CD11b, allophycocyanin (APC)-major histocompatibility complex class II (MHC-II), and PE-Cy7-Gr1 (eBiosciences); PE-SiglecF, PE-Ly6G, and APC-Cy7-Ly6C (BD); and fluorescein isothiocyanate (FITC)-CD45.2 (UCSF). The antibodies used to identify T cells and NK cells were as follows: APC-CD4, PerCp-Cy5.5-CD8, PE-Cy7-CD69, and PE-CD69 (BD) and PerCp-Cy5.5-NK1.1, FITC-CD3ε, and APC-gamma delta T cell receptor (γδ TCR) (eBiosciences). Flow cytometry data were analyzed using FlowJo version 7.6.5.
Intracellular cytokine staining.
Dissociated lung cells were prepared as described for the flow cytometry experiments, except that 1 μl/ml BD GolgiPlug (brefeldin A) was added to all media until the cells were stained with antibodies. Extracellular staining proceeded as described above. After staining, samples were fixed in Cytofix/Cytoperm Buffer (BD) for 20 min and then stained with intracellular cytokine antibodies in 1× Perm/Wash buffer (BD) for 30 min and stored at 4°C until analysis. BV786–IFN-γ (BD) antibody was used to stain IFN-γ-producing cells.
BrdU staining.
Mice were given 1 mg bromodeoxyuridine (BrdU) in 100 μl PBS intraperitoneally (i.p.) on day 5 and again on day 6 postinfection (p.i.). Lungs and mediastinal lymph nodes from BrdU-treated mice were harvested 12 h after the last injection (day 7). Single-cell suspensions from total lung tissue and lymph nodes were stained for surface CD3, CD4, and CD8. The cells were fixed, processed, and stained for BrdU using the BrdU Flow Kit (BD Pharmingen) according to the manufacturer's protocol.
Generation of BMDCs.
Bone-marrow-derived dendritic cells (BMDCs) were derived by culturing bone marrow freshly isolated from femurs of 6- to 8-week-old female mice in DC medium consisting of RPMI 1640 (Gibco), 2 mM glutaMAX (Life Technologies), penicillin-streptomycin, 50 μM 2-mercaptoethanol (Sigma), 10 mM HEPES, 10% heat-inactivated FBS, 10 ng/ml recombinant GM-CSF (Peprotech), and 1 ng/ml recombinant IL-4 (Life Technologies). Nonadherent cells were harvested on day 6, and CD11c+ cells were purified using magnetic columns and anti-mouse CD11c microbeads (Miltenyi) according to the manufacturer's instructions. According to flow cytometry analysis, all samples were at least 90% CD11c+ cells. A total of 4 × 105 cells/well were seeded into 24-well plates in DC medium and cultured 48 h before infection.
Isolation of alveolar macrophages.
Alveolar macrophages were harvested by bronchiolar lavage of naive mouse lungs with 10 ml PBS containing 1 mM EDTA, and 5 × 105 cells/well were plated in 24-well tissue culture plates in Dulbecco modified Eagle medium (DMEM) containing 20% heat-inactivated FBS, 2 mM glutaMAX, and penicillin-streptomycin. Nonadherent cells were removed from the wells via gentle washing with 1 ml DMEM before infection with Histoplasma.
Isolation of CD4+ T cells.
Spleens from mice infected with a sublethal dose (1 × 104 cells) of wild-type Histoplasma (G217B-AC) were harvested 15 days postinfection and dissociated in HBSS using the GentleMacs Dissociator. Leukocytes were enriched via Lympholyte-M separation (Cedarlane), and B cells were depleted by incubating the cells on anti-IgM and anti-IgG antibody-coated plates for 2 h. Nonadherent cells were collected, and T cells were purified by negative selection using a mouse Pan-T Cell Isolation Kit (Miltenyi). CD4+ T cells were then isolated by positive selection using anti-mouse CD4 microbeads (Miltenyi).
BMDC-T cell cocultures.
BMDCs were seeded in 96-well plates at a density of 3 × 104 cells/well and infected with wild-type Histoplasma (G217B-AC) at a multiplicity of infection (MOI) of 1. Immediately after infection with Histoplasma, 1 × 105 purified CD4+ T cells were added to the coculture. At various time points postinfection, the medium was removed from each well and 200 μl of double-distilled H2O (ddH2O) was added. The cells were mechanically lysed by vigorous pipetting after a 5-min incubation at room temperature. The cell lysate was diluted in PBS, plated on HMM agarose, and grown for 10 days at 37°C to enumerate CFU.
Isolation of CD11c+ cells from lungs.
Dissociated lung cells were incubated with anti-mouse CD11c microbeads and purified by magnetic separation on a MACS column (Miltenyi) according to the manufacturer's instructions. The purity of lung cell isolates was determined by flow cytometry using the antibodies described above. All samples were at least 90% CD11c+ cells.
RNA isolation and qRT-PCR.
Immediately after CD11c+ purification, cells were lysed with Qiazol (Qiagen), flash-frozen in liquid nitrogen, and stored at −80°C. RNA was harvested using a Purelink RNA minikit (Life Technologies) following the manufacturer's instructions for TRIzol reagent-based purification of RNA. cDNA was synthesized using Superscript II (Life Technologies) and a 1:1 mixture of oligo(dT) and random primers. The reaction mixture was diluted 1:50 in ddH2O prior to analysis by quantitative reverse transcription (qRT)-PCR, which was performed using SYBR green Universal master mix (Roche) and 200 nM primers on an Mx3000P QPCR system (Stratagene). The relative expression (ΔCT) compared to an endogenous control, hypoxanthine-guanine phosphoribosyltransferase (HPRT), was determined. Primer sequences were obtained from the Harvard Primer Bank database (http://pga.mgh.harvard.edu/primerbank/index.html) (26–28). The primer identifiers (IDs) are as follows: HPRT, 7305155a1; IL-1β, 118130747b1; IL-6, 13624310b2; TNF-α, 133892368b2; and KC, 6680109a1.
Statistical analysis.
Statistical analysis for experiments was performed using Prism (GraphPad Software, San Diego, CA). Analysis of variance (ANOVA) with Tukey's posttest was used to analyze the significance of cytokine expression and flow cytometry experiments. The log rank sum test was used to analyze survival. Statistically significant differences were indicated by P values of <0.05.
