

Abstract
Homologous recombination requires nucleolytic degradation (resection) of DNA double-strand break (DSB) ends. In Saccharomyces cerevisiae, the MRX complex and Sae2 are involved in the onset of DSB resection, whereas extensive resection requires Exo1 and the concerted action of Dna2 and Sgs1. Here, we show that the checkpoint protein Rad9 limits the action of Sgs1/Dna2 in DSB resection by inhibiting Sgs1 binding/persistence at the DSB ends. When inhibition by Rad9 is abolished by the Sgs1-ss mutant variant or by deletion of RAD9, the requirement for Sae2 and functional MRX in DSB resection is reduced. These results provide new insights into how early and long-range resection is coordinated.
Keywords: double-strand break, Rad9, resection, Saccharomyces cerevisiae, Sgs1
Introduction
DNA double-strand breaks (DSBs) can be repaired by homologous recombination (HR), which uses undamaged homologous DNA sequences as a template for repair in a mostly error-free manner. The first step in HR is the processing of DNA ends by 5′ to 3′ nucleolytic degradation (resection) to generate 3′-ended single-stranded DNA (ssDNA) that can invade a homologous template 1. This ssDNA generation also induces activation of the DNA damage checkpoint, whose key players are the protein kinases ATM and ATR in mammals as well as their functional orthologs Tel1 and Mec1 in Saccharomyces cerevisiae 2.
Initiation of DSB resection requires the conserved MRX/MRN complex (Mre11/Rad50/Xrs2 in yeast; Mre11/Rad50/Nbs1 in mammals) that, together with Sae2, catalyses an endonucleolytic cleavage of the 5′ strands 3, 4, 5. More extensive resection of the 5′ strands depends on two pathways, which require the 5′ to 3′ double-stranded DNA exonuclease Exo1 and the nuclease Dna2 working in concert with the 3′ to 5′ helicase Sgs1 4, 5.
Double-strand break resection is controlled by the activity of cyclin-dependent kinases (Cdk1 in yeast) 6, which promotes DSB resection by phosphorylating Sae2 7 and Dna2 8, as well as by ATP-dependent nucleosome remodelling complexes 9. Recently, the chromatin remodeler Fun30 has been shown to be required for extensive resection 10, 11, 12, possibly because it overcomes the resection barrier exerted by the histone-bound checkpoint protein Rad9 10, 13, 14.
The MRX/Sae2-mediated initial endonucleolytic cleavage becomes essential to initiate DSB resection when covalent modifications or bulky adducts are present at the DSB ends and prevent the access of the long-range Exo1 and Dna2/Sgs1 resection machinery. For example, Sae2 and the MRX nuclease activity are essential during meiosis to remove Spo11 from the 5′-ended strand of the DSBs 15, 16. Furthermore, both sae2Δ and mre11 nuclease-defective (mre11-nd) mutants exhibit a marked sensitivity to methyl methanesulfonate (MMS) and ionizing radiation (IR), which can generate chemically complex DNA termini, and to camptothecin (CPT), which extends the half-life of topoisomerase I (Top1)–DNA cleavable complexes 17. CPT-induced DNA lesions need to be processed by Sae2 and MRX unless the Ku heterodimer is absent. In fact, elimination of Ku restores partial resistance to CPT in both sae2Δ and mre11-nd cells 18, 19. This suppression requires Exo1, indicating that Ku increases the requirement for MRX/Sae2 activities in DSB resection by inhibiting Exo1.
To identify other possible mechanisms regulating MRX/Sae2 requirement in DSB resection, we searched for extragenic mutations that suppressed the sensitivity to DNA damaging agents of sae2Δ cells. This search allowed the identification of the SGS1-ss allele, which suppresses the resection defect of sae2Δ cells by escaping Rad9-mediated inhibition of DSB resection. The Sgs1-ss variant is robustly associated with the DSB ends both in the presence and in the absence of Rad9 and resects the DSB more efficiently than wild-type Sgs1. Moreover, we found that Rad9 limits the binding at the DSB of Sgs1, which is in turn responsible for rapid resection in rad9Δ cells. We propose that Rad9 limits the activity in DSB resection of Sgs1/Dna2 and the escape from this inhibition can reduce the requirement of Sae2 and functional MRX in DSB resection.
Results and Discussion
Sgs1-ss suppresses the sensitivity to DNA damaging agents of sae2Δ and mre11-nd mutants
SAE2 deletion causes hypersensitivity to CPT, which creates replication-associated DSBs. The lack of Ku suppresses CPT hypersensitivity of sae2Δ mutants, and this rescue requires Exo1 18, 19, indicating that Ku prevents Exo1 from initiating DSB resection. To identify other possible pathways bypassing Sae2 function in DSB resection, we searched for extragenic mutations that suppress the CPT sensitivity of sae2Δ cells. CPT-resistant sae2Δ candidates were crossed to each other and to the wild-type strain to identify, by tetrad analysis, 15 single-gene suppressor mutants that fell into 11 distinct allelism groups. Genome sequencing of the five non-allelic suppressor clones that stood from the others for the best suppression phenotype identified single-base pair substitutions either in the TOP1 gene, encoding the CPT target topoisomerase I, or in the PDR3,PDR10 and SAP185 genes, which encode for proteins involved in multi-drug resistance. The mutation responsible for the suppression in the fifth clone was a single-base pair substitution in the SGS1 gene (SGS1-ss), causing the amino acid change G1298R in the HRDC domain that is conserved in the RecQ helicase family. The identity of the genes that are mutated in the six remaining suppressor clones remained to be determined.
The SGS1-ss allele suppressed the sensitivity of the sae2Δ mutant not only to CPT, but also to phleomycin (phleo) and MMS, resulting in almost wild-type survival of sae2Δ SGS1-ss cells treated with these drugs (Fig1A). The ability of Sgs1-ss to suppress the sensitivity of sae2Δ to genotoxic agents was dominant, as sae2Δ/sae2Δ SGS1/SGS1-ss diploid cells were less sensitive to CPT, phleomycin and MMS compared to sae2Δ/sae2Δ SGS1/SGS1 diploid cells (Fig1B).
