

Abstract
Background. We developed a 2-step approach to screen molecules that prevent and/or reverse Plasmodium falciparum–infected erythrocyte (IE) binding to host receptors. IE adhesion and sequestration in vasculature causes severe malaria, and therefore antiadhesion therapy might be useful as adjunctive treatment. IE adhesion is mediated by the polymorphic family (approximately 60 members) of P. falciparum EMP1 (PfEMP1) multidomain proteins.
Methods. We constructed sets of PfEMP1 domains that bind ICAM-1, CSA, or CD36, receptors that commonly support IE binding. Combinations of domain-coated beads were assayed by Bio-Plex technology as a high-throughput molecular platform to screen antiadhesion molecules (antibodies and small molecules). Molecules identified as so-called hits in the screen (first step) then could be assayed individually for inhibition of binding of live IE to receptors (second step).
Results. In proof-of-principle studies, the antiadhesion activity of several antibodies was concordant in Bio-Plex and live IE assays. Using this 2-step approach, we identified several molecules in a small molecule library of 10 000 compounds that could inhibit and reverse binding of IEs to ICAM-1 and CSA receptors.
Conclusion. This 2-step screening approach should be efficient for identification of antiadhesion drug candidates for falciparum malaria.
Keywords: malaria, Plasmodium falciparum, sequestration of parasites, antiadhesion therapy, high-throughput screening, PfEMP1 proteins, host receptors, CD36, ICAM-1, CSA, small molecules
Malaria is one of the most devastating diseases in the world, with approximately 0.5 billion cases and 0.5 million deaths, mostly among young children, each year. Most deaths result from severe malaria syndromes, including cerebral malaria, respiratory distress, and severe anemia, or from sequelae of placental malaria. Although effective antimalarial drugs can rapidly kill parasites, significant mortality (10%–15%) ensues in severe malaria cases, particularly within 24 hours of hospital admission and possibly because Plasmodium falciparum–infected erythrocytes (IEs) remain sequestered in deep vascular beds after the parasite has been killed [1].
Adjunctive therapy with drugs that reverse and/or prevent parasite adhesion and sequestration may significantly improve the outcomes for patients with severe malaria by decreasing local and systemic inflammation associated with severe disease [2] and reestablishing the microvascular blood flow. While standard parasiticidal drugs need time to pass through 2 or 3 membranes of the IE to be effective, antiadhesion drugs would be expected to be immediately effective at the interface of the IE and the host receptor. These drugs would be used as an adjunct to parasiticidal drugs to reduce mortality from severe malaria cases. IE that have been desequestered or blocked from adhesion would be cleared by the spleen.
Parasite adhesion is mediated by the P. falciparum EMP1 (PfEMP1) family (approximately 60 members) of clonally variant, surface-expressed erythrocyte transmembrane proteins [3]. In malaria-endemic areas, severe malaria accounts for <2% of total malaria cases in young children and only occurs once or twice in a lifetime [4], with a marked reduction of the number of cases after infancy in areas of high transmission. This indicates that broad immunity against severe malaria develops quickly in early childhood [5] despite variation of IE surface proteins, suggesting that diversity of severe malaria-relevant variant epitopes might be significantly restricted. A monoclonal antibody that inhibits binding of several malaria parasite strains to the host ligand chondroitin sulfate A (CSA) during pregnancy [6] indicates that conserved features of PfEMP1 protein variants might be sufficient to provide cross-strain effectiveness of antiadhesion molecules. Additionally, molecules that mimic binding motifs of host receptors might be effective against multiple variants of the relevant PfEMP1 proteins, as demonstrated by the small molecule that mimics an ICAM-1 loop and inhibits binding of 2 parasite variants to the ICAM-1 receptor [7]. Thus, a single adhesion-inhibiting molecule might exhibit cross-strain and multi-PfEMP1 activity for a particular host receptor.
Because of technical limitations, live parasite isolates are not amenable to high-throughput adaptation for direct screening for antiadhesion molecules. We have developed a 2-step approach to identify antiadhesion molecules that overcomes these limitations. First, molecules are screened in a high-throughput manner for inhibition of interactions between known host receptors that are involved in IE adhesion and various PfEMP1 domains that bind these receptors. Second, the molecules identified in the first step are tested for adhesion-inhibitory activity using live IE. As the number of hit molecules to test with IE is low, the 2-step procedure will provide high-throughput identification of antiadhesion molecules that can be further developed into antiadhesion drugs.
To validate this approach, we selected 3 human receptors that commonly support binding to IE (CD36, CSA, and ICAM-1) and a number of PfEMP1 domains and parasite strains that interact with these receptors [8–10]. We demonstrate that adhesion-inhibitory molecules identified in high-throughput in vitro assays have similar adhesion-inhibitory activity against live IE binding to the corresponding receptor.
MATERIALS AND METHODS
PfEMP1 Domains
The following domains of the 3D7 parasite line were cloned and expressed as previously described: Duffy-binding-like (DBL) domain 2β from PF11_0521 in pHisAdEx vector and COS-7 cells [9] and in pET28b vector and Escherichia coli cells [11]; DBL1 through DBL6 single domains from VAR2CSA and AMA1 in pET28b vector and E. coli cells [12]; NTF-DBL1-CIDR1 from PFD0005w and PFF1595c, CIDR1 from PF08_0106 and PFD0995c in pHisAdEx vector and COS-7 cells [13]. Constructs NTF-DBL1-CIDR1 from PFF1580c (amino acids [aa] 2–842) and PF07_0049 (aa 2–847), and CIDR1 from MAL7P1.56 (aa 414–842) were cloned and expressed in the pHisAdEx vector and COS-7 cells. The full-length extracellular region of 3D7 VAR2CSA (DBL1 through DBL6) was expressed and purified as previously described [14].