RESULTS
MyD88−/− mice are more susceptible to Histoplasma infection than Dectin-1−/− or IL-1R−/− mice.
MyD88 is a critical downstream mediator of key innate signaling pathways, including TLR signaling, but the requirement for MyD88-dependent signaling in the host response to Histoplasma has not been explored. To determine the role of MyD88-dependent signaling in the immune response during respiratory Histoplasma infection, whole-body MyD88-deficient (MyD88−/−) and wild-type mice were inoculated intranasally with a sublethal dose of Histoplasma. The mice were monitored daily for symptoms of infection, which included weight loss and signs of respiratory distress. All the mice began to show symptoms of infection by day 6. Wild-type mice were able to resolve Histoplasma infection and recovered within 14 to 16 days; however, MyD88−/− mice experienced progressively worsening symptoms during this time and ultimately succumbed to disease 14 to 20 days after infection (Fig. 1A). MyD88 is required for TLR, IL-18, and IL-1 signaling; the last is already known to contribute to host protection against Histoplasma (17). However, we found that MyD88−/− mice were significantly more susceptible to Histoplasma infection than IL-1R−/− mice (Fig. 1A), implying that deficient TLR and/or IL-18 signaling could contribute to the increased sensitivity of the MyD88−/− mutant. Additionally, we compared the sensitivity of MyD88−/− mice to that of mice lacking the pattern recognition receptor Dectin-1, which can recognize Histoplasma in vitro (18, 29–32). Recently, Dectin-1−/− mice have been reported to have a higher pulmonary fungal burden than wild-type mice during Histoplasma infection (18). In our infection model, we found that Dectin-1, unlike MyD88, was dispensable for host survival (Fig. 1A). Overall, these results indicate that MyD88 is required for host survival and suggest that other receptor signaling pathways upstream of MyD88, such as TLR and/or IL-18 signaling, may also be critical for host recognition of and response to Histoplasma.
FIG 1.
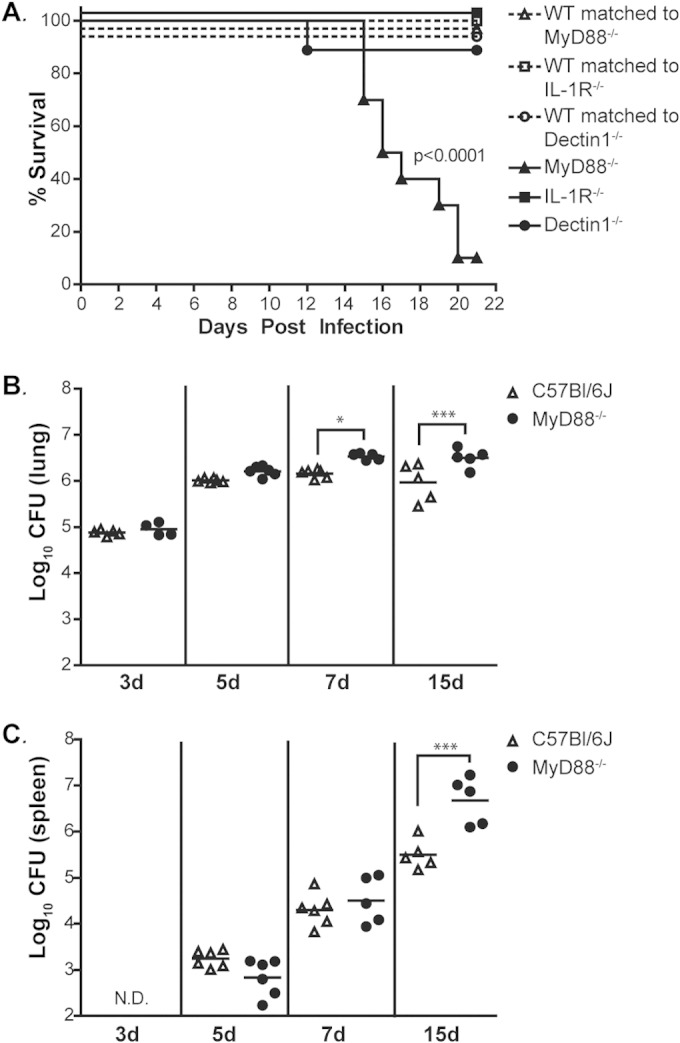
MyD88 is required for host survival after infection with Histoplasma. (A) Gender- and age-matched MyD88−/−, Dectin-1−/−, and IL-1R−/− mice and their respective wild-type counterparts (n = 6 to 10) were infected intranasally with 1.8 × 104 Histoplasma cells and monitored for survival. Differences in survival were determined using a log-rank test. (B and C) Lungs (B) and spleens (C) of infected MyD88−/− and C57BL/6J mice were harvested, homogenized, and plated for CFU at the indicated time points (n = 5 or 6 mice/time point for each strain; d, days). All results are representative of at least three independent experiments. *, P < 0.05; ***, P < 0.001; N.D., not detectable. P values were determined using ANOVA. Horizontal bars are means of analyzed samples.
To further investigate how MyD88 signaling affects the progression of disease, we determined the fungal burdens in the lungs and spleens of infected mice at 3, 5, 7, and 15 days p.i. In wild-type mice, fungal growth in the lungs peaked at day 7 and then declined as the mice recovered (Fig. 1B). In contrast, the fungal burden in the lungs of MyD88−/− mice was significantly higher than in wild-type mice at 7 days p.i. (Fig. 1B) and continued to increase until the time of death. We also determined that the fungal burden in the spleen was significantly higher in MyD88−/− animals than in wild-type mice at 15 days p.i. (Fig. 1C). By this time point, the MyD88−/− animals displayed disease symptoms, such as inactivity, hunching, panting, and weight loss (data not shown), and succumbed to infection. Collectively, these results demonstrate that MyD88 is required for responding to and resolving Histoplasma infection.
MyD88 is required for the early innate immune response to Histoplasma.
To determine the role of MyD88 in the immune response to Histoplasma, we next investigated cytokine production in the lungs of infected mice during the first 7 days of infection. Analysis of cytokines harvested from infected lungs revealed that MyD88−/− mice did not produce normal levels of the inflammatory cytokines IL-6, IL-1β, CCL2, and KC at 5 days p.i. (Fig. 2), despite displaying a fungal burden equivalent to that of wild-type mice. Additionally, at 5 days p.i., we also observed that MyD88−/− mice produced lower levels of IL-12p70, a key cytokine in the initiation of the Th1 response. Notably, not all early cytokine production required MyD88-dependent signaling, as TNF-α levels were unaffected by MyD88 deficiency during infection (3 and 5 days p.i.). Interestingly, by 7 days p.i., cytokine levels were higher in MyD88−/− mouse lungs than in wild-type mouse lungs, correlating with the significant increase in fungal burden in the mice at this time point.