Figure 1.

- A, B Exponentially growing cells were serially diluted (1:10), and each dilution was spotted out onto YEPD plates with or without camptothecin (CPT), phleomycin or MMS.
- C, D Meiotic tetrads were dissected on YEPD plates that were incubated at 25°C, followed by spore genotyping.
- E Exponentially growing cells were serially diluted (1:10), and each dilution was spotted out onto YEPD plates with or without CPT, phleomycin or MMS.
- F, G Meiotic tetrads were dissected on YEPD plates that were incubated at 25°C, followed by spore genotyping.
Besides providing the endonuclease activity to initiate DSB resection, MRX also promotes stable association of Exo1, Sgs1 and Dna2 at the DSB ends 20, thus explaining the severe resection defect of cells lacking the MRX complex compared to cells lacking either Sae2 or the Mre11 nuclease activity. Sgs1-ss suppressed the hypersensitivity to genotoxic agents of mre11-H125N cells, which were specifically defective in Mre11 nuclease activity (Fig1A). By contrast, mre11Δ SGS1-ss double-mutant cells were as sensitive to genotoxic agents as the mre11Δ single mutant (Fig1A). Altogether, these findings indicate that Sgs1-ss can bypass the requirement of Sae2 or MRX nuclease activity for survival to genotoxic agents, but it still requires the physical integrity of the MRX complex to exert its function.
Sgs1 promotes DSB resection by acting as a helicase 4, 5, prompting us to investigate whether Sgs1-ss requires its helicase activity to exert the suppression effect. Both the lack of Sgs1 and its helicase-dead Sgs1-hd variant, carrying the K706A amino acid substitution 21, impaired viability of sae2Δ cells 5 (Fig1C). This synthetic sickness is likely due to poor DSB resection, as it is known to be alleviated by making DNA ends accessible to the Exo1 nuclease 18, 19. The K706A substitution was therefore introduced in Sgs1-ss, thus generating the Sgs1-hd-ss variant, and meiotic tetrads from diploid strains double heterozygous for sae2Δ and sgs1-hd-ss were analysed for spore viability on YEPD plates. All sae2Δ sgs1-hd-ss double-mutant spores formed much smaller colonies than each single-mutant spore (Fig1D), with a colony size similar to that obtained from sae2Δ sgs1-hd double-mutant spores (Fig1C). Thus, Sgs1-ss appears to require its helicase activity to suppress the lack of Sae2 function.
Suppression of sae2Δ by Sgs1-ss requires Dna2, but not Exo1
The ssDNA formed by Sgs1 unwinding is degraded by the nuclease Dna2, which acts in DSB resection in a parallel pathway with respect to Exo1 5. Thus, we asked whether the suppression of sae2Δ hypersensitivity to DNA damaging agents by Sgs1-ss requires Exo1 and/or Dna2. Although the lack of Exo1 exacerbated the sensitivity of sae2Δ cells to some DNA damaging agents (Fig1E), the SGS1-ss allele was still capable to suppress the sensitivity to CPT, phleomycin and MMS of sae2Δ exo1Δ double-mutant cells (Fig1E), indicating the suppression of sae2Δ by Sgs1-ss is independent of Exo1.
As DNA2 is essential for cell viability, dna2Δ cells were kept viable by the pif1-M2 mutation, which impairs the ability of Pif1 to promote formation of long flaps that are substrates for Dna2 22. Diploids homozygous for the pif1-M2 mutation and heterozygous for sae2Δ,dna2Δ and SGS1-ss were generated, followed by sporulation and tetrads dissection. No viable sae2Δ dna2Δ pif1-M2 cells could be recovered, and the presence of the SGS1-ss allele did not restore viability of sae2Δ dna2Δ pif1-M2 triple-mutant spores (Fig1F). By contrast, tetrads from a diploid homozygous for the pif1-M2 mutation and heterozygous for sae2Δ, dna2Δ and ku70Δ showed that the lack of Ku70, which relieved Exo1 inhibition 18, 19, restored viability of sae2Δ dna2Δ pif1-M2 spores (Fig1G). These findings indicate that Sgs1-ss requires Dna2 to bypass Sae2 requirement.
Sgs1-ss suppresses the adaptation defect of sae2Δ cells
A single irreparable DSB triggers a checkpoint-mediated cell cycle arrest. Yeast cells can escape an extended checkpoint arrest and resume cell cycle progression even with an unrepaired DSB (adaptation) 23, 24. Sae2 lacking cells, like other resection deficient mutants, fail to turn off the checkpoint triggered by an unrepaired DSB and remain arrested at G2/M as large budded cells 12, 25, 26, 27. To investigate whether Sgs1-ss suppresses the adaptation defect of sae2Δ cells, we used JKM139 derivative strains carrying the HO endonuclease gene under the control of a galactose-inducible promoter. Galactose addition leads to generation at the MAT locus of a single DSB that cannot be repaired by HR, because the homologous donor loci HML or HMR are deleted 23. When G1-arrested cell cultures were spotted on galactose-containing plates, sae2Δ SGS1-ss cells formed microcolonies with more than two cells more efficiently than sae2Δ cells, which were still arrested at the two-cell dumbbell stage after 24 h (Fig2A). Checkpoint activation was monitored also by following Rad53 phosphorylation, which is required for Rad53 activation and is detectable as a decrease of its electrophoretic mobility. When galactose was added to exponentially growing cell cultures of the same strains, sae2Δ and sae2Δ SGS1-ss mutant cells showed similar amounts of phosphorylated Rad53 after HO induction (Fig2B), indicating that Sgs1-ss did not affect checkpoint activation. However, Rad53 phosphorylation decreased in sae2Δ SGS1-ss double-mutant cells within 12–14 h after galactose addition, whereas it persisted longer in sae2Δ cells that were defective in re-entering the cell cycle (Fig2B). Thus, Sgs1-ss suppresses the inability of sae2Δ cells to turn off the checkpoint in the presence of an unrepaired DSB.
