

Abstract
Gamma interferon (IFN-γ) drives antiparasite responses and immunopathology during infection with Plasmodium species. Immunity-related GTPases (IRGs) are a class of IFN-γ-dependent proteins that are essential for cell autonomous immunity to numerous intracellular pathogens. However, it is currently unknown whether IRGs modulate responses during malaria. We have used the Plasmodium berghei ANKA (PbA) model in which mice develop experimental cerebral malaria (ECM) to study the roles of IRGM1 and IRGM3 in immunopathology. Induction of mRNA for Irgm1 and Irgm3 was found in the brains and spleens of infected mice at times of peak IFN-γ production. Irgm3−/− but not Irgm1−/− mice were completely protected from the development of ECM, and this protection was associated with the decreased induction of inflammatory cytokines, as well as decreased recruitment and activation of CD8+ T cells within the brain. Although antigen-specific proliferation of transferred CD8+ T cells was not diminished compared to that of wild-type recipients following PbA infection, T cells transferred into Irgm3−/− recipients showed a striking impairment of effector differentiation. Decreased induction of several inflammatory cytokines and chemokines (interleukin-6, CCL2, CCL3, and CCL4), as well as enhanced mRNA expression of type-I IFNs, was found in the spleens of Irgm3−/− mice at day 4 postinfection. Together, these data suggest that protection from ECM pathology in Irgm3−/− mice occurs due to impaired generation of CD8+ effector function. This defect is nonintrinsic to CD8+ T cells. Instead, diminished T cell responses most likely result from defective initiation of inflammatory responses in myeloid cells.
INTRODUCTION
Cerebral malaria (CM) is the most severe manifestation of Plasmodium falciparum malaria infection in humans, and it is responsible for more than half a million deaths annually, predominantly in sub-Saharan Africa (1). Gamma interferon (IFN-γ) production by leukocytes is a prominent feature of malarial infection. Typically, this IFN-γ contributes to parasite clearance; however, it may also drive pathology (2). Clinically, the brain dysfunction that occurs during CM manifests as seizures and coma, with progression to death occurring in the absence of treatment. While a definitive understanding of the pathological events underlying CM remains elusive, considerable evidence supports a role for IFN-γ (3).
Infection of C57BL/6 mice with blood-stage Plasmodium berghei ANKA (PbA) leads to experimental cerebral malaria (ECM), which reproduces many features of human CM (4). IFN-γ, produced either by NK cells or by CD4+ T cells prior to end-stage disease, markedly increases the expression of major histocompatibility complex I (MHC-I) molecules, ICAM-1 cell adhesion molecules, and CXCR3 ligands in endothelial cells (3, 5). Together, these changes contribute to the recruitment of leukocytes, particularly CD8+ T cells, to the brain microvasculature (3, 6). Current evidence indicates that CD8+ T cell-derived IFN-γ itself does not contribute to pathology (7). Instead, cross-presentation of malaria antigen on central nervous system (CNS) microvascular endothelial cells and recognition by CD8+ cytotoxic T cells (8) leads to endothelial damage in a granzyme B- and perforin-dependent manner (9, 10). Despite the accumulation of knowledge of the effects of IFN-γ in Plasmodium infection, its actions are highly pleiotropic; therefore, it is likely that IFN-γ-dependent pathways that influence disease progression are yet to be identified.
Among the nearly 2,000 genes that are known to be modulated by IFN-γ (11), the p47 immunity-related GTPases (IRGs) are critical for protection against a range of intracellular bacteria, protozoa, and viruses in diverse cell types (12, 13). A subset of IRGs (IRGM1-IRGM3 in mice and the constitutively expressed IRGMa-IRGMd, resulting from alternative splicing, in humans) has received much attention. IRGM1 and IRGM3, in particular, have been argued to act by modulating the positioning of effector molecules, including other IRG family members, to intracellular vacuoles that contain pathogens (14–19). This leads to breakdown of the vacuole and release of the pathogen into the cytosol. Subsequently, this results in either necroptosis or autophagy, depending upon the cell type (20, 21). Alternatively, other studies have argued that IRM1 and IRGM3 play roles in pathogen sensing. For example, IRGM1 may act as a pathogen sensor by binding to the autophagy signaling lipids PtdIns(3,4)P2 and PtdIns(3,4,5)P3 on the membrane of mycobacterial phagosomes, where it also may exert effector activity by accelerating phagosome-lysosome fusion (14, 22, 23). In addition, since IRGM proteins can inhibit effector IRGs from becoming activated on membranes, and since parasitophorous vacuole membranes may lack IRGM proteins, it has been proposed that IRGM proteins also act as a “missing-self” signal on pathogen-containing vacuoles (17, 24). Finally, it has been reported that IRGM3 plays a role in cross-presentation through its ability to control the formation of lipid bodies (25).
Given the strong IFN-γ dependence of anti-Plasmodium immunity, as well as the requirement for IFN-γ in ECM pathology, we hypothesized that the IRG family members IRGM1 and IRGM3 contribute to these processes during blood-stage PbA infection. We found that both Irgm1 and Irgm3 were induced following infection, but neither strain exhibited any deficiency in the control of peripheral parasitemia. However, strikingly, Irgm3−/− but not Irgm1−/− mice were fully protected from ECM. This protection was associated with decreased cytokine induction and recruitment of CD8+ T cells to the brains of infected mice. Furthermore, decreased induction of inflammatory cytokine expression was seen within the spleen at early stages of infection. Finally, parasite-specific CD8+ T cell effector differentiation was found to be impaired in Irgm3−/− mice in a non-T cell-intrinsic manner. Together, these data suggest that defective sensing of parasites and subsequent blunted inflammatory responses contribute to impairment of the pathogenic CD8+ T cell effector response in Irgm3−/− mice.
MATERIALS AND METHODS
Mice.
Irgm3 knockout (Irgm3−/−) (26) and Irgm1 knockout (Irgm1−/−) (27) mice originally were generated on a 129sv background. These strains were backcrossed to a C57Bl/6Ncr background for 11 and 12 generations, respectively, prior to the start of experiments (28) and were bred in-house. For these strains, cohoused C57Bl/6Ncr mice were used as wild-type (WT) controls. C57BL/6J IFN-γ knockout (Ifng−/−) (29) and OT-I transgenic mice expressing the T cell receptor (TCR) specific for ovalbumin peptide SIINFEKL/H-2 Kb (OVA257–264) on a C57Bl/6J.SJL-Ptprc congenic (CD45.1+) background (Ptprca-OT-I) (30, 31) were bred in-house. C57BL/6J mice, serving as controls for Ifng−/− animals, were purchased from the Australian Animal Research Centre (Perth, Australia) and cohoused for 3 weeks prior to commencement of experiments. Mice were housed in the Medical Foundation Building Animal House, University of Sydney (Sydney, Australia), in group cages under a 12-h light-dark cycle with food and water ad libitum. Male and female mice, age 6 to 9 weeks, were used in separate experiments. Experimental procedures were performed under the guidelines of the University of Sydney Animal Care and Ethics Committee, with the Committee's approval.
Parasite infection.
Unless otherwise stated, PbA infection was established by infecting naive mice with 1 × 106 PbA-infected red blood cells (pRBC) via intraperitoneal (i.p.) injection (32). Two transgenic parasite lines, one expressing green fluorescent protein (GFP) (PbG) and the other expressing MHC-I-restricted chicken OVA257–264 epitopes (SIINFEKL; H-2Kb restricted) fused to GFP under the control of the ef1-α promoter (PbTG) (33), were used in adoptive transfer studies. Mice infected with PbTG or PbG were treated with 10 mg/kg of body weight pyrimethamine in drinking water, as a selectable marker for transgenic parasites, throughout the experiment (34, 35). Blood parasitemia was monitored by thin tail-blood smears stained with 30% (vol/vol) Giemsa. Tail blood smears were examined by light microscopy, and the percentage of pRBC was estimated by counting per 1,000 erythrocytes. The infected mice were closely monitored and classified as ECM if they had a clinical score of 3 or 4 (36). Mice that did not develop ECM by day 6 to 8 postinfection were monitored continuously up to 18 days postinfection (p.i.), as most mice developed acute anemia and hyperparasitemia by then.
Blood-brain barrier (BBB) integrity.
Mice were injected i.p. with 200 μl of 2% (wt/vol) Evans blue solution in phosphate-buffered saline (PBS) at day 6 p.i. Three hours later, mice were perfused intracardially with 20 ml cold PBS under deep isoflurane anesthesia. Whole brains were removed and split into the two hemispheres. Each hemisphere was weighed and submerged in 500 μl N,N-dimethyl formamide. Tissues were kept at 4°C for 72 h protected from light. After extraction, 200 μl formamide from each sample was plated in a 96-well flat-bottom plate. The optical density of Evans blue was measured at 620 nm (OD620) (37, 38) using a Spectramax 190 spectrophotometer (Molecular Devices, USA). The amount of Evans blue per sample was estimated from the regression of serial dilutions of Evans blue prepared in formamide. The amount of Evans blue in each sample was divided by the weight of tissue and is presented as mean Evans blue weight/tissue weight of the two hemispheres.
RT-qPCR.
Reverse transcription-quantitative PCR (RT-qPCR) was performed as described previously (39), except that total RNA was isolated from brain and spleen homogenates using the Isolate II RNA minikit (Bioline). PCR was performed with a Rotor-Gene Q system (Qiagen) using Kapa SYBR Fast qPCR mix (Kapa Biosystems). Forty cycles of amplification were performed using a 95°C (15-s) denaturation step, followed by a 60°C (20-s) annealing/extension step. Relative quantitation was calculated using the ΔΔCT method (where CT indicates threshold cycle), with normalization to the RPL13a reference gene. Amplification efficiencies of different primer sets were compared using serial dilutions of cDNA, and the purity of amplified products was assessed by melting curve analysis. Fold changes in the gene expression of infected mice relative to those of naive mice were calculated. The primers are listed in Table 1.
TABLE 1.