FIG 2.

MyD88 is essential for the kinetics of normal cytokine production in the lungs of mice infected with Histoplasma. Lungs of infected MyD88−/− and C57BL/6J mice were harvested, homogenized, and evaluated for cytokine levels at the indicated time points. *, P < 0.05; ***, P < 0.001; P values were determined by ANOVA. All the results are representative of at least three independent experiments. The bars represent means ± standard deviations (SD) for 5 mice.
Since MyD88−/− mice were deficient in the production of cytokines known to recruit monocytes and neutrophils, such as CCL2 and KC, we next investigated the recruitment of inflammatory cells to the lungs of MyD88−/− mice. Flow cytometry analysis of single-cell suspensions from the lung revealed a significant block in the overall recruitment of CD45+ inflammatory cells to MyD88−/− lungs by 7 days p.i. (Fig. 3A and B). In particular, we found that MyD88−/− mice displayed a decrease in the numbers of recruited inflammatory neutrophils and dendritic cells by 5 days p.i. and a significant decrease in the numbers of monocytes and alveolar macrophages by 7 days p.i. (Fig. 3C to F). A similar recruitment defect was observed in IL-1R−/− mice 5 days postinfection, demonstrating that recruitment is at least partially mediated by IL-1 signaling (see Fig. S1 in the supplemental material). Thus, MyD88-dependent signaling is required for cytokine production early in infection and for the subsequent recruitment of inflammatory cells known to be important for the host response to and control of Histoplasma infection.
FIG 3.

Inflammatory cell recruitment to the lung requires MyD88 signaling. Infected lungs were dissociated into single-cell suspensions and analyzed via flow cytometry to determine the numbers of specific cell populations. (A) Total lung cell counts. (B) CD45+ cell counts. (C) Alveolar macrophage count as defined by CD11c+ SiglecF+ autofluorescent cells. (D) Dendritic cell count as defined by CD11c+ CD11b+ MHC class IIhigh cells. (E) Neutrophil count as defined by CD11c− CD11b+ Ly6C+ Ly6G+ cells. (F) Monocyte count as defined by CD11c− CD11b+ Ly6C+ Ly6G− cells. The bars represent the means ± SD of 5 or 6 mice. All results are representative of at least three independent experiments. **, P < 0.01; ***, P < 0.001; P values were determined using ANOVA.
MyD88 deficiency results in delayed activation of T and NK cells.
A robust adaptive T cell response is critical to resolution of Histoplasma infection (13, 33, 34). Since the production of key regulators of the Th1 and Th17 response, namely, IL-12, IL-1β, and IL-6, by MyD88−/− mice was delayed, we asked if an impaired T cell response might underlie their survival defect. First, we examined the levels of the cytokines IFN-γ and IL-17A, which are produced by activated NK cells, γδ TCR+ cells, and CD4+ and CD8+ T cells (35, 36). Whereas wild-type mice consistently produced high levels of these cytokines during Histoplasma infection, MyD88−/− mice failed to do so; IFN-γ and IL-17A remained low throughout the course of infection (Fig. 4A). Next, we examined NK and T cell numbers in the lung during infection, as well as the expression of CD69, an early marker of activation (37). The lungs of infected WT and MyD88−/− mice contained roughly equivalent numbers of these cells (with the exception of γδ TCR+ cells at day 7, which were more numerous in wild-type mice). In contrast, CD69 expression was significantly decreased for γδ TCR+ cells (Fig. 4B), NK cells (Fig. 4C), and CD8+ T cells (Fig. 4D) at both day 5 and day 7 in MyD88−/− mice compared to wild-type controls, indicating a defect in activation of these cells. Similarly, CD69 expression in CD4+ T cells was decreased in MyD88−/− lungs at 7 days p.i. (Fig. 4E; see Fig. S2 in the supplemental material). In addition, we found that that while no difference in T-cell proliferation was observed in the lungs at 7 days p.i., there was a significant decrease in T cell proliferation in the mediastinal lymph nodes, as measured by BrdU incorporation (Fig. 5A). Importantly, γδ TCR+ cells, NK cells, CD8+ T cells, and CD4+ T cells are all major sources of IFN-γ, which is required for activation of macrophages and control of intracellular fungal growth (36, 38). We found that the percentage of IFN-γ-producing cells was significantly decreased in MyD88-deficient mice for γδ TCR+ cells, NK cells, and CD4+ T cells (Fig. 5B and C). Thus, MyD88 is required for the activation of both innate and adaptive immune cells to produce IFN-γ, a cytokine critical for control of fungal growth and resolution of Histoplasma infection (8, 9, 11).
FIG 4.

MyD88 is necessary for timely recruitment and activation of T cells and NK cells. (A) Analysis of global cytokine production in the lungs of Histoplasma-infected mice for total IFN-γ and IL-17A. (B to E) Analysis of lung cell counts and percent cells expressing CD69 for γδ TCR+ cells (B), NK cells (C), CD8+ T cells (D), and CD4+ T cells (E). CD4+ T cells are defined as CD45+ CD3ε+ CD4+ cells. CD8+ T cells are defined as CD45+ CD3ε+ CD8+ cells. γδ TCR+ cells are defined as CD45+ CD3ε+ γδ TCR+ cells. NK cells are defined as CD45+ CD3ε− NK1.1+ cells. *, P < 0.05; **, P < 0.01; ***, P < 0.001; P values were determined by ANOVA.
FIG 5.