Figure 2.
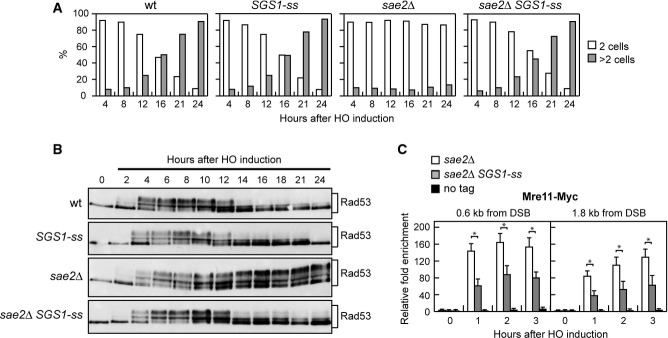
- A YEPR G1-arrested cell cultures of wild-type JKM139 and otherwise isogenic derivative strains were plated on galactose-containing plates (time zero). At the indicated time points, 200 cells for each strain were analysed to determine the frequency of large budded cells and of cells forming microcolonies of more than two cells. The mean values from three independent experiments are represented (n + 3).
- B Exponentially growing YEPR cultures of the strains in (A) were transferred to YEPRG (time zero), followed by Western blot analysis with anti-Rad53 antibodies.
- C ChIP analysis. Exponentially growing YEPR cell cultures of JKM139 derivative strains were transferred to YEPRG, followed by ChIP analysis of the recruitment of Mre11–Myc at the indicated distance from the HO-cut compared to untagged Mre11 (no tag). In all diagrams, the ChIP signals were normalized for each time point to the corresponding input signal. The mean values are represented with error bars denoting s.d. (n + 3). *P < 0.01, t-test.
The adaptation defect of sae2Δ cells has been proposed to be due to an increased persistence at DSBs of the MRX complex, which in turn causes unscheduled Tel1 activation 26, 27. We then asked by chromatin immunoprecipitation (ChIP) and quantitative real-time PCR (qPCR) analysis whether Sgs1-ss can reduce the binding of MRX to the DSB ends in sae2Δ cells. When HO was induced in exponentially growing cells, the amount of Mre11 bound at the HO-induced DSB end was lower in sae2Δ SGS1-ss than in sae2Δ cells (Fig2C). As MRX persistence at the DSB in sae2Δ cells has been proposed to be due to defective DSB resection, this finding suggests that Sgs1-ss suppresses the resection defect of sae2Δ cells.
Sgs1-ss suppresses the resection defect of sae2Δ cells
To investigate whether Sgs1-ss suppresses the sensitivity to genotoxic agents and the adaptation defect of sae2Δ cells by restoring DSB resection, we used JKM139 derivative strains to monitor directly generation of ssDNA at the DSB ends 23. Because ssDNA is resistant to cleavage by restriction enzymes, we directly monitored ssDNA formation at the irreparable HO-cut by following the loss of SspI restriction fragments after galactose addition by Southern blot analysis under alkaline conditions, using a single-stranded probe that anneals to the 3′ end at one side of the break (Fig3A). Resection in sae2Δ SGS1-ss cells was markedly increased compared to sae2Δ cells, indicating that Sgs1-ss suppresses the resection defect caused by the lack of Sae2 (Fig3B and C).
Figure 3.

- A Method to measure double-strand break (DSB) resection. Gel blots of SspI-digested genomic DNA separated on alkaline agarose gel were hybridized with a single-stranded MAT probe (ss probe) that anneals to the unresected strand. 5′–3′ resection progressively eliminates SspI sites (S), producing larger SspI fragments (r1 through r7) detected by the probe.
- B DSB resection. YEPR exponentially growing cell cultures of JKM139 derivative strains were transferred to YEPRG at time zero. Genomic DNA was analysed for ssDNA formation at the indicated times after HO induction as described in (A).
- C Densitometric analyses. The experiment as in (B) has been independently repeated three times, and the mean values are represented with error bars denoting s.d. (n + 3).
- D DSB repair by single-strand annealing (SSA). In YMV45 strain, the HO-cut site is flanked by homologous leu2 sequences that are 4.6 kb apart. HO-induced DSB formation results in generation of 12- and 2.5-kb DNA fragments (HO-cut) that can be detected by Southern blot analysis with a LEU2 probe of KpnI-digested genomic DNA. DSB repair by SSA generates an 8-kb fragment (product).
- E Densitometric analysis of the product band signals. The intensity of each band was normalized with respect to a loading control (not shown). The mean values are represented with error bars denoting s.d. (n + 3).
Repair of a DSB flanked by direct repeats occurs primarily by single-strand annealing (SSA), which requires nucleolytic degradation of the 5′ DSB ends to reach the complementary DNA sequences that can then anneal 28. To assess whether the Sgs1-ss-mediated suppression of the resection defect caused by the lack of Sae2 was physiologically relevant, we asked whether Sgs1-ss suppresses the SSA defect of sae2Δ cells. To this end, we introduced the SGS1-ss allele in YMV45 strain, which carries two tandem leu2 repeats located 4.6 kb apart, with a HO recognition site adjacent to one of the repeats 28. This strain also harbours a GAL-HO construct for galactose-inducible HO expression. As expected, accumulation of the repair product was reduced in sae2Δ compared to wild-type cells, whereas it occurred with almost wild-type kinetics in sae2Δ SGS1-ss double-mutant cells (Fig3D and E), indicating that Sgs1-ss improves SSA-mediated DSB repair in the absence of Sae2.
Altogether, these findings indicate that Sgs1-ss suppresses both the sensitivity to genotoxic agents of sae2Δ cells and the MRX persistence at DSBs by restoring DSB resection. Interestingly, the effects of the SGS1-ss mutation are opposite to those of the separation-of-function sgs1-D664Δ allele, which specifically impairs viability of sae2Δ cells and DSB resection without affecting other Sgs1 functions 29.