Receptors
Human CD36-Fc (1955-CD-050) and ICAM-1-Fc (720-IC-050) chimeras were purchased from R&D Systems; CSA was purchased from Sigma (C9819).
Antibodies
We used anti-CD36 monoclonal antibody (mAb; FA6-152) from Abcam (ab17044), anti–ICAM-1 mAb (My-13) from Life Technologies (07-5403), goat polyclonal antibodies (pAb) against human CD36 from R&D Systems (AF1955), rat pAb against DBL2PF11_0521 (as previously described [11]), and rat pAb against VAR2CSA domains (single and multi) from the 3D7 parasite line (as previously described [12]). Phycoerythrin (PE)–labeled molecules (Jackson Immunoresearch) were used for detection of the receptors bound to the Bio-Plex bead-immobilized domains: donkey anti–human immunoglobulin G (IgG)-PE (709-116-149) for ICAM-1–Fc and CD36-Fc receptors and streptavidin-PE (116-110-084) for biotinylated CD36 and CSA receptors. Goat anti–mouse immunoglobulin G (IgG)–PE (115-116-146) and anti–rat IgG-PE (112-116-143) were used for detection of IgG bound to PfEMP1 domains in experiments with antidomain antibodies.
Measurement of Receptor Binding and Inhibition of Receptor Binding to PfEMP1 Domains Immobilized on Bio-Plex Beads
Binding of ICAM-1–Fc and CD36-Fc chimeras and inhibition of ICAM-1–Fc binding was previously described [9, 11, 13]. Binding of biotinylated CSA (Bio-CSA) and biotinylated CD36 (Bio-CD36) and inhibition of their binding was performed similarly, using the concentrations indicated in the text and figure legends. Biotinylation of CSA and CD36 was performed using the EZ-Link Sulfo-NHS-Biotinylation Kit (Pierce; 21 425). Reversal of receptor binding was done by incubating bead-immobilized domains with receptors, washing away unbound receptor, incubating with inhibitory molecules at indicated concentrations, and detecting bound receptor, as previously described [9].
Small-Molecule Library Screening
A total of 10 000 small drug-like molecules (a subset of DIVERSet Library; 10 mM in dimethyl sulfoxide [DMSO]) were purchased from ChemBridge. We prepared pools of 80 compounds for initial screening (125 µM of each compound in DMSO; 125 pools). Bio-Plex beads with immobilized domains and negative-control constructs (a 52-kDa product of empty pHisAdEx vector [9, 15], for COS-7–expressed domains; and AMA1, a non-PfEMP1 merozoite surface protein, for E. coli–expressed domains) were resuspended in phosphate-buffered saline with 0.05% tween-20 (PBST) and incubated with receptors and compounds at room temperature in 96-well MultiScreen filter plates (Millipore), as follows: 50-µL compound pools were prepared at 1:5 in PBS added to the protein-immobilized beads and incubated for 1 hour. A total of 50 µL of receptor prepared in PBST plus 0.1% bovine serum albumin (BSA; concentrations are specified in figure legends) was added to each pool and incubated for additional 1 hour. The beads were then washed 3 times with PBST and resuspended in 50 µL of detection molecules prepared in PBST plus 0.1% BSA (1:250 donkey anti–human IgG-PE or 1:5000 streptavidin-PE) and incubated for 30 minutes. Beads were then washed 3 times with PBST, 1 time with PBS, re-suspended in 125 µL of PBS and analyzed by a BioPlex 200 (BioRad). The mean fluorescence (in the PE channel) of beads incubated in buffer with 20% DMSO (100% binding) was compared to the fluorescence of beads incubated with compounds or controls, to calculate inhibition. Positive controls for inhibition were as follows: unlabeled CSA, for the Bio-CSA receptor (300 µg/mL; >90% inhibition); rat anti–DBL2PF11_0521 antiserum, for the ICAM-1 receptor (1:20 in PBST-0.1% BSA; >90% inhibition); and anti–CD36 mAb FA6–152, for the CD36 receptor (5 µg/mL in PBST-0.1% BSA; 60%–95% inhibition, depending on the domain). Domains used for library screening were as follows: CIDR1 from PF08_0106, PFD0995c, and MAL7P1.56 expressed in COS-7, for the CD36 receptor; DBL3, DBL4, and DBL5 from VAR2CSA expressed in E. coli, for the CSA receptor; and DBL2β from PF11_0521 expressed in COS-7, for the ICAM-1 receptor. Any pool that was able to inhibit receptor binding to PfEMP1 by ≥75% was deconvoluted for identification of the inhibitory compound(s).