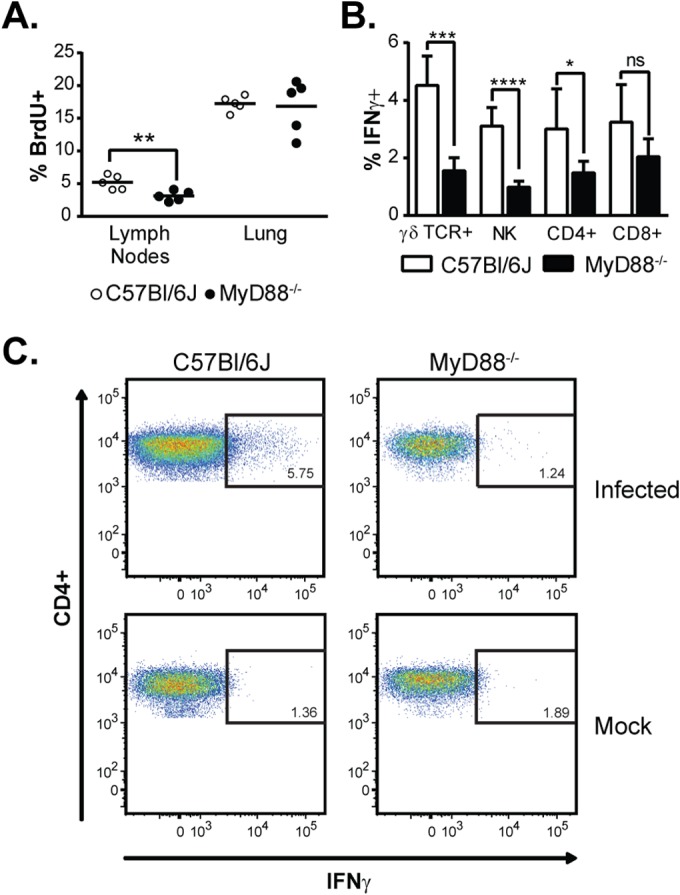
MyD88 is required for T cell proliferation and production of IFN-γ by T cells and NK cells. (A) T cell proliferation in the lungs and mediastinal lymph nodes 7 days p.i. as measured by in vivo BrdU incorporation. (B) Ex vivo intracellular cytokine staining on lung cells 7 days p.i. to detect IFN-γ production in γδ TCR+ cells, NK cells, and CD4+ and CD8+ T cells isolated from the lung. The bars represent the means ± SD of 5 mice. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; ns, not significant; P values were determined by ANOVA. (C) Representative fluorescence-activated cell sorter (FACS) plots for IFN-γ-producing CD4+ gated populations.
MyD88 mediates intrinsic responses of alveolar macrophages and dendritic cells in vitro.
During pulmonary infection, Histoplasma is found primarily in resident innate immune cells, including macrophages and dendritic cells (39). We hypothesized that MyD88 would be required intrinsically for these innate immune cells to recognize and respond to Histoplasma. To test this, we first evaluated the abilities of MyD88-deficient BMDCs (Fig. 6A) and freshly isolated alveolar macrophages (Fig. 6B) to respond to Histoplasma infection in vitro. We found a substantial delay in the production of inflammatory cytokines at 48 h postinfection for both cell types, suggesting that MyD88 is intrinsically required for the innate immune response to Histoplasma. In contrast, IL-1R−/− BMDCs were fully competent for cytokine production in response to Histoplasma infection (see Fig. S3 in the supplemental material), indicating that the cytokine defect we observed in the MyD88−/− mutant is not solely due to a failure in IL-1 signaling.
FIG 6.

MyD88 is required for appropriate cytokine signaling in alveolar macrophages and bone-marrow-derived dendritic cells in vitro. (A and B) BMDCs (A) and alveolar macrophages (AvMs) (B) from WT and MyD88−/− mice were harvested and infected in vitro with Histoplasma at an MOI of 1. Supernatants from infected cells were collected in triplicate at 48 h and evaluated for cytokine levels. (C) BMDCs infected with wild-type Histoplasma (MOI = 1) were cocultured with purified splenic WT CD4+ T cells. The cells were lysed, and CFU were counted at the indicated time points. *, P < 0.05; ***, P < 0.001; P values were determined by ANOVA. The bars represent the means ± SD of 3 or 4 samples. All the results are representative of at least three independent experiments.
We then used a coculture model of Histoplasma-infected BMDCs and T cells to determine if MyD88 deficiency affected the interaction between these cell types (40). We first infected WT and MyD88−/− BMDCs with Histoplasma and monitored intracellular fungal growth by lysing host cells at multiple time points over the course of infection and enumerating intracellular yeasts by plating. We observed equivalent fungal growth in WT and MyD88−/− BMDCs when cultured in the absence of T cells (see Fig. S4 in the supplemental material). However, when we cultured infected BMDCs with CD4+ T cells purified from the spleens of infected wild-type mice, we observed that wild-type BMDCs showed greater restriction of fungal growth than MyD88−/− BMDCs (Fig. 6C). These data indicate that MyD88 signaling contributes to the response of infected BMDCs to primed T cells.
MyD88 is required for the normal response of dendritic cells to Histoplasma in vivo.
To extend our in vitro observations to in vivo infections, we purified CD11c+ cells (consisting of alveolar macrophages and dendritic cells) from the lungs of infected WT and MyD88−/− mice. Transcriptional analysis of these cells revealed a requirement for MyD88 in cytokine production as early as 3 days p.i. (Fig. 7A). To determine the relative contribution of MyD88 signaling in these immune cells to the overall host response, we utilized mice carrying a floxed MyD88 allele (MyD88fl/fl) and expressing a cell-specific Cre recombinase to delete MyD88 specifically in dendritic cells and alveolar macrophages (CD11c-MyD88) or in macrophages and neutrophils (LysM-MyD88) (24). As we found with global deletion of MyD88, alveolar CD11c+ cells (alveolar macrophages and lung dendritic cells) from infected CD11c-MyD88 mice were deficient in the early transcription of key cytokines during Histoplasma infection (Fig. 7B). Surprisingly, however, CD11c-MyD88 and LysM-MyD88 mice did not display any significant defects in survival or control of fungal growth upon infection with a sublethal dose of Histoplasma (Fig. 8A and B), nor did they demonstrate a robust in vivo cytokine deficiency, as observed upon global deletion of MyD88 in mice (Fig. 8C and D). These data suggest that MyD88 signaling in other hematopoietic cell types can compensate for the defect in alveolar macrophages and dendritic cells. This hypothesis is consistent with the observation that Vav-MyD88 mice, which are MyD88 deficient in all hematopoietic cells, displayed a significant survival defect when infected with a sublethal dose of Histoplasma (Fig. 8E). In sum, MyD88 is required intrinsically in both dendritic cells and alveolar macrophages for their normal response to Histoplasma infection, but defects in these cell populations alone are insufficient to fully recapitulate the effects of global MyD88 deficiency. These data imply a role for MyD88-dependent signaling in additional hematopoietic cells during Histoplasma infection.