Primers used for RT-qPCR
| Target | Primer sequence |
|---|---|
| Perforin | 5′-GGTGGAGTGGAGGTTTTTGTACC-3′ (sense) |
| 5′-CAGAATGCAAGCAGAAGCACAAG-3′ (antisense) | |
| Granzyme B | 5′-CCTGAAGGAGGCTGTGAAAGAATC-3′ (sense) |
| 5′-CCCTGCACAAATCATGTTTAGTCC-3′ (antisense) | |
| IFN-γ | 5′-CAGCAACAGCAAGGCGAAA-3′ (sense) |
| 5′-GCTGGATTCCGGCAACAG-3′ (antisense) | |
| IFN-α | 5′-GTGAGGAAATACTTCCACAG-3′ (sense) |
| 5′-GGCTCTCCAGACTTCTGCTC-3′ (antisense) | |
| IFN-β | 5′-CAGCTCCAAGAAAGGACGAAC-3′ (sense) |
| 5′-GGCAGTGTAACTCTTCTGCAT-3′ (antisense) | |
| Irgm1 | 5′-CTCTGACACCGAGAGAAT-3′ (sense) |
| 5′-GGAGAAAGTGAAGTACCC-3′ (antisense) | |
| Irgm3 | 5′-CATCTCCAGTCTCCCTGTA-3′ (sense) |
| 5′-CAGCTACTGCAGAGTATC-3′ (antisense) | |
| RPL13a | 5′-CTTAGGCACTGCTCCTGTGGAT-3′ (sense) |
| 5′-GGTGCGCTGTCAGCTCTCTAAT-3′ (antisense) |
CBA.
Cytokine levels in brain and spleen homogenates were quantified using a cytometric bead array (CBA; Becton Dickinson Biosciences) as described previously (39). The CBA kit was used according to the manufacturer's instructions, with the modification that volumes of all reagents and samples were either 50% or 10% of those in the original protocol. Data were collected using a Beckman Coulter cytomics FC500 MLP flow cytometer (Beckman Coulter) and analyzed with FlowJo software (TreeStar). The concentration of cytokine was calculated by standard curve regression.
ELISA.
Mouse CXCL10 was measured by a mouse CXCL10/IP-10 DuoSet enzyme-linked immunosorbent assay (ELISA) development kit (R&D Systems) used according to the manufacturer's instructions. Cytokine concentrations were calculated by standard curve regression.
Preparation of tissue leukocytes for flow cytometry.
Brain leukocytes were prepared as previously described (39) after intracardial perfusion. Briefly, brain hemispheres were mashed between frosted slides and treated with 0.5 mg/ml collagenase type IV (Sigma-Aldrich) and 28 U/ml DNase I (Sigma-Aldrich) at room temperature for 1 h. The brain homogenate was mixed with 7 ml of isotonic 30% Percoll in PBS (GE Healthcare) and centrifuged for 10 min at 500 × g. Erythrocytes in the cell pellet were lysed with Tris-ammonium chloride (Tris-NH4Cl) lysis buffer. Splenocytes for analysis of costimulatory molecules were prepared as described previously (40). Briefly, minced splenic tissue was incubated with 0.5 mg/ml collagenase type IV (Sigma-Aldrich) and 28 U/ml DNase I (Sigma-Aldrich) at room temperature for 20 min, triturated through a Pasteur pipette, and incubated for a further 20 min. Erythrocytes were lysed with Tris-NH4Cl lysis buffer.
Adoptive transfer and intracellular cytokine staining.
Proliferation and differentiation of adoptively transferred OT-I cells was performed as described previously (33, 35, 41, 42), with minor modifications. Pooled spleen and lymph node cells from Ptprca-OT-I mice, containing ∼20% CD8+ T cells, were labeled with 1 μmol/liter carboxyfluorescein succinimidyl ester (CFSE; eBioscience) at 37°C for 10 min in the dark. Cells were washed 3 times in RPMI 1640 medium supplemented with 5% (vol/vol) fetal calf serum (FCS). The labeled Ptprca-OT-I cells (0.75 × 106 to 1 × 106 CD8+ T cells/100 μl) were adoptively transferred into naive WT and Irgm3−/− mice via intravenous (i.v.) injection. Recipient mice were infected with PbTG or PbG (1 × 106 pRBC i.p.) 24 h later. For determination of OT-I proliferation, spleens of infected recipient mice were harvested at day 3 p.i. OT-I cells were identified and CFSE dilution was determined by flow cytometry. For determination of effector function, splenocytes were harvested from infected WT and Irgm3−/− recipient mice at day 6 p.i. and plated at 4 × 106 splenocytes per sample in a 96-well round-bottom plate. Cells were pulsed with 10 μg/ml brefeldin A and 2 pg/ml recombinant human interleukin-2 (IL-2) in RPMI 1640 containing 5% (vol/vol) FCS with or without 10 μg/ml OVA257–264 (SIINFEKL) peptide for 5 h at 37°C with 5% CO2. After standard surface staining, cells were fixed in 4% (vol/vol) paraformaldehyde fixation buffer (BioLegend) for 20 min at room temperature. Fixed cells were permeabilized by washing 3 times in 1× permeabilization washing buffer containing 0.1% (wt/vol) saponin (BioLegend). Permeabilized cells were stained with phycoerythrin (PE)/Cy7-anti-mouse-IFN-γ for 20 min at room temperature.
Flow cytometry.
Cell staining for flow cytometry was performed using standard protocols as previously described (40). Briefly, cell suspensions (typically 1 × 106 to 2 × 106 cells) were preblocked with anti-CD16/32 (Fc-block) and then stained with antibody cocktails on ice for 60 min in the dark. Intracellular IFN-γ and granzyme B staining were performed essentially as described previously (39), with the modification that prior to staining, brain-sequestered leukocytes were incubated for 3 h at 37°C, 5% CO2 in Dulbecco's modified Eagle medium (DMEM) containing 10% fetal calf serum in the presence of a 1/1,000 dilution of stock brefeldin A (EBioscience). Data were acquired on either a 10-laser BD fluorescence-activated cell sorter (FACS) LSR II SORP (Becton Dickinson Biosciences), BD Fortessa (Becton Dickinson Biosciences), or FC500 or Gallios flow cytometer (Beckman Coulter). Data were analyzed with FlowJo software (TreeStar, Ashland, OR). Cell populations were defined as the following: CD4+ T cells, CD45hi CD3+ CD4+; CD8+ T cells, CD45hi CD3+ CD8+; NK cells, CD45hi NK1.1+ NKp46+; monocytes, CD45hi CD11b+ Ly6C+; and Ptprca-OT-1 CD8+ T cells, CD45.1+ CD45.2− CD3+ CD8+ Vα2+. For analysis of costimulatory molecule expression on spleen populations, autofluorescence (AF) was incorporated into gating strategies to facilitate identification of splenic myeloid subsets (40). Populations were defined as the following: CD8+ dendritic cells (DC), Lin− AFlo CD11chi CD11b− CD8+; CD11b+ DC, Lin− AFlo CD11chi CD11b+ CD8−; PDC, Lin− AFlo CD11cint CD11b− B220+ Ly6C+ Siglec-h+; Ly6Chi monocytes, Lin− AFlo CD11b+ Ly6Chi SSClo; RPM, Lin− AFhi F4/80hi.
Antibodies.
Fluorochrome-conjugated antibodies were purchased from Becton Dickinson, EBioscience, or BioLegend: biotin- or fluorescein isothiocyanate (FITC)-anti-CD3e (clone 145-2C11); violet 450-anti-CD3e (clone 500A2); biotin- or FITC-anti-CD19 antibody (clone 6D5); biotin- or FITC-anti-NK1.1 (clone PK136); biotin- or FITC-anti-Ly6G (clone 1A8); FITC-, PE-, V500-, and allophycocyanin (APC)-Cy7-anti-CD8 (clone 53-6.7); FITC-anti-CD4 (clone H129.19); PE-anti-CD4 (clone GK1.5); PE-anti-Ly6C (clone HK1.4); horizon V450-anti-Ly6C (clone AL-21); PE-anti-Vα2 (clone B20.1); PE-Cy7-anti-CD45 (clone 30-F11); APC-anti-CD3e antibody (clone 145-2C11); APC-anti-CD11b or APCCy7-anti-CD11b (clone M1/70); APC-anti-CD44 antibody (clone 1M7); Alexa Fluor 647-anti-NKp46 (clone 29A1.4); peridinin chlorophyll protein (PerCP)-Cy5.5-anti-CD45.1 (clone A20); horizon 500-anti-CD45.2 (clone 104); PE-anti-CD40 (clone 1C10); PE-anti-CD80 (clone 10-10A1); PE-anti-CD86 (clone GL1); PE-anti-4-1BBL (clone TKS-1); PE-anti-CD70 (clone FR70); PE-Cy7-anti-F4/80 (clone BM8); A647-anti-Siglec-h (clone 551); Alexa Fluor 700-anti-CD11c (clone N418); PE-anti-IFN-γ (clone XMG1.2); Alexa Fluor 647-anti-granzyme B (clone GB11); PerCP-streptavidin.
Data and statistical analysis.
All cytometric assays were analyzed using FlowJo software (TreeStar, USA). All standard curve regression analyses were performed by GraphPad Prism, version 5.01 for Windows (GraphPad Software, La Jolla, CA), and concentrations of cytokines in tissue homogenates were normalized to total protein content for each sample. Data are presented as means ± standard errors of the means (SEM) with n > 3 from at least two independent experiments unless otherwise specified. For comparison between two groups, unpaired t tests were used. For multigroup comparisons, one-way analysis of variance (ANOVA) and Tukey's test or two-way ANOVA and Bonferroni's test were used. Survival curves were generated in GraphPad Prism 5.01, and the significance of differences was calculated by Mantel-Cox log-rank test. Statistical significance was defined as P < 0.05.
RESULTS
Induction of Irgm1 and Irgm3 mRNA during PbA infection.
As an initial step in investigating any function of Irgm1 and Irgm3 during PbA infection, we determined whether mRNA for the genes was induced in the brains of mice at the typical phase of infection during which mice develop ECM (days 6 to 7 p.i.) and in the spleens of infected mice at the peak of the systemic IFN-γ response (day 4 p.i.). There was an average 80- ± 25-fold induction of Irgm1, as well as a 125- ± 44-fold induction of Irgm3 expression in the brains of PbA-infected mice at days 6 to 7 p.i. This induction was completely abrogated in Ifng−/− mice (Fig. 1A). In the spleens of infected mice, at day 4 p.i., both Irgm1 and Irgm3 were induced compared to the levels in naive mice (average, 4.4- ± 0.8-fold and 5.4- ± 0.6-fold, respectively), but, as seen in the brain, no induction was seen in Ifng−/− mice (Fig. 1B).