Inhibition of IE Binding and Reversal of Binding to Various Receptors
Human O+ blood and AB serum (HP1022) for growth of P. falciparum parasites was purchased from Valley Biomedical. Parasite cultures for these experiments had 2% hematocrit and 5%–8% parasitemia. Receptors were immobilized on plastic Petri dishes, and binding assays were performed as previously described [16]. For inhibition of binding, 1 volume of inhibitory agents (10 times the final concentration of antibody or small molecule in PBS or PBS plus 1% DMSO, respectively) or control buffer (PBS or PBS plus 1% DMSO) were mixed with 9 volumes of IE, incubated for 30 minutes at 37°C, and applied to the plastic-immobilized receptors. For reversal of bound IEs, the inhibitory agents (1 times the final concentration in PBS or PBS plus 0.1% DMSO) or control buffer were added to IEs prebound to the plastic-immobilized receptors and incubated for 30 minutes at 37°C. In experiments using DMSO as a vehicle, the concentration of DMSO during incubation with IEs was 0.1%. This concentration of DMSO does not affect the binding, integrity, or viability of IEs (data not shown). Unbound IEs were removed by washing plates as previously described [16]. Inhibition was evaluated by comparison of the mean number of bound IEs per field (counted in 20 fields each in duplicate spots) between control (buffer) and experiment (buffer plus inhibitory agent). Concentrations of inhibitory molecules are indicated in the text and figure legends. Parasite lines used in these experiments include CSA-binding CS2 [17], CD36-binding NF54-E3 (described below), and ICAM-1–binding 3G8 [18].
Isolation of NF54 Cell Lines Expressing Single var Genes
Cell lines were obtained by limiting dilution [18, 19]. Transcribed var genes in each line were measured as follows. Total RNA was isolated by the RNeasy Micro Kit (Qiagen) and converted into complementary DNA (cDNA), using Superscript III (Life technologies). This cDNA was amplified by polymerase chain reaction, using a set of degenerate primers (Supplementary Figure 1) that amplify DBL1 domain fragments of all var genes with similar efficiency (tested using 3D7 genomic DNA; data not shown). After cloning cDNA fragments into topoTA vector (Life Technologies), 10–20 clones for each line were sequenced to identify transcribed var genes. We identified 2 lines used for this work, E3 and E9, which express the single var genes PF08_0106 and PFL2665c, respectively.
RESULTS AND DISCUSSION
Antireceptor and Anti-PfEMP1 Domain Antibodies Can Efficiently Inhibit Binding Between PfEMP1 Domains and Receptors and Between Live IEs and Receptors In Vitro
Interactions With ICAM-1
We previously demonstrated that binding of the DBL2βPF11_0521 domain (from the 3D7 strain) to ICAM-1 can be efficiently blocked by an anti–ICAM-1 mAb, My-13 [9]. However, reversal of binding by using the same mAb is not efficient (Figure 1A). This mAb was previously shown to inhibit ICAM-1 binding of the A4 parasite line [20], and here we demonstrate that it can also inhibit ICAM-1 binding of the 3G8 parasite line that expresses a different ICAM-1–binding PfEMP1 protein [18, 21] (Figure 1B).
Figure 1.

Antireceptor (A and B) and antidomain (C and D) antibodies as antiadhesion molecules for ICAM-1 binding. A and B, Testing anti–ICAM-1 monoclonal antibody (Ab) My-13 as an antiadhesion molecule. A, Reversal of ICAM-1 binding to Duffy-binding-like (DBL) 2PF11_0521 domain on Bio-Plex beads. Error bars are standard deviations. B, Inhibition of ICAM-1 binding of live 3G8 parasite–infected erythrocytes. Error bars are standard errors of the mean. C and D, Reversal and blocking of ICAM-1 binding to DBLPF11_0521 domain on Bio-Plex beads by rat antidomain Abs. C, Reversal is inefficient, while blocking is efficient. D, Amounts of domain-bound Abs are similar in blocking and reversal experiments. Immune and preimmune sera were used at a 1:20 dilution. Error bars are standard deviations. Abbreviation: IE, Plasmodium falciparum–infected erythrocyte.
Polyclonal antibodies raised in rats and mice against full-length (aa 1–520) DBL2βPF11_0521 domain constructs [11] inhibited ICAM-1 binding by this domain in vitro [11], but they were not able to reverse the interaction (Figure 1C). The amount of antidomain antibody bound to the domain was similar in both blocking assays (antibodies were added before receptor) and reversal assays (antibodies were added after receptor; Figure 1D). This suggests that inhibitory antibodies only represent a small fraction of the total antidomain polyclonal antibodies. These antidomain antibodies were strain specific and did not inhibit ICAM-1 binding or recognize the heterologous 3G8 strain (data not shown).
Similar behavior was observed for naturally acquired antibodies from adults immune to malaria parasite infection, which can block but not reverse ICAM-1 binding to both an ICAM-1–binding domain [9] and an ICAM-1–binding isolate [22]. The results indicate that neither antidomain nor antireceptor antibodies may be efficient for reversal of ICAM-1–bound IEs, possibly because of the high avidity (KD, approximately 2–4 nM [11]) of these interactions. While reversal of ICAM-1 binding by antibodies is not efficient, this may not necessarily be the case for reversal by small molecules.