FIG 7.

Alveolar macrophages and dendritic cells require MyD88 for cytokine gene expression in vivo. Single-cell suspensions were harvested from infected wild-type, MyD88−/−, and CD11c-MyD88 mice at 3 days p.i. CD11c+ cells (alveolar macrophages and lung dendritic cells) were purified from lung cells via magnetic-bead separation and subjected to RNA isolation followed by analysis of cytokine transcription. (A) C57BL/6J (wild-type) and MyD88−/− (mutant) CD11c+ cells. (B) MyD88fl/fl (wild-type) and CD11c-MyD88 (mutant) CD11c+ cells. **, P < 0.01; ***, P < 0.001; P values were determined by ANOVA. The bars represent the means ± SD of 3 samples harvested from 2 or 3 pooled mice. All the results are representative of at least three independent experiments.
FIG 8.

Loss of MyD88-dependent signaling in dendritic cells, alveolar macrophages, or neutrophils is not sufficient to impair host survival, control of fungal growth, or global cytokine production in the lungs. MyD88fl/fl mice carrying the CD11c-Cre (CD11c-MyD88) or LysM-Cre (LysM-MyD88) transgene were infected with 1.6 × 104 Histoplasma cells and monitored for survival (n = 8 to 10 mice/strain) (A) or fungal burden (B) at 7 days p.i. (C) Whole-lung homogenates of infected CD11c-MyD88 mice were analyzed for cytokine production at 5 days p.i. (n = 5 or 6 mice/strain). (D) RNA harvested from whole lungs of infected LysM-MyD88 mice was analyzed via qRT-PCR for transcription of cytokines at 4 days p.i. (n = 3 to 5 mice/strain). (E) MyD88fl/fl mice or MyD88fl/fl mice carrying the Vav-Cre transgene (Vav-MyD88) were infected with Histoplasma and monitored for survival (n = 6 to 9 mice/strain). n.s., not significant; P values were determined by ANOVA.
DISCUSSION
In this study, we demonstrate that MyD88 signaling is a crucial component in the defense against Histoplasma. Using MyD88−/− mice, we showed that MyD88 is required for control of the fungal burden in both the lung and spleen and demonstrated that MyD88−/− mice fail to resolve infection by Histoplasma, instead succumbing to disseminated disease. During early infection of the lung, MyD88−/− mice fail to produce inflammatory cytokines, including CCL2 and IL-1β, that are known to trigger critical events for controlling Histoplasma infection, such as the recruitment and activation of neutrophils and monocytes (16, 17). This early innate immune response appears to be partly mediated by a cell-intrinsic requirement for MyD88 in the recognition of and response to infection with Histoplasma by alveolar macrophages and dendritic cells. Upon detailed analysis of dendritic cell responses in vitro, we found that MyD88 signaling contributes to cytokine production and control of intracellular fungal growth. In addition to early cytokine responses and recruitment of inflammatory cells, other key events during the host response to Histoplasma infection include an increase in CD4+ T cell activation and IFN-γ/IL-17 production. Recruitment of inflammatory cells to the site of infection is associated with control of fungal growth, and production of IFN-γ/IL-17 is necessary for the restriction of Histoplasma growth by activated macrophages (11, 16, 41). In the MyD88−/− mouse, production of the cytokines involved in inducing both a Th1 and a Th17 response is delayed. Despite a higher fungal burden in the MyD88−/− mouse, the Th1 response is muted, as IFN-γ production by both innate (γδ TCR+ and NK cells) and adaptive (CD4+ T cells) cells is decreased, indicating that MyD88-dependent pathways contribute to these responses.
The requirements for MyD88 vary widely for different fungal pathogens in murine infection models. MyD88 is required for host survival, inflammatory cytokine production, and T cell activation during infection with Candida, Cryptococcus, and Paracoccidioides (42–45). In contrast, during respiratory infection with Aspergillus, MyD88 is important for production of TNF-α and early pulmonary responses but is required for survival only if the host is immunosuppressed (21, 44). Importantly, multiple studies have demonstrated a requirement for MyD88 in directing T cell responses to fungal pathogens, which is critical, since a robust Th1 and Th17 response is often important to achieve a productive resolution to fungal infection. Indeed, while MyD88 plays only a moderate role in the recruitment and proliferation of T cells during infection with A. fumigatus, it is critical for full IFN-γ production by these cells (46). Interestingly, in this context, MyD88 does not seem to be intrinsically required within T cells, but rather is important for the ability of resident lung cells, such as dendritic cells, to influence the T cell response. Correspondingly, MyD88 has been shown to mediate both T cell-dependent IFN-γ production and the T cell-dendritic cell interaction during infection with C. albicans (21). Here, we show that MyD88 signaling is important for both T cell activation and dendritic cell cytokine production during Histoplasma infection, suggesting that during MyD88 deficiency, the interaction between these two cell types may be impaired. In line with this, we show that the ability of infected dendritic cells to restrict fungal growth when cocultured with primed T cells is MyD88 dependent. However, the precise molecular mechanisms that drive this interaction remain unknown. Accordingly, future studies to clarify the role of MyD88 during the T cell response to Histoplasma may indicate that MyD88 signaling in dendritic cells is an important mediator of T cell activation.
Numerous dendritic cell responses were MyD88 dependent in our infection models, including optimal growth restriction of Histoplasma and timely cytokine responses. Studies have shown that dendritic cells are activated in response to pathogen infection, both directly through pathogen-associated molecular pattern (PAMP) recognition and indirectly through exposure to the cytokine milieu (47). Both recognition of PAMPs and exposure to cytokines, such as TNF-α, IFN-γ, and IFN-α/β, increases the upregulation of T cell activation ligands, such as MHC-II and CD86; the production of inflammatory cytokines; and the processes involved in the control of pathogen growth, such as the production of reactive oxygen and nitrogen species (48–50). The delayed cytokine production and increased fungal growth we see in MyD88−/− dendritic cells may be a reflection of a failure to recognize Histoplasma. Alternatively, MyD88−/− dendritic cells may fail to respond appropriately to the cytokine milieu created by primed T cells, thus causing defects in dendritic cell maturation, activation, and fungal killing.