FIG 1.
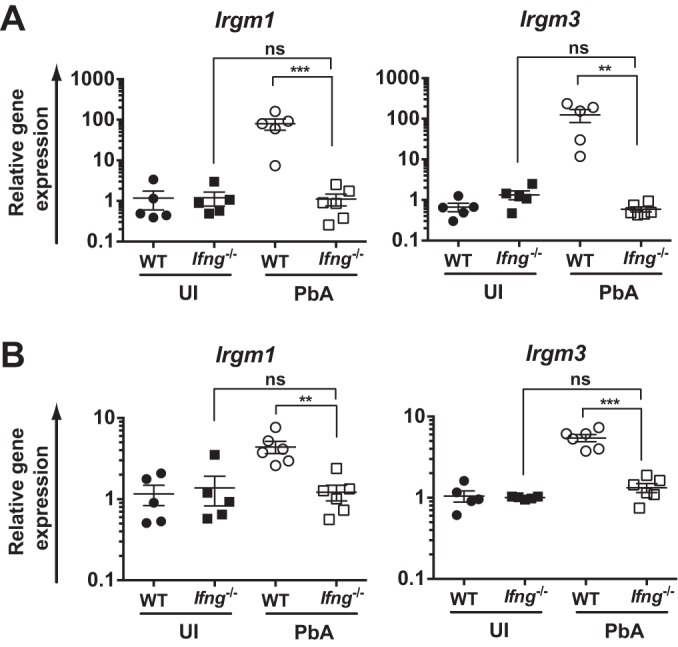
IFN-γ-dependent induction of Irgm1 and Irgm3 mRNA in the brain and spleen during PbA infection. C57BL/6 (WT) and Ifng−/− mice were infected with PbA. Mice were assessed for Irgm1 and Irgm3 mRNA induction by RT-qPCR in the brain at days 6 to 7 p.i. (A) and spleen at day 4 p.i. (B). Data are presented as means ± SEM. Results are derived from a single experiment with n = 5 to 6 per group. Statistical analysis was performed via one-way ANOVA with Tukey's test. **, P < 0.01; ***, P < 0.005; ns, P > 0.05. UI, uninfected.
Irgm3 deficiency protects mice from ECM but does not modify peripheral parasitemia.
The effect of deletion of Irgm1 and Irgm3 on disease course during PbA infection was investigated in C57Bl/6Ncr (WT), Irgm1−/−, and Irgm3−/− mice. Animals were infected with blood-stage PbA and were monitored for up to 18 days p.i. As was expected, 80% of WT mice showed neurological signs on days 6 to 9 p.i., when parasitemia reached 10 to 20%, and were euthanized within 24 h. While the development of ECM in Irgm1-deficient mice showed a modest delay compared to that in WT mice, strikingly, the Irgm3-deficient mice did not develop ECM by day 6 p.i. (P < 0.001 by Mantel-Cox log-rank test) (Fig. 2A). All Irgm3−/− mice and the remaining 20% of the WT mice that did not become moribund with ECM developed hyperparasitemia and anemia and were euthanized at day 18 p.i. (Fig. 2A). There were no differences in the levels of peripheral parasitemia between infected WT, Irgm1−/−, and Irgm3−/− mice at any stage of infection. This indicated that neither IRGM1 nor IRGM3 was involved in clearance of circulating parasites, and that the ECM-resistant phenotype of Irgm3−/− mice was not due to differences in the control of blood-stage parasite expansion. Given the minimal changes to disease course in Irgm1-deficient mice but the striking protection seen in Irgm3−/− animals, we focused subsequent studies on investigating the role of Irgm3 in protection from ECM.
FIG 2.

Irgm3 deficiency protects mice from PbA-induced ECM. (A) Irgm1−/−, Irgm3−/−, and wild-type (WT) mice aged 6 to 8 weeks were infected with 1 × 106 PbA pRBC and monitored for 18 days p.i. or until they became moribund. Parasitemia was determined from Giemsa-stained blood smears and is shown as means ± SEM. Data are derived from one experiment with WT (n = 11) and Irgm1−/− (n = 6) mice and from three independent experiments with WT (n = 27) and Irgm3−/− (n = 28) mice. The Mantel-Cox log-rank test was used to compare the survival rate between WT and gene knockout mice. Significance was defined as P < 0.05. (B) Effect of Irgm3 deficiency on brain vascular permeability to Evans blue/protein in PbA-infected mice. Mice were injected with 0.2 ml 2% (wt/vol) Evans blue on day 6 p.i. Two hours after injection, following intracardiac perfusion, brains were harvested and Evans blue content was extracted in formamide. Data are presented as means ± SEM. Results are from one experiment with n ≥ 4 per group. Statistical analysis was performed via one-way ANOVA with Tukey's posttest test. *, P < 0.05. UI, uninfected.
To assess the degree of cerebral involvement in WT and Irgm3−/− mice inoculated with PbA, histological parameters were examined in postmortem brain sections. In a blinded study, WT mouse brains at day 6 p.i. showed typical histopathological features of ECM, such as hemorrhages in the olfactory bulb and monocyte adherence to the microvascular endothelium. These features were absent from infected Irgm3−/− mice and uninfected controls (see Fig. S1 in the supplemental material). To extend these findings, we measured Evans blue extravasation as an indicator of blood-brain barrier (BBB) integrity in the brains of WT and Irgm3−/− mice at the peak of disease, when ECM had developed in the PbA-infected WT mice. Evans blue was administered 2 h before euthanasia on day 6 or 7 p.i. (Fig. 2B). PbA-infected WT mice with symptomatic ECM had elevated brain Evans blue content compared to that of the WT uninfected controls. Although Evans blue, which binds to plasma proteins, was found in the brains of PbA-infected Irgm3−/− mice, microvascular leakage of plasma proteins into the brain parenchyma was significantly reduced in the PbA-infected Irgm3−/− mice compared to that in infected WT controls as measured by Evans blue extravasation.
Reduced induction of cytokines, chemokines, and effector T cell marker genes in the brains of Irgm3−/− mice.
Since end-stage pathology in ECM is mediated by IFN-γ-dependent recruitment of effector CD8+ T cells (36, 43), we quantified the levels of IFN-γ and relevant chemokines in the brains of infected WT and Irgm3−/− mice. Figure 3A shows that IFN-γ, CCL2, CXCL9, and CXCL10 proteins were elevated in the brains of infected WT mice, compared to levels for naive mice, during the peak of disease at days 6 to 7 p.i. Levels of these mediators also were raised in infected Irgm3−/− mice. However, IFN-γ protein levels were approximately 2-fold higher (P = 0.0005) in infected WT animals (2.8 ± 0.3 pg/mg) than in Irgm3−/− mice (1.4 ± 0.2 pg/mg) (Fig. 3A). This pattern also was seen at the mRNA level, where the induction of Ifng mRNA was around twice as great in infected WT mice (40- ± 8-fold) as in Irgm3−/− mice (18- ± 3-fold) (Fig. 3B). The levels of the IFN-γ-inducible chemokine protein CCL2 also was higher (P < 0.0001) in brains of infected WT mice (155.0 ± 9.9 pg/mg) than in Irgm3−/− mice (50.7 ± 3.5 pg/mg). However, there were no significant differences in the production of CXCL9 and CXCL10 proteins in brains of PbA-infected Irgm3−/− mice compared to that of WT mice. This suggested that ECM protection in Irgm3−/− mice was not associated with differences in CXCL9 and CXCL10 cytokine production in endothelial cells and/or astrocytes. Finally, perforin and granzyme B are cytotoxic proteins present at high levels in the granules of effector CD8+ T cells and are required for the development of ECM (9, 10, 44). Therefore, we measured changes in their expression within the brain at day 6 p.i. (Fig. 3B). Infected WT mice showed greater inductions of both perforin (28- ± 6-fold) and granzyme B (1,344- ± 215-fold) mRNA than infected Irgm3−/− mice (perforin, 13- ± 2-fold; granzyme B, 491- ± 46-fold).
FIG 3.

Cytokine levels and effector CD8+ T cell marker mRNA expression in brains of Irgm3−/− mice during acute PbA infection. WT and Irgm3−/− (KO) mice were infected with 1 × 106 PbA pRBC, and brains were collected following intracardiac PBS perfusion on day 6 or 7 p.i. (A) Half brains from individual mice were homogenized in protease inhibitor cocktail in PBS and were assessed for IFN-γ, CCL2, and CXCL9 using a cytometric bead array. CXCL10 protein was measured by ELISA. Cytokine protein was normalized to total protein and is presented as pg/mg. (B) Expression of mRNA for IFN-γ (Ifng), granzyme B (Gzmb), and perforin (Prf1) in brain homogenates was determined by RT-qPCR. Symbols represent individual animals, and horizontal lines and error bars represent means ± SEM. Data are from two or three pooled experiments with a total of n = 5 to 23/group. Statistical analysis was performed using one-way ANOVA with Tukey's test. *, P < 0.05; ***, P < 0.005; n.s, P > 0.05. UI, uninfected.
Decreased recruitment and activation of brain-sequestered CD8+ T cells in Irgm3−/− mice.
The reduced induction of IFN-γ, perforin, and granzyme B within the brains of infected Irgm3−/− mice suggested either that there was diminished recruitment of CD8+ T cells or that the activation state of recruited cells was impaired. Therefore, we quantified the absolute numbers of sequestered leukocyte subsets, as well as expression of activation markers on T cells, on day 6 or 7 p.i. and compared them to those of naive mice (Fig. 4A). Numbers of CD8+ T cells (CD45hi, CD3+, CD8+) in the brains of PbA-infected WT mice were increased ∼65-fold compared to those for naive mice. Although the brains of PbA-infected Irgm3−/− mice also had a substantial increase in the number of CD8+ T cells compared to that of naive controls, the average CD8+ T cell number was reduced by 48% compared to that for infected WT mice (Fig. 4A). Fewer CD4+ T cells (CD45hi, CD3+, CD4+) were recruited to the brains of infected mice than CD8+ T cells. In addition, there was no significant difference between the number of brain-sequestered CD4+ T cells in infected Irgm3−/− mice and that in infected WT mice (Fig. 4A). Likewise, the numbers of brain-sequestered NK cells were raised in PbA-infected mice, but there was no significant difference in the numbers of sequestered NK cells between PbA-infected WT and Irgm3−/− mice (Fig. 4A). Inflammatory monocytes were increased by ∼4-fold in the brains of PbA-infected WT mice compared with that of naive mice, while infected Irgm3−/− mice had significantly fewer sequestered monocytes than the infected WT (Fig. 4A).