Interactions With CSA
Several DBL domains of PfEMP1 VAR2CSA individually bind CSA though different binding profiles have been reported, depending on the laboratory and the strain [23]. We expressed all DBL domains of VAR2CSA (PFL0030c) from the 3D7 line in E. coli and refolded them as previously described [12, 24]. We found that all of these domains were able to bind Bio-CSA (Figure 2A). Differences in the observed binding profiles between laboratories may be due to the genetic background of the strains but seem more likely to be a result of the different domain boundaries used for cloning the recombinant proteins, location of protein purification tags, and different refolding schemes.
Figure 2.
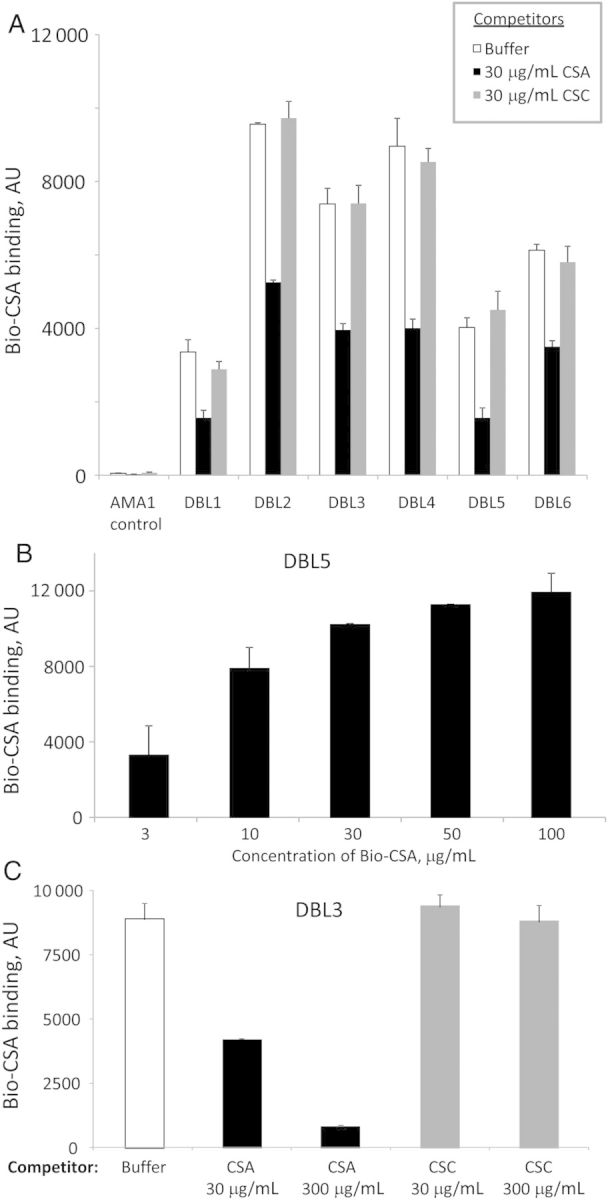
All Duffy-binding-like (DBL) domains of VAR2CSA can bind Bio-CSA specifically. A, Inhibition of binding of Bio-CSA (30 µg/mL) by nonlabeled CSA and chondroitin sulfate C (CSC). B, Binding of Bio-CSA is concentration dependent (shown for DBL5). C, Inhibition of binding of Bio-CSA is concentration dependent with unlabeled CSA but not with unlabeled CSC (shown for DBL3). Error bars are standard deviations.
Binding of Bio-CSA (30 µg/mL) to all DBL domains is inhibited by preincubation with 30 µg/mL of nonlabeled CSA but not chondroitin sulfate C (CSC), a similarly charged but differently sulfated molecule (Figure 2A). Binding (shown for DBL5 in Figure 2B), as well as inhibition (shown for the DBL3 domain in Figure 2C), is concentration dependent. Similarly, parasite binding to CSA is competitively inhibited by CSA but not by other chondroitin sulfates [25].
Rat antibodies to various VAR2CSA domains [12] specifically block CSA binding to corresponding domains (Figure 3A–C), as well as to the full-length extracellular part of 3D7 VAR2CSA (Figure 3D). The same anti-DBL domain antibodies can variably inhibit binding of different CSA-binding field isolates to both placental sections and CSA immobilized on plastic [12]. Antibodies prepared against similarly designed VAR2CSA DBL domain constructs from the FCR3 and 7G8 laboratory strains were also shown to recognize and inhibit CSA binding of some laboratory and field isolates [26–29].
Figure 3.

Antidomain antibodies (Abs) as antiadhesion molecules for CSA binding in molecular assay. A–D, Inhibition of Bio-CSA (30 µg/mL) binding to VAR2CSA domains (A–C) and to full-length VAR2CSA extracellular part (D) by various antidomain Abs. Duffy-binding-like (DBL) 4/5 and DBL5/6 are double-domain constructs [12]. Inhibition by positive (unlabeled CSA) and negative (unlabeled chondroitin sulfate C [CSC]) controls is also shown (30 µg/mL each). Rat sera dilutions were 1:100. Error bars are standard deviations. Abbreviation: PI, preimmune serum.
Interactions With CD36
We selected 5 CD36-binding PfEMP1 domain constructs for use in binding inhibition assays [13]. Using the anti-CD36 mAb FA6-152 that inhibits binding of the FCR3-CD36 IE line to CD36 [28], we found that the inhibition efficiency varied substantially for CIDR1 domains from different PfEMP1 proteins (Figure 4A). This suggests that CD36-binding domains from different proteins do not contact precisely the same sites of the CD36 receptor surface. Reversal of CD36 binding was not efficient with this mAb (Figure 4A).