The ability of the host to mount an appropriate immune response to Histoplasma depends on its capacity to recognize and respond to the pathogen; however, the panel of immune receptors responsible for the initiation of this response to Histoplasma in vivo is still being uncovered. Indeed, the plethora of pattern recognition receptors and downstream signaling pathways engaged by distinct fungal organisms is an area of active inquiry (51, 52). Recent work has demonstrated the importance of C-type lectin receptors in vaccine immunity, as well as primary immune responses to Histoplasma and other, related fungi (18). The contribution of Toll-like receptors in the primary in vivo host response to Histoplasma, however, has not been elucidated, and by uncovering the role of MyD88, our work suggests that TLR signaling could play a key role in the corresponding host response. Importantly, MyD88 is also a mediator of IL-1 and IL-18 cytokine receptor signaling. Previous work has demonstrated that IL-1R deficiency resulted in an increase in host susceptibility to Histoplasma, as well as a deficiency in IFN-γ production in the lungs of infected mice at later time points (17), similar to the MyD88−/− results presented here. However, we found that IL-1R−/− mice display no defects in overall host survival when infected with a dose of Histoplasma that was lethal to MyD88−/− mice. Interestingly, the IL-1R−/− mouse did display a defect in cell recruitment to the lungs similar to that of the MyD88−/− mouse, suggesting that impaired cellular recruitment by itself is not sufficient to decrease host survival. These results demonstrate that IL-1 signaling alone cannot explain the defects we observe during MyD88 deficiency and thus suggest that TLR recognition and/or IL-18 signaling is also important for the host immune response. In several pathogen infection models, single TLR deficiencies are frequently not sufficient to produce a measurable difference in immune response, as the effects of recognition by individual TLRs are redundant. Future studies to determine the contribution of TLR recognition to Histoplasma in vivo may require the investigation of the response in mice containing multiple TLR knockouts, such as mice lacking TLR2, -4, and -9 (53). Alternatively, since IL-18, in cooperation with IL-12, is known to induce a strong IFN-γ response from T cells, it may be that the reduced ability of MyD88−/− mice to produce IFN-γ reflects a failure of MyD88-dependent IL-18 signaling (54, 55).
A critical point to emerge from these experiments is the discovery that the requirement for MyD88 signaling in the response to Histoplasma was not confined to narrow immune cell subsets. Our work is in contrast to studies of infection responses to other pathogens, where the use of in vivo cell-specific deletion of MyD88 has elucidated the roles of distinct cell types in the host response. For example, during Toxoplasma infection, deletion of MyD88 only in the CD11c+ cell compartment (alveolar macrophages and dendritic cells) leads to a defect in IL-12 production and an increased susceptibility to infection (25). Since Histoplasma interacts primarily with phagocytic cell populations in the lung, we expected that deletion of MyD88 signaling in these specific cell types would recapitulate the phenotype we observed in the global MyD88 knockout mouse. In vitro, this was indeed the case, with both alveolar macrophages and dendritic cells showing delayed cytokine production after infection with Histoplasma. Surprisingly, whereas deletion of MyD88 in the entire hematopoietic compartment caused a significant decrease in host survival, specific deletion of MyD88 in either (i) alveolar macrophages and dendritic cells or (ii) macrophages and neutrophils failed to affect survival or the fungal burden in vivo. These data suggest that intact MyD88 signaling in either of these two subsets may be sufficient for host protection. Alternatively, MyD88 may contribute to host protection through other immune cell types, such as T cells, and future studies will be required to elucidate the role of MyD88 signaling in additional hematopoietic cell types. Collectively, these results highlight the multifaceted roles of MyD88 in the host response to Histoplasma infection and underscore the multiple strategies used by mammalian hosts to combat the ability of a primary fungal pathogen to cause disease.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to Anthony DeFranco, Richard Locksley, Joanne Engel, Kirk Jones, and members of the Sil laboratory for useful discussion as this work progressed. We thank Davina Hocking Murray and Johnny Tse for technical assistance. We thank Sinem Beyhan, Bevin English, Sarah Gilmore, and Nancy Van Prooyen for comments on the manuscript.
This work was supported by an NSF Graduate Research Fellowship to A.C., NIH grants (PO1AI063302 and RO1AI093640) to A.S., and an HHMI Early Career Scientist Award (http://www.hhmi.org/research/ecs/) to A.S.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.02619-14.
REFERENCES
- 1.Kauffman CA. 2007. Histoplasmosis: a clinical and laboratory update. Clin Microbiol Rev 20:115–132. doi: 10.1128/CMR.00027-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Assi MA, Sandid MS, Baddour LM, Roberts GD, Walker RC. 2007. Systemic histoplasmosis: a 15-year retrospective institutional review of 111 patients. Medicine 86:162–169. doi: 10.1097/md.0b013e3180679130. [DOI] [PubMed] [Google Scholar]
- 3.Chu JH, Feudtner C, Heydon K, Walsh TJ, Zaoutis TE. 2006. Hospitalizations for endemic mycoses: a population-based national study. Clin Infect Dis 42:822–825. doi: 10.1086/500405. [DOI] [PubMed] [Google Scholar]
- 4.Gutierrez ME, Canton A, Sosa N, Puga E, Talavera L. 2005. Disseminated histoplasmosis in patients with AIDS in Panama: a review of 104 cases. Clin Infect Dis 40:1199–1202. doi: 10.1086/428842. [DOI] [PubMed] [Google Scholar]
- 5.Smith JA, Kauffman CA. 2009. Endemic fungal infections in patients receiving tumour necrosis factor-alpha inhibitor therapy. Drugs 69:1403–1415. doi: 10.2165/00003495-200969110-00002. [DOI] [PubMed] [Google Scholar]