FIG 4.
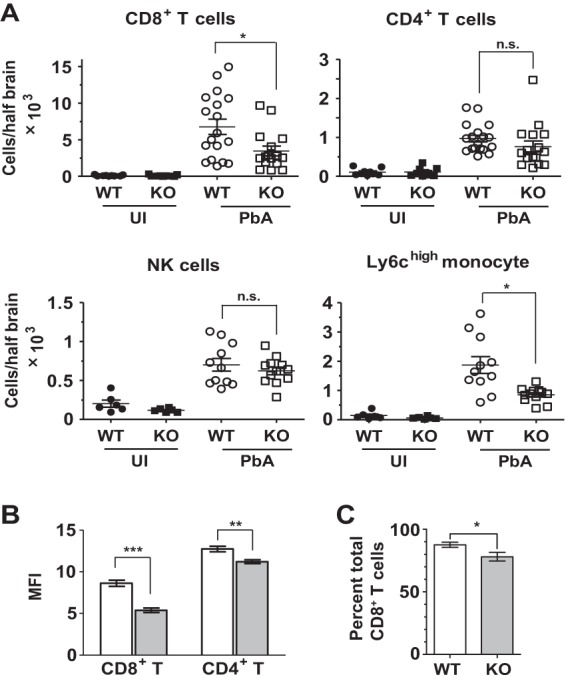
Irgm3 deletion affects leukocyte recruitment to the brain. WT and Irgm3−/− (KO) mice were infected with 1 × 106 PbA pRBC, and brains were collected following intracardiac PBS perfusion at day 6 or 7 p.i. (A) Flow-cytometric analysis of brain-sequestered leukocytes was performed. Symbols represent individual animals, and horizontal lines and error bars represent means ± SEM. Statistical analysis was performed via one-way ANOVA with Tukey's test. (B) Analysis of cell surface CD44 expression on brain-sequestered CD8+ and CD4+ T cells from PbA-infected WT and Irgm3−/− mice on day 6 or 7 p.i. Open bars represent the WT, and shaded bars represent Irgm3−/− mice. (C) Granzyme B intracellular staining in brain-sequestered CD8+ T cells from infected mice on day 7 p.i. The percentage of granzyme B+ cells is shown. Data presented are means ± SEM. Data are derived from two to three experiments with total n = 6 to 18/group (A and B) or a single experiment with 4 to 8/group (C). Statistical analysis was performed via unpaired t test (C) or two-way ANOVA with Bonferroni test (A and B). *, P < 0.05; **, P < 0.01; ***, P < 0.005; n.s, P > 0.05. UI, uninfected.
As an initial step to investigate whether recruited T cells in infected WT and Irgm3−/− mice had functional differences, the expression of CD44 on brain-sequestered CD4+ and CD8+ T cells was examined by flow cytometry. In PbA-infected WT and Irgm3−/− mice, CD44 expression was upregulated on both CD4+ and CD8+ T cells. However, the median level of CD44 expression on CD8+ T cells from the brains of PbA-infected Irgm3−/− mice was significantly lower than that of CD8+ T cells isolated from the brains of infected WT animals at corresponding times (Fig. 4B). The expression of both intracellular granzyme B and IFN-γ protein in brain-sequestered CD8+ T cells also was examined. Although there was no difference in the expression of IFN-γ by brain-sequestered CD8+ T cells between infected WT and Irgm3−/− mice (data not shown), there was a modest but significant decrease in the percentage of brain-sequestered CD8+ T cells that expressed granzyme B in infected gene-deficient animals (Fig. 4C). Taken together, these data suggest that Irgm3 deficiency leads both to decreased CD8+ T cell recruitment to the brain and to reduced activation during PbA infection.
Irgm3 deficiency impairs effector CD8+ T cell differentiation, but not proliferation, in a cell-extrinsic manner.
Given that generation of antigen-specific CD8+ T cells is required for development of ECM and that Irgm3 deficiency has been reported to impair MHC-I cross-presentation by DCs (25), it was important to determine whether Irgm3−/− mice could generate specific CD8+ T cell responses to blood-stage P. berghei infection. To address this question, we used the OT-I transgenic mouse system in which transgenic CD8+ T cells recognize ovalbumin residues 257 to 264 (SIINFEKL) in the context of H-2Kb. Irgm3-sufficient Ptprca-OT-I splenocytes were labeled with CFSE and injected intravenously into WT and Irgm3−/− mice. Recipient mice were infected 1 day later with parasites expressing SIINFEKL (PbTG) or with a control strain that did not express the peptide (PbG) (33). On day 3 p.i., spleens were harvested and proliferation of the Ptprca-OT-I population responding to the parasite-origin SIINFEKL epitope was determined. The Ptprca-OT-I CD8+ T cells in WT and Irgm3−/− mice showed similar levels of proliferation on day 3 following PbTG infection (see Fig. S2 in the supplemental material). These findings indicated that antigens expressed during the blood-stage P. berghei infection can be efficiently presented by APC from Irgm3−/− mice to induce parasite-specific CD8+ T cell proliferation.
These data show that a robust antigen-specific CD8+ T cell-proliferative response to P. berghei blood-stage infection occurred in Irgm3−/− mice. They did not, however, elucidate whether there were any defects in CD8+ T cell differentiation. Therefore, we performed a similar assay using adoptively transferred CFSE-labeled Ptprca-OT-I cells as responders in WT and Irgm3−/− mice but used IFN-γ production as a measure of effector differentiation. Mice were infected with PbTG or PbG 1 day after receiving Ptprca-OT-I cells. After 6 days of infection, spleens of recipient mice were harvested and then stimulated in vitro with SIINFEKL-pulsed APC. OT-I cells were stained for intracellular IFN-γ as a measure of effector T cell differentiation. Consistent with data from day 3 p.i., on day 6 p.i. OT-I cells that were adoptively transferred into either WT or Irgm3−/− recipients showed similar levels of proliferation, as evidenced by the comparable intensity of CFSE staining (Fig. 5A). Strikingly, however, OT-I cells that were transferred into WT recipients showed much greater production of IFN-γ at day 6 p.i. than OT-I cells transferred into Irgm3−/− recipients (Fig. 5A and B). Together, these findings suggest that IRGM3, expressed in cells other than antigen-specific CD8+ T cells, plays a role in supporting CD8+ effector T cell differentiation.
FIG 5.
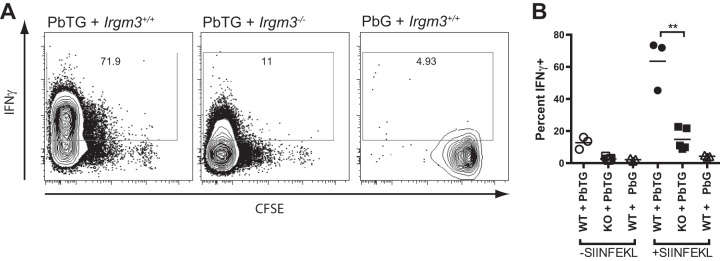
Irgm3−/− mice do not efficiently generate IFN-γ-producing effector CD8+ T cells. CFSE-labeled Ptprca-OT-I splenocytes were adoptively transferred into WT or Irgm3−/− (KO) recipient mice 1 day prior to infection with SIINFEKL-expressing (PbTG) or control (PbG) parasite strains. On day 6 p.i., splenocytes harvested from recipient mice were stimulated with recombinant IL-2 for 5 h with or without SIINFEKL peptide, and donor OT-I cells (CD45.1+, CD45.2−, CD3+, CD8+, Vα2+) were assessed for production of IFN-γ by intracellular staining. (A) Flow-cytometric analysis of splenocytes showing CFSE dilution and intracellular IFN-γ staining from SIINFEKL-stimulated cultures. (B) Quantification of IFN-γ staining in Ptprca-OT-I CD8+ T cells. Symbols represent expression of IFN-γ+ OT-I T cells isolated from individual animals. Data are derived from a single experiment with n = 3 to 5/group. Statistical analysis was performed via one-way ANOVA with Tukey's test. **, P < 0.01.
Irgm3 deficiency perturbs inflammatory cytokine production and myeloid costimulatory molecule expression.
Differentiation of effector functions in CD8+ T cells requires the provision of signal 3, which typically is provided by inflammatory cytokines and/or by costimulatory molecules on APCs (45). To investigate whether Irgm3 deficiency could influence signals contributing to early effector CD8+ T cell differentiation during PbA infection, we examined both cytokine production within the spleen and expression of costimulatory molecules by splenic myeloid populations.
Preliminary experiments revealed that levels of several inflammatory cytokines and chemokines within the plasma peaked around day 4 p.i. (data not shown). Therefore, we quantified inflammatory cytokine and chemokine levels in spleen homogenates from WT or Irgm3−/− mice infected either 3 or 4 days previously with PbA. Multiple cytokines, including IFN-γ, tumor necrosis factor (TNF), IL-6, and IL-12p70, showed increased levels in the spleen at day 3 p.i. in both WT and Irgm3−/− mice; however, no significant differences were found between infected groups (see Fig. S3 in the supplemental material). Similarly, the inflammatory chemokines CCL2, CCL3, and CCL4 were induced within the spleen at day 3 p.i., but there were no differences in chemokine levels between infected WT and Irgm3-deficient mice (see Fig. S3). When the same inflammatory cytokines and chemokines were quantified in spleen homogenates from day 4 p.i., all remained upregulated in WT animals, with the exception of IL-12p70 (Fig. 6A). However, in contrast to the pattern seen at day 3 p.i., infected Irgm3−/− mice showed lower levels of IL-6, CCL2, CCL3, and CCL4 at day 4 p.i. than infected WT mice. In addition, we investigated Ifna and Ifnb mRNA expression by RT-qPCR on days 3 and 4 p.i. (Fig. 6B; also see Fig. S3). These data were difficult to interpret, as no statistically significant induction of either Ifna or Ifnb was seen in infected mice compared to the respective uninfected genotype controls on either day 3 or 4 p.i. (see Fig. S3). Nevertheless, on day 4 p.i., infected Irgm3−/− mice showed increased mRNA expression for both type I IFN species compared to infected WT (Fig. 6B).