Figure 4.

Antireceptor antibodies (Abs) as antiadhesion molecules for CD36 binding in the molecular assay (A and C) and with live IE (B). A. Inhibition of binding of CD36 to various CD36-binding, bead-immobilized domains by anti-CD36 monoclonal Ab (mAb) FA6-152. Black bars, blocking; gray bars, reversal (4 hours). Concentrations: CD36 at 1 µg/mL, mAb FA6-152 at 5 µg/mL. B, Binding of E3 line to plastic-immobilized CD36. One hundred percent binding was 152 Plasmodium falciparum–infected erythrocytes (IEs) per microscope field. C, CD36 binding to bead-immobilized CIDR1PF08_0106 domain. Error bars are standard deviations. Abbreviation: pAb, goat polyclonal antibody.
Using limiting dilution [18, 30], we obtained a CD36-binding NF-54 IE line (E3) that solely expresses the var gene PF08_0106, which encodes a CD36-binding CIDR1 domain [10, 13]. Binding of E3 to CD36 was inhibited by both mAb FA6-152 and by anti-CD36 pAb (Figure 4B), although inhibition with the pAb was less efficient. Similar results were obtained with line E9 that solely expresses CD36-binding var gene PFL2665c (data not shown). Likely only a fraction of the polyclonal antibodies inhibits the interaction of the CD36 receptor with the CIDR1PF08_0106 domain, similar to pAbs against the ICAM-1–binding PfEMP1 domain (described above). We obtained similar results when using these antibodies to test inhibition of CD36 binding in the Bio-Plex format (Figure 4C), indicating the consistency of our 2-step screening approach.
The concordance between the in vitro molecular Bio-Plex assays and the assays with live IEs by using antibodies as inhibitory agents for all of the receptors in this work supported the feasibility of our 2-step approach for screening antiadhesion molecules for these receptors. As the physical principles and outcomes of the assay are the same for both large and small antiadhesion molecules, we expected to get similarly concordant results with the same 2-step approach, using a small-molecule library at the first step.
Small Molecules That Inhibit Binding Between PfEMP1 Domains and Human Receptors Identified in Our Molecular Platform Can Inhibit Binding of Live IE to the Same Receptors
As further proof of principle for our 2-step screening system, we tested a 10 000-compound library (ChemBridge) of small drug-like molecules for inhibition of binding of each of the 3 receptors to the corresponding PfEMP1 domains. The binding interactions of all 3 selected receptors and corresponding PfEMP1 domains can tolerate up to 20% DMSO by volume (Supplementary Figure 2).
Interactions With CD36
We used the strongest 3 CD36-binding PfEMP1 CIDR1 domains [13] for CD36-binding inhibition screening. None of the small-molecule pools inhibited CD36 binding to any domain by ≥75%, so we did not proceed with deconvolution to single compounds at this stage. We anticipate repeating this screen with a larger chemical library in future.
Interactions With CSA
We selected the DBL3, DBL4, and DBL5 domains of VAR2CSA for screening compounds that inhibit Bio-CSA binding. Initial inhibiting mixtures were deconvoluted to single compounds, using the DBL3 domain. Three compounds inhibited binding of DBL3 to CSA (Supplementary Figure 3A), of which 2 (5210644 and 5210653) share the same sulfonyl-containing primary scaffold (Figure 5A), which may compete with the charged sulfated residues of CSA for VAR2CSA binding. Charge is unlikely to be the main reason for the binding inhibition effect, as other sulfonyl-containing compounds in the library did not have a similar inhibitory effect (data not shown). The third compound (6917839) has a different scaffold (Figure 5A) but some structural similarities to the other 2 compounds. All 3 compounds exhibited concentration-dependent inhibition (Figure 5B and 5C and Supplementary Figure 3B) with low or submicromolar median inhibitory concentrations (IC50) for all domains and the full-length (FL) extracellular part of the VAR2CSA. Note that the level of binding of Bio-CSA to FL VAR2CSA was lower than that for any domain, owing to its approximately 8-fold greater molecular weight and proportionally lower molar occupancy on the beads (10 µg of protein is coupled to 100 µL of each bead kind). Compound 5210653 was able to reverse CSA binding to DBL3 domain (Figure 5C), although reversal was not as efficient as blocking activity (IC50, 4 µM and 0.65 µM, respectively).
Figure 5.

Characterization of CSA-binding inhibitory compounds. A, Chemical structures. B, Concentration-dependent inhibition. Error bars are standard deviations. C, Calculation of the median inhibitory concentration (IC50) for binding of VAR2CSA Duffy-binding-like (DBL) 2, DBL3, and DBL6 domains and the full-length extracellular part (FL), and for reversal of DBL3 with compound 5210653. Error bars are standard deviations. D, Inhibition and reversal of CS2 Plasmodium falciparum–infected erythrocyte binding to CSA immobilized on surface of plastic dish by two compounds. The last 3 digits of the tested inhibitory compound numbers are shown. Bars are means of independent experiments; error bars are standard errors of the mean. Numbers below the x-axis indicate the number of independent experiments. Concentrations used for inhibition/reversal: CSA, 50 µg/mL; compounds, 100 µM. Average binding (without inhibition) to CSA was about 150 IE per microscope field. Abbreviations: Conc, concentration; Inh, inhibition; Rev, reversal.