- 6.Hoebe K, Janssen E, Beutler B. 2004. The interface between innate and adaptive immunity. Nat Immunol 5:971–974. doi: 10.1038/ni1004-971. [DOI] [PubMed] [Google Scholar]
- 7.Martin TR, Frevert CW. 2005. Innate immunity in the lungs. Proc Am Thorac Soc 2:403–411. doi: 10.1513/pats.200508-090JS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Allendoerfer R, Deepe GS. 1997. Intrapulmonary response to Histoplasma capsulatum in gamma interferon knockout mice. Infect Immun 65:2564–2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Clemons K, Darbonne W, Curnutte J, Sobel R, Stevens D. 2000. Experimental histoplasmosis in mice treated with anti-murine interferon-gamma antibody and in interferon-gamma gene knockout mice. Microbes Infect 2:997–1001. doi: 10.1016/S1286-4579(00)01253-3. [DOI] [PubMed] [Google Scholar]
- 10.Deepe GS, Gibbons RS. 2009. Interleukins 17 and 23 influence the host response to Histoplasma capsulatum. J Infect Dis 200:142–151. doi: 10.1086/599333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wu-Hsieh BA, Howard DH. 1987. Inhibition of the intracellular growth of Histoplasma capsulatum by recombinant murine gamma interferon. Infect Immun 55:1014–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhou P, Sieve MC, Bennett J, Kwon-Chung KJ, Tewari RP, Gazzinelli RT, Sher A, Seder RA. 1995. IL-12 prevents mortality in mice infected with Histoplasma capsulatum through induction of IFN-gamma. J Immunol 155:785–795. [PubMed] [Google Scholar]
- 13.Gomez AM, Bullock WE, Taylor CL, Deepe GS. 1988. Role of L3T4+ T cells in host defense against Histoplasma capsulatum. Infect Immun 56:1685–1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Deepe GS, Gibbons R, Woodward E. 1999. Neutralization of endogenous granulocyte-macrophage colony-stimulating factor subverts the protective immune response to Histoplasma capsulatum. J Immunol 163:4985–4993. [PubMed] [Google Scholar]
- 15.Allendoerfer R, Deepe GS. 1998. Blockade of endogenous TNF-alpha exacerbates primary and secondary pulmonary histoplasmosis by differential mechanisms. J Immunol 160:6072–6082. [PubMed] [Google Scholar]
- 16.Szymczak WA, Deepe GS. 2009. The CCL7-CCL2-CCR2 axis regulates IL-4 production in lungs and fungal immunity. J Immunol 183:1964–1974. doi: 10.4049/jimmunol.0901316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Deepe GS, McGuinness M. 2006. Interleukin-1 and host control of pulmonary histoplasmosis. J Infect Dis 194:855–864. doi: 10.1086/506946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang H, LeBert V, Hung CY, Galles K, Saijo S, Lin X, Cole GT, Klein BS, Wüthrich M. 2014. C-type lectin receptors differentially induce Th17 cells and vaccine immunity to the endemic mycosis of North America. J Immunol 192:1107–1119. doi: 10.4049/jimmunol.1302314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Adachi O, Kawai T, Takeda K, Matsumoto M, Tsutsui H, Sakagami M, Nakanishi K, Akira S. 1998. Targeted disruption of the MyD88 gene results in loss of IL-1- and IL-18-mediated function. Immunity 9:143–150. doi: 10.1016/S1074-7613(00)80596-8. [DOI] [PubMed] [Google Scholar]
- 20.Medzhitov R, Preston-Hurlburt P, Kopp E, Stadlen A, Chen C, Ghosh S, Janeway CA. 1998. MyD88 is an adaptor protein in the hToll/IL-1 receptor family signaling pathways. Mol Cell 2:253–258. doi: 10.1016/S1097-2765(00)80136-7. [DOI] [PubMed] [Google Scholar]
- 21.Bellocchio S, Montagnoli C, Bozza S, Gaziano R, Rossi G, Mambula SS, Vecchi A, Mantovani A, Levitz SM, Romani L. 2004. The contribution of the Toll-like/IL-1 receptor superfamily to innate and adaptive immunity to fungal pathogens in vivo. J Immunol 172:3059–3069. doi: 10.4049/jimmunol.172.5.3059. [DOI] [PubMed] [Google Scholar]
- 22.Bretz C, Gersuk G, Knoblaugh S, Chaudhary N, Randolph-Habecker J, Hackman RC, Staab J, Marr KA. 2008. MyD88 signaling contributes to early pulmonary responses to Aspergillus fumigatus. Infect Immun 76:952–958. doi: 10.1128/IAI.00927-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Worsham PL, Goldman WE. 1988. Quantitative plating of Histoplasma capsulatum without addition of conditioned medium or siderophores. J Med Vet Mycol 26:137–143. doi: 10.1080/02681218880000211. [DOI] [PubMed] [Google Scholar]
- 24.Hou B, Reizis B, DeFranco AL. 2008. Toll-like receptors activate innate and adaptive immunity by using dendritic cell-intrinsic and -extrinsic mechanisms. Immunity 29:272–282. doi: 10.1016/j.immuni.2008.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hou B, Benson A, Kuzmich L, DeFranco AL, Yarovinsky F. 2011. Critical coordination of innate immune defense against Toxoplasma gondii by dendritic cells responding via their Toll-like receptors. Proc Natl Acad Sci U S A 108:278–283. doi: 10.1073/pnas.1011549108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang X, Seed B. 2003. A PCR primer bank for quantitative gene expression analysis. Nucleic Acids Res 31:e154. doi: 10.1093/nar/gng154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Spandidos A, Wang X, Wang H, Dragnev S, Thurber T, Seed B. 2008. A comprehensive collection of experimentally validated primers for polymerase chain reaction quantitation of murine transcript abundance. BMC Genomics 9:633. doi: 10.1186/1471-2164-9-633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Spandidos A, Wang X, Wang H, Seed B. 2010. PrimerBank: a resource of human and mouse PCR primer pairs for gene expression detection and quantification. Nucleic Acids Res 38:D792–D799. doi: 10.1093/nar/gkp1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rappleye CA, Eissenberg LG, Goldman WE. 2007. Histoplasma capsulatum α-(1,3)-glucan blocks innate immune recognition by the β-glucan receptor. Proc Natl Acad Sci U S A 104:1366–1370. doi: 10.1073/pnas.0609848104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Edwards JA, Alore E, Rappleye CA. 2011. The yeast-phase virulence requirement for α-glucan synthase differs among Histoplasma capsulatum chemotypes. Eukaryot Cell 10:87–97. doi: 10.1128/EC.00214-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lin JS, Huang JH, Hung LY, Wu SY, Wu-Hsieh BA. 2010. Distinct roles of complement receptor 3, Dectin-1, and sialic acids in murine macrophage interaction with Histoplasma yeast. J Leukoc Biol 88:95–106. doi: 10.1189/jlb.1109717. [DOI] [PubMed] [Google Scholar]