Sgs1-ss accelerates DSB resection by escaping Rad9 inhibition
The Sgs1-ss mutant variant can bypass Sae2 requirement in initiation of DSB resection either because it allows Dna2 to substitute for Sae2/MRX endonuclease activity or because it increases the resection efficiency. To distinguish between these two possibilities, we asked whether Sgs1-ss could bypass Sae2 requirement in resecting meiotic DSBs, where the Sae2/MRX-mediated endonucleolytic cleavage is absolutely required to initiate DSB resection by allowing the removal of Spo11 from the DSB ends 15, 16. A sae2Δ/sae2Δ SGS1-ss/SGS1-ss diploid strain was constructed and its kinetics of processing/repair of meiotic DSBs generated at the THR4 hotspot was compared to those of a sae2Δ/sae2Δ diploid. DSBs disappeared in both wild-type and SGS1-ss/SGS1-ss cells about 4 h after transfer to sporulation medium, while they persisted until the end of the experiment in both sae2Δ/sae2Δ and sae2Δ/sae2Δ SGS1-ss/SGS1-ss diploid cells (Supplementary Fig S1). Thus, Sgs1-ss cannot substitute the endonucleolytic clipping by Sae2/MRX when this is absolutely required to initiate DSB resection.
Interestingly, the Sgs1-ss mutant variant accelerates both DSB resection and SSA compared to wild-type Sgs1 (Fig3B–E), suggesting that Sgs1-ss might increase the resection efficiency by escaping the effect of negative regulators of this process. In particular, Rad9 provides a barrier to resection through an unknown mechanism 13, 14. As shown in Fig4A and B, both SGS1-ss and rad9Δ mutant cells accumulated the resection products more efficiently than wild-type cells, and the presence of Sgs1-ss did not accelerate further the generation of ssDNA in rad9Δ cells. Thus, the lack of Rad9 and the presence of Sgs1-ss appear to increase the efficiency of DSB resection through the same mechanism. Furthermore, cells lacking Rad9 displayed sensitivity to CPT and phleomycin (Fig4C). Consistent with the finding that the SGS1-ss and rad9Δ alleles affect the same process, rad9Δ was epistatic to SGS1-ss with respect to the survival to genotoxic agents, as sae2Δ rad9Δ SGS1-ss cells were as sensitive to CPT and phleomycin as sae2Δ rad9Δ and rad9Δ cells (Fig4C).
Figure 4.
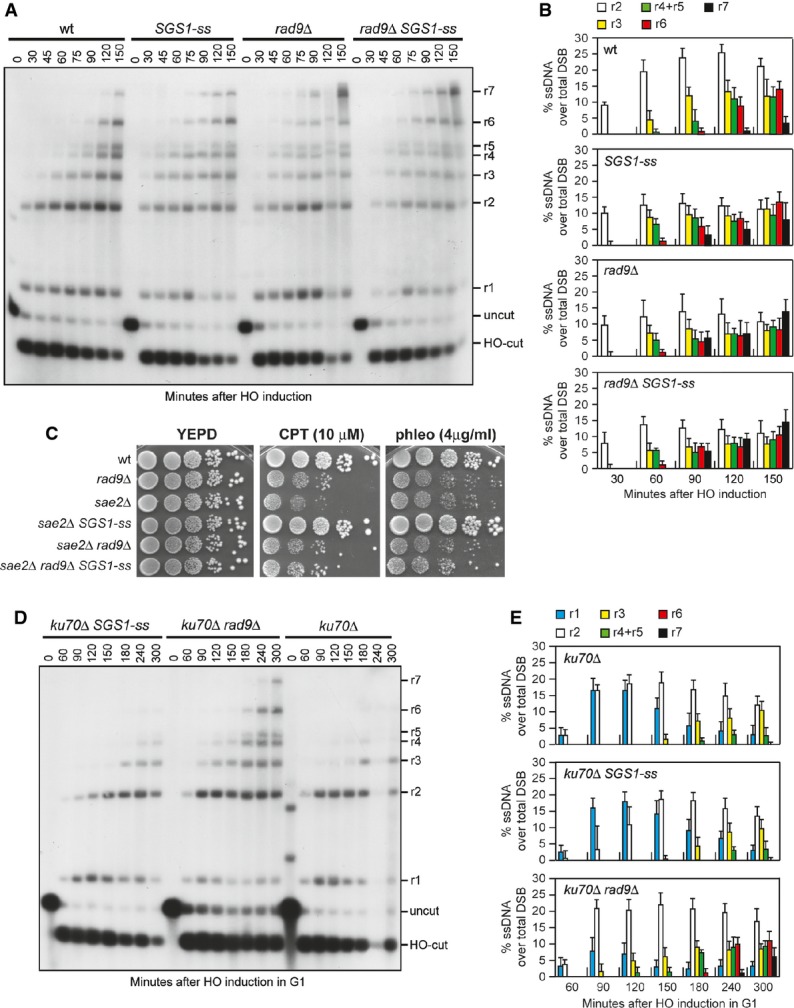
- A DSB resection. YEPR exponentially growing cell cultures of JKM139 derivative strains were transferred to YEPRG at time zero. Genomic DNA was analysed for ssDNA formation as described in Fig3A.
- B Densitometric analyses. The experiment as in (A) has been independently repeated three times, and the mean values are represented with error bars denoting s.d. (n + 3).
- C Exponentially growing cells were serially diluted (1:10), and each dilution was spotted out onto YEPD plates with or without camptothecin (CPT) or phleomycin.
- D DSB resection. HO was induced at time zero in α-factor-arrested JKM139 derivative cells that were kept arrested in G1 with α-factor throughout the experiment. Genomic DNA was analysed for ssDNA formation as described in Fig3A.