To test the compounds with live IE, we used the CS2 laboratory parasite line (originally selected from the ITG laboratory isolate) that efficiently binds to CSA [17] and is commonly used in CSA binding experiments. We tested compounds 5210653 and 6917839 at 100 µM and demonstrated that both compounds inhibit and reverse IE binding to the same degree as CSA (Figure 5D). Thus, the compounds identified in our experiments with CSA-binding domains from 3D7 immobilized on beads are cross-inhibitory for CSA binding and reversal of parasites from a different genetic background.
Interactions With ICAM-1
We used the DBL2β domain from PF11_0521 to test binding inhibition of the ICAM-1 receptor. Two compounds inhibited binding to ICAM-1 (Supplementary Figure 3C), each (5306995 and 9121948) with a unique scaffold (Figure 6A). Inhibition of ICAM-1 binding to DBL2βPF11_0521 domain by these compounds was found to be concentration dependent (Figure 6B and 6C). Neither compound was able to reverse binding to ICAM-1 (data for 5306995 are shown in Figure 6C). IC50 for blocking of binding with compound 5306995 was determined to be 18 µM.
Figure 6.

Characterization of ICAM-1–binding inhibitory compounds. A, Chemical structures. B, Concentration-dependent inhibition using bead-immobilized COS7-expressed Duffy-binding-like (DBL) 2PF11_0521 domain. Error bars are standard deviations. C, Blocking (black) and reversal (gray) of ICAM-1 binding by compound 5306995, using bead-immobilized Escherichia coli–expressed DBL2PF11_0521 domain. Error bars are standard deviations. D, Both compounds bind to domain, with compound 5306995 having a higher affinity. Compounds at various concentrations were incubated with bead-immobilized DBL2PF11_0521 domain (E. coli expressed and refolded), and unbound compound was removed by washing. Beads were further incubated with ICAM-1 and washed, and binding of ICAM-1 was measured as described in Materials and Methods. Black bars, DBL2PF11_0521 domain; white bars, control protein AMA1 expressed in E. coli. Error bars are standard deviations. Abbreviations: Conc, concentration; IC50, median inhibitory concentration.
To determine whether the inhibitory molecule binds to the PfEMP1 domain or the ICAM-1 receptor, we incubated DBL2βPF11_0521 domain-immobilized beads with each compound, washed and incubated the beads with ICAM-1-Fc, and used anti-human IgG for detection. Both compounds blocked ICAM-1 binding in this assay (Figure 6D). Compound 5306995 blocked with an efficiency similar to that determined in the screening assay (Figure 6C). This indicates that both compounds bind to the PfEMP1 domain, although compound 5306995 binds with higher affinity.
To test compound 5306995 activity against IE binding, we used 3G8, a heterologous strain (IT4 genetic background [21]) that has been selected for ICAM-1 binding [18]. 3G8 parasite binding to ICAM-1 can be inhibited (Figure 7) with an efficiency similar to that of ICAM-1 binding to the bead-immobilized DBL2βPF11_0521 domain at the same concentration (Figure 6B and 6C). These experiments indicate that ICAM-1–binding inhibitory compounds identified using one genetic background are cross-inhibitory for ICAM-1 binding of parasites of different genetic background, similar to our findings for CSA-binding inhibitors.
Figure 7.

Inhibition of binding of 3G8 line ICAM-1–binding Plasmodium falciparum–infected erythrocytes (IEs) to surface-immobilized ICAM-1 receptor by compound 5306995. Compound was dissolved in phosphate-buffered saline plus 0.1% dimethyl sulfoxide. Control was phosphate-buffered saline plus 0.1% dimethyl sulfoxide. Error bars are standard errors of mean.
Implications for Antiadhesion Drugs
We have demonstrated that the 2-step approach using molecular binding assays followed by parasite binding assays may lead to the characterization of large and small molecules that inhibit binding of IEs to host receptors. This approach establishes a foundation for efficient identification of antiadhesion compounds that can be further developed for use as adjunctive therapy in cases of severe malaria complications associated with the ability of IEs to sequester in host vasculature.
Targeting antiadhesion therapies to PfEMP1 domains is preferred as agents that target host receptors may produce undesirable off-target effects. However, PfEMP1 proteins have considerable interspecies and intraspecies diversity, and it has not been clear whether a single small molecule might inhibit various alleles of the same adhesive class from binding to host receptors. The results we present here indicate that single compounds can inhibit binding of parasite lines with different genetic backgrounds to host receptors.
Compounds that can reverse and block IE adhesion may be particularly valuable for severe malaria treatment. This might reverse the local inflammation in patients with severe malaria that is thought to be a major contributing cause of pathology [31]. Here, we described compounds that block IE binding to ICAM-1 or CSA, or that reverse IE binding to CSA, identified by screening a 10 000-compound library. While these proof-of-principle studies provide evidence for the usefulness of the 2-step platform, significant challenges remain for the development of highly effective drugs that act by blocking (or reversing) cytoadherence. We anticipate that screening larger libraries with 100 000–500 000 compounds may identify additional active compounds and, possibly, compounds with lower IC50 than those identified in this work and/or with the ability to both block and reverse IE adhesion to various receptors. Identification of such compounds by screening larger libraries or by chemical modification/optimization of compounds identified in this study is feasible with our 2-step platform.