FIG 6.

Effect of Irgm3 deficiency on spleen cytokine levels on day 4 p.i. WT and Irgm3−/− (KO) mice were infected with PbA on day 0. On day 4 p.i., groups of uninfected (UI) and PbA-infected mice were euthanized and cytokine protein (A) or mRNA (B) levels were determined. (A) The indicated cytokines/chemokines were quantified by cytometric bead array. Data show cytokine concentration normalized to total protein within the homogenate. (B) Relative levels of the indicated cytokine genes were determined by RT-qPCR. Data are from two pooled experiments in which samples were processed and assayed together. Statistical significance was determined using one-way ANOVA with Tukey's posttest. Symbols represent individual mice, and horizontal bars are the geometric mean of groups. *, P < 0.05; **, P < 0.01; ns, P > 0.05.
The relative abundance of myeloid populations within the spleen, as well as the expression levels of a range of costimulatory molecules on the same populations, were determined on day 4 p.i. (see Fig. S4 in the supplemental material). Following infection with PbA, there was a distinct drop in the percentage of CD8+ DC with respect to total nucleated cells. Similar changes were not seen with other myeloid subsets. Moreover, there were no significant differences between infected WT and Irgm3−/− mice in terms of the abundance of any myeloid population (see Fig. S4A). The expression of a number of costimulatory molecules by major splenic myeloid populations was examined (CD40, CD80, CD86, CD70, OX40-L, B7-H3, ICOS-L, 4-1BBL, B7-H4, PDL1, and PDL2). However, in general, minimal perturbation of costimulatory pathways associated with Irgm3 deletion in infected mice was seen, with the possible exception of decreased expression of CD86 by PDC and 4-1BBL by Ly6Chi monocytes (see Fig. S4B and data not shown).
DISCUSSION
IRG family members play important roles in IFN-γ-dependent cell autonomous immunity during infection with many organisms. However, with the exception of a single report that demonstrated lack of involvement of IRGA6 during liver-stage PbA infection (46), they have not been investigated in the context of malaria. In the current study, we have determined the effect of Irgm1 and Irgm3 gene deletion on the disease course of blood-stage PbA infection. Deletion of neither Irgm1 nor Irgm3 impacted circulating parasite levels, indicating that IRGM1- or IRGM3-dependent processes are not involved in clearance of circulating parasites. When development of severe malarial pathology was investigated, Irgm1−/− mice showed a minor but significant delay in the development of neurological disease. Much more striking was the observation that PbA-infected Irgm3−/− mice did not show any evidence of neurological impairment up to 18 days of infection, and they did not show vascular hemorrhage in postmortem brain histopathology or breakdown of the BBB. This complete protection from ECM pathology was unexpected, and subsequent efforts focused on understanding the mechanisms behind this phenomenon.
IFN-γ is known to have a range of effects during ECM, including activation of endothelial cells and astrocytes, CD8+ T cell accumulation through modulation of chemokines, pRBC sequestration, and BBB permeabilization to protein (4, 5, 36, 47–52). Furthermore, Irgm3 mRNA expression was found to be upregulated in the brain at day 6 p.i., which was consistent with a role for IRGM3 protein at late stages in the immunopathological response to Plasmodium infection. We initially investigated whether Irgm3 deficiency led to modulation in the levels of relevant IFN-γ-dependent mediators. An increase in the proinflammatory chemokines CCL2, CXCL9, and CXCL10 is associated with ECM (36, 53); therefore, these chemokines were quantified in brains collected from PbA-infected mice on day 6 or 7 p.i. We found that levels of CCL2, but not CXCL9 and CXCL10, in brains of infected Irgm3−/− mice were lower than those of the infected WT animals.
CCL2 is produced by endothelial cells, astrocytes, monocytes, and microglial cells and is the major attractant for the recruitment of CCR2-expressing inflammatory monocytes to sites of inflammation (54). As such, it is involved in a range of diseases characterized by monocytic infiltration (55–57). PbA-infected Irgm3−/− mice had reduced CCL2 protein as well as decreased monocyte sequestration in the brain. The presence of monocytes is obvious in ECM brain lesions, and they have been shown to enhance recruitment of CD8+ T cells to the brain during ECM, probably via production of T cell-attracting chemokines (58). However, inflammatory monocytes are nonessential for the development of ECM pathogenesis (58, 59). Therefore, although Irgm3 deficiency may contribute to the decreased numbers of CD8+ T cells found in the brains of infected mice through decreased recruitment of inflammatory chemokine-producing monocytes, it is unlikely that this pathway ultimately is responsible for protection from ECM.
A number of studies have described a critical role for the recruitment of antigen-specific CD8+ effector T cells in mediation of the end stages of ECM pathology. The recruitment of these cells is largely dependent upon the CXCR3 ligands CXCL9 and CXCL10, which are highly upregulated in the brains of mice during ECM (36). Expression of these chemokine and chemokine receptor genes, i.e., CXCL9/CXCR3 and CXCL10/CXCR3, is important in recruitment of human and murine TH1-polarized CD4+ T cells and CD8+ cytotoxic T cells (60). They also may stimulate T cell proliferation and effector function (61). Furthermore, elevated plasma levels of CXCL10 strongly correlate with human CM mortality (62, 63). In the ECM model, immunohistological staining with in situ hybridization has revealed that CXCL9 and CXCL10 are produced by brain endothelial cells and astrocytes in response to IFN-γ stimulation (36). This upregulation of CXCL9/10 is argued to be stimulated by IFN-γ, which is produced either by NK cells (64) or CD4+ T cells (7). Sequestration of CD8+ T cells within the brain vasculature of PbA-infected mice was shown to be dependent on CXCR3 and CXCL10 (10, 36, 41, 53, 65). Importantly, Campanella et al. (53) and Miu et al. (36) demonstrated that blockade of the CXCR3-CXCL10/CXCL9 signaling axis reduced recruitment of CD8+ T cells to the brain and resulted in partial, but significant, protection from ECM. Despite this clear link between CXCR3 ligands and ECM pathology, in the current study Irgm3−/− mice did not show altered levels of CXCL9 or CXCL10 within the brain on days 6 and 7 p.i. compared to those of infected WT mice. It is perhaps surprising that changes in CXCL9 and CXCL10 did not mirror the reduction of IFN-γ and CD8+ T cells. We did note that at days 6 and 7 p.i., Irgm3 deficiency was not associated with a decrease in NK and CD4+ T cell recruitment to the brain. Thus, one possibility is that brain CXCL9 and CXCL10 levels generally reflect the inflammatory environment created by functional CD4+ T cells and NK cells in Irgm3−/− mice during PbA infection. Taken together, these results indicate that the ECM-resistant phenotype in Irgm3−/− mice is not due solely to modulation of the CXCR3 pathway.
During ECM, parasite-specific CD8+ T cells mediate pathology in a granzyme B- and perforin-dependent manner (9, 10). It has been proposed that antigen cross-presented by brain endothelium is recognized by CD8+ cytotoxic T lymphocytes (CTL) (8), which causes endothelial apoptosis and BBB disruption by granzyme B- and perforin-mediated cytotoxicity (9, 10, 44). Despite an absence of the impact of Irgm3 deletion on CXCR3 ligand expression compared to that of infected WT controls, Irgm3−/− mice showed decreased sequestration of CD8+ T cells, as well as decreased induction of IFN-γ, perforin, and granzyme B. This was consistent with a scenario in which protection from ECM results from decreased CD8+ T cell recruitment. However, the effect was modest (∼2- to 3-fold) for all parameters. Decreases in T cell recruitment of similar magnitude, resulting from depletion of inflammatory monocytes, do not result in protection from ECM (58). Therefore, it seems unlikely that the observed diminution in the absolute number of recruited CD8+ T cells could account for the dramatically divergent clinical outcomes. Supporting this notion, diminished levels of expression of CD44 and granzyme B were seen in sequestered CD8+ T cells, which suggested that CTL that were recruited somehow were functionally impaired. With this in mind, we further investigated the impact of Irgm3 deficiency on CD8+ T cell effector differentiation.
Optimal expansion of antigen-specific CD8+ T cell clones and acquisition of effector functions depends on the coordinated provision of signals from APCs, in particular cross-presenting DC populations. These signals include TCR-MHC-I engagement (signal 1), costimulatory molecules (signal 2), which primarily drive T cell proliferation, and cytokines and/or costimulatory molecules (signal 3), which largely dictate differentiation of effector functions (45, 66). Additionally, production of chemokines such as CCL3, CCL4, CXCL9, and CXCL10 within responding lymphoid tissues may coordinate interactions between cross-priming DCs and naive CD8+ T cells (66, 67). Cross presentation of parasite antigen by splenic CD8+, Clec9A+ DCs has been shown to be essential for CD8+ T cell priming during PbA infection and ultimately for the development of ECM (33, 68–70). Therefore, we systematically investigated the impact of Irgm3 deficiency on signals 1 to 3, as well as chemokine production, during PbA infection. Such a pathway was felt to be particularly relevant, since IFN-γ stimulates Irgm3 expression in DCs and Irgm3−/− DCs have been reported to have impaired capacity for cross-presentation, a phenomenon that is mediated through modulation of lipid body formation (25). To isolate the effect of Irgm3 deficiency on myeloid cells, we transferred naive, Irgm3-sufficient CD8+ OT-I T cells into WT or Irgm3−/− mice prior to infection with SIINFEKL-expressing (PbTG) or control (PbG) parasites. CFSE dilution in vivo, and antigen-driven production of IFN-γ ex vivo, were used as readouts of proliferation and effector differentiation, respectively.
At the whole-tissue level, Irgm3 mRNA was found to be upregulated within the spleen at day 4 p.i., which is consistent with a potential role for the protein in myeloid cells at this time point. The CCR5 ligands CCL3 and CCL4 also were found to be produced at high levels in the spleens of infected mice. These chemokines may be critical for guiding rare naive CD8+ T cells to cross-presenting DCs within lymphoid organs (71), so impairment in production in Irgm3−/− animals could explain poor cross-priming. Indeed, at the tissue level, concentrations of CCL3 and CCL4 were significantly lower in the spleens of infected Irgm3−/− mice than in infected WT mice. However, importantly, infection of either WT or Irgm3−/− recipients with PbTG led to comparable levels of OT-I proliferation between the two strains. In contrast, when the development of an effector phenotype was examined based on the capacity of transferred OT-I cells to produce IFN-γ in an antigen-dependent manner, Irgm3−/− recipients were found to have a marked defect in their ability to support effector differentiation. These data supported a model in which Irgm3 deficiency had no impact, at least at a gross level, on initial priming of naive T cells (signal 1 or signal 2). However, impaired effector function in Irgm3−/− mice strongly suggested that IRGM3 somehow was affecting signal 3.