- E Densitometric analyses. The experiment as in (D) has been independently repeated three times, and the mean values are represented with error bars denoting s.d. (n + 3).
Double-strand break resection in the G1 phase of the cell cycle is specifically inhibited by the Ku complex, whose lack allows nucleolytic processing in G1 cells independently of Cdk1 activity 30. RAD9 deletion does not allow DSB resection in G1, but it enhances resection in G1-arrested kuΔ cells 31, indicating that Rad9 inhibits DSB resection in G1, but this function becomes apparent only when Ku is absent. To investigate whether Sgs1-ss was capable to counteract the inhibitory function of Rad9 in G1, we monitored DSB resection in SGS1-ss and ku70Δ SGS1-ss cells that were kept arrested in G1 by α-factor during HO induction. Consistent with the requirement of Cdk1 activity for efficient DSB resection, the 3′-ended resection products were barely detectable in wild-type G1 cells, whereas their amount increased in ku70Δ G1 cells that, as previously reported 30, accumulated mostly 1.7-, 3.5- and 4.7-kb ssDNA products (r1, r2, r3) (Supplementary Fig S2). By contrast, DSB resection in SGS1-ss cells was undistinguishable from that observed in wild-type cells (Supplementary Fig S2), indicating that Sgs1-ss does not allow DSB resection in G1. Furthermore, while RAD9 deletion enhanced the resection efficiency of ku70Δ G1 cells, G1-arrested ku70Δ and ku70Δ SGS1-ss cells accumulated resection products with similar kinetics (Fig4D and E). Altogether, these findings indicate that Sgs1-ss is not capable to allow DSB resection in G1 either in the presence or in the absence of Ku. As Sgs1-ss function in DSB resection depends on Dna2, whose activity requires Cdk1-mediated phosphorylation 8, the inability of Sgs1-ss to overcome both Ku- and Rad9-mediated inhibition in G1 may be due to the requirement of Cdk1 activity to support Dna2 and therefore Sgs1-ss function in DSB resection.
Rapid DSB resection in rad9Δ cells depends mainly on Sgs1
Generation of ssDNA at uncapped telomeres in rad9Δ cells has been shown to be more dependent on Dna2/Sgs1 than on Exo1 32. This observation, together with the finding that SGS1-ss does not accelerate further the generation of ssDNA in rad9Δ cells (Fig4A and B), raises the possibility that Rad9 inhibits DSB resection by limiting Sgs1 activity and that the Sgs1-ss variant can escape this inhibition. We tested this hypothesis by investigating the contribution of Sgs1 and Exo1 to the accelerated DSB resection displayed by rad9Δ cells. As shown in Fig5A and B, sgs1Δ was epistatic to rad9Δ with respect to DSB resection, as sgs1Δ rad9Δ double-mutant and sgs1Δ single-mutant cells resected the HO-induced DSB with similar kinetics. By contrast, DSB resection in exo1Δ rad9Δ cells was more efficient than in exo1Δ cells, although it was delayed compared to rad9Δ cells (Fig5C and D). Thus, the rapid resection in the absence of Rad9 depends mainly on Sgs1, although also Exo1 contributes to resect the DSB in the absence of Rad9. Consistent with the finding that Sgs1-ss overrides Rad9 inhibition, SGS1-ss exo1Δ cells resected the DSB with kinetics similar to that of rad9Δ exo1Δ cells (Supplementary Fig S3).
Figure 5.
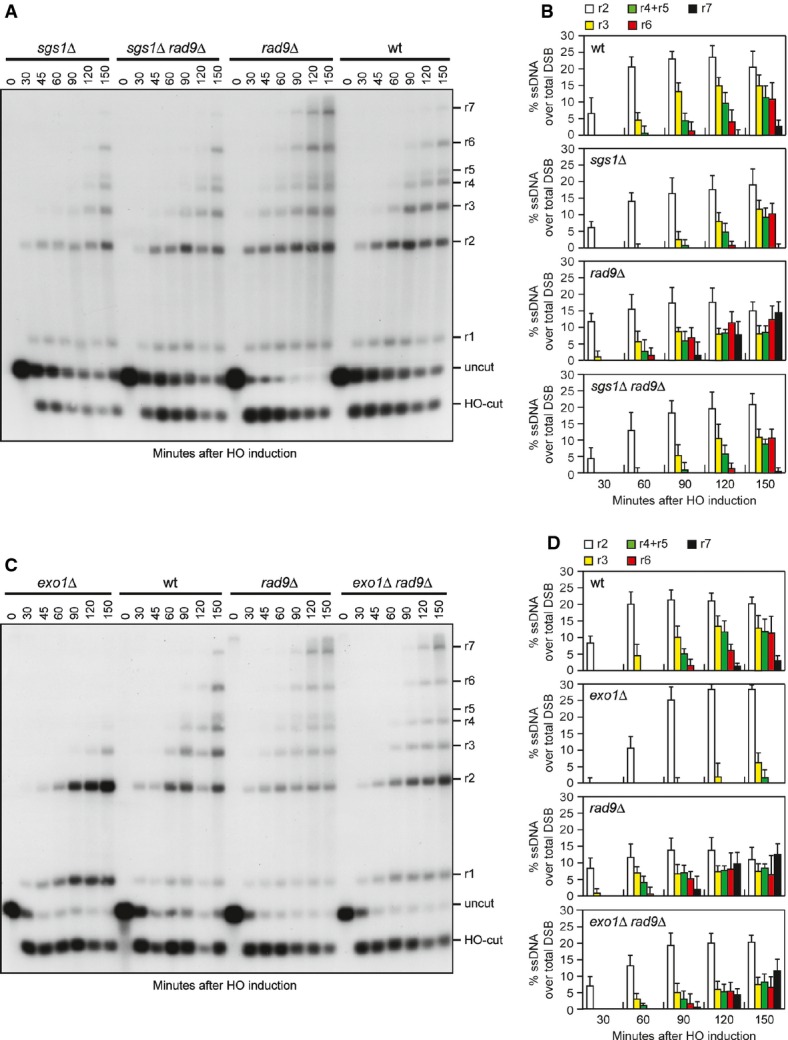
- A Double-strand break (DSB) resection. YEPR exponentially growing cell cultures of JKM139 derivative strains were transferred to YEPRG at time zero. Genomic DNA was analysed for ssDNA formation as described in Fig3A.