Supplementary Data
Supplementary materials are available at The Journal of Infectious Diseases online (http://jid.oxfordjournals.org). Supplementary materials consist of data provided by the author that are published to benefit the reader. The posted materials are not copyedited. The contents of all supplementary data are the sole responsibility of the authors. Questions or messages regarding errors should be addressed to the author.
Notes
Acknowledgments. We thank Eddie Rossnagle and Isaak Tyler Frey, for cloning several PfEMP1 domains used in this work; and Dr Joe Smith, for kindly providing parasite ICAM-1–binding strain 3G8.
Financial support. This work was supported by the Foundation for the National Institutes of Health through the Grand Challenges in Global Health Initiative, which is funded by the Bill and Melinda Gates Foundation (grant 1364 to P. E. D.) and by the National Institutes of Health (grants 1R56AI083668, 5R01HD058005, and 5R01AI092120 to A. V. O.).
Potential conflicts of interest. All authors: No reported conflicts.
All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
- 1.Hughes KR, Biagini GA, Craig AG. Continued cytoadherence of Plasmodium falciparum infected red blood cells after antimalarial treatment. Mol Biochem Parasitol. 2010;169:71–8. doi: 10.1016/j.molbiopara.2009.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dondorp AM. Clinical significance of sequestration in adults with severe malaria. Transfus Clin Biol. 2008;15:56–7. doi: 10.1016/j.tracli.2008.04.013. [DOI] [PubMed] [Google Scholar]
- 3.Smith JD, Rowe JA, Higgins MK, Lavstsen T. Malaria's deadly grip: cytoadhesion of Plasmodium falciparum-infected erythrocytes. Cell Microbiol. 2013;15:1976–83. doi: 10.1111/cmi.12183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Goncalves BP, Huang CY, Morrison R, Holte S, Kabyemela E, et al. Parasite burden and severity of malaria in Tanzanian children. N Engl J Med. 2014;370:1799–808. doi: 10.1056/NEJMoa1303944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hogh B. Clinical and parasitological studies on immunity to Plasmodium falciparum malaria in children. Scand J Infect Dis Suppl. 1996;102:1–53. [PubMed] [Google Scholar]
- 6.Soerli J, Barfod L, Lavstsen T, Bernasconi NL, Lanzavecchia A, et al. Human monoclonal IgG selection of Plasmodium falciparum for the expression of placental malaria-specific variant surface antigens. Parasite Immunol. 2009;31:341–6. doi: 10.1111/j.1365-3024.2009.01097.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dormeyer M, Adams Y, Kramer B, Chakravorty S, Tse MT, et al. Rational design of anticytoadherence inhibitors for Plasmodium falciparum based on the crystal structure of human intercellular adhesion molecule 1. Antimicrob Agents Chemother. 2006;50:724–30. doi: 10.1128/AAC.50.2.724-730.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gamain B, Trimnell AR, Scheidig C, Scherf A, Miller LH, et al. Identification of multiple chondroitin sulfate A (CSA)-binding domains in the var2CSA gene transcribed in CSA-binding parasites. J Infect Dis. 2005;191:1010–3. doi: 10.1086/428137. [DOI] [PubMed] [Google Scholar]
- 9.Oleinikov AV, Amos E, Frye IT, Rossnagle E, Mutabingwa TK, et al. High throughput functional assays of the variant antigen PfEMP1 reveal a single domain in the 3D7 Plasmodium falciparum genome that binds ICAM1 with high affinity and is targeted by naturally acquired neutralizing antibodies. PLoS Pathog. 2009;5:e1000386. doi: 10.1371/journal.ppat.1000386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Robinson BA, Welch TL, Smith JD. Widespread functional specialization of Plasmodium falciparum erythrocyte membrane protein 1 family members to bind CD36 analysed across a parasite genome. Mol Microbiol. 2003;47:1265–78. doi: 10.1046/j.1365-2958.2003.03378.x. [DOI] [PubMed] [Google Scholar]
- 11.Gullingsrud J, Saveria T, Amos E, Duffy PE, Oleinikov AV. Structure-function-immunogenicity studies of PfEMP1 domain DBL2betaPF11_0521, a malaria parasite ligand for ICAM-1. PLoS One. 2013;8:e61323. doi: 10.1371/journal.pone.0061323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Saveria T, Oleinikov AV, Wiliamson K, Chaturvedi R, Lograsso J, et al. Antibodies to Escherichia coli-expressed C-terminal domains of Plasmodium falciparum variant surface antigen 2-chondroitin sulfate A (VAR2CSA) inhibit binding of CSA-adherent parasites to placental tissue. Infect Immun. 2013;81:1031–9. doi: 10.1128/IAI.00978-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Oleinikov AV, Voronkova VV, Frye IT, Amos E, Morrison R, et al. A Plasma Survey Using 38 PfEMP1 Domains Reveals Frequent Recognition of the Plasmodium falciparum Antigen VAR2CSA among Young Tanzanian Children. PLoS One. 2012;7:e31011. doi: 10.1371/journal.pone.0031011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Srivastava A, Gangnard S, Round A, Dechavanne S, Juillerat A, et al. Full-length extracellular region of the var2CSA variant of PfEMP1 is required for specific, high-affinity binding to CSA. Proc Natl Acad Sci U S A. 2010;107:4884–9. doi: 10.1073/pnas.1000951107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Oleinikov AV, Rossnagle E, Francis S, Mutabingwa TK, Fried M, et al. Effects of sex, parity, and sequence variation on seroreactivity to candidate pregnancy malaria vaccine antigens. J Infect Dis. 2007;196:155–64. doi: 10.1086/518513. [DOI] [PubMed] [Google Scholar]