The best-characterized stimuli that support differentiation of CD8+ T cells into effectors are inflammatory cytokines, most notably IL-12 and type-I IFNs; however, IFN-γ and IL-2, as well as other inflammation-associated cytokines, also may contribute (72, 73). In addition, a subset of costimulatory molecules, including 4-1BBL, OX40L, CD70, CD40, CD80, and CD86, may augment or, in some cases, substitute for cytokine signaling depending on the infection model (73). Therefore, we investigated whether the modulation of these signals in Irgm3−/− mice could account for inefficient T cell effector differentiation. When the expression of costimulatory molecules on spleen myeloid subsets was examined, gross defects were not seen in Irgm3−/− mice. Irgm3 deficiency potentially was associated with decreased induction of CD86 on PDC and 4-1BBL on inflammatory monocytes. An effect of Irgm3 deficiency on PDC function would be particularly intriguing, since these cells are prominent producers of type-I IFNs and have been argued to be the only nucleated cells to be infected by PbA (74). However, it has been reported that neither PDC (70) nor inflammatory monocytes (58) are required for ECM development. While Irgm3 deficiency did not affect splenic levels of IL-1β, IL-2, IL-12, TNF, or IFN-γ at the peak of systemic cytokine production, levels of IL-6 were significantly lower in infected Irgm3−/− mice than in infected WT controls. Consistent with a potential role in CD8+ effector differentiation during ECM, IL-6 has been reported to enhance CTL differentiation (75), in particular when in combination with IL-7 and/or IL-15 (76). Elevated levels of IL-6 in serum have been correlated with CM (52, 77). However, mice treated with anti-IL-6 antibodies still developed ECM (78). Therefore, we suspect the decrease of IL-6 levels in Irgm3−/− mice is not the only reason for dramatic changes in ECM pathology. Infected Irgm3−/− mice also showed an unexpected increase in expression of type I IFN genes compared to levels for infected WT controls. It has been reported recently that type I IFN signaling in CD8− DCs inhibits development of ECM (79). However, this resulted from inhibition of IFN-γ production by CD4+ T cells via suboptimal expression of costimulatory molecules on CD8− DCs. Since we did not see decreased expression of these costimulatory molecules on CD8− (CD11b+) DCs in Irgm3−/− mice, this mechanism appears unlikely to contribute to protection. Nevertheless, taken together, these data suggest that protection from ECM is due to decreased inflammatory cytokine signaling. However, the exact pathways by which this occurs remain to be established in detail.
In summary, we have demonstrated that Irgm3 deficiency leads to protection from ECM pathology following infection with PbA. This protection is associated with decreased recruitment of CTL to the brain; however, more obviously, we noted a strikingly diminished capacity of Irgm3−/− mice to support antigen-specific CD8+ T cell effector differentiation. The mechanisms underlying this defect in CTL generation currently are unclear, but blunted production of inflammatory cytokines, including IL-6, which can contribute to CTL differentiation, may be involved.
Supplementary Material
ACKNOWLEDGMENTS
The work in the Molecular Immunopathology Unit was supported by a grant from the National Health and Medical Research Council of Australia to N.H.H. and H.J.B. J.G. was the recipient of an Australian National Health and Medical Research Council Biomedical Scholarship. This work was supported in part by the Intramural Research Program of NIAID, NIH.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.02701-14.
REFERENCES
- 1.Idro R, Marsh K, John CC, Newton CRJ. 2010. Cerebral malaria: mechanisms of brain injury and strategies for improved neurocognitive outcome. Pediatr Res 68:267–274. doi: 10.1203/00006450-201011001-00524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McCall MBB, Sauerwein RW. 2010. Interferon-gamma-central mediator of protective immune responses against the preerythrocytic and blood stage of malaria. J Leukoc Biol 88:1131–1143. doi: 10.1189/jlb.0310137. [DOI] [PubMed] [Google Scholar]
- 3.Hunt NH, Ball HJ, Hansen AS, Khaw LT, Guo J, Bakmiwewa S, Mitchell AJ, Combes V, Grau GER. 2014. Cerebral malaria: gamma-interferon redux. Front Cell Infect Microbiol 4:113. doi: 10.3389/fcimb.2014.00113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hunt NH, Grau GE. 2003. Cytokines: accelerators and brakes in the pathogenesis of cerebral malaria. Trends Immunol 24:491–499. doi: 10.1016/S1471-4906(03)00229-1. [DOI] [PubMed] [Google Scholar]
- 5.Miu J, Hunt NH, Ball HJ. 2008. Predominance of interferon-related responses in the brain during murine malaria, as identified by microarray analysis. Infect Immun 76:1812–1824. doi: 10.1128/IAI.01650-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Renia L, Howland SW, Claser C, Gruner AC, Suwanarusk R, Teo TH, Russell B, Ng LFP. 2012. Cerebral malaria mysteries at the blood-brain barrier. Virulence 3:193–201. doi: 10.4161/viru.19013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Villegas-Mendez A, Greig R, Shaw TN, de Souza JB, Findlay EG, Stumhofer JS, Hafalla JCR, Blount DG, Hunter CA, Riley EM, Couper KN. 2012. IFN-gamma-producing CD4(+) T cells promote experimental cerebral malaria by modulating CD8(+) T cell accumulation within the brain. J Immunol 189:968–979. doi: 10.4049/jimmunol.1200688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Howland SW, Poh CM, Gun SY, Claser C, Malleret B, Shastri N, Ginhoux F, Grotenbreg GM, Renia L. 2013. Brain microvessel cross-presentation is a hallmark of experimental cerebral malaria. EMBO Mol Med 5:984–999. doi: 10.1002/emmm.201202273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Haque A, Best SE, Unosson K, Amante FH, de Labastida F, Anstey NM, Karupiah G, Smyth MJ, Heath WR, Engwerda CR. 2011. Granzyme B expression by CD8(+) T cells is required for the development of experimental cerebral malaria. J Immunol 186:6148–6156. doi: 10.4049/jimmunol.1003955. [DOI] [PubMed] [Google Scholar]
- 10.Nitcheu J, Bonduelle O, Combadiere C, Tefit M, Seilhean D, Mazier D, Combadiere B. 2003. Perforin-dependent brain-infiltrating cytotoxic CD8+ T lymphocytes mediate experimental cerebral malaria pathogenesis. J Immunol 170:2221–2228. doi: 10.4049/jimmunol.170.4.2221. [DOI] [PubMed] [Google Scholar]
- 11.Schroder K, Hertzog PJ, Ravasi T, Hume DA. 2004. Interferon-gamma: an overview of signals, mechanisms and functions. J Leukoc Biol 75:163–189. doi: 10.1189/jlb.0603252. [DOI] [PubMed] [Google Scholar]
- 12.Kim BH, Shenoy AR, Kumar P, Bradfield CJ, MacMicking JD. 2012. IFN-inducible GTPases in host cell defense. Cell Host Microbe 12:432–444. doi: 10.1016/j.chom.2012.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.MacMicking JD. 2012. Interferon-inducible effector mechanisms in cell-autonomous immunity. Nat Rev Immunol 12:367–382. doi: 10.1038/nri3210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tiwari S, Choi HP, Matsuzawa T, Pypaert M, MacMicking JD. 2009. Targeting of the GTPase Irgm1 to the phagosomal membrane via PtdIns(3,4)P-2 and PtdIns(3,4,5)P-3 promotes immunity to mycobacteria. Nat Immunol 10:907–917. doi: 10.1038/ni.1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hunn JP, Koenen-Waisman S, Papic N, Schroeder N, Pawlowski N, Lange R, Kaiser F, Zerrahn J, Martens S, Howard JC. 2008. Regulatory interactions between IRG resistance GTPases in the cellular response to Toxoplasma gondii. EMBO J 27:2495–2509. doi: 10.1038/emboj.2008.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Khaminets A, Hunn JP, Konen-Waisman S, Zhao YO, Preukschat D, Coers J, Boyle JP, Ong YC, Boothroyd JC, Reichmann G, Howard JC. 2010. Coordinated loading of IRG resistance GTPases on to the Toxoplasma gondii parasitophorous vacuole. Cell Microbiol 12:939–961. doi: 10.1111/j.1462-5822.2010.01443.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Haldar AK, Saka HA, Piro AS, Dunn JD, Henry SC, Taylor GA, Frickel EM, Valdivia RH, Coers JR. 2013. IRG and GBP host resistance factors target aberrant, “non-self” vacuoles characterized by the missing of“self” IRGM proteins. PLoS Pathog 9:e1003414. doi: 10.1371/journal.ppat.1003414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Al-Zeer MA, Al-Younes HM, Braun PR, Zerrahn J, Meyer TF. 2009. IFN-gamma-inducible Irga6 mediates host resistance against Chlamydia trachomatis via autophagy. PLoS One 4:e4588. doi: 10.1371/journal.pone.0004588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lapaquette P, Glasser AL, Huett A, Xavier RJ, Darfeuille-Michaud A. 2010. Crohn's disease-associated adherent-invasive E. coli are selectively favoured by impaired autophagy to replicate intracellularly. Cell Microbiol 12:99–113. doi: 10.1111/j.1462-5822.2009.01381.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ling YM, Shaw MH, Ayala C, Coppens I, Taylor GA, Ferguson DJP, Yap GS. 2006. Vacuolar and plasma membrane stripping and autophagic elimination of Toxoplasma gondii in primed effector macrophages. J Exp Med 203:2063–2071. doi: 10.1084/jem.20061318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhao YO, Khaminets A, Hunn JP, Howard JC. 2009. Disruption of the Toxoplasma gondii parasitophorous vacuole by IFN gamma-inducible immunity-related GTPases (IRG proteins) triggers necrotic cell death. PLoS Pathog 5:e1000288. doi: 10.1371/journal.ppat.1000288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shenoy AR, Kim BH, Choi HP, Matsuzawa T, Tiwari S, MacMicking JD. 2007. Emerging themes in IFN-gamma-induced macrophage immunity by the p47 and p65 GTPase families. Immunobiology 212:771–784. doi: 10.1016/j.imbio.2007.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.MacMicking JD, Taylor GA, McKinney JD. 2003. Immune control of tuberculosis by IFN-gamma-inducible LRG-47. Science 302:654–659. doi: 10.1126/science.1088063. [DOI] [PubMed] [Google Scholar]