- B Densitometric analyses. The experiment as in (A) has been independently repeated three times, and the mean values are represented with error bars denoting s.d. (n + 3).
- C DSB resection. The experiment was performed as in (A).
- D Densitometric analyses. The experiment as in (C) has been independently repeated three times, and the mean values are represented with error bars denoting s.d. (n + 3).
Rad9 inhibits DSB resection by limiting Sgs1 association at DNA breaks
If loss of end protection by Rad9 allowed Sgs1 to initiate DSB resection, which normally requires Sae2, then RAD9 deletion, like Sgs1-ss, should suppress the resection defect of sae2Δ cells. Indeed, DSB resection in sae2Δ rad9Δ cells was as fast as in rad9Δ cells, which resected the DSB more efficiently than wild-type and sae2Δ cells (Fig6A and B), indicating that the lack of Rad9 bypasses Sae2 function in DSB resection.
Figure 6.

- A DSB resection. YEPR exponentially growing cell cultures of JKM139 derivative strains were transferred to YEPRG at time zero. Genomic DNA was analysed for ssDNA formation as described in Fig3A.
- B Densitometric analyses. The experiment as in (A) has been independently repeated three times, and the mean values are represented with error bars denoting s.d. (n + 3).
- C ChIP analysis. Exponentially growing YEPR cell cultures of JKM139 derivative strains were transferred to YEPRG, followed by ChIP analysis of the recruitment of Sgs1-HA and Sgs1-ss-HA at the indicated distance from the HO-cut compared to untagged Sgs1 (no tag). In all diagrams, the ChIP signals were normalized for each time point to the corresponding input signal. The mean values are represented with error bars denoting s.d. (n + 3). *P < 0.01, t-test.
- D ChIP analysis in G1-arrested cells. As in (C), but showing ChIP analysis of the recruitment of Sgs1-HA and Sgs1-ss-HA in cells that were kept arrested in G1 by α-factor. The mean values are represented with error bars denoting s.d. (n + 3). *P < 0.01, t-test.
- E ChIP analysis in G1-arrested cells. As in (C), but showing ChIP analysis of the recruitment of Exo1–Myc in cells that were kept arrested in G1 by α-factor. The mean values are represented with error bars denoting s.d. (n + 3). *P < 0.01, t-test.
We then asked by ChIP and qPCR analysis whether Rad9 limits Sgs1 activity by regulating Sgs1 binding/persistence to the DSB ends. When HO was induced in exponentially growing cells, the amount of Sgs1 bound at the HO-induced DSB was higher in rad9Δ than in wild-type cells (Fig6C), indicating that Rad9 counteracts Sgs1 recruitment to the DSB. Interestingly, the Sgs1-ss variant was recruited at the DSB with equivalent efficiencies in both exponentially growing wild-type and rad9Δ cells (Fig6C). These differences were not due to different resection kinetics, as we obtained similar results also when the HO-induced DSB was generated in G1-arrested cells (Fig6D), which resected the DSB very poorly due to the low Cdk1 activity 6. Interestingly, the amount of Sgs1-ss bound to the DSB was higher than the amount of wild-type Sgs1 in rad9Δ cells (Fig6C and D), suggesting that Sgs1-ss has a higher intrinsic ability to bind/persist at the DSB. Altogether, these results indicate that Rad9 limits the association of Sgs1 to the DSB ends and that the Sgs1-ss variant escapes this inhibition possibly because it binds more tightly the DSB. Interestingly, the robust association of Sgs1-ss to the DSB in G1-arrested cells (low Cdk1 activity) did not result in DSB resection (Supplementary Fig S2) possibly because Sgs1 acts in DSB resection together with Dna2, whose activity requires Cdk1-mediated phosphorylation 8. Consistent with a contribution of Exo1 in promoting DSB resection in the absence of Rad9, rad9Δ cells showed an increased Exo1 recruitment to the DSB compared to wild-type cells (Fig6E).
In summary, we show that Rad9 increases the requirement for the MRX/Sae2 activities in DSB resection by inhibiting the action of the Sgs1/Dna2 long-range resection machinery. Extensive resection in Rad9-deficient cells is mainly dependent on Sgs1, whose recruitment at DSBs is inhibited by Rad9. By contrast, Sgs1-ss, which suppresses the resection defect of sae2Δ cells, is robustly associated with the DSB ends both in the presence and in the absence of Rad9 and resects the DSB more efficiently than wild-type Sgs1. These findings indicate that Rad9 inhibits the activity of Sgs1/Dna2 by limiting Sgs1 binding/persistence at DSB ends and that the Sgs1-ss mutant variant escapes this inhibition possibly because it is more tightly bound to DNA. Thus, while Ku increases the requirement for the MRX/Sae2 activities in DSB resection by inhibiting preferentially Exo1 20, Rad9 mainly restricts the action of Sgs1/Dna2. As MRX and Sae2 are especially important for initial processing of DNA ends that contain adducts, the Rad9- and Ku-mediated inhibitions of Sgs1/Dna2 and Exo1 activities in initiating DSB resection ensure that all DSBs are processed in a similar manner independently of their nature.
Materials and Methods
Yeast strains
The yeast strains used in this study are derivatives of W303, JKM139 or SK1 (Supplementary Table S1). Cells were grown in YEP medium (1% yeast extract, 2% peptone) supplemented with 2% glucose (YEPD), 2% raffinose (YEPR) or 2% raffinose and 3% galactose (YEPRG).