- 16.Beeson JG, Chai W, Rogerson SJ, Lawson AM, Brown GV. Inhibition of binding of malaria-infected erythrocytes by a tetradecasaccharide fraction from chondroitin sulfate A. Infect Immun. 1998;66:3397–402. doi: 10.1128/iai.66.7.3397-3402.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rogerson SJ, Chaiyaroj SC, Ng K, Reeder JC, Brown GV. Chondroitin sulfate A is a cell surface receptor for Plasmodium falciparum-infected erythrocytes. J Exp Med. 1995;182:15–20. doi: 10.1084/jem.182.1.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Janes JH, Wang CP, Levin-Edens E, Vigan-Womas I, Guillotte M, et al. Investigating the host binding signature on the Plasmodium falciparum PfEMP1 protein family. PLoS Pathog. 2011;7:e1002032. doi: 10.1371/journal.ppat.1002032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maher SP, Balu B, Shoue DA, Weissenbach ME, Adams JH. A highly sensitive, PCR-based method for the detection of Plasmodium falciparum clones in microtiter plates. Malar J. 2008;7:222. doi: 10.1186/1475-2875-7-222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Berendt AR, McDowall A, Craig AG, Bates PA, Sternberg MJ, et al. The binding site on ICAM-1 for Plasmodium falciparum-infected erythrocytes overlaps, but is distinct from, the LFA-1-binding site. Cell. 1992;68:71–81. doi: 10.1016/0092-8674(92)90207-s. [DOI] [PubMed] [Google Scholar]
- 21.Horrocks P, Pinches R, Christodoulou Z, Kyes SA, Newbold CI. Variable var transition rates underlie antigenic variation in malaria. Proc Natl Acad Sci U S A. 2004;101:11129–34. doi: 10.1073/pnas.0402347101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gardner JP, Pinches RA, Roberts DJ, Newbold CI. Variant antigens and endothelial receptor adhesion in Plasmodium falciparum. Proc Natl Acad Sci U S A. 1996;93:3503–8. doi: 10.1073/pnas.93.8.3503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dahlback M, Nielsen MA, Salanti A. Can any lessons be learned from the ambiguous glycan binding of PfEMP1 domains? Trends Parasitol. 2010;26:230–5. doi: 10.1016/j.pt.2010.02.002. [DOI] [PubMed] [Google Scholar]
- 24.Oleinikov AV, Francis SE, Dorfman JR, Rossnagle E, Balcaitis S, et al. VAR2CSA domains expressed in E.coli induce cross-reactive antibodies to native protein. J Infect Dis. 2008;197:1119–23. doi: 10.1086/529526. [DOI] [PubMed] [Google Scholar]
- 25.Fried M, Duffy PE. Adherence of Plasmodium falciparum to chondroitin sulfate A in the human placenta. Science. 1996;272:1502–4. doi: 10.1126/science.272.5267.1502. [DOI] [PubMed] [Google Scholar]
- 26.Fernandez P, Petres S, Mecheri S, Gysin J, Scherf A. Strain-transcendent immune response to recombinant Var2CSA DBL5-epsilon domain block P. falciparum adhesion to placenta-derived BeWo cells under flow conditions. PLoS One. 2010;5:e12558. doi: 10.1371/journal.pone.0012558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fried M, Avril M, Chaturvedi R, Fernandez P, Lograsso J, et al. Multilaboratory approach to preclinical evaluation of vaccine immunogens for placental malaria. Infect Immun. 2013;81:487–95. doi: 10.1128/IAI.01106-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Magistrado PA, Minja D, Doritchamou J, Ndam NT, John D, et al. High efficacy of anti DBL4varepsilon-VAR2CSA antibodies in inhibition of CSA-binding Plasmodium falciparum-infected erythrocytes from pregnant women. Vaccine. 2011;29:437–43. doi: 10.1016/j.vaccine.2010.10.080. [DOI] [PubMed] [Google Scholar]
- 29.Nielsen MA, Pinto VV, Resende M, Dahlback M, Ditlev SB, et al. Induction of adhesion-inhibitory antibodies against placental Plasmodium falciparum parasites by using single domains of VAR2CSA. Infect Immun. 2009;77:2482–7. doi: 10.1128/IAI.00159-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Horrocks P, Pinches R, Kyes S, Kriek N, Lee S, et al. Effect of var gene disruption on switching in Plasmodium falciparum. Mol Microbiol. 2002;45:1131–41. doi: 10.1046/j.1365-2958.2002.03085.x. [DOI] [PubMed] [Google Scholar]
- 31.Mackintosh CL, Beeson JG, Marsh K. Clinical features and pathogenesis of severe malaria. Trends Parasitol. 2004;20:597–603. doi: 10.1016/j.pt.2004.09.006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.