- 24.Coers J. 2013. Self and non-self discrimination of intracellular membranes by the innate immune system. PLoS Pathog 9:e1003538. doi: 10.1371/journal.ppat.1003538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bougneres L, Helft J, Tiwari S, Vargas P, Chang BHJ, Chan L, Campisi L, Lauvau G, Hugues S, Kumar P, Kamphorst AO, Dumenil AML, Nussenzweig M, MacMicking JD, Amigorena S, Guermonprez P. 2009. A role for lipid bodies in the cross-presentation of phagocytosed antigens by MHC class I in dendritic cells. Immunity 31:232–244. doi: 10.1016/j.immuni.2009.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Taylor GA, Collazo CM, Yap GS, Nguyen K, Gregorio TA, Taylor LS, Eagleson B, Secrest L, Southon EA, Reid SW, Tessarollo L, Bray M, McVicar DW, Komschlies KL, Young HA, Biron CA, Sher A, Van de Woude GF. 2000. Pathogen-specific loss of host resistance in mice lacking the IFN-gamma-inducible gene IGTP. Proc Natl Acad Sci U S A 97:751–755. doi: 10.1073/pnas.97.2.751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Collazo CM, Yap GS, Sempowski GD, Lusby KC, Tessarollo L, Woude GFV, Sher A, Taylor GA. 2001. Inactivation of LRG-47 and IRG-47 reveals a family of interferon gamma-inducible genes with essential, pathogen-specific roles in resistance to infection. J Exp Med 194:181–187. doi: 10.1084/jem.194.2.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Coers J, Bernstein-Hanley I, Grotsky D, Parvanova I, Howard JC, Taylor GA, Dietrich WF, Starnbach MN. 2008. Chlamydia muridarum evades growth restriction by the IFN-gamma-inducible host resistance factor Irgb10. J Immunol 180:6237–6245. doi: 10.4049/jimmunol.180.9.6237. [DOI] [PubMed] [Google Scholar]
- 29.Dalton DK, Pittsmeek S, Keshav S, Figari IS, Bradley A, Stewart TA. 1993. Multiple defects of immune cell-function in mice with disrupted interferon-gamma genes. Science 259:1739–1742. doi: 10.1126/science.8456300. [DOI] [PubMed] [Google Scholar]
- 30.Clarke SRM, Barnden M, Kurts C, Carbone FR, Miller JF, Heath WR. 2000. Characterization of the ovalbumin-specific TCR transgenic line OT-I: MHC elements for positive and negative selection. Immunol Cell Biol 78:110–117. doi: 10.1046/j.1440-1711.2000.00889.x. [DOI] [PubMed] [Google Scholar]
- 31.Hogquist KA, Jameson SC, Heath WR, Howard JL, Bevan MJ, Carbone FR. 1994. T-cell receptor antagonist peptides induce positive selection. Cell 76:17–27. doi: 10.1016/0092-8674(94)90169-4. [DOI] [PubMed] [Google Scholar]
- 32.Ma NL, Hunt NH, Madigan MC, Chan Ling T. 1996. Correlation between enhanced vascular permeability, up-regulation of cellular adhesion molecules and monocyte adhesion to the endothelium in the retina during the development of fatal murine cerebral malaria. Am J Pathol 149:1745–1762. [PMC free article] [PubMed] [Google Scholar]
- 33.Lundie RJ, de Koning-Ward TF, Davey GM, Nie CQ, Hansen DS, Lau LS, Mintern JD, Belz GT, Schofield L, Carbone FR, Villadangos JA, Crabb BS, Heath WR. 2008. Blood-stage Plasmodium infection induces CD8(+) T lymphocytes to parasite-expressed antigens, largely regulated by CD8 alpha(+) dendritic cells. Proc Natl Acad Sci U S A 105:14509–14514. doi: 10.1073/pnas.0806727105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Janse CJ, Ramesar J, Waters AP. 2006. High-efficiency transfection and drug selection of genetically transformed blood stages of the rodent malaria parasite Plasmodium berghei. Nat Protoc 1:346–356. doi: 10.1038/nprot.2006.53. [DOI] [PubMed] [Google Scholar]
- 35.Ryg-Cornejo V, Nie CQ, Bernard NJ, Lundie RJ, Evans KJ, Crabb BS, Schofield L, Hansen DS. 2013. NK cells and conventional dendritic cells engage in reciprocal activation for the induction of inflammatory responses during Plasmodium berghei ANKA infection. Immunobiology 218:263–271. doi: 10.1016/j.imbio.2012.05.018. [DOI] [PubMed] [Google Scholar]
- 36.Miu J, Mitchell AJ, Muller M, Carter SL, Manders PM, McQuillan JA, Saunders BM, Ball HJ, Lu B, Campbell LL, Hunt NH. 2008. Chemokine gene expression during fatal murine cerebral malaria and protection due to CXCR3 deficiency. J Immunol 180:1217–1230. doi: 10.4049/jimmunol.180.2.1217. [DOI] [PubMed] [Google Scholar]
- 37.van der Heyde HC, Bauer P, Sun G, Chang WL, Yin LJ, Fuseler J, Granger DN. 2001. Assessing vascular permeability during experimental cerebral malaria by a radiolabeled monoclonal antibody technique. Infect Immun 69:3460–3465. doi: 10.1128/IAI.69.5.3460-3465.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Warnick RE, Fike JR, Chan PH, Anderson DK, Ross GY, Gutin PH. 1995. Measurement of vascular-permeability in spinal-cord using Evans-blue spectrophotometry and correction for turbidity. J Neurosci Methods 58:167–171. doi: 10.1016/0165-0270(94)00172-D. [DOI] [PubMed] [Google Scholar]
- 39.Mitchell AJ, Yau B, McQuillan JA, Ball HJ, Too LK, Abtin A, Hertzog P, Leib SL, Jones CA, Gerega SK, Weninger W, Hunt NH. 2012. Inflammasome-dependent IFN-gamma drives pathogenesis in Streptococcus pneumoniae meningitis. J Immunol 189:4970–4980. doi: 10.4049/jimmunol.1201687. [DOI] [PubMed] [Google Scholar]
- 40.Mitchell AJ, Pradel LC, Chasson L, Van Rooijen N, Grau GE, Hunt NH, Chimini G. 2010. Technical advance: autofluorescence as a tool for myeloid cell analysis. J Leukoc Biol 88:597–603. doi: 10.1189/jlb.0310184. [DOI] [PubMed] [Google Scholar]
- 41.Nie CQ, Bernard NJ, Norman MU, Amante FH, Lundie RJ, Crabb BS, Heath WR, Engwerda CR, Hickey MJ, Schofield L, Hansen DS. 2009. IP-10-mediated T cell homing promotes cerebral inflammation over splenic immunity to malaria infection. PLoS Pathog 5:e1000369. doi: 10.1371/journal.ppat.1000369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Miyakoda M, Kimura D, Yuda M, Chinzei Y, Shibata Y, Honma K, Yui K. 2008. Malaria-specific and nonspecific activation of CD8(+) T cells during blood stage of Plasmodium berghei infection. J Immunol 181:1420–1428. doi: 10.4049/jimmunol.181.2.1420. [DOI] [PubMed] [Google Scholar]
- 43.Belnoue E, Potter SM, Rosa DS, Mauduit M, Gruner AC, Kayibanda M, Mitchell AJ, Hunt NH, Renia L. 2008. Control of pathogenic CD8(+) T cell migration to the brain by IFN-gamma during experimental cerebral malaria. Parasite Immunol 30:544–553. doi: 10.1111/j.1365-3024.2008.01053.x. [DOI] [PubMed] [Google Scholar]
- 44.Potter S, Chan-Ling T, Ball HJ, Mansour H, Mitchell A, Maluish L, Hunt NH. 2006. Perforin mediated apoptosis of cerebral microvascular endothelial cells during experimental cerebral malaria. Int J Parasitol 36:485–496. doi: 10.1016/j.ijpara.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 45.Curtsinger JM, Mescher MF. 2010. Inflammatory cytokines as a third signal for T cell activation. Curr Opin Immunol 22:333–340. doi: 10.1016/j.coi.2010.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liesenfeld O, Parvanova I, Zerrahn J, Han SJ, Heinrich F, Munoz M, Kaiser F, Aebischer T, Buch T, Waisman A, Reichmann G, Utermohlen O, von Stebut E, von Loewenich FD, Bogdan C, Specht S, Saeftel M, Hoerauf A, Mota MM, Konen-Waisman S, Kaufmann SHE, Howard JC. 2011. The IFN-gamma-inducible GTPase, Irga6, protects mice against Toxoplasma gondii but not against Plasmodium berghei and some other intracellular pathogens. PLoS One 6:e20568. doi: 10.1371/journal.pone.0020568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Grau GE, Taylor TE, Molyneux ME, Wirima JJ, Vassalli P, Hommel M, Lambert PH. 1989. Tumor necrosis factor and disease severity in children with falciparum-malaria. N Engl J Med 320:1586–1591. doi: 10.1056/NEJM198906153202404. [DOI] [PubMed] [Google Scholar]