Search for suppressors of sae2Δ sensitivity to CPT
To search for suppressor mutations of the CPT sensitivity of sae2Δ mutant, 5 × 106 sae2Δ cells were plated on YEPD in the presence of 30 μM CPT. Survivors were recovered and crossed to wild-type cells to identify by tetrad analysis the suppression events that were due to single-gene mutations. Subsequent genetic analyses allowed grouping the single-gene suppression events in 11 classes. The five classes that showed the most efficient suppression were chosen and the suppressor genes were identified by genome sequencing and genetic analyses. To confirm that the SGS1-ss mutation was responsible for the suppression, a URA3 gene was integrated downstream of the SGS1-ss stop codon and the resulting strain was crossed to wild-type cells to verify by tetrad dissection that the suppression of the sae2Δ CPT sensitivity co-segregated with the URA3 allele.
DSB resection
Double-strand break end resection at the MAT locus was analysed on alkaline agarose gels as described in Clerici et al 30. Quantitative analysis of DSB resection was performed by calculating the ratio of band intensities for ssDNA and total amount of DSB products.
Synchronous meiotic time course and detection of meiotic DSBs
To obtain synchronous G1/G0 cell population, overnight liquid YEPD cell cultures were diluted to a final concentration of 1 × 107 cells/ml in 200 ml YPA (1% yeast extract, 2% bactopeptone, 1% potassium acetate) and grown for 13 h at 30°C. Cells were then washed and transferred into the same volume of SPM (0.3% potassium acetate, 0.02% raffinose) to induce meiosis. Genomic DNA was digested with EcoRI and separated on native agarose gels. DSBs at the THR4 hotspot were detected with a 1.6-kb DNA fragment spanning the 5′ region of THR4.
Other techniques
ChIP assays were performed as described in Viscardi et al 33. Data are expressed as fold enrichment at the HO-induced DSB over that at the non-cleaved ARO1 locus, after normalization of each ChIP signals to the corresponding input for each time point. Fold enrichment was then normalized to the efficiency of DSB induction. Rad53 was detected by using anti-Rad53 (ab104232) polyclonal antibodies from Abcam.
Acknowledgments
We thank J. Haber for strains. Michela Clerici and Giovanna Lucchini are acknowledged for critical reading of the manuscript. This work was supported by grants from Associazione Italiana per la Ricerca sul Cancro (AIRC) (Grant No. IG15210) and Cofinanziamento 2010-2011 MIUR/Università di Milano-Bicocca to MPL.
Author contributions
Conceived and designed the experiments: DB, MV, MPL. Performed the experiments: DB, MV, EG, CC, GT. Analysed the data: DB, MV, EG, MPL. Wrote the manuscript: MPL.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting Information
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Table S1
Review Process File
References
- Symington LS, Gautier J. Double-strand break end resection and repair pathway choice. Annu Rev Genet. 2011;45:247–271. doi: 10.1146/annurev-genet-110410-132435. [DOI] [PubMed] [Google Scholar]
- Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell. 2010;40:179–204. doi: 10.1016/j.molcel.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannavo E, Cejka P. Sae2 promotes dsDNA endonuclease activity within Mre11-Rad50-Xrs2 to resect DNA breaks. Nature. 2014;514:122–125. doi: 10.1038/nature13771. [DOI] [PubMed] [Google Scholar]
- Mimitou EP, Symington LS. Sae2, Exo1 and Sgs1 collaborate in DNA double-strand break processing. Nature. 2008;455:770–774. doi: 10.1038/nature07312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Z, Chung WH, Shim EY, Lee SE, Ira G. Sgs1 helicase and two nucleases Dna2 and Exo1 resect DNA double-strand break ends. Cell. 2008;134:981–994. doi: 10.1016/j.cell.2008.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ira G, Pellicioli A, Balijja A, Wang X, Fiorani S, Carotenuto W, Liberi G, Bressan D, Wan L, Hollingsworth NM, et al. DNA end resection, homologous recombination and DNA damage checkpoint activation require CDK1. Nature. 2004;431:1011–1017. doi: 10.1038/nature02964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huertas P, Cortés-Ledesma F, Sartori AA, Aguilera A, Jackson SP. CDK targets Sae2 to control DNA-end resection and homologous recombination. Nature. 2008;455:689–692. doi: 10.1038/nature07215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Niu H, Chung WH, Zhu Z, Papusha A, Shim EY, Lee SE, Sung P, Ira G. Cell cycle regulation of DNA double-strand break end resection by Cdk1-dependent Dna2 phosphorylation. Nat Struct Mol Biol. 2011;18:1015–1019. doi: 10.1038/nsmb.2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeber A, Hauer M, Gasser SM. Nucleosome remodelers in double-strand break repair. Curr Opin Genet Dev. 2013;23:174–184. doi: 10.1016/j.gde.2012.12.008. [DOI] [PubMed] [Google Scholar]
- Chen X, Cui D, Papusha A, Zhang X, Chu CD, Tang J, Chen K, Pan X, Ira G. The Fun30 nucleosome remodeller promotes resection of DNA double-strand break ends. Nature. 2012;489:576–580. doi: 10.1038/nature11355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costelloe T, Louge R, Tomimatsu N, Mukherjee B, Martini E, Khadaroo B, Dubois K, Wiegant WW, Thierry A, Burma S, et al. The yeast Fun30 and human SMARCAD1 chromatin remodellers promote DNA end resection. Nature. 2012;489:581–584. doi: 10.1038/nature11353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eapen VV, Sugawara N, Tsabar M, Wu WH, Haber JE. The Saccharomyces cerevisiae chromatin remodeler Fun30 regulates DNA end resection and checkpoint deactivation. Mol Cell Biol. 2012;32:4727–4740. doi: 10.1128/MCB.00566-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lydall D, Weinert T. Yeast checkpoint genes in DNA damage processing: implications for repair and arrest. Science. 1995;270:1488–1491. doi: 10.1126/science.270.5241.1488. [DOI] [PubMed] [Google Scholar]