- 32.Adachi Y, Ishii T, Ikeda Y, Hoshino A, Tamura H, Aketagawa J, Tanaka S, Ohno N. 2004. Characterization of beta-glucan recognition site on C-type lectin, Dectin 1. Infect Immun 72:4159–4171. doi: 10.1128/IAI.72.7.4159-4171.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Allendoerfer R, Magee DM, Deepe GS, Graybill JR. 1993. Transfer of protective immunity in murine histoplasmosis by a CD4+ T-cell clone. Infect Immun 61:714–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Allendörfer R, Brunner GD, Deepe GS. 1999. Complex requirements for nascent and memory immunity in pulmonary histoplasmosis. J Immunol 162:7389–7396. [PubMed] [Google Scholar]
- 35.Vantourout P, Hayday A. 2013. Six-of-the-best: unique contributions of γδ T cells to immunology. Nat Rev Immunol 13:88–100. doi: 10.1038/nri3384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cain JA, Deepe GS Jr. 1998. Evolution of the primary immune response to Histoplasma capsulatum in murine lung. Infect Immun 66:1473–1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sancho D, Gómez M, Sánchez-Madrid F. 2005. CD69 is an immunoregulatory molecule induced following activation. Trends Immunol 26:136–140. doi: 10.1016/j.it.2004.12.006. [DOI] [PubMed] [Google Scholar]
- 38.Chen K, Kolls JK. 2013. T cell-mediated host immune defenses in the lung. Annu Rev Immunol 31:605–633. doi: 10.1146/annurev-immunol-032712-100019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Deepe GS, Gibbons RS, Smulian AG. 2008. Histoplasma capsulatum manifests preferential invasion of phagocytic subpopulations in murine lungs. J Leukoc Biol 84:669–678. doi: 10.1189/jlb.0308154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lin JS, Yang CW, Wang DW, Wu-Hsieh BA. 2005. Dendritic cells cross-present exogenous fungal antigens to stimulate a protective CD8 T cell response in infection by Histoplasma capsulatum. J Immunol 174:6282–6291. doi: 10.4049/jimmunol.174.10.6282. [DOI] [PubMed] [Google Scholar]
- 41.Fermin Lee A, Chen HY, Wan L, Wu SY, Yu JS, Huang AC, Miaw SC, Hsu DK, Wu-Hsieh BA, Liu FT. 2013. Galectin-3 modulates Th17 responses by regulating dendritic cell cytokines. Am J Pathol 183:1209–1222. doi: 10.1016/j.ajpath.2013.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Villamón E, Gozalbo D, Roig P, Murciano C, O'Connor JE, Fradelizi D, Gil ML. 2004. Myeloid differentiation factor 88 (MyD88) is required for murine resistance to Candida albicans and is critically involved in Candida-induced production of cytokines. Eur Cytokine Netw 15:263–271. [PubMed] [Google Scholar]
- 43.Loures FV, Pina A, Felonato M, Feriotti C, de Araújo EF, Calich VLG. 2011. MyD88 signaling is required for efficient innate and adaptive immune responses to Paracoccidioides brasiliensis infection. Infect Immun 79:2470–2480. doi: 10.1128/IAI.00375-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Marr KA, Balajee SA, Hawn TR, Ozinsky A, Pham U, Akira S, Aderem A, Liles WC. 2003. Differential role of MyD88 in macrophage-mediated responses to opportunistic fungal pathogens. Infect Immun 71:5280–5286. doi: 10.1128/IAI.71.9.5280-5286.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yauch LE, Mansour MK, Shoham S, Rottman JB, Levitz SM. 2004. Involvement of CD14, Toll-like receptors 2 and 4, and MyD88 in the host response to the fungal pathogen Cryptococcus neoformans in vivo. Infect Immun 72:5373–5382. doi: 10.1128/IAI.72.9.5373-5382.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rivera A, Ro G, Van Epps HL, Simpson T, Leiner I, Sant'Angelo DB, Pamer EG. 2006. Innate immune activation and CD4+ T cell priming during respiratory fungal infection. Immunity 25:665–675. doi: 10.1016/j.immuni.2006.08.016. [DOI] [PubMed] [Google Scholar]
- 47.Spörri R, Reis e Sousa C. 2005. Inflammatory mediators are insufficient for full dendritic cell activation and promote expansion of CD4+ T cell populations lacking helper function. Nat Immunol 6:163–170. doi: 10.1038/ni1162. [DOI] [PubMed] [Google Scholar]
- 48.Gallucci S, Lolkema M, Matzinger P. 1999. Natural adjuvants: endogenous activators of dendritic cells. Nat Med 5:1249–1255. doi: 10.1038/15200. [DOI] [PubMed] [Google Scholar]
- 49.Trevejo JM, Marino MW, Philpott N, Josien R, Richards EC, Elkon KB, Falck-Pedersen E. 2001. TNF-alpha-dependent maturation of local dendritic cells is critical for activating the adaptive immune response to virus infection. Proc Natl Acad Sci U S A 98:12162–12167. doi: 10.1073/pnas.211423598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Aline F, Bout D, Dimier-Poisson I. 2002. Dendritic cells as effector cells: gamma interferon activation of murine dendritic cells triggers oxygen-dependent inhibition of Toxoplasma gondii replication. Infect Immun 70:2368–2374. doi: 10.1128/IAI.70.5.2368-2374.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Brown GD. 2011. Innate antifungal immunity: the key role of phagocytes. Annu Rev Immunol 29:1–21. doi: 10.1146/annurev-immunol-030409-101229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hardison SE, Brown GD. 2012. C-type lectin receptors orchestrate antifungal immunity. Nat Immunol 13:817–822. doi: 10.1038/ni.2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Arpaia N, Godec J, Lau L, Sivick KE, McLaughlin LM, Jones MB, Dracheva T, Peterson SN, Monack DM, Barton GM. 2011. TLR signaling is required for Salmonella typhimurium virulence. Cell 144:675–688. doi: 10.1016/j.cell.2011.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nakanishi K, Yoshimoto T, Tsutsui H, Okamura H. 2001. Interleukin-18 regulates both Th1 and Th2 responses. Annu Rev Immunol 19:423–474. doi: 10.1146/annurev.immunol.19.1.423. [DOI] [PubMed] [Google Scholar]
- 55.Akira S. 2000. The role of IL-18 in innate immunity. Curr Opin Immunol 12:59–63. doi: 10.1016/S0952-7915(99)00051-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.