- 48.Grau GE, Heremans H, Piguet PF, Pointaire P, Lambert PH, Billiau A, Vassalli P. 1989. Monoclonal-antibody against interferon-gamma can prevent experimental cerebral malaria and its associated overproduction of tumor necrosis factor. Proc Natl Acad Sci U S A 86:5572–5574. doi: 10.1073/pnas.86.14.5572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mitchell AJ, Hansen AM, Hee L, Ball HJ, Potter SM, Walker JC, Hunt NH. 2005. Early cytokine production is associated with protection from murine cerebral malaria. Infect Immun 73:5645–5653. doi: 10.1128/IAI.73.9.5645-5653.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hunt NH, Golenser J, Chan-Ling T, Parekh S, Rae C, Potter S, Medana IM, Miu J, Ball HJ. 2006. Immunopathogenesis of cerebral malaria. Int J Parasitol 36:569–582. doi: 10.1016/j.ijpara.2006.02.016. [DOI] [PubMed] [Google Scholar]
- 51.Day NPJ, Hien TT, Schollaardt T, Loc PP, Van Chuong L, Chau TTH, Mai NTH, Phu NH, Sinh DX, White NJ, Ho M. 1999. The prognostic and pathophysiologic role of pro- and antiiflammatory cytokines in severe malaria. J Infect Dis 180:1288–1297. doi: 10.1086/315016. [DOI] [PubMed] [Google Scholar]
- 52.Lyke KE, Burges R, Cissoko Y, Sangare L, Dao M, Diarra I, Kone A, Harley R, Plowe CV, Doumbo OK, Sztein MB. 2004. Serum levels of the proinflammatory cytokines interleukin-1 beta (IL-1 beta), IL-6, IL-8, IL-10, tumor necrosis factor alpha, and IL-12(p70) in Malian children with severe Plasmodium falciparum malaria and matched uncomplicated malaria or healthy controls. Infect Immun 72:5630–5637. doi: 10.1128/IAI.72.10.5630-5637.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Campanella GSV, Tager AM, El Khoury JK, Thomas SY, Abrazinski TA, Manice LA, Colvin RA, Lustert AD. 2008. Chemokine receptor CXCR3 and its ligands CXCL9 and CXCL10 are required for the development of murine cerebral malaria. Proc Natl Acad Sci U S A 105:4814–4819. doi: 10.1073/pnas.0801544105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Deshmane SL, Kremlev S, Amini S, Sawaya BE. 2009. Monocyte chemoattractant protein-1 (MCP-1): an overview. J Interferon Cytokine Res 29:313–326. doi: 10.1089/jir.2008.0027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rollins BJ, Yoshimura T, Leonard EJ, Pober JS. 1990. Cytokine-activated human endothelial-cells synthesize and secrete a monocyte chemoattractant, MCP-1/JE. Am J Pathol 136:1229–1233. [PMC free article] [PubMed] [Google Scholar]
- 56.Getts DR, Terry RL, Getts MT, Muller M, Rana S, Shrestha B, Radford J, Van Rooijen N, Campbell IL, King NJC. 2008. Ly6c(+) “inflammatory monocytes” are microglial precursors recruited in a pathogenic manner in West Nile virus encephalitis. J Exp Med 205:2319–2337. doi: 10.1084/jem.20080421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Palframan RT, Jung S, Cheng CY, Weninger W, Luo Y, Dorf M, Littman DR, Rollins BJ, Zweerink H, Rot A, von Andrian UH. 2001. Inflammatory chemokine transport and presentation in HEV: a remote control mechanism for monocyte recruitment to lymph nodes in inflamed tissues. J Exp Med 194:1361–1373. doi: 10.1084/jem.194.9.1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pai S, Qin J, Cavanagh L, Mitchell A, El-Assaad F, Jain R, Combes V, Hunt NH, Grau GER, Weninger W. 2014. Real-time imaging reveals the dynamics of leukocyte behaviour during experimental cerebral malaria pathogenesis. PLoS Pathog 10:e1004236. doi: 10.1371/journal.ppat.1004236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Belnoue E, Costa FTM, Vigario AM, Voza T, Gonnet F, Landau I, van Rooijen N, Mack M, Kuziel WA, Renia L. 2003. Chemokine receptor CCR2 is not essential for the development of experimental cerebral malaria. Infect Immun 71:3648–3651. doi: 10.1128/IAI.71.6.3648-3651.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Groom JR, Luster AD. 2011. CXCR3 ligands: redundant, collaborative and antagonistic functions. Immunol Cell Biol 89:207–215. doi: 10.1038/icb.2010.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Whiting D, Hsieh G, Yun JJ, Banerji A, Yao W, Fishbein MC, Belperio J, Strieter RM, Bonavida B, Ardehali A. 2004. Chemokine monokine induced by IFN-gamma/CXC chemokine ligand 9 stimulates T lymphocyte proliferation and effector cytokine production. J Immunol 172:7417–7424. doi: 10.4049/jimmunol.172.12.7417. [DOI] [PubMed] [Google Scholar]
- 62.Wilson NO, Jain V, Roberts CE, Lucchi N, Joel PK, Singh MP, Nagpal AC, Dash AP, Udhayakumar V, Singh N, Stiles JK. 2011. CXCL4 and CXCL10 predict risk of fatal cerebral malaria. Dis Markers 30:39–49. doi: 10.1155/2011/828256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Armah HB, Wilson NO, Sarfo BY, Powell MD, Bond VC, Anderson W, Adjei AA, Gyasi RK, Tettey Y, Wiredu EK, Tongren JE, Udhayakumar V, Stiles JK. 2007. Cerebrospinal fluid and serum biomarkers of cerebral malaria mortality in Ghanaian children. Malar J 6:147. doi: 10.1186/1475-2875-6-147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hansen DS, Bernard NJ, Nie CQ, Schofield L. 2007. NK cells stimulate recruitment of CXCR3(+) T cells to the brain during Plasmodium berghei-mediated cerebral malarial. J Immunol 178:5779–5788. doi: 10.4049/jimmunol.178.9.5779. [DOI] [PubMed] [Google Scholar]
- 65.Belnoue E, Kayibanda M, Vigario AM, Deschemin JC, van Rooijen N, Viguier M, Snounou G, Renia L. 2002. On the pathogenic role of brain-sequestered alpha beta CD8(+) T cells in experimental cerebral malarial. J Immunol 169:6369–6375. doi: 10.4049/jimmunol.169.11.6369. [DOI] [PubMed] [Google Scholar]
- 66.Kurts C, Robinson BWS, Knolle PA. 2010. Cross-priming in health and disease. Nat Rev Immunol 10:403–414. doi: 10.1038/nri2780. [DOI] [PubMed] [Google Scholar]
- 67.Munoz MA, Biro M, Weninger W. 2014. T cell migration in intact lymph nodes in vivo. Curr Opin Cell Biol 30:17–24. doi: 10.1016/j.ceb.2014.05.002. [DOI] [PubMed] [Google Scholar]
- 68.Piva L, Tetlak P, Claser C, Karjalainen K, Renia L, Ruedl C. 2012. Cutting edge: Clec9a(+) dendritic cells mediate the development of experimental cerebral malaria. J Immunol 189:1128–1132. doi: 10.4049/jimmunol.1201171. [DOI] [PubMed] [Google Scholar]
- 69.Lau LS, Ruiz DF, Davey GM, de Koning-Ward TF, Papenfuss AT, Carbone FR, Brooks AG, Crabb BS, Heath WR. 2011. Blood-stage Plasmodium berghei infection generates a potent, specific CD8(+) T-cell response despite residence largely in cells lacking MHC I processing machinery. J Infect Dis 204:1989–1996. doi: 10.1093/infdis/jir656. [DOI] [PubMed] [Google Scholar]
- 70.deWalick S, Amante FH, McSweeney KA, Randall LM, Stanley AC, Haque A, Kuns RD, MacDonald KPA, Hill GR, Engwerda CR. 2007. Cutting edge: conventional dendritic cells are the critical APC required for the induction of experimental cerebral malaria. J Immunol 178:6033–6037. doi: 10.4049/jimmunol.178.10.6033. [DOI] [PubMed] [Google Scholar]
- 71.Castellino F, Huang AY, Altan-Bonnet G, Stoll S, Scheinecker C, Germain RN. 2006. Chemokines enhance immunity by guiding naive CD8(+) T cells to sites of CD4 T cell-dendritic cell interaction. Nature 440:890–895. doi: 10.1038/nature04651. [DOI] [PubMed] [Google Scholar]
- 72.Cox MA, Kahan SM, Zajac AJ. 2013. Anti-viral CD8 T cells and the cytokines that they love. Virology 435:157–169. doi: 10.1016/j.virol.2012.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kaech SM, Cui WG. 2012. Transcriptional control of effector and memory CD8(+) T cell differentiation. Nat Rev Immunol 12:749–761. doi: 10.1038/nri3307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wykes MN, Kay JG, Manderson A, Liu XQ, Brown DL, Richard DJ, Wipasa J, Jiang SHH, Jones MK, Janse CJ, Waters AP, Pierce SK, Miller LH, Stow JL, Good MF. 2011. Rodent blood-stage Plasmodium survive in dendritic cells that infect naive mice. Proc Natl Acad Sci U S A 108:11205–11210. doi: 10.1073/pnas.1108579108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Okada M, Kitahara M, Kishimoto S, Matsuda T, Hirano T, Kishimoto T. 1988. IL-6/BSF-2 functions as a killer helper factor in the in vitro induction of cytotoxic T-cells. J Immunol 141:1543–1549. [PubMed] [Google Scholar]
- 76.Gagnon J, Ramanathan S, Leblanc C, Cloutier A, McDonald PP, Ilangumaran S. 2008. IL-6, in synergy with IL-7 or IL-15, stimulates TCR-independent proliferation and functional differentiation of CD8(+) T lymphocytes. J Immunol 180:7958–7968. doi: 10.4049/jimmunol.180.12.7958. [DOI] [PubMed] [Google Scholar]
- 77.Wenisch C, Linnau KF, Looaresuwan S, Rumpold H. 1999. Plasma levels of the interleukin-6 cytokine family in persons with severe Plasmodium falciparum malaria. J Infect Dis 179:747–750. doi: 10.1086/314630. [DOI] [PubMed] [Google Scholar]
- 78.Grau GE, Frei K, Piguet PF, Fontana A, Heremans H, Billiau A, Vassalli P, Lambert PH. 1990. Interleukin-6 production in experimental cerebral malaria–modulation by anticytokine antibodies and possible role in hypergammaglobulinemia. J Exp Med 172:1505–1508. doi: 10.1084/jem.172.5.1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Haque A, Best SE, de Oca MM, James KR, Ammerdorffer A, Edwards CL, Rivera FD, Amante FH, Bunn PT, Sheel M, Sebina I, Koyama M, Varelias A, Hertzog PJ, Kalinke U, Gun SY, Renia L, Ruedl C, MacDonald KPA, Hill GR, Engwerda CR. 2014. Type I IFN signaling in CD8(-) DCs impairs Th1-dependent malaria immunity. J Clin Investig 124:2483–2496. doi: 10.1172/JCI70698. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.