- Lazzaro F, Sapountzi V, Granata M, Pellicioli A, Vaze M, Haber JE, Plevani P, Lydall D, Muzi-Falconi M. Histone methyltransferase Dot1 and Rad9 inhibit single-stranded DNA accumulation at DSBs and uncapped telomeres. EMBO J. 2008;27:1502–1512. doi: 10.1038/emboj.2008.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keeney S, Kleckner N. Covalent protein-DNA complexes at the 5′ strand termini of meiosis-specific double-strand breaks in yeast. Proc Natl Acad Sci USA. 1995;92:11274–11278. doi: 10.1073/pnas.92.24.11274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usui T, Ohta T, Oshiumi H, Tomizawa J, Ogawa H, Ogawa T. Complex formation and functional versatility of Mre11 of budding yeast in recombination. Cell. 1998;95:705–716. doi: 10.1016/s0092-8674(00)81640-2. [DOI] [PubMed] [Google Scholar]
- Deng C, Brown JA, You D, Brown JM. Multiple endonucleases function to repair covalent topoisomerase I complexes in Saccharomyces cerevisiae. Genetics. 2005;170:591–600. doi: 10.1534/genetics.104.028795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mimitou EP, Symington LS. Ku prevents Exo1 and Sgs1-dependent resection of DNA ends in the absence of a functional MRX complex or Sae2. EMBO J. 2010;29:3358–3369. doi: 10.1038/emboj.2010.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster SS, Balestrini A, Petrini JH. Functional interplay of the Mre11 nuclease and Ku in the response to replication-associated DNA damage. Mol Cell Biol. 2011;31:4379–4389. doi: 10.1128/MCB.05854-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shim EY, Chung WH, Nicolette ML, Zhang Y, Davis M, Zhu Z, Paull TT, Ira G, Lee SE. Saccharomyces cerevisiae Mre11/Rad50/Xrs2 and Ku proteins regulate association of Exo1 and Dna2 with DNA breaks. EMBO J. 2010;29:3370–3380. doi: 10.1038/emboj.2010.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullen JR, Kaliraman V, Brill SJ. Bipartite structure of the Sgs1 DNA helicase in Saccharomyces cerevisiae. Genetics. 2000;154:1101–1114. doi: 10.1093/genetics/154.3.1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budd ME, Reis CC, Smith S, Myung K, Campbell JL. Evidence suggesting that Pif1 helicase functions in DNA replication with the Dna2 helicase/nuclease and DNA polymerase delta. Mol Cell Biol. 2006;26:2490–2500. doi: 10.1128/MCB.26.7.2490-2500.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SE, Moore JK, Holmes A, Umezu K, Kolodner RD, Haber JE. Saccharomyces Ku70, Mre11/Rad50 and RPA proteins regulate adaptation to G2/M arrest after DNA damage. Cell. 1998;94:399–409. doi: 10.1016/s0092-8674(00)81482-8. [DOI] [PubMed] [Google Scholar]
- Pellicioli A, Lee SE, Lucca C, Foiani M, Haber JE. Regulation of Saccharomyces Rad53 checkpoint kinase during adaptation from DNA damage-induced G2/M arrest. Mol Cell. 2001;7:293–300. doi: 10.1016/s1097-2765(01)00177-0. [DOI] [PubMed] [Google Scholar]
- Usui T, Ogawa H, Petrini JH. A DNA damage response pathway controlled by Tel1 and the Mre11 complex. Mol Cell. 2001;7:1255–1266. doi: 10.1016/s1097-2765(01)00270-2. [DOI] [PubMed] [Google Scholar]
- Clerici M, Mantiero D, Lucchini G, Longhese MP. The Saccharomyces cerevisiae Sae2 protein negatively regulates DNA damage checkpoint signalling. EMBO Rep. 2006;7:212–218. doi: 10.1038/sj.embor.7400593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clerici M, Trovesi C, Galbiati A, Lucchini G, Longhese MP. Mec1/ATR regulates the generation of single-stranded DNA that attenuates Tel1/ATM signaling at DNA ends. EMBO J. 2014;33:198–216. doi: 10.1002/embj.201386041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaze MB, Pellicioli A, Lee SE, Ira G, Liberi G, Arbel-Eden A, Foiani M, Haber JE. Recovery from checkpoint-mediated arrest after repair of a double-strand break requires Srs2 helicase. Mol Cell. 2002;10:373–385. doi: 10.1016/s1097-2765(02)00593-2. [DOI] [PubMed] [Google Scholar]
- Bernstein KA, Mimitou EP, Mihalevic MJ, Chen H, Sunjaveric I, Symington LS, Rothstein R. Resection activity of the Sgs1 helicase alters the affinity of DNA ends for homologous recombination proteins in Saccharomyces cerevisiae. Genetics. 2013;195:1241–1251. doi: 10.1534/genetics.113.157370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clerici M, Mantiero D, Guerini I, Lucchini G, Longhese MP. The Yku70-Yku80 complex contributes to regulate double-strand break processing and checkpoint activation during the cell cycle. EMBO Rep. 2008;9:810–818. doi: 10.1038/embor.2008.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trovesi C, Falcettoni M, Lucchini G, Clerici M, Longhese MP. Distinct Cdk1 requirements during single-strand annealing, noncrossover, and crossover recombination. PLoS Genet. 2011;7:e1002263. doi: 10.1371/journal.pgen.1002263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngo GH, Balakrishnan L, Dubarry M, Campbell JL, Lydall D. The 9-1-1 checkpoint clamp stimulates DNA resection by Dna2-Sgs1 and Exo1. Nucleic Acids Res. 2014;42:10516–10528. doi: 10.1093/nar/gku746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viscardi V, Bonetti D, Cartagena-Lirola H, Lucchini G, Longhese MP. MRX-dependent DNA damage response to short telomeres. Mol Biol Cell. 2007;18:3047–3058. doi: 10.1091/mbc.E07-03-0285. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Table S1
Review